

Abstract
Elucidation of the mechanism of dopamine signaling to ERK that underlies plasticity in dopamine D1 receptor-expressing neurons leading to acquired cocaine preference is incomplete. NCS-Rapgef2 is a novel cAMP effector, expressed in neuronal and endocrine cells in adult mammals, that is required for D1 dopamine receptor-dependent ERK phosphorylation in mouse brain. In this report, we studied the effects of abrogating NCS-Rapgef2 expression on cAMP-dependent ERK→Egr-1/Zif268 signaling in cultured neuroendocrine cells; in D1 medium spiny neurons of NAc slices; and in either male or female mouse brain in a region-specific manner. NCS-Rapgef2 gene deletion in the NAc in adult mice, using adeno-associated virus-mediated expression of cre recombinase, eliminated cocaine-induced ERK phosphorylation and Egr-1/Zif268 upregulation in D1-medium spiny neurons and cocaine-induced behaviors, including locomotor sensitization and conditioned place preference. Abrogation of NCS-Rapgef2 gene expression in mPFC and BLA, by crossing mice bearing a floxed Rapgef2 allele with a cre mouse line driven by calcium/calmodulin-dependent kinase IIα promoter also eliminated cocaine-induced phospho-ERK activation and Egr-1/Zif268 induction, but without effect on the cocaine-induced behaviors. Our results indicate that NCS-Rapgef2 signaling to ERK in dopamine D1 receptor-expressing neurons in the NAc, but not in corticolimbic areas, contributes to cocaine-induced locomotor sensitization and conditioned place preference. Ablation of cocaine-dependent ERK activation by elimination of NCS-Rapgef2 occurred with no effect on phosphorylation of CREB in D1 dopaminoceptive neurons of NAc. This study reveals a new cAMP-dependent signaling pathway for cocaine-induced behavioral adaptations, mediated through NCS-Rapgef2/phospho-ERK activation, independently of PKA/CREB signaling.
SIGNIFICANCE STATEMENT ERK phosphorylation in dopamine D1 receptor-expressing neurons exerts a pivotal role in psychostimulant-induced neuronal gene regulation and behavioral adaptation, including locomotor sensitization and drug preference in rodents. In this study, we examined the role of dopamine signaling through the D1 receptor via a novel pathway initiated through the cAMP-activated guanine nucleotide exchange factor NCS-Rapgef2 in mice. NCS-Rapgef2 in the NAc is required for activation of ERK and Egr-1/Zif268 in D1 dopaminoceptive neurons after acute cocaine administration, and subsequent enhanced locomotor response and drug seeking behavior after repeated cocaine administration. This novel component in dopamine signaling provides a potential new target for intervention in psychostimulant-shaped behaviors, and new understanding of how D1-medium spiny neurons encode the experience of psychomotor stimulant exposure.
Keywords: cAMP, cocaine reward, D1 dopaminergic signaling, Egr-1, MAPK, NCS-Rapgef2
Introduction
Behavioral plasticity, including locomotor sensitization and development of drug preference, is associated with psychomotor stimulant-induced neuroadaptation in brain reward circuits linked to addiction (Robinson and Berridge, 2000; Laakso et al., 2002; Fernando and Robbins, 2011; Uhl et al., 2019). Neurons in striatum, PFC, amygdala, and hippocampus that receive dopaminergic input from the VTA are central to psychomotor stimulant-induced neuroadaptation (Rebec and Sun, 2005; Nestler and Luscher, 2019). Amphetamine and cocaine increase the concentration of dopamine released from nerve terminals in these brain regions (Heien et al., 2005; Nestler and Luscher, 2019). Dopamine acts at receptors promoting either an increase (D1 type) or a decrease (D2 type) in cAMP production in distinct dopaminoceptive neurons. A balance between these two processes contributes to the behavioral sequelae, including addiction, of repeated psychomotor stimulant use (Lobo and Nestler, 2011). Cellular mechanisms for the behavioral effects of psychomotor stimulant exposure have been sought by examining dopamine signaling through the Gs-coupled D1 receptor in various brain areas. One of these is the NAc, which contains GABAergic medium spiny neurons (MSNs) whose activity is modulated by dopamine and represents the afferent targets of the reward pathway from VTA to NAc in mammalian brain (Hikida et al., 2010; Russo and Nestler, 2013).
The MAP kinase ERK is activated in vivo on treatment with D1 agonists, cocaine or amphetamine (Valjent et al., 2000; Gerfen et al., 2002; Gutierrez-Arenas et al., 2014; Jin et al., 2019) in brain regions innervated by dopaminergic neurons. Pharmacological and genetic manipulations have further implicated ERK activation in transcriptional effects of psychomotor stimulants, notably expression of the immediate-early gene egr1/zif268, linked to psychomotor stimulant-induced behaviors (Valjent et al., 2006c). However, genetic and pharmacological tools have limited cellular and anatomic specificity for analysis of these pathways. For example, generalized effects on locomotor activity and coregulatory effects of ERK1 and ERK2 complicate interpretation of ERK1/2 KO effects on both behavior and cell signaling (Mazzucchelli et al., 2002; Ferguson et al., 2006; Hutton et al., 2017). Consequently, it has been difficult to identify a signaling pathway arising from D1 receptor activation leading to the specific ERK activation and Egr-1/Zif268 induction that mediates cellular plasticity within dopaminoceptive neurons, in defined brain circuits, responsible for the altered behaviors typical of repeated psychomotor stimulant administration.
In particular, the role of protein kinase A (PKA) as the downstream effector of cAMP in the D1-dependent activation of Egr-1/Zif268 by ERK (Funahashi et al., 2019), leading to cocaine-dependent locomotor sensitization, has been difficult to confirm. This suggests that a second cAMP-dependent pathway for ERK regulation of Egr-1/Zif268 induction, and subsequent behavioral effects of cocaine mediated through dopamine, may exist in D1-MSNs.
We have previously identified a novel pathway for ERK activation via the novel cAMP effector NCS-Rapgef2 in NS-1 neuroendocrine cells in culture (Emery and Eiden, 2012; Emery et al., 2013, 2014, 2017a). NCS-Rapgef2 is activated by cAMP binding to facilitate GDP-GTP exchange on the Rap1 molecule, allowing Rap1 to further stimulate the MAP kinase cascade culminating in ERK phosphorylation/activation (see Jiang et al., 2017). Using this assay as a readout for the NCS-Rapgef2-dependent cAMP pathway, we have shown that the dopamine D1 receptor couples to NCS-Rapgef2 to activate ERK in neuroendocrine cells, and further demonstrated that this pathway is required for D1-dependent activation of ERK in hippocampus and other forebrain regions of the adult mouse brain in vivo (Jiang et al., 2017).
These data open the possibility that a direct pathway for activation of ERK by the Gs/olf-coupled D1 receptor leading to Egr-1/Zif268 induction exists in D1 dopaminoceptive neurons involved in the behavioral effects of repeated cocaine administration. Here, we have asked whether the NCS-Rapgef2-mediated activation of ERK after cAMP elevation by the Gs-coupled D1 receptor is responsible for the behavioral effects of psychomotor stimulant administration, and if so, in which neuronal ensembles or circuits it might be relevant.
Materials and Methods
Animals and drugs administered in vivo
Male or female mice (WT or transgenic) on C57BL/6J background were housed 2-5 per cage and acclimatized to 12 h light/12 h dark cycle with food and water ad libitum. Animal care, drug treatment, and surgeries were approved by the National Institute of Mental Health Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health guidelines. The floxed Rapgef2 mouse strain Rapgef2cko/cko and corticolimbic Rapgef2 KO mouse strain Camk2α-cre+/−::Rapgef2cko/cko were generated as described previously (Jiang et al., 2017). The transgenic Drd1a-tdTomato BAC mouse (The Jackson Laboratory, #016204), expressing tdTomato selectively in D1-dopaminoceptive neurons (Shuen et al., 2008), was obtained from The Jackson Laboratory. The Drd1a BAC-cre driver line (FK150, Drd1-cre+/−) and Drd1a-cre induced EGFP expression produced with the Rosa26_EGFP reporter (Drd1-cre+/−:: Rosa-eGFP) have been described previously (Gerfen et al., 2013). Pharmaceutical grade D1 dopamine receptor agonist SKF81297, D2 antagonist eticlopride, and cocaine were purchased from Millipore Sigma and dissolved in saline. All drugs used in vivo were administrated by intraperitoneal injection.
Brain slice pharmacology
The 250-µm-thick coronal slices of mouse brain containing the NAc were prepared exactly as described previously for electrophysiological examination under perfusion (Tejeda et al., 2017), except that they were incubated in a static bath of continuously aerated ACSF, total volume 10 ml per slice, to which various drugs and drug combinations were added to examine the parameters of dopamine and cAMP-dependent signaling in dopaminoceptive cells of core and shell of this brain region. Inhibitors or vehicle were added for a 10 min period before addition of 10 μm SKF81297, or vehicle. Incubation at 34°C was continued for 30 min, and slices then quickly removed to cold 10% formalin in PBS for immunohistochemical staining and microscopic analysis as described below. N6-phenyl, 9-tetrahydrofuranyl adenine (EL1101), a preferential inhibitor of the Rap guanine nucleotide exchange activity of NCS-Rapgef2 compared with cAMP effectors PKA and Epac, was synthesized and purified as described previously (Emery et al., 2017b). All drugs were initially dissolved in DMSO, present to a final concentration of 0.1% in all experiments. Final concentration of U0126 was 10 μm, and final concentration of EL1101 was 100 μm, in slice experiments.
Cell lines and cell culture
NS-1 cells (Emery et al., 2013) were cultured in Roswell Park Memorial Institute 1640 medium supplemented with 10% HyClone horse serum (GE Healthcare), 5% heat-inactivated FBS, 2 mm L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The established NS-1 cell line with NCS-Rapgef2 knocked-out was described previously (Jiang et al., 2017). The cell-permeable cAMP analog 8-chlorophenylthio-cAMP (8-CPT-cAMP) was purchased from Biolog Life Science Institute, and the stock solution was prepared at 100 mm in DMSO, diluted in culture media to a final concentration of 100 μm.
Viral injection
Modified adeno-associated virus (AAV) vectors were obtained from Penn Vector Core. Rapgef2cko/cko mice were bilaterally injected with AAV9.hSynap.HI.eGFP-Cre.WPRE.SV40, encoding eGFP-fused Cre recombinase under the control of the human synapsin promoter, to knock out Rapgef2 expression in NAc on both sides of the brain. AAV9.hSynap.eGFP.WPRE.SV40, encoding eGFP without Cre, also under the control of the synapsin promoter, was used as a control. Surgeries and viral injection were conducted according to the National Institutes of Health ARAC guidelines for survival rodent surgery. Briefly, animals were anesthetized, shaved, and mounted into the stereotaxic apparatus. A small midline incision was then made, and the pericranial tissue was teased away from the skull with an ethanol-soaked swab to enable identification of the bregma and λ areas. Using a small hand-held drill, a very small hole was made in the skull according to the calculated coordinates for NAc (bregma 1.5 mm, ML 0.7 mm, DV 4.4 mm). A Hamilton syringe (preloaded with viral particles) was then slowly lowered, penetrating the dura to the determined depth. A volume of 0.3 μl of the virus (∼0.5 × 109 infectious particle per microliter) was slowly (∼0.1 μl/min) injected into the brain. A 2-3 min wait is performed before the needle is very slowly retracted from the brain to prevent backflow of the viral vector. The animals were allowed to recover and subjected to behavioral tests 4 weeks after viral injection. Brain injection sites were histologically verified with counterstaining from postmortem sections and plotted on standardized sections from the stereotaxic atlas.
Immunohistochemistry
Immunohistochemistry was conducted as previously described (Jiang et al., 2017) after animal perfusion with 4% paraformaldehyde. Mouse brains were sectioned by Vibratome at a 40 μm thickness. Free-floating sections were washed in TBS containing 0.5% Triton X-100 (TBST; 3 washes, 15 min), incubated at room temperature in blocking solution (10% normal goat or donkey serum in TBST; 1 h), and then incubated in primary antibody diluted in blocking solution overnight at 4°C. The following day, sections were washed in TBST (3 washes, 15 min), incubated in the dark in Alexa-555-conjugated goat anti-rabbit-IgG, Alexa-488-conjugated goat anti-mouse IgG, Alexa-488-conjugated goat anti-rabbit-IgG, Alexa-555-conjugated donkey anti-goat-IgG, or Alexa-488-conjugated donkey anti-rabbit-IgG (1:300; Invitrogen) for 2 h following primary antibody incubation. Sections were mounted in Vectashield (Vector Laboratories). The primary antibodies used were rabbit anti-phospho-ERK (1:1500, Cell Signaling), anti-phospho-CREB (1:1000, Cell Signaling), mouse anti-PSD95 (1:1000, NeuroMab), mouse anti-Bassoon (1:250, Enzo Life Sciences), goat anti-tdTomato (1:1000, MyBiosource), rabbit anti-Egr-1 (588) (1:7500, Santa Cruz Biotechnology), and rabbit anti-Rapgef2 (NNLE-2, custom-made by Anaspec) (see Jiang et al., 2017). Confocal images were obtained on a Carl Zeiss LSM 510 confocal microscope at the National Institute of Neurologic Disorders and Stroke Light Imaging Facility. Immunoreactive (IR) signals of NCS-Rapgef2, phospho-ERK, and Egr-1 from different brain areas as indicated were quantified by National Institutes of Health ImageJ using the mean gray values of integrated density after being converted to grayscale. To quantify the NCS-Rapgef2, phospho-ERK, and phospho-CREB IR signals in AAV-targeted neurons in the NAc, confocal images with double fluorescence channels were imported into Fiji ImageJ and split into different channels. Images in GFP channel were inverted and made into binary mask after adjusting threshold. GFP-positive neurons were selected as ROI following particle analysis. The ROI selections were overlaid to the NCS-Rapgef2, phospho-ERK, or phospho-CREB channel. The mean gray value of IR signal intensity for NCS-Rapgef2, phospho-ERK, or phospho-CREB IR in ROIs created from GFP-positive neurons were measured. Cell number for phospho-ERK-positive neurons was counted in ROIs created from GFP-positive neurons as well.
Western blot for cultured cell and mouse tissues
NS-1 cells were seeded in collagen 1-coated 12-well plates and collected in ice-cold RIPA lysis buffer (Thermo Fisher Scientific) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Tissues were punched from 0.5 mm coronal sections of mouse brains, weighed, and snap-frozen. Samples were sonicated on ice with RIPA buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail. RIPA-insoluble fractions were removed by centrifugation (3000 rpm for 10 min at 4°C). Supernatants were retained, and protein concentration was determined by MicroBCA Protein Assay kit (Thermo Fisher Scientific) according to the protocol provided by the manufacturer. Western blot was performed as described previously (Jiang et al., 2017). The rabbit monoclonal antibodies used included anti-ERK (Cell Signaling Technology, catalog #9102, 1:1000), rabbit anti-phospho-ERK (Cell Signaling Technology, catalog #9101, 1:1000), rabbit anti-Egr-1 (15F7) (Cell Signaling Technology, catalog #4153; 1:1000), and anti-GAPDH (D16H11) (Cell Signaling Technology, catalog #5174; 1:1000). Rabbit anti-Rapgef2 (NNLE-2, 1:1000) was used for NCS-Rapgef2 Western blot. IR bands were visualized with a SuperSignal West Dura Chemiluminescent Substrate (Thermo Fisher Scientific), photographed with a ChemiDoc Imaging system, and quantified with National Institutes of Health ImageJ.
Behavioral tests
Animal behavioral tests were conducted in the National Institute of Mental Health Rodent Behavioral Core Facility.
Elevated zero maze
The elevated zero maze for anxiety was performed by placing the mouse into an open quadrant of a continuous circular 5.5-cm-wide track elevated 65 cm above the floor and divided into alternating walled and open quadrants in a dimly lit room, for 6 min. Video recording from above in the last 5 min was automatically scored for time spent in each quadrant (Top Scan software suite, Clever System).
Locomotor sensitization
The mice were tested for sensitized responses to cocaine using a two-injection protocol (Valjent et al., 2010). Briefly, mice were placed in an Accuscan open field chamber (20 × 20 cm2, 8 beam sensors for each dimension) to monitor locomotor activity. Room light was set up as 30-50 lux. For the first 4 d, mice were placed for 30 min in the activity chamber (habituation phase), received an intraperitoneal injection of saline, then placed back to the chamber for 1 h. Then locomotor sensitization was tested by replacing injection of saline with cocaine (15 mg/kg, i.p.) for 2 continuous days in the same environment.
Conditioned place preference (CPP)
Place-conditioning boxes consisted of two chambers with balanced contexts. A partition with an opening separated the two chambers but allowed animals access to either side. This partition was closed off during the pairing days. The walls, floors, and partition of chambers were cleaned with CLIDOX-S and dried after each session. The CPP paradigm was performed using a biased strategy as follows: On day 1, mice were allowed to explore both sides for 15 min, and time spent in each side was recorded. Animals were separated into groups with approximately equal biases for the sides to start from. The side on which an animal spent most time was defined as the “preferred side” for this mouse. On day 2, starting from the unpreferred side, animals were allowed to explore both sides for 15 min, and data were collected to calculate place preference for the preconditioning phase. Beginning on day 3, the animals were paired for 8 d (30 min per day), with the saline group receiving injections (0.9% sodium chloride) on both sides of the boxes; whereas the drug-paired group received cocaine (10 mg/kg) on the unpreferred side and saline on the preferred side. On day 11, all the animals were given a saline injection, placed on the unpreferred side, and allowed to run freely between the two sides for 15 min, and time spent on each side was recorded (Top Scan software suite, Clever System).
Experimental design and statistical analysis
Mice of both sexes were used in all studies. The sample size (n) per group is described in the figure legends for each experiment. Statistical analyses were conducted using Sigma Plot 14.0 (Systat Software). Student's t tests and factorial model ANOVAs were used where appropriate. Post hoc analyses were performed using the Bonferroni test. Data were reported using histograms and scatter plots to represent mean ± SEM and individual data in each group. Differences were considered to be significant when p < 0.05.
Results
NCS-Rapgef2 dependency of phospho-ERK activation and Egr-1 induction in response to cAMP elevation in NS-1 cells
The immediate early gene Egr-1/Krox-24/Zif268 is induced by cocaine treatment in vivo, and implicated in the behavioral actions of cocaine (Valjent et al., 2006c). We have previously shown that Egr-1 and NCS-Rapgef2 are both required for neuritogenesis induced by Gs-coupled GPCRs, including the D1 receptor, or elevation of cAMP itself, in neuroendocrine cells (Ravni et al., 2008; Emery et al., 2013, 2017a; Jiang et al., 2017). In order to link cAMP-dependent activation of NCS-Rapgef2 to signaling to ERK resulting in Egr-1 protein induction, we treated NS-1 rat pheochromocytoma cells, and an NS-1 cell line in which Rapgef2 expression was deleted with CRISPR-mediated gene mutation (Jiang et al., 2017), with 100 μm of the cell-permeant cAMP analog 8-CPT-cAMP, and measured levels of phospho-ERK and Egr-1. ERK phosphorylation and Egr-1 induction in response to 8-CPT-cAMP were abolished in the absence of NCS-Rapgef2 (Fig. 1; two-way ANOVA by genotype of cell lines and drug treatment: for phospho-ERK, gene effect F(1,8) = 3.099, p = 0.116; drug effect F(1,8) = 10.643, p = 0.011; gene × drug F(1,8) = 15.356, p = 0.004; post hoc Bonferroni test, 8-CPT-cAMP vs vehicle, p < 0.001 for NS-1 (Rapgef2+/+), p = 0.655 for NS-1 (Rapgef2−/−); for Egr-1, gene effect F(1,8) = 26.22, p < 0.001; drug effect F(1,8) = 19.62, p = 0.002; gene × drug: F(1,8) = 10.393, p = 0.012; post hoc Bonferroni test, 8-CPT-cAMP vs vehicle, p < 0.001 for NS-1 (Rapgef2+/+), p = 0.419 for NS-1 (Rapgef2−/−)). This prompted us to examine whether elevation of both phospho-ERK and Egr-1 after cocaine administration, in dopaminoceptive neurons in vivo, might also be linked by the cAMP-dependent action of NCS-Rapgef2.
Figure 1.

NCS-Rapgef2-dependent phospho-ERK activation and Egr-1 upregulation in neuroendocrine cell line NS-1 induced by 8-CPT-cAMP. A, NS-1 cells (NS-1(Rapgef2+/+)) or NS-1 cells with NCS-Rapgef2 KO by CRISPR (NS-1 (Rapgef2−/−)) were treated with 100 μm 8-CPT-cAMP or 0.01% DMSO (8-CPT-cAMP (–) group) for 1 h; then the cell lysate was collected for Western blot analysis of pan-ERK, phospho-ERK, and Egr-1. Equal amount of total protein (20 µg) was loaded in each lane. B, Phospho-ERK and Egr-1 protein levels were quantified with ImageJ and compared with NS1 (Rapgef2+/+)/8-CPT-cAMP (–) group (n = 3 per group). *p < 0.05; **p < 0.001; post hoc Bonferroni test following two-way ANOVA. Columns and bars represent mean ± sem and scattered points represent individual values. ns, not significant.
Postsynaptic localization of NCS-Rapgef2 in dopaminoceptive regions of mPFC, BLA, and NAc
NCS-Rapgef2 is highly expressed in corticolimbic areas and striatum in mouse brain, including cell types and brain regions (cortical pyramidal cells; hippocampal granule cells of dentate gyrus and pyramidal cells of CA1-3; basolateral amygdalar excitatory neurons; central amygdalar inhibitory neurons; and NAc MSNs) (Jiang et al., 2017), which receive innervation from dopaminergic neurons of the VTA and substantia nigra. Modulation of neurons in these brain regions by dopamine is implicated in several aspects of learning, memory, stress responding, and reward. Dysfunction of these neurons is similarly implicated in the pathogenesis of a broad range of neuropsychiatric and neurologic disorders, including Parkinson's disease, schizophrenia, and drug addiction. We wished to determine whether NCS-Rapgef2 was localized within dopaminoceptive neurons at an intracellular location consistent with postsynaptic responses to dopamine receptor activation on cells in these regions. For this purpose, we examined the colocalization and differential localization of NCS-Rapgef2 and both presynaptic and postsynaptic markers in these regions. NCS-Rapgef2 IR signal is not colocalized with Bassoon, a scaffolding protein of presynaptic active zone, but rather is in close apposition to the Bassoon puncta in mPFC, NAc, and BLA (Fig. 2A). However, NCS-Rapgef2 IR signal is colocalized with postsynaptic marker PSD95 IR signal in the soma and dendrites of neurons in mPFC pyramidal cells, NAc MSNs, and excitatory neurons in BLA (Fig. 2B). These results suggest a postsynaptic location of NCS-Rapgef2 in neurons, including those responsive to dopamine.
Figure 2.
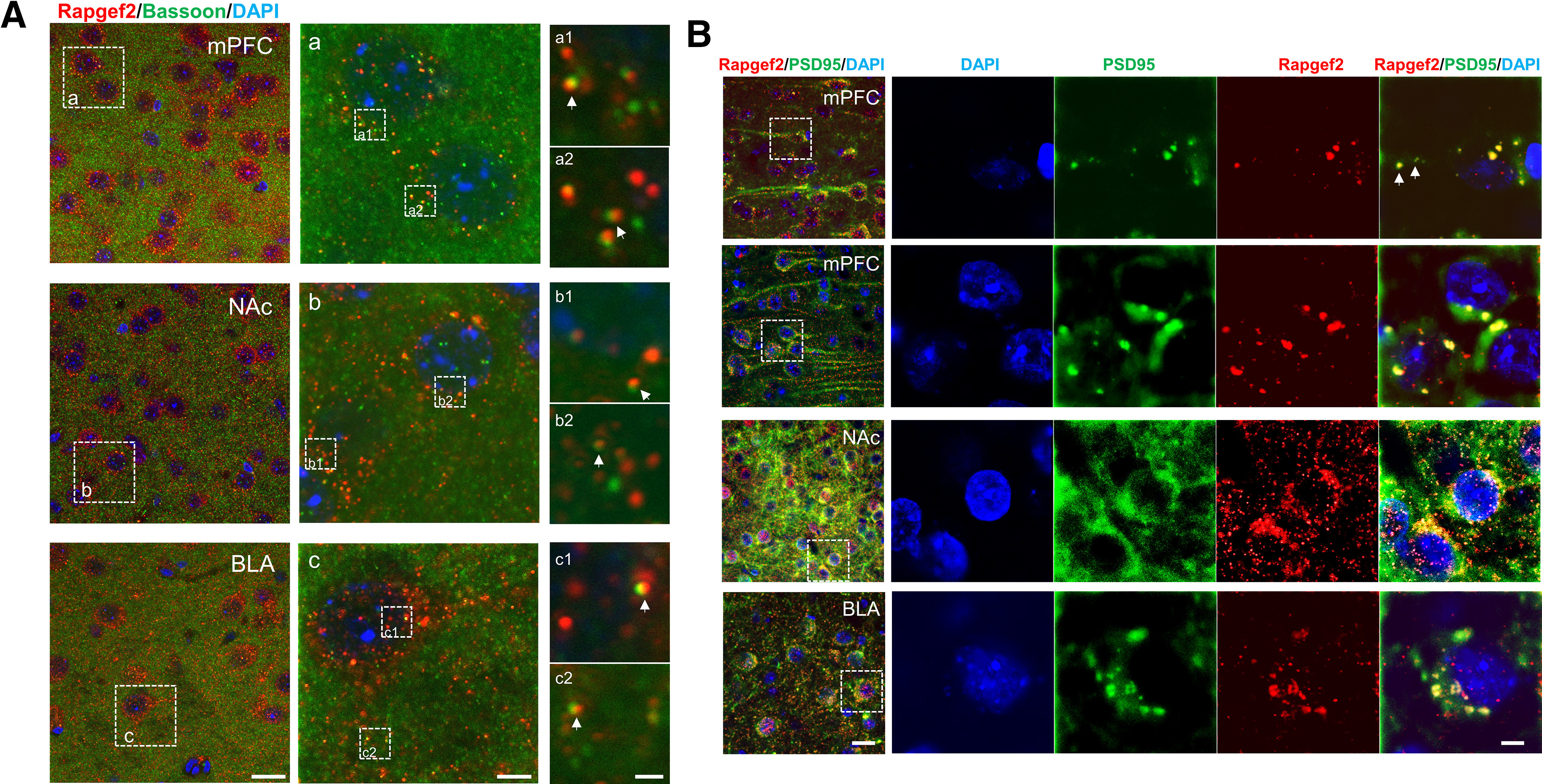
Postsynaptic localization of NCS-Rapgef2 in dopaminoceptive neurons in mPFC, NAc, and BLA. A, NCS-Rapgef2 IR signal (red) is not colocalized with Bassoon (green), a scaffolding protein of presynaptic active zone, but in close apposition to the Bassoon puncta, indicated by white arrows in a1 & a2 (mPFC), b1 & b2 (NAc), and c1 & c2 (BLA). Scale bars: Left, 20 µm; Middle, 5 µm; Right, 1 µm. B, NCS-Rapgef2 IR signal (red) is colocalized with postsynaptic marker PSD95 IR signal (green) in the soma and dendrites (indicated by white arrows) of neurons in mPFC pyramidal cells, NAc MSNs, and excitatory neurons in BLA. Right, At higher magnification, the images outlined by white boxes on the left. Scale bars: Left, 20 µm; Right, 5 µm.
ERK activation by D1 receptor stimulation in mouse NAc ex vivo: dependence on NCS-Rapgef2
Given the existence of a cAMP→ERK→Egr-1/Zif268 pathway dependent on NCS-Rapgef2 in neuroendocrine cells in culture, we next inquired about the existence of this pathway in neurons receiving dopaminergic input within the mouse CNS, as already implied by our previous observations of Rapgef2-dependent ERK activation in various brain regions after systemic administration of SKF81297 (Jiang et al., 2017). Accordingly, we examined the effects of SKF81297 on phospho-ERK activation after its direct application to NAc-containing brain slices, in the presence and absence of the NCS-Rapgef2 inhibitor EL1101; 10 μm SKF81297 produced a robust increase in phospho-ERK in both core and shell of NAc slices in a pattern similar to that seen after SKF81297 administration in vivo (Fig. 3A,C). Furthermore, this activation was completely confined to D1-expressing neurons (i.e., >95% phospho-ERK-positive neurons are D1 receptor/tdTomato-positive; see Fig. 3B). SKF81297-stimulated ERK phosphorylation was blocked by the MEK1 inhibitor U0126, or in the presence of 100 μm of the NCS-Rapgef2 inhibitor EL1101 (Fig. 3A,C; one-way ANOVA by drug treatment: F(2,8) = 49.435, p < 0.001; post hoc Bonferroni test, SKF81297 vs vehicle or SKF81297+EL1101, p < 0.001), indicating that the effects of D1 agonist treatment on ERK activation are mediated directly at D1 dopaminoceptive MSNs of the NAc, and are mediated via NCS-Rapgef2 and MEK.
Figure 3.

NCS-Rapgef2 inhibitor EL1101 attenuated phospho-ERK activation in the NAc of mouse brain slice induced by D1 receptor agonist SKF81297. A, Brain slices (coronal sections in 250 µm) were incubated vehicle (DMSO final concentration 0.1%), 10 μm SKF81297, 10 μm U0126 plus 10 μm SKF81297, or 100 μm EL1101 plus 10 μm SKF81297 for 30 min. After fixation with 4% PFA, slices were stained with phospho-ERK antibody. Phospho-ERK activation in the NAc of mouse brain slice can be induced by D1 receptor agonist SKF81297, which was blocked by the MEK1 inhibitor U0126 or attenuated by NCS-Rapgef2 inhibitor EL1101. Right, At higher magnification, the images outlined by white boxes on the left. Scale bars: Left, 500 µm; Right, 100 µm. B, D1-specific activation of phospho-ERK in the NAc of D1d1a-tdTomato mouse indicated by colocalization of phospho-ERK IR signal (green) and D1 MSNs (tdTomato, red). Scale bar, 50 µm. C, Phospho-ERK IR signals in the NAc of mouse brain slices from multiple C57BL/6J animals in multiple experiments were quantified with National Institutes of Health ImageJ using the mean gray values of integrated density after being converted to grayscale. Relative phospho-ERK IR from 10 μm SKF81297-treated or 100 μm EL1101- plus 10 μm SKF81297-treated slices for each animal was normalized by phospho-ERK IR from vehicle (DMSO final concentration 0.1%)-treated slices. N = 3 or 4 animals per group. *p < 0.001 (post hoc Bonferroni test following one-way ANOVA). Columns and bars represent mean ± sem and scattered points represent individual values.
Cocaine-induced upregulation of ERK and Egr-1/Zif268 in mouse brain requires expression of NCS-Rapgef2
We next sought to specifically manipulate Rapgef2 levels in dopaminoceptive brain regions, the mPFC, BLA, and NAc, closely associated with behaviors affected by cocaine. NCS-Rapgef2 KO specifically in corticolimbic areas, but not NAc, was achieved by crossing floxed Rapgef2 (Rapgef2cko/cko) with Camk2α-cre mice. NCS-Rapgef2 expression was decreased at least 85% in mPFC and BLA in these mice, whereas NCS-Rapgef2 expression in NAc was unaffected (Fig. 4A,B; Student's t test, p < 0.001 for mPFC and BLA, p = 0.470 for NAc). Knockdown of NCS-Rapgef2 expression in D1 MSNs in NAc is not achievable by crossing a D1 receptor cre driver line with Rapgef2cko/cko (Drd1-cre+/−:: Rapgef2cko/cko) (Fig. 4C-E; Student's t test, p = 0.598 for NCS-Rapgef2 expression in NAc from Rapgef2cko/cko vs Drd1-cre+/−:: Rapgef2cko/cko; however, p = 0.015 for NCS-Rapgef2 expression in mPFC from Rapgef2cko/cko vs Camk2α-cre+/−::Rapgef2cko/cko). Therefore, an alternative method was used to achieve the ablation of NCS-Rapgef2 expression in NAc, that is, by bilateral injection of AAV9-hSyn-Cre-eGFP into the ventral striatum of Rapgef2cko/cko mice (Fig. 4F-H). Expression of NCS-Rapgef2 in the NAc was not affected by injection of AAV9-hSyn-eGFP control virus into Rapgef2cko/cko mice, or by injection of AAV9-hSyn-cre-eGFP into C57BL/6J WT mice. However, NCS-Rapgef2 in NAc was significantly reduced by injection of AAV9-hSyn-Cre-eGFP into Rapgef2cko/cko mice (Fig. 4G,H; one-way ANOVA: F(2,8) = 24.533, p < 0.001).
Figure 4.

Region-specific ablation of NCS-Rapgef2 expression. A, Camk2α-cre+/−::Rapgef2cko/cko (cKO) was generated by crossing Rapgef2cko/cko (flox) with Camk2α-cre mice. NCS-Rapgef2 expression was largely ablated in mPFC and BLA of cKO but was unaffected in NAc of cKO. Scale bars: mPFC and BLA, 100 µm; NAc, 50 and 10 µm. B, NCS-Rapgef2 IR signals in mPFC, BLA, and NAc from cKO mice were quantified with National Institutes of Health ImageJ and compared with NCS-Rapgef2 IR signals from flox mice. N = 4-7 animals per group. *p < 0.001 (Student's t test for each region). C-E, Cre expression in D1d1a-cre mouse line is not strong enough to KO NCS-Rapgef2 in D1 MSNs in NAc. NCS-Rapgef2 expression is largely intact in the NAc (C, top panels), especially in D1-MSNs (bottom panels, green) of Drd1-cre+/−:: Rosa-eGFP:: Rapgef2cko/cko compared with Drd1-cre+/−::Rosa-eGFP, indicated by Rapgef2 immunohistochemistry (C). Phospho-ERK activation in the NAc of Drd1-cre+/−:: Rapgef2cko/cko was normally induced by D1 receptor agonist SKF81297 (2 mg/kg, i.p., 15 min) (D). Western blots using protein lysates from the NAc of Drd1-cre+/−:: Rapgef2cko/cko (Drd1-cre+/−::flox) further confirmed that NCS-Rapgef2 expression was not significantly decreased, compared with flox controls (n = 5 for each group; Student's t test). However, NCS-Rapgef2 expression in the mPFC from Camk2α-cre+/−::Rapgef2cko/cko (Camk2α-cre+/−::flox) was largely reduced (n = 3 or 4 for each group). *p < 0.05 (Student's t test) (E). F-H, NCS-Rapgef2 in NAc was abrogated by bilateral injection of AAV9-hSyn-cre-eGFP (cre virus) into the ventral striatum of Rapgef2cko/cko (flox) mice. Representative image of brain slice 4 weeks after AAV bilateral viral injection in NAc (F). NCS-Rapgef2 can be efficiently eliminated in NAc by injection of AAV9-hSyn-cre-eGFP (cre virus) into the ventral striatum of Rapgef2cko/cko (flox) mice. However, expression of NCS-Rapgef2 in the NAc was not affected by injection of AAV9-hSyn-eGFP (ctrl virus) into Rapgef2cko/cko (flox) mice or by injection of AAV9-hSyn-cre-eGFP (cre virus) into C57BL/6J WT mice. Bottom panels, At higher magnification, the images outlined by white boxes in top panels. Scale bars: Top, 20 µm; Bottom, 10 µm (G). NCS-Rapgef2 IR signals in GFP-positive neurons were quantified with ImageJ. N = 3 or 4 animals per group (H). *p < 0.05 (post hoc Bonferroni test following one-way ANOVA). Columns and bars represent mean ± sem and scattered points represent individual values. ns, not significant.
Thus, NCS-Rapgef2 expression can be efficiently and specifically eliminated in NAc, by stereotaxic AAV9-hSyn-Cre-eGFP injection in Rapgef2cko/cko mice, and in BLA and mPFC, by genetic cross of Camk2α-cre+/− and Rapgef2cko/cko mice.
Cocaine treatment is known to induce phospho-ERK elevation and trigger ERK-dependent Egr-1/Zif268 expression in D1 medium-sized spiny neurons (Bertran-Gonzalez et al., 2008), implicated in locomotor and drug preference responses to cocaine (Valjent et al., 2006c). To confirm this, and to also confirm the existence of the ERK→Egr-1/Zif268 pathway in D1-dopaminoceptive neurons in mPFC and BLA, we treated mice in which D1 neurons are labeled with tdTomato, with cocaine. Phospho-ERK neuronal induction after cocaine occurs predominantly in D1-dopaminoceptive (tdTomato-positive) neurons in NAc, BLA, and mPFC (Fig. 5A). We further quantified the percentage of phospho-ERK+ cells that were also D1 receptor-expressing (tdTomato-positive) after treatment with cocaine. In PFC, 56.7 ± 2.8% of phospho-ERK+ neurons in layer II/III, 78.1 ± 2.4% of pERK+ neurons in layer V, and 97.3 ± 0.7% phospho-ERK+ neurons in layer VI were also tdTomato-positive. In BLA, 72.2 ± 2.8% of phospho-ERK+ neurons were D1 receptor-positive; and in shell and core of NAc, almost all (98.6 ± 0.1%) phospho-ERK+ neurons were D1-MSNs.
Figure 5.
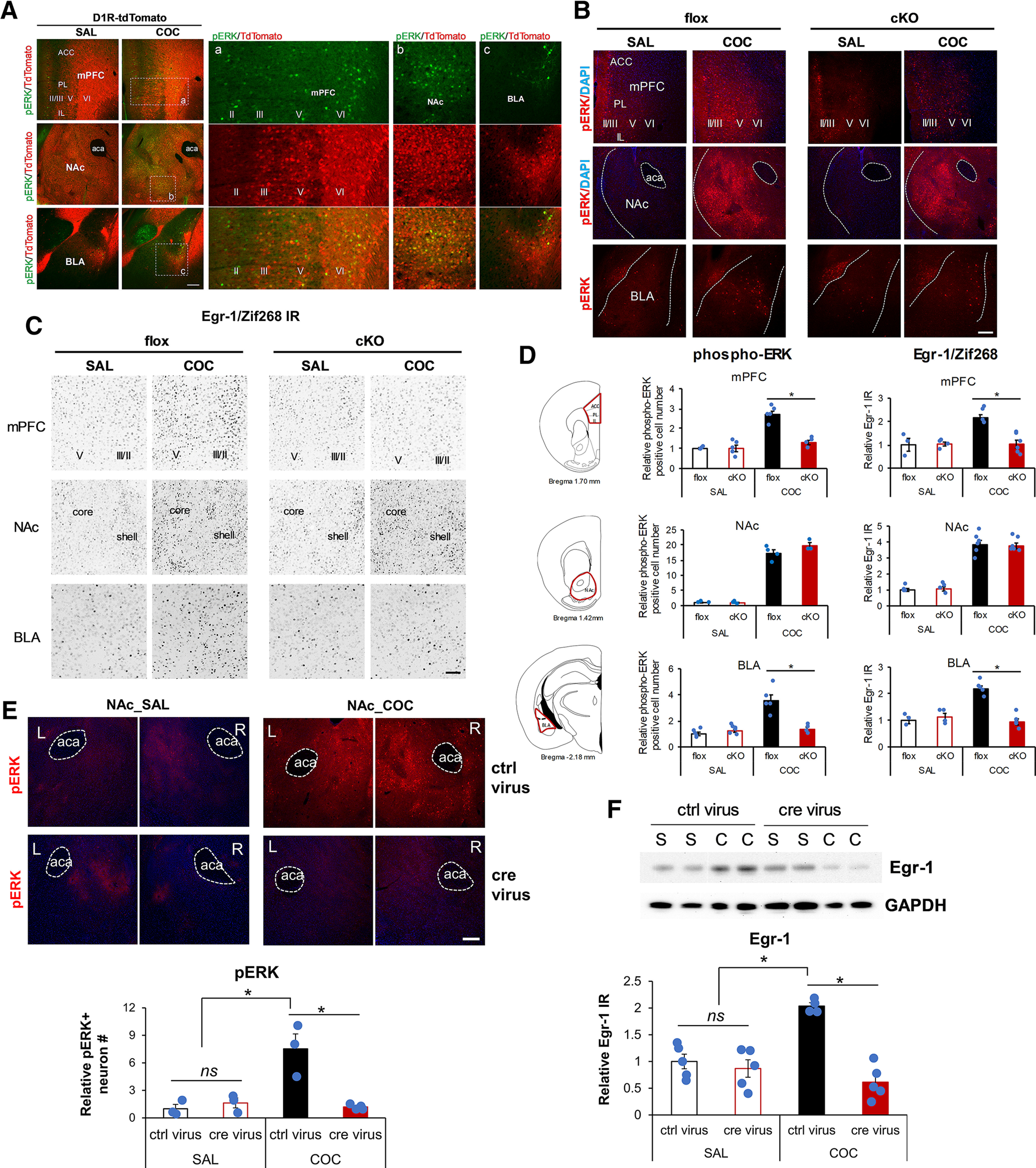
Expression of NCS-Rapgef2 is required for cocaine-induced phosphorylation of ERK and upregulation of Egr-1/Zif268 in mouse brain. A, A majority of cocaine-induced phospho-ERK (pERK) activation (10 min after administration of 20 mg/kg cocaine, i.p.) was in D1 receptor-positive neurons in mPFC, NAc, and BLA. In PFC, 56.7 ± 2.8% of pERK+ neurons in layer II/III, 78.1 ± 2.4% of pERK+ neurons in layer V, and 97.3 ± 0.7% pERK+ neurons in layer VI were also tdTomato-positive. In BLA, 72.2 ± 2.8% of pERK+ neurons were D1 receptor-positive; and in shell and core of NAc almost all (98.6 ± 0.1%) pERK+ neurons were D1-MSNs. Right panels (Aa,Ab,Ac) show the images at higher magnification, outlined by white boxes on the left. Scale bar, 200 µm. B, Phospho-ERK elevation observed 10 min after intraperitoneal administration of 20 mg/kg cocaine in Rapgef2cko/cko (flox) mice is attenuated in mPFC and BLA, but not in NAc, of Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice. Scale bar, 200 µm. C, Egr-1/Zif268 elevation observed 1 h after 20 mg/kg cocaine treatment in Rapgef2cko/cko (flox) mice is attenuated in mPFC and BLA, but not in NAc, of Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice. Scale bar, 100 µm. D, Quantification of phospho-ERK and Egr-1/Zif268 in Rapgef2cko/cko (flox) or Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice after saline or cocaine treatment. Relative number of phospho-ERK-positive neurons or Egr-1 IR from mPFC, NAc, and BLA of each animal was normalized by the average value from age-matched saline-treated Rapgef2cko/cko (flox) mice. N = 3-6 animals per group. *p < 0.001 (post hoc Bonferroni test following two-way ANOVA). E, Bilateral ablation of NCS-Rapgef2 in NAc impaired phospho-ERK elevation in response to cocaine. AAV9-hSyn-eGFP (ctrl virus) or AAV9-hSyn-cre-eGFP (cre virus) was injected into the NAc of Rapgef2cko/cko mice. Four weeks later, phospho-ERK activation in both sides of NAc was examined by immunohistochemistry after acute cocaine (20 mg/kg, i.p., 10 min) injection. N = 3 or 4 animals per group. *p < 0.001 (post hoc Bonferroni test following two-way ANOVA). F, Cocaine-induced Egr-1/Zif268 increase in the NAc of Rapgef2cko/cko mice with ctrl virus was impaired in the NAc with cre virus. Four weeks after viral infusion, mice were killed 1 h after saline or cocaine (20 mg/kg) injection. The tissue from NAc was punched out from 0.5 mm coronal sections of mouse brains and protein lysate was used for Western blot with Egr-1/Zif268 antibody. Total protein loading of each sample was first normalized by GAPDH immunoreactivity (IR). Relative Egr-1/Zif268 IR was obtained by comparing to average of that from NAc of saline-treated Rapgef2cko/cko bilaterally injected with AAV9-hSyn-eGFP (ctrl virus). N = 4 or 5 animals per group. *p < 0.001 (post hoc Bonferroni test following two-way ANOVA). Columns and bars represent mean ± sem and scattered points represent individual values. ns, not significant.
We examined the requirement for NCS-Rapgef2 in cocaine-induced ERK phosphorylation/activation, and subsequent Egr-1/Zif268 induction, in two sets of experiments. First, the effects of cocaine injection were compared in Rapgef2cko/cko control mice, and in Camk2α-cre+/−::Rapgef2cko/cko mice in which Rapgef2 expression is eliminated in mPFC and BLA but not in NAc. ERK phosphorylation and Egr-1/Zif268 induction occurred in all three areas in control mice at 10 and 60 min, respectively, after intraperitoneal cocaine administration (20 mg/kg). Induction of both ERK phosphorylation (Fig. 5B & 5D; two-way ANOVA by genotype and drug treatment, phospho-ERK in mPFC: genotype effect F(1,13) = 21.312, p < 0.001, drug effect F(1,13) = 43.108, p < 0.001, interaction F(1,13) = 21.502, p < 0.001; phospho-ERK in BLA: genotype effect F(1,16) = 14.395, p = 0.002, drug effect F(1,16) = 26.668, p < 0.001, interaction F(1,16) = 23.691, p < 0.001; post hoc Bonferroni test, flox vs cKO within COC, p < 0.001 in mPFC or BLA) and Egr-1/Zif268 (Fig. 5C & 5D; two-way ANOVA by genotype and drug treatment, Egr-1 in mPFC: genotype effect F(1,15) = 10.348, p = 0.006, drug effect F(1,15) = 11.435, p = 0.004, interaction F(1,15) = 11.62, p = 0.004; Egr-1 in BLA: genotype effect F(1,13) = 19.872, p < 0.001, drug effect F(1,13) = 15.786, p = 0.002, interaction F(1,13) = 29.799, p < 0.001; post hoc Bonferroni test, flox vs cKO within COC, p < 0.001 in mPFC or BLA) was abolished in mPFC and BLA, where Rapgef2 expression was absent; but not in NAc (Fig. 5B-D; two-way ANOVA by genotype and drug treatment, phospho-ERK in NAc, genotype effect F(1,10) = 2.131, p = 0.175, drug effect F(1,10) = 433.739, p < 0.001, interaction F(1,10) = 2.427, p = 0.150; Egr-1 in NAc: genotype effect F(1,16) = 0.00125, p = 0.972, drug effect F(1,16) = 169.616, p < 0.001, interaction F(1,16) = 0.11, p = 0.744; post hoc Bonferroni test, flox vs cKO within COC, p = 0.352 for phospho-ERK, p = 1 for Egr-1), where Rapgef2 expression was spared. Next, the effects of cocaine injection were compared in Rapgef2cko/cko mice receiving AAV9-Syn-Cre-eGFP (to eliminate Rapgef2 expression in NAc) or AAV9-Syn-eGFP (controls). ERK phosphorylation and Egr-1/Zif268 induction by cocaine were eliminated in NAc after loss of NCS-Rapgef2 expression in mice receiving intra-NAc AAV9-Syn-Cre-eGFP injection (Fig. 5E,F; two-way ANOVA by genotype and drug treatment, phospho-ERK in NAc: genotype effect F(1,9) = 12.262, p = 0.007, drug effect F(1,9) = 13.901, p = 0.005, interaction F(1,9) = 18.216, p = 0.002; Egr-1 in NAc: genotype effect F(1,15) = 31.507, p < 0.001; drug effect F(1,15) = 7.995, p = 0.013; genotype × drug effect F(1,15) = 21.792, p < 0.001; post hoc Bonferroni test, ctrl virus vs cre virus within COC, p < 0.001 for phospho-ERK or Egr-1) with no effect on ERK phosphorylation or Egr-1/Zif268 induction in mPFC or BLA (data not shown). Thus, expression of NCS-Rapgef2 is required for cocaine-induced upregulation of ERK and Egr-1/Zif268 in D1-expressing neurons of NAc, of mPFC, and of BLA.
NCS-Rapgef2 in D1 MSNs of ventral striatum, but not excitatory neurons in mPFC or BLA, contributes to cocaine-induced psychomotor sensitization and CPP
Neither Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice nor Rapgef2cko/cko (flox) mice infused with AAV-Syn-Cre-eGFP in NAc, compared with corresponding control mice, showed altered locomotor activity and anxiety in the elevated zero maze, indicated by similar total distance traveled (flox vs cKO: 7488 ± 545 mm vs 8006 ± 715 mm, p = 0.575; ctrl virus vs cre virus: 5407 ± 620 mm vs 5494 ± 539 mm, p = 0.918; Student's t test) and percentage of time spent in open arms (flox vs cKO: 23.3 ± 2.3% vs 28.4 ± 4.5%, p = 0.262; ctrl virus vs cre virus: 16.0 ± 3.2% vs 15.9 ± 2.7%; p = 0.984; Student's t test). Mice were then subjected to testing for locomotor sensitization to cocaine using the two-injection protocol (Valjent et al., 2010) (Fig. 6A): For the first 4 d, animals were habituated for 30 min in an activity box, then injected with saline, and allowed free activity for 1 h. On day 5, after 30 min of habituation in the same box in the same room, animals received their first dose of cocaine (15 mg/kg, i.p.). All four groups of animals showed hyperactivity after cocaine injection (Fig. 6B,C; one-way repeated-measures ANOVA followed by post hoc Bonferroni test, COC1 vs SAL, p < 0.001). On day 6, in the same context, animals received a second dose of cocaine (15 mg/kg, i.p.) after the habituation trial. Both control groups (Rapgef2cko/cko mice, and Rapgef2cko/cko mice injected with AAV9-Syn-eGFP control virus in the NAc) exhibited a comparable increase in locomotor activity compared with their response to the first dose of cocaine (Fig. 6B,C; one-way repeated-measures ANOVA between treatments, for Rapgef2cko/cko mice: F(2,22) = 44.407, p < 0.001; post hoc Bonferroni test, COC2 vs COC1, p = 0.002; for Rapgef2cko/cko mice injected with control virus: F(2,22) = 36.907, p < 0.001; post hoc Bonferroni test COC2 vs COC1, p = 0.009). Similar locomotor sensitization in response to cocaine was observed in another control group: C57BL/6J WT mice injected with AAV9-Syn-cre-eGFP virus in the NAc (Fig. 6C; one-way repeated-measures ANOVA by repeated treatments of SAL, COC1, and COC2: F(2,22) = 24.389, p < 0.001; post hoc Bonferroni test, COC2 vs COC1, p = 0.006). Locomotor sensitization was similarly unaffected in Camk2α-cre+/−::Rapgef2cko/cko mice (one-way repeated-measures of ANOVA between treatments, F(2,20) = 69.99, p < 0.001; post hoc Bonferroni test, COC2 vs COC1, p = 0.001), similar to its relative control Rapgef2cko/cko mice. However, cocaine-induced locomotor sensitization was abolished in Rapgef2cko/cko mice with AAV9-Syn-cre-eGFP bilateral infusion in the NAc (Fig. 6C,D; one-way repeated-measures of ANOVA between treatments: F(2,22) = 17.965, p < 0.001; post hoc Bonferroni test, COC2 vs COC1, p = 0.899), although COC1 induced increased locomotor activity compared with saline injection (COC1 vs SAL Avg, p < 0.001).
Figure 6.
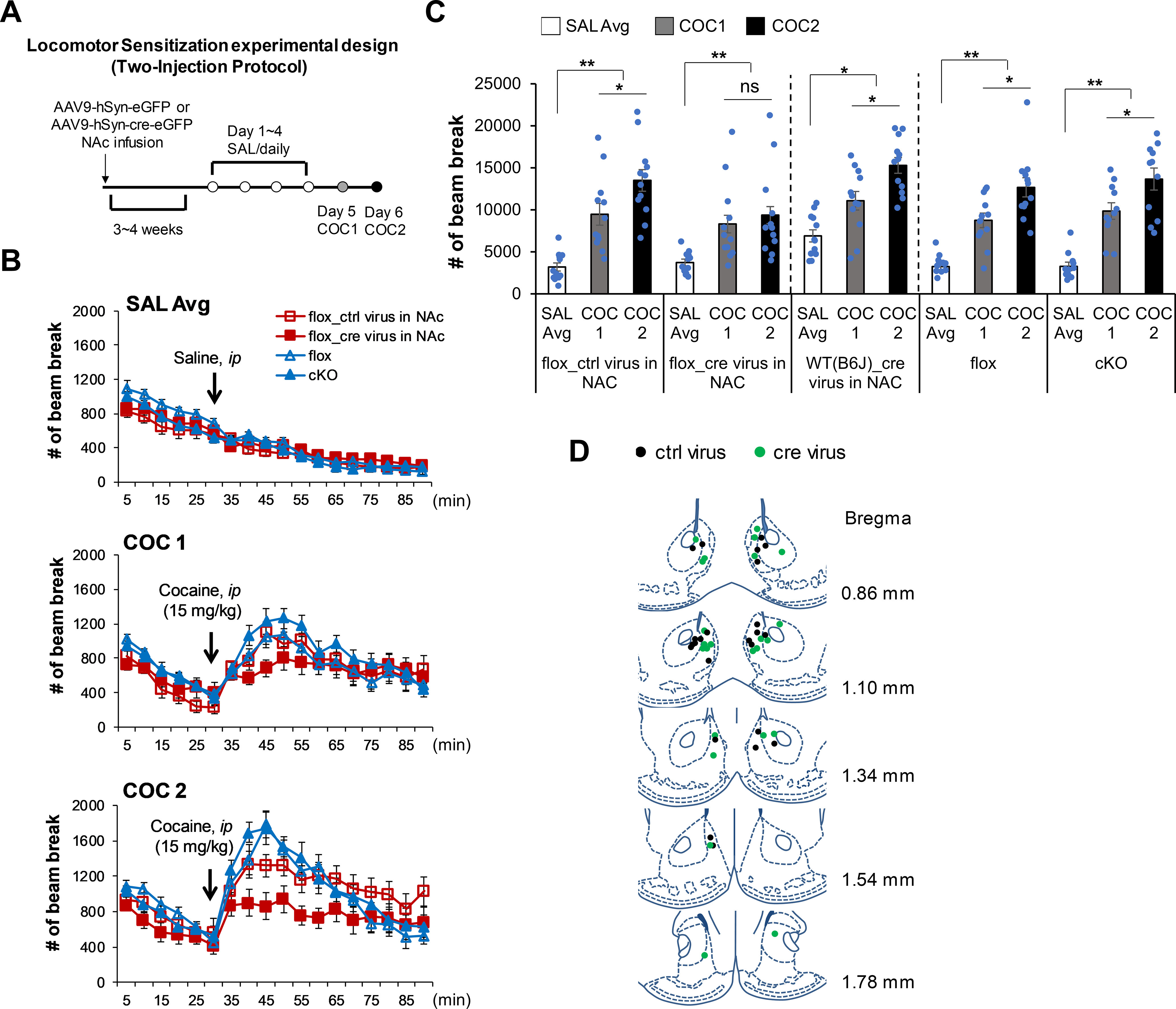
Locomotor sensitization induced by cocaine was abolished in the mice with NCS-Rapgef2 ablation in the NAc. A, Experimental design for locomotor sensitization. Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice, Rapgef2cko/cko (flox) mice with bilateral NAc AAV9-hSyn-cre-eGFP (cre virus) injection, and their corresponding controls were subjected to a two-injection protocol for cocaine locomotor sensitization. For the first 4 d, animals were habituated for 30 min in an activity chamber, then injected with saline, and allowed free movement within the chamber for 60 min. On day 5, after 30 min of habituation in the same box in the same room, animals received the first dose of cocaine. On day 6, in the same context (same box in same room), animals received a second dose of cocaine after a habituation trial. B, Animal locomotor activity in response to saline or cocaine administration. Activity was monitored in two trials each day: 30 min free running and 1 h after saline or cocaine (15 mg/kg) injection. C, Locomotor activity (total number of beam breaks) for 1 h following saline or cocaine administration. SAL Avg, Average of activity during 4 d of saline injection; COC1, activity after the first dose of cocaine (15 mg/kg) on day 5; COC2, activity after the second dose of cocaine (15 mg/kg) on day 6. N = 11 or 12 animals per group. Repeated treatments of SAL, COC1, and COC2 on each animal in each group: *p < 0.05; **p < 0.001; one-way repeated-measures ANOVA followed by post hoc Bonferroni test. Columns and bars represent mean ± sem and scattered points represent individual values. D, Schematic representation of the injection sites in the NAc for the mice (Rapgef2cko/cko (flox) with bilateral injection of AAV9-hSyn-eGFP (ctrl virus) or AAV9-hSyn-cre-eGFP (cre virus)) used for locomotor sensitization test. N = 12 animals for each group. ns, not significant.
We also studied the effects of region-specific NCS-Rapgef2 KO on the effects of repeated administration of cocaine on drug preference, using the CPP test (Fig. 7A). Ablation of NCS-Rapgef2 in NAc by viral injection also abolished cocaine-induced CPP (Fig. 7B,D; three-way ANOVA by context conditioning, genotype, and drug treatment: preconditioning vs postconditioning F(1,58) = 36.937, p < 0.001; genotype effect F(1,58) = 3.059, p = 0.086; drug effect F(1,58) = 5.687, p = 0.02; conditioning × genotype interaction F(1,58) = 4.875, p = 0.031; conditioning × drug interaction F(1,58) = 4.5, p = 0.038; conditioning × genotype × drug interaction F(1,58) = 0.443, p = 0.508. Post hoc all pairwise multiple comparison Bonferroni test for postconditioning: COC versus SAL, p = 0.002 for ctrl virus, p = 0.211 for cre virus; ctrl virus versus cre virus within COC, p = 0.005). These results indicate that two major behavioral consequences of repeated psychomotor stimulant (cocaine) administration are dependent on ERK→Egr-1/Zif268 signaling in D1-MSNs, and further suggest cellular mechanistic linkage between the two behaviors. CPP was not affected in Camk2α-cre+/−::Rapgef2cko/cko mice (Fig. 7C; three-way ANOVA by context conditioning, genotype and drug treatment: preconditioning vs postconditioning F(1,90) = 67.764, p < 0.001; genotype effect F(1,90) = 0.315, p = 0.576; drug effect F(1,90) = 1.629, p < 0.001; conditioning × genotype interaction F(1,90) = 0.969, p = 0.328; conditioning × drug interaction F(1,90) = 12.82, p < 0.001; genotype × drug interaction F(1,90) = 0.0099, p = 0.921; conditioning × genotype × drug interaction F(1,90) = 5.332, p = 0.023. Post hoc all pairwise multiple comparison Bonferroni test for postconditioning: COC versus SAL, p = 0.022 for flox, p < 0.001 for cKO; flox versus cKO within COC, p = 0.652), indicating that the development of CPP inheres in cellular changes in dopaminoceptive neurons of the NAc, but not the BLA or mPFC.
Figure 7.
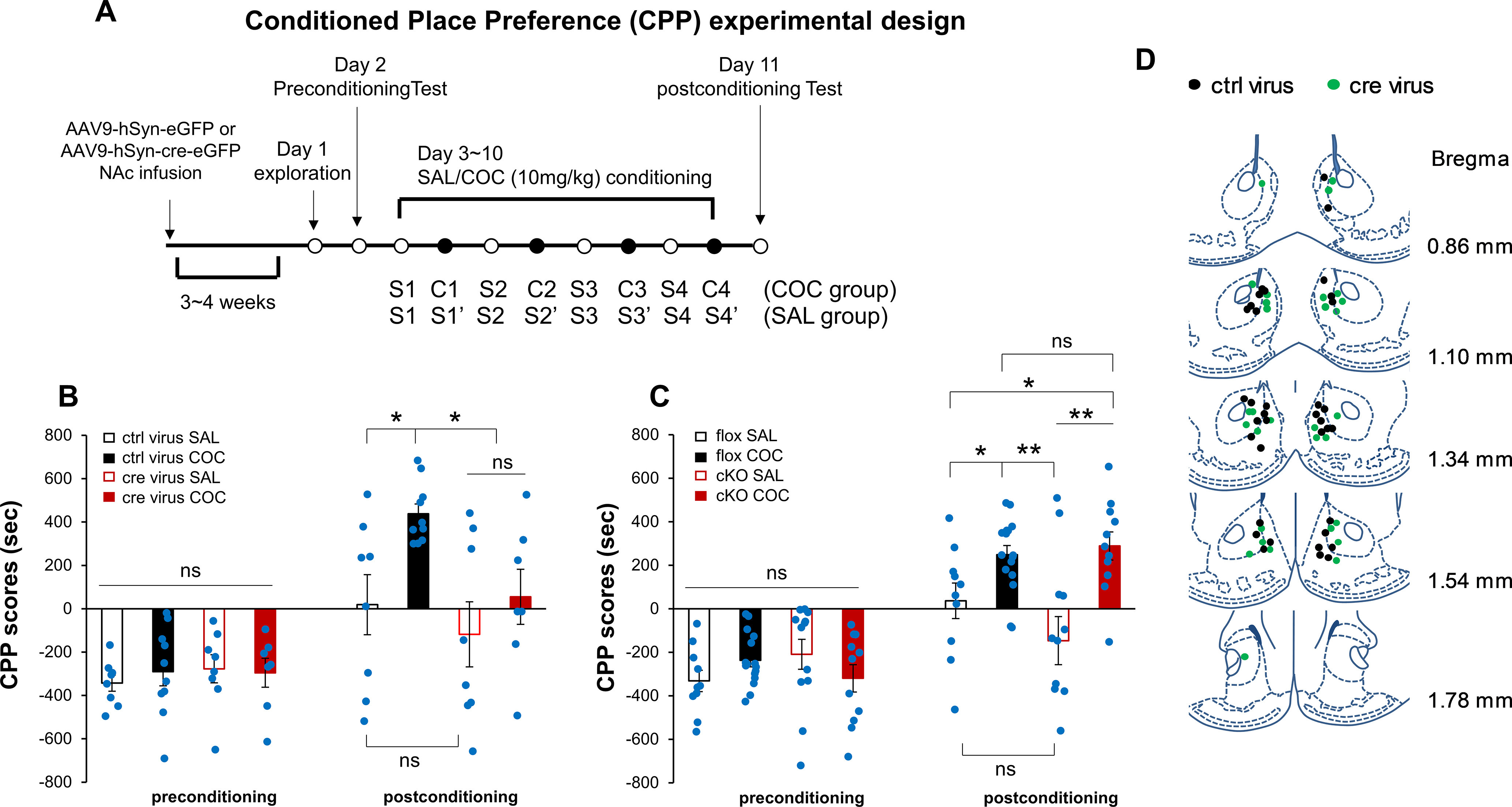
Conditioned Place Preference (CPP) induced by cocaine was impaired in the mice with NCS-Rapgef2 KO in the NAc. A, Experimental design for CPP. A shuttle box with a door that can be opened to allow the animal free access to the two chambers was used for CPP. The two chambers differed on several sensory and environmental properties. Animals received 8 d/four sets of cocaine and saline-pairing: for cocaine group, pair one chamber with cocaine, pair opposite one with saline; for saline group, pair both sides with saline. Animals were tested for the place preference before drug pairing and postconditioning by recording the amount of time they spent in each chamber. B, CPP scores for mice with NCS-Rapgef2 KO in the NAc compared with the corresponding controls. N = 7-10 animals per group. *p < 0.05 (three-way ANOVA followed by post hoc all pairwise multiple comparison Bonferroni test). C, CPP scores for Camk2α-cre+/−::Rapgef2cko/cko (cKO) mice compared with the corresponding controls Rapgef2cko/cko (flox). N = 10-16 animals per group. *p < 0.05; **p < 0.001; three-way ANOVA followed by post hoc all pairwise multiple comparison Bonferroni test. Columns and bars represent mean ± sem and scattered points represent individual values. D, Schematic representation of the injection sites in the NAc for the mice used for CPP test. N = 18 animals for ctrl virus injection: n = 8 animals for saline group, n = 10 animals for cocaine group. N = 15 animals for cre virus injection: n = 8 animals for saline group, n = 7 animals for cocaine group. ns, not significant.
Parcellation of cAMP signaling to ERK and CREB in NAc
Several models for intracellular signaling underlying dopamine-dependent ERK activation in D1 dopaminoceptive neurons emphasize that the sole cAMP effector for this process is PKA, both because inhibition of ambient ERK phosphatase activity can be achieved through PKA-dependent D1 signaling and because previously no direct pathway for ERK activation by cAMP elevation has been identified in neurons. To test whether or not the activation of ERK associated with upregulation of Egr-1/Zif268 and locomotor sensitization/place preference conditioning by cocaine involved activation of both PKA and NCS-Rapgef2, we quantified phospho-ERK and phospho-CREB, as surrogates for NCS-Rapgef2 and PKA activation, respectively, after cocaine treatment in Rapgef2cko/cko control and NCS-Rapgef2-deficient mice. As shown in Figure 8A, B, NCS-Rapgef2 KO in the NAc caused loss of ERK phosphorylation after cocaine treatment, whereas cocaine-dependent CREB phosphorylation was preserved (two-way ANOVA by genotype and drug treatment, for phospho-ERK: drug effect F(1,9) = 27.067, p < 0.001; genotype effect F(1,9) = 17.114, p = 0.003; drug × genotype interaction F(1,9) = 22.667, p = 0.001; phospho-CREB induction only showed drug effect F(1,9) = 84.582, p < 0.001; no genotype effect F(1,9) = 0.173, p = 0.687; no genotype × drug interaction F(1,9) = 0.058, p = 0.815). Thus, PKA signaling is apparently intact in mice in which both D1-MSN ERK activation, and acquisition of cocaine locomotor sensitization and CPP are lost after deletion of NCS-Rapgef2 in NAc.
Figure 8.
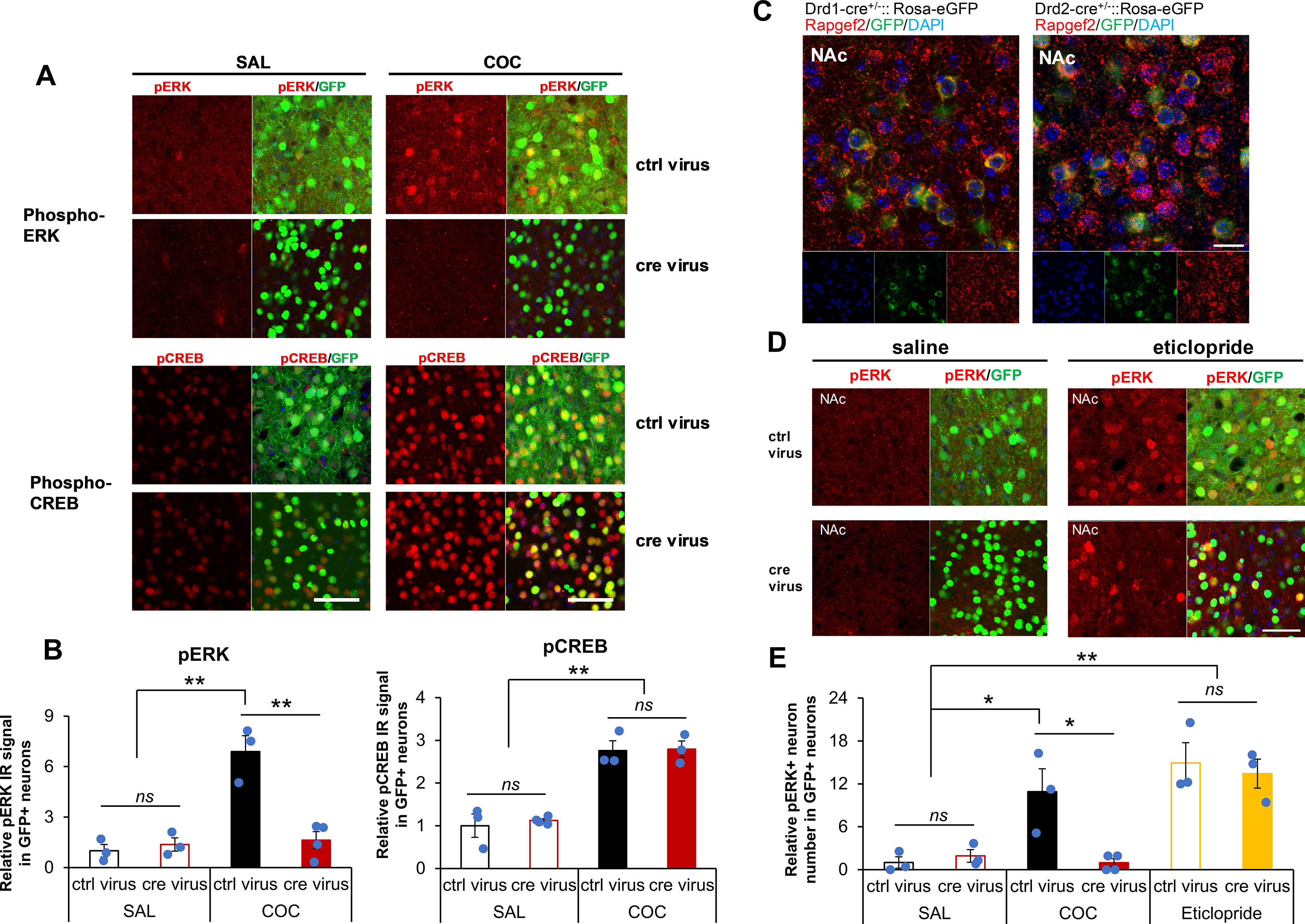
Cocaine treatment resulted in ERK phosphorylation in an NCS-Rapgef2-dependent manner, whereas CREB phosphorylation was unaffected by NCS-Rapgef2 ablation. A, AAV9-hSyn-eGFP (ctrl virus) or AAV9-hSyn-cre-eGFP (cre virus) was injected into both sides of NAc of Rapgef2cko/cko mice. After 4 weeks, saline or cocaine (20 mg/kg) was injected (i.p.), and animals were perfused 10 min later for phospho-ERK and phospho-CREB immunohistochemistry. The phospho-ERK activation was significantly increased in the NAc with ctrl virus after cocaine treatment, but not in the NAc with cre virus; phospho-CREB elevation induced by cocaine treatment was not affected by NCS-Rapgef2 ablation in the NAc. Scale bars: 50 µm. B, Phospho-ERK and phospho-CREB IR signals in AAV-targeted neurons in the NAc were quantified using Fiji ImageJ (detailed information in Methods and Materials, n = 3 or 4 animals per group). Mean gray value of phospho-ERK and phospho-CREB IR intensity in GFP-positive neurons was measured. Relative phospho-ERK IR signal of each animal was normalized by the average value from the control virus-injected group that was administered saline. **p < 0.001 (two-way ANOVA followed by post hoc Bonferroni test). C, NCS-Rapgef2 is expressed in both D1 and D2 MSNs in NAc. NCS-Rapgef2 immunohistochemistry was performed using brain sections from Drd1-cre+/−::Rosa-eGFP or Drd2-cre+/−::Rosa-eGFP mice. NCS-Rapgef2 IR signal (red) was seen in either D1 MSNs, indicated by GFP IR signal (green) in the NAc of Drd1-cre+/−::Rosa-eGFP mice, or in D2 MSNs indicated by GFP IR signal (green) in the NAc of Drd2-cre+/−::Rosa-eGFP mice. Scale bars, 20 µm. D, Phospho-ERK induction in presumptive D2-MSNs in NAc after in vivo eticlopride (eti) administration is not NCS-Rapgef2-dependent. Phospho-ERK activation in NAc was measured in NAc of Rapgef2cko/cko mice in which either AAV9-hSyn-eGFP (ctrl virus) or AAV9-hSyn-cre-eGFP (cre virus) was injected into NAc; and 4 weeks later, mice were treated with either saline or the D2R antagonist eticlopride (2 mg/kg, i.p., 15 min). Scale bars, 50 µm. E, Phospho-ERK-positive neurons with GFP-positive (pERK+ and GFP+) in NAc were quantified after saline or eticlopride treatment and compared with cocaine treatment. Relative number of phospho-ERK-positive neurons in NAc of each animal was normalized by the average value from saline-treated Rapgef2cko/cko (flox) mice with ctrl virus. N = 3 or 4 animals per group. *p < 0.05; **p <0.001; two-way ANOVA followed by post hoc Bonferroni test. Columns and bars represent mean ± sem and scattered points represent individual values. ns, not significant.
We note that both D1 and D2 MSNs express NCS-Rapgef2 (Fig. 8C). To determine whether ERK activation in D2 MSNs requires NCS-Rapgef2, Rapgef2cko/cko mice with NCS-Rapgef2 ablation in the NAc by AAV-Syn-cre-eGFP infusion were treated with the D2 antagonist eticlopride, which caused phospho-ERK induction in D2-MSN unaffected by ablation of NCS-Rapgef2 expression (Fig. 8D,E; two-way ANOVA indicated significant drug effect (F(2,13) = 22.128, p < 0.001), genotype effect (F(1,13) = 5.09, p = 0.042), and drug × genotype interaction (F(2,13) = 4.672, p = 0.03. Post hoc Bonferroni test: ctrl virus versus cre virus within eticlopride treatment, p = 0.594; ctrl virus versus cre virus within cocaine treatment, p = 0.002. For control virus, COC versus SAL, p = 0.01; eticlopride versus SAL, p < 0.001; eticlopride versus COC, p = 0.497. For cre virus, COC versus SAL, p = 1; eticlopride versus SAL, p = 0.003; eticlopride versus COC, p < 0.001).
Discussion
Activation of ERK in vivo by D1 agonists, or the psychomotor stimulants cocaine and amphetamine, in D1 neurons is well established (Valjent et al., 2000; Gutierrez-Arenas et al., 2014; Jin et al., 2019). Expression of locomotor sensitization and CPP after cocaine administration is ERK-dependent (Brami-Cherrier et al., 2005; Miller and Marshall, 2005; Ferguson et al., 2006; Valjent et al., 2006a,b; Fasano et al., 2009), and is linked to ERK-dependent transcription of the egr1 gene and expression of Egr-1/Zif268 protein in dopaminoceptive neurons in striatum and cerebral cortex (Valjent et al., 2006c; Gangarossa et al., 2011).
Whether dopamine signaling to ERK in D1 neurons is direct, or indirect via modulation of ERK activation by other neurotransmitters including glutamate (Valjent et al., 2005), has been an unresolved question. It has been hypothesized that glutamatergic activation of a Ras-signaling pathway for ERK phosphorylation is augmented by D1-dependent inhibition of ERK dephosphorylation, shifting the balance between ERK activation/phosphorylation and dephosphorylation toward phospho-ERK elevation and stimulation of ERK-dependent cellular activation events, including egr1 gene transcription (Valjent et al., 2005; Lu et al., 2006; Santini et al., 2007, 2012; Zhang et al., 2010). This mechanism has been favored because striatal dopaminoceptive neurons possess a PKA- and dopamine responsive protein phosphatase (DARPP-32), which disinhibits the striatal-enriched phosphatase STEP, leading to inhibition of the putative proximate ERK phosphatase PP1 (Valjent et al., 2005; Sun et al., 2007; Gutierrez-Arenas et al., 2014). However, differences between D1 signaling to ERK in ventral and dorsal striatum (Gerfen et al., 2008) have occasioned a search for a direct intracellular signaling pathway for ERK phosphorylation on dopamine-stimulated elevation of intracellular cAMP. Indeed, G protein-mediated cAMP pathway upregulation has been hypothesized as a common mechanism of adaptation to chronic administration of drugs of abuse (Nestler, 2016), and ERK activation in dopamine D1 receptor-expressing neurons in mesocorticolimbic projection areas, including both medial PFC and NAc, has been associated with drug-induced neuroadaptations (Valjent et al., 2000, 2005; Adams and Sweatt, 2002).
We have recently identified intracellular cAMP-dependent signaling downstream of Gs-coupled receptors in neurons and endocrine cells via parallel and insulated (parcellated) pathways initiated by the cAMP effectors PKA, Epac2, and NCS-Rapgef2. These effectors activate distinct but coordinated cellular tasks. Thus, in neuroendocrine NS-1 cells, PKA signaling to CREB mediates cell survival and neuron-specific gene expression; Epac2 signaling to the MAP kinase p38 mediates growth arrest; and NCS-Rapgef2 signaling to the MAP kinase ERK mediates neuritogenesis (Emery et al., 2013, 2014, 2017a). As the ERK pathway is associated with neuritogenesis in NS-1 cells, and similar morphologic changes occur via D1 signaling in NAc after cocaine (Lee et al., 2006), we hypothesized that the cAMP→NCS-Rapgef2→ERK pathway might link D1 receptor activation in response to cocaine, to ERK activation and downstream events involved in cellular plasticity causing behavioral effects of repeated psychomotor stimulant administration.
The demonstration that NCS-Rapgef2 is required for Egr-1/Zif268 induction by cAMP in neuroendocrine cells, coupled with our previous finding of NCS-Rapgef2 dependence for dopamine/D1 activation of neuritogenesis in these cells, suggests the existence of a direct dopamine→D1→cAMP→NCS-Rapgef2→ERK→Egr-1/Zif268 signaling pathway with potential relevance to cellular signaling in D1-MSNs in vivo. A role for NCS-Rapgef2 in mediating the effects of increased synaptic dopamine after cocaine administration is further supported by the localization of NCS-Rapgef2 to postsynaptic elements at dopaminergic synapses, and by NCS-Rapgef2-dependent activation of ERK by the D1 agonist SKF81297 in NAc brain slices ex vivo, as reported here. Finally, we have studied the behavioral effects of cocaine in mice in which NCS-Rapgef2 expression was eliminated in various brain regions (mPFC, BLA, and NAc) in which dopamine acts at D1 receptors. Loss of NCS-Rapgef2 expression in mPFC and BLA excitatory neurons eliminated ERK activation by both SKF81297 and cocaine in these brain regions, without affecting cocaine-induced locomotor sensitization, or CPP. Elimination of NCS-Rapgef2 expression in NAc correspondingly abolished ERK activation by cocaine or D1 agonist, and also cocaine-induced locomotor sensitization and CPP. This occurred without affecting cocaine-induced activation of CREB phosphorylation in MSNs. It remains to be tested whether or not NCS-Rapgef2-dependent signaling in either mPFC or BLA is required for cocaine-dependent behaviors not studied here, such as relapse (Rebec and Sun, 2005).
These findings have several implications for understanding the role of cAMP in mediating the postsynaptic actions of dopamine and subsequent dopamine-dependent behaviors, and for defining the brain circuits in which dopamine signaling is critical for the various behavioral consequences of acute and chronic cocaine ingestion.
Cocaine reinstatement depends on different cAMP-dependent pathways as a function of the reinstatement stimulus: it is CREB-dependent for cocaine itself, and ERK-dependent for restraint stress (Kreibich and Blendy, 2004; Briand and Blendy, 2013). These findings accord well with our observation of parcellation of cAMP signaling to ERK and CREB through NCS-Rapgef2 and PKA, respectively, in cultured neuroendocrine cell lines (Emery et al., 2014). The present report demonstrates that parcellation extends to D1-dependent activation of Egr-1/Zif268 via ERK independently of CREB in NS-1 cells. Dopamine-dependent activation of ERK is likewise NCS-Rapgef2-dependent, whereas signaling to CREB is not. This observation has allowed us to determine that activation of ERK, apparently independently of PKA/CREB, and in dopaminoceptive neurons of the NAc rather than mPFC or BLA, is required for the behavioral effects of repeated cocaine administration.
ERK signaling itself is required for the sensitized locomotor, but not the initial locomotor effects of cocaine administration (Valjent et al., 2006b). NCS-Rapgef2, however, may be an even more practical target for mitigation of the neuroplastic effects of cocaine that underlie its addictive potential, since it represents a single conditional input to ERK activation (see model, Fig. 9). A related member of the protein family present in brain, Rapgef6, mediates a different set of memory-related functions in amygdala and hippocampus (Levy et al., 2015); and interestingly, both Rapgef2 and Rapgef6 are genetically linked to susceptibility to schizophrenia (Maeta et al., 2018). The specialized behavioral functions of Rapgefs in the CNS, and the specific pharmacological and behavioral effects of NCS-Rapgef2 demonstrated here, suggest that NCS-Rapgef2 merits consideration as a target for other reward-related psychopathology in addition to addiction.
Figure 9.

Proposed direct model for dopamine-dependent ERK activation in D1-dopaminoceptive neurons. Cocaine acts by increasing synaptic dopamine leading to ERK activation in NAc, required for locomotor sensitization and CPP. It has previously been proposed that D1 receptor activation affects ERK activity only indirectly, via PKA- and DARPP-32/STEP-mediated inhibition of ERK dephosphorylation (Svenningsson et al., 2004; Valjent et al., 2005), whereas direct ERK activation itself occurs in the D1 MSNs only via NMDAR-dependent glutamatergic signaling (Pascoli et al., 2011; Cahill et al., 2014) through calcium-sensitive Ras-guanine nucleotide releasing factor (Ras-GRF1) (Farnsworth et al., 1995; Fasano et al., 2009). We propose here a more parsimonious mechanism for ERK-dependent cocaine-induced dopaminergic signaling, in which cAMP elevation by dopamine in D1-MSNs results in parcellation of signaling between ERK and CREB with separate cellular consequences under the control of each pathway. An indirect modulatory role for PKA-dependent ERK phosphatase inhibition after psychomotor stimulant administration (Valjent et al., 2005; Sun et al., 2007), and additional ERK regulation by glutamatergic input to D1 dopaminoceptive neurons in the context of cellular plasticity underlying cocaine addiction (Valjent et al., 2000; Park et al., 2013; Cahill et al., 2014; Pascoli et al., 2014) is not contradicted by this model. We posit that D1 receptor activation, and cAMP elevation, in D1 MSNs likely results in parallel effects on ERK, both directly via NCS-Rapgef2 and indirectly via PKA with the effects of cocaine requiring multiple necessary, but perhaps individually insufficient inputs activated by dopamine, that converge on D1-MSN ERK phosphorylation. These include PKA/DARPP/STEP/PP1 (Svenningsson et al., 2004), PKA/RasGRP2/Rap1 (Nagai et al., 2016a,b), a NMDAR-dependent ras activation (Fasano et al., 2009; Pascoli et al., 2011), and NCS-Rapgef2/Rap1/B-Raf/MEK.
Convergent activation of ERK activation in D1-MSNs through direct (NCS-Rapgef2-mediated) and indirect (PKA-mediated) dopaminergic signaling, and via glutamatergic signaling, provides a mechanistically complete picture of D1-MSN cellular plasticity during repeated exposure to psychomotor stimulants (Fig. 9). It has implications for understanding, and intervening in, psychomotor stimulant addiction. It is known that complete blockade of D1 signaling, as occurs in D1 receptor-deficient (Xu et al., 1994) or D1-MSN ERK-deleted mice (Brami-Cherrier et al., 2005; Ferguson et al., 2006) abolishes cocaine-induced locomotor activation altogether. Inhibition of either the NCS-Rapgef2 or PKA parcellated signaling pathways may selectively block different subsets of learned behaviors occurring during chronic cocaine administration.
Cocaine administration causes ERK activation predominantly, if not exclusively, in D1- compared with D2-MSNs in the ventral striatum (Valjent et al., 2005; Bertran-Gonzalez et al., 2008), and D1 receptor-expressing neurons play a predominant role in cocaine sensitization and reward learning (see Baik, 2013, and references therein). Recent studies using optogenetic manipulation and in vivo calcium imaging confirm that activation of D1 neurons promotes the formation of cocaine reward (Lobo et al., 2010; Kravitz et al., 2012; Chandra et al., 2013; Calipari et al., 2016). NCS-Rapgef2 is expressed in both D1 and D2 neurons. ERK activation does not occur in D2-MSNs after cocaine administration but is activated in this neuronal population by the D2 receptor antagonist eticlopride, a response that is not impaired in NCS-Rapgef2-deficient mice (Fig. 8). Thus, the NCS-Rapgef2-dependent activation of ERK in D1-MSN signaling documented here is likely to be the major action of this signaling molecule in mediating the cocaine-dependent behaviors examined here. D1- and D2-MSNs in NAc can drive both reward and aversion, depending on their stimulation pattern (Soares-Cunha et al., 2020): it remains to be determined whether or not NCS-Rapgef2 signaling in D2 neurons contributes to these other aspects of NAc function.
Additional questions remain: what is the role of the NCS-Rapgef2 pathway in D1 signaling outside of the NAc? Might signaling through NCS-Rapgef2 mediate cocaine relapse associated with dopamine neurotransmission in mPFC (Miller and Marshall, 2005; Lu et al., 2006; Girault et al., 2007)? What might be the role(s) of CREB-dependent signaling (Shaywitz and Greenberg, 1999), spared on abrogation of NCS-Rapgef2, in the D1-MSN response to cocaine? Finally, might it be possible to develop brain-permeant NCS-Rapgef2 antagonists of sufficient potency and selectivity for use in vivo? If dendritic spinogenesis in D1-MSNs is the morphologic correlate of Rapgef2-dependent ERK signaling to Egr-1/Zif268 resulting in neuritogenesis in NS-1 cells, it might be supposed that blocking this process could affect not only short-term effects of repeated cocaine administration, including locomotor sensitization and cocaine preference, but also longer-term effects of cocaine-seeking behavior associated with altered D1-MSN spine density (Lee et al., 2006; Ren et al., 2010).
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institute of Mental Health Intramural Research Program, Project 1ZIAMH002386 to L.E.E. and 1ZIAMH002497 to C.R.G. We thank Haiying Zhang for mouse colony management and genotyping; the NIMH Rodent Behavior Core for expert guidance with behavioral experiments; and Dr. Kazu Nakazawa (Southern Research Institute) for helpful comments.
References
- Adams JP, Sweatt JD (2002) Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol 42:135–163. 10.1146/annurev.pharmtox.42.082701.145401 [DOI] [PubMed] [Google Scholar]
- Baik JH. (2013) Dopamine signaling in reward-related behaviors. Front Neural Circuits 7:152 10.3389/fncir.2013.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Herve D, Valjent E, Girault JA (2008) Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci 28:5671–5685. 10.1523/JNEUROSCI.1039-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J (2005) Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci 25:11444–11454. 10.1523/JNEUROSCI.1711-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand LA, Blendy JA (2013) Not all stress is equal: CREB is not necessary for restraint stress reinstatement of cocaine-conditioned reward. Behav Brain Res 246:63–68. 10.1016/j.bbr.2013.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill E, Salery M, Vanhoutte P, Caboche J (2014) Convergence of dopamine and glutamate signaling onto striatal ERK activation in response to drugs of abuse. Front Pharmacol 4:172 10.3389/fphar.2013.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Pena CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, Deisseroth K, Nestler EJ (2016) In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci USA 113:2726–2731. 10.1073/pnas.1521238113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra R, Lenz JD, Gancarz AM, Chaudhury D, Schroeder GL, Han MH, Cheer JF, Dietz DM, Lobo MK (2013) Optogenetic inhibition of D1R containing nucleus accumbens neurons alters cocaine-mediated regulation of Tiam1. Front Mol Neurosci 6:13. 10.3389/fnmol.2013.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Eiden LE (2012) Signaling through the neuropeptide GPCR PAC1 induces neuritogenesis via a single linear cAMP- and ERK-dependent pathway using a novel cAMP sensor. FASEB J 26:3199–3211. 10.1096/fj.11-203042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery A, Eiden MV, Mustafa T, Eiden LE (2013) GPCR-Gs signaling to ERK is controlled by the cAMP-sensing guanine nucleotide exchange factor NCS/Rapgef2 in neuronal and endocrine cells. Sci Signal 6:ra51. 10.1126/scisignal.2003993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Eiden MV, Eiden LE (2014) Separate cyclic AMP sensors for neuritogenesis, growth arrest, and survival of neuroendocrine cells. J Biol Chem 289:10126–10139. 10.1074/jbc.M113.529321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Xu W, Eiden MV, Eiden LE (2017a) Guanine nucleotide exchange factor Epac2-dependent activation of the GTP-binding protein Rap2A mediates cAMP-dependent growth arrest in neuroendocrine cells. J Biol Chem 292:12220–12231. 10.1074/jbc.M117.790329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AC, Alvarez RA, Eiden MV, Xu W, Simeon FG, Eiden LE (2017b) Differential pharmacophore definition of the cAMP binding sites of neuritogenic cAMP sensor-Rapgef2, protein kinase A, and exchange protein activated by cAMP in neuroendocrine cells using an adenine-based scaffold. ACS Chem Neurosci 8:1500–1509. 10.1021/acschemneuro.6b00462 [DOI] [PubMed] [Google Scholar]
- Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376:524–527. 10.1038/376524a0 [DOI] [PubMed] [Google Scholar]
- Fasano S, D'Antoni A, Orban PC, Valjent E, Putignano E, Vara H, Pizzorusso T, Giustetto M, Yoon B, Soloway P, Maldonado R, Caboche J, Brambilla R (2009) Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) controls activation of extracellular signal-regulated kinase (ERK) signaling in the striatum and long-term behavioral responses to cocaine. Biol Psychiatry 66:758–768. 10.1016/j.biopsych.2009.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE (2006) Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology 31:2660–2668. 10.1038/sj.npp.1301014 [DOI] [PubMed] [Google Scholar]
- Fernando AB, Robbins TW (2011) Animal models of neuropsychiatric disorders. Annu Rev Clin Psychol 7:39–61. 10.1146/annurev-clinpsy-032210-104454 [DOI] [PubMed] [Google Scholar]
- Funahashi Y, Ariza A, Emi R, Xu Y, Shan W, Suzuki K, Kozawa S, Ahammad RU, Wu M, Takano T, Yura Y, Kuroda K, Nagai T, Amano M, Yamada K, Kaibuchi K (2019) Phosphorylation of Npas4 by MAPK regulates reward-related gene expression and behaviors. Cell Rep 29:3235–3252.e3239. 10.1016/j.celrep.2019.10.116 [DOI] [PubMed] [Google Scholar]
- Gangarossa G, Di Benedetto M, O'Sullivan GJ, Dunleavy M, Alcacer C, Bonito-Oliva A, Henshall DC, Waddington JL, Valjent E, Fisone G (2011) Convulsant doses of a dopamine D1 receptor agonist result in Erk-dependent increases in Zif268 and Arc/Arg3.1 expression in mouse dentate gyrus. PLoS One 6:e19415 10.1371/journal.pone.0019415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, Brown P (2002) D1 dopamine receptor hypersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP Kinase. J Neurosci 22:5042–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, Worley P (2008) Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J Neurosci 28:7113–7120. 10.1523/JNEUROSCI.3952-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, Heintz N (2013) GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80:1368–1383. 10.1016/j.neuron.2013.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Herve D (2007) ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol 7:77–85. 10.1016/j.coph.2006.08.012 [DOI] [PubMed] [Google Scholar]
- Gutierrez-Arenas O, Eriksson O, Kotaleski JH (2014) Segregation and crosstalk of D1 receptor-mediated activation of ERK in striatal medium spiny neurons upon acute administration of psychostimulants. PLoS Comput Biol 10:e1003445 10.1371/journal.pcbi.1003445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien ML, Khan AS, Ariansen JL, Cheer JF, Phillips PE, Wassum KM, Wightman RM (2005) Real-time measurement of dopamine fluctuations after cocaine in the brain of behaving rats. Proc Natl Acad Sci USA 102:10023–10028. 10.1073/pnas.0504657102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, Nakanishi S (2010) Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66:896–907. 10.1016/j.neuron.2010.05.011 [DOI] [PubMed] [Google Scholar]
- Hutton SR, Otis JM, Kim EM, Lamsal Y, Stuber GD, Snider WD (2017) ERK/MAPK signaling is required for pathway-specific striatal motor functions. J Neurosci 37:8102–8115. 10.1523/JNEUROSCI.0473-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang SZ, Xu W, Emery AC, Gerfen CR, Eiden MV, Eiden LE (2017) NCS-Rapgef2, the protein product of the neuronal Rapgef2 gene, is a specific activator of D1 dopamine receptor-dependent ERK phosphorylation in mouse brain. eNeuro 4:e0248-17.2017 10.1523/ENEURO.0248-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DZ, Mao LM, Wang JQ (2019) Amphetamine activates non receptor tyrosine kinase Fyn and stimulates ERK phosphorylation in the rat striatum in vivo. Eur J Pharmacol 843:45–54. 10.1016/j.ejphar.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, Kreitzer AC (2012) Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 15:816–818. 10.1038/nn.3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreibich AS, Blendy JA (2004) cAMP response element-binding protein is required for stress but not cocaine-induced reinstatement. J Neurosci 24:6686–6692. 10.1523/JNEUROSCI.1706-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laakso A, Mohn AR, Gainetdinov RR, Caron MG (2002) Experimental genetic approaches to addiction. Neuron 36:213–228. 10.1016/S0896-6273(02)00972-8 [DOI] [PubMed] [Google Scholar]
- Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P (2006) Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci USA 103:3399–3404. 10.1073/pnas.0511244103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RJ, Kvajo M, Li Y, Tsvetkov E, Dong W, Yoshikawa Y, Kataoka T, Bolshakov VY, Karayiorgou M, Gogos JA (2015) Deletion of Rapgef6, a candidate schizophrenia susceptibility gene, disrupts amygdala function in mice. Transl Psychiatry 5:e577 10.1038/tp.2015.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Nestler EJ (2011) The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat 5:41 10.3389/fnana.2011.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ (2010) Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330:385–390. 10.1126/science.1188472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y (2006) Role of ERK in cocaine addiction. Trends Neurosci 29:695–703. 10.1016/j.tins.2006.10.005 [DOI] [PubMed] [Google Scholar]
- Maeta K, Hattori S, Ikutomo J, Edamatsu H, Bilasy SE, Miyakawa T, Kataoka T (2018) Comprehensive behavioral analysis of mice deficient in Rapgef2 and Rapgef6, a subfamily of guanine nucleotide exchange factors for Rap small GTPases possessing the Ras/Rap-associating domain. Mol Brain 11:27 10.1186/s13041-018-0370-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R (2002) Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34:807–820. 10.1016/S0896-6273(02)00716-X [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF (2005) Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron 47:873–884. 10.1016/j.neuron.2005.08.006 [DOI] [PubMed] [Google Scholar]
- Nagai T, Yoshimoto J, Kannon T, Kuroda K, Kaibuchi K (2016a) Phosphorylation signals in striatal medium spiny neurons. Trends Pharmacol Sci 37:858–871. 10.1016/j.tips.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Nagai T, Nakamuta S, Kuroda K, Nakauchi S, Nishioka T, Takano T, Zhang X, Tsuboi D, Funahashi Y, Nakano T, Yoshimoto J, Kobayashi K, Uchigashima M, Watanabe M, Miura M, Nishi A, Kobayashi K, Yamada K, Amano M, Kaibuchi K (2016b) Phosphoproteomics of the dopamine pathway enables discovery of Rap1 activation as a reward signal in vivo. Neuron 89:550–565. 10.1016/j.neuron.2015.12.019 [DOI] [PubMed] [Google Scholar]
- Nestler EJ. (2016) Reflections on: “A general role for adaptations in G-Proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function.” Brain Res 1645:71–74. 10.1016/j.brainres.2015.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Luscher C (2019) The molecular basis of drug addiction: linking epigenetic to synaptic and circuit mechanisms. Neuron 102:48–59. 10.1016/j.neuron.2019.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Hu JH, Milshteyn A, Zhang PW, Moore CG, Park S, Datko MC, Domingo RD, Reyes CM, Wang XJ, Etzkorn FA, Xiao B, Szumlinski KK, Kern D, Linden DJ, Worley PF (2013) A prolyl-isomerase mediates dopamine-dependent plasticity and cocaine motor sensitization. Cell 154:637–650. 10.1016/j.cell.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Besnard A, Herve D, Pages C, Heck N, Girault JA, Caboche J, Vanhoutte P (2011) Cyclic adenosine monophosphate-independent tyrosine phosphorylation of NR2B mediates cocaine-induced extracellular signal-regulated kinase activation. Biol Psychiatry 69:218–227. 10.1016/j.biopsych.2010.08.031 [DOI] [PubMed] [Google Scholar]
- Pascoli V, Cahill E, Bellivier F, Caboche J, Vanhoutte P (2014) Extracellular signal-regulated protein kinases 1 and 2 activation by addictive drugs: a signal toward pathological adaptation. Biol Psychiatry 76:917–926. 10.1016/j.biopsych.2014.04.005 [DOI] [PubMed] [Google Scholar]
- Ravni A, Vaudry D, Gerdin MJ, Eiden MV, Falluel-Morel A, Gonzalez B, Vaudry H, Eiden LE (2008) A cAMP-dependent, PKA-independent signaling pathway mediating neuritogenesis through Egr1 in PC12 cells. Mol Pharmacol 73:1688–1708. 10.1124/mol.107.044792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Sun W (2005) Neuronal substrates of relapse to cocaine-seeking behavior: role of prefrontal cortex. J Exp Anal Behav 84:653–666. 10.1901/jeab.2005.105-04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Sun WL, Jiao H, Zhang D, Kong H, Wang X, Xu M (2010) Dopamine D1 and N-methyl-D-aspartate receptors and extracellular signal-regulated kinase mediate neuronal morphological changes induced by repeated cocaine administration. Neuroscience 168:48–60. 10.1016/j.neuroscience.2010.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (2000) The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95 [Suppl 2]:S91–S117. 10.1046/j.1360-0443.95.8s2.19.x [DOI] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ (2013) The brain reward circuitry in mood disorders. Nat Rev Neurosci 14:609–625. 10.1038/nrn3381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Feyder M, Gangarossa G, Bateup HS, Greengard P, Fisone G (2012) Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J Biol Chem 287:27806–27812. 10.1074/jbc.M112.388413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Herve D, Greengard P, Fisone G (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci 27:6995–7005. 10.1523/JNEUROSCI.0852-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861. 10.1146/annurev.biochem.68.1.821 [DOI] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, Calakos N (2008) Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J Neurosci 28:2681–2685. 10.1523/JNEUROSCI.5492-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares-Cunha C, de Vasconcelos NA, Coimbra B, Domingues AV, Silva JM, Loureiro-Campos E, Gasper R, Sotiropoulos I, Sousa N, Rodrigues AJ (2020) Nucleus accumbens medium spiny neurons subtypes signal both reward and aversion. Mol Psychiatry. 25:3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun WL, Zhou L, Hazim R, Quinones-Jenab V, Jenab S (2007) Effects of acute cocaine on ERK and DARPP-32 phosphorylation pathways in the caudate-putamen of Fischer rats. Brain Res 1178:12–19. 10.1016/j.brainres.2007.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P (2004) DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol 44:269–296. 10.1146/annurev.pharmtox.44.101802.121415 [DOI] [PubMed] [Google Scholar]
- Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, Lowell BB, Carlezon WA Jr, Bonci A (2017) Pathway- and cell-specific kappa-opioid receptor modulation of excitation-inhibition balance differentially gates D1 and D2 accumbens neuron activity. Neuron 93:147–163. 10.1016/j.neuron.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl GR, Koob GF, Cable J (2019) The neurobiology of addiction. Ann NY Acad Sci 1451:5–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J (2000) Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci 20:8701–8709. 10.1523/JNEUROSCI.20-23-08701.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102:491–496. 10.1073/pnas.0408305102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Herve D (2006a) Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci 7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corbille AG, Bertran-Gonzalez J, Herve D, Girault JA (2006b) Inhibition of ERK pathway or protein synthesis during reexposure to drugs of abuse erases previously learned place preference. Proc Natl Acad Sci USA 103:2932–2937. 10.1073/pnas.0511030103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Aubier B, Corbille AG, Brami-Cherrier K, Caboche J, Topilko P, Girault JA, Herve D (2006c) Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine. J Neurosci 26:4956–4960. 10.1523/JNEUROSCI.4601-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Bertran-Gonzalez J, Aubier B, Greengard P, Hervé D, Girault JA (2010) Mechanisms of locomotor sensitization to drugs of abuse in a two-injection protocol. Neuropsychopharmacology 35:401–415. 10.1038/npp.2009.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S (1994) Elimination of cocaine-induced hyperactivity and dopamine-mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell 79:945–955. 10.1016/0092-8674(94)90026-4 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kurup P, Xu J, Carty N, Fernandez SM, Nygaard HB, Pittenger C, Greengard P, Strittmatter SM, Nairn AC, Lombroso PJ (2010) Genetic reduction of striatal-enriched tyrosine phosphatase (STEP) reverses cognitive and cellular deficits in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA 107:19014–19019. 10.1073/pnas.1013543107 [DOI] [PMC free article] [PubMed] [Google Scholar]