

Abstract
Although Tau accumulation is clearly linked to pathogenesis in Alzheimer's disease and other Tauopathies, the mechanism that initiates the aggregation of this highly soluble protein in vivo remains largely unanswered. Interestingly, in vitro Tau can be induced to form fibrillar filaments by oxidation of its two cysteine residues, generating an intermolecular disulfide bond that promotes dimerization and fibrillization. The recently solved structures of Tau filaments revealed that the two cysteine residues are not structurally equivalent since Cys-322 is incorporated into the core of the fibril, whereas Cys-291 projects away from the core to form the fuzzy coat. Here, we examined whether mutation of these cysteines to alanine affects differentially Tau mediated toxicity and dysfunction in the well-established Drosophila Tauopathy model. Experiments were conducted with both sexes, or with either sex. Each cysteine residue contributes differentially to Tau stability, phosphorylation status, aggregation propensity, resistance to stress, learning, and memory. Importantly, our work uncovers a critical role of Cys-322 in determining Tau toxicity and dysfunction.
SIGNIFICANCE STATEMENT Cysteine-291 and Cysteine-322, the only two cysteine residues of Tau present in only 4-Repeat or all isoforms, respectively, have competing functions: as the key residues in the catalytic center, they enable Tau auto-acetylation; and as residues within the microtubule-binding repeat region are important not only for Tau function but also instrumental in the initiation of Tau aggregation. In this study, we present the first in vivo evidence that their substitution leads to differential consequences on Tau's physiological and pathophysiological functions. These differences raise the possibility that cysteine residues play a potential role in determining the functional diversity between isoforms.
Keywords: aggregation, cysteines, dysfunction, Tau protein, toxicity, ubiquitination
Introduction
Tau is a microtubule-associated protein that occurs mainly in neurons, crucial for the elongation of microtubule labile domains (Qiang et al., 2018) among several other physiological functions at the synapse and in the nucleus, as well as interactions with the plasma membrane and the actin cytoskeleton (Sotiropoulos et al., 2017). In the adult human brain, alternative splicing of a single-copy gene generates six Tau isoforms that differ by the number of N-terminal insertions and microtubule-binding repeats (MBRs). Tau has 85 potential phosphorylation sites that regulate its affinity for microtubules, with excess steady-state phosphorylation being associated with disease (Wang and Mandelkow, 2016).
Intracellular Tau aggregates are the defining feature of a group of neurodegenerative dementias called Tauopathies, presenting varying clinical symptoms (Arendt et al., 2016). In addition to serving as markers for differential diagnosis, Tau aggregates can mediate disease propagation (Peng et al., 2020) and serve as direct drivers of toxicity (Ballatore et al., 2007). In recent years, studies in animal models have shown that Tau oligomers correlate with synapse loss and behavioral deficits much better than the appearance of mature fibrils and prefibrillar events have gained considerable attention with respect to their toxic potential (Santacruz et al., 2005; Berger et al., 2007).
Tau presents conformational flexibility, rendering it highly soluble; and it is therefore surprising that it assembles into filaments (Wang and Mandelkow, 2016). It has been proposed that Tau fibrillization may be associated with folding events, which allow Tau monomers to acquire partially folded, β-sheet-enriched structures that further self-assemble to form highly ordered fibrils (Jeganathan et al., 2008), potentially mediated by two hexapeptide motifs in the MBR (von Bergen et al., 2005). In addition, cysteines appear capable of forming disulfide-linked dimers that accelerate the conformational conversion of Tau into fibrillary aggregates in vitro (Schweers et al., 1995; Bhattacharya et al., 2001) and compounds that target these residues prevent Tau aggregation (Soeda et al., 2015). However, the specific events leading in Tau aggregation in vivo are not yet established.
Of the two Tau cysteines, Cys-322 is present in all isoforms, whereas Cys-291 is found only in 4R species. A recent study revealed the ability of Tau to perform thiol/disulfide exchanges with tubulin and brought new insights into the role of these two residues in the correct localization of Tau on microtubules (Martinho et al., 2018). In the two C-shaped protofilaments constituting the Tau filaments from AD brains, Cys-322 is incorporated into the core of the fibril, whereas Cys-291 is in the disordered portion of the protein forming the “fuzzy coat” (Fitzpatrick et al., 2017). This was evidence that two Tau cysteines are not structurally equivalent. Based on this distinction, we aimed to determine whether they were equivalent or contributed differentially to Tau-mediated neuronal toxicity and dysfunction in vivo using the established Drosophila melanogaster Tauopathy model (Papanikolopoulou and Skoulakis, 2011).
Materials and Methods
Drosophila culture and strains
Flies were cultured in standard sugar-wheat flour food supplemented with soy flour and CaCl2 (Acevedo et al., 2007). Pan-neuronal transgene expression was achieved using the ElavC155-Gal4;Ras2-Gal4 driver (Robinow and White, 1988; Walker et al., 2006; Keramidis et al., 2020). The double driver was constructed using standard genetic crosses. The gstD-ARE:GFP (antioxidant response element [ARE] of the gstD gene) reporter transgenic line was a kind gift from Prof. D. Bohmann (University of Rochester). Experiments were performed at 25°C unless noted otherwise. To generate the new equivalently expressing transgenes within the same attp site, we used the UAS-htauFLAG-2N4R, which has been described previously (Kosmidis et al., 2010). The same cDNA was subcloned into pUAS.attB (Bischof et al., 2007) as EcoRI/XbaI fragment. The Cys mutants were generated by replacing Cys-291 and Cys-322 with Ala using the QuikChange XL site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. The mutagenic oligonucleotides for each mutant are shown below. The silent restriction site BamHI introduced for effective screening of positive clones appears underlined in italics, whereas the amino acid substitution is shown in bold. The mutagenic oligonucleotides for C291A 5′-GCAACGTCCAGTCCAAGGCTGGATCCAAGGATAATATC-3′ 5′- GATATTATCCTTGGATCCAGCCTTGGACTGGACGTTGC-3′ and for C322A 5′-CCTGAGCAAGGTGACCTCCAAGGCTGGATCCTTAGGCAACATCC-3′ 5′- GGATGTTGCCTAAGGATCCAGCCTTGGAGGTCACCTTGCTCAGG-3′ were annealed onto the pUAS.attB UAS-htauFLAG-2N4R plasmid template. For the generation of the double Cys mutant, the pUAS.attB UAS-htauC291A FLAG-2N4R served as a template for cloning with the C322A primers. The sequence of the mutants was confirmed by dsDNA sequencing (VBC-Biotech). Transgenic flies were generated by phiC31-mediated transgenesis by BestGene. DNAs were injected into genomic landing site attP2 on the third chromosome (BDSC #8622).
RNA extraction and RT-PCR
Total RNA was extracted from Drosophila heads using TRI Reagent (Sigma Millipore) following the manufacturer's instructions. Reverse transcription reaction from DNase I-treated total RNA was conducted using SuperScript II Reverse Transcriptase (Invitrogen). Aliquots of 1 μg cDNA from each RT reaction were then subjected to PCR using the Go Taq Flexi DNA Polymerase (Promega). Semiquantitative PCR analyses were run using the following conditions: a denaturation step at 95°C for 10 min, followed by 28 cycles of denaturation at 95°C for 1 min, primer annealing at 62°C for 40 s, and primer extension at 72°C for 1 min. The ribosomal gene rp49 was used as a normalizer. The primers used were as follows: Tau-F:5′-CCCGCACCCCGTCCCTTCC-3′; Tau-R:5′-GATCTCCGCCCCGTGGTCTGTCTT-3′; rp49-F:5′-GATCGTGAAGAAGCGCAC-3′; and rp49-R: 5′-CTTCTTGAATCCGGTGGG-3′. Four independent experiments were performed. PCR products were analyzed by agarose gel electrophoresis, and quantification of gels was performed by scanning densitometry for the digital image analysis of PCR amplicons using the freely available ImageJ software.
Western blotting and antibodies
For Western blotting, adult female at 1-3 d after eclosion were homogenized in 1× Laemmli buffer (50 mm Tris, pH 6.8, 100 mm DTT, 5% 2-mercaptoethanol, 2% SDS, 10% glycerol, and 0.01% bromophenol blue), the extracts heated for 3 min at 95°C, centrifuged at 11,000 × g for 5 min, and separated in 10% SDS-acrylamide gels. For Western blotting with phospho-antibodies, adult male heads at 1-3 d after eclosion have equally been used. Proteins were transferred to PVDF membranes and probed with mouse monoclonal anti-Tau (5A6, Developmental Studies Hybridoma Bank), anti-Ub (P4D1, Santa Cruz Biotechnology), anti-GFP (B2, Santa Cruz Biotechnology), AT270, AT100, and AT8 from Thermo Fisher Scientific, and the polyclonal antibodies anti-pT212 (BioSource), anti-pS214 (BioSource), anti-pS262 (ProSci), and anti-pS396 (Cell Signaling). All Tau antibodies were used at 1:1000, whereas the appropriate anti-mouse or anti-rabbit HRP-conjugated secondary antibody was applied at 1:5000 dilution. Anti-Ub was used at 1:5000 dilution and anti-GFP at 1:1000 dilution. To normalize for sample loading, the membranes were concurrently probed with an anti-syntaxin (Syx) primary antibody (8C3, Developmental Studies Hybridoma Bank) at a 1:3000 dilution or anti-tubulin (E7, Developmental Studies Hybridoma Bank) at a 1:500 dilution. Proteins were visualized with chemiluminescence (Immobilon Crescendo, Millipore), and signals were quantified by densitometry with the Image Lab 5.2 program (Bio-Rad). Results were plotted as mean ± SEM from at least three independent experiments. The data were analyzed by standard parametric statistics (t tests) as indicated in the figure legends.
Proteasome inhibitor treatment
Young transgenic flies (2 d after eclosion) were cultured for 2 d at 25°C in standard fly food with or without 20 μm bortezomib (Sigma Millipore). After that, treated flies were transferred in normal food for 5 d (Tsakiri et al., 2017). Four female heads from each group were homogenized in 1× Laemmli sample buffer and analyzed by Western blot.
LC-MS/MS analysis
Fly heads were homogenized in lysis buffer consisting of 50 mm Tris HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100 supplemented with protease and phosphatase inhibitors. After centrifugation at 11,000 × g for 10 min, supernatants were incubated overnight at 4°C with anti-FLAG-coated agarose beads (Sigma Millipore) using a roller shaker. After washing the beads 3 times with 50 mm Tris HCl, 150 mm NaCl, pH 7.4, elution was performed under acidic conditions using 0.1 m glycine HCl, pH 3.5, following immediate neutralization of the sample according to the manufacturer's instructions. The eluent samples were subjected to the sp3 protein purification and digestion protocol (Hughes et al., 2019). Briefly, the immunoprecipitated eluates were subjected to alkylation with iodoacetamide at a final concentration of 100 mm and then mixed with 20 μg of a hydrophilic paramagnetic bead mixture at a final concentration of 50% ethanol. The samples were placed on a magnetic rack, and the supernatant was discarded. The beads were washed 3 times with 80% ethanol. The proteins were digested with trypsin/LysC mix (Promega) in 25 mm ammonium bicarbonate solution at an enzyme ratio of 1:50 overnight at 37 C on a shaking incubator. The next day, the supernatant was kept and vacuum-dried. Finally, the samples were solubilized in 0.1% formic acid (FA) in water.
LC conditions
The samples were preconcentrated on a PepMap C18 trapping column at a rate of 30 μl of Buffer A (0.1% FA in water) for 1 min. The samples were injected on a 50-cm-long PepMap column, and the LC gradient used was 8% Buffer B (0.1% FA in acetonitrile) to 40% in 81.5 min followed by an increase to 90% in 0.5 min. The gradient was kept for 2 min, and the column was equilibrated for 15 min before next injection.
MS conditions
A full MS scan was acquired on a Q Exactive Orbitrap HF-X mass spectrometer operated in the range of 390-1400 m/z using 60K resolving power with an AGC of 3 × 106 and max IT of 45 ms, followed by MS/MS scans of the 15 most abundant ions, using 15K resolving power with an Automatic Gain Control (AGC) of 2 × 105 and max Injection Time (IT) of 22 ms. All survey scans were internally calibrated using the 445.1200 background ion mass. Selected ions were then dynamically excluded from further selection during 30 s.
Data analysis
The spectra were analyzed by MaxQuant 1.6.14.0 (Tyanova et al., 2016) using the wild-type Tau 2N4R FASTA sequence and the mutated 2N4R Tau at positions 291 and 322. The search was looking for fully tryptic peptides potentially modified by ubiquitination of lysine residues, deamidation of asparagines and glutamines, oxidation of methionines, and with FDR filtering at 5% (peptide, PSM and protein level).
Microtubule-binding assay
Endogenous microtubules
Microtubule-binding experiments were based on established methods (Feuillette et al., 2010; Gorsky et al., 2017; Papanikolopoulou et al., 2019) that use Taxol-stabilized microtubules isolated from head extracts (50 fly heads) by ultracentrifugation at 100,000 × g for 1 h. Soluble and insoluble fractions were probed with anti-Tau 5A6 and E7 β-tubulin 97EF at 1:1000 dilution from Developmental Studies Hybridoma Bank.
Exogenous microtubules
Determination of Tau binding to exogenously added bovine microtubules was performed according to the manufacturer's instructions (Cytoskeleton). Briefly, 50 heads were homogenized in 175 μl lysis buffer consisting of 80 mm PIPES, pH 7.0, 2 mm MgCl2, and 0.5 mm EGTA, including protease and phosphatase inhibitors. Extracts were clarified with a low spin at 4000 × g for 5 min followed by a high spin of 7500 × g for 20 min at 4°C. Upon addition of Taxol at 20 μm, the supernatant was incubated with 10 μl of paclitaxel-stabilized in vitro polymerized microtubules (1 mg/ml) at room temperature for 30 min. Following incubation, fractions were loaded onto 300 μl of 50% glycerol gradient in 80 mm PIPES, pH 7.0, 2 mm MgCl2, 0.5 mm EGTA, 20 μm Taxol, and centrifuged at 110,000 × g for 45 min. The supernatant and pellet fractions were collected and analyzed by immunoblotting with 5A6 to estimate the amount of Tau bound to microtubules.
F-actin precipitation assay
Total F-actin has been isolated as in Papanikolopoulou et al. (2019). Briefly, after dissection, eight brains from each genotype were homogenized in 25 μl of 100 mm Na2HPO4–NaH2PO4 at pH 7.2, 2 mm ATP, and 2 mm MgCl2 buffer supplemented with phosphatase (Sigma Millipore) and protease (Thermo Fisher Scientific) inhibitor cocktails. Biotinylated phalloidin (Invitrogen) was added to a final concentration of 0.15 units per brain followed by precipitation with streptavidin-coupled Dynabeads (Invitrogen). The precipitated material and supernatant were probed with 5A6 (1:1000) and anti-actin (1:1000, Sigma Millipore).
Tau solubility assay
As described by Sealey et al. (2017), 10 fly heads were homogenized in 100 μl buffer (50 mm Tris-HCl, pH 7.4, 175 mm NaCl, 1 m sucrose, and 5 mm EDTA supplemented with protease and phosphatase inhibitors). The samples were then spun for 2 min at 1000 × g, and the supernatant was centrifuged at 186,000 × g for 2 h at 4°C. The supernatants were retained and regarded as the soluble S1 fraction. The resulting pellets were then resuspended in SDS buffer (50 mm Tris-HCl, pH 7.4, 175 mm NaCl, 5% SDS) and centrifuged for 2 h at 200,000 × g (25°C). The supernatants were collected as the SDS-soluble (S2). S1 and S2 fractions were diluted in 2× Laemmli buffer, boiled for 5 min, separated by SDS-PAGE, and analyzed by immunoblotting as described previously using 5A6 and 8C3 antibodies.
For the extraction of aggregates with Formic Acid (FA) as described previously (Feuillette et al., 2010; Papanikolopoulou and Skoulakis, 2015), flies were raised at 25°C and then aged for 10 d at 30°C. Thirty fly heads were homogenized in 100 μl RIPA buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 20 mm EDTA, 1% Nonidet-P40 supplemented with protease and phosphatase inhibitors). Samples were agitated for 1 h at 4°C and then centrifuged at 11,300 × g for 20 min at 4°C. The supernatants were retained and regarded as the RIPA fraction. Pellets were resuspended in RIPA buffer as a washing step and recentrifuged. The resulting pellets were then re-extracted with 50 μl of 70% FA. The supernatants were collected as the FA fraction after centrifugation at 11,300 × g for 20 min at 4°C. FA was removed by Speed-Vac centrifugation, and 50 μl of SDS buffer (240 mm Tris, pH 6.8, 6% SDS, 30% glycerol, and 0.06% bromophenol blue) was then added to the samples. RIPA and FA fractions were separated by SDS-PAGE and analyzed by immunoblotting as described previously (Papanikolopoulou and Skoulakis, 2015). MC1 antibody was a kind gift from P. Davies and used at 1:100.
Lifespan determination
Animals accumulating wt and mutant Tau protein under the control of the pan-neuronal ElavC155-Gal4;Ras2-Gal4 driver were raised at 25°C together with control driver heterozygotes. Groups of 20 young male flies (1-3 d old) were collected and were maintained at 29°C until they expired. Flies were transferred to fresh vials every 3 d. Two independent experiments with a total of 300 flies were assessed per genotype. Survival curves were compared using log-rank tests (JMP 7.1 statistical software package, SAS Institute Inc).
Paraquat and H2O2 sensitivity
Animals accumulating wt and mutant Tau protein under the control of the pan-neuronal ElavC155-Gal4;Ras2-Gal4 driver were raised at 25°C together with control driver heterozygotes. Oxidative stress tests were performed at 25°C using medium containing 10% sucrose, 1× PBS, 0.8% low melt agarose, and 5% H2O2. Twenty young males (1-3 d old) were transferred to fresh medium every 24 h (Svensson and Larsson, 2007). Paraquat feeding has been performed as described by Dias-Santagata et al. (2007). Groups of 20 male flies (1-3 d old) were fed 30 mm of methyl viologen (Acros Organics) in standard fly food. At least 300 flies were assessed per genotype.
Heat stress assay
Animals accumulating wt and mutant Tau protein under the control of the pan-neuronal ElavC155-Gal4;Ras2-Gal4 driver were raised at 25°C together with control driver heterozygotes. Groups of 30 young male flies were collected in vials containing fresh medium. The next day, flies were incubated at 40°C in empty vials, and the time until all individuals were paralyzed was recorded. Assays were videotaped for scoring, and at least 150 flies were assessed per genotype.
Behavioral analyses
Mixed sex population flies were trained with classical olfactory aversive conditioning protocols (Tully and Quinn, 1985) as previously described (Sealey et al., 2017; Papanikolopoulou et al., 2019). The aversive odors used for conditioning were benzaldehyde (5% v/v) and 3-octanol (50% v/v) diluted in isopropylmyristate (Fluka). Training and testing were conducted at 25°C and 75% relative humidity under dim red light. To assess learning, flies were tested immediately after a single training cycle consisting of 30 s Odor A with six 90 V shocks at 5 s interstimulus interval, 30 s air, and 30 s Odor B without reinforcement. For limited training, flies were tested immediately after a single training cycle consisting of 15 s Odor A with three 90 V shocks at 5 s interstimulus interval, 30 s air, and 15 s Odor B without reinforcement. For long-term memory (LTM), flies underwent five training cycles spaced at 15 min rest intervals, and tested 24 h later. For LTM–anesthesia-resistant memory (ARM), flies were submitted to five massed conditioning cycles, and memory was tested after 24 h. In each cycle, flies were exposed for 1 min to Odor A paired with twelve 90 V electric shocks at 5 s interstimulus interval, followed by 30 s of air and 1 min Odor B without reinforcement. To obtain animals for both learning and memory assays, UAS-hTau transgenes were crossed en masse with ElavC155-Gal4;Ras2-Gal4 driver at 25°C. Upon eclosion, they were separated in groups of 50-70 animals in vials and placed at 30°C overnight. All experiments were conducted in a balanced manner, where all genotypes involved in an experiment were tested per day. Data were analyzed parametrically with the JMP statistical package (SAS Institute) as described previously (Sealey et al., 2017; Papanikolopoulou et al., 2019).
Experimental design and statistical analysis
For all experiments, controls and experimental genotypes were tested in the same session in balanced design. The order of training and testing these genotypes was randomized. We required an experimental result to be significantly different from both genetic controls. Data are mean ± SEM. Data were analyzed parametrically with the JMP 7.1 statistical software package (SAS Institute) as described previously. Following initial ANOVA, planned multiple comparisons were performed using p = 0.05. The level of significance was adjusted for the experiment-wise error rate. Detailed results of all ANOVA and planned comparisons are reported in the text. Detailed results of all ANOVA and planned comparisons using the Least Squares Means (LSM) approach are reported in the text.
Results
Cys-322 plays a dominant role in regulating tau stability
To unravel the effect of each cysteine mutation in vivo, we used a site-directed integration strategy to generate transgenic fly lines expressing the WT human Tau 2N4R isoform, as well as its single (C291A and C322A) and double (C291A/C322A, hereafter called Cd) cysteine mutants. The attP/attB-targeted integration system has previously been shown to ensure comparable expression levels among transgenes (Markstein et al., 2008). However, despite comparable levels of transgene-encoded messenger RNAs (Fig. 1A, for C291A, C322A, and Cd, respectively: p = 0.931, p = 0.994, p = 0.996, n = 4), their protein levels appeared significantly altered compared with wt Tau (Fig. 1A, for C291A, C322A, and Cd, respectively: p = 0.839, p = 0.00053, p = 6.72e-12, n = 4). Mutation of Cys-322, but not of Cys-291, resulted in decreased Tau levels, suggesting that Cys-322 is one key residue involved in stability of the protein. Noteworthy, mutation of both cysteines led to a dramatic decrease of total Tau levels.
Figure 1.

Cys-322 substitution alters Tau stability. A, Left, Representative blot from head lysates of flies accumulating wt Tau pan-neuronally compared with similar lysates from site-specific Cys mutants. For the quantifications, total Tau levels (revealed with the 5A6 antibody) were normalized using Syx and shown as ratios of their means ± SEM relative to their respective levels in fly heads expressing wt Tau alone, which is arbitrarily set to 1. *p < 0.05, significantly reduced levels of the indicated mutant from wt Tau (Dunnett's). Right, Representative RT-PCR of Tau mRNA levels in flies expressing pan-neuronally the indicated Tau transgenes. The RP49 RNA served as an internal reference gene for the reaction and has been used as a normalization control for the quantifications. The normalized level of wt Tau for each quantification was fixed to 1. Error bars indicate mean ± SEM relative mRNA levels of mutant transgenes, over that of the wt Tau. B, Flies expressing pan-neuronally the Tau transgenes were treated with 20 μΜ of the proteasomal inhibitor bortezomib for 2 d and after transferred in normal food for 5 d to allow recovery of proteasome function. Lysates were immunoblotted with anti-ubiquitin to probe the accumulation of ubiquitinated proteins on inhibition of the proteasome. C, Representative Western blots demonstrate the Tau levels for each condition as described in B (untreated, treated with the inhibitor, and after recovery of proteasome function). For the quantifications below, levels of the protein were normalized using the Syx loading control and shown as a ratio of their means ± SEM relative to their respective levels in fly heads treated with the inhibitor, which is set to 1. *p < 0.05, significantly altered levels compared with +inh. #p < 0.05, significant differences between levels after the drug is removed and predrug levels (rec vs –inh) (each pair Student's t test). Detailed statistics can be found in the text.
It has previously been reported that mutation of both cysteines abolished the auto-acetylation activity of Tau (Cohen et al., 2013). Since Lys-directed modifications play key regulatory roles with respect to rates of Tau turnover through the ubiquitin-proteasome system (Min et al., 2010), we hypothesized that cysteine-deficient Tau may be more prone to ubiquitination, which subsequently could enhance its proteasomal degradation (Morris et al., 2015). To test our hypothesis, we first treated flies with the proteasome inhibitor bortezomib (Tsakiri et al., 2017) for 2 d, to block proteasome function as indicated by the accumulation of ubiquitinated proteins (Fig. 1B, +inh). This resulted in a twofold increase in the levels of all four Tau lines (Fig. 1C, –inh vs +inh, WT p = 0.00105, C291A p = 0.00798, C322A p = 5.66e-05, Cd p = 7.22e-05, n = 4). Five days after removing the inhibitor and hence reactivation of the proteasome, we measured the amount of Tau present in the cell. We detected persistently elevated wt Tau levels over the pretreated Tau levels (Fig. 1C, rec vs –inh, p = 0.0006, n = 4) with only 20% being degraded (+inh vs rec, p = 5e-10, n = 4). Similarly, a 20% reduction was observed for the C291A mutant (+inh vs rec, p = 9.64e-06, rec vs –inh, p = 0.0008, n = 4), 40% reduction for C322A (+inh vs rec, p = 1.8e-07, rec vs –inh, p = 0.0051, n = 4), and 50% reduction for the double mutant (+inh vs rec, p = 5.9e-05, n = 4), which reached its initial expression levels before addition of the inhibitor (rec vs –inh, p = 0.8805, n = 4).
To explore the biochemical mechanism underlying Tau destabilization induced by Cys substitution, we decided to perform mass spectrometry analysis for potential differences in the ubiquitination profile among the different Tau lines. The approach for the present investigation was to use FLAG immunoprecipitation to enrich for Tau and perform LC-MS/MS to localize ubiquitin modification to specific lysine residues. When the conjugated ubiquitin is cleaved with trypsin, it leaves two Gly residues on the modified lysine residues, generating a “ubiquitin signature peptide” (Peng et al., 2003). The LC-MS/MS analyses from four biological replicas yielded tryptic peptides from the Tau protein with a sequence coverage of ∼80%. It should be noted here that it is particularly challenging to identify ubiquitination sites because of the low abundance of ubiquitinated proteins under normal conditions in the cell mostly because they are rapidly degraded by the proteasome (Wilkinson et al., 1995; Mann and Jensen, 2003). In congruence, as shown in Table 1, for each biological replicate, the ratio of ubiquitinated/unmodified peptide for Lys-87, Lys-254, Lys-281, Lys-290, Lys-311, Lys-369, and Lys-370 is particularly low in all samples. In that table, zero denotes that peptides containing the ubiquitinated residue were not captured in the proteomic experiments. Interestingly, ubiquitinated peptides for Lys-331, which is the first lysine residue downstream Cys-322 (Fig. 2D), have been detected only for C322A and Cd mutants with relatively high ratios. Representative spectra of detected modified and unmodified peptide species for Lys-331 are shown in Figure 2B, E. To further validate this result, we decided to include in our analysis three additional biological and two technical replicas for wt Tau reaching 93% coverage of the protein sequence. Only in one sample, we have detected ubiquitinated peptides for Lys-331 (Fig. 2A) with the ratio of ubiquitinated/unmodified peptide being equal to 0.07 and 0.05 in the two technical replicas of this sample. Moreover, peptides of the sequence 322C(A)GSLGNIHHK331PGGGQVEVK340 containing two diglycine-modified lysines have been positively identified only in the samples of the C322A mutant (Table 1, Lys340; Fig. 2C). A final striking difference is the particularly high ratio of Lys280 ubiquitination observed only for the Cd mutant (Table 1; Fig. 2F) and not for the other Tau lines. Given the critical role of ubiquitination in Tau turnover (Petrucelli et al., 2004; Tan et al., 2008), the above differences in the ubiquitination profile likely account for the decreased stability of the C322A and Cd mutants. Because we focus on the potentially differential contribution of each Cys to Tau properties and pathogenicity and also because of the decreased stability and particularly low Cd protein levels, the double mutant was not included in subsequent analyses.
Table 1.
Relative ubiquitin occupancy ratios for the indicated lysine residues from each one of the four biological replicas per genotype
| Ratio ubiquitinated/unmodified peptide | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| K87 | K254 | K280 | K281 | K290 | K311 | K331 | K340 | K369 | K370 | |
| Tau | 0.01408 | 0.06801 | 0 | 0.12651 | 0.01316 | 0 | 0 | 0 | 0.080482 | 0.090157 |
| Tau | 0 | 0.0788 | 0 | 0.20541 | 0 | 0 | 0 | 0 | 0.13288 | 0.10792 |
| Tau | 0 | 0 | 0 | 0.09234 | 0.00753 | 0.52086 | 0 | 0 | 0.07066 | 0.27483 |
| Tau | 0 | 0 | 0 | 0.15705 | 0.01631 | 0.07942 | 0 | 0 | 0.10758 | 0.2004 |
| C291A | 0.01695 | 0.06085 | 0 | 0.16463 | 0.01826 | 0 | 0 | 0 | 0.089301 | 0.066477 |
| C291A | 0.01131 | 0 | 0 | 0.2876 | 0 | 0.20959 | 0 | 0 | 0.060651 | 0.053265 |
| C291A | 0 | 0 | 0 | 0.09248 | 0 | 0.5864 | 0 | 0 | 0.084816 | 0 |
| C291A | 0 | 0 | 0 | 0.04654 | 0 | 0.29649 | 0 | 0 | 0.04709 | 0.19023 |
| C322A | 0.01756 | 0 | 0 | 0 | 0 | 0.04257 | 0.73966 | 0.28838 | 0.10684 | 0.0087613 |
| C322A | 0.02865 | 0.0649 | 0 | 0.20924 | 0.01769 | 0 | 0.7793 | 0.31489 | 0.13978 | 0.22778 |
| C322A | 0 | 0 | 0 | 0 | 0 | 0.04826 | 0.81804 | 0.35677 | 0.11204 | 0.084214 |
| C322A | 0 | 0 | 0 | 0.06676 | 0 | 0 | 0.30261 | 0 | 0.0855 | 0.061038 |
| Cd | 0.02242 | 0.07693 | 0.11869 | 0.13441 | 0.02257 | 0 | 0.38529 | 0 | 0.099263 | 0.072858 |
| Cd | 0 | 0 | 3.4277 | 0.09718 | 0.0036 | 0.24817 | 0.3703 | 0 | 0.085122 | 0.064483 |
| Cd | 0 | 0 | 4.6141 | 0.2954 | 0.00292 | 0.03007 | 1.6025 | 0 | 0.10357 | 0 |
| Cd | 0 | 0 | 0.67599 | 0.16907 | 0.00284 | 0.02692 | 0.19647 | 0 | 0.083227 | 0.046969 |
Figure 2.
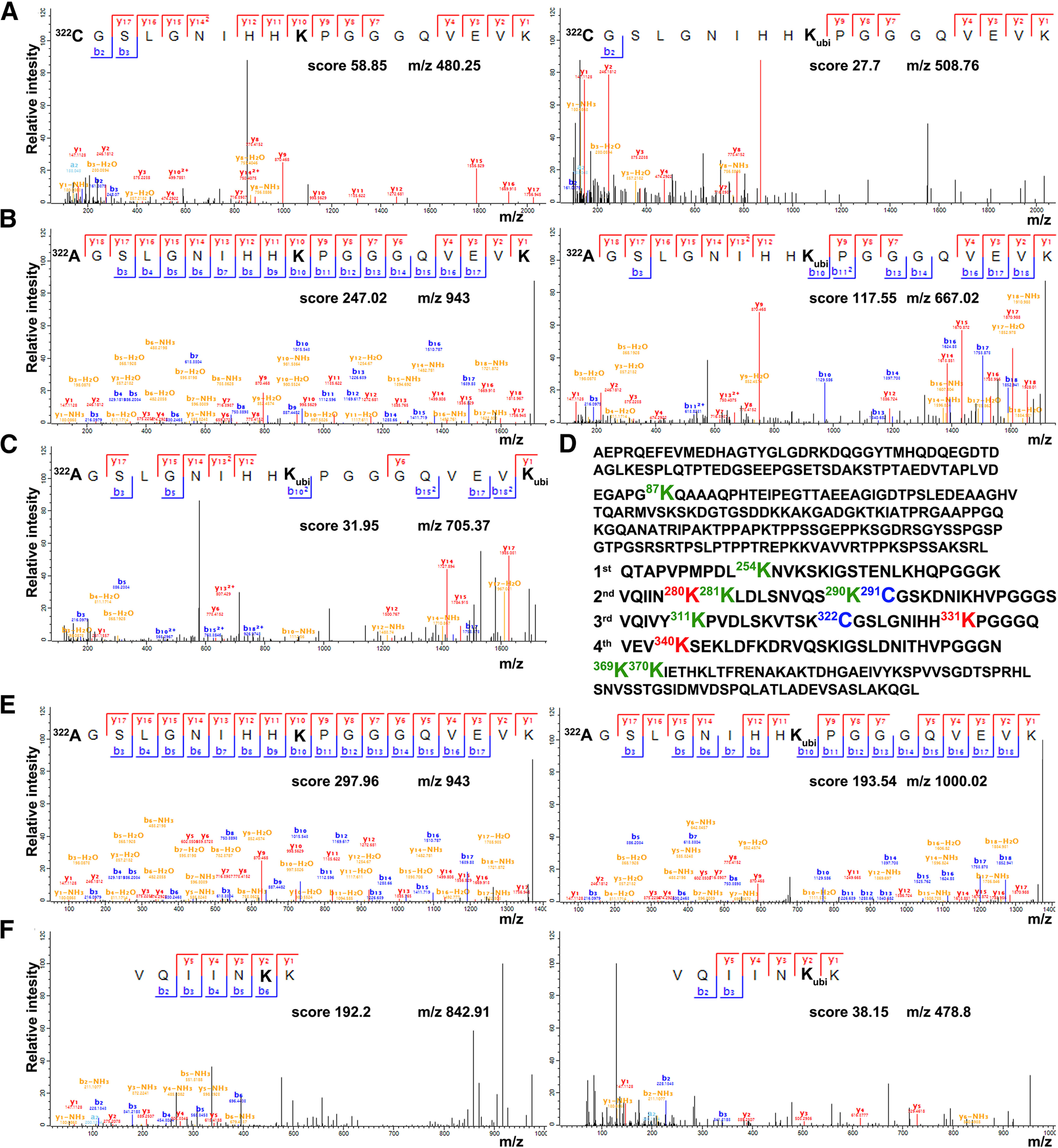
Cys-322 substitution affects the ubiquitination of Tau. A-C, E, F, Annotated spectrum of the indicated peptides unmodified or carrying the motif GlyGly (114.0429 Da) on the highlighted in bold lysine residues, characteristic of the ubiquitin remnant after tryptic cleavage. The annotation is done in the MaxQuant viewer, and the MaxQuant score is displayed together with the precursor ion mass. A, Modified Lys331 identified in wt Tau samples. B, Modified Lys331 identified in C322A samples. C, Modified Lys340 identified in C322A samples. E, Modified Lys331 identified in Cd samples. F, Modified Lys280 identified in Cd samples. D, Amino acid sequence of the Tau protein with the mutated cysteine residues shown in blue. Lys-87, Lys-281, Lys-290, Lys-254, Lys-311, Lys-369, and Lys-370 display low ubiquitination in all Tau variants and are highlighted in green. Red represents the lysine residues with high ubiquitinated/unmodified ratios identified in C322A and Cd samples.
Both cysteines impact the interaction of tau with microtubules, but only cys-322 affects tau actin-binding properties
One of the most important physiological functions of the Tau protein is to regulate assembly, dynamic behavior, and the spatial organization of microtubules (Barbier et al., 2019). Therefore, microtubule-binding affinity was tested for the three Tau constructs using two different assays. First, endogenous microtubules were sedimented by ultracentrifugation from fly head lysates in the presence of the microtubule stabilizing agent Taxol (Fig. 3, top) (Papanikolopoulou et al., 2019). The second assay was conducted by incubating lysates of wt or mutant Tau with preformed Taxol stabilized bovine microtubules (Fig. 3, middle). The pellet and supernatant fractions were subsequently probed for Tau and tubulin. Quantification of at least four independent experiments representing the relative level of Tau in the supernatant and pellet fractions showed that, in contrast to wt Tau, both cysteine-mutated proteins remained mostly in the supernatant and did not coprecipitate with microtubules (Fig. 3, top; C291A p = 2.35e-06, C322A p = 0.0005, n = 4; Fig. 3, middle; C291A p = 0.01,466, C322A p = 8.59e-06, n = 5). In congruence with the recently published Tau-binding mode to Taxol-stabilized microtubules (Martinho et al., 2018) suggesting that Cys-291 in R2 interacts with Cys-347 of α-tubulin and Cys-322 in R3 with Cys-131 of β-subunit, substitution of any of these two residues dramatically decreases the affinity of Tau for microtubules in vivo. Interestingly, these tubulin cysteines are conserved in vertebrate and invertebrate species (Little and Seehaus, 1988), and both Drosophila and bovine microtubules possess them.
Figure 3.

Effect of individual cysteine mutations on the association of Tau with the cytoskeleton. Top, Endogenous microtubules were purified from fly head lysates expressing pan-neuronally the indicated transgenes in the presence of Taxol. Pellet (p) and supernatant (s) fractions were analyzed by Western blotting using antibodies against Tau (5A6) and tubulin (E7). The ratio of the relative level of Tau in the supernatant to the Tau in the pellet fractions was used for quantification and shown as ratios of their means ± SEM relative to the ratio of wt Tau, which is arbitrarily set to 1. *p < 0.05, significantly reduced levels of Tau coprecipitated with microtubules for both Cys mutants (Dunnett's) compared with wt Tau. Middle, Preformed bovine microtubules have been added to fly head lysates expressing pan-neuronally the indicated transgenes and have been probed and quantified as above. *p < 0.05, significantly reduced levels of microtubule-bound Tau for both Cys mutants (Dunnett's) compared with wt Tau. Bottom, Phalloidin-bound F-actin was isolated from fresh brain extracts of flies expressing pan-neuronally the indicated transgenes, and pellet (p) and supernatant (s) fractions have been probed for Tau and actin. The ratio of the relative level of Tau in the supernatant to the Tau in the pellet fractions was used for quantification and shown as ratios of their means ± SEM relative to the ratio of wt Tau, which is arbitrarily set to 1. *p < 0.05, significantly increased levels of Tau coprecipitated with actin for C322A mutant (Dunnett's) compared with wt Tau.
In addition to microtubules, Tau is also implicated in the regulation of actin cytoskeleton (Elie et al., 2015), and in vivo human Tau interacts with endogenous filamentous actin in Drosophila neurons (Fulga et al., 2007). Therefore, we tested the potential effects of cysteine substitution on actin-binding affinity. F-actin was isolated from fresh brain extracts using biotinylated-phalloidin, and the precipitated material was probed for Tau. As shown in Figure 3 (bottom), quantification of three independent experiments representing the relative ratio of Tau in the supernatant and pellet fractions did not reveal differences between wt and the C291A mutant (p = 0.3929, n = 3), but a striking fraction of the C322A protein coprecipitated with F-actin (Fig. 3, bottom; p = 0.0009, n = 3). These results indicate that, although both cysteine residues are equally important for the interaction of Tau with microtubules, Cys-322 seems to be a differential negative regulator of Tau binding to actin filaments.
Cysteines regulate phosphorylation of tau at disease-associated epitopes
Since the cytoskeletal interactions of Tau depend critically on the phosphorylation status of the protein (Wang and Mandelkow, 2016), we hypothesized that, apart from making critical contacts with tubulin, cysteine substitution could alter the phosphorylation state of Tau, and therefore its ability to interact with the microtubules. To address this, we examined whether cysteine residues affect the overall phosphorylation pattern of Tau protein. All variants, of both sexes, were probed with a panel of antibodies targeting specific phosphorylated sites, and phospho-Tau levels were normalized against total Tau levels (Fig. 4). These phospho-antibodies recognize sites that are reported to be highly enriched AD brains and include pT181 (AT270), pS202/pT205 (AT8), pT212/pS214 (AT100), pT212, pS214, pS262, and pS396. Among these epitopes, only occupation of pS396 follows total Tau levels (Fig. 4, pS396: females, C291A p = 0.9584, C322A p = 0.4894; and males, C291A p = 0.4009, C322A p = 0.9971, n = 6), indicating no specific effects of loss of either Cys. However, all other sites appeared significantly suppressed in both mutants regardless of the fly sex (Fig. 4, pT212: females, C291A p = 9.16e-06, C322A p = 1.73e-06; and males, C291A p = 5.37e-05, C322A p = 3.03e-05, n = 4; AT8: females, C291A p = 3.77e-09, C322A p = 2.32e-09; and males, C291A p = 1.25e-08, C322A p = 2.32e-09, n = 6; AT270: females, C291A p = 5.22e-08, C322A p = 1.48e-09; and males, C291A p = 3.19e-06, C322A p = 1.22e-06, n = 4; and pS262: females, C291A p = 7.19e-06, C322A p = 5.63e-09; and males, C291A p = 6.91e-05, C322A p = 3.38e-07, n = 4). An exception was phosphorylation at pS214, which appears elevated in the C291A mutant (Fig. 4, pS214: females, C291A p = 0.0004, C322A p = 0.0167; and males, C291A p = 0.0002, C322A p = 0.0073, n = 5). Such quantitative effects may be mediated by local conformational changes precipitated by cysteine substitution potentially constraining access of kinases or phosphatases to these epitopes.
Figure 4.

Effect of individual cysteine mutations on Tau phosphorylation at disease-associated sites. Schematic representation of human microtubule-associated protein Tau and the serine (S) and threonine (T) residues targeted by the phosphor antibodies used in this study. Most of the Ser and Thr are located in the proline-rich region flanking the four microtubule-binding repeats (R1-R4), and include T181, S202, T205, T212, S214, S262, and S396. Representative Western blots from head lysates of flies accumulating wt Tau pan-neuronally compared with similar lysates from single Cys mutants probed with the antibodies indicated on the right are shown below together with quantifications of at least four independent biological replicates for both female (♀) and male (♂) flies. Levels of the phosphorylated protein were first normalized using the Syx loading control and then against total Tau levels. The normalized level of wt Tau for each quantification was fixed to 1. Error bars indicate mean ± SEM relative levels of mutants phosphorylated at the given sites, over that of the wt Tau. *p < 0.05, significantly altered levels (Dunnett's).
Phosphorylation at pT212 and pS214 in the proline-rich region creates a phosphoepitope recognized specifically by the AT100 antibody, which is prominent in the paired helical filamentous form of Tau, and often used in histopathological assessments (Zheng-Fischhofer et al., 1998; Regalado-Reyes et al., 2019). Interestingly, both mutants lack AT100 immunoreactivity, but apparently for different reasons. Reduced Ser-214 and Thr-212 occupation probably abolishes the AT100 signal in the C322A mutant, whereas a skewed ratio of phosphorylated pT212 to pS214 (Fig. 4, pS214 is highly occupied but not pT212) is apparently not conducive to recognition by the AT100 antibody (Papanikolopoulou and Skoulakis, 2015) in the case of C291A mutant. In addition to ubiquitination, this finding reveals further subtle differences in the biochemical profile of the mutants.
Cys-322 affects tau aggregation propensity
It has been reported that formation of intermolecular disulfide bonds accelerates Tau aggregation in vitro (Schweers et al., 1995; Bhattacharya et al., 2001; Sahara et al., 2007; Furukawa et al., 2011). Hence, it is anticipated that Cys substitution would affect Tau aggregation propensity in vivo, so we assessed the solubility profile of the mutants. Head lysates from animals expressing pan-neuronally these proteins were fractionated into soluble and insoluble materials and were probed for Tau. As shown in Figure 5A, Cys-291 has no effect on Tau solubility, whereas the ratio of soluble/insoluble fraction greatly increases on Cys-322 substitution (Fig. 5A, C291A p = 0.6181, C322A p = 3.30e-07, n = 5). Similar results were obtained with a different method on RIPA fractionation and extraction of the insoluble material with 70% FA (Fig. 5B, FA insoluble fraction). These results clearly differentiate the role of each cysteine residue in regulating Tau aggregation propensity.
Figure 5.

Effect of individual cysteine mutations on Tau conformation and aggregation propensity. A, Representative Western blots of aqueous soluble and insoluble fractions generated from adult heads following pan-neuronal expression of the indicated transgenes. Syx was used as loading control. The Syx-normalized level of wt Tau in the soluble fraction over the insoluble fraction was fixed to 1. Error bars indicate mean ± SEM relative sol/ins ratios of the mutants, over that of the wt Tau. *p < 0.05, significantly low amount of the C322A mutant in the insoluble fraction (Dunnett's). Detailed statistics can be found in the text. B, Transgenic flies expressing wt Tau or the single cysteine mutants under the control of the Elav;Ras-Gal4 driver were aged for 10 d. Proteins from adult heads were sequentially extracted with RIPA buffer and 70% FA and probed for Tau (5A6). Soluble RIPA fraction has also been probed with the conformation-specific MC1 antibody, and all transgenes displayed positive immunoreactivity. Syx was used as loading control.
Finally, to evaluate potential conformational alterations induced by the mutation of cysteines, we probed the immunoreactivity of the mutants with the MC1 antibody (Fig. 5B). MC1 targets a disease-specific conformational modification of Tau (Weaver et al., 2000) formed when discontinuous residues in the amino terminus become proximal to the MBRs (Jicha et al., 1997). Interestingly, both mutants were immunoreactive with the MC1 antibody, suggesting no differences in folding or pathology-related structure with wt Tau. Given that all Tau variants share common structural elements, this implies that other factors are likely modulating Tau self-assembly in vivo affecting the stability of the aggregates (i.e., post-translational modifications, such as ubiquitination, formation of disulfide bonds, etc.) (Dickey et al., 2006; Park et al., 2018).
Differential contribution of cysteine residues to tau-dependent toxicity
Does blocking these two cysteines have phenotypic consequences to Tau-induced toxicity? To address this, age-dependent mortality as well as fly survival to heat and oxidative stress were assessed. Neuronal homeostatic responses to environmental stressors are essential for maintaining robust behavioral outputs, and Tau-induced toxicity has been shown to be accompanied by impaired neuronal fitness and function (Papanikolopoulou and Skoulakis, 2011). Flies experiencing increased oxidative stress because of pathologic hTau accumulation have increased susceptibility to oxidative injury (Dias-Santagata et al., 2007; Keramidis et al., 2020). To that end, we first tested whether Tau mutant transgenic flies manifest altered responses to oxidative stress. Compared with the nontransgenic controls, flies pan-neuronally expressing wt Tau and the C291A mutant presented significantly higher sensitivity to the exogenous reactive oxygen species inducers H2O2 and paraquat (Fig. 6A, H2O2: WT p = 2.7e-08, C291A p = 0.000176 and paraquat: WT p = 1.39e-08, C291A p = 9.27e-05). It should be noted, however, that the sensitivity of the C291A mutants was intermediate between control and wt Tau-expressing flies (each pair Student's t test, C291A vs WT, H2O2: p = 0.011 and paraquat: p = 5.15e-05). In contrast, administration of H2O2 over 52 h and 30 mm paraquat over 28 h induced the same lethality in control and C322A mutant flies (Fig. 6A, H2O2: C322A p = 0.7181 and paraquat: C322A p = 0.2908).
Figure 6.

Cysteine substitution affects Tau neurotoxicity in vivo. A, Response of flies expressing pan-neuronally the indicated transgenes to oxidant molecules. Flies have been treated either with 5% H2O2 or with 30 mm paraquat, and mortality was scored after 52 and 28 h, respectively. Data are mean ± SEM from two independent experiments, with a total of 300 flies assessed per genotype. *p < 0.05, significant difference (Dunnett's) from control driver heterozygote flies (Elav/+; Ras/+). B, Representative Western blot of gstD1 (ARE)-GFP reporter expression in nontransgenic control flies and flies overexpressing pan-neuronally the indicated human Tau transgenes. For the quantifications below, levels of GFP expression were normalized using the tubulin (Tub) loading control and shown as a ratio of their means ± SEM relative to their respective levels in nontransgenic fly heads, which is set to 1. *p < 0.05, significantly altered levels (Dunnett's). C, Flies expressing pan-neuronally the indicated Tau transgenes and control driver heterozygotes Elav/+; Ras/+ were subjected to a sustained 40°C heat shock. The rates at which flies of different genotypes succumb to heat shock are shown. Data are mean ± SEM from two independent experiments, with a total of 150 flies assessed per genotype. Increased susceptibility to heat shock was revealed for wt Tau and C291A-expressing flies (p < 0.05, Dunnett's). D, Survival curves for animals expressing pan-neuronally the indicated transgenes at 30°C, in comparison with Elav/+; Ras/+ control heterozygotes. Data are mean ± SEM from two independent experiments, with a total of 300 flies assessed per genotype. Statistical analysis using the log rank test indicated significant differences in longevity after accumulation of wt Tau and C291A mutant (day 18 and until day 29 prob χ2 < 0.01). Detailed statistics can be found in the text.
One of the defense mechanisms against oxidative stress as well as other stressors is the activation of the Nrf2 pathway (Sykiotis and Bohmann, 2008). Upon oxidative injury, the transcription factor Nrf2 is released from its repressor Keap1 and is transferred to the nucleus where it binds to AREs and activates its targets (antioxidant genes, mitochondrial biosynthesis genes, etc.). Because the mutants present differential resistance to oxidative stress, we investigated the status of the Keap1/Nrf2 antioxidant pathway in Tau-expressing flies by crossing them with a GFP reporter line bearing the ARE of the gstD1 gene (Sykiotis and Bohmann, 2008). Detection of ARE-dependent GFP expression by Western blot revealed that, compared with nontransgenic control flies, wt Tau caused potent induction of Keap1/Nrf2 signal transduction system (Fig. 6B, WT: p = 7.46e-07, n = 4). Interestingly, the antioxidant response was significantly suppressed in C291A-expressing flies (Fig. 6B, C291A: p = 2.11e-05, n = 4), suggesting that the wt and this mutant protein decrease the resistance to paraquat and H2O2-mediated oxidation via different mechanisms. Significantly, in the C322A-expressing flies, activation of genes under the regulatory control of the ARE promoter sequence was similar to that of control flies (Fig. 6B, C322A: p = 0.6181, n = 4), indicating that the presence of this Cys contributes to Tau-mediated oxidative stress.
We next analyzed fly survival under constant conditions of hyperthermia. To this end, Tau-expressing adult flies were subjected to a sustained 40°C heat shock to assess neuronal robustness by determining their ability to withstand environmental stress. Nontransgenic control flies were gradually affected by the thermal stress such that, by 30 min, almost 50% of the population was unresponsive (Fig. 6C). By comparison, wt Tau-expressing flies succumb more rapidly to heat stress starting at 18 min of continuous stress, with nearly 90% of the animals affected after 30 min (Fig. 6C, WT at 18 min p = 0.0024 and at all time points p < 0.001). Flies expressing the C291A mutant Tau also became catatonic and unresponsive more rapidly than control flies (Fig. 6C, C291A at 18 min p = 0.2287, at 22 min p = 0.0074, and at all time points p < 0.01). Finally, flies expressing C322A presented similar sensitivity to heat shock with control flies (Fig. 6C, C322A at all time points p > 0.05).
Tau toxicity ultimately results in the death of the organism, which can easily be scored by counting the number of surviving flies over time (Wittmann et al., 2001; Colodner and Feany, 2010; Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017). Assaying lifespan at 30°C, wt Tau-expressing animals start to expire around day 18 (Fig. 6D, WT day 18 prob χ2 = 0.0005 and until day 29 prob χ2 < 0.0001) with 50% attrition occurring at day 24 (prob χ2 = 1e-07). In contrast, 50% attrition in controls required at least 4 additional days (Fig. 6D, day 28). A reduction in lifespan was also observed for C291A starting at day 18 (Fig. 6D, C291A day 18 prob χ2 = 0.00315 and until day 29 prob χ2 < 0.01) with 50% attrition occurring at day 26 (prob χ2 = 0.001), yet the effect of C291A on decreasing lifespan was more modest than that of wt Tau (Fig. 6D, C291A vs WT day 21 prob χ2 = 0.0236, day 25 prob χ2 = 5.68e-06, day 27 prob χ2 = 0.001, and day 28 prob χ2 = 0.0405). Finally, survival of C322A-expressing animals was statistically indistinguishable from that of control flies (Fig. 6D, C322A prob χ2 > 0.2 at all time points).
Collectively, these results indicate that Cys substitution is sufficient to alleviate Tau neurotoxicity in vivo. Abnormal Tau phosphorylation has traditionally been implicated in mediating Tau-induced toxicity (Wittmann et al., 2001; Nishimura et al., 2004), and the reduced toxic effects of both cysteine mutants compared with wt Tau could in part be correlated to their phosphorylation profile (Fig. 4). Drosophila models (Chatterjee et al., 2009; Feuillette et al., 2010; Talmat-Amar et al., 2011) provided further evidence that an optimal level of Tau phosphorylation is required to achieve a balance in the level of “free” and “microtubule bound” Tau. Both free cytosolic hyperphosphorylated forms of Tau with low affinity for microtubules and hypophosphorylated forms of Tau with abnormally high affinity for microtubule-binding cause an imbalance in the function of microtubules, eventually leading to toxicity. In the case of the cysteine mutants, it appears that the presence of unbound hypophosphorylated forms of Tau (Fig. 3) supports their lower toxicity compared with wt Tau. However, the mutants possess distinct biochemical characteristics that lead to differential ARE response (Fig. 6B) and actin-binding properties (Fig. 3), although it is not documented if and how the binding of hypophosphorylated forms of Tau to actin filaments affects its toxicity. Finally, differences in the biochemical profile of the mutants, such as ubiquitination (Table 1, Lys331 and Lys340), which leads to reduced Tau stability and expression levels, may also account for the benign character of C322A mutant compared with C291A.
Both cysteine residues impact hTau-associated dysfunction
Insights into Tau-mediated neuronal dysfunction can be assayed in the Drosophila model systems through investigation of learning and memory (Kosmidis et al., 2010; Papanikolopoulou and Skoulakis, 2015; Sealey et al., 2017). Interestingly, phosphorylation at Ser-262 was proposed to be necessary for and predict learning deficits (Papanikolopoulou and Skoulakis, 2015; Keramidis et al., 2020); and given that this site is hypophosphorylated in the two mutants, we hypothesized that cysteine substitution could impact the cognitive performance of animals. To examine associative learning and memory, we used the olfactory classical conditioning paradigm (Tully and Quinn, 1985) and tested control and Tau-expressing animals immediately after training to assess 3 min memory and 24 h later to probe consolidated memories. Two forms of consolidated memories can be assayed in Drosophila: the protein synthesis-dependent LTM induced after multiple rounds of spaced training; and the protein synthesis-independent ARM, elicited after repeated massed training cycles (Tully et al., 1994).
Animals accumulating pan-neuronally the Tau transgenes were first subjected to an olfactory-associative learning task in which flies were presented with either 3 or 6 odor/shock pairings (Fig. 7A). In both conditions, wt Tau-expressing flies presented impaired performance compared with controls (Fig. 7A, 6 pairings ANOVA: F(6104) = 3.7120, p = 0.0023; subsequent LSM: p = 0.00007876 and p = 0.003932 vs controls, respectively; and 3 pairings ANOVA: F(6,95) = 4.8434, p = 0.0003; subsequent LSM: p = 0.001984 and p = 0.001123 vs controls, respectively). In contrast, expression of the C322A mutant did not precipitate learning deficits (Fig. 7A; 6 pairings ANOVA: F(6,104) = 3.7120, p = 0.0023; subsequent LSM: p = 0.9312 and p = 0.2107 vs controls, respectively; and 3 pairings ANOVA: F(6,95)= 4.8434, p = 0.0003; subsequent LSM: p = 0.8718 and p = 0.1268 vs controls, respectively). Interestingly, flies expressing the C291A mutant performed equally well with controls at 6 pairings but limited training with just 3 pairings resulted in deficient learning (Fig. 7A, 6 pairings ANOVA: F(6,104) = 3.7120, p = 0.0023; subsequent LSM: p = 0.0758 and p = 0.4481 vs controls, respectively; and 3 pairings ANOVA: F(6,95) = 4.8434, p = 0.0003; subsequent LSM: p = 0.001 and p = 0.0106 vs controls, respectively). In agreement with previous reports (Sealey et al., 2017), pan-neuronal expression of wt human Tau resulted specifically in impaired LTM (Fig. 7B, ANOVA: F(2,39) = 17.9823, p < 0.0001; subsequent LSM: p = 2.37e-06 and p = 0.000031 vs controls, respectively), whereas ARM remained unaffected (Fig. 7C, ANOVA: F(2,35) = 3.8899, p = 0.0304; subsequent LSM: p = 0.9170 and p = 0.0257 vs controls, respectively). In contrast, the memory performance of flies expressing the two cysteine mutants was not significantly different from controls (Fig. 7B, C291A ANOVA: F(2,31) = 6.0886, p = 0.0062; subsequent LSM: p = 0.0015 and p = 0.1429 vs controls, respectively; and C322A ANOVA: F(2,39) = 14.5887, p < 0.0001; subsequent LSM: p = 0.2914 and p = 7.52e-06 vs controls, respectively). These results indicate that blocking cysteines of Tau confers a crucial role for this protein in neuronal dysfunction and support the proposal that elevated occupation of phosphor-Ser262 is indeed requisite for the learning deficits (Papanikolopoulou and Skoulakis, 2015; Keramidis et al., 2020).
Figure 7.

Cysteine substitution affects learning and memory performance. A, Learning performance of animals at 6 and 3 pairings accumulating pan-neuronally the indicated Tau variants (black bars) compared with driver and transgene heterozygotes (white and gray bars). The genotypes of all animals are indicated below each bar. Data are mean ± SEM. *p < 0.05, significant differences from both controls. The number of experimental replicates (n) is indicated within the bars. Expression of wt Tau caused impairment in learning in both conditions, whereas expression of C291A mutant caused impairment in learning only under limited training with 3 pairings. B, Memory performance of animals accumulating pan-neuronally the indicated Tau transgenes (black bar), compared with driver and transgene heterozygotes (white and gray bars). The genotypes of all animals are indicated below each bar. Data are mean ± SEM. *p < 0.05, significant differences from both controls. The number of experimental replicates (n) is indicated within the bars. Only expression of wt Tau caused impairment in LTM. C, Pan-neuronal expression of wt Tau (black bar) does not affect memory after massed training (ARM, p > 0.05), compared with driver and transgene heterozygotes (white and gray bars). The number of experimental replicates (n) is indicated within the bars. Data are mean ± SEM. Detailed statistics can be found in the text. Trs, Transgene.
Discussion
This work highlights the importance of cysteines to protein structure stabilization and to specific physiological and pathophysiological functions of Tau, including association with the cytoskeleton, neuronal toxicity, and dysfunction in vivo. How these residues influence such a range of functions is quite remarkable and is probably related to their redox-active nature and a complex array of intermolecular interactions with a variety of proteins (Bechtel and Weerapana, 2017). Our results point toward a synergistic interplay between cysteines to regulate Tau stability. Elimination of Cys-291 by itself cannot destabilize Tau, whereas Cys-322 substitution led to a significant reduction of Tau protein levels. This effect on stability was further exacerbated on concomitant mutation of Cys-291, as a consequence of differential protein degradation involving the ubiquitin-proteasome system (Fig. 1; Table 1).
Intriguingly, the elimination of cysteines seems to trigger perturbations to their local structural environment since blocking Cys-322 markedly influenced the ubiquitination of only its two proximal downstream lysines, namely, Lys-331 and Lys-340 (Table 1; Fig. 2D). It has also been reported that mutation of both cysteines abolishes Tau's intrinsic enzymatic acetyltransferase activity at lysines located only in the microtubule-binding domain (Cohen et al., 2013). Consistent with this observation, cysteine-deficient Tau was the only to present enhanced ubiquitination on Lys-280 located on the second MBR upstream of Cys-291, probably promoting proteasome-mediated Tau degradation and accounting for the extreme low protein levels of the mutant (Min et al., 2010; Tracy et al., 2016).
Tau protein is known to undergo highly regulated phosphorylation, which negatively regulates its affinity for microtubules (Morris et al., 2011). Interestingly, despite their decreased phosphorylation at disease-associated epitopes (Fig. 4), both single cysteine mutants bind very poorly to microtubules (Fig. 3), which, in agreement with structural studies (Kellogg et al., 2018; Martinho et al., 2018), highlights the role of these residues in making critical contacts with tubulin. It is worth mentioning, however, that Drosophila Tau does not possess any cysteine residues within its microtubule-binding repeats (Bouleau and Tricoire, 2015), indicating that it is not a conserved mechanism of interaction of Tau proteins with tubulin. That difference between Drosophila and human Tau could account for the observation that the two proteins do not compete in vivo for the binding to Drosophila microtubules (Feuillette et al., 2010). How phosphorylation impacts the interaction of Tau with F-actin remains to be elucidated; however, hyperphosphorylated forms of Tau have been shown to stabilize F-actin promoting Tau neurotoxicity in vivo (Fulga et al., 2007; Bardai et al., 2018). Strikingly, Cys-322 appears to be a negative regulator of the Tau-actin interaction (Fig. 3), in contrast to Cys-291, which appears to play no role, suggesting that, although the binding of Tau to both actin and microtubules is mediated by the MBRs (Elie et al., 2015), it clearly involves different mechanisms.
The recently identified cysteine-modifying MAPT mutation C291R in a patient diagnosed with corticobasal degeneration (Marshall et al., 2015) further highlights the importance of Cys residues in Tau physiology and pathology. Although this genetic form of corticobasal degeneration has not yet been studied postmortem, in vitro studies revealed that a C291R mutant Tau construct containing only the four MBRs aggregated into different kind of protofibrils than wt Tau construct (Karikari et al., 2020). In our in vivo model system, although all Tau variants adopt the MC1 conformation (Fig. 5B), a well-established marker that labels otherwise normal-appearing neurons early in disease progression and before neurofibrillary tangle formation (Hyman et al., 1988; Jicha et al., 1997, 1999), they present significant changes in their solubility. Both wt Tau and C291A mutant accumulated in the insoluble fraction, suggesting that modifying Cys-291 is not sufficient to change Tau aggregation propensity in vivo. In contrast, Cys-322 played an inhibitory role in the process since its elimination greatly reduced the formation of aggregates (Fig. 5).
Collectively, our results imply that, for the mechanism of Tau aggregation in vivo, several contributing factors may be envisaged, including a complex cross-talk of post-translational modifications (PTMs; i.e., ubiquitination, phosphorylation, acetylation, oxidation) that may not necessarily alter the conformation of Tau but reverse the stability of aggregates by weakening the Tau-Tau binding interactions needed to maintain the structure of the core of the aggregate and probably facilitating their degradation (Arakhamia et al., 2020). Still, it remains to be elucidated which combinations of PTMs can be considered as the rate-limiting step of Tau fibrillization, although considering the enormous heterogeneity of Tau PTMs, it is unlikely that there are only one or two triggering combinations.
Importantly, cysteines modulate Tau-induced neurotoxicity in vivo mainly because of their impact on Tau phosphorylation and microtubule-binding properties. What should be noted from the work herein is that targeting these residues may be one way to modify the early steps of Tau neurotoxicity. Blocking Cys-322 mutation significantly abolishes Tau toxicity as revealed by the increased resistance to oxidative injury, thermal stress, and longevity (Fig. 6). Upon Cys-291 substitution, Tau retains its toxicity, yet flies are not as severely affected as flies expressing wt Tau. It seems that a delicate balance between Tau modifications exists (i.e., ubiquitination), any deviation from which may lead to toxic effects of Tau. These subtle distinctions affecting Tau stability could contribute to the differences between the mutants. Interestingly, wt Tau and C291A mutant seem to follow different pathways to eventually trigger neurotoxicity, as suggested by their opposing effects on the activation of the Nrf2/Keap1 pathway (Fig. 6B). Nrf2 enables the cell to maintain redox balance under conditions of oxidative injury, whereas both pathway inhibition and overactivation can induce detrimental effects. For example, Nrf2 is inhibited in AD (Ramsey et al., 2007) as well as in animal models of AD (Kanninen et al., 2009; Kerr et al., 2017), while its unregulated activation shortens lifespan in Drosophila (Tsakiri et al., 2013) and promotes cancer development in humans (Ohta et al., 2008; Shibata et al., 2008). Strikingly, it has been reported that expression of the hTau 0N3R isoform in the Drosophila CNS suppressed Nrf2 signaling similarly to hTau-2N4R C291A mutant (Fig. 6B). These results point toward a dominant role of Cys-322 in regulating the response of Nrf-2 and on its substitution Nrf-2 activity remained at control levels (Fig. 6B). However, the outcome of the response depends critically on Cys-291, since its absence (mutation or lack of the second MBR in 0N3R) suppressed the Nrf2 signaling, whereas its presence led to activation and upregulation of Nrf2 target genes. Therefore, our collective data strongly support the notion that cysteine-mediated interactions could define isoform-specific differences (3R vs 4R) (Goode et al., 2000; Sealey et al., 2017; Kadas et al., 2019).
In addition to dissecting mechanisms of Tau toxicity, research efforts are also focused on understanding aspects of Tau-mediated dysfunction mostly manifested as behavioral phenotypes, which have been key in dissociating Tau-mediated toxicity from Tau-mediated dysfunction (Mudher et al., 2004; Santacruz et al., 2005; Ubhi et al., 2007; Papanikolopoulou and Skoulakis, 2015). Along this line, we observed that, while wt Tau expression impaired associative learning (Fig. 7A) and LTM (Fig. 7B) in agreement with prior results (Kosmidis et al., 2010; Papanikolopoulou et al., 2010; Sealey et al., 2017), eliminating cysteines did not affect either of these processes. Only after limited training with three odor/shock pairings, the C291A mutant presented learning deficits (Fig. 7A), suggesting that this mutation potentially affects the efficiency of learning, but not learning ability per se.
It is interesting that, although blocking Cys-291 does not appear to affect the conformation or aggregation propensity of Tau, the mutant is less toxic than wt protein, and its accumulation in the insoluble fraction does not result in memory deficits. A possible explanation will be that the C291A mutation leads to aggregated forms of the protein with distinct structural characteristics and reduced toxicity compared with wt Tau (Cowan et al., 2015; Shammas et al., 2015). This again demonstrates the complex interplay of Tau PTMs (i.e., differences in phosphorylation) underpinning the differences in toxicity and dysfunction described here. Similar observations were made for the 0N3R isoform that, although it assumed an MC1 conformation with concomitant accumulation of aggregates, its overexpression in the Drosophila CNS did not precipitate impairments in learning or LTM (Sealey et al., 2017).
In addition to the structural information of Tau aggregates (Scheres et al., 2020), elucidating what defines isoform-specific molecular and physiological differences and what triggers the transition of Tau from a highly soluble form to insoluble toxic forms is imperative to identify an underlying pathogenic mechanism that occurs during the onset or progression of Tauopathies. It is a challenge of future work to elucidate, for example, the mechanisms mediating the effect of Tau isoforms on Nrf2 activity. Such studies could help to elucidate the apparent differential involvement of Tau isoforms in different Tauopathies and contribution to neuronal dysfunction and toxicity.
Footnotes
This work was supported by Stavros Niarchos Foundation to the Biomedical Sciences Research Center “Alexander Fleming,” as part of the Foundation's initiative to support the Greek research center ecosystem, Fondation Sante, and the Hellenic Foundation for Research and Innovation. We thank Dr. Efthimios Skoulakis for fly strains and for critical reading of the manuscript; and Dr. Jean Y Gouzi for performing pilot LTM experiments.
The authors declare no competing financial interests.
References
- Acevedo SF, Froudarakis EI, Kanellopoulos A, Skoulakis EM (2007) Protection from premature habituation requires functional mushroom bodies in Drosophila. Learn Mem 14:376–384. 10.1101/lm.566007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN, Seyfried NT, Petrucelli L, Fitzpatrick AW (2020) Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 180:633–644.e612. 10.1016/j.cell.2020.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126:238–292. 10.1016/j.brainresbull.2016.08.018 [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8:663–672. 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, Tsvetkov PO, Devred F, Landrieu I (2019) Role of tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci 11:204. 10.3389/fnagi.2019.00204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, Feany MB (2018) A conserved cytoskeletal signaling cascade mediates neurotoxicity of FTDP-17 tau mutations in vivo. J Neurosci 38:108–119. 10.1523/JNEUROSCI.1550-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel TJ, Weerapana E (2017) From structure to redox: the diverse functional roles of disulfides and implications in disease. Proteomics 17:1600391 10.1002/pmic.201600391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C (2007) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci 27:3650–3662. 10.1523/JNEUROSCI.0587-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya K, Rank KB, Evans DB, Sharma SK (2001) Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem Biophys Res Commun 285:20–26. 10.1006/bbrc.2001.5116 [DOI] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K (2007) An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA 104:3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouleau S, Tricoire H (2015) Drosophila models of Alzheimer's disease: advances, limits, and perspectives. J Alzheimers Dis 45:1015–1038. 10.3233/JAD-142802 [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Sang TK, Lawless GM, Jackson GR (2009) Dissociation of tau toxicity and phosphorylation: role of GSK-3beta, MARK and Cdk5 in a Drosophila model. Hum Mol Genet 18:164–177. 10.1093/hmg/ddn326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen TJ, Friedmann D, Hwang AW, Marmorstein R, Lee VM (2013) The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat Struct Mol Biol 20:756–762. 10.1038/nsmb.2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colodner KJ, Feany MB (2010) Glial fibrillary tangles and JAK/STAT-mediated glial and neuronal cell death in a Drosophila model of glial tauopathy. J Neurosci 30:16102–16113. 10.1523/JNEUROSCI.2491-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Quraishe S, Hands S, Sealey M, Mahajan S, Allan DW, Mudher A (2015) Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci Rep 5:17191. 10.1038/srep17191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB (2007) Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Invest 117:236–245. 10.1172/JCI28769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Yue M, Lin WL, Dickson DW, Dunmore JH, Lee WC, Zehr C, West G, Cao S, Clark AM, Caldwell GA, Caldwell KA, Eckman C, Patterson C, Hutton M, Petrucelli L (2006) Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci 26:6985–6996. 10.1523/JNEUROSCI.0746-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elie A, Prezel E, Guerin C, Denarier E, Ramirez-Rios S, Serre L, Andrieux A, Fourest-Lieuvin A, Blanchoin L, Arnal I (2015) Tau co-organizes dynamic microtubule and actin networks. Sci Rep 5:9964. 10.1038/srep09964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillette S, Miguel L, Frebourg T, Campion D, Lecourtois M (2010) Drosophila models of human tauopathies indicate that Tau protein toxicity in vivo is mediated by soluble cytosolic phosphorylated forms of the protein. J Neurochem 113:895–903. 10.1111/j.1471-4159.2010.06663.x [DOI] [PubMed] [Google Scholar]
- Fitzpatrick AW, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SH (2017) Cryo-EM structures of tau filaments from Alzheimer's disease. Nature 547:185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulga TA, Elson-Schwab I, Khurana V, Steinhilb ML, Spires TL, Hyman BT, Feany MB (2007) Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat Cell Biol 9:139–148. 10.1038/ncb1528 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Nukina N (2011) Tau protein assembles into isoform- and disulfide-dependent polymorphic fibrils with distinct structural properties. J Biol Chem 286:27236–27246. 10.1074/jbc.M111.248963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Chau M, Denis PE, Feinstein SC (2000) Structural and functional differences between 3-repeat and 4-repeat tau isoforms: implications for normal tau function and the onset of neurodegenerative disease. J Biol Chem 275:38182–38189. 10.1074/jbc.M007489200 [DOI] [PubMed] [Google Scholar]
- Gorsky MK, Burnouf S, Sofola-Adesakin O, Dols J, Augustin H, Weigelt CM, Grönke S, Partridge L (2017) Pseudo-acetylation of multiple sites on human Tau proteins alters Tau phosphorylation and microtubule-binding, and ameliorates amyloid beta toxicity. Sci Rep 7:9984. 10.1038/s41598-017-10225-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CS, Moggridge S, Muller T, Sorensen PH, Morin GB, Krijgsveld J (2019) Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc 14:68–85. 10.1038/s41596-018-0082-x [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Wolozin BL, Davies P, Kromer LJ, Damasio AR (1988) Alz-50 antibody recognizes Alzheimer-related neuronal changes. Ann Neurol 23:371–379. 10.1002/ana.410230410 [DOI] [PubMed] [Google Scholar]
- Jeganathan S, von Bergen M, Mandelkow EM, Mandelkow E (2008) The natively unfolded character of tau and its aggregation to Alzheimer-like paired helical filaments. Biochemistry 47:10526–10539. 10.1021/bi800783d [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P (1997) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48:128–132. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Berenfeld B, Davies P (1999) Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res 55:713–723. [DOI] [PubMed] [Google Scholar]
- Kadas D, Papanikolopoulou K, Xirou S, Consoulas C, Skoulakis EM (2019) Human Tau isoform-specific presynaptic deficits in a Drosophila central nervous system circuit. Neurobiol Dis 124:311–321. 10.1016/j.nbd.2018.12.004 [DOI] [PubMed] [Google Scholar]
- Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, Yla-Herttuala S, Tanila H, Levonen AL, Koistinaho M, Koistinaho J (2009) Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 106:16505–16510. 10.1073/pnas.0908397106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikari TK, Thomas R, Moffat KG (2020) The C291R Tau variant forms different types of protofibrils. Front Mol Neurosci 13:39. 10.3389/fnmol.2020.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EH, Hejab NM, Poepsel S, Downing KH, DiMaio F, Nogales E (2018) Near-atomic model of microtubule-tau interactions. Science 360:1242–1246. 10.1126/science.aat1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keramidis I, Vourkou E, Papanikolopoulou K, Skoulakis EM (2020) Functional interactions of Tau phosphorylation sites that mediate toxicity and deficient learning in Drosophila melanogaster. Front Mol Neurosci 13:569520. 10.3389/fnmol.2020.569520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr F, Sofola-Adesakin O, Ivanov DK, Gatliff J, Gomez Perez-Nievas B, Bertrand HC, Martinez P, Callard R, Snoeren I, Cocheme HM, Adcott J, Khericha M, Castillo-Quan JI, Wells G, Noble W, Thornton J, Partridge L (2017) Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer's disease. PLoS Genet 13:e1006593. 10.1371/journal.pgen.1006593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmidis S, Grammenoudi S, Papanikolopoulou K, Skoulakis EM (2010) Differential effects of Tau on the integrity and function of neurons essential for learning in Drosophila. J Neurosci 30:464–477. 10.1523/JNEUROSCI.1490-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little M, Seehaus T (1988) Comparative analysis of tubulin sequences. Comp Biochem Physiol B 90:655–670. 10.1016/0305-0491(88)90320-3 [DOI] [PubMed] [Google Scholar]
- Mann M, Jensen ON (2003) Proteomic analysis of post-translational modifications. Nat Biotechnol 21:255–261. 10.1038/nbt0303-255 [DOI] [PubMed] [Google Scholar]
- Markstein M, Pitsouli C, Villalta C, Celniker SE, Perrimon N (2008) Exploiting position effects and the gypsy retrovirus insulator to engineer precisely expressed transgenes. Nat Genet 40:476–483. 10.1038/ng.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Guerreiro R, Thust S, Fletcher P, Rohrer JD, Fox NC (2015) A novel MAPT mutation causing corticobasal syndrome led by progressive apraxia of speech. J Alzheimers Dis 48:923–926. 10.3233/JAD-150477 [DOI] [PubMed] [Google Scholar]
- Martinho M, Allegro D, Huvent I, Chabaud C, Etienne E, Kovacic H, Guigliarelli B, Peyrot V, Landrieu I, Belle V, Barbier P (2018) Two Tau binding sites on tubulin revealed by thiol-disulfide exchanges. Sci Rep 8:13846. 10.1038/s41598-018-32096-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67:953–966. 10.1016/j.neuron.2010.08.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70:410–426. 10.1016/j.neuron.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris M, Knudsen GM, Maeda S, Trinidad JC, Ioanoviciu A, Burlingame AL, Mucke L (2015) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18:1183–1189. 10.1038/nn.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A, Shepherd D, Newman TA, Mildren P, Jukes JP, Squire A, Mears A, Drummond JA, Berg S, MacKay D, Asuni AA, Bhat R, Lovestone S (2004) GSK-3beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol Psychiatry 9:522–530. 10.1038/sj.mp.4001483 [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, Lu B (2004) PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116:671–682. 10.1016/S0092-8674(04)00170-9 [DOI] [PubMed] [Google Scholar]
- Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S (2008) Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 68:1303–1309. 10.1158/0008-5472.CAN-07-5003 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Skoulakis EM (2011) The power and richness of modelling tauopathies in Drosophila. Mol Neurobiol 44:122–133. 10.1007/s12035-011-8193-1 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Skoulakis EM (2015) Temporally distinct phosphorylations differentiate Tau-dependent learning deficits and premature mortality in Drosophila. Hum Mol Genet 24:2065–2077. 10.1093/hmg/ddu726 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Kosmidis S, Grammenoudi S, Skoulakis EM (2010) Phosphorylation differentiates tau-dependent neuronal toxicity and dysfunction. Biochem Soc Trans 38:981–987. 10.1042/BST0380981 [DOI] [PubMed] [Google Scholar]
- Papanikolopoulou K, Roussou IG, Gouzi JY, Samiotaki M, Panayotou G, Turin L, Skoulakis EM (2019) Drosophila Tau negatively regulates translation and olfactory long-term memory, but facilitates footshock habituation and cytoskeletal homeostasis. J Neurosci 39:8315–8329. 10.1523/JNEUROSCI.0391-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Lee JH, Jeon JH, Lee MJ (2018) Degradation or aggregation: the ramifications of post-translational modifications on tau. BMB Rep 51:265–273. 10.5483/bmbrep.2018.51.6.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Trojanowski JQ, Lee VM (2020) Protein transmission in neurodegenerative disease. Nat Rev Neurol 16:199–212. 10.1038/s41582-020-0333-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21:921–926. 10.1038/nbt849 [DOI] [PubMed] [Google Scholar]
- Petrucelli L, Dickson D, Kehoe K, Taylor J, Snyder H, Grover A, De Lucia M, McGowan E, Lewis J, Prihar G, Kim J, Dillmann WH, Browne SE, Hall A, Voellmy R, Tsuboi Y, Dawson TM, Wolozin B, Hardy J, Hutton M (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet 13:703–714. 10.1093/hmg/ddh083 [DOI] [PubMed] [Google Scholar]
- Qiang L, Sun X, Austin TO, Muralidharan H, Jean DC, Liu M, Yu W, Baas PW (2018) Tau does not stabilize axonal microtubules but rather enables them to have long labile domains. Curr Biol 28:2181–2189.e2184. 10.1016/j.cub.2018.05.045 [DOI] [PubMed] [Google Scholar]
- Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66:75–85. 10.1097/nen.0b013e31802d6da9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regalado-Reyes M, Furcila D, Hernández F, Ávila J, DeFelipe J, León-Espinosa G (2019) Phospho-tau changes in the human CA1 during Alzheimer's disease progression. J Alzheimers Dis 69:277–288. 10.3233/JAD-181263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S, White K (1988) The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol 126:294–303. 10.1016/0012-1606(88)90139-x [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Murayama M, Suzuki T, Dohmae N, Yen SH, Takashima A (2007) Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur J Neurosci 25:3020–3029. 10.1111/j.1460-9568.2007.05555.x [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. 10.1126/science.1113694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Zhang W, Falcon B, Goedert M (2020) Cryo-EM structures of tau filaments. Curr Opin Struct Biol 64:17–25. 10.1016/j.sbi.2020.05.011 [DOI] [PubMed] [Google Scholar]
- Schweers O, Mandelkow EM, Biernat J, Mandelkow E (1995) Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci USA 92:8463–8467. 10.1073/pnas.92.18.8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S, Skoulakis EM, Mudher A (2017) Distinct phenotypes of three-repeat and four-repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 105:74–83. 10.1016/j.nbd.2017.05.003 [DOI] [PubMed] [Google Scholar]
- Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, Mandelkow E, Knowles TP, Mandelkow E, Klenerman D (2015) A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun 6:7025. 10.1038/ncomms8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S (2008) Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci USA 105:13568–13573. 10.1073/pnas.0806268105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda Y, Yoshikawa M, Almeida OF, Sumioka A, Maeda S, Osada H, Kondoh Y, Saito A, Miyasaka T, Kimura T, Suzuki M, Koyama H, Yoshiike Y, Sugimoto H, Ihara Y, Takashima A (2015) Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat Commun 6:10216. 10.1038/ncomms10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulos I, Galas MC, Silva JM, Skoulakis E, Wegmann S, Maina MB, Blum D, Sayas CL, Mandelkow EM, Mandelkow E, Spillantini MG, Sousa N, Avila J, Medina M, Mudher A, Buee L (2017) Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun 5:91. 10.1186/s40478-017-0489-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson MJ, Larsson J (2007) Thioredoxin-2 affects lifespan and oxidative stress in Drosophila. Hereditas 144:25–32. 10.1111/j.2007.0018-0661.01990.x [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D (2008) Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell 14:76–85. 10.1016/j.devcel.2007.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talmat-Amar Y, Arribat Y, Redt-Clouet C, Feuillette S, Bougé AL, Lecourtois M, Parmentier ML (2011) Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum Mol Genet 20:3738–3745. 10.1093/hmg/ddr290 [DOI] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17:431–439. 10.1093/hmg/ddm320 [DOI] [PubMed] [Google Scholar]
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, Cong X, Schilling B, Ellerby LM, Huganir RL, Gan L (2016) Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 90:245–260. 10.1016/j.neuron.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiri EN, Sykiotis GP, Papassideri IS, Terpos E, Dimopoulos MA, Gorgoulis VG, Bohmann D, Trougakos IP (2013) Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 12:802–813. 10.1111/acel.12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiri EN, Terpos E, Papanagnou ED, Kastritis E, Brieudes V, Halabalaki M, Bagratuni T, Florea BI, Overkleeft HS, Scorrano L, Skaltsounis AL, Dimopoulos MA, Trougakos IP (2017) Milder degenerative effects of Carfilzomib vs. Bortezomib in the Drosophila model: a link to clinical adverse events. Sci Rep 7:17802. 10.1038/s41598-017-17596-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T, Quinn WG (1985) Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol A 157:263–277. 10.1007/BF01350033 [DOI] [PubMed] [Google Scholar]
- Tully T, Preat T, Boynton SC, Del Vecchio M (1994) Genetic dissection of consolidated memory in Drosophila. Cell 79:35–47. 10.1016/0092-8674(94)90398-0 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319. 10.1038/nprot.2016.136 [DOI] [PubMed] [Google Scholar]
- Ubhi KK, Shaibah H, Newman TA, Shepherd D, Mudher A (2007) A comparison of the neuronal dysfunction caused by Drosophila tau and human tau in a Drosophila model of tauopathies. Invert Neurosci 7:165–171. 10.1007/s10158-007-0052-4 [DOI] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E (2005) Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta 1739:158–166. 10.1016/j.bbadis.2004.09.010 [DOI] [PubMed] [Google Scholar]
- Walker JA, Tchoudakova AV, McKenney PT, Brill S, Wu D, Cowley GS, Hariharan IK, Bernards A (2006) Reduced growth of Drosophila neurofibromatosis 1 mutants reflects a non-cell-autonomous requirement for GTPase-Activating Protein activity in larval neurons. Genes Dev 20:3311–3323. 10.1101/gad.1466806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E (2016) Tau in physiology and pathology. Nat Rev Neurosci 17:5–21. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- Weaver CL, Espinoza M, Kress Y, Davies P (2000) Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging 21:719–727. 10.1016/S0197-4580(00)00157-3 [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Tashayev VL, O'Connor LB, Larsen CN, Kasperek E, Pickart CM (1995) Metabolism of the polyubiquitin degradation signal: structure, mechanism, and role of isopeptidase T. Biochemistry 34:14535–14546. 10.1021/bi00044a032 [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293:711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E (1998) Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 252:542–552. 10.1046/j.1432-1327.1998.2520542.x [DOI] [PubMed] [Google Scholar]