

Abstract
Small extracellular vesicles (sEVs) encompass a variety of distinct vesicles that are secreted to the extracellular space. Many methodologies currently used for EV isolation (e.g., differential ultracentrifugation concluding in a high-speed pellet, precipitation by macromolecular crowding agents or size excusion chromatography–SEC) do not fractionate distinct sEV sub-populations. Samples obtained by the aforementioned methods are usually used for characterization and physiological studies. However the fraction that contains the molecule of interest or is the carrier of a specific activity is unknown. Therefore isolating distinct sEV sub-populations is critical to understand EV function. The goal of this procedure is to purify distinct sEV sub-populations based on slight differences in their buoyant density. Moreover, this technique also allows sEVs purification from vesicle-free RNA-protein complexes co-isolating in the high-speed pellet or by the use of crowding agents. This protocol describes cultivation of mammalian cells for sEV collection, sEV sedimentation, buoyant density fractionation of sEV sub-populations and immunoblots for sEV markers. This protocol can be used to fractionate distinct sEV sub-populations produced by a variety of mammalian cells.
Keywords: Extracellular vesicles, Differential ultracentrifugation, Buoyant density, Exosomes, RNA-protein complexes, OptiPrep
Background
Small extracellular vesicles (sEVs) are released by virtually all mammalian cells. They have been proposed to play a variety of roles ranging from immunity to cancer metastasis ( Pegtel et al., 2010 ; Mittelbrunn et al., 2011 ; Montecalvo et al., 2012 ; Fong et al., 2015 ; Dickman et al., 2017 ). Most of the current methodologies used to isolate sEVs however do not distinguish among different sEV sub-population nor do they purify sEVs from co-isolating protein and RNA-protein complexes (Temoche- Diaz et al., 2019 ; Kowal et al., 2016 ; Willms et al., 2016 ; Jeppesen et al., 2019 ). Therefore the sedimentable fraction or specific vesicle species that promotes a particular physiological response remains unknown ( Shurtleff et al., 2018 ). A methodology that allows purification of sEVs away from contaminants and fractionation of distinct sEV sub-populations is of broad utility. The procedure explained below uses differential ultracentrifugation followed by buoyant density flotation to allow: 1) Purification of particles associated with a lipidic membrane (sEVs) from non-vesicular particulate material and 2) Fractionation of distinct sEV sub-populations based on slight differences in their buoyant densities. The purified sEV fractions are of suitable purity for use in physiological and/or characterization experiments.
Materials and Reagents
15 cm dishes (Tissue Culture Dish) (Corning, Falcon, catalog number: 08-772-6)
Transfer pipettes (Fisherbrand, catalog number: 13-711-7M)
Syringe 3 ml (BD, catalog number: 309657)
Syringe 10 ml (BD, catalog number: 302995)
Stainless steel 304 syringe needle (Sigma, catalog number: Z117021-1EA)
PrecisionGlide Needle 22G (BD, catalog number: 305159)
Immbilion-P PVDF membrane (Millipore Sigma, catalog number: IPVH10010)
50 ml conical tube
-
Open-Top Thinwall Ultra-Clear Tube (38.5 ml), 25 x 89 mm (Beckman Coulter, catalog number: 344058)
Note: On text “38.5 ml ultra-clear tube”.
-
Open-Top Thinwall Ultra-Clear Tube (13.2 ml), 14 x 89 mm (Beckman Coulter, catalog number: 344059)
Note: On text “13.2 ml ultra-clear tube”.
4-20% Criteron Tris-HCl Protein Gel, 26 well (Bio-Rad, catalog number: 3450034)
OptiPrep (Sigma, catalog number: D1556-250ml)
Sucrose (Fisher Chemical, catalog number: S5-3)
HEPES (Sigma, catalog number: H4034-1Kg)
Sodium chloride (Fisher Chemical, catalog number: S271-3)
Fetal bovine serum (FBS) (VWR, catalog number: 89510-194)
FBS exo-depleted (System Biosciences, catalog number: EXO-FBS-250A-1)
RPMI + Glutamax (Gibco, catalog number: 10566-016)
Bovine serum albumin (BSA) (Sigma, catalog number: A3294-100G)
-
Antibodies
CD9 (Cell Signaling Technology, catalog number: 13174S)
MFG-E8 (R&D Systems, catalog number: MAB2767)
ITGA3 (Abcam, catalog number: ab190731)
CD63 (Fisher Scientific, catalog number: BDB556019)
Dicer (Santa Cruz, catalog number: sc30226)
Flot-2 (BD Biosciences, catalog number: 610383)
IgG-anti rabbit HRP linked (Sigma Millipore, catalog number: GENA934-1ml)
IgG-anti rabbit HRP linked (Sigma Millipore, catalog number: NXA931-1ml)
Dithiothreitol (DTT) (Goldbio, catalog number: DTT25)
Sodium dodecyl sulfate (SDS) (J.T. Baker, catalog number: 4095-02)
Tween-20 (Sigma, catalog number: P7949-500ml)
Tris-buffered saline (TBS, pH 7.6)
Bromophenol-blue (Bio-Rad, catalog number: 161-0404)
Glycerol (AMRESCO, catalog number: M152-4L)
Glycine
Methanol (Fisher Chemical, catalog number: A452-4)
-
Mammalian cell culture media (see Recipes)
Cell culture growing medium
Exosome-depleted growing medium
-
Stock solutions (see Recipes)
1 M HEPES pH 7.4
Concentrated OptiPrep buffer
1 M DTT
1 M Tris pH 6.8
4x Laemmli buffer
4x Laemmli buffer no DTT
10x Transfer buffer
OptiPrep working solution
-
Buffer solutions (see Recipes)
EV buffer
Sucrose cushion
Concentrated sucrose cushion
OptiPrep working solutions 25%, 20%, 15%, 10%, 5%
TBS-T
Equipment
Refractometer (Fisher Scientific)
Sorvall RC 6+ centrifuge (Thermo Scientific, model: 46910)
Fixed angle rotor F14S-6X250y FiberLite (Thermo Scientific, catalog number: 78500)
Ultracentrifuge (Beckman Coulter, model: Optima XE-90, catalog number: A94471)
Swinging-bucket rotor SW 32 Ti and bucket set (Beckman Coulter, catalog number: 369694)
Swinging-bucket rotor SW 41 Ti and bucket set (Beckman Coulter, catalog number: 311362)
ChemiDoc MP Imaging Sytem (Bio-Rad Laboratories, model: ChemiDoc MP 10)
Software
ImageLab software v4.0
ImageJ
GraphPad Prism
Procedure
-
Cell culture
-
Grow MDA-MB-231 cells in four 15 cm dishes in 30 ml cell culture growth medium (see Recipe A1) until cells reach 80% confluency (the cell number at this point is ~16 x 106 cells per dish).
Notes:
Other model mammalian cells may be used. This methodology has been successful at fractionating sEV sub-populations of several mammalian cells tried to date: HEK-293T, MCF7, MCF10A, MDA-MB-231, PC12 and N2A cells.
The surface available for cell growth in these dishes is 151.9 sq. cm. The total volume of cell culture growth medium used for these dishes is 30 ml.
-
Expand the four 15 cm dishes to a total of fourteen 15 cm dishes each containing an approximate of 4 x 106 cells at moment of seeding in 30 ml of exosome-depleted growth medium (see Recipe A2).
Note: The maximum volume of medium that can be processed in the steps below is 420 ml.
-
Incubate cells for 3 d or until cells reach 80% confluency. Proceed with medium collection.
Note: Do not allow cells to become overconfluent, we have observed a dramatic decrease in sEVs from overconfluent cultures.
-
-
Conditioned medium collection and extracellular vesicle sedimentation
Collect conditioned medium from fourteen 15 cm plates by pouring it off in adequate containers. The total amount of conditioned medium collected is ~420 ml.
Centrifuge the conditioned medium at 1,000 × g for 15 min at 4 °C in a Sorvall RC 6+ centrifuge with fixed angle rotor F14S-6X250y FiberLite (or equivalent) to sediment floating cells (Low speed spin).
Immediately after the centrifuge stops, gently pour the supernatant into a new container. Be careful not to disturb the pellet.
Centrifuge the supernatant at 10,000 × g for 15 min at 4 °C in a Sorvall RC 6+ centrifuge with fixed angle rotor F14S-6X250y FiberLite to sediment cellular debris and large extracellular vesicles (Medium speed spin).
Immediately after the centrifuge stops, gently pour the supernatant into a new container. Be careful not to disturb the pellet.
-
Add 32 ml of the collected supernatant into a single 38.5 ml ultra-clear tube. Repeat this step 5 times to fill a total of six 38.5 ml ultra-clear tubes.
Note: This step will consume ~200 ml of conditioned medium. Save the remaining ~220 at 4 °C for a second round of ultracentrifugation (Step B11).
Attach the stainless steel 304 needle to a 10 ml syringe. Fill the 10 ml syringe with sucrose cushion (see Recipe C2).
Quickly introduce the stainless steel needle into the conditioned medium-filled 38.5 ml ultra-clear tube until it reaches the bottom of the tube. Add 2 ml of sucrose cushion and remove the stainless steel needle immediately. Repeat this step 5 times for a total of six 38.5 ml ultra-clear tubes.
Centrifuge at ~100,000 × g (29,500 RPM) for 1.5 h using a SW32 Ti rotor at 4 °C at maximum acceleration and brake.
Immediately after, aspirate the supernatant from the top to just above the sucrose cushion using an aspirating pipet fixed to a vacuum system. Be careful not to aspirate the interface at the cushion and the conditioned medium (this is where the sEVs have sedimented). Aspirate until leaving ~4-5 ml of conditioned medium resting on top of the sucrose cushion. Repeat this step 5 times, for a total of six 38.5 ml ultra-clear tubes.
Very carefully add the remaining conditioned medium (from Step B6) to each 38.5 ml ultra-clear tube. Do so by using transfer pipettes (see materials) and adding the conditioned medium very slowly and smoothly along the tube wall. Disturbance of the sucrose cushion must be avoided. Any fast pouring can disturb the sucrose cushion.
Repeat Steps B9 and B10.
-
Using a 1 ml micropipette carefully collect and discard ~2-3 ml of the ~4-5 ml remaining on top of the sucrose cushion after aspiration. Do so until there is ~2 ml of conditioned medium remaining on top of the sucrose cushion.
Note: For this step, the use of a 1ml micropipette is recommended over vacuum aspiration. Using a 1 ml micropipette at this step allows a more controlled/accurate medium collection.
Collect the ~2 ml of remaining conditioned medium, plus the top-most 1 ml of sucrose cushion and save the 3 ml into a 50 ml conical tube. Repeat this step 5 times with the 5 remaining tubes and pool them together in a single 50 ml conical tube (final volume ~18 ml).
-
Measure the sucrose concentration of the solution from Step B14 by using a refractometer. Make sure the concentration does not exceed 21% sucrose (w/v). If the concentration exceeds 21% sucrose dilute with EV buffer. Generally an adequate sucrose concentration ranges from 15 to 20%.
Note: If the sucrose concentration from B15 exceeds 21% sucrose (w/v), sEVs will not sediment in Step B18 and instead they will equilibrate throughout the 13.2 ml ultra-clear tubes.
Fill two 13.2 ml ultra-clear tubes with the collected sEVs from Step B15. Add ~9-11 ml per tube.
Add 1 ml of sucrose cushion on the bottom of each 13.2 ml ultra-clear tube as in Step B8.
Centrifuge at ~130,000 × g (32,500 RPM) for 15 h using a SW41 Ti rotor at 4 °C at maximum acceleration and brake.
Immediately after, use a vacuum system to aspirate the supernatant from top to bottom. Be careful not to aspirate the layer in between the cushion and the conditioned medium (this is where the sEVs have sedimented). Aspirate until leaving ~2 ml of conditioned medium resting on top of the sucrose cushion. Repeat this step 1 time with the remaining 13.2 ml ultra-clear tube.
-
Using a 1 ml micropipette collect and discard an extra ~1.2 ml of conditioned medium remaining on top. Repeat this step 1 time with the remaining 13.2 ml ultra-clear tube.
Note: For this final step, the use of a 1 ml micropipette is recommended over vacuum aspiration to have a more controlled/accurate collection of medium.
Collect the remaining content of the 13.2 ml ultra-clear tube. This includes 800 μl of conditioned medium remaining on top of the sucrose cushion, plus 1 ml of the sucrose cushion. Pool the content of the two 13.2 ml ultra-clear tubes together into a 50 ml conical tube (the total volume at this step is ~3.6 ml).
Add 1.9 ml of concentrated sucrose cushion (see Recipe C2) and mix properly.
-
Use the refractometer to measure sucrose concentration. The sucrose concentration after this step is generally higher than 42% sucrose (w/v). Values equal or higher than 42% are adequate.
Note: If the sucrose concentration does not exceed 42% add more concentrated sucrose cushion until it is equal or exceeds this value. If the concentration is lower, the OptiPrep layer from Step C3 will have higher density and sink, impeding the proper linear gradient formation.
Save 10% from B23 (500 μl) to use as “100,000 × g pellet” in Procedures D and E.
-
Extracellular vesicle flotation into an OptiPrep linear gradient and fraction collection
Add the remaining 5 ml from Step B23 to the bottom of a 13.2 ml ultra-clear tube by using a 1 ml micropipette.
Attach the 22G needle to a 3 ml syringe. Then fill the 3 ml syringe with OptiPrep solution 25% (see Recipe C4).
-
Extend the 22G needle to the top-most layer in the 13.2 ml ultra-clear tube. Once the 22G needle is within a few millimeters from the top layer, very slowly and carefully add 1.5 ml of OptiPrep solution 25%.
Note: This step and Step C4 require extreme caution. Adding the OptiPrep layers must be done very slowly and smoothly. Failure to do so can cause the layers to mix and subsequent failure of gradient formation.
Repeat Step C3 with the OptiPrep solutions 20%, 15%, 10% and 5% (see Recipe C4) in this respective order.
-
Centrifuge at ~160,000 × g (36,000 RPM) for 15 h using a SW41 Ti rotor at 4 °C at minimum acceleration and NO brake.
Note: It is important to have no brake during deceleration. Having the brake on can cause disruption of the gradient and mixture of sEV sub-populations.
-
Once stopped, collect 400 μl fractions from top to bottom using of a 1 ml micropipette. A schematic summarizing Steps B1 to C6 is shown in Figure 1.
Note: Two different sEV sub-populations can be observed against a black background and with the appropriate lighting (application of an artificial or natural source of light can aid visualization). The 2 distinct sEV sub-populations will appear as 2 separate white bands contrasting with the black background.
-
Mix each 400 μl fraction by the use of a vortexer and measure their refractive indices to confirm linearity. The formation of an OptiPrep linear gradient is required for the success of this protocol in order to fractionate sEVs with slight differences in their densities. Disruption of linearity would cause failure of an adequate sEV fractionation.
Notes:
Linearity is often observed from fraction 1 to 25. The remaining 6 bottom fractions have little to no change in their density as the fraction number increases.
The low density sEV fractions generally range from fractions 9-11. The high density sEV fractions generally float to fractions 15-17. However there can be slight variations in different linear gradients and the fraction number where sEVs equilibrate can slightly change. Therefore it is recommended to note the refractive index of each fraction and match this refractive index to sEV presence (by immunoblots, Procedure E below). Once the density corresponding to sEV sub-populations has been empirically determined in this way, only refractive index measurements are necessary in future experiments to identify sEV containing fractions.
-
Fractions can now be mixed with 4x Laemmli buffer (see Recipe B5) or be concentrated as in Procedure D.
Note: The detection of certain sEV markers can be done successfully without the need for concentrating by additional ultracentrifugation, however concentration is recommended to detect most of the sEV markers. Antibodies that show sufficient sensitivity to detect marker proteins without concentration in our experience are anti-CD9, anti-ITGA3 and anti-dicer.
-
Concentration of fractions
Mix each fraction with 10 ml of EV buffer.
Centrifuge at ~130,000 × g (32,500 RPM) for 1.5 h using a SW41 Ti rotor at 4 °C at maximum acceleration and deceleration.
Decant the supernatant.
-
Add 50-100 μl of 1x Laemmli buffer. Pipette up and down 3-7 times. Incubate for 10 min at room temperature.
Note: If the detection of CD63 is desired, resuspend as specified above in 1x Laemmli buffer without DTT. The addition of reducing reagents causes failure of the detection of CD63 with the antibody BDB556019.
Collect the material and freeze at -20 °C or use immediately.
-
Immunoblot
Incubate samples at 95 °C for 5 min.
-
Load 10ul of samples per lane in a 4-20% Criteron 26-well gel.
Note: Pooling samples 1-3; 4, 5; 23-25; 26-28 and 29, 30 is advised in order to fit all fractions in the same gel. The aforementioned fractions generally do not contain sEVs in our observations (although we cannot exclude variation in other cell lines).
Run at constant voltage, 150 V, for 1.5 h.
Transfer into a PVDF membrane with constant amps, 0.4 A, for 2.5 h.
Block membrane(s) for 1 h with 5% BSA in TBS-T (see Recipes C5).
Incubate membrane(s) with the desired primary antibodies overnight in the cold room (4 °C). Working dilution for anti-CD9 is 1:3,000. All other antibodies 1:1,000.
Wash 3 times with TBS-T (see Recipes C5) for 8 min each wash.
Incubate with horseradish peroxidase (HRP) conjugated secondary antibody (anti-mouse or anti-rabbit) for 1 h at room temperature. Working dilutions 1:10,000.
Wash 3 times with TBS-T (see Recipes C5) for 8 min each wash.
-
Add the HRP substrate following the manufacturer specifications. Develop using Chemidoc Imaging System.
Note: We regularly use Pierce ECL Plus western blotting substrate (Thermo Scientific) though similar chemiluminescent substrates may be substituted.
Figure 1. Schematic showing the two-step purification methodology.
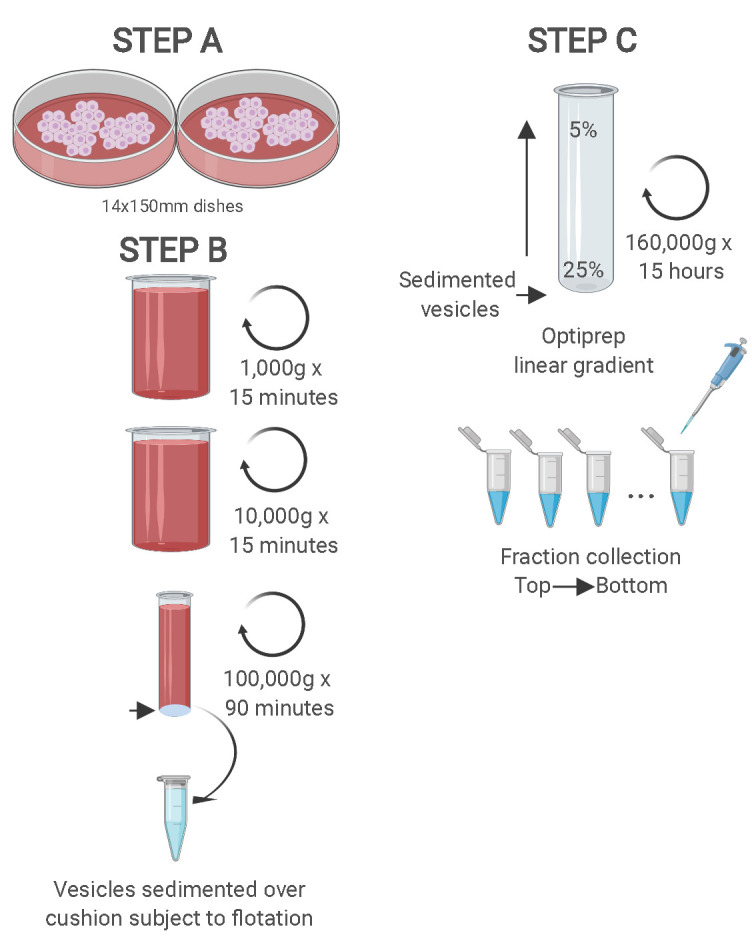
Differential ultracentrifugation is followed by buoyant density flotation in a linear OptiPrep gradient. Figure taken from Temoche-Diaz et al. (2019) .
Data analysis
Export the .tif images using the ImageLab software v4.0.
-
Open images in ImageJ, use a rectangle tool to highlight bands and quantify the intensity of bands by choosing Analyze → Gels → Label peaks.
Note: For a detailed explanation on how to analyze immunoblot bands by ImageJ, please access to this link: https://www.sybil-fp7.eu/node/95.
Use GraphPad Prism or preferred program to plot the sEV markers distributions (Figure 2B).
Collide Stack and align images using Adobe Illustrator or preferred program (Figure 2A).
Figure 2. Two biochemically distinct sEV sub-populations are released by MDA-MB-231.

A. Representative image of immunoblots for sEV markers across the gradient. B. Quantification of the intensity of each sEV marker across the gradient. Black line delineates the maximum signal across the gradient showing three distinctive areas. The first, second and third peak represent the low density EVs, high density EVs and non vesicular protein-RNA complexes respectively. In order to follow the International Society of Extracellular Vesicle (ISEV) guidelines ( Thery et al., 2018 ), electron microscope images and nanoparticle analysis are also shown in C, D and E. C. Representative electron micrographs of negative stained high density and low-density EV samples. D. Nanoparticle tracking analysis showing the size distribution of the high-density and low-density sub-populations. The high-speed pellet is also shown. E. Quantification of the particle number per sEV subpopulation using Nanoparticle tracking analysis. Data plotted are from two independent experiments; error bars represent standard deviation from independent samples. Figure taken from Temoche-Diaz et al. (2019)
Notes
This protocol assumes the users have knowledge of how to perform immunoblots and analyze the data.
Recipes
-
Mammalian cell culture media
-
Cell culture growing medium
Add 10% FBS to DMEM-Glutamax
-
Exosome-depleted growing medium
Add 10% exo-depleted FBS to DMEM-Glutamax
-
-
Stock solutions
-
1 M HEPES pH 7.4 (500 ml)
Dissolve 119.2 g of HEPES in 400 ml of ddH2O
Add 10 N NaOH until pH reaches 7.4
Add ddH2O to 500 ml
Store at 4 °C
-
Concentrated OptiPrep buffer (100 ml)
Dissolve 0.85 g of NaCl in 70 ml of ddH2O
Add 6 ml of 1 M HEPES pH 7.4
Add ddH2O to 100 ml
-
1 M DTT (10 ml)
Dissolve 1.5 g of DTT in 6 ml of ddH2O
Add ddH2O to 10 ml
Make 1 ml aliquots
Store at -20 °C
-
1 M Tris pH 6.8 (100 ml)
Dissolve 12.1 g of Tris Base in 80 ml of ddH2O
Adjust pH with 10 N HCl to 6.8
Add ddH2O to 100 ml
-
4x Laemmli buffer (20 ml)
Dissolve 1.6 g of SDS in 6 ml of ddH2O
Add 5 ml of 1 M Tris-HCl pH 6.8
Add 1.23 g of DTT. Dissolve
Add 8 mg of bromophenol blue
Add 8 ml of glycerol
Add ddH2O to 20 ml
Make 1 ml aliquots
Store at -20 °C
-
4x Laemmli buffer no DTT (20 ml)
Follow as Recipe B5, but do not include DTT.
-
10x Transfer buffer (1 L)
Dissolve 114.2 g of glycine in 400 ml of ddH2O
Add 24.2 g of Tris-base. Dissolve
Add ddH2O to 1 L
Store at 4 °C
-
OptiPrep working solution
Mix 10 ml of OptiPrep with 2 ml of concentrated OptiPrep buffer
Store at 4 °C
-
-
Buffers/Solutions
-
EV buffer (100 ml)
Dissolve 0.85 g of NaCl in 70 ml of ddH2O
Add 1 ml of 1 M HEPES pH 7.4
Add ddH2O to 100 ml
Store at 4 °C
-
Sucrose cushion (40 ml)
Add 27 g of sucrose in a 50 ml conical tube
-
Add EV buffer up to 40 ml. Dissolve the sucrose
Note: It takes ~10 min with constant mixing to dissolve the sucrose completely.
Measure the refractive index and calculate the sucrose concentration
-
Adjust to 60% (w/v) of sucrose by adding more sucrose or diluting with EV buffer
Note: Adding 24 g of sucrose for a 40 ml solution does not yield a 60% sucrose solution (as measured by refractive index). Therefore it is advised to start step 2a with 27 g of sucrose instead.
Store at 4°C
-
Concentrated sucrose cushion (40 ml)
Add 31 g of sucrose in a 50 ml conical tube
Add EV buffer to 40 ml. Dissolve the sucrose completely
-
Take the refractive index and calculate the sucrose concentration
Note: This solution will have an approximate 65% sucrose concentration.
Store at 4 °C
-
OptiPrep working solutions (5 ml)
Follow the Table 1 below to prepare the OptiPrep working solutions.
-
TBS-T
0.1% Tween 20 (v/v) in 1x TBS
-
Table 1. Table showing calculations of different concentrations of OptiPrep working media.
| OptiPrep Working Solution (ml) | EV buffer (ml) | |
| OptiPrep 25% | 2.5 | 2.5 |
| OptiPrep 20% | 2 | 3 |
| OptiPrep 15% | 1.5 | 3.5 |
| OptiPrep 10% | 1 | 4 |
| OptiPrep 5% | 0.5 | 4.5 |
Acknowledgments
We would like to thank current and former Schekman lab members who provided technical advice in the development of this protocol. We also acknowledge the Howard Hughes Medical Institute for funding. This protocol has been adapted from Temoche- Diaz et al., 2019 .
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Dickman C. T., Lawson J., Jabalee J., MacLellan S. A., LePard N. E., Bennewith K. L. and Garnis C.(2017). Selective extracellular vesicle exclusion of miR-142-3p by oral cancer cells promotes both internal and extracellular malignant phenotypes. Oncotarget 8(9): 15252-15266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fong M. Y., Zhou W., Liu L., Alontaga A. Y., Chandra M., Ashby J., Chow A., O'Connor S. T., Li S., Chin A. R., Somlo G., Palomares M., Li Z., Tremblay J. R., Tsuyada A., Sun G., Reid M. A., Wu X., Swiderski P., Ren X., Shi Y., Kong M., Zhong W., Chen Y. and Wang S. E.(2015). Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol 17(2): 183-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jeppesen D. K., Fenix A. M., Franklin J. L., Higginbotham J. N., Zhang Q., Zimmerman L. J., Liebler D. C., Ping J., Liu Q., Evans R., Fissell W. H., Patton J. G., Rome L. H., Burnette D. T. and Coffey R. J.(2019). Reassessment of Exosome Composition. Cell 177(2): 428-445 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kowal J., Arras G., Colombo M., Jouve M., Morath J. P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M. and Thery C.(2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 113(8): E968-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mittelbrunn M., Gutierrez-Vazquez C., Villarroya-Beltri C., Gonzalez S., Sanchez-Cabo F., Gonzalez M. A., Bernad A. and Sanchez-Madrid F.(2011). Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Montecalvo A., Larregina A. T., Shufesky W. J., Stolz D. B., Sullivan M. L., Karlsson J. M., Baty C. J., Gibson G. A., Erdos G., Wang Z., Milosevic J., Tkacheva O. A., Divito S. J., Jordan R., Lyons-Weiler J., Watkins S. C. and Morelli A. E.(2012). Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 119(3): 756-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pegtel D. M., Cosmopoulos K., Thorley-Lawson D. A., van Eijndhoven M. A., Hopmans E. S., Lindenberg J. L., de Gruijl T. D., Wurdinger T. and Middeldorp J. M.(2010). Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107(14): 6328-6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shurtleff M. J., Temoche-Diaz M. M. and Schekman R.(2018). Extracellular Vesicles and Cancer: Caveat Lector. Annu Rev Cancer Biol 2(1): 395-411. [Google Scholar]
- 9. Thery C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G. K., et al. (2018). Minimal information for studies of extracellular vesicles 2018(MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7(1): 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Temoche-Diaz M. M., Shurtleff M. J., Nottingham R. M., Yao J., Fadadu R. P., Lambowitz A. M. and Schekman R.(2019). Distinct mechanisms of microRNA sorting into cancer cell-derived extracellular vesicle subtypes. Elife 8: 47544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willms E., Johansson H. J., Mager I., Lee Y., Blomberg K. E., Sadik M., Alaarg A., Smith C. I., Lehtio J., El Andaloussi S., Wood M. J. and Vader P.(2016). Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci Rep 6: 22519. [DOI] [PMC free article] [PubMed] [Google Scholar]