

Abstract
Transcriptome analysis can provide clues to biological processes affected in different genetic backgrounds or/and under various conditions. The price of RNA sequencing (RNA-seq) has decreased enough so that medium- to large-scale transcriptome analyses in a range of conditions are feasible. However, the price and variety of options for library preparation of RNA-seq can still be daunting to those who would like to use RNA-seq for their first time or for a single experiment. Among the criteria for selecting a library preparation protocol are the method of RNA isolation, nucleotide fragmentation to obtain desired size range, and library indexing to pool sequencing samples for multiplexing. Here, we present a high-quality and a high-throughput option for preparing libraries from polyadenylated mRNA for transcriptome analysis. Both high-quality and high-throughput protocol options include steps of mRNA enrichment through magnetic bead-enabled precipitation of the poly-A tail, cDNA synthesis, and then fragmentation and adapter addition simultaneously through Tn5-mediated ‘tagmentation’. All steps of the protocols have been validated with Arabidopsis thaliana leaf and seedling tissues and streamlined to work together, with minimal cost in money and time, thus intended to provide a beginner-friendly start-to-finish RNA-seq library preparation for transcriptome analysis.
Keywords: Transcriptomics, RNA-seq, Tagmentation, Multiplexing, Plant, Arabidopsis thaliana
Background
The ability to assay the transcriptional landscape revolutionized the study of biology, through Southern blotting, expressed sequence tag (EST) analysis, microarrays, and finally RNA sequencing (RNA-seq). RNA-seq in particular, through its unprecedented range and sensitivity, allows investigation of otherwise intractable questions in gene regulation ( Lister et al., 2008 ; Mortazavi et al., 2008 ; Wang et al., 2009 ). Advanced applications of RNA-seq include expanding the sample size for study across time or tissue, allowing gene regulatory network inference (de Luis Balaguer et al., 2017 ), and refining understanding of precision responses in a finer scale, such as in single- or few-cell transcriptomics experiments (Efroni and Birnbaum, 2016). The range of biological questions that can be addressed by RNA-seq, and its use, have risen dramatically since its introduction ( Hrdlickova et al., 2017 ). However, many factors have limited its use more broadly, particularly in non-specialist labs.
One of the chief restricting factors of RNA-seq is its overall cost. While the cost of sequencing itself has fallen greatly, the cost of preparing libraries for sequencing has not experienced such a dramatic shift (Rohland and Reich, 2012; Hou et al., 2015 ). The prohibitive cost of library preparation is compounded by the growing understanding of biological variability in transcription ( Busby et al., 2013 ), hence requiring larger numbers of biological replicates and finer dissection of tissue-specific responses which, combined, lead to a requirement for greater numbers of libraries to address any single question ( Yuan et al., 2018 ). This greater number of libraries is not only more expensive but can cost greater amounts of time invested in sample preparation, especially as not all library preparation protocols are easily scalable. Recent advances in RNA-seq library preparation have the potential to ameliorate these issues, increasing speed and decreasing cost of sample preparation for RNA-seq experiments.
Originally, preparation of sequencing libraries typically involved fragmentation of nucleic acids via enzyme digestion, sonication, or divalent cations and heat with RNA, followed by ligation with adapter sequences for both library identification and amplification. The use of a hyperactive Tn5 transposase allows for fragmentation and adapter tagging in one step, termed ‘tagmentation’ ( Picelli et al., 2014 ). This simple dual-step procedure requires no hardware beyond a thermocycler, is amenable to large-scale multiplexing, and requires as little as picogram amounts of input nucleic acid ( Adey et al., 2010 ; Brouilette et al., 2012 ). The Nextera kits available from Illumina use Tn5-based tagmentation technology, but several recent studies have shown that user-prepared Tn5 transposasome and associated buffers perform as well as commercially available versions ( Picelli et al., 2014 ; Pisupati et al., 2017 ; Hennig et al., 2018 ).
While Tn5 presents a library preparation technique suitable for multiplexing, the extraction of RNA and preparation of cDNA remain a bottleneck. RNA extraction may be performed by traditional Trizol extraction or through the use of RNA extraction kits, both of which are traditionally not easily scalable and can become prohibitively expensive when preparing large numbers of samples. Most papers exploring user-prepared Tn5 have focused on small numbers of samples with many technical replicates, but techniques for RNA extraction with many samples and cDNA synthesis from plant tissue are available ( Kumar et al., 2012 ; Townsley et al., 2015 ).
Here, we present a tissue-to-library RNA-seq protocol, amenable to multiplexing with complete preparation of 96 samples in three days, with costs of less than £10 per sample. Moreover, we include notes on optimization to accommodate end-user modifications, and finally novel insights, such as the use of 9-bp indices, which facilitate downstream analysis.
Materials and Reagents
Notes:
Store all reagents at room temperature unless otherwise noted.
Reagents used in multiple sections are only mentioned once here.
-
For total RNA and mRNA isolation
2-ml SafeSeal microcentrifuge tubes (Sarstedt. catalog number: 72.695.500)
1.5-ml TubeOne® microcentrifuge natural tubes, free from RNase, DNase, DNA and Pyrogen (STARLAB, catalog number: S1615-5550)
Tungsten carbide beads (Qiagen, catalog number: 69997)
10-µl Graduated Tip, Refill, RNase, DNase, DNA and Pyrogen free (STARLAB, catalog number: S1111-3700-C)
200-µl Yellow Tip, Refill, RNase, DNase, DNA and Pyrogen free (STARLAB, catalog number: S1111-0706-C)
1,250-µl XL Graduated Tip, Refill, RNase, DNase, DNA and Pyrogen free (STARLAB, catalog number: S1112-1720-C)
Plant tissue: 5-week-old and 2-week-old Arabidopsis thaliana plants were used successfully
Liquid nitrogen
RNA Clean & Concentrator-25 with Zymo-SpinTM IIC Column (Zymo Research, catalog number: R1017 or R1018)
-
TRI Reagent® (Sigma-Aldrich, catalog number: T9424)
Note: Store at 4 °C.
-
DEPC-Treated Water (InvitrogenTM, catalog number: 11597065)
Note: RNase-free water (e.g., Sigma-Aldrich, catalog number: W4502) works equally well.
≥ 99.8% ethanol/EtOH (Sigma-Aldrich, catalog number: 32221-M)
RNaseZapTM RNase Decontamination Solution (InvitrogenTM, catalog number: AM9780, AM9782 or AM9784)
-
For high-throughput total RNA isolation
Conical tubes (Corning, Falcon, catalog numbers: 352096 and 352070)
Glass beads for tissue homogenization (Sigma-Aldrich, catalog number: Z250473)
Tris-HCl pH 8.0, 1 M (InvitrogenTM, catalog number: AM9855G)
LiCl, 8 M (Sigma-Aldrich, catalog number: L7026-500 ml)
EDTA pH 8.0, 0.5 M (Sigma-Aldrich, catalog number: 03690)
SDS (InvitrogenTM, catalog number: AM9820)
Dithiothreitol (DTT, Sigma-Aldrich, catalog number: 43815)
Antifoam A (Sigma-Aldrich, catalog number: A5633-100G)
5 M NaCl (InvitrogenTM, catalog number: AM9760G)
-
2-mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
Note: Store in hazardous chemical cabinet.
RNase-free water (Sigma-Aldrich, catalog number: W4502-1L)
-
Streptavidin-conjugated magnetic beads (NEB, catalog number: S1420S)
Note: Store at 4 °C.
-
Biotinylated poly-dT oligo (Thermo Fisher, catalog number: NA)
Notes:
5′-bio-ACAGGACATTCGTCGCTTCCTTTTTTTTTTTTTTTTTTTT-3′.
Store at -20 °C.
RNase-free PCR strips (Brooks Life Sciences, 4titude, catalog number: 4ti-0781)
Optional: Tween-20 (Sigma-Aldrich, catalog number: P1379-25ML)
Optional Sera-mag speedbeads (Thermo Fisher, catalog number: 09-981-123)
Optional: SYBR green I (Thermo Fisher, catalog number: S7563)
Lysis/Binding Buffer (LBB) (see Recipes)
htLBB (see Recipes)
WBA (see Recipes)
WBB (see Recipes)
LSB (see Recipes)
1 M betamercaptoethanol (see Recipes)
5 mM DTT (see Recipes)
Optional: Serapure (see Recipes)
-
For RNA cleanup and cDNA preparation
Notes:
All reagents in Procedures C and D should be stored at -20 °C;
Wherever possible, we recommend purchasing reagents as solutions rather than powders to minimize manipulation prior to use and facilitate prevention of RNase contamination.
Dynabeads® Oligo (dT)25 (Thermo ScientificTM, catalog number: 61002)
-
Random Decamers (50 µM) (InvitrogenTM, catalog number: AM5722G)
Note: The Random Primer Mix (New England BioLabs, catalog number: S1330S) can also be used as an alternative. The random primer mix may give slightly better coverage of the entire coding sequence.
RNaseOUTTM Recombinant Ribonuclease Inhibitor (InvitrogenTM, catalog number: 10777019)
-
DNase I recombinant, RNase-free (Roche, catalog number: 04716728001)
Note: To remove large amounts of DNA contamination, alternatively TURBO DNA-freeTM Kit (InvitrogenTM, catalog number: AM1907) may be used.
High-fidelity polymerase (Thermo ScientificTM, Phusion, catalog number: F534L)
-
High-fidelity reverse transcriptase (InvitrogenTM, SuperScript III, catalog number: 18080093)
Optional: For some applications, a less high-fidelity reverse transcriptase (e.g., RevertAid, Thermo ScientificTM, catalog number: K1622) may be used at a great cost savings.
dNTP mix, 25 mM each (Thermo ScientificTM, catalog number: R1121)
E. coli DNA Ligase and buffer (New England BioLabs, catalog number: M0205S/L)
E. coli DNA Polymerase I (New England BioLabs, catalog number: M0209S/L)
RNaseH (New England BioLabs, catalog number: M0297S/L)
E. coli Exonulease I (New England BioLabs, catalog number: M0293S/L)
RNase A, DNase and protease-free (10 mg/ml) (Thermo ScientificTM, catalog number: EN0531)
Ampure XP beads (Beckman Coulter, catalog number: A63880)
Qubit dsDNA HS Assay Kit (InvitrogenTM, catalog number: Q32851 or Q32854)
-
For Tagmentation library preparation
-
Nextera DNA Library Prep Kit (Illumina, catalog number: FC-121-1030)
Note: This has been discontinued, and listed reagent in item 2 (below) is the replacement.
Illumina Tagment DNA TDE1 Enzyme and Buffer Kits (Illumina, catalog number: 20034197 or 20034198)
NEBNext® High-Fidelity 2× PCR Master Mix (New England BioLabs, catalog number: M0541S/L)
KAPA SYBR FAST qPCR Master Mix (2×) Universal (Roche, catalog number: KK4600, KK4601, KK4602 or KK4618)
TAPS-NaOH (Sigma-Aldrich, catalog number: T5130)
MgCl2 (Sigma-Aldrich, catalog number: M1028)
PEG 8000 (Sigma-Aldrich, catalog number: 1546605)
Homemade 5× tagmentation buffer (see Recipes)
-
-
For library indexing
Custom designed Illumina sequencing compatible index primers and synthesized by Sigma (Supplemental)
Equipment
-
Tissue homogenizer (SPEX Sample Prep, Geno/Grinder)
Note: Qiagen TissueLyser also used successfully: sample plates should be rotated half-way through grinding to ensure even grinding.
Vortex mixer (any brand)
NanoDropTM 8000 Spectrophotometer (Thermo ScientificTM, catalog number: ND-8000-GL)
2100 BioAnalyzer System (Agilent, catalog number: G2939BA)
4150 TapeStation System, optional (Agilent, catalog number: G2992AA)
MagnetoPURE BIG SIZE (Chemicell, catalog number: MP-20)
1.5-ml tube rotator mixer (SoCal BioMed, Waverly, catalog number: RT1-E)
Magwell 96-well magnetic separator (EdgeBio, catalog number: 57624)
Plate mixer (Scientific Industries, Genie, catalog number: Si-0400A)
Microcentrifuge (Thermo ScientificTM, MicroCL 17, catalog number: 75002450)
Strip centrifuge (any brand)
PCR machine (any programmable thermo-cycler)
-
96-well microplate heat block, optional (any brand)
Note: Preferred for single temperature incubation steps.
Multichannel pipette, 0.5-10 µl (Eppendorf, catalog number: ES-12-10)
Multichannel pipette, 30-300 µl (Eppendorf, catalog number: ES-12-300)
QubitTM Flex Fluorometer (InvitrogenTM, catalog number: Q33327)
Real-time quantitative PCR machine, here we use CFX96 Touch Real-Time PCR Detection System C1000 TouchTM (BioRad)
Procedure
Notes:
Procedures A-D are standard protocols for mRNA-seq library preparation using Tn5. Details on which steps can be paused or customized are also indicated as additional notes. Procedure E is for high-throughput RNA-seq library preparation. Users who are already familiar with all steps in Procedures A-D and confident with handling large amount of RNA-seq samples can start their library preparation from the Procedure E.
For all steps here involving RNA or mRNA, please make sure all surfaces are clean (e.g., use RNaseZapTM RNase Decontamination Solution to clean the bench surface). All tubes and tips that will touch nucleic acids should be nuclease-free grade as indicated.
-
Total RNA isolation
Here we are using 5-week-old Arabidopsis thaliana plants growing under short-day (10-h light/14-h dark) at 22 °C as an example for demonstration.
-
To harvest material, quickly and gently fold tissue into pre-labelled 2-ml tube containing 2 homogenization carbide beads, and snap freeze tissue in liquid nitrogen. Two fully-expanded leaves from each plant and 3 plants from each genotype and treatment (total of 6 leaves) were used as one sample ( Ding et al., 2020 ).
Notes:
To reduce the sample size and working load, 2 leaves per sample was tested and also works well with this protocol.
If working with younger Arabidopsis, approximately 2 seedlings or 20-100 mg (fresh weight) plant tissue per sample works well.
All the following steps can be scaled up in proportion if more starting material is desired.
Always use eye protection when working with liquid nitrogen.
-
Grind tissue in Geno/Grinder blocks pre-cooled in liquid nitrogen: a single 30-s pulse at 30/s frequency is sufficient for leaf material. After getting the powder of the finely ground tissue, re-freeze in liquid nitrogen.
Notes:
It is helpful to keep blocks in liquid nitrogen while loading samples for grinding.
Grinding conditions may vary depending on tissue. 0.5-1 min grinding at 25-30/s frequency is generally enough, but samples should be visually evaluated for homogenization to a fine powder.
-
Add 800 ml pre-cooled TRI Reagent® solution into each tube, close the lid and quickly resuspend the tissue powder with a vortex mixer. Incubate the samples on ice for 3 min.
Note: Take care when opening tubes to avoid explosions from expanding warming samples.
Vortex samples again from slurry into a homogenized solution.
-
Incubate all samples on ice for another 5 min. From this step onwards, work at room temperature.
Note: If working with multiple samples, work with 3 to 4 samples in one turn for the two-step vortexing, to minimize RNA degradation.
Spin all tubes at 10,000 × g in a benchtop centrifuge for 8 min.
While waiting for the centrifugation, prepare the same number of fresh 2-ml tubes as in Step A1. Label and add 400 µl ≥ 99.8% EtOH into each tube.
-
Take 400 µl supernatant from each tube into the corresponding new 2-ml tube prefilled with EtOH. Mix the samples by inverting the tubes gently for several times until all samples are homogenized.
Notes:
Left-over supernatant can be stored at -70 or -80 °C as backup samples.
Volume of supernatant can be adjusted; volume of EtOH should be adjusted to remain equivalent to supernatant volume.
Bind RNA: transfer the mixed solution to a Zymo-SpinTM IIC Column in a Collection Tube and centrifuge at 10,000 × g for 30 s. Discard the flow-through.
Wash the column: add 250 µl RNA Wash Buffer and centrifuge at 10,000 × g for 30 s. Discard the flow-through.
-
Remove genomic DNA (gDNA): for each sample to be treated, prepare DNase I Reaction Mix in an RNase-free tube. Mix well by gentle inversion. Transfer 80 µl mix onto each column, close the lids and incubate at room temperature (20-30 °C) for 15 min.
Component Volume (1 reaction)
DEPC-Treated Water 69 µl
RNaseOUTTM (40 U/µl) 1 µl
DNaseI Incubation Buffer (10×) 8 µl
Recombinant DNase I, RNase-free (10 U/µl) 2 µl
80 µl/each tube
Notes:
If you have multiple tubes of RNA samples, make a master mix by multiplexing the standard DNase I Reaction Mix; make with 10% extra.
If large amounts of gDNA contamination are consistently found after Procedure B (e.g., by PCR), TURBO DNase may be used for higher efficiency DNA removal, according to manufacturer’s protocol.
Wash the column: Add 400 µl RNA Wash Buffer and centrifuge at 10,000 × g for 30 s. Skip the step of discarding the flow-through.
Final wash: Add 200 µl RNA Wash Buffer and centrifuge at 10,000 × g for 2 min to ensure complete removal of the wash buffer. Transfer the column carefully into a 1.5-ml tube and discard the flow-through collection tube.
-
RNA Elution: Add 50 µl DEPC-Treated Water directly to the column matrix and centrifuge at 10,000 × g for 30 s.
Note: Alternatively, for highly concentrated RNA, use ≥ 25 µl Nuclease-free Water or DEPC-treated water.
-
Measure RNA concentration with a NanoDrop spectrophotometer.
Notes:
Eluted RNA should be kept on ice.
Optional break point: RNA can be stored at -70~-80 °C for up to several weeks;
Optional: whenever it is possible, the quality of total RNA can be assessed on high-sensitivity device with corresponding compatible RNA chips/reagents and program that is specifically designed for plant RNA, e.g., Bioanalyzer 2100 or TapeStation 4150 from Agilent. The RNA electropherogram results from Aglient machines provide an RNA integrity number (RIN). RNA with RIN greater than 6 are desirable for constructing high quality NGS libraries.
-
-
mRNA isolation from total RNA
Note: Perform all steps on ice unless stated otherwise, particularly those affecting mRNA.
-
Prepare Dynabeads® Oligo (dT)25: Resuspend Dynabeads® Oligo(dT)25 by pipetting or gentle vortexing, thoroughly before use.
Note: No clumps of beads should be visible in base of container and solution should appear homogenous in pipet tip. From the storage container this should take several seconds of gentle mixing; for later steps with small volumes generally 5-10 complete cycles of pipetting up and down will result in complete bead resuspension.
-
For each sample, transfer 10 µl well-suspended beads (suitable for 2-10 µg total Arabidopsis RNA, adjust volume if desired) to an RNase-free 1.5-ml microcentrifuge tube and place the tube on a magnet (e.g., MagnetoPURE).
Note: This step can also be performed in PCR strips on a magnet that is compatible.
After 30 s (or when the suspension is clear), remove the supernatant.
Wash beads: Remove the tube from the magnet and resuspend beads in 50-100 µl fresh Lysis/Binding Buffer provided by Dynabeads® Oligo (dT)25 kit. Place suspension back on magnet.
After 30 s (or when the suspension is clear), remove the supernatant (Lysis/Binding Buffer) from the washed Dynabeads.
Remove the microcentrifuge tube from the magnet.
Add up to 10 µg isolated total RNA in 300 µl DEPC-Treated Water (up to 33 ng/µl) onto the bead pellet and resuspend beads completely by pipetting.
Place tubes in a rotator mixer and incubate with continuous mixing for 3-5 min at room temperature to allow the polyA tail of the mRNA to hybridize to the oligo (dT)25 on the beads.
Place the suspension back on the magnet for 2 min to collect beads/mRNA and remove the supernatant.
Wash the beads/mRNA complex two times with 600 µl Washing Buffer A provided by Dynabeads® Oligo (dT)25 kit at room temperature. Resuspend beads completely and use the magnet to separate the beads from the solution for each washing step.
Wash the beads/mRNA complex once with 300 µl Washing Buffer B provided by Dynabeads® Oligo (dT)25 kit. Resuspend beads completely and use the magnet to separate the beads from the solution.
-
Elute mRNA: add 20 µl of 10 mM Tris-HCl pH 7.5 (Elution Buffer) and incubate at 65-80 °C (water bath or heat block) for 2 min. Immediately place the tube on the magnet, transfer the supernatant containing the mRNA to a new RNase-free tube and place this tube on ice. Do not discard the beads.
Notes:
In this step, the heat melts the mRNA from the Oligo(dT)25. If the sample cools down before transfer, the mRNA re-binds onto the Oligo(dT)25 and yield is reduced. It is possible to re-heat the sample to re-release the mRNA;
At this step, usually the majority of ribosomal RNA (rRNA) will be removed. However, for the downstream cDNA library construction for NGS, any traceable amount of rRNA contamination should be avoided. The following steps ensure better purity of cDNA libraries for RNA-seq.
Reuse of Dynabeads® Oligo(dT)25: wash the beads two times in Washing Buffer B (same as Step B11). Resuspend beads completely in each wash.
Dilute eluted mRNA with 80 µl Lysis/Binding Buffer (4× volume).
Start second mRNA purification: remove second wash of Washing Buffer B from the beads and add the diluted mRNA.
Incubate with mixing at room temperature for 3-5 min.
-
Repeat Steps B4 to B12, eluting RNA finally in 10 µl 10 mM Tris-HCl pH 7.5.
Notes:
Optional break point: mRNA can be stored at -70~-80 °C for up to several weeks;
Keep the other 10 µl of mRNA in the -70~-80 °C freezer as backups.
-
-
Synthesis of double stranded (ds) cDNA
Note: Perform all steps on ice unless stated otherwise, particularly those affecting mRNA.
-
Add the following components to a PCR strip as Mix I:
Component Volume (1 reaction)
Random Decamers (50 µM) or Random Primer Mix (60 µM) 1 µl
Purified mRNA (from Procedure B or Procedure E) 10 µl
Denature RNA to remove secondary structures: heat mixture to 70 °C for 5 min, then transfer immediately onto ice for at least 1 min.
-
Collect the contents of the tube by brief centrifugation and add Mix II:
Component Volume (1 reaction)
DEPC-Treated Water 1 µl
5× SuperScript III First-Strand Buffer 4 µl
0.1 M DTT 1 µl
10 mM dNTPs Mix 1 µl
RNaseOUTTM (40 U/µl) 1 µl
SuperScriptTM III RTase (200 U/µl) 1 µl
9 µl/each tube
Note: The recipes provided are for a final volume of 20 µl. If the yield of mRNA is adequate, all volumes can be halved for a 10 µl reaction with no damage to yield or quality of first-strand cDNA.
-
After mixing by pipetting and brief centrifugation, load the PCR strip into a thermocycler with the following program:
Temperature Time
25 °C 5 min
50 °C 60 min
70 °C 15 min
Notes:
Optional break point: first strand cDNA synthesized from mRNA can be stored at -20 °C for several months.
Only use 10 µl of first strand cDNA for the following steps and keep remainder in -20 °C freezer as backup.
-
Prepare following second strand cDNA synthesis mix:
Component Volume (1 reaction)
DEPC-Treated Water 28 µl
10× E. coli DNA Ligase Buffer 5 µl
25× additive (500 mM Tris-HCl, 25 mM MgCl2) 2 µl
RNaseH (0.5 U/µl) 1 µl
E. coli DNA Polymerase I (10 U/µl) 2 µl
E. coli DNA Ligase (10 U/µl) 1 µl
10 mM dNTPs 1 µl
40 µl/each tube
The final volume of the mix is 50 µl.
Incubate reaction at 16 °C for 2.5 h.
Add 1 µl of Exonulease (Exo) I (20 U/µl) to each well of the PCR strip to deplete excess random decamers/primers.
Incubate at 37 °C for 30 min.
Add 5 µl of 0.5 M EDTA (pH 8.0) to terminate reaction.
Add 1 µl of 10 mg/ml RNase A.
Incubate at 37 °C for 30 min to degrade all residual RNA.
-
Transfer 20-µl ds cDNA into 1.5-ml tubes (DNase-free).
Notes:
Optional break point: ds cDNA can be stored at -20 °C for several months.
Store the remainder at -20 °C as backup.
-
Gently vortex AMPure XP to resuspend settled magnetic beads. Add 36 µl AMPure XP beads (1.8× volume of the input cDNA) to the cDNA.
Note: AMPure XP beads bind nucleic acids size-selectively based on the concentration of the ABR buffer (that they are stored in), so following the volume ratios is crucial.
Mix the AMPure XP and sample thoroughly by pipetting (mixing >10 times).
Incubate at room temperature 5 min.
-
Place the reaction tube onto a magnetic rack for 2 min to separate beads from the solution and remove the supernatant.
Note: To avoid loss of cDNA bound on beads, leave 5 µl volume behind (e.g., remove 51 µl).
EtOH wash: add 200 µl 70% EtOH on the magnet rack. Incubate at room temperature for 30 s, no need to resuspend beads. Fully remove and discard ethanol.
Repeat for a total of two washes. Remove the tubes from the magnet rack.
-
Elution: add ≥ 25 µl of 10 mM Tris-HCl pH 7.5 (Elution Buffer), mix thoroughly by pipetting.
Notes:
Recommend pipetting up and down 20 times for complete mixing;
Application of less than 25 µl of Elution Buffer may result in decreased efficiency of elution.
Incubate at room temperature for 2 min.
Place the reaction plate onto a magnet rack for 1 min to separate beads from solution.
Transfer purified ds cDNA to fresh 1.5-ml tubes or strips (DNase-free).
-
Measure the cDNA concentration with dsDNA HS Assay Kit in a Qubit Flex Fluorometer by following the commercial recommended protocol.
Note: Optional break point. purified ds cDNA can be stored at -20 °C for several months.
-
-
Library preparation with cDNA tagmentation
Notes:
Keep all samples and products on ice all the time except when they are in the thermocycler.
Protocol can be paused after each step in this section; store products at -20 °C.
-
Dilute cDNA to desired concentration
Note: The tagmentation reaction has at least four parameters (starting cDNA, Tn5 amount, incubation time, and optional PEG in reaction) which can be optimized to obtain an acceptable range of library size with minimal PCR cycles. We present here two possibilities: one below (more labor-intensive to minimize amplification steps) and one for high-throughput library prep (Procedure E, generally acceptable yields with reduced time required per-library, Figure 1). These should provide reasonable starting points, but cannot fully replace optimization for desired conditions. For a more complete exploration of parameters see Picelli et al. (2014).
-
Make master mix for tagmentation and aliquot in each well of PCR strip or plate as shown in the table below:
Component Volume (1 reaction) Nuclease-free water 3.4 µl Illumina Tagment DNA TDE1 Enzyme 0.1 µl Illumina Tagment DNA TDE1 Buffer 0.5 µl 4 µl/each well (Optional break point) Incubate the reaction mix at 55 °C for 5 min. At the end of the reaction, place the tube immediately on ice.
-
Amplify transposed cDNA fragments: add 10 µl of 2× NEBNext PCR master mix to each cDNA tagmentation product (5 µl) into each well of PCR strip or plate. Add barcoded paired primers P7-i and P5-j into each tube in the strips.
Component Volume (1 reaction) tagmentation reaction 5 µl NEBNext PCR master mix 10 µl 10 µM P7-i (i=1,2,...) 2.5 µl 10 µM P5-j (j=1,2, …) 2.5 µl -
Load the reaction mix into a thermocycler with the program for pre-PCR cycles:
Time Temperature 1 cycle 5 min 72 °C (gap filling) 30 s 98 °C 5 cycles 10 s 98 °C 30 s 63 °C 1 min 72 °C (Optional break point)
-
To determine how many cycles for optimal library yield with minimal PCR duplications, using real-time quantitative PCR (RT-qPCR). Prepare the qPCR mix as shown in the table:
Component Volume (1 reaction) Nuclease-Free Water 4 µl 2× KAPA fast qPCR mix 5 µl 10 µM P5-i 0.25 µl 10 µM P7-j 0.25 µl Pre-PCR cDNA library mix (each) 1 µl Notes:
P5-i and P7-j are the same primers as those used in each pre-PCR cDNA library mix;
Other quality control methods can also be applied to determine the additional cycles that are required for generating the final cDNA NGS libraries.
-
Load the reaction mix on a RT-qPCR machine with the program below. Based on the relative fluorescence unit (RFU) curves of each sample, draw a horizontal line at the 1/3 of maximum RFU, and at the cross-over draw a vertical line to cross the axis indicating cycle number (Figure 2, High-quality pre-amplification step). This number is the number of additional cycles that should be applied to the pre-PCR products to generate the final cDNA libraries ( Buenrostro et al., 2015 ).
Time Temperature 40 cycles 10 s 98 °C 30 s 63 °C 1 min 72 °C -
Perform secondary library amplification with determined cycle number.
Time Temperature Determined cycles (5-15 cycles in our experience) 10 s 98 °C 30 s 63 °C 1 min 72 °C Purify the cDNA libraries following Steps C13-C22 using AMPure XP beads. (Optional break point)
Analyse the quantity and quality of the libraries using Bioanalyzer High Sensitivity DNA Analysis, following the standard commercial protocol. Multiplex the cDNA libraries with equal molar ratio and similar size distribution and use appropriate amounts for NGS on desired Illumina sequencing platforms (following instructions from your sequencing facility).
-
Alternative protocol for mRNA isolation
When more than 24 samples are to be prepared, it is beneficial to use a multiplexed RNA extraction. The following protocol is adapted from the HT-Seq and BrAD-seq protocols (Kumar et al. 2012; Townsley et al., 2015 ) and can easily accommodate 48 samples from tissue preparation through ds cDNA synthesis in one day. Figure 2 provides a schematic guide to the mRNA extraction.
-
Harvest tissue into 2-ml tubes containing grinding beads, snap-freeze in liquid nitrogen. Tissue recommendations are as in Procedure A.
Note: Two to four 4-mm glass beads also work well for grinding.
Prepare htLBB: allow approximately 1 ml htLBB per sample if using up to 50 mg tissue. Let htLBB warm to room temperature if prepared beforehand and add 5 µg/µl 2-mercaptoethanol before adding to sample–do not store htLBB with 2-mercaptoethanol.
Grind in liquid nitrogen-cooled Geno/Grinder blocks: grinding conditions as in Procedure A.
-
Re-grind samples with htLBB: add similar volume room-temperature htLBB to plant powder, move samples to a room temperature block and grind again for 1 min. Do not allow samples to freeze.
Notes:
For two Arabidopsis seedlings, add ~450 µl htLBB: excess htLBB is not a problem, but too little htLBB will interfere with mRNA extraction;
If multiple sets of Geno/Grinder blocks are available, changing to a fresh set can help avoid sample freezing.
Incubate at room temperature for approximately 5 min.
Pellet plant debris in room temperature microcentrifuge, ~16,000 × g, 10 min. During spin, label five sets of 8-well PCR strips: two for lysate, one for magnetic beads, and two for sequential RNA elutions.
-
Aliquot supernatant to PCR strips: 2× 200 µl. One set of strips will move forward, keep the second at -70 to -80 °C as backup.
Note: Pellet is not completely solid: it’s recommended that the first, cleaner supernatant aliquot is for immediate use.
Add 1 µl 12.5 µM biotinylated oligo-dT to active strips. Aliquot and store biotin-dT in a master PCR strip to facilitate using multi-channel pipette to dispense to sample tubes.
Incubate 10 min at room temperature.
-
During incubation, prepare strips (third set) with streptavidin magnetic beads. Dispense 20 µl streptavidin bead slurry to each tube and place on magnet. When beads have settled, remove bead storage buffer and resuspend fully in 100 µl htLBB.
Notes:
It is helpful to again use a master mix strip and multichannel pipette.
Beads settle easily: resuspend stock by vortexing at moderate speed after every few tubes dispensed, and with pipet in individual tubes.
When biotin incubation is complete, place htLBB-magnetic bead strips on magnet, remove the supernatant (htLBB), and add the 200 µl lysate-oligo dT mixture onto the beads.
-
Incubate 10 min at room temperature with mixing.
Note: For mixing a plate mixer is recommended, but plates can also be taped to a 1.5-ml tube incubator-mixer set at 1,400/min, or to a vortex at minimum speed.
When mixing is complete, spin strips briefly to remove lysate/bead mixture from lids, place strips in magnetic rack and collect beads for 2 min.
-
Remove supernatant and add 200 µl ice-cold WBA, pipetting well to resuspend beads completely.
Notes:
Supernatant may be discarded or kept for DNA extraction if desired.
It may improve mRNA stability to keep wash buffers, dispensing reservoirs, and magnetic rack on ice, but this is not necessary.
Place strips back in the magnetic rack to collect beads, remove supernatant (WBA), add 200 µl ice-cold WBB, and pipet well to resuspend beads completely.
Place strips back in the magnetic rack to collect beads, remove supernatant (WBB), add 200 µl ice-cold LSB, and pipet well to resuspend beads completely.
Place strips back in a magnetic rack to collect beads, remove supernatant (LSB), add 15.5 µl room temperature 10 mM Tris-HCl, and pipet well to resuspend beads completely.
-
Incubate beads at 80 °C for 2 min.
Note: During heating, warm magnetic rack, by placing in a warm water bath or under warm flowing water.
-
Remove strips from the heating block, place on the magnetic rack, and immediately remove 15 µl to fresh strips (fourth set).
Note: In this step, the heat melts the mRNA from the Oligo(dT)25. If the sample cools down before transfer, the mRNA re-binds onto the Oligo(dT)25 and yield is reduced. It is possible to re-heat the sample to re-release the mRNA.
Resuspend beads in 100 µl WBB.
-
Make DNase master mix:
Component Volume (1 reaction)
DEPC-Treated Water 5 µl
RNaseOUTTM (40 U/µl) 0.5 µl
DNAseI Incubation Buffer (10×) 2.5 µl
Recombinant DNase I, RNase-free (10 U/µl) 2 µl
10 µl/each tube
-
DNase I digestion: add 10 µl reaction mix to 15 µl mRNA, incubate at 37 °C for 20 min.
Note: DNase incubation is not required for all tissues. When optimizing, consider skipping DNase incubation and testing for DNA contamination in RNA (e.g., through a PCR specific to gDNA). If no significant gDNA contamination is detected, DNase treatment may be skipped; a second round of purification is still recommended to remove rRNA contamination.
DNase I inactivation: add 1.5 µl EDTA to each reaction, mix thoroughly by tapping, and incubate at 75 °C for 10 min.
During DNase inactivation, discard WBB from beads (Step E20) and resuspend beads in fresh WBB.
Transfer strips immediately to ice; incubate for at least 2 min.
Add 175 µl htLBB to 25 µl DNase reaction.
Discard second WBB wash and resuspend beads in 200 µl DNAse-treated RNA in htLBB.
-
Repeat Steps E12-E19, performing final elution in 10 µl 10 mM Tris-HCl into the fifth set of labeled strips.
Note: mRNA yield can be estimated by using nanodrop spectrophotometer. Yields should be 20-30 ng/µl, with clean peaks, 260/280 in the range of 2-2.2, 260/230 approximately 2. Testing has shown essentially no rRNA contamination, and gDNA contamination typically in the 0.1-1 ng range, with most samples closer to 0.1 ng.
-
Remaining steps of library preparation (first and second strand cDNA synthesis, tagmentation) can proceed approximately as in Procedures C and D, with the adjustments shown below and in Figure 3.
First and second strand cDNA synthesis can be performed multiplexed in PCR strips. Exonuclease and RNase A steps can be skipped as these contaminants will be largely removed through subsequent AMPure/Serapure cleanup steps.
High-throughput dsDNA quantification: rather than ds cDNA quantification in Qubit, concentration can be estimated using SYBR green with a standard curve in a black 96-well plate using a standard plate reader ( Leggate et al., 2006 ). Briefly: mix 1-2 µl ds cDNA with 1 µl 100x SYBR green in a 96-well plate. Incubate in the dark > 1 min, then excite at 485 nm and measure fluorescence at 535 nm. A standard curve from 0-10 ng/µl can be made from commercially available DNA preps with known concentration (e.g., DNA ladder, calf thymus DNA) and allows estimation of sample concentration.
-
High-throughput AMPure cleanup: for earlier steps, AMPure can be replaced with homemade ‘Serapure’ ( DeAngelis et al., 1995 ; Fisher et al., 2011 ; Lundin et al., 2010 ; Rohland and Reich, 2012).
Note: ‘Serapure’ costs significantly less than AMPure but loses stability after about 1-2 weeks and is not recommended for final purification steps.
-
To minimize PCR cycles needed for library amplification without optimizing for each library, increase starting amounts of ds cDNA, and add PEG to reaction mixture.
Component Volume (1 reaction)
ds cDNA (2 ng/µl) 1 µl
5× homemade tagmentation buffer 1 µl
Illumina Tagment DNA TDE1 Enzyme 0.1 µl
40% PEG 8000 1 µl
RNase-free H2O 1.9 µl
5 µl/each tube
Tagment for 7 min, not 5 min. With these conditions, 10 cycles of amplification will be sufficient for most libraries without over-amplification. Any samples identified without sufficient amplification can be amplified via the two-step procedure outlined in Procedure D. Homemade tagmentation buffer is prepared as in Picelli et al., 2014 with TAPS and MgCl2 (see Recipes).
For library QC, visualize on standard agarose gel and quantify using plate reader and SYBR dye. Use these results to ascertain which libraries need to be re-prepared and to design pooling. Test only final pooled libraries for sequencing on BioAnalyzer.
-
Figure 1. Various reaction conditions can result in acceptable library size distributions.
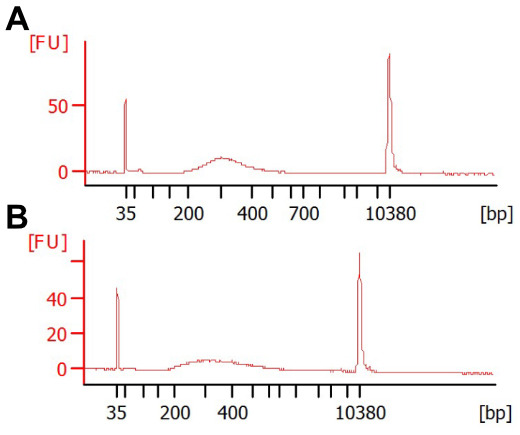
Shown are BioAnalyzer traces from two extremes of DNA concentration, (A) 0.1 ng or (B) 2 ng tagmented cDNA, with 16 or 10 cycles PCR, respectively. Each reaction was performed at 55 °C for 7 min with 0.1 μl Tn5. The reaction in (B) was performed in the presence of 8% PEG. The majority of libraries should be between 300-500 bp in size, as fragments larger than this do not sequence well and fragments smaller than this risk preferential sequencing.
Figure 2. Schematic view of library preparation with tagmentation.

Figure 3. Schematic view of high-throughput mRNA isolation.

Recipes
-
Lysis/Binding Buffer (LBB)
Solution components Stock solutions Volumes for 50 ml 100 mM Tris-HCl 1 M pH 7.5 5 ml 0.5 M LiCl 8 M 6.25 ml 10 mM EDTA 500 mM pH 8 1 ml 1% LiDS 20% w/v 2.5 ml 5 mM DTT 1 M 250 µl RNase-free H2O to 50 ml -
htLBB
Notes:
1 M beta-mercaptoethanol (Recipe 6) should be added to LBB immediately before use: 5 µl/ml.
If using pre-prepared LBB, allow at least 30 min at room temperature to allow antifoam A to warm.
Add LBB at approximately 9 µl/mg, or 450 µl for recommended sample size.
Solution components Stock solutions Volumes for 50 ml 100 mM Tris-HCl 1 M pH 8 5 ml 1 M LiCl 8 M 6.25 ml 10 mM EDTA 500 mM pH 8 1 ml 1% SDS 20% w/v 2.5 ml 5 mM DTT 1 M 250 µl Antifoam A 750 µl RNase-free H2O to 50 ml -
WBA
Solution components Stock solutions Volumes for 50 ml 10 mM Tris-HCl 1 M pH 8 500 µl 150 mM LiCl 8 M 940 µl 1 mM EDTA 500 mM pH 8 100 µl 0.1% SDS 20% (w/v) 125 µl RNase-free H2O to 50 ml -
WBB
Solution components Stock solutions Volumes for 50 ml 1 M pH 8 500 µl 1 M pH 8 500 µl 150 mM LiCl 8 M 940 µl 1 mM EDTA 500 mM pH 8 100 µl RNase-free H2O to 50 ml -
LSB
Solution components Stock solutions Volumes for 50 ml 10 mM Tris-HCl 1 M pH 8 500 µl 150 mM NaCl 5 M 1.5 ml 1 mM EDTA 500 mM pH 8 100 µl RNase-free H2O to 50 ml -
1 M betamercaptoethanol
Solution components Stock solutions Volumes for 100 µl 1 M 2-Mercaptoethanol 14.3 M 7 µl RNase-free H2O 93 µl -
5 mM DTT
Solution components Stock Volumes for 4 ml Dithiothreitol (powder) -- 2.08 g RNase-free H2O to 4 ml -
Serapure
Solution components Stock Volumes for 50 µl PEG 8000 -- 9 g NaCl 5 M 10 ml Tris-HCl, pH 8.0 1 M 500 µl EDTA, pH 8.0 5 M 100 µl RNase-free H2O -- to 49 ml Tween 20 -- 27.5 µl Sera-mag SpeedBeads -- 1 ml (wash twice with TE buffer) -
5× tagmentation buffer ( Picelli et al., 2014 )
Solution components Stock Volumes for 5 ml 50 mM TAPS NaOH pH 8.5 1 M 250 µl 25 mM MgCl2 5 M 10 ml Filter sterilize, aliquot and freeze
Acknowledgments
MB acknowledges the support from the European Union’s Horizon 2020 Research and Innovation Program under Marie Skłodowska-Curie Actions (grant agreement: 703954). KK acknowledges the support from the European Union’s Horizon 2020 Research and Innovation Program under Marie Skłodowska-Curie Reintegration fellowship (grant agreement: 790057). Research in the CZ laboratory is supported by the Gatsby Charitable Foundation, the University of Zurich, the European Research Council (grant agreement: 773153) and the Swiss National Science Foundation (grant agreement: 31003A_182625). PD acknowledges the support from the European Union’s Horizon 2020 Research and Innovation Program under Marie. Skłodowska-Curie Actions (grant agreement: 656243) and a Future Leader Fellowship from the Biotechnology and Biological Sciences Research Council (grant agreement: BB/R012172/1).
The high throughput protocol presented here was modified from published HTR and BrADseq protocols ( Kumar et al., 2012 ; Townsley et al., 2015 ).
Competing interests
The authors have no competing interests to declare.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Supplementary Data.
References
- 1. Adey A., Morrison H. G., Asan, Xun X., Kitzman J. O., Turner E. H., Stackhouse B., MacKenzie A. P., Caruccio N. C., Zhang X. and Shendure J.(2010). Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition . Genome Biol 11(12): R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brouilette S., Kuersten S., Mein C., Bozek M., Terry A., Dias K. R., Bhaw-Rosun L., Shintani Y., Coppen S., Ikebe C., Sawhney V., Campbell N., Kaneko M., Tano N., Ishida H., Suzuki K. and Yashiro K.(2012). A simple and novel method for RNA-seq library preparation of single cell cDNA analysis by hyperactive Tn5 transposase. Dev Dyn 241(10): 1584-1590. [DOI] [PubMed] [Google Scholar]
- 3. Buenrostro J. D., Wu B., Chang H. Y. and Greenleaf W. J.(2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109: 21 29 21-21 29 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Busby M. A., Stewart C., Miller C. A., Grzeda K. R. and Marth G. T.(2013). Scotty: a web tool for designing RNA-Seq experiments to measure differential gene expression. Bioinformatics 29(5): 656-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeAngelis M. M., Wang D. G. and Hawkins T. L.(1995). Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res 23(22): 4742-4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ding P., Ngou B. P. M., Furzer O. J., Sakai T., Shrestha R. K., MacLean D. and Jones J. D. G.(2020). High-resolution expression profiling of selected gene sets during plant immune activation. Plant Biotechnol J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Efroni I. and Birnbaum K. D.(2016). The potential of single-cell profiling in plants. Genome Biol 17: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fisher S., Barry A., Abreu J., Minie B., Nolan J., Delorey T. M., Young G., Fennell T. J., Allen A., Ambrogio L., Berlin A. M., Blumenstiel B., Cibulskis K., Friedrich D., Johnson R., Juhn F., Reilly B., Shammas R., Stalker J., Sykes S. M., Thompson J., Walsh J., Zimmer A., Zwirko Z., Gabriel S., Nicol R. and Nusbaum C.(2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol 12(1): R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hennig B. P., Velten L., Racke I., Tu C. S., Thoms M., Rybin V., Besir H., Remans K. and Steinmetz L. M.(2018). Large-scale low-cost NGS library preparation using a robust Tn5 purification and tagmentation protocol. G3(Bethesda) 8(1): 79-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou Z., Jiang P., Swanson S. A., Elwell A. L., Nguyen B. K., Bolin J. M., Stewart R. and Thomson J. A.(2015). A cost-effective RNA sequencing protocol for large-scale gene expression studies. Sci Rep 5: 9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hrdlickova R., Toloue M. and Tian B.(2017). RNA-Seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kumar R., Ichihashi Y., Kimura S., Chitwood D. H., Headland L. R., Peng J., Maloof J. N. and Sinha N. R.(2012). A high-throughput method for Illumina RNA-Seq library preparation. Front Plant Sci 3: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leggate J., Allain R., Isaac I. and Blais B. W.(2006). Microplate fluorescence assay for the quantification of double stranded DNA using SYBR Green I dye. Biotechnol Lett 28(19):1587-94. [DOI] [PubMed] [Google Scholar]
- 14. Lister R., O'Malley R. C., Tonti-Filippini J., Gregory B. D., Berry C. C., Millar A. H. and Ecker J. R.(2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis . Cell 133(3): 523-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Luis Balaguer M. A., Fisher A. P., Clark N. M., Fernandez-Espinosa M. G., Moller B. K., Weijers D., Lohmann J. U., Williams C., Lorenzo O. and Sozzani R.(2017). Predicting gene regulatory networks by combining spatial and temporal gene expression data in Arabidopsis root stem cells . Proc Natl Acad Sci U S A 114(36): E7632-E7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lundin S., Stranneheim H., Pettersson E., Klevebring D. and Lundeberg J.(2010). Increased throughput by parallelization of library preparation for massive sequencing. PLoS One 5(4): e10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mortazavi A., Williams B. A., McCue K., Schaeffer L. and Wold B.(2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5(7): 621-628. [DOI] [PubMed] [Google Scholar]
- 18. Picelli S., Björklund A. K., Reinius B., Sagasser S., Winberg G. and Sandberg R.(2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24(12): 2033-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pisupati R., Reichardt I., Seren Ü., Korte P., Nizhynska V., Kerdaffrec E., Uzunova K., Rabanal F. A., Filiault D. L. and Nordborg M.(2017). Verification of Arabidopsis stock collections using SNPmatch, a tool for genotyping high-plexed samples . Sci Data 4(1): 170184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rohland N. and Reich D.(2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22(5): 939-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Townsley B. T., Covington M. F., Ichihashi Y., Zumstein K. and Sinha N. R.(2015). BrAD-seq: Breath Adapter Directional sequencing: a streamlined, ultra-simple and fast library preparation protocol for strand specific mRNA library construction. Front Plant Sci 6: 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Z., Gerstein M. and Snyder M.(2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10(1): 57-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yuan Y., Lee H., Hu H., Scheben A. and Edwards D.(2018). Single-Cell Genomic Analysis in Plants. Genes(Basel) 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.