

Abstract
The aggregation of the copper−zinc superoxide dismutase (SOD1) protein is linked to familial amyotrophic lateral sclerosis, a progressive neurodegenerative disease. A recent experimental study has shown that the 147GVIGIAQ153 SOD1 C-terminal segment not only forms amyloid fibrils in isolation but also accelerates the aggregation of full-length SOD1, while substitution of isoleucine at site 149 by proline blocks its fibril formation. Amyloid formation is a nucleation−polymerization process. In this study, we investigated the oligomerization and the nucleus structure of this heptapeptide. By performing extensive replica-exchange molecular dynamics (REMD) simulations and conventional MD simulations, we found that the GVIGIAQ hexamers can adopt highly ordered bilayer β-sheets and β-barrels. In contrast, substitution of I149 by proline significantly reduces the β-sheet probability and results in the disappearance of bilayer β-sheet structures and the increase of disordered hexamers. We identified mixed parallel−antiparallel bilayer β-sheets in both REMD and conventional MD simulations and provided the conformational transition from the experimentally observed parallel bilayer sheets to the mixed parallel−antiparallel bilayer β-sheets. Our simulations suggest that the critical nucleus consists of six peptide chains and two additional peptide chains strongly stabilize this critical nucleus. The stabilized octamer is able to recruit additional random peptides into the β-sheet. Therefore, our simulations provide insights into the critical nucleus formation and the smallest stable nucleus of the 147GVIGIAQ153 peptide.
Keywords: Replica exchange method, molecular dynamics simulations, oligomers, bilayer β-sheet, nucleus, free energy landscape
Graphical Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal disease characterized by degeneration of motor neurons in the cortex, brainstem, and spinal cord, often causing death within 2−5 years.1 The hallmark of this disease is the selective death of motor neurons in the brain and spinal cord, leading to paralysis of voluntary muscles.2 Among ALS cases, 10% are inherited familial ALS (fALS), ∼20% of which are attributed to mutations in the copper−zinc superoxide dismutase (SOD1) protein.3,4
SOD1 is a metalloenzyme that lies in the intracellular cytoplasmic space of aerobic organisms.5 It can catalyze the dismutation of superoxide into molecular oxygen and hydrogen peroxide.5,6 An earlier study showed that the SOD1-encoding gene mutation was one of the pathogenies in fALS, and 11 different SOD1 missense mutations were identified in 13 different fALS families, opening up molecular genetics studies of fALS.7 SOD1 aggregation was first found in SOD1 mutant in the motor neurons of mice.8 SOD1 fibrillation kinetics in vitro mirrors with remarkable accuracy the spinal cord aggregate buildup and disease progression in transgenic mice.9 Accumulation of misfolded proteins plays an important role in the process of the ALS. Mounting experimental studies have focused on elucidating the mechanism for how mutations promote SOD1 misfolding and aggregation.10−12 On the computational side, a growing number of molecular dynamics (MD) studies have been carried out to investigate the local unfolding and structural properties of full-length SOD1 protein and its various mutants.13−18 These experimental and computational studies have greatly enhanced our understanding of the role of different mutations on the aggregations and structural stability of SOD1 protein, while the aggregation property at the atomic level is not well understood.
Often, the aggregation of a protein domain could be triggered by a short protein stretch within the domain, typically a hexapeptide fragment.19,20 The nucleation process of the triggering short amyloidogenic stretches could be self-aggregation or interaction with other cooperative regions.21−31 Amyloid formation has been characterized as nucleation−polymerization processes. Numerous studies aimed at understanding the nucleation processes.32−46 Preventing the nucleation processes could be an effective way to inhibit amyloid formation. Recent experimental studies by Ivanova et al.47 have identified two SOD1 fragments (101DSVISLS107 and147GVIGIAQ153) that are likely to trigger the aggregation of full-length SOD1, and they also solved their crystal structures with fibril-like assembly. Particularly, the 147GVIGIAQ153 segment not only forms fibril-like aggregates but also accelerates fibril formation of full-length SOD1, metal-free (apo) SOD1, and G93A mutant. More than 100 SOD1 gene mutations have been found, and their sites are almost across the whole protein sequence, existing in each domain in the protein.48,49 Several mutations are within the 147GVIGIAQ153 segment, for example, V148G, V148I, I149T, and I151T,49 and it is found that most of these in-segment disease mutations are compatible with aggregate formation.47 However, the substitution of isoleucine 149 by proline blocks fibril formation of the full-length protein and leads to rod-shaped aggregates.47
The experimental identification of the structure and biological significance of the 147GVIGIAQ153 segment to accelerate the amyloid formation of full-length SOD1 and one of the best characterized familial mutants of SOD1, G93A, prompted us to study the dynamic aspects of self-assembly of 147GVIGIAQ153 segment. Proline is a known secondary structure breaker, and it would disrupt the aggregation of the peptide. Therefore, the I149P mutation is an excellent comparison for the wild-type 147GVIGIAQ153 segment. We have investigated the nucleation/oligomerization of the GVIGIAQ peptide and its I149P mutant by conducting two independent 200 ns atomistic replica exchange molecular dynamics (REMD) simulations with explicit solvent. As a first step to understand the early nucleation stage of aggregation of GVIGIAQ peptide, we simulated the self-assembly of six peptide chains. Our REMD simulations show that six GVIGIAQ peptide chains self-assemble into β-sheet-rich hexameric aggregates that are highly structurally heterogeneous. These β-sheet-rich hexamers are predominantly disordered but also comprise low-populated ordered bilayer β-sheets and β-barrels. The bilayer β-sheets display the cross-β structural feature of fibrils. The monolayer β-sheets consisting of four strands are found to be the critical β-sheet for the formation of bilayer β-sheets and β-barrels. Calculation of interpeptide residue−residue contact probability shows that I149 plays a key role in the formation of ordered structures. Substitution of I149 with proline results in a dramatic decrease of the β-sheet content and a significant conformational shift to disordered aggregates, consistent with the experimental results.47 Conventional MD simulations for two different sizes of experiment-derived bilayer sheets, each sheet made of three or four parallel β-strands, show that six GVIGIAQ peptide chains have a preference to form mixed parallel−antiparallel β-sheets, while the four-stranded bilayer β-sheet (octamer) with each sheet made of parallel β-strands remains stable within the 200 ns simulation. Mixed parallel−antiparallel bilayer β-sheets were observed in both REMD and conventional MD simulations, thus providing the conformational conversion pathway from the mixed parallel−antiparallel β-sheets to the experiment-derived parallel bilayer β-sheets. While hexameric parallel bilayer β-sheets are not stable, two additional peptides strongly stabilize the critical nucleus for GVIGIAQ fibril elongation.
Although proline is a known secondary structure breaker, it is not clear whether it disrupts peptide aggregation, which involves the tertiary structure formation of peptides. It was reported by Ivanova et al. that the full-length SOD1 protein with I18P and I35P substitutions formed fibrils similar in morphology to the wild-type protein, while I104P and I149P substitutions inhibited aggregation of full-length protein.47 Similarly, the Aβ1−42 E22P mutant was reported to form amyloid fibril even faster than the wild-type Aβ1−42 peptide.50 These findings indicate that proline does not necessarily disrupt 3D-structure-involved peptide aggregation although it is a secondary structure breaker. Moreover, even though the I149P mutation was found to block 147GVIGIAQ153 fibrillization, how this mutation affects the 3D structures of oligomers is unclear at the atomic level.
RESULTS AND DISCUSSION
We first checked the convergence of the REMD simulations at 310 K, by comparing the probability of hydrogen bond (H-bond) number of the hexamer and the end-to-end distance of each chain using the 100−150 and 150−200 ns data for the wild-type and I149P mutant systems. As shown in Figure S1, the two curves in these independent time intervals overlap very well both for wild-type and I149P mutant. Figure S2 plots the dominant secondary structure contents of each amino acid residue and the average probability of each dominant secondary structure over all residues within these two different time intervals. The dominant secondary structures include coil, β-sheet, β-bridge, and bend (turn and helix contents are negligible in both systems). Both wild-type and I149P mutant have well overlapped curves within the two time intervals. The residue-based secondary structure contents are also similar between the two time intervals. The time evolution of the first replica is given in Figure S3, showing that the whole temperature space has been visited sufficiently for both the wild-type and I149P systems. The number of times for a replica passing from the lowest to the highest temperature during the 200 ns REMD simulation is 4. These results suggest that our REMD simulations are reasonably converged for both systems. Unless specified, all the REMD simulation results presented below are based on the last 100 ns (t = 100−200 ns) of simulation data generated at 310 K.
Secondary Structure Contents and β-Sheet-Size Distributions in Wild-Type and I149P Hexamers.
The probabilities of the dominant secondary structures (β-sheet, coil, bend, and β-bridge) were calculated for both systems. As shown in Figure 1a, the GVIGIAQ peptide predominantly adopts β-sheet (with a probability of 55.8%) and to a slightly lesser extent coil (41.9%) conformations, while the β-bridge and bend conformations are insignificant. As expected, substituting isoleucine at position 149 by proline leads to a significant decrease of β-sheet content (29.1%) and an increase of coil content (59.2%), indicating that the I149P mutation shifts the conformations of the GVIGIAQ hexamers from a β-sheet-rich structure to a collapsed coil-rich state.
Figure 1.

Calculated secondary structure probability in the REMD runs during 100−200 ns for wild-type and I149P mutant systems at 310 K for (a) average secondary structures of all residues and residue-based (b) β-sheet, (c) coil, (d) β-bridge, and (e) bend conformations.
The I149P mutation-induced conformational change can be also seen from the secondary structure probability of each amino acid residue. As seen in Figure 1b, the residues in the wild-type system have the probability of 62.7%−88.4% to adopt β-sheet states, with residues V148, I149, G150, I151, and A152 having a probability of 65.5%, 86.8%, 88.4%, 84.2%, and 62.7% respectively. However, in the I149P mutant hexamers, all of the residues have a reduced β-sheet probability, and the β-sheet probability for residues close to the mutation site decrease most significantly (V148, P149, and G150 having, respectively, a probability of 19.4%, 28.9%, and 50.4%). The wild-type system has a coil percentage of 7.1%−31.8% in the above-mentioned residues, while an increased coil percentage of 23.9%−70.8% appears in the I149P mutant hexamers. The percentage of β-bridge structure in wild-type is lower (0.4%−4.6%) than that of in I149P mutant (3.2%−15.9%) (Figure 1d) and so is the bend structure (0.9%−3.0% vs 4.4%−19.8%). The I149P mutation reduces not only the β-sheet probability of the residue at position 149 (from 86.8% to 28.9%) but also that of its adjacent residues V148 and G150 (from 65.5% to 19.4% for V148 and from 88.4% to 50.4% for G150). All of these data demonstrate that I149 plays an important role in the β-sheet conformation of GVIGIAQ hexamers.
The probability of the β-sheet sizes at 310 K was calculated to probe the β-structures in a possible GVIGIAQ aggregation nucleus (Figure 2a). The two-stranded β-sheets in both systems have approximate probabilities (22.8% and 24.4%), while the probability of three-stranded β-sheets is enormously reduced from 43.4% in wild-type to 9.6% in I149P mutant. The I149P mutant has a significantly decreasing probability for the larger sizes of β-sheets relative to the wild-type. The probabilities of three-, four-, five-, and six-stranded β-sheets are, respectively, 43.5%, 8.2%, 3.3%, and 3.9% in wild-type, whereas they drop to 9.0%, 3.3%, 0.6% and 0.1% in I149P mutant. These data indicate that the I149P mutation dramatically impedes the formation of larger sizes of β-sheets. We use the probability of the angle between two neighboring strands in a β-sheet to examine their orientation preference. Figure 2b shows that the peptide in hexamers can form both parallel and antiparallel β-sheets, with a higher probability to form parallel β-sheets. It is interesting to see that proline mutation not only decreases β-sheet size, but also unexpectedly changes the β-sheet orientation. The I149P mutation shifts the dominant β-sheet from parallel strand alignment to antiparallel alignment.
Figure 2.

Probability of different sizes of β-sheet (a) and the probability of the angle between two adjacent β-strands in all sizes of β-sheet in wild-type and I149P hexamers at 310 K from the REMD simulations.
GVIGIAQ Hexamers Can Adopt Ordered Bilayer β-Sheets and β-Barrels, While I149P Substitution Leads to the Disappearance of Bilayer β-Sheets and an Increase of Disordered Hexamers.
With a Cα-root-mean-square-deviation (Cα-RMSD) cutoff of 0.3 nm, the 50000 conformations of wild-type hexamers at 310 K are separated into 261 clusters. The representative conformations of the top nine most-populated clusters are shown in Figure 3a, which represent 42.14% of all conformations. We noticed that GVIGIAQ hexamers in these clusters display various β-sheet-rich conformations. The first cluster contains four-stranded open β-barrel with mixed parallel−antiparallel strands. The second, third, and fourth clusters all consist of 3 + 3 or 3 + 2 β-sheet bilayers, explaining the high probability of the three-stranded β-sheets. Cluster-7 contains 2 + 2 β-sheet bilayers, with a population of 3.54%. The bilayer β-sheets and β-barrels in these clusters have populations of 26.49% and 15.65%, respectively.
Figure 3.

Representative conformations of the first nine most-populated clusters for wild-type (a) and I149P mutant (b) hexamers at 310 K from the REMD simulations. The corresponding population of each cluster is given in parentheses.
There are 594 clusters for I149P hexamers. Figure 3b shows the top nine most-populated conformations, which represent 23.61% of all conformations. This large cluster number reflects a decrease in the nucleation propensity by the I149P mutation. The I149P hexamers are mostly in amorphous states, and β-sheet bilayers are not observed in the top nine most-populated clusters. As expected, I149P hexamers seem to be more disordered structurally than wild-type at 310 K, and most conformations consist of oligomers with randomly associated four, five, and six β-strands or random coil chains.
Recently, Laganowsky et al.51 found that a six-stranded antiparallel β-barrel formed by an 11-residue K-11-V (with sequence KVKVLGDVIEV) fragment is cytotoxic. The formation of β-barrel structures by the amyloidogenic peptide NHVTLSQ from the β2 microglobulin protein was also reported in earlier MD and REMD simulation studies by Derreumaux et al.52,53 While there is no experimental result for the cytotoxicity of the GVIGIAQ peptide, it is still interesting to check a possible β-barrel conformer. We found that there are several β-barrel clusters, which mostly consist of mixed parallel and antiparallel β-sheets with a dominant parallel β-strand alignment, while β-barrels with antiparallel β-strand alignment are frequently found in I149P hexamers.
From the top nine most-populated clusters of GVIGIAQ hexamers in Figure 3a, we found that most of the bilayer β-sheet structures are 3 + 3 β-sheets, indicating a high population of a bilayer β-sheet structure for GVIGIAQ hexamers. The four-stranded β-sheets mainly form two kinds of conformations: bilayer β-sheet consisting of a four-stranded β-sheet and a two-stranded β-sheet, and open β-barrels consisting of a curved four-stranded β-sheet and two chains in random coil conformation. The β-sheets with a size of 5 or 6 both form β-barrels. These results indicate that for wild-type hexamers, monolayer β-sheets of size 4 are the critical β-sheet that can form both bilayer β-sheet and β-barrels, and bilayer β-sheets are formed when size <4, while they transit into β-barrels when size >4.
The Importance of Hydrophobic Interactions in Shaping the Free Energy Landscape of GVIGIAQ Hexamers.
To have an overall view of the conformational space of wild-type and I149P hexamers, Figure 4a,b draws the potential of mean force (PMF) as a function of the number of inter-peptide H-bonds and radius of gyration (Rg). The free energy minimum basin of I149P hexamers is much broader than that of the wild-type. There is one single deep basin located at (H-bonds, Rg) value of (25, 0.93) in wild-type and similarly centered at (20, 0.92) in I149P hexamers. The existence of a large number of H-bonds is the outcome of the formation of β-sheet-rich conformation in the GVIGIAQ hexamers. The amide nitrogen in proline is not able to form a H-bond, so it is expected that I149P mutant will have fewer H-bonds. The decreased number of inter-peptide H-bonds in I149P mutant is consistent with the I149P mutation reducing the β-sheet content and leading to the formation of disordered hexamers.
Figure 4.

Free-energy landscape (kcal/mol) for wild-type (a) and I149P (b) hexamers at 310 K from the REMD simulations, plotted as a function of the number of interpeptide H-bonds and Rg.
To find well-organized assemblies in wild-type and I149P hexamers, we calculated the probability of different species consisting of bilayer β-sheets and β-barrels using all conformations generated within the 100−200 ns duration. Representative structures of the bilayer β-sheet and β-barrel in both systems are shown in Figure 5. In wild-type hexamers (Figure 5a), bilayer β-sheets include mixed parallel−antiparallel 3 + 3, 3 + 2, and 2 + 2 bilayer β-sheet structures and β-barrels include four-, five-, and six-stranded open/closed β-barrels. The 3 + 3 and 3 + 2 bilayer β-sheet structures all have a high probability, consistent with the high probability of three-stranded β-sheet seen in Figure 2a. We found that the I149P mutant (Figure 5b) can only form four-, five-, and six-stranded open β-barrel conformations, and the bilayer β-sheet structures disappear, again showing that I149P changes β-sheet organization. The percentages of four-, five-, and six-stranded open β-barrels are 5.49%, 2.13%, and 2.13% in wild-type hexamers, while they decrease to 3.09%, 0.57%, and 0.05% in I149P mutant. The closed β-barrel structures even vanish after mutation. All of the data show that the I149P mutation prevents the bilayer β-sheet conformation and reduces the population of ordered assemblies.
Figure 5.

Representative structures of bilayer β-sheets and β-barrels of wild-type (a) and I149P mutant (b) hexamers at 310 K from the REMD simulations, shown in two different views: side view and top view. Different sizes of bilayer β-sheets are denoted by m + n, where m and n represent, respectively, the m- and n-stranded β-sheets forming the bilayer β-sheets. The four different β-barrels include four-, five-, and six-stranded open β-barrels (open B4, open B5, open B6) and six-stranded closed β-barrel (closed B6).
The contact probabilities of the interpeptide side-chain−side-chain (SC-SC) contacts between all pairs of residues were calculated in order to identify the residue contribution for the formation of bilayer β-sheet and β-barrel structures. Figure 6 a,b shows that I149−I149 (14.96%), I149−I151 (14.64%), and V148−I149 (14.44%) pairs have the highest SC-SC contact probabilities compared with other residue pairs, while their contact probabilities are dramatically reduced (9.43%, 10.48%, and 11.66% for P149−P149, P149−I151, and V148−P149, respectively) after residue isoleucine 149 is substituted by proline. These data, together with the structural differences in Figures 3 and 4, reveal the crucial role of I149 in the bilayer β-sheet and β-barrel formation. After mutation, the contact probability of the V148−V148 pair increases from 8.20% to 13.91% to compensate for the loss of hydrophobic contributions from isoleucine 149.
Figure 6.

Side-chain−side-chain contact probability map (a,b) and main chain H-bond number (c,d) averaged over all 100−200 ns REMD-generated conformations at 310 K for wild-type and I149P hexamers.
The important role of Ile149 in β-sheet formation can also be seen from the backbone−backbone (MC-MC) H-bond network of wild-type and I149P hexamers (Figure 6 c,d). In wild-type hexamers, the I149−I151 pair has the largest number of H-bonds (2.50) with respect to other pairs. The I149P mutation dramatically reduces the number of H-bonds (0.76), partially due to the nitrogen atom in proline not being a H-bond acceptor. The average H-bond numbers of I149−I150 and I149−I149 are also reduced (1.57 versus 0.35 for the I149−I150 pair and 0.97 versus 0 for the I149−I149 pair). The H-bond number for G150-I151 pair increases from 1.24 to 1.71, becoming the pair that has the largest number of H-bond. These results indicate that the I149P mutation alters the H-bond network, reflecting the key role of I149 in the backbone H-bond formation. We also checked the hexamer stabilities of two mutants (I149P and I149A) to illustrate the importance of I149 to maintain fibril structures. As indicated in Figures 7, S6, and S9, both mutants cannot maintain stable β-sheet hexamers (Supporting Information).
Figure 7.

Analysis of the structural stability of 3 + 3 bilayer β-sheet for fibril-like wild-type and I149P mutant hexamers: time evolution of Cα-RMSD in conventional MD simulations with respect to the 3 + 3 bilayer β-sheet at t = 0 ns (a, b) and snapshots at five different time points for (c) wild-type and (d) I149P hexamers.
Hexamers Are the Critical Nucleus for Fibril Formation and Octamers Are the Smallest Stable Nucleus for GVIGIAQ Fibril Elongation.
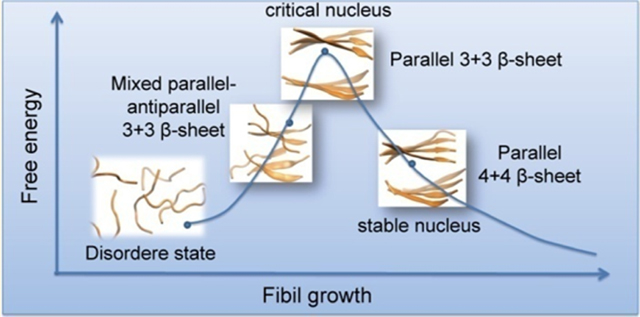
The critical nucleus may be defined as the number of proteins or peptides at which the free energy reaches maximum. In order to rigorously estimate the critical nucleus size, one has to study the dependence of the free energy,46,54 lag phase times,55 or times to add a nascent polypeptide chain to the preformed template56 as a function of the number of monomers in studied systems. From our REMD simulations, as can be seen in Figure 2, we have shown that the most abundant β-sheet size for the GVIGIAQ hexamers is 3, which counts for more than 40% of the populations. The counting of β-sheet size in Figure 2 refers to the continuous β-sheet in one layer. One should note that most of the trimeric β-sheets form a double layer, which is equivalent to hexamer as indicated in Figures 3 and 5. Figure 5 shows that the GVIGIAQ hexamers are rich in mixed parallel−antiparalleled β-sheet structures. However, the GVIGIAQ crystal structure has only parallel β-sheets.47 In this sense, the missing of parallel only β-sheets in our REMD simulated hexamer system indicated that the crystal-structure-like parallel β-sheet is in the maximum free energy state. Then we examine whether the hexamer can be qualified as a critical nucleus. To this end, we need to understand how the mixed parallel−antiparalleled β-sheet nucleus relates to the parallel β-sheet fibril and why the fibril-like 3 + 3 bilayer β-sheet is rarely populated in our REMD simulations. To address these questions, we performed two MD simulations of 200 and 400 ns initiated from the fibril-like 3 + 3 bilayer β-sheet. The time evolution of Cα-RMSD with respect to the initial state in Figure 7a shows that the Cα-RMSD gradually increases to 1.0 nm within the first 100 ns and then decreases to 0.8 nm at t = 150 ns, fluctuating around this value during the rest of this simulation. The large Cα-RMSD indicates that the initial bilayer β-sheet hexamer is not stable. Figure 7c highlights dynamic structural transitions at five different time points during the 400 ns MD simulation. Initially, three chains, A, B, and C, in the bottom layer are parallel. The chains A and B dissociate from the β-sheet and turn around, shifting from fibril-like assembly to a 3 + 3 mixed parallel−antiparallel bilayer β-sheet at t = 100 ns. Then, all of the chains turn around along the horizontal direction during the 100−200 ns. This new 3 + 3 mixed parallel−antiparallel bilayer β-sheet structure remains stable during the last 200 ns, though the rotation between the two sheets is seen during the simulations. Similar structures of the 3 + 3 mixed parallel−antiparallel bilayer β-sheet are obtained from the other independent 200 ns MD simulation (Figure S5). All of these data indicate that six GVIGIAQ peptide chains have a preference to adopt mixed parallel−antiparallel β-sheet.
It would be interesting to know whether the mixed parallel−antiparallel bilayer β-sheet can transit into the parallel bilayer β-sheet. To this aim, we have conducted two additional MD simulations starting from the mixed parallel−antiparallel bilayer β-sheet generated at t = 200 ns using different velocity distributions (Figure S8). However, conformational transition was not observed, indicating that the transition energy barrier is much higher than that of the opposite transition.
Clearly, the conventional MD simulation from the GVIGIAQ crystal structure provides a pathway connecting the 3 + 3 mixed parallel−antiparallel bilayer β-sheet to the fibril-like 3 + 3 parallel bilayer β-sheet structure. To probe whether the mixed parallel−antiparallel 3 + 3 bilayer β-sheet, which equilibrated in the MD run (Figure 7b), is sampled in our REMD simulation, we calculated the Cα-RMSD with respect to the representative structures of each cluster of WT hexamers in the 400 ns MD trajectory. We found that three clusters (clusters 25, 51, and 81) generated in the REMD runs are quite similar to the mixed parallel−antiparallel bilayer β-sheet observed in the MD run. The structural similarity can be seen from the time evolution of Cα-RMSD of conformations generated in MD runs with respect to the REMD clusters 25, 51, and 81 (see Figure 8). A Cα-RMSD value of ≤0.4 nm is seen from t = 80 to 150 ns and 360 to 400 ns in the 400 ns MD simulation, indicating that the mixed parallel−antiparallel bilayer β-sheet conformations were sampled in both REMD and conventional MD simulations. These data together with the results seen in Figure 7a,b elucidate that GVIGIAQ peptide is able to form a 3 + 3 mixed parallel−antiparallel bilayer β-sheet, which can be transformed to the fibril-like 3 + 3 parallel β-sheet. The remaining question is how the 3 + 3 fibril-like parallel β-sheet can be further stabilized.
Figure 8.

Time evolution of Cα-RMSD of wild-type 3 + 3 parallel bilayer β-sheet hexamer in conventional MD simulations with respect to the center structure of three ordered clusters generated in the REMD run of wild-type hexamers. The inset is the snapshot of the center structure of cluster-25 (a), cluster-51 (b), and cluster-81 (c).
To answer the above question, we conducted two 200 ns MD simulations starting from a fibril-like 4 + 4 parallel bilayer β-sheet (Figures 9 and S7). The Cα-RMSD with respect to the initial state as a function of time shows that the Cα-RMSD increases rapidly to 0.4 nm in the first 10 ns and then stays at this value during the remaining simulation period (Figure 9a). The increase of the Cα-RMSD in the initial 10 ns mainly results from the twisting of the β-sheet in two layers (see Figure 9b). The small Cα-RMSD value of 0.4 nm during the full 200 ns MD simulation indicates that the 4 + 4 parallel bilayer β-sheet is stable within this 200 ns simulation time, which can be seen clearly from the snapshots in Figure 9b. Similar results are obtained from the other independent 200 ns MD simulation (Figure S7), indicating the structural stability of the 4 + 4 parallel bilayer β-sheet. These data, together with the results in Figure 7 a,c, reveal that the hexamer is the critical nucleus for GVIGIAQ fibril formation and the octamer can be the smallest nucleus for GVIGIAQ fibril growth.
Figure 9.

Analysis of the structural stability of the 4 + 4 fibril-like parallel bilayer β-sheet of GVIGIAQ peptide in conventional MD simulations: time evolution of Cα-RMSD with respect to the 4 + 4 bilayer β-sheet at t = 0 ns (a) and snapshots at four different time points (b).
To further demonstrate that the nucleus size is six, we have performed an additional MD simulation starting from a state in which two random coil chains (labeled as chain 1 and 2) are placed on one side of a 3 + 3 parallel bilayer β-sheet (see Figure 10). It can be seen from Figure 10 that the two chains come together to form a short two-stranded parallel β-sheet at their N-terminal region on top of the preformed 3 + 3 bilayer β-sheet at t = 25 ns. This β-sheet extends to the C-terminal residues to form a longer two-stranded parallel β-sheet at t = 50 ns (Figure 10a,c). This two-stranded β-sheet interacts with the 3 + 3 bilayer β-sheet via side chain steric zipper and forms a 3 + 3 + 2 trilayer β-sheet (having a sheet−sheet distance of ∼1.0 nm with the top layer β-sheet, Figure 10b). Strands in the newly formed two-stranded β-sheet have a parallel orientation with those in the top layer of the 3 + 3 bilayer β-sheet in the remaining 450 ns of the simulation. It is noted that during the 500 ns MD simulation, strands between the two three-stranded β-sheets have different orientations such as antiparallel (t = 50 ns), perpendicular (t = 75, 150, and 400 ns), and parallel orientations (t = 100 and 450 ns) (Figure 10c).
Figure 10.

Analysis of the MD trajectory on the system consisting of a 3 + 3 bilayer β-sheet and two random chains in conventional MD simulations. Time evolution of (a) the backbone H-bond number and the main-chain distance between chain 1 and chain 2. (b) The main chain center-of-mass distance between chain 1 + 2 and the upper layer β-sheet of the 3 + 3 bilayer β-sheet. (c) Representative snapshots at 12 different time points.
We have also conducted a MD simulation starting from a state where two extended peptide chains (labeled as chain 1 and 2) are placed on one side of a 4 + 4 parallel bilayer β-sheet (see Figure 11). There are no H-bonds between the two chains and the preformed 4 + 4 β-sheet at t = 0 ns. The two chains become collapsed within the initial 25 ns, and they remain in an collapsed state during t = 25−75 ns. At t = 100 ns, chain 2 docks to the top layer β-sheet and adopts an extended state. It interacts with the main chain of the edge strand in the bottom layer at t = 133 ns. Through some time of rearrangement, this chain forms an in-register five-stranded parallel β-sheet with the bottom layer β-sheet (see the snapshot at t = 200 ns). This five-stranded β-sheet remains stable during the period of t = 200−400 ns. Chain 2 starts to dissociate from the bottom layer β-sheet and interact with the edge strand in the top layer β-sheet at t = 450 ns and forms an antiparallel β-sheet at t = 500 ns. This coil-to-β-sheet transition process follows the dock−lock fibril growth mechanism proposed by Thirumalai et al.57 It is noted that the strands in the two different β-sheets remain antiparallel during the full period of MD simulation. These results further indicate that the 3 + 3 hexamer is the critical nucleus and the 4 + 4 octamer is the smallest stable nucleus for GVIGIAQ fibril formation and growth.
Figure 11.

Analysis of the MD simulation on the system consisting of a 4 + 4 bilayer β-sheet and two random chains in conventional MD simulations. The time evolution of (a) backbone H-bond number, the minimum distance between chain 1 and 4 + 4 bilayer β-sheet, and the main-chain distance between chain 1 and chain 2. (b) Backbone H-bond number and the minimum distance between chain 2 and chain 8. (c) Representative snapshots at 12 different time points.
CONCLUSIONS
Amyloid formation relates to biological functions and is a responsible agent in human disease. The nature and molecular origin of protein aggregation process, especially the lag-phase, has been a central element in the quest for a mechanism of amyloid formation.36 By combining extensive REMD simulations with conventional MD simulations, we investigated oligomeric structures of GVIGIAQ peptide and the critical nucleus for fibril formation. While nonspecific interactions in amyloid nucleation are very important,33 we found that the GVIGIAQ hexamers are enriched with highly ordered β-sheets.
The strands in different conformations are predominantly parallel. The monolayer β-sheets with a size of 4 are the critical β-sheet that can form bilayer β-sheet when the size <4 and β-barrels when the size >4. In contrast, the I149P mutant does not form bilayer β-sheets. As expected, when isoleucine 149 is substituted by proline, the average β-sheet content is greatly reduced. The I149P hexamers display a higher propensity to visit disordered states, mostly consisting of some amorphous structures, consistent with the experiment showing that the I149P mutation prevents fibril formation. It is interesting to see that proline mutation not only decreases β-sheet size but also unexpectedly changes the β-sheet orientation.
It is hard to capture and characterize amyloid nucleus experimentally, since nucleation typically creates transient conformations that get stabilized as the oligomer grows.33,46 Computational approaches have added important insights into the critical nucleus for amyloid formation, with nuclei sizes ranging from 6 to 18 peptide chains.22,46,55,56,58−64 Using 147GVIGIAQ153 as an example, our simulations provided important atomic details of the critical nucleus formation and stabilization. We found that the stabilized octamer is able to recruit additional random peptides into the β-sheet. We revealed the pathway connecting the dynamic hexamer nucleus to fibril structure and the further stabilization of the nucleus at the octamer level. While it is important to link the GVIGIAQ fibril formation to the full-length SOD1, nevertheless, our findings provide significant insight into the oligomeric structures and aggregation mechanism of GVIGIAQ peptide and clues to ALS pathogenic mechanisms.
METHODS
GVIGIAQ Peptide and Its I149P Mutant.
The GVIGIAQ peptide and its I149P mutant (amino acid sequence: GVPGIAQ) are both capped by the ACE (CH3CO) group at the N-terminus and the NH2 group at the C-terminus. For the REMD simulations, we used a 10 ns MD simulation at 420 K to generate a random coil conformation. The reason that we chose a random coil conformation as the starting state is to remove a bias of ordered secondary structures (such as helix and β-sheet). In brief, we use wild-type and I149P mutant to denote the two hexameric systems. Each system was fully solvated with SPC water molecules65 in a cubic box with a size of 6 × 6 × 6 nm3, with a minimum distance of 0.9 nm from the peptides to the edges of water box. The total numbers of atoms for GVIGIAQ hexamers and I149P mutant are 21159 and 21177, respectively.
Two different sizes of preformed fibril-like GVIGIAQ oligomers (3 + 3 hexamer and 4 + 4 octamer) were built using the crystal structure deposited in protein data bank (PDB ID 4NIP).47 The GVPGIAQ fibril revealed by X-ray crystallography consists of in-register parallel β-sheets packed in a face-to-face steric zipper,47 where strands in the same β-sheet are parallel and those in the two different sheets are antiparallel. According to the atomic fibril structure (PDB ID 4NIP),47 we constructed a 3 + 3 bilayer sheet (i.e., each sheet consists of three β-strands) using a single β-strand as a building block. First, a three-stranded in-register parallel β-sheet was generated by translating one β-strand by 0.47 nm and −0.47 nm along the backbone H-bond direction. A copy of this three-stranded β-sheet was made to obtain another monolayer β-sheet. Then we constructed the two β-sheets in a face-to-face steric zipper pattern. The strands in the two β-sheets are antiparallel and those in the same sheet are parallel. The initial distance between the two sheets is 1.14 nm. The fibril-like 3 + 3 hexamers of I149P and I149A mutants were obtained by substituting I149 with proline and alanine. A 4 + 4 fibril-like GVIGIAQ bilayer β-sheet was constructed using the same strategy as done for a 3 + 3 bilayer β-sheet.
REMD Simulations.
The REMD method is an enhanced sampling protocol,66−68 in which the temperature of each replica can go up and down with certain exchange ratios, so it can easily avoid getting trapped in local-minimum free energy states. Due to the efficiency of REMD method in simulating complex problems such as peptide aggregation, we have performed a 200 ns REMD simulation on the hexamer of the wild-type and I149P mutant using the GROMACS-4.5.3 software package.69 Gromos53a6 force field70 was employed to describe the two peptide systems. The reason for choosing Gromos53a6 force field is given in Supporting Information. The REMD simulations have been conducted in the isothermal−isobaric (NPT) ensemble. There are 48 replicas with temperatures ranging from 310 to 430.79 K for both systems, each of 200 ns duration (temperature list: 310, 312.26, 314.54, 316.82, 319.12, 321.4, 323.73, 326.07, 328.42, 330.78, 333.16, 335.54, 337.94, 340.36, 342.79, 345.24, 347.7, 350.17, 352.65, 354.9, 357.42, 359.94, 362.48, 365.03, 367.6, 370.19, 372.79, 375.4, 378.03, 380.67, 383.33, 385.99, 388.68, 391.38, 394.1, 396.83, 399.58, 402.34, 405.12, 407.91, 410.72, 413.55, 416.39, 419.25, 422.06, 424.94, 427.87, and 430.79 K). The average acceptance ratios for both systems are ∼22.62%. We used the LINCS71 and SETTLE algorithms72 to constrain the bond lengths of peptides and those of water molecules, respectively. This allows an integration time step of 2 fs. A cutoff of 1.4 nm was used for the nonbonded interactions. The pressure (1 bar) was maintained by weak coupling to temperature and pressure baths,73 with coupling constants of 0.1 and 1.0 ps, respectively. Nonbonded pair lists were updated every five integration steps. The protein and nonprotein groups were separately coupled to an external heat bath with a relaxation time of 0.1 ps using a velocity rescaling coupling method.74
MD Simulations.
To examine the structural stability of different sizes of GVIGIAQ fibril-like oligomers and to identify the smallest stable fibril-like GVIGIAQ nucleus, we performed two independent MD simulations for each system in NPT ensemble at 310 K starting from the constructed 3 + 3 or 4 + 4 fibril-like bilayer β-sheet (hexamer or octamer). To further examine the role of I149P in the stability of β-sheet bilayers, we performed 100 and 200 ns MD runs on the 3 + 3 fibril-like bilayer β-sheet of I149P mutant. We also performed two 200 ns MD simulations of the 3 + 3 fibril-like bilayer β-sheet of the I149A mutant to examine the importance of I149 to maintain fibril structures. There are 6928, 6938, 6860, and 6946 water molecules in the four different systems, respectively. We did not include salt ions in our simulations, as done previously by many MD simulation studies on the aggregation of several peptides.62,75−78
Analysis Methods.
We used the facilities implemented in the GROMACS software package and our in-home-developed codes to analyze the trajectories. The first 100 ns of data of each REMD simulation was discarded in order to remove the bias of the initial states. Therefore, the structural properties of each system are based on the last 100 ns. Several parameters are used to analyze the REMD trajectories, including the secondary structure of the peptide, the percentage of various sizes of β-sheet, the probability of the angle between two neighboring strands in each β-sheet, the free energy landscape, the number of hydrogen bonds, and the residue−residue contact probability for both side-chain−side-chain (SC-SC) and main-chain−main-chain (MC-MC). The secondary structure is identified using the dictionary of secondary structure prediction (DSSP).79 The size of a β-sheet is the number of strands in an n-stranded β-sheet, for example, the β-sheet size of a four-stranded β-sheet is four. Two chains are considered to form a β-sheet if (1) at least two consecutive residues in each chain visit the β-strand state and (2) they have at least two H-bonds. One H-bond is taken as formed if the N···O distance is less than 0.35 nm and the angle of N−H···O is greater than 150°. Two chains are considered as forming an antiparallel (parallel) β-sheet if the scalar product of their normalized end-to-end unit vectors is less than −0.7 (greater than 0.7) and at least two consecutive residues of each chain visit the β-strand state. The free energy surfaces are constructed using −RT ln H(x, y), where H(x, y) is the histogram of two selected parameters x and y. In this study, the x coordinate is the radius of gyration (Rg) of the hexamer and the y coordinate is the number of interpeptide hydrogen bonds (H-bonds). The Daura cluster analysis method80 is used to cluster the conformations sampled in the REMD simulations with a Cα-RMSD cutoff of 0.3 nm using all the residues. In the coordinate file of GVIGIAQ hexamers, the six chains are topologically identical. In the coordinate files, each chain has a fixed identifier. We calculated the RMSD by ignoring the chain identifier. A closed β-barrel is considered to be formed if (1) the β-sheet size is ≥4 and (2) the number of H-bonds between the first and the last strand in the β-sheet is ≥2. All representations of wild-type and I149P hexamers are displayed using the VMD program.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Luogang Xie and Dr. Zhenyuan Qian for setting up the system and Mr. Jiangtao Lei for helpful discussion. Simulations were performed at the High Performance Computing Server (PowerEdge T710) of Shanghai University of Sport.
Funding
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract Number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneur-o.5b00242.
Additional methods and supplemental figures (PDF)
REFERENCES
- (1).Rowland LP, and Shneider NA (2001) Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700. [DOI] [PubMed] [Google Scholar]
- (2).Pasinelli P, and Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. [DOI] [PubMed] [Google Scholar]
- (3).Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, and Brown RH (1997) Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 41, 210–221. [DOI] [PubMed] [Google Scholar]
- (4).Cleveland DW, and Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2, 806–819. [DOI] [PubMed] [Google Scholar]
- (5).Fridovich I. (1975) Superoxide dismutases. Annu. Rev. Biochem. 44, 147–159. [DOI] [PubMed] [Google Scholar]
- (6).D’Alessandro M, Paci M, Di Nola A, Amadei A, and Aschi M. (2004) Theoretical Modeling of Enzyme Reaction Chemistry: The Electron Transfer of the Reduction Mechanism in CuZn Superoxide Dismutase. J. Phys. Chem. B 108, 16255–16260. [Google Scholar]
- (7).Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62. [DOI] [PubMed] [Google Scholar]
- (8).Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, and Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281, 1851–1854. [DOI] [PubMed] [Google Scholar]
- (9).Lang L, Zetterstrom P, Brannstrom T, Marklund SL, Danielsson J, and Oliveberg M. (2015) SOD1 aggregation in ALS mice shows simplistic test tube behavior. Proc. Natl. Acad. Sci. U. S. A. 112, 9878–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O’Halloran TV, and Siddique T. (2006) Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. U. S. A. 103, 7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Prudencio M, Durazo A, Whitelegge JP, and Borchelt DR (2009) Modulation of mutant superoxide dismutase 1 aggregation by co-expression of wild-type enzyme. J. Neurochem. 108, 1009–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Prudencio M, Hart PJ, Borchelt DR, and Andersen PM (2009) Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum. Mol. Genet. 18, 3217–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Strange RW, Yong CW, Smith W, and Hasnain SS (2007) Molecular dynamics using atomic-resolution structure reveal structural fluctuations that may lead to polymerization of human Cu-Zn superoxide dismutase. Proc. Natl. Acad. Sci. U. S. A. 104, 10040–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Muneeswaran G, Kartheeswaran S, Muthukumar K, Dharmaraj CD, and Karunakaran C. (2014) Comparative structural and conformational studies on H43R and W32F mutants of copper-zinc superoxide dismutase by molecular dynamics simulation. Biophys. Chem. 185, 70–78. [DOI] [PubMed] [Google Scholar]
- (15).Galaleldeen A, Strange RW, Whitson LJ, Antonyuk SV, Narayana N, Taylor AB, Schuermann JP, Holloway SP, Hasnain SS, and Hart PJ (2009) Structural and biophysical properties of metal-free pathogenic SOD1 mutants A4V and G93A. Arch. Biochem. Biophys. 492, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ding F, Furukawa Y, Nukina N, and Dokholyan NV (2012) Local unfolding of Cu, Zn superoxide dismutase monomer determines the morphology of fibrillar aggregates. J. Mol. Biol. 421, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Milardi D, Pappalardo M, Grasso DM, and La Rosa C. (2010) Unveiling the unfolding pathway of FALS associated G37R SOD1 mutant: a computational study. Mol. BioSyst. 6, 1032–1039. [DOI] [PubMed] [Google Scholar]
- (18).Keerthana SP, and Kolandaivel P. (2015) Study of mutation and misfolding of Cu-Zn SOD1 protein. J. Biomol. Struct. Dyn. 33, 167–183. [DOI] [PubMed] [Google Scholar]
- (19).Pastor MT, Esteras-Chopo A, and Serrano L. (2007) Hacking the code of amyloid formation: the amyloid stretch hypothesis. Prion 1, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Esteras-Chopo A, Serrano L, and Lopez de la Paz M. (2005) The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. U. S. A. 102, 16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hu L, Cui W, He Z, Shi X, Feng K, Ma B, and Cai YD (2012) Cooperativity among short amyloid stretches in long amyloidogenic sequences. PLoS One 7, e39369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ma B, and Nussinov R. (2002) Molecular dynamics simulations of alanine rich beta-sheet oligomers: Insight into amyloid formation. Protein science: a publication of the Protein Society 11, 2335–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zanuy D, Ma B, and Nussinov R. (2003) Short peptide amyloid organization: stabilities and conformations of the islet amyloid peptide NFGAIL. Biophys. J. 84, 1884–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Haspel N, Zanuy D, Ma B, Wolfson H, and Nussinov R. (2005) A comparative study of amyloid fibril formation by residues 15−19 of the human calcitonin hormone: a single beta-sheet model with a small hydrophobic core. J. Mol. Biol. 345, 1213–1227. [DOI] [PubMed] [Google Scholar]
- (25).Zheng J, Ma B, Tsai CJ, and Nussinov R. (2006) Structural stability and dynamics of an amyloid-forming peptide GNNQQNY from the yeast prion sup-35. Biophys. J. 91, 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang M, Hu R, Chen H, Chang Y, Ma J, Liang G, Mi J, Wang Y, and Zheng J. (2015) Polymorphic cross-seeding amyloid assemblies of amyloid-beta and human islet amyloid polypeptide. Phys. Chem. Chem. Phys. 17, 23245–23256. [DOI] [PubMed] [Google Scholar]
- (27).Lu Y, Wei G, and Derreumaux P. (2012) Structural, thermodynamical, and dynamical properties of oligomers formed by the amyloid NNQQ peptide: insights from coarse-grained simulations. J. Chem. Phys. 137, 025101. [DOI] [PubMed] [Google Scholar]
- (28).Wei G, Song W, Derreumaux P, and Mousseau N. (2008) Self-assembly of amyloid-forming peptides by molecular dynamics simulations. Front. Biosci., Landmark Ed. 13, 5681–5692. [DOI] [PubMed] [Google Scholar]
- (29).Wei G, Jewett AI, and Shea JE (2010) Structural diversity of dimers of the Alzheimer amyloid-beta(25−35) peptide and polymorphism of the resulting fibrils. Phys. Chem. Chem. Phys. 12, 3622–3629. [DOI] [PubMed] [Google Scholar]
- (30).Shea JE, and Urbanc B. (2012) Insights into Abeta aggregation: a molecular dynamics perspective. Curr. Top. Med. Chem. 12, 2596–2610. [DOI] [PubMed] [Google Scholar]
- (31).Viet MH, Nguyen PH, Derreumaux P, and Li MS (2014) Effect of the English familial disease mutation (H6R) on the monomers and dimers of Abeta40 and Abeta42. ACS Chem. Neurosci. 5, 646–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liang C, Ni R, Smith JE, Childers WS, Mehta AK, and Lynn DG (2014) Kinetic intermediates in amyloid assembly. J. Am. Chem. Soc. 136, 15146–15149. [DOI] [PubMed] [Google Scholar]
- (33).Saric A, Chebaro YC, Knowles TP, and Frenkel D. (2014) Crucial role of nonspecific interactions in amyloid nucleation. Proc. Natl. Acad. Sci. U. S. A. 111, 17869–17874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Szavits-Nossan J, Eden K, Morris RJ, MacPhee CE, Evans MR, and Allen RJ (2014) Inherent variability in the kinetics of autocatalytic protein self-assembly. Phys. Rev. Lett. 113, 098101. [DOI] [PubMed] [Google Scholar]
- (35).Eden K, Morris R, Gillam J, MacPhee CE, and Allen RJ (2015) Competition between primary nucleation and autocatalysis in amyloid fibril self-assembly. Biophys. J. 108, 632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Arosio P, Knowles TP, and Linse S. (2015) On the lag phase in amyloid fibril formation. Phys. Chem. Chem. Phys. 17, 7606–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dovidchenko NV, Finkelstein AV, and Galzitskaya OV (2014) How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of aggregation. I. Modeling the amyloid protofibril formation. J. Phys. Chem. B 118, 1189–1197. [DOI] [PubMed] [Google Scholar]
- (38).Straub JE, and Thirumalai D. (2011) Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Annu. Rev. Phys. Chem. 62, 437–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nguyen P, and Derreumaux P. (2014) Understanding amyloid fibril nucleation and abeta oligomer/drug interactions from computer simulations. Acc. Chem. Res. 47, 603–611. [DOI] [PubMed] [Google Scholar]
- (40).Carulla N, Zhou M, Giralt E, Robinson CV, and Dobson CM (2010) Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc. Chem. Res. 43, 1072–1079. [DOI] [PubMed] [Google Scholar]
- (41).Cabriolu R, and Auer S. (2011) Amyloid fibrillation kinetics: insight from atomistic nucleation theory. J. Mol. Biol. 411, 275–285. [DOI] [PubMed] [Google Scholar]
- (42).Auer S. (2014) Amyloid fibril nucleation: effect of amino acid hydrophobicity. J. Phys. Chem. B 118, 5289–5299. [DOI] [PubMed] [Google Scholar]
- (43).Kashchiev D, Cabriolu R, and Auer S. (2013) Confounding the paradigm: peculiarities of amyloid fibril nucleation. J. Am. Chem. Soc. 135, 1531–1539. [DOI] [PubMed] [Google Scholar]
- (44).Castello F, Casares S, Ruedas-Rama MJ, and Orte A. (2015) The First Step of Amyloidogenic Aggregation. J. Phys. Chem. B 119, 8260–8267. [DOI] [PubMed] [Google Scholar]
- (45).Auer S. (2015) Nucleation of polymorphic amyloid fibrils. Biophys. J. 108, 1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hills RD Jr., and Brooks CL 3rd. (2007) Hydrophobic cooperativity as a mechanism for amyloid nucleation. J. Mol. Biol. 368, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ, and Eisenberg DS (2014) Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. U. S. A. 111, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Valentine JS, Doucette PA, and Zittin Potter S. (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593. [DOI] [PubMed] [Google Scholar]
- (49).Turner BJ, and Talbot K. (2008) Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 85, 94–134. [DOI] [PubMed] [Google Scholar]
- (50).Morimoto A, Irie K, Murakami K, Ohigashi H, Shindo M, Nagao M, Shimizu T, and Shirasawa T. (2002) Aggregation and neurotoxicity of mutant amyloid beta (A beta) peptides with proline replacement: importance of turn formation at positions 22 and 23. Biochem. Biophys. Res. Commun. 295, 306–311. [DOI] [PubMed] [Google Scholar]
- (51).Laganowsky A, Liu C, Sawaya MR, Whitelegge JP, Park J, Zhao M, Pensalfini A, Soriaga AB, Landau M, Teng PK, Cascio D, Glabe C, and Eisenberg D. (2012) Atomic view of a toxic amyloid small oligomer. Science 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).De Simone A, and Derreumaux P. (2010) Low molecular weight oligomers of amyloid peptides display beta-barrel conformations: a replica exchange molecular dynamics study in explicit solvent. J. Chem. Phys. 132, 165103. [DOI] [PubMed] [Google Scholar]
- (53).Song W, Wei G, Mousseau N, and Derreumaux P. (2008) Self-assembly of the beta2-microglobulin NHVTLSQ peptide using a coarse-grained protein model reveals a beta-barrel species. J. Phys. Chem. B 112, 4410–4418. [DOI] [PubMed] [Google Scholar]
- (54).Ferrone F. (1999) Analysis of protein aggregation kinetics. Methods Enzymol. 309, 256–274. [DOI] [PubMed] [Google Scholar]
- (55).Pellarin R, and Caflisch A. (2006) Interpreting the aggregation kinetics of amyloid peptides. J. Mol. Biol. 360, 882–892. [DOI] [PubMed] [Google Scholar]
- (56).Co NT, and Li MS (2012) New method for determining size of critical nucleus of fibril formation of polypeptide chains. J. Chem. Phys. 137, 095101. [DOI] [PubMed] [Google Scholar]
- (57).Nguyen PH, Li MS, Stock G, Straub JE, and Thirumalai D. (2007) Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. U. S. A. 104, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Nasica-Labouze J, and Mousseau N. (2012) Kinetics of amyloid aggregation: a study of the GNNQQNY prion sequence. PLoS Comput. Biol. 8, e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wu C, Lei H, and Duan Y. (2004) Formation of partially ordered oligomers of amyloidogenic hexapeptide (NFGAIL) in aqueous solution observed in molecular dynamics simulations. Biophys. J. 87, 3000–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete NV, Cote S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, and Derreumaux P. (2015) Amyloid beta Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 115, 3518–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Nguyen PH, and Derreumaux P. (2013) Conformational ensemble and polymorphism of the all-atom Alzheimer’s Abeta(37−42) amyloid peptide oligomers. J. Phys. Chem. B 117, 5831–5840. [DOI] [PubMed] [Google Scholar]
- (62).Baftizadeh F, Pietrucci F, Biarnes X, and Laio A. (2013) Nucleation process of a fibril precursor in the C-terminal segment of amyloid-beta. Phys. Rev. Lett. 110, 168103. [DOI] [PubMed] [Google Scholar]
- (63).Baftizadeh F, Biarnes X, Pietrucci F, Affinito F, and Laio A. (2012) Multidimensional view of amyloid fibril nucleation in atomistic detail. J. Am. Chem. Soc. 134, 3886–3894. [DOI] [PubMed] [Google Scholar]
- (64).Matthes D, Gapsys V, and de Groot BL (2012) Driving forces and structural determinants of steric zipper peptide oligomer formation elucidated by atomistic simulations. J. Mol. Biol. 421, 390–416. [DOI] [PubMed] [Google Scholar]
- (65).Berendsen HJC, Postma JPM, van Gunsteren WF, and Hermans J. (1981) Interaction Models for Water in Relation to Protein Hydration. Intermolecular Force.The Jerusalem Symposia on Quantum Chemistry and Biochemistry 14, 331–342. [Google Scholar]
- (66).Sugita Y, and Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- (67).Okamoto Y. (2004) Generalized-ensemble algorithms: enhanced sampling techniques for Monte Carlo and molecular dynamics simulations. J. Mol. Graphics Modell. 22, 425–439. [DOI] [PubMed] [Google Scholar]
- (68).Nadler W, and Hansmann UH (2008) Optimized explicit-solvent replica exchange molecular dynamics from scratch. J. Phys. Chem. B 112, 10386–10387. [DOI] [PubMed] [Google Scholar]
- (69).Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJ (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- (70).Oostenbrink C, Villa A, Mark AE, and van Gunsteren WF (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676. [DOI] [PubMed] [Google Scholar]
- (71).Hess B, Bekker H, Berendsen HJC, and Fraaije JGEM (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- (72).Miyamoto S, and Kollman PA (1992) Settle: An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 13, 952–962. [Google Scholar]
- (73).Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, and Haak JR (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- (74).Bussi G, Donadio D, and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101. [DOI] [PubMed] [Google Scholar]
- (75).Larini L, and Shea JE (2012) Role of beta-hairpin formation in aggregation: the self-assembly of the amyloid-beta(25−35) peptide. Biophys. J. 103, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Mu X, Eckes KM, Nguyen MM, Suggs LJ, and Ren P. (2012) Experimental and computational studies reveal an alternative supramolecular structure for fmoc-dipeptide self-assembly. Biomacromolecules 13, 3562–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Levine ZA, Larini L, LaPointe NE, Feinstein SC, and Shea JE (2015) Regulation and aggregation of intrinsically disordered peptides. Proc. Natl. Acad. Sci. U. S. A. 112, 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Sun Y, Qian Z, Guo C, and Wei G. (2015) Amphiphilic Peptides A6K and V6K Display Distinct Oligomeric Structures and Self-Assembly Dynamics: A Combined All-Atom and Coarse-Grained Simulation Study. Biomacromolecules 16, 2940–2949. [DOI] [PubMed] [Google Scholar]
- (79).Kabsch W, and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (80).Daura X, Gademann K, Jaun B, Seebach D, van Gunsteren WF, and Mark AE (1999) Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed. 38, 236–240. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.