

Abstract
Proteolytic complexes in Mycobacterium tuberculosis (Mtb), the deadliest bacterial pathogen, are major foci in tuberculosis drug development programs. The Clp proteases, which are essential for Mtb viability, are high priority targets. These proteases function through the collaboration of ClpP1P2, a barrel-shaped heteromeric peptidase, with associated ATP-dependent chaperones like ClpX and ClpC1 that recognize and unfold specific substrates in an ATP-dependent fashion. The critical interaction of the peptidase and its unfoldase partners is blocked by the competitive binding of acyldepsipeptide antibiotics (ADEPs) to the interfaces of the ClpP2 subunits. The resulting inhibition of Clp protease activity is lethal to Mtb. Here, we report the surprising discovery that a fragment of the ADEPs retains anti-Mtb activity, yet stimulates rather than inhibits the ClpXP1P2-catalyzed degradation of proteins. Our data further suggest that the fragment stabilizes the ClpXP1P2 complex and binds ClpP1P2 in a fashion distinct from the intact ADEPs. A structure-activity relationship study of the bioactive fragment defines the pharmacophore and points the way towards the development of new drug leads for the treatment of tuberculosis.
Keywords: ClpP peptidase, ClpX, ClpC1, acyldepsipeptide, antibacterial, tuberculosis, proteolysis, protein-ligand interaction, agonist
INTRODUCTION
The proteolytic complexes formed by ClpP and its AAA+ (ATPases associated with diverse cellular activities) partners are among the most captivating antibacterial drug targets to emerge in the past decade.1–3 ClpP is a highly conserved self-compartmentalized peptidase whose physiological function is contingent on physical association with partner AAA+ unfoldases like ClpX, ClpA, or ClpC. Clp proteases are involved in the turnover of a variety of cellular proteins, including proteins arising from aberrant synthesis and transcription factors that regulate virulence-factor production and stress responses.4–9 In its catalytically active state, two heptameric ClpP rings stack “face-to-face” forming a barrel-shaped tetradecamer with a solvent-filled proteolytic chamber large enough to accommodate proteins consisting of several hundred amino acids. The interior surface of the barrel is decorated with 14 serine peptidase active sites, each having a conventional Ser-His-Asp catalytic triad. Because narrow axial pores at each end of the barrel prevent entry of natively folded proteins10–12, degradation of folded proteins by the peptidase requires its association with AAA+ partners that bind, unfold, and translocate substrates into the hydrolytic chamber via conformational changes driven by ATP hydrolysis.7–9,13,14 Complexes of ClpP and its AAA+ partners typically degrade proteins having targeting motifs known as “degrons.” For example, ClpXP can degrade almost any protein bearing a C-terminal ssrA tag.7–9 Without AAA+ partners, ClpP can only degrade small peptides that can diffuse through the pores into the proteolytic chamber.
The genes encoding ClpP and its AAA+ partners are essential for virulence in some pathogenic bacteria (e.g., Staphylococcus aureus15,16, Streptococcus pneumonia17–19, and Listeria monocytogenes20) and for viability in others (e.g., Mycobacterium tuberculosis21–23), making these enzymes compelling targets for drug development. In M. tuberculosis (Mtb), a globally important human pathogen, ClpP and its AAA+ partners are attractive both as drug targets and as subjects for basic research. Mtb is distinguished from many bacteria in that it has two co-transcribed genes called clpP1 and clpP2.24–28 The cognate gene products, Mtb ClpP1 and ClpP2, form discrete heptameric rings that are individually inactive, but conditionally associate to form an active heterotetradecamer.26,28,29 The ClpP1P2 assembly is stabilized by: interaction with ClpX or ClpC1, active translocation of protein substrates into the degradation chamber, and binding N-blocked peptide “agonists” that mimic substrate contacts within the active site (Fig. 1a).25–28 We determined the crystal structure of Mtb ClpP1P2 stabilized by an ADEP and a peptide agonist,28 and others have determined the structure stabilized by an agonist.30 The ClpP1 and ClpP2 rings share an overall fold, yet have numerous structural differences;28,30 some of which explain the selective interaction of Mtb ClpX and ClpC1 with the ClpP2 ring of ClpP1P2.29
Figure 1.
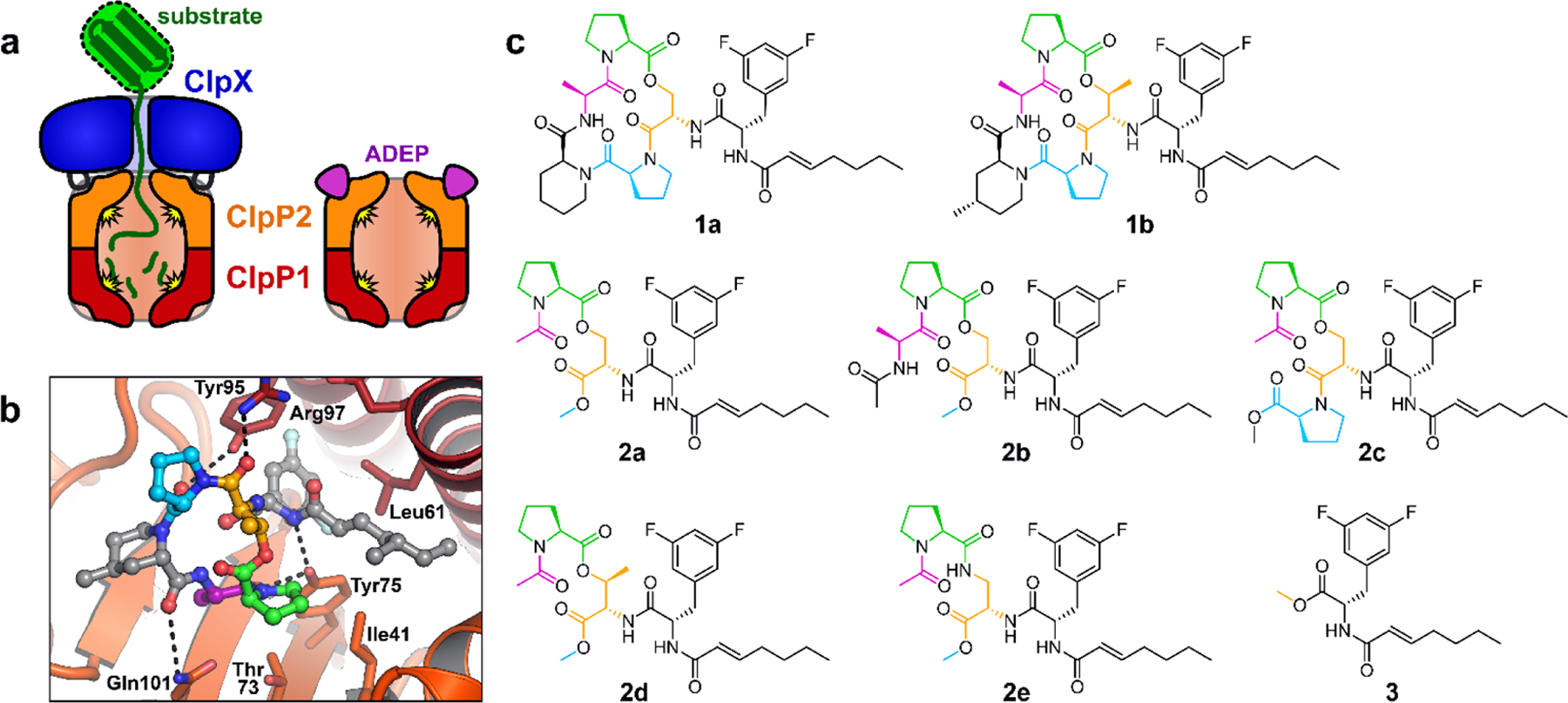
ADEPs bind to the ClpP2 LGF-pockets. (a) Mtb Clp peptidases consist of heptameric rings of ClpP1 and ClpP2 that interact to form a ClpP1P2 heteromer, together with a ring hexamer of ClpX or ClpC1 (not shown) that binds to the surface of ClpP2 via flexible LGF-loops. ADEPs bind to the LGF-pockets of ClpP2, compete with ClpX/ClpC1 binding, and stabilize peptidolytically active ClpP1P2 in the absence of a partner ATPase. (b) ADEP-2B-5Me (ball-and-sticks) is shown bound to a ClpP2 LGF-pocket (ClpP2 subunits shown as dark red and orange cartoons; key residues shown as sticks) from crystal structure 4U0G. The difluorophenylalanine side chain projects into the pocket and the N-acyl chain extends to the right (gray carbons). The macrocycle rests on the surface of ClpP2, to the left. Macrocycle residues shown with colored carbons are present on some of the ADEP fragments described in this study. (c) Structures of ADEPs (1a & 1b), ADEP fragments bearing portions of the macrocycle (2a – 2e), and the N-heptenoyl-difluorophenylalanine methyl ester (3) are colored as in b.
The catalytic activity of ClpP is known to be affected by several molecules that were discovered in unbiased screens for antibacterial agents and in mechanistic investigations of natural products with antibacterial activity.2,31–33 Some are covalent inhibitors, like the β-lactones that form a covalent adduct with the active site serine of Mtb ClpP2.34 By contrast, ADEPs alter ClpP activity by binding to the surface of the peptidase in the same hydrophobic pockets that are involved in binding of AAA+ partners.23,35 This mode of ClpP binding is believed to be achieved via the ADEPs’ mimicry of the LGF loops of the AAA+ partners that participate in their binding to the peptidase.22,23,28 When in complex with the acyldepsipeptides (ADEPs), ClpP can degrade larger peptides and even some unstructured proteins lacking degrons due to induced widening of its axial pores. The indiscriminate degradation of small and unstructured cytoplasmic proteins by an activated ClpP has been proposed as the mechanism by which the ADEPs kill most bacteria.21–23,36–42 Indeed, there are multiple reports of activation of bacterial ClpP enzymes by the ADEPs. Intriguingly, we found that the antibiotics inhibit proteolysis of a degron-tagged protein substrate by Mtb ClpXP1P2 in vitro.28 Subsequent studies established that the lethality of ADEPs in Mtb is the result of inhibition of essential proteolytic activities.43
Here, we report that a fragment of the ADEPs exhibits potent anti-mycobacterial activity against Mtb strain H37Rv, especially in the presence of efflux-pump inhibitors. Unexpectedly, this fragment enhances the rate of the ATP-dependent degradation of protein substrates by ClpXP1P2. Further experiments show that the fragment stabilizes the active ClpXP1P2 complex, and does so by binding to a site on the ClpP1P2 peptidase that is distinct from that to which the ADEP binds. The anti-bacterial activity of the fragment motivated a limited structure-activity relationship study which defined a simple pharmacophore that could be elaborated into drug leads for the treatment of tuberculosis.
RESULTS AND DISCUSSION
An ADEP fragment retains anti-Mtb activity.
We previously reported that ADEP fragments lacking a macrocycle retain the ability to dysregulate ClpP and kill Bacillus subtilis, although the macrocycle contributed ~1000-fold to ClpP binding affinity and antibacterial potency in this organism.44 As an extension of that study, we evaluated the antibacterial activity of a conventional ADEP (1a; Fig. 1c), an ADEP with a structurally optimized macrocycle and side chain (1b),28 an ADEP fragment having some constituents of the macrocycle (2a),44 and a fragment lacking the macrocycle entirely (3)44 against Mtb strain H37Rv in culture (Table 1). ADEP 1a inhibited growth with a MIC of 50 µg/mL (~66 µM). The optimized structure of 1b exhibited improved potency, as expected,45 resulting in a four-fold lower MIC of 12.5 µg/mL (~16 µM). Surprisingly, compound 2a exhibited ~70% the molar potency of 1b, with a MIC of 12.5 µg/mL (~23 µM) against the same Mtb strain (Table 1). Compound 3 did not have a measureable MIC below 200 µg/mL.46,47
Table 1.
Antibacterial activity of ADEPs and fragment
| compound | MIC vs M. tuberculosis H37Rv µg mL−1 (µM) |
|
|---|---|---|
| without verapamil | verapamil (50 µg/mL) | |
| 1a | 50 (66) | 25 (33) |
| 1b | 12.5 (16) | 3.1 (3.9) |
| 2a | 12.5 (23) | 0.78 (1.4) |
Because the activities of ADEPs against Mtb H37Rv are potentiated by efflux-pump inhibitors like verapamil,46,47 we tested the effect of verapamil on the anti-Mtb activity of 1b and 2a. At 50 µg/mL, verapamil potentiated the activities of 1b and 2a against Mtb H37Rv by 4- and 16-fold, respectively (Table 1). Remarkably, fragment 2a had ~4-fold greater molar potency than ADEP 1b under those conditions. There are a few credible explanations for the differences in the degrees of potentiation by verapamil. For instance, fragment 2a’s activity may be more strongly potentiated than that of the ADEP because fragments are inherently better substrates for verapamil-sensitive pumps.47 Conversely, there may be verapamil-insensitive pumps that act preferentially on the ADEP. Alternatively, assuming that the fragment and ADEP are equally effluxed by verapamil-sensitive pumps, the fragment could either more potently and/or critically perturb the essential activity of Mtb ClpP1P2. We also cannot rule out a ClpP-independent mechanism by which the fragment kills Mtb. In any case, the efflux and bioactivity of fragment 2a is especially interesting in light of our report that a simpler fragment of the ADEP (3) is inactive, yet it potentiates the anti-Mtb activity of the ADEPs via preferential efflux.46,47
The ADEP macrocycle is dispensable for peptidase activation of ClpP1P2.
Given the potent antibacterial activity of 2a and its structural similarity to the ADEP, we next carried out experiments to determine if the ADEP fragment interacts with ClpP1P2. Since ADEP binding is known to stimulate the assembly of ClpP1P2 and its peptidolytic activity in vitro,28,43 we assayed these phenomena in experiments using the unoptimized ADEP (1a) and two fragments lacking a full macrocycle: 2a and 3 (Fig. 1c). Interestingly, compounds 1a and 2a each stimulated Mtb ClpP1P2 peptidase activity with a EC50 of ~40 µM, while the EC50 for 3 was ~90 µM (Fig. 2a, Table 2). The comparable affinities of ADEP 1a and fragment 2a results correlate well with observations in the ADEP•ClpP1P2 crystal structure: moieties corresponding to the fragment could make extensive complimentary contacts at the interfaces of ClpP2 subunits including those of the macrocycle engaged in hydrogen bonds with Tyr75 and Tyr95 (Fig. 1b); moreover, components of the macrocycle not included in the fragment are largely solvent exposed and do not bind ClpP1P2.28
Figure 2.
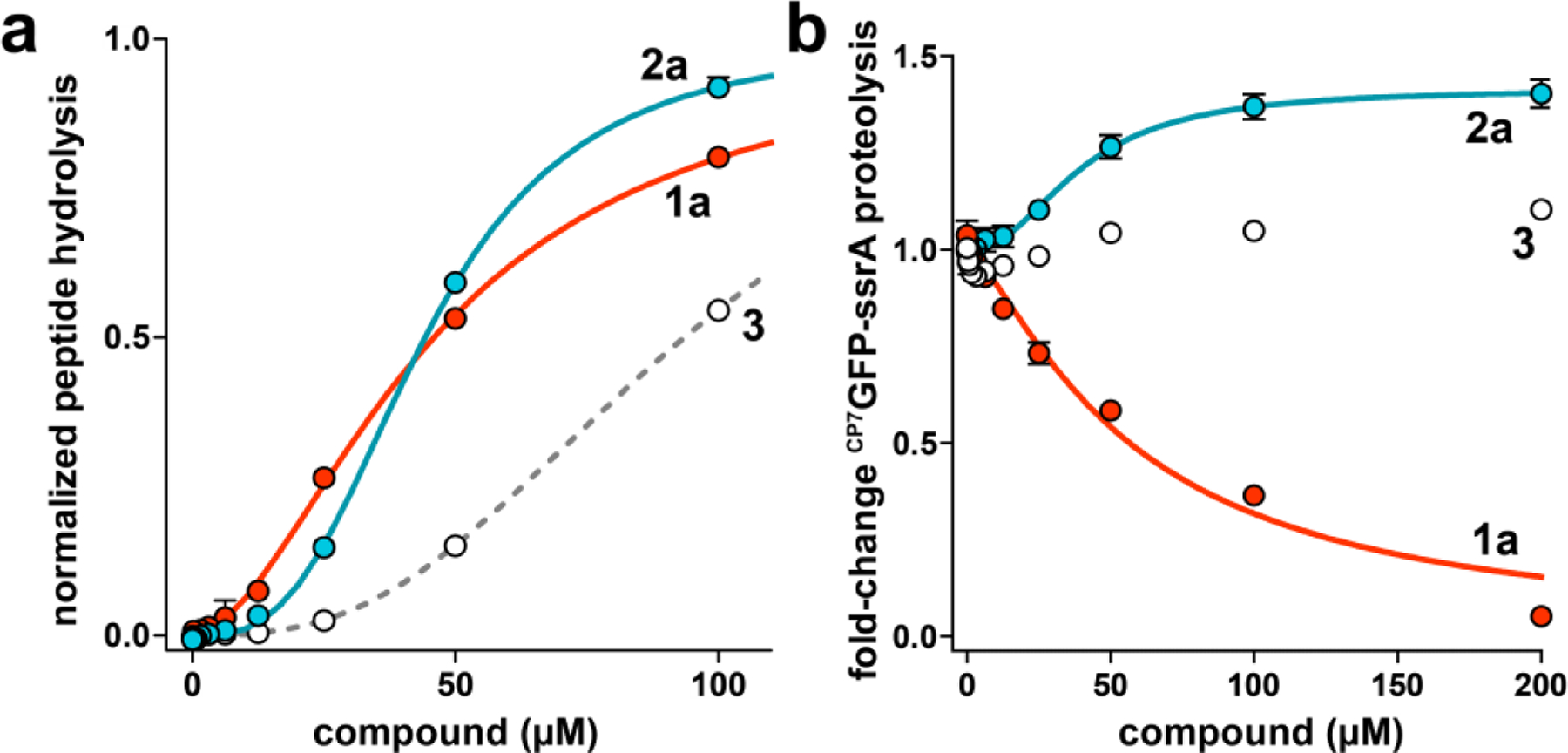
ADEP and fragments stimulate ClpP1P2 peptidase activity, but have different effects on ClpXP1P2 proteolysis activity. (a) Hydrolysis of a fluorogenic decapeptide (Abz-KASPVSLGYNO2D; 15 μM) by 0.5 μM ClpP1P2 was stimulated by ADEP 1a (red circles), fragment 2a (blue circles), and fragment 3 (white circles). Data were fit to a Hill equation. 1a stimulated peptidase activity with EC50 = 38 ± 2 μM, Hill constant (n) = 1.9 ± 0.07 (solid red line); 2a with EC50 = 46 ± 0.4 μM, n = 3.3 ± 0.2 (solid blue line); and 3 with EC50 = 94 ± 4 μM, n = 2.7 ± 0.1 (dashed gray line). (b) 1a inhibited proteolysis of 10 μM CP7GFP-ssrA by 1 μM ClpXP1P2 with IC50 = 59 ± 9 μM, n = 1.3 ± 0.2 μM. 2a stimulated with EC50 = 40 ± 5 μM, n = 2.1 ± 0.4, to a maximum activity 1.4-fold above the basal proteolysis rate. As fragment 3 had little effect on proteolysis over the tested concentration range, data were not fit to a binding equation. Values in all panels are averages of three biological replicates (N = 3) ± 1 standard deviation (SD).
Table 2.
Fit parameters for peptidase activation of ClpP1P2 by ADEPs and fragments
| compound | Peptidase stimulation | ||
|---|---|---|---|
| EC50 (µM)a | Hill coefficienta | R2 | |
| 1a b | 38 ± 2 | 1.9 ± 0.07 | 0.998 |
| 1b | 6.7 ± 0.2 | 2.1 ± 0.1 | 0.997 |
| 2a b | 46 ± 0.4 | 3.3 ± 0.2 | 0.999 |
| 2b | 67 ± 0.5 | 3.4 ± 0.07 | 0.999 |
| 2c | 16 ± 0.3 | 3.7 ± 0.2 | 0.998 |
| 2d | 58 ± 2 | 3.5 ± 0.3 | 0.998 |
| 2e | 35 ± 0.3 | 3.2 ± 0.07 | 0.999 |
| 3 b | 94 ± 4 | 2.7 ± 0.1 | 0.998 |
fit value ± standard error
average of three biological replicates
Fragment 2a enhances canonical protein degradation by ClpXP1P2.
Although ADEPs kill many bacterial species by dysregulating the activity of ClpP such that numerous proteins are indiscriminately degraded,21–23,38 they kill Mtb by inhibiting essential ClpP-catalyzed proteolysis.28,43 We compared the effects of 1a, 2a, and 3 on the ClpXP1P2-catalyzed proteolysis of a circularly permuted GFP variant ending in β-strand 7, and bearing a C-terminal ssrA degradation tag (CP7GFP-ssrA; Fig. 2b).48 The terminal β-strand in this variant is more easily extracted from the GFP β-barrel than the native terminal strand 11,48 making this an easier proteolysis substrate. Surprisingly, fragment 2a enhanced the rate of proteolysis by ~40% at saturation with an EC50 of 40 µM. In control experiments, ADEP 1a inhibited CP7GFP-ssrA degradation with an IC50 of 59 µM while the fragment having no antibacterial activity and low affinity for ClpP1P2 (3) had relatively small effects on the rate of proteolysis. Intriguingly, the enhancement of ClpXP1P2 proteolysis by fragment 2a suggested that it had a different mode of protease binding than the ADEPs, which exclusively bind ClpP2 within the heterotetradecamer and inhibit proteolysis by blocking the interaction of ClpP1P2 with the chaperone ClpX.
The enhancement of proteolysis by fragment 2a was reminiscent of the canonical ADEP mode of action,21–23,36–42 wherein binding of the molecules to homotetradecameric ClpPs enables the degradation of proteins lacking degrons without the intervention of ATP of the AAA+ partners. Thus, we assessed whether or not the 2a-simulated degradation of CP7GFP by ClpP1P2 required ClpX, ATP, and a ssrA tag. The enhanced rate of CP7GFP-ssrA degradation by ClpP1P2 was only observed when ClpX, ATP, and fragment 2a were all present (Fig. 3a), which stood in sharp contrast with the ATP and chaperone-independent activation of ClpP effected by the ADEPs. Proteolysis was not observed in the absence of either ClpP1 or ClpP2 (Fig. 3b), indicating that stimulation requires the heteromeric ClpP1P2 peptidase. Curiously, the stimulatory effects of 2a were not observed when proteolysis by ClpP1P2 was directed by ClpC1, the alternative AAA+ partner in Mtb. 2a inhibited proteolysis of CP7GFP-ssrA by ClpC1P1P2 to about 20% the activity of a DMSO control (Fig. 3c). The apparent ATP-dependence of the fragment-promoted stimulation of ClpXP1P2 proteolysis was further supported by the observation of very low levels of degradation in the presence of ATPγS (Fig. 3a), which is more slowly hydrolyzed by ClpX than ATP.49 The other departure from the canonical ADEP mechanism was that fragment 2a did not stimulate degradation of a GFP substrate lacking the ssrA degron (Fig. S1a). The fragment’s stimulatory effect was however not exclusive to CP7GFP-ssrA. Indeed, fragment 2a enhanced the capacity of ClpXP1P2 to degrade a non-permuted GFP-ssrA, an unrelated folded protein substrate (TAMRAHalo-ssrA),50 and an unfolded protein substrate (fl-V15Ptitin-ssrA)51 (Fig. S1a, S1b), which indicates that the chemo-activated protease can act on proteins irrespective of their tertiary structure or stability.
Figure 3.
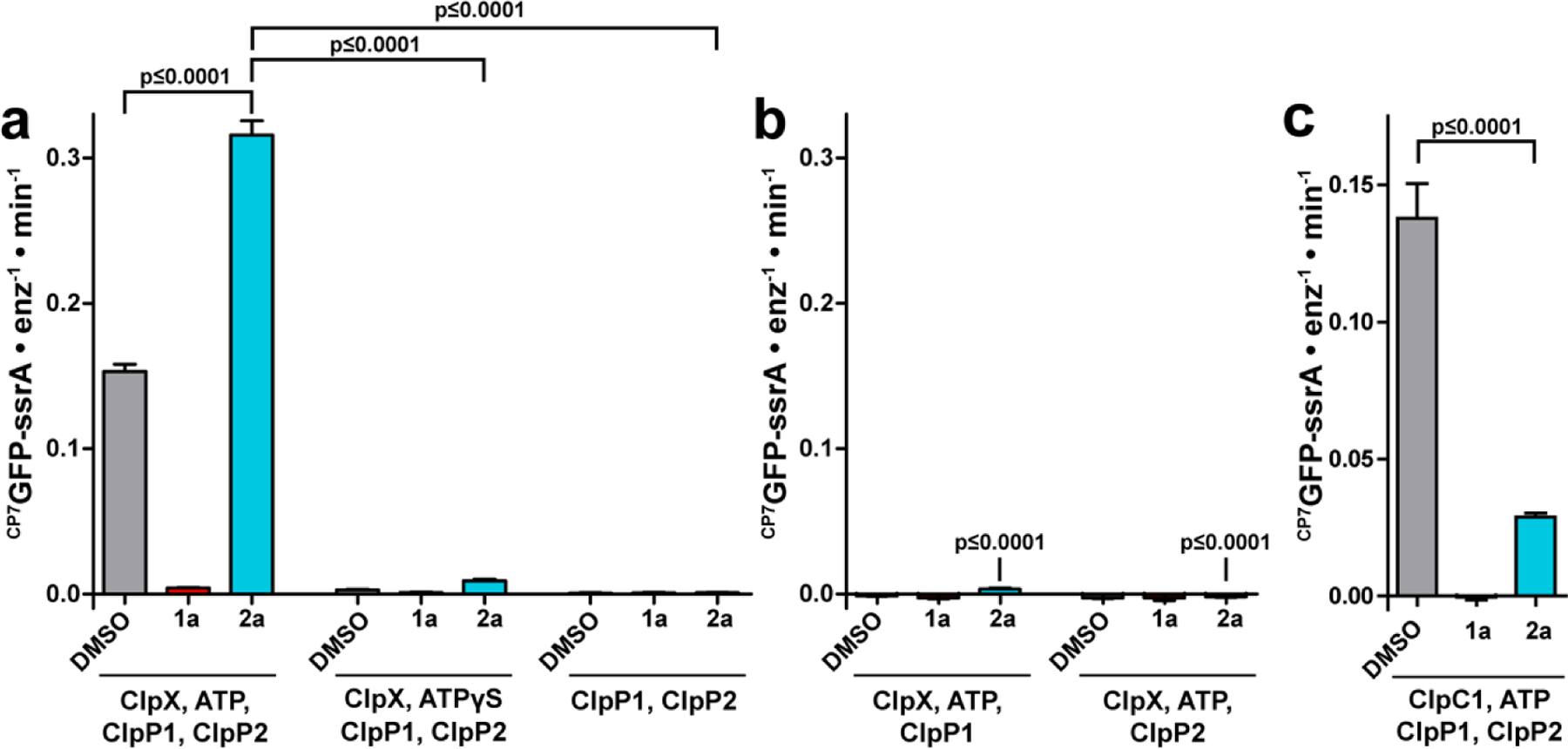
Fragment 2a stimulates canonical ATP-dependent proteolysis by ClpXP1P2. Proteolysis of 10 μM of the indicated substrate was assayed in the presence of DMSO vehicle, 200 μM 1a, or 200 μM 2a. (a) Proteolysis and stimulation by 2a requires ClpX and ATP. Degradation of CP7GFP-ssrA was observed in the presence of 2 μM ClpP1P2, 1 μM ClpX, and 2.5 mM ATP together with an ATP regeneration system. Proteolysis was inhibited by 1a and stimulated by 2a. Weak proteolysis occurred in the presence of 2.5 mM ATPγS, and this activity was stimulated by 2a. No activity was detected in the presence of ClpP1P2 alone. (b) Proteolytic stimulation by 2a requires both ClpP1 and ClpP2. Degradation of CP7GFP-ssrA was not observed when 1 μM ClpX, 2.5 mM ATP and an ATP regeneration system were combined with ClpP1 or ClpP2 individually. (c) Degradation of CP7GFP-ssrA by 1 μM ClpC1, 2 μM ClpP1P2, and an ATP regeneration system was partially inhibited by 2a. Values in all panels are averages of three technical replicates (N = 3) ± 1 SD. p-values were calculated by unpaired two-tailed Student’s t-test. The p-values in panel b are compared to the leftmost condition in panel a.
Collectively, these results indicate that fragment 2a, unlike the ADEP, does not impart any new functions to ClXP1P2; it simply enhances the native functions of the chaperone-dependent protease.
Fragment 2a binds ClpP1P2 differently than ADEP 1a.
Because 2a stimulates ClpXP1P2’s proteolytic activity, we reasoned that it could not bind competitively with ClpX at the ClpP2 interfaces. The observations could only be explained by binding of the fragment to unique binding sites in the ClpXP1P2 complex. To clarify the binding site and mechanism by which 2a stimulates proteolysis, we first examined the effects of ADEP 1a and fragment 2a on ATP hydrolysis by ClpX because its rate is strongly influenced by both the chaperone’s interaction with ClpP1P2 and by protein substrate turnover by proteolytic complex. In agreement with prior studies,27 addition of ClpP1P2 suppressed ClpX ATP hydrolysis by ~20% via native association of the chaperone and peptidase. Additionally, the presence of a protein substrate enhanced ClpXP1P2 ATPase activity by ~50% (Fig. 4a, middle and right triplets). Compared to a vehicle control, neither 1a nor 2a altered the ATPase activity of ClpX alone (Fig. 4a, leftmost triplet), making it unlikely that the effects of 2a arise through direct interactions with ClpX. In concordance with reports that ADEPs prevent binding of ClpX to ClpP1P2,28 1a blocked both the capacity of ClpP1P2 to suppress ATP hydrolysis by ClpX and the stimulation of ClpX-catalyzed ATP hydrolysis in the presence of ClpP1P2 and a protein degradation substrate (Fig. 4a; red bars, middle and right triplets). Surprisingly, in the presence of 2a, we observed greater suppression of the ATPase activity of ClpX upon the addition of ClpP1P2 (~45%) and greater simulation of ATP hydrolysis by ClpXP1P2 in the presence of a degradation substrate (~120%) (Fig. 4a, blue bars, middle and right triplets). As evidenced by the disparate effects of the compounds on the rates of ATP hydrolysis by ClpX in the presence of ClpP1P2 alone and with a protein substrate, we surmised that ADEP 1a blocks the interaction of ClpX and ClpP1P2 while fragment 2a stabilizes the proteolytic complex.
Figure 4.
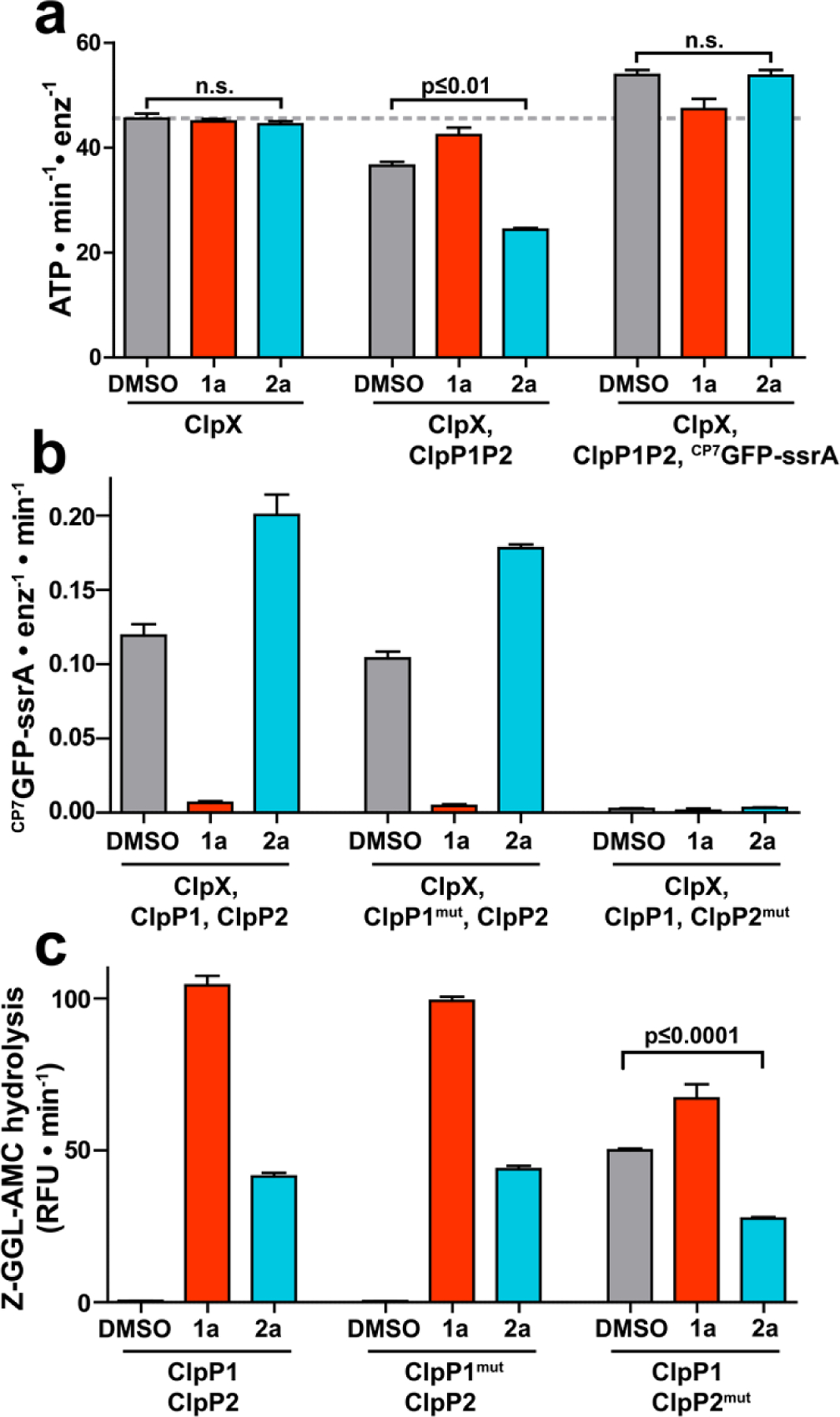
Fragment 2a binds to a second site on ClpP1P2. (a) The ATPase activity of 1 μM ClpX (left group), 1 μM ClpX with 2 μM ClpP1P2 (center group), or 1 μM ClpX with both 2 μM ClpP1P2 and 10 µM CP7GFP-ssrA (right group) was measured in the presence of DMSO vehicle, 200 μM 1a, or 200 μM 2a. Neither 1a nor 2a altered the basal ClpX ATPase activity. 1a, but not 2a, blocked ATPase suppression by ClpP1P2 and ATPase stimulation by substrate. (b) Proteolysis of CP7GFP-ssrA by 1 µM ClpX and 2 µM ClpP1P2 incorporating LGF-pocket substitutions in the indicated subunits was measured in the presence of DMSO control, 200 µM compound. 1a inhibited and 2a stimulated proteolysis by wild-type enzyme and enzyme incorporating ClpP1mut. No proteolysis was observed by enzyme incorporated ClpP2mut, which weakens ClpX binding. (c) Hydrolysis of a fluorogenic tripeptide (Z-Gly-Gly-Leu-AMC; 50 μM) by 0.5 μM ClpP1P2 incorporating wild-type subunits or LGF-pocket substitutions was assayed in the presence of 200 μM 1a or 2a. Peptidase incorporating ClpP2mut exhibited peptidase activity in the absence of compound, which was stimulated by 1a and suppressed by 2a. p-values were calculated by unpaired two-tailed Student’s t-test; n.s. = not significant. Values in all panels are averages of three technical replicates (N = 3) ± 1 SD.
The apparent stabilization of ClpXP1P2 by 2a suggested that the fragment did not bind at the ClpP2 interfaces like the ADEPs and ClpX. One plausible location for the 2a binding site is the surface of ClpP1,28,30 which has hydrophobic pockets similar to those on ClpP2 where both the ADEPs and the LGF loops of the AAA+ partners bind exclusively.28,29 Our analysis of the ADEP•ClpP1P2 crystal structures suggested that clashes with the macrocycle prevent ADEP binding to the cognate sites on ClpP1.28 Thus, we anticipated that fragments lacking the intact macrocycle might evade the steric constraints and bind the interfaces of ClpP1 in a fashion analogous to ADEP binding those in ClpP2. To test this, we introduced previously described substitutions near the LGF-pockets in ClpP1 (S61A, Y63V, L83A, and Y91V; ClpP1mut) and ClpP2 (Y75V and Y95V; ClpP2mut) that are expected to weaken small molecule and Clp unfoldase binding.29 Indeed, no proteolysis was observed in complexes incorporating the ClpP2mut variant that cannot bind ClpX. We tested the effects of 1a and 2a on ClpXP1P2 protease activity in the context of ClpP1 and ClpP2 LGF-pocket substitutions (Fig. 4b). Interestingly, the compounds had similar effects on both wild-type ClpXP1P2 and on complexes incorporating ClpP1mut, with 1a inhibiting and 2a stimulating proteolysis. The limited impact of mutations at the ClpP1 interfaces on the perturbations of proteolytic activity by compounds 1a and 2a was not consistent with binding of the fragment to ClpP1 in a manner reminiscent of ADEP binding to ClpP2.
In a separate set of experiments, we assessed how the aforementioned substitutions influenced the peptidolytic activity of ClpP1P2 and its enhancement by compounds 1a and 2a. We made intriguing observations concerning the degradation of a Z-Gly-Gly-Leu-AMC model peptide, which is predominantly cleaved by ClpP1 in the heterotetradecamer.26 We found that wild-type ClpP1P2 and complexes incorporating ClpP1mut responded equivalently to 1a and 2a (Fig. 4c, left and middle triplets, respectively). Complexes incorporating ClpP2mut exhibited substantial peptidase activity even in the absence of compound, suggesting that these substitutions have pleiotropic effects on ClpP1P2 stability and activity. Nevertheless, the activity of ClpP1P2mut was still modestly stimulated by ADEP 1a. In contrast, fragment 2a unexpectedly inhibited baseline peptidase activity (Fig. 4c, right triplet). We were further surprised that 2a had little influence on the hydrolysis of a different peptide (Z-Tyr-Val-Ala-Asp-AFC), which is preferentially cleaved by ClpP2 in the heterotetradecamer (Fig S2). Further experiments revealed that 2a exhibits dose-dependent inhibition of Z-GGL-AMC degradation by ClpP1P2mut with a IC50 of 120 µM, while having little effect on Z-YVAD-AFC cleavage (Fig. S3). Though we cannot rule out other binding events that explain both the modest suppression of the ClpP1-specific peptide cleavage and the stimulation of ClpXP1P2 proteolysis, our favored interpretation of the observations is that compound 2a occupies the active site of the ClpP1 subunits. Support for the active site occupancy model can be derived from the crystal structures of Mtb ClpP1P2 in which the fourteen active sites are occupied by the N-blocked peptide agonists that are required absolutely for reconstitution of a catalytically active peptidase in vitro.25–28,30 By extension, we propose that the ADEP fragment selectively binds the ClpP1 active site, presumably with greater affinity than the N-blocked peptide agonists that are obligate components in all of the peptidolysis and proteolysis assays.
Fragment binding stabilizes ClpXP1P2.
The catalytically active ClpXP1P2 complex exists in equilibrium with inactive and/or disassociated species.27,29,30,46 We sought to establish whether 2a induces formation of a novel hyperactive state of the protease, or drives equilibrium towards the known active complex. To address this question, we monitored CP7GFP-ssrA proteolysis using four concentrations of ClpXP1P2 and increasing concentrations of 2a (Fig. 5a). In the absence of fragment, the lowest enzyme concentration exhibited negligible proteolytic activity, whereas increasing enzyme concentration led to higher levels of proteolysis, likely by driving equilibrium towards the active complex. Titration of ClpXP1P2 with 2a induced a large stimulation of proteolysis at low enzyme concentrations (16-fold at 0.25 µM protease), but progressively smaller stimulation as enzyme concentration increased (only 1.2-fold stimulation at 2 µM protease). Notably, fragment 2a stimulated almost the same maximum activity at 1 µM and 2 µM enzyme, illustrating that 2a binding shifts equilibrium towards the normal active state of the protease rather than a hyperactive state. Consistent with the thermodynamic linkage expected for this behavior, fragment 2a stimulated proteolysis with a lower EC50 at higher concentrations of enzyme (Fig. 5a).
Figure 5.
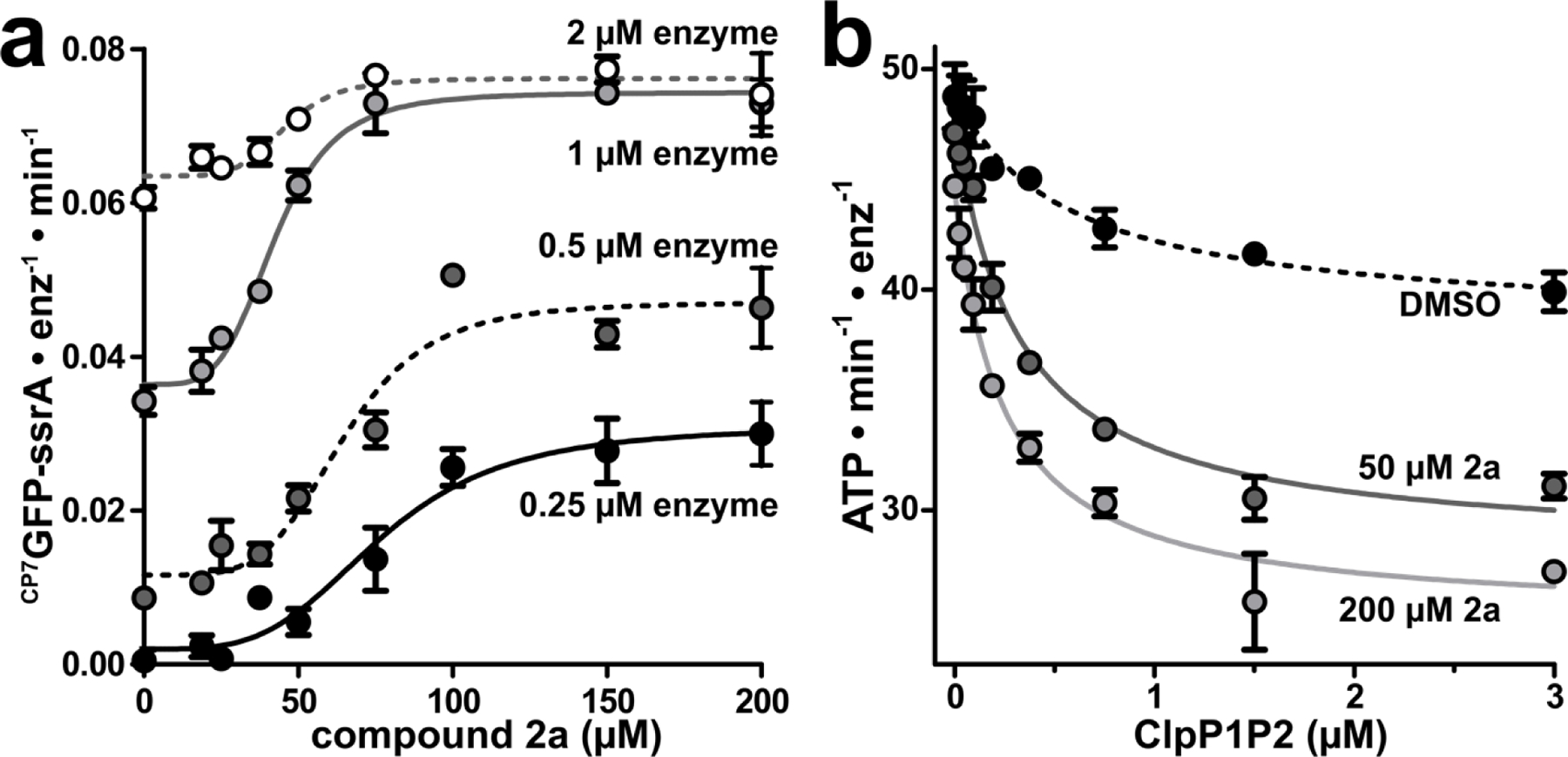
Fragment 2a stabilizes ClpXP1P2 assembly. (a) CP7GFP-ssrA proteolysis was measured as a function of 2a concentration for four concentrations of ClpXP1P2. Data were fit to a Hill equation. 0.25 μM ClpXP1P2 stimulated activity ~16-fold with EC50 = 77 ± 6 μM, n = 3.7 ± 1; 0.5 μM enzyme stimulated ~4-fold with EC50 = 65 ± 4 μM, n = 4.7 ± 1; 1 μM enzyme stimulated 2-fold with EC50 = 42 ± 2 μM, n = 4.4 ± 0.7; and 2 μM stimulated 1.2-fold with EC50 = 46 ± 4 μM, n = 5.5 ± 3. (b) The ATPase activity of 0.25 μM ClpX was measured as a function of ClpP1P2 concentration in the presence of DMSO mock (black circles), 50 μM 2a (dark gray circles), or 200 μM 2a (light gray circles). Data were fit to a non-cooperative binding model. ClpP1P2 suppressed ClpX ATP hydrolysis to ~80% of basal activity in the presence of DMSO with a Kapp of 0.57 ± 0.2 μM; to ~60% in the presence of 50 μM 2a with Kapp 0.31 ± 0.04 μM; and to ~55% in the presence of 200 µM 2a with Kapp 0.24 ± 0.03 μM. Fragments with KD-ClpP1 << KD-ClpP2 may stimulate proteolysis at concentrations below KD-ClpP2. p-values were calculated by unpaired two-tailed Student’s t-test; n.s. = not significant. Values in all panels are averages of three technical replicates (N = 3) ± 1 SD.
Additionally, we tested the effects of 2a on ClpXP1P2 stability by measuring changes in ClpX ATPase activity as a function of ClpP1P2 concentration. The inclusion of 50 µM 2a lowered the Kapp for ClpX/ClpP1P2 binding from 0.57 µM to 0.31 µM; at 200 µM, 2a further tightened Kapp to 0.24 µM (Fig. 5b). Together, these data suggest that fragment 2a stimulates proteolysis by binding to and stabilizing ClpXP1P2, driving higher occupancy of the active complex under the assay conditions.
Activities of other ADEP fragments.
We prepared a series of fragment 2a analogs having structural similarity to ADEPs 1a and/or 1b (Fig. 1c), in order to define the structure-activity relationships. Fragments were designed to incorporate additional moieties from the intact macrocycle on either the proline or serine residues of fragment 2a or to be more rigid by replacing serine residue of 2a with more conformationally restricted amino acids. All fragments stimulated ClpP1P2 peptidase activity, albeit with EC50 values that varied over a 4-fold range (Table 2). Interestingly, the apparent Hill coefficients of the fragments are greater than 3, which suggests cooperativity in their binding (Table 2). We made notable observations for fragments 2b and 2c, which have more constituents of the ADEP macrocycle than fragment 2a. Fragment 2b, in which N-acetyl alanine is appended to the proline residue of 2a, exhibited a higher EC50 for activation of ClpP1P2 peptidase activity (67 µM) and stimulated proteolysis similarly to 2a (Fig. 6a). In contrast, fragment 2c, with a proline methyl ester appended to the serine residue of 2a, stimulated peptidase activity with a lower EC50 than the parent fragment (16 µM) and robustly inhibited CP7GFP-ssrA degradation (Fig. 6a). The tighter apparent affinity of 2c could result formation of a hydrogen bond between its additional proline and Tyr95 in ClpP2 (Fig. 1b), although the capacity for 2b to make an additional hydrogen bond to Gln101 evidently does not improve its binding. We examined the effects of 2c on proteolysis over a range of concentrations and observed biphasic behavior (Fig. 6c), characterized by stimulation at low concentrations with a EC50 of 7.8 µM and inhibition at higher concentrations with a EC50 of ~50 µM. These results suggest that 2c binds more tightly than 2a to binding sites in ClpP2 and ClpP1, and that its competition with ClpX for ClpP2 binding overrides the stimulatory effect at high concentrations.
Figure 6.
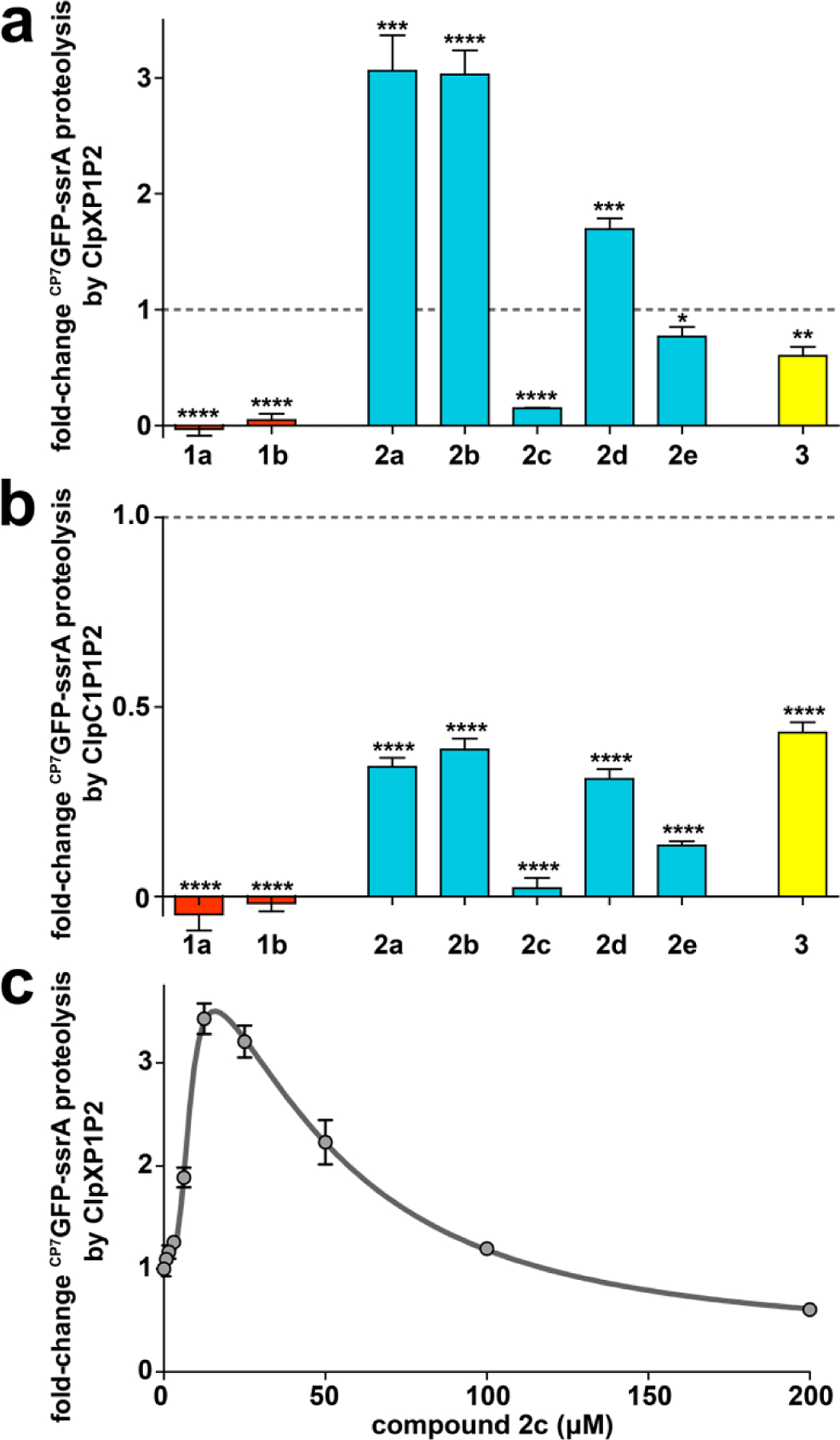
ADEP fragments may stimulate or inhibit proteolysis. (a) Distinct fragments have different effects on proteolysis by ClpXP1P2. Proteolysis of 10 μM CP7GFP-ssrA by 0.75 μM ClpX, 2 μM ClpP1P2, and an ATP regeneration system was assayed in the presence of 200 μM of the indicated compound (except for 1b, which was assayed at 50 μM). Activities were normalized to that of a DMSO vehicle control (dashed horizontal line). (b) All fragments inhibit proteolysis by ClpC1P1P2 to varying extents. Proteolysis of CP7GFP-ssrA (10 μM) by 0.75 μM ClpC1, 2 μM ClpP1P2, and an ATP regeneration system was assayed as in panel a. (c) Proteolysis of CP7GFP-ssrA (10 μM) by 1 μM ClpX, 2 μM ClpP1P2, 2.5 mM ATP and a regeneration system was measured as a function of 2c concentration. Data were fit to a double Hill equation (see Supporting Information). 2c stimulated proteolysis at low concentrations with EC50–1 = 7.8 ± 0.8 μM, n1 = 3.8 ± 1, and inhibited proteolysis at high concentrations with EC50–2 = 50 ± 10 μM, n2 = 1.8 ± 0.6. Values in all panels are averages of three technical replicates (N = 3) ± 1 SD. p-values were calculated by unpaired two-tailed Student’s t-test: * p≤0.05; ** p≤0.01; *** p≤0.001; **** p≤0.0001.
Based on our findings that replacement of serine residue of the ADEP natural products with a more conformationally restricted allo-threonine residue dramatically improved the compounds’ affinity for ClpP and antibacterial activity, we prepared analogs of 2a that had rigidifying elements (i.e., an ester of a secondary alcohol or a secondary amide). Compounds 2d and 2e had either allo-threonine or diaminopropionate, respectively, in place of the serine residue in fragment 2a. Compound 2d was a weaker activator of ClpP1P2 than the parent in peptidolysis assays (EC50 = 58 µM), while 2e was slightly more potent in the same assay (EC50 = 35 µM) (Table 2). The lower affinity ligand 2d was also a weaker stimulator of proteolysis by ClpXP1P2 (Fig. 6a, Table 2). The observations concerning 2d were surprising given that ADEPs having allo-threonine are more rigid and bind ClpP with much higher affinity than those with serine at the same position.40,45 In contrast, compound 2e, which exhibited slightly higher affinity than 2a, was an inhibitor of proteolysis – albeit a weak one (Fig. 6a, Table 2). Arguably, replacement of the ester with an amide either imparts improved binding at same sites on ClpP2 wherein the inhibitory ADEPs bind or enable binding in another fashion that inhibits the proteolytic activity of ClpXP1P2 (e.g., the active sites of both subunits).
Interestingly, all ADEP fragments inhibited proteolysis by the complex constituted by ClpP1P2 and the other AAA+ partner ClpC1 (Fig. 6b). Fragment 2c, which strongly inhibited ClpXP1P2 proteolysis, also strongly inhibited proteolysis directed by ClpC1. Fragments that stimulated ClpXP1P2 proteolysis (2a, 2b, and 2d), actually decreased proteolysis by ClpC1P1P2 to about ~40% the activity of a DMSO control. The inability of fragments to stimulate ClpC1P1P2 may be a consequence of the lower affinity of ClpC1 for ClpP1P2 (~4.5-fold weaker than that of ClpX),27 which is sufficiently low that the fragments can compete with ClpC1 for binding at the ClpP2 interfaces. The observed inhibition of ClpC1P1P2 suggests that even fragments that strongly stimulate ClpXP1P2 have some significant binding affinity for ClpP2. These results, and the biphasic behavior of 2c, suggest that the influence of compound on proteolysis depend on the specific conditions of the assay, including compound and protease concentration.
Overall, we found that analogs of 2a with lower EC50 inhibited protein degradation by ClpXP1P2 (e.g., 2c and 2e), whereas analogs with higher EC50 stimulated protein degradation (e.g., 2b and 2d). It is possible that the relatively high EC50 of 2c and 2e specifically arise from tighter binding to the ClpP2 interfaces, thus enhancing inhibition via competition with ClpX. By extension, we predict that a hypothetical compound that binds secondary sites more tightly than 2a would be superior as a stimulator of protein degradation by ClpXP1P2. Interestingly, the relationships between fragment structure, ClpP1P2 affinity, and activity could not be clearly rationalized based on the ADEP binding mode to ClpP2 observed in the crystal structure.28 This may be because macrocycle truncation reduces pre-organization of the compound and increases the entropic cost of binding to ClpP2. It may also reflect the constraints of binding to a second interaction site, that has not yet been structurally defined.
Model for stimulatory effects by fragment.
Our data support a model in which stimulatory ADEP fragments, like 2a, thermodynamically stabilize ClpXP1P2 by binding to a second site on the active form of the peptidase (Fig. 7). The active sites of ClpP1 are a plausible location for this secondary interaction. There is precedent for thermodynamic stimulation via binding to the ClpP1P2 peptidase active sites, as a variety of N-terminally blocked peptide agonists (e.g., Z-Leu-Leu) have been shown to stimulate assembly and catalytic activity via binding at some or all of the fourteen active sites in the ClpP1P2 heterotetradecamer.26,27,30 Nevertheless, the observed effects of ADEP fragments on Clp proteases occur in the context of a complex interaction landscape, where N-terminally blocked peptide agonists and AAA+ partners both influence complex stability. Although the ADEP fragments in this study presumably retain the ability to bind the ClpP2 interfaces, their binding is too weak to effectively compete with ClpX. We expect that any fragment capable of binding the ClpP2 docking-pockets would inhibit AAA+ partner binding at a sufficiently high concentration and thus inhibit proteolysis, as exemplified by the biphasic effects of 2c. In our model (Figure 7), the net effect on proteolytic activity of a specific fragment is determined by i) the fragment concentration, ii) its relative affinities for sites on ClpP1P2, and iii) the affinity of the AAA+ unfoldase present. We cannot currently deconvolute the individual interactions between fragment and ClpP1P2, and further structural information or biophysical data will be required to unambiguously define the site(s) to which fragment 2a binds.
Figure 7.
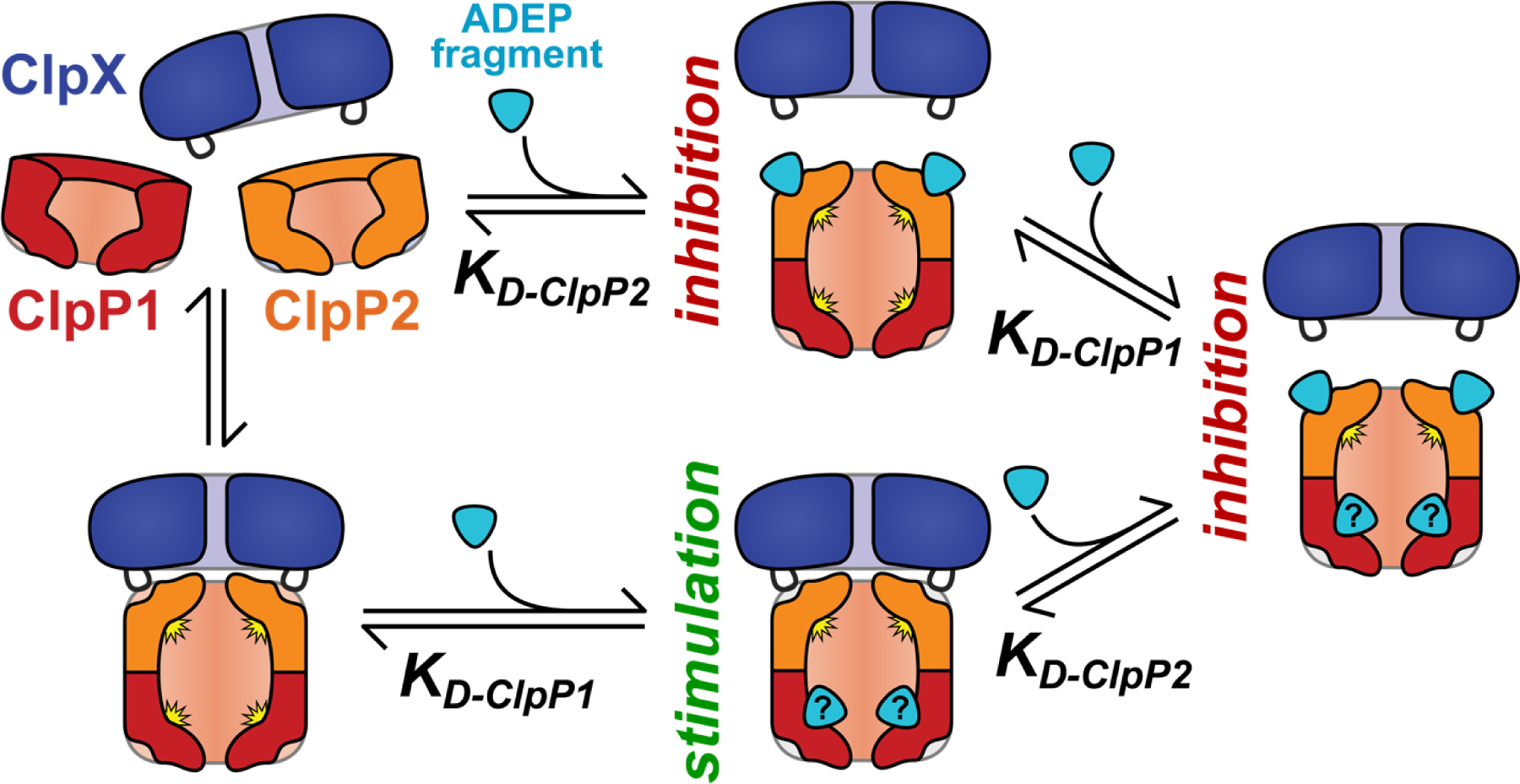
A simple model of the effects of ADEP fragments on ClpXP1P2 activity. Binding of fragment to ClpP2 LGF-pockets leads to competition for ClpX binding and proteolysis inhibition, while fragment binding to a second set of sites stabilizes the active protease. (The second site is illustrated as the ClpP1 active site, but could be elsewhere on the complex.) The net effect of ADEP fragment on proteolysis depends on the relative affinities for the two classes of binding sites. Fragments with KD-ClpP2 << KD-ClpP1 are always inhibitory.
The work reported here was initially inspired by our findings that the N-heptenoyl-3,5-difluorophenylalanine moiety appended to the ADEP macrocycle was necessary and sufficient for ClpP binding and antibacterial activity in B. subtilis.44 Based on the concept of fragment-based ligand development,52 we anticipated that the simple pharmacophore could be elaborated into a novel, high affinity ClpP ligand having potent activity against Mtb. Our efforts yielded a few surprises. For instance, we found that the ADEP fragment having the most potent activity against B. subtilis, N-heptenoyl-3,5-difluorophenylalanine methyl ester (3) (MIC= 8 μg/mL), was inactive against M. tuberculosis. Intriguingly, fragment 2a, having the N-heptenoyl-3,5-difluorophenylalanine pharmacophore coupled to a partial mimic of the ADEP macrocycle constituted by an ester linked N-acetyl proline and serine methyl ester, was less potent than 3 against B. subtilis (MIC= 8 vs. 32 μg/mL);44 yet, it was active against Mtb (MIC= 12.5 μg/mL). Other curious observations about fragment 2a was that its EC50 for activation of peptidolysis by the B. subtilis ClpP was nearly equivalent to that of 3 (~6 μM);44 whereas, the EC50 for activation of peptidolysis by Mtb ClpP1P2 by 2a was significantly higher (~44 μM) yet less than half that calculated for 3 (~100 μM). Most surprisingly, 2a enhanced the ATP-dependent degradation of degron-tagged proteins by ClpXP1P2 and appeared to stabilize the interaction of the peptidase with the AAA+ partner. These findings stand in sharp contrast to those concerning the ADEPs wherein they inhibit Mtb ClpP1P2 via their occupancy of the sites on ClpP2 to which the AAA+ partners (i.e., ClpX and ClpC1) bind.28 Consistent with those differences, our experiments indicate that 2a has a different binding site on ClpP1P2 than the ADEPs. In total, our observations suggest a new mode of lethal perturbation of Mtb ClpP activity and may serve as useful starting points for the development of compounds with improved potency and target selectivity. More broadly speaking, our observations are serendipitously representative of a newly expanding strategy in drug discovery wherein the focus is not on inhibitors of target function but on small molecules that alter the binding partners of their targets, influence the dynamic properties of their targets, and/or effect gain-of-function of the targets in ways that critically affect cellular physiology.53
The essentiality of ClpP activity in Mtb provides a simple explanation for the toxicity of the inhibitory ADEPs.43 It is possible that 2a kills Mtb through an identical inhibitory mechanism in the context of the mycobacterial cytosol, irrespective of the stimulation observed in vitro. Alternatively, the lethality of 2a may arise from its ability to stimulate the proteolytic activity of ClpXP1P2, in which case more nuanced explanations are required. We can envision three models where lethality arises through protease stabilization. In the first model, chemo-activated ClpXP1P2 aberrantly degrades an essential protein whose copy number is tightly controlled for viability – for example, a protein that must accumulate at some point in the cell cycle. We previously speculated that the unusual assembly characteristics of Mtb Clp proteases create an opportunity for mycobacteria to regulate protease activity.27 Aberrant stabilization by fragment 2a could thus disrupt a dynamic regulatory process. In a second model, the lethality of 2a arises entirely through disruption of the ClpC1•ClpP1P2 interaction, thus inhibiting essential proteolytic processes specifically carried out by ClpC1P1P2. A third possible model posits that stabilization of ClpX binding to ClpP1P2 by the ADEP fragment may effectively sequester the tetradecamer away from ClpC1 with which it must interact for other essential proteolytic functions. This possibility is relevant because ClpP1P2 is thought to dynamically and conditionally associate with different accessory ATPases like ClpC1 to effect the degradation of distinct populations of substrates.27,29 The ClpP1P2 sequestration model is consistent with observations that clpC1 is essential and that clpX overexpression reduces Mtb viability.54 The anti-bacterial activities of an ADEP fragment like 2c may be more complicated. Such ADEP fragments may either stimulate or inhibit ClpP proteolysis within the cell, depending on parameters such as binding affinities and the intracellular concentration of compound. Those key parameters may be difficult to assess because the compounds appear to accumulate in Mtb by passive partitioning or active transport, as we note that the MICs of the ADEPs and ADEP fragments in the presence of verapamil are lower than the measured EC50 values determined in vitro (Tables 1 and 2). Efforts to optimize ADEP fragment binding further and to define unambiguously the mechanism of killing are underway in our laboratories.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge D. Carney for synthesis of the ADEPs used in this study; R. Neunuebel and K. Gupta for the use of instrumentation; C. Presloid and M. Prorok for technical assistance. This work was supported by NIH grant AI123400 to J.K.S. and NIH grant GM-101988 to R.T.S. It was further supported by funds from the Lura Cook Hull Trust and from Brown University to J.K.S. K.R.S. was supported by a Charles A. King Trust Postdoctoral Research Fellowship, Bank of America, N.A., Co-Trustee and startup funds from the University of Delaware.
Footnotes
Supporting Information: The Supporting Information is available free of charge via the internet at http://pubs.acs.org.
• Supplemental biochemical and bacteriological methods, enzymatic data, chemical synthesis details and spectra
REFERENCES
- (1).Brötz-Oesterhelt H; Sass P (2014) Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol 304, 23–30. [DOI] [PubMed] [Google Scholar]
- (2).Culp E; Wright GD (2017) Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot 70, 366–377. [DOI] [PubMed] [Google Scholar]
- (3).Raju RM; Goldberg AL; Rubin EJ (2012) Bacterial proteolytic complexes as therapeutic targets. Nat. Rev. Drug Discovery 11, 777–789. [DOI] [PubMed] [Google Scholar]
- (4).Maurizi MR; Thompson MW; Singh SK; Kim SH (1994) Endopeptidase Clp: ATP-dependent Clp protease from Escherichia coli. Methods Enzymol 244, 314–331. [DOI] [PubMed] [Google Scholar]
- (5).Gottesman S (2003) Proteolysis in Bacterial Regulatory Circuits. Annu. Rev. Cell Dev. Biol 19, 565–587. [DOI] [PubMed] [Google Scholar]
- (6).Gottesman S; Wickner S; Maurizi MR (1997) Protein quality control: Triage by chaperones and proteases. Genes Dev 11, 815–823. [DOI] [PubMed] [Google Scholar]
- (7).Baker TA; Sauer RT (2006) ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem. Sci 31, 647–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yu AYH; Houry WA (2007) ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett 581, 3749–3757. [DOI] [PubMed] [Google Scholar]
- (9).Sauer RT; Baker TA (2011) AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem 80, 587–612. [DOI] [PubMed] [Google Scholar]
- (10).Wang J; Hartling JA; Flanagan JM (1997) The Structure of ClpP at 2.3 Å Resolution Suggests a Model for ATP-Dependent Proteolysis. Cell 91, 447–456. [DOI] [PubMed] [Google Scholar]
- (11).Wang J; Hartling JA; Flanagan JM (1998) Crystal Structure Determination of Escherichia coli ClpP Starting from an EM-Derived Mask. J. Struct. Biol 124, 151–163. [DOI] [PubMed] [Google Scholar]
- (12).Szyk A; Maurizi MR (2006) Crystal structure at 1.9Å of E. coli ClpP with a peptide covalently bound at the active site. J. Struct. Biol 156, 165–174. [DOI] [PubMed] [Google Scholar]
- (13).Singh SK; Grimaud R; Hoskins JR; Wickner S; Maurizi MR (2000) Unfolding and internalization of proteins by the ATP-dependent proteases ClpXP and ClpAP. Proc. Natl. Acad. Sci. U. S. A 97, 8898–8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Alexopoulos JA; Guarné A; Ortega J (2012) ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol 179, 202–210. [DOI] [PubMed] [Google Scholar]
- (15).Frees D; Qazi SNA; Hill PJ; Ingmer H (2003) Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol. Microbiol 48, 1565–1578. [DOI] [PubMed] [Google Scholar]
- (16).Frees D; Sørensen K; Ingmer H (2005) Global virulence regulation in Staphylococcus aureus: Pinpointing the roles of ClpP and ClpX in the sar/agr regulatory network. Infect. Immun 73, 8100–8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kwon H-Y; Ogunniyi AD; Choi M-H; Pyo S-N; Rhee D-K; Paton JC (2004) The ClpP Protease of Streptococcus pneumoniae Modulates Virulence Gene Expression and Protects against Fatal Pneumococcal Challenge. Infect. Immun 72, 5646–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kwon HY; Kim SW; Choi MH; Ogunniyi AD; Paton JC; Park SH; Pyo SN; Rhee DK (2003) Effect of heat shock and mutations in ClpL and ClpP on virulence gene expression in Streptococcus pneumoniae. Infect. Immun 71, 3757–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Robertson GT; Ng WL; Foley J; Gilmour R; Winkler ME (2002) Global transcriptional analysis of clpP mutations of type 2 Streptococcus pneumoniae and their effects on physiology and virulence. J. Bacteriol 184, 3508–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gaillot O; Pellegrini E; Bregenholt S; Nair S; Berche P (2000) The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol. Microbiol 35, 1286–1294. [DOI] [PubMed] [Google Scholar]
- (21).Kirstein J; Hoffmann A; Lilie H; Schmidt R; Rübsamen-Waigmann H; Brötz-Oesterhelt H; Mogk A; Turgay K (2009) The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol. Med 1, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lee BG; Park EY; Lee KE; Jeon H; Sung KH; Paulsen H; Rübsamen-Schaeff H; Brötz-Oesterhelt H; Song HK (2010) Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat. Struct. Mol. Biol 17, 471–478. [DOI] [PubMed] [Google Scholar]
- (23).Li DHS; Chung YS; Gloyd M; Joseph E; Ghirlando R; Wright GD; Cheng Y-Q; Maurizi MR; Guarn A; Ortega J (2010) Acyldepsipeptide Antibiotics Induce the Formation of a Structured Axial Channel in ClpP: A Model for the ClpX/ClpA-Bound State of ClpP. Chem. Biol 17, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Raju RM; Unnikrishnan M; Rubin DHF; Krishnamoorthy V; Kandror O; Akopian TN; Goldberg AL; Rubin EJ (2012) Mycobacterium tuberculosis ClpP1 and ClpP2 Function Together in Protein Degradation and Are Required for Viability in vitro and During Infection. PLoS Pathog 8, e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Personne Y; Brown AC; Schuessler DL; Parish T (2013) Mycobacterium tuberculosis ClpP Proteases Are Co-transcribed but Exhibit Different Substrate Specificities. PLoS One 8, e60228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Akopian T; Kandror O; Raju RM; UnniKrishnan M; Rubin EJ; Goldberg AL (2012) The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J 31, 1529–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schmitz KR; Sauer RT (2014) Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol 93, 617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schmitz KR; Carney DW; Sello JK; Sauer RT (2014) Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc. Natl. Acad. Sci. U. S. A 111, E4587–E4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Leodolter J; Warweg J; Weber-Ban E (2015) The Mycobacterium tuberculosis ClpP1P2 Protease Interacts Asymmetrically with Its ATPase Partners ClpX and ClpC1. PLoS One 10, e0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li M; Kandror O; Akopian T; Dharkar P; Wlodawer A; Maurizi MR; Goldberg AL (2016) Structure and Functional Properties of the Active Form of the Proteolytic Complex, ClpP1P2, from Mycobacterium tuberculosis. J. Biol. Chem 291, 7465–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ye F; Li J; Yang C-G (2017) The development of small-molecule modulators for ClpP protease activity. Mol. BioSyst 13, 23–31. [DOI] [PubMed] [Google Scholar]
- (32).Stahl M; Korotkov VS; Balogh D; Kick LM; Gersch M; Pahl A; Kielkowski P; Richter K; Schneider S; Sieber SA (2018) Selective Activation of Human Caseinolytic Protease P (ClpP). Angew. Chem., Int. Ed 57, 14602–14607. [DOI] [PubMed] [Google Scholar]
- (33).Moreno-Cinos C; Sassetti E; Salado IG; Witt G; Benramdane S; Reinhardt L; Cruz CD; Joossens J; Van Der Veken P; Brötz-Oesterhelt H; Tammela P; Winterhalter M; Gribbon P; Windshügel B; Augustyns K (2019) α-Amino Diphenyl Phosphonates as Novel Inhibitors of Escherichia coli ClpP Protease. J. Med. Chem 62, 774–797. [DOI] [PubMed] [Google Scholar]
- (34).Compton CL; Schmitz KR; Sauer RT; Sello JK (2013) Antibacterial Activity of and Resistance to Small Molecule Inhibitors of the ClpP Peptidase. ACS Chem. Biol 8, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kim YI; Levchenko I; Fraczkowska K; Woodruff RV; Sauer RT; Baker TA (2001) Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat. Struct. Biol 8, 230–233. [DOI] [PubMed] [Google Scholar]
- (36).Brötz-Oesterhelt H; Beyer D; Kroll H-P; Endermann R; Ladel C; Schroeder W; Hinzen B; Raddatz S; Paulsen H; Henninger K; Bandow JE; Sahl H-G; Labischinski H (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med 11, 1082–1087. [DOI] [PubMed] [Google Scholar]
- (37).Sass P; Josten M; Famulla K; Schiffer G; Sahl H-G; Hamoen L; Brotz-Oesterhelt H (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc. Natl. Acad. Sci. U. S. A 108, 17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Conlon BP; Nakayasu ES; Fleck LE; LaFleur MD; Isabella VM; Coleman K; Leonard SN; Smith RD; Adkins JN; Lewis K (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gersch M; Famulla K; Dahmen M; Göbl C; Malik I; Richter K; Korotkov VS; Sass P; Rübsamen-Schaeff H; Madl T; Brötz-Oesterhelt H; Sieber SA (2015) AAA+ chaperones and acyldepsipeptides activate the ClpP protease via conformational control. Nat. Commun 6, 6320. [DOI] [PubMed] [Google Scholar]
- (40).Goodreid JD; Janetzko J; Santa Maria JP; Wong KS; Leung E; Eger BT; Bryson S; Pai EF; Gray-Owen SD; Walker S; Houry WA; Batey RA (2016) Development and Characterization of Potent Cyclic Acyldepsipeptide Analogues with Increased Antimicrobial Activity. J. Med. Chem 59, 624–646. [DOI] [PubMed] [Google Scholar]
- (41).Leung E; Datti A; Cossette M; Goodreid J; McCaw S; Mah M; Nakhamchik A; Ogata K; El?Bakkouri M; Cheng Y-Q; Wodak S; Eger B; Pai E; Liu J; Gray-Owen S; Batey R; Houry W (2011) Activators of Cylindrical Proteases as Antimicrobials: Identification and Development of Small Molecule Activators of ClpP Protease. Chem. Biol 18, 1167–1178. [DOI] [PubMed] [Google Scholar]
- (42).Lavey NP; Coker JA; Ruben EA; Duerfeldt AS (2016) Sclerotiamide: The First Non-Peptide-Based Natural Product Activator of Bacterial Caseinolytic Protease P. J. Nat. Prod 79, 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Famulla K; Sass P; Malik I; Akopian T; Kandror O; Alber M; Hinzen B; Ruebsamen-Schaeff H; Kalscheuer R; Goldberg AL; Brötz-Oesterhelt H (2016) Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol. Microbiol 101, 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Carney DW; Compton CL; Schmitz KR; Stevens JP; Sauer RT; Sello JK (2014) A Simple Fragment of Cyclic Acyldepsipeptides Is Necessary and Sufficient for ClpP Activation and Antibacterial Activity. ChemBioChem 15, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Carney DW; Schmitz KR; Truong JV; Sauer RT; Sello JK (2014) Restriction of the Conformational Dynamics of the Cyclic Acyldepsipeptide Antibiotics Improves Their Antibacterial Activity. J. Am. Chem. Soc 136, 1922–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Ollinger J; O’Malley T; Kesicki EA; Odingo J; Parish T (2012) Validation of the Essential ClpP Protease in Mycobacterium tuberculosis as a Novel Drug Target. J. Bacteriol 194, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Compton CL; Carney DW; Groomes PV; Sello JK (2015) Fragment-Based Strategy for Investigating and Suppressing the Efflux of Bioactive Small Molecules. ACS Infect. Dis 1, 53–58. [DOI] [PubMed] [Google Scholar]
- (48).Nager AR; Baker TA; Sauer RT (2011) Stepwise Unfolding of a Beta Barrel Protein by the AAA+ ClpXP Protease. J. Mol. Biol 413, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Burton RE; Baker TA; Sauer RT (2003) Energy-dependent degradation: Linkage between ClpX-catalyzed nucleotide hydrolysis and protein-substrate processing. Protein Sci 12, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Iosefson O; Olivares A; Baker T; Sauer R; Coffino P; Martin A; Bustamante C; Fernandez JM; Bustamante C; Sauer RT (2015) Dissection of Axial-Pore Loop Function during Unfolding and Translocation by a AAA+ Proteolytic Machine. Cell Rep 12, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kenniston JA; Baker TA; Fernandez JM; Sauer RT (2003) Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell 114, 511–520. [DOI] [PubMed] [Google Scholar]
- (52).Hajduk PJ; Greer J (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery 6, 211–219. [DOI] [PubMed] [Google Scholar]
- (53).Schreiber SL (2019) A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines. Isr. J. Chem 59, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Dziedzic R; Kiran M; Plocinski P; Ziolkiewicz M; Brzostek A; Moomey M; Vadrevu IS; Dziadek J; Madiraju M; Rajagopalan M (2010) Mycobacterium tuberculosis ClpX interacts with FtsZ and interferes with FtsZ assembly. PLoS One 5, e11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.