

Summary
While the extracellular matrix (ECM) is known to regulate neural stem cell quiescence in the adult subventricular zone (SVZ), the function of ECM in the developing SVZ remains unknown. Here, we report that the ECM receptor dystroglycan regulates a unique developmental restructuring of ECM in the early postnatal SVZ. Dystroglycan is furthermore required for ependymal cell differentiation and assembly of niche pinwheel structures, at least in part by suppressing Notch activation in radial glial cells, which leads to the increased expression of MCI, Myb, and FoxJ1, transcriptional regulators necessary for acquisition of the multiciliated phenotype. Dystroglycan also regulates perinatal radial glial cell proliferation and transition into intermediate gliogenic progenitors, such that either acute or constitutive dystroglycan loss-of-function results in increased oligodendrogenesis. These findings reveal a role for dystroglycan in orchestrating both the assembly and function of the SVZ neural stem cell niche.
eTOC Paragraph
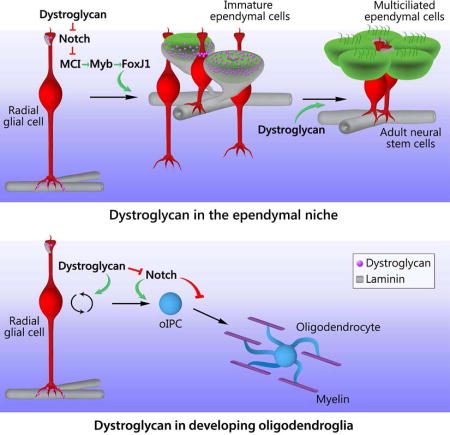
McClenahan and Sharma et al. uncover dystroglycan’s role in driving extracellular matrix reorganization and cellular development during the emergence of the adult subventricular zone neural stem cell niche. During this process dystroglycan helps tune Notch signaling, promoting ependymal cell maturation and the regulated production of glia by niche stem cells.
Introduction
Embryonic radial glial neural stem cells span the entire cortical plate of the developing brain, terminating apically at the surface of the lateral ventricle and basally at the pial basement membrane. Perinatally, radial glia detach from the pial surface and transform into both adult neural stem cells and multiciliated ependymal cells, which then arrange into “pinwheels”, rings of ependymal cells that surround the apical processes of adult neural stem cells (Merkle et al., 2004; Mirzadeh et al., 2008; Spassky et al., 2005). These pinwheels are central to the function of the adult ventricular/subventricular zone (V/SVZ), a specialized microenvironment that supports stem cell quiescence and the controlled production of neurons and glia. Concurrent with V/SVZ stem cell niche construction, neural stem and progenitor cells engender the principal wave of dorsal gliogenesis. However while factors regulating these processes and their coordinated coupling are likely critical to proper brain development, they remain poorly understood.
The extracellular matrix (ECM) has recently been identified as a regulator of proliferation in the adult SVZ niche, where neural stem cell basal processes attach to the laminin-rich basal lamina of the SVZ vascular network (Mirzadeh et al., 2008; Shen et al., 2008; Tavazoie et al., 2008). Activated neural stem cells and transit-amplifying progenitors express higher levels of the laminin receptor α6β1 integrin than more lineage-restricted cells, and blocking α6β1 results in detachment and aberrant stem/progenitor proliferation (Kazanis et al.; Shen et al., 2008). The adult SVZ also has a unique extra-vascular ECM, featuring ECM aggregates near the ventricular surface as well as “fractones”; thin, highly-branched ECM structures that appear to emanate from the vascular basal lamina. Fractones contain the heparan sulfate proteoglycan perlecan, which sequesters the mitogen FGF-2 thereby enhancing proliferation in fractone-contacting stem cells (Douet et al., 2013; Kerever et al., 2014; Kerever et al., 2007; Mercier et al., 2002). ECM aggregates near the ventricle surface, on the other hand, remain of unknown function. Despite the recent attention the specialized ECM of the adult V/SVZ has garnered, roles for cell-ECM interactions during SVZ stem cell niche assembly, and the concurrent process of postnatal SVZ gliogenesis, remain unknown.
Dystroglycan is a transmembrane ECM receptor that mediates cell interactions with multiple ECM ligands, including laminins. Dystroglycan is best known as a member of the dystrophin-glycoprotein complex that bridges ECM and cytoskeleton in skeletal myocytes. Yet dystroglycan is also critical for brain development, as “dystroglycanopathies”, muscular dystrophies arising from loss of dystroglycan expression or defective dystroglycan glycosylation, result in profound deficits in brain structure and function. In the developing brain, dystroglycan is found on the basal endfeet of embryonic radial glia (Myshrall et al., 2012) and the loss of radial glial attachment to the pial basement membrane is thought to underlie neuronal migration defects in these dystroglycanopathies (Moore et al., 2002). In the adult brain, dystroglycan is found on the perivascular endfeet of astrocytes, where it mediates their adhesion to the vascular basal lamina at the blood-brain barrier and regulates Kir4.1 and aquaporin-4 localization (Hawkins et al., 2013; Hirrlinger et al., 2011; Menezes et al., 2014; Nico et al., 2010; Noell et al., 2011). However there is limited understanding of dystroglycan’s function in the developing postnatal brain.
Here, we identify a role for brain dystroglycan as an essential regulator of postnatal SVZ niche development. We find that dystroglycan regulates neural stem and progenitor cell proliferation, suppresses Notch activation in neural stem cells to promote ependymal cell maturation, and promotes the formation of stem cell niche pinwheels in the early postnatal V/SVZ. Additionally, we reveal that dystroglycan modulates oligodendrogenesis (i.e. niche output) and may also regulate Notch activity in oligodendrocyte lineage cells to promote the timely oligodendrocyte differentiation and myelination.
Results
Dystroglycan organizes laminin into hubs and tethers during early postnatal VZ/SVZ niche assembly
Extracellular matrix (ECM) proteins in the adult SVZ neural stem cell niche are uniquely arranged, existing in both vascular basal lamina and extra-vascular structures (Shen et al., 2008; Tavazoie et al., 2008). However, how niche ECM organization is adopted and the factors regulating its development remain unknown. We therefore examined laminin immunoreactivity in wild type SVZ wholemounts during the period when the SVZ niche is first established. At birth, the vascular plexus is morphologically similar to, but denser than, that of the adult mouse (Fig. 1A). Fractones, thin ECM structures projecting from the vascular basal lamina (Mercier et al., 2002), are present at birth (arrowheads and inset, Fig. 1B), whereas laminin-rich aggregates begin to appear at the ventricular surface between P3 and P8 (arrows, Fig. 1B). Z-projections of optical stacks at P8 indicate that these laminin aggregates are distinct from the vascular basal lamina (arrows, ventricle surface at the top). At P3, laminin is diffusely distributed on or near the apical (ventrical-adjacent) surface of immature ependymal cells, which have a apical surface area several-fold larger than neural stem cells (Fig. 1C). However, by P8, large laminin aggregates, or “hubs” begin to coalesce at the ventricular surface of these cells, concurrent with a reduction in diffuse ependymal cell-associated laminin (P8, arrowheads). Finally, by P21, ventricular surface laminin is now largely restricted to hubs (P21, arrowheads) with many such hubs found at the interface of ependymal cells and adult neural stem cells within niche pinwheels (P21, arrows).
Figure 1. Laminin-rich ECM Structures in the Early Postnatal SVZ.

(A) Laminin immunoreactivity defines the vascular basal lamina in a SVZ wholemount from a WT postnatal day 0 (P0) mouse. Box denotes anterior-dorsal area used for wholemount analysis. A, anterior; P, posterior. (B) Confocal stack projections from WT wholemounts highlighting vascular and extra-vascular laminin. Extensive blood vessel-fractone networks (arrowheads) and ECM “hubs” (arrows) are visible by P8. Bottom panels: XZ projections, with ventricular surface at top. (C) En face views of Laminin and β-catenin immunoreactivity in WT wholemounts illustrate the dramatic ECM reorganization from P3 to P21. (D) 3D reconstructions of confocal stacks showing laminin immunoreactivity in the wild-type V/SVZ. At P3, ventricular surface cell-associated laminin is contiguous with the blood vessel basal lamina either directly (arrowheads) or via “tethers” (arrows). At P8, ventricular surface “hubs”, (arrowheads) and “bulbs”, deeper in the SVZ at fractone termini (arrows), are also observed. (E) En face views of the SVZ ventricular surface at P21. 3D reconstructions of confocal stacks show laminin+ hubs in conjunction with β-catenin. In some instances, small tethers appear to bridge adjacent hubs (arrow). (F) Laminin and GFP IHC in wholemounts from FoxJ1-GFP mice. Left panels: En face view of cell-associated laminin in developing ependymal cells. Top right: 3D reconstructions of confocal stacks showing laminin “tethers”. Bottom right: 2D orthogonal view of top right. (G) Laminin and GFP IHC in a P21 FoxJ1-GFP mouse wholemount. Left panel: En face views of the ventricular surface, with dashed lines delineating individual pinwheels. Right panel: 3D reconstructions of confocal stacks from the same field. At P21, most laminin hubs remain associated with ependymal cell surfaces (arrow), but many are now found at the center of pinwheels (closed arrowhead) or appear to be in the process of relocating (open arrowhead). (H) Laminin and GFAP IHC in an adult FoxJ1-GFP wholemount. Left panel: En face view, with dashed lines delineating individual pinwheels. Right panel: 3D reconstruction. Scale bars: 50 µm (B), 25 µm (C), 20 µm (F, G, H, I).
3D reconstructions of V/SVZ laminin revealed that at P3, ventricular surface laminin is, in fact, contiguous with the vascular basal lamina, either directly (Fig. 1D, P3, arrowheads) or via “tethers” (arrows). By P8, however, two distinct classes of laminin aggregates emerge: laminin hubs, visible at the surface (P8, arrowheads) and fractone “bulbs” that appear to be fractone termini, which lie beneath the developing ependymal layer (P8, arrow). These reconstructions confirm that by P21, most laminin hub structures localize to ependymal cell borders, often near pinwheel centers, and are not contiguous with the vascular basal lamina. Additionally, many hubs appear to be connected via tethers, even across cell borders (Fig. 1E, inset, arrow), suggestive of coordinated grouping and/or merging of hub structures.
To confirm that the laminin-positive cells we observed were indeed ependymal cells, we used FoxJ1-GFP reporter mice to label ependymal cells during development. Both en face views and 3D reconstructions over the first postnatal week confirmed that (i) cell-associated laminin is almost exclusively restricted to immature ependymal cells, indicating an ependymal cell origin of surface laminin, and (ii) laminin tethers beneath the apical surface serve as bridges, connecting immature ependymal cells to the vascular basal lamina during niche construction (Fig. 1F). We additionally found that niche pinwheel arrangement is nearly complete by P21. At this point, many laminin hubs are central on the apical surfaces of ependymal cells (Fig. 1G, arrow), while some are transposed to the center of pinwheels (closed arrowhead), and others appear to be in transit, protruding into the center of a pinwheel while retaining attachment to an ependymal cell (open arrowhead). By adulthood, however, virtually all pinwheels contain one or more laminin hubs, localized to the interface between ependymal cells and neural stem cell apical processes (Fig. 1H). Taken together, our observations indicate that in the early postnatal V/SVZ, extravascular laminin is concentrated around immature ependymal cells, becoming restricted with niche maturation, and eventually achieving an adult configuration of discrete laminin hubs at the interfaces between neural stem cells and ependymal cells.
The extensive reorganization of V/SVZ laminin during niche development and location of laminin hubs within niche pinwheels suggested that laminin-receptor interactions regulates aspects of niche construction and function. In several types of cells, the laminin receptor dystroglycan is required for initial laminin clustering on cell surfaces (Cohen et al., 1997; Colognato et al., 1999; Henry and Campbell, 1998; Henry et al., 2001; Montanaro et al., 1999; Weir et al., 2006). Examination of laminin and dystroglycan in the V/SVZ using wholemount IHC (Fig. 2) revealed that laminin and dystroglycan are localized mainly to radial glial cell (RGC) cell junctions in the newborn VZ (Fig. 2A). Emerging ependymal cells, identified by their large apical surface areas, have more cell surface laminin by P3, paralleled by an increase in surface dystroglycan. At P8, cell-associated laminin is more restricted, and dystroglycan immunoreactivity is even more limited, with dual laminin- and dystroglycan-positive hubs now appearing at the ventricular surface. To more carefully examine the relationship between dystroglycan and ependymal cell maturation, we turned to SVZ-derived ependymal cell cultures (Fig. 2B). Using SVZ cells from P0 FoxJ1-GFP mice (wild type for dystroglycan), we found that dystroglycan level is tightly correlated with ependymal cell maturation (Fig. 2C), recapitulating our in vivo observations, and is highest in mature, multicilliated ependymal cells relative to immature ependymal cells or non-ependymal cells (Fig. 2D).
Figure 2. Dystroglycan Regulates Laminin Organization in the Developing Ependymal Niche.

(A) En face views of laminin and dystroglycan IHC in wild-type SVZ wholemounts reveals developmental upregulation on ependymal cell surfaces. By P8, laminin- and dystroglycan-positive hubs are apparent (arrowheads, inset). (B) Schematic of SVZ ependymal cell cultures. A typical ependymal cell cluster is outlined (far right; GFP, green; β-catenin, red; γ-tubulin, white) (C) Dystroglycan IHC in developing SVZ ependymal cell cultures; mature ependymal cells are identified by ciliary acetylated-α-tubulin immunoreactivity. (D) Quantification of dystroglycan intensity in CD24-, CD24+ and CD24+ multi-ciliated cells. Error bars, SEM; n=3. (E) Laminin and β-catenin IHC in SVZ wholemounts. (F) Laminin and β-catenin IHC in P21 SVZ wholemounts to detect laminin+ ECM hubs. Images with matched intensity thresholds (for quantification; see Fig. 2G,H) are shown in left panels; enhanced laminin intensity (“max intensity”) to reveal weakly stained laminin hubs is shown in right panels. (G) Quantification of laminin hubs per 1000 µm2. ***p < 0.001, Student’s t-test; error bars, SEM; n=4. (H) Quantification of mean hub size. Error bars, SEM; n=4. (I) Laminin and GFP IHC in FoxJ1-GFP SVZ ependymal cultures with control or dystroglycan blocking antibodies added during differentiation. Scale bars: 25 µm (A,C, E), 50 µm (I).
We next investigated dystroglycan’s role in the developing SVZ neural stem cell niche by comparing nestin-cre;DAGFl/Fl (DAG cKO) mice, which lack dystroglycan in neural stem cells and their progeny, to nestin-cre−/−;DAGFl/Fl (WT) littermates (Fig. S1A). Having confirmed the loss of dystroglycan protein by IHC (Fig. S1B), and western blot (Fig. S1C), we examined SVZ RGC density at birth and found that DAG cKO and WT mice have similar BLBP+ cell densities (45.1 ± 4.2 × 105 cells/mm3 vs. 44.6 ± 2.3 × 105 in WT; Fig. S2A,B). The apical processes of nestin+ RGCs make contact with laminin-positive puncta at the ventricular surface at birth (Fig. S2C), but fewer VZ laminin puncta are present in DAG cKO mice and the association of RGC apical processes with laminin puncta is diminished in the dystroglycan-deficient SVZ. It should be noted that laminin immunoreactivity in the vascular basal lamina appears normal in DAG cKO mice, with typical contacts between RGC basal processes and blood vessel basal lamina (Fig. S2C). Additionally, qRT-PCR revealed that the expression of laminin genes in the DAG cKO SVZ is largely unchanged (not shown), indicating that large deficits in apical surface laminin (i.e., laminin hubs) reflect either a failure of laminin recruitment/organization on cell surfaces, or, a highly localized loss of expression.
To gain a more detailed spatiotemporal understanding of VZ laminin, we examined en face views of the ventricular surface from SVZ wholemounts during early niche development (Fig. 2E). As expected, at P0 the density of RGCs at the apical surface of the lateral ventricle appears normal in DAG cKO mice, with laminin immunoreactivity diminished from that in wild type. By P3, developing ependymal cells are readily apparent in both WT and DAG cKO mice, but the dystroglycan-deficient VZ has a marked decrease in pericellular laminin associated with developing ependymal cells (Fig. 2E). Interestingly, by P8, cells at the ventricular surface of DAG cKO mice have increased cell-associated laminin, surpassing levels seen in wildtype littermates (Fig. 2E). At P21, however, when laminin immunoreactivity is largely confined to hubs in the WT VZ, DAG cKO mice display a more than 30-fold reduction in the number of laminin hubs per 1000 µm2 (Fig. 2F, G). Additionally, we found a trend towards a reduction in mean hub size in DAG cKO wholemounts (Fig. 2H; Fig. 1.45±0.19 µm2 in WT vs. 0.54±0.25 µm2 in DAG cKO). As we noted that some weakly immunoreactive hubs were eliminated by intensity thresh-holding, we also adjusted the image intensity to visualize some very weakly immunoreactive hubs in the DAG cKO (Fig. 2F “max intensity”). However even upon inclusion of all weakly positive laminin hubs, DAG cKO wholemounts contained almost two-fold fewer laminin hubs.
We next turned to SVZ ependymal cell cultures (Fig. 2B) to test whether blocking dystroglycan’s ability to bind ECM is sufficient to disrupt ependymal cell surface laminin. We allowed RGCs from P0 FoxJ1-GFP mice to proliferate for 5 days, and added dystroglycan blocking (DG block) or IgM control antibodies during the ependymal differentiation stage. Thin, fibrillary laminin structures appeared in on FoxJ1-GFP ependymal cells, but these cell surface laminin clusters appeared less abundant and more disorganized in the presence of dystroglycan-blocking antibodies (Fig. 2I). These data indicate that laminin recruitment and/or retention at ependymal cell surfaces is aberrant in the absence of dystroglycan, and, even in ependymal cells of the same genotype, laminin surface organization is perturbed by blocking dystroglycan’s ability to bind ECM proteins.
Dystroglycan is required for ependymal cell maturation and SVZ stem cell niche assembly
Early in SVZ stem cell niche construction, a subset of RGCs transform into ependymal cells (Merkle et al., 2004; Spassky et al., 2005). As ECM interactions can regulate cell differentiation in the adult stem cell niche (Douet et al., 2012; Douet et al., 2013; Shen et al., 2008; Tavazoie et al., 2008) we wondered if dystroglycan might regulate the maturation of RGCs into ependymal cells. Despite having equivalent RGC densities at birth (Fig. S2B), P3 DAG cKO mice have fewer CD24+ ependymal cells than wildtype littermates (3.01±0.23 × 103 cells/mm2 vs. 4.82±0.34 × 103 in WT; Fig. 3A,B) and slightly more BLBP+ RGCs than WT littermates (44.2±2.16 × 105 cells/mm3 vs. 38.2±2.05 × 105 in WT; Fig. 3C,D). Indeed, while the BLBP+ population in WT mice decreases from birth to P3, it does not change in the DAG cKO SVZ (Fig. S2B and Fig. 3D). Together, our findings of decreased ependymal cell densities concurrent with elevated RGC densities indicate a delayed differentiation of RGCs into ependymal cells. As ependymal cells mature, they increase their apical surface area and acquire large patches of multiple cilia that can be visualized using γ-tubulin IHC to detect ciliary basal bodies (Delgehyr et al., 2015). By these measures, DAG cKO SVZ also exhibit lower levels of ependymal cell maturation through the third postnatal week (Fig. 3E, H), with a significantly lower percentage of apical surface coverage by multiciliated ependymal cells both at P8 (39.8±2.3% of total area vs. 62.0±3.1% in WT; Fig. 3F) and P21 (41.5±3.0% of total area vs. 61.8% ± 5.0% in WT; Fig. 3I). Dystroglycan loss also disrupts the ability of ependymal cells to arrange into pinwheels. At P8 (Fig. 3G) and P21 (Fig. 3J), ependymal-NSC pinwheel clusters are smaller in the DG cKO SVZ (P8: 7.5±0.3 × 102 µm2 vs. 11.3±0.6 × 102 in WT; P21: 8.3±1.0 × 102 µm2 vs. 13.9±1.4 × 102 in WT), and have a disorganized appearance (Fig. 3E,H).
Figure 3. Dystroglycan is Required for Ependymal Cell Maturation and Niche Assembly.

(A) β-catenin, CD24, and laminin IHC in SVZ wholemounts from P3 mice. (B) Quantification of ventricular surface CD24+ cells at P3. *p < 0.05, Student’s t-test; error bars, SEM; n=3. (C) BLBP IHC in SVZ coronal sections at P3. (D) Quantification of BLBP+ cells in the dorsal SVZ at P3 . *p < 0.05, Student’s t-test; error bars, SEM; n=3. (E) β-catenin and γ-tubulin IHC in SVZ wholemounts from P8 WT and DAG cKO mice. Right panels show examples of measured clusters. (F) Quantification of relative multicilliated cell cluster coverage in P8 SVZ wholemounts. *p<0.05, Student’s t-test; error bars, SEM; n=3. (G) Quantification of average cluster size in P8 SVZ wholemounts. **p<0.01, Student’s t-test; error bars, SEM; n=3. (H) β-catenin and γ-tubulin IHC in SVZ wholemounts. (I) Quantification of relative surface area coverage of multiciliated cells in P21 WT and DAG cKO wholemounts (**p<0.01, Student’s t-test; error bars, SEM; n=5). (J) Quantification of average cluster area in P21 WT and DAG cKO wholemounts. (K) Higher magnification view of β-catenin and γ-tubulin IHC in P21 wholemounts highlights basal body (BB) patches. (L) Schematic depicting BB patch analysis on ependymal cell apical surfaces. BB displacement is the length from the apical surface center to the BB patch center (“a”), over the length from the apical surface center to the cell perimeter (“b”), with b being measured along the vector of “a”. (M) Quantification of fractional displacement of BB patches (p=0.0007; n=337 cells from 3 mice per genotype). (N) Absolute distance measurements of BB patch displacement and cell radii (p=0.432; n=3). (O) Quantification of fractional patch area (n=315 (WT) or n=337 (KO) cells from 3 mice per genotype; p<0.001). (P) Distribution of BB patch angles (each BB patch angle normalized to the average BB patch angle per field). (n=315 (WT) or n=337 (DAG cKO) cells from 3 mice per genotype, p=0.0860, Watson’s U2 test). Scale bars: 25µm (A, E, H), 50µm (C).
The size and polarity of ependymal cell ciliary basal body patches, normally tightly regulated and essential for proper CSF flow and neural stem cell function (Delgehyr et al., 2015), are also abnormal in the DAG cKO SVZ at P21 (Fig. 3K). The fractional basal body (BB) patch displacement (Fig. 3L; also Methods), is significantly increased in DAG cKO ependymal cells (Fig. 3M; 0.42±0.01 vs. 0.30±0.01 in WT). This change reflects an increase in BB patch displacement itself, as the absolute distance of BB patch displacement increases (Fig. 3N), while the average cell radius does not (Fig. 3N).
Additionally, BB patches are smaller in DAG cKO ependymal cells (Fig. 3O; 0.111±0.004 vs. 0.156±0.004 in WT). And, while the BB patch angle, a readout for ependymal cell translational polarity (with each cell’s BB patch angle normalized to the average BB patch angle per field), displays a trend towards a greater variability in DAG cKO ependymal cells relative to that in WT, this trend is not statistically significant.
Dystroglycan suppresses Notch to promote ependymal cell maturation
FoxJ1 is a transcription factor essential for regulating ciliary biogenesis in all multiciliated cells (Brody et al., 2000; Jacquet et al., 2009). To explore whether dystroglycan’s effects on ependymal development are due to modulation of FoxJ1 expression, we applied dystroglycan-blocking (IIH6) or control antibodies to SVZ cultures of FoxJ1-GFP neonates (as in Fig. 2B), allowing us to assess FoxJ1 activation within each cell, rather than as a global change (Fig. 4A). In agreement with ependymal cell deficits seen in DAG cKO mice, dystroglycan blockade significantly reduces the percentage of GFP+ ependymal cells after 7 days in differentiation-promoting medium (49.4±5.5% of total cells vs. 71.5±5.1% in controls; Fig. 4B), as well as the proportion of GFP+ cells that became multiciliated (17.5±2.2% vs. 31.8±3.1% in controls; Fig. 4C). Additionally, dystroglycan-blocking antibodies antagonize the ability of GFP+ cells to arrange into pinwheel-like clusters, reducing cluster areas by over 50% (2.48±0.08 × 103 µm2 vs. 5.62±0.75 × 103 in controls; Fig. 4D).
Figure 4. Dystroglycan suppresses Notch in promoting ependymal cell maturation.

(A) IHC to detect GFP and β-catenin (upper panel) or γ-tubulin, FoxJ1, and phalloidin (middle and lower panels) in differentiating FoxJ1-GFP SVZ ependymal cultures in the presence of control or DG-blocking antibodies. (B) Quantification of GFP+ ependymal cells following differentiation with control or dystroglycan-blocking antibodies (*p<0.05, Student’s t-test; error bars, SEM; n=3). (C) Quantification of multiciliated GFP+ cells after differentiation with control or dystroglycan-blocking antibodies (*p<0.05, Student’s t-test; error bars, SEM; n=3). (D) Quantification of ependymal cell/RGC cluster area in control or DG-blocked cultures. (*p<0.05, Student’s t-test; error bars, SEM; n=3). (E) Fold change in mRNA levels of multiciliogenic program genes at maturation onset or after 5 days in medium to promote ependymal cell maturation (*p<0.05, Student’s t-test; error bars, SEM; n=4). (F) Fold change in mRNA levels of Hes1 and HeyL in SVZ ependymal cultures at 6 and 24 hours post-switch to maturation medium in the presence or absence of dystroglycan-blocking antibodies (*p<0.05, **p<0.01 Student’s t-test; error bars, SEM; n=6). (G) Fold change in mRNA levels of MCI and Myb following 24 hours in dystroglycan blocking antibodies, the γ-seretase inhibitor, DAPT, or a combination of both (*p<0.05, **p<0.01, ***p<0.001, Student’s t-test; error bars, SEM; n=6). (H) Ependymal cell cultures at day 3 of differentiation, with IHC to detect either FoxJ1 and cortical actin (phalloidin; upper panels), or FoxJ1 and γ-tubulin (lower panels) in cultures treated with DAPT, dystroglycan-blocking antibodies (DG block), or a combination thereof. (I) Quantification of the percentage of SVZ cells acquiring a multiciliated phenotype in SVZ ependymal cultures treated as in (H) (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=4). (J) Quantification of the percentage of FoxJ1+ cells in SVZ ependymal cultures treated as in (H) (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=4). (K) Analysis of solo and clustered FoxJ1+ cells in SVZ ependymal cultures treated as in (H) (*p<0.05, Student’s t-test; error bars, SEM; n=3). Scale bars, 50 microns (D), 20 microns (I).
To further probe dystroglycan’s effects on the ependymal differentiation program, we assessed FoxJ1 expression along with that of its upstream regulators in ependymal cells - Mcidas (MCI) and Myb - in SVZ cultures treated with dystroglycan-blocking antibodies (Fig. 4E). While MCI, Myb, and FoxJ1 mRNA levels remain unchanged by acute treatment during the cell expansion phase (6 or 24 hours; not shown), by maturation onset, Myb and FoxJ1 are significantly suppressed (0.08±0.03 fold and 0.43±0.09 fold control, respectively). At this time point, MCI was not reliably detectable making the determination of ΔCT difficult (not shown). By day 5 in ependymal cell maturation medium, however, MCI levels are significantly lower (0.73±0.07 fold) in the dystroglycan-blocking condition compared to control. FoxJ1 mRNA levels also remain significantly reduced at this time (0.71±0.04 fold control), while Myb expression is indistinguishable from controls (0.91±0.10 fold control), in keeping with other studies in which Myb is only transiently upregulated during ciliogeneis (Tan et al., 2013).
Known to suppress the expression of MCI, Myb, and FoxJ1 in ependymal (Kyrousi et al., 2015) and bronchial airway cells (Stubbs et al., 2012; Tan et al., 2013), the signaling receptor Notch is a master cell fate regulator in many tissues including the brain (Ables et al., 2011). Thus Notch activity suppresses acquisition of a multiciliated phenotype (Deblandre et al., 1999; Liu et al., 2007; Marcet et al., 2011; Morimoto et al., 2010; Tsao et al., 2009). We therefore asked whether dystroglycan regulates ependymal cell development by influencing Notch activation. Acute application of dystroglycan-blocking or control antibodies at the stage when cultures are switched to a low serum medium to induce ependymal cell maturation leads to small but significant increases in canonical Notch-regulated gene expression, as measured by Notch target genes Hes1 (1.15±0.09 fold control) and HeyL (1.46±0.17 fold control) at 6 hours post-dystroglycan blocking antibody treatment (Fig. 4F). We therefore wondered whether blocking Notch activation could rescue the deficiencies in ependymal cell differentiation genes (e.g., MCI) seen with dystroglycan loss-of-function, and therefore used the γ-secretase inhibitor DAPT, which prevents Notch cleavage and therefore its activation. First, we confirmed that DAPT treatment significantly blocks canonical Notch gene expression, and does so both in the presence and absence of dystroglycan-blocking antibodies (Fig. S3). At 24 hours, as with longer-term cultures (Fig. 4E), we found that dystroglycan block decreases MCI expression (0.81±0.02 fold control, Fig. 4G). Conversely, DAPT alone increases MCI expression (1.27±0.05 fold), and the addition of DAPT to dystroglycan blocking antibodies fully rescues the dystroglycan loss-of-function effect (1.03±0.05 fold control), indicating that Notch activation is required for the dystroglycan loss-of-function phenotype. While dystroglycan blockade for 24 hours does not significantly affect Myb expression, (0.94±0.04 fold control), we did find that DAPT treatment significantly increases Myb and MCI expression and that, interestingly, the addition of dystroglycan blocking antibodies returns Myb and MCI expression in DAPT-treated cells to control levels, indicating that dystroglycan may also regulate MCI/Myb expression independent of Notch.
We next examined whether DAPT could rescue the ability of dystroglycan blocking antibodies to suppress ependymal cell differentiation and FoxJ1 expression. Dystroglycan-blocking antibodies significantly reduce the percentage of multiciliated cells at day 3 (3.20±0.76% vs. 6.80±0.55% in control; Fig. 4H, I), consistent with the results of longer-term blockade (Fig. 4B). Conversely, DAPT treatment to block Notch activation increases the percentage of multiciliated cells (9.59±0.61% vs. 6.80±0.55 in control). Finally, treating with both DAPT and dystroglycan blocking antibody engenders a “rescue” of the dystroglycan loss-of-function phenotype such that the percentage of multiciliated cells is no longer different from that in control cultures (6.43±0.8% vs. 6.80±0.55% in control). Additionally, DAPT plus dystroglycan block (6.43±0.80%) is significantly different from DAPT treatment alone (9.59±0.61). We also used the peptide inhibitor SAHM1, to specifically disrupt the Notch-RBPJ-MAML1 transcriptional activator complex, and observed similar results (Fig. S4A,B), indicating that these effects are mediated specifically by modulation of canonical Notch signaling.
We also found that DAPT enhances the percentage of FoxJ1+ cells (35.46±3.95% versus 22.56±1.46% in control; Fig. 4J), an effect that is blocked by concurrent administration of DAPT and dystroglycan blocking antibodies (13.64±5.78%; Fig. 4J). As we performed DAPT rescue experiments on SVZ ependymal cultures that were not fully mature (day 3 in maturation medium), we modified our cluster analysis criteria (Fig. 4D) and scored the percentage of solo FoxJ1+ cells versus those that were adjacent to one or more FoxJ1+ cells, as a rough indicator of the degree of nascent clustering. Dystroglycan blocking antibody significantly enhances the percentage of solo (i.e., non-clustered) FoxJ1+ cells at day 3 (9.06±1.39% versus 2.680.37% in the control; Fig. 4K). Interestingly, however, DAPT alone does not alter the fraction of solo FoxJ1+ cells (4.33±0.98%), and has no effect on the ability of the dystroglycan block to increase the percentage of solo FoxJ1+ cells. These data indicate that dystroglycan regulates pinwheel/cluster formation through a Notch-independent mechanism. In summary, Notch activation is necessary for dystroglycan loss-of-function’s effects on many aspects of ependymal cell maturation, indicating that dystroglycan normally acts to suppress Notch activation and promote ependymal cell maturation in the early postnatal SVZ. However dystroglycan may also act via Notch-independent mechanisms to affect ependymal cell maturation, for instance, in regulating ependymal cell spatial organization into pinwheels.
Gliogenesis is dysregulated in the dystroglycan-deficient SVZ stem cell niche
As perinatal DAG cKO RGCs are defective in their ability to transform into ependymal cells and establish a normal SVZ stem cell niche, we wanted to determine if these disturbances impacted the niche’s ability to produce progenitors. The principal wave of dorsal oligodendrogenesis begins perinatally and remains high into the second postnatal week. During this time, oligodendrogenic intermediate progenitor cells (oIPCs) are specified from neural stem- and transit amplifying cells. oIPCs, as they transition into oligodendrocyte progenitor cells (OPCs), proliferate and migrate out of the SVZ into the overlying white matter. We examined oIPC/OPC production and proliferation in DAG cKO mice and their WT littermates at P0 and P3 (Fig 5A). At birth, oligodendrogenesis has begun in DAG cKO mice, although to a slightly lesser extent than seen in WT littermates (10.6±1.8 × 105 PDGFRα+ cells/mm3 vs. 15.1±3.6 × 105 in WT; Fig. 5B). By P3, however, DAG cKO mice catch up and far surpass WT, with an SVZ oIPC/OPC density of more than double that of their WT littermates (30.6±3.1 × 105 cells/mm3 vs. 14.0±1 × 105 in WT; Fig. 5B). DAG cKO SVZ oIPC/OPC densities remain elevated over WT levels until the beginning of the second postnatal week (not shown). We did not, however, observe similar disturbances in neurogenesis, with the density of Pax6+ neuronal IPCs being unchanged (44.4±3.7 × 105 cells/mm3 vs. 44.2±4.3 × 105 in WT; not shown).
Figure 5. Dysregulated Gliogenesis in the Dystroglycan cKO SVZ.

(A) PDGFRa IHC in dorsal SVZ coronal sections from P0 and P3 WT and DAG cKO mice. OPC proliferation is indicated by PCNA co-IHC (insets). (B) Quantification of PDGFαR+ cell densities in the SVZ of P0 and P3 WT and DAG cKO mice (*p<0.05, Student’s t-test; error bars, SEM; n=3). (C) Quantification of BLBP+ cell proliferation (i.e., PCNA+) at P0 and P3 in WT and DAG cKO mice (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=3). (D) Quantification of PDGFRα+ cell proliferation (i.e., PCNA+) in P0 and P3 WT and DAG cKO mice (*p<0.05, Student’s t-test; error bars, SEM; n=3). (E) Quantification of BLBP+ and PDGFαR+ cells within the PCNA+ population in P3 WT and DAG cKO mice (*p<0.05, Student’s t-test; error bars, SEM; n=3). (F) Time line of tamoxifen injections and cellular analysis for inducible dystroglycan conditional knockout mice (iDAG cKO). (G) PDGFRα IHC in the dorsal-lateral SVZ in coronal sections from P8 WT and iDAG cKO mice. (H) Quantification of PDGFRα+ cell density in the SVZ of P8 WT and iDAG cKO mice (*p<0.05, Student’s t-test; error bars, SEM; n=3). (I) IHC to detect Notch Intracellular Domain (NICD), GFAP, and γ-tubulin in coronal sections from P8 WT and iDAG cKO SVZ. GFAP+ radial glial endfeet are indicated by white asterisks. Regions of maturing ependymal cells are indicated by apical surface γ-tubulin immunoreactivity. Lower panels depict higher magnification views of GFAP+ radial glial endfeet. (J) Quantification of the percent area coverage by GFAP immunoreactivity in the WT and iDAG cKO SVZ at P8 (*p<0.05, Student’s t-test; error bars, SEM; n=3 (K) Time line of tamoxifen injections and gene expression analysis for iDAG cKO. (L)
Quantification of mRNA levels of Notch-regulated genes, Hes1, Hes5, Hey1, and HeyL, in the WT and iDAG-cKO SVZ (*p<0.05, Student’s t-test; error bars, SEM; n=4). (M) Dissociated neurospheres from WT SVZ were plated in the presence of control or dystroglycan blocking antibodies (DG block) and evaluated at 6 hours to determine mRNA levels of Notch-regulated genes, Hes1, Hey1, Hes5, HeyL, and Sox9 (*p<0.05, **p<0.01 Student’s t-test; error bars, SEM; n=6). (N) Control or dystroglycan (DG)-blocking antibodies were injected into the lateral ventricles of rats at P2 followed by analysis at 6 and 24 hours post-injection (hpi). IHC to detect laminin (Lm) and β-catenin in wholemounts was used to track laminin+ hubs. (O) Upper panels: IHC to detect PDGFRα in the dorsal SVZ in coronal sections taken from control or DG=blocking antibody-injected rats at 6 hours post-injection (hpi). Lower panels: IHC to detect Sox2 and PCNA at 24 hpi. (P) Quantification of PDGFRα+ OPC densities in the SVZ 6 hours post-injection of control or DG-blocking antibodies (*p< 0.05, Student’s t-test; error bars, SEM; n=3). (Q) Quantification of Pax6+ neuronal progenitor densities in the SVZ 6 hours post-injection of control or DG-blocking antibodies (error bars, SEM; n=3). (R) Quantification of PDGFRα+ OPC proliferation in the SVZ at 6 hours post-injection with control or DG-blocking antibodies (error bars, SEM; n=3). (S) Quantification of PDGFRα expression within Sox2+ cells (NSC/IPCs) at 6 and 24 hours post antibody injection (*p<0.05, Student’s t-test; error bars, SEM; n=3). (T) Quantification of Sox2+ cell proliferation (PCNA+) at 6 and 24 hours post antibody injection (*p < 0.05, Student’s t-test; error bars, SEM; n=3). Scale bars: 100µm (I, upper panels), 50µm (A, G, N, O), 25µm (insets in A), 20µm (lower panel in I).
We next sought to determine if dysregulated proliferation contributes to increased numbers of oIPC/OPCs in the DAG cKO SVZ. At birth, DAG cKO mice exhibit significantly decreased SVZ cell proliferation (38.8±3.6 × 105 PCNA+ cells/mm3 vs. 63.1±2.1 × 105 in WT littermates; Fig. S5A,B). While proliferation sharply declines in the WT SVZ between P0 and P3, proliferation in the DG cKO SVZ remains relatively constant between P0 and P3, resulting in a slight elevation over WT levels by P3 (44.2±8.4 × 105 cells/mm3 vs. 28.7±4.2 × 105 in WT; Fig. S5A,B). Additionally, while WT NSC/NPC proliferation is concentrated mainly in the VZ, proliferative Sox2+ cells (NSC/IPCs) are mislocalized throughout the DAG cKO SVZ (Fig. S5C). To determine which cell types contribute to this altered proliferation, we examined the radial glial cell (RGC) marker BLBP in conjunction with PCNA. At P0 the percentage of proliferating RGCs is significantly lower in DAG cKO mice (36.1% ± 3.5% vs. 64.8% ± 1.9% in WT; Fig. 5C), mirroring the phenotype observed in the general PCNA+ population (Fig. S5B). However, while WT mice undergo the typical decrease in RGC proliferation between P0 and P3, RGC proliferation levels remain constant in DAG cKO mice between P0 and P3, again resulting in RGC proliferation levels being slightly higher by P3 (34.4% ± 1.9% vs. 27.8% ± 1.8% in WT littermates; Fig. 5C). Dystroglycan loss also significantly increases oIPC/OPC proliferation at P3 (57.7% ± 2.8% vs. 39.3% ± 5.3% in WT; Fig. 5D), although not at P0 (65.6% ± 8.6% vs. 67.6% ± 5.7% in WT; Fig. 5D). Analysis of overall proliferation at P3 confirms that while RGCs contribute to the increase in SVZ proliferation, oIPC/OPCs contribute most to the high levels of proliferation in the dystroglycan-deficient SVZ (Fig. 5E).
To rule out the possibility that embryonic loss of dystroglycan leads to global alterations in the NSC developmental profile, which could indirectly alter oIPC/OPC production, we used nestin-creER to generate an inducible dystroglycan conditional knockout mouse in which postnatal tamoxifen injections drive the loss of dystroglycan gene expression in NSCs and their progenitors (iDAG cKO; Fig. 5F). Following 3 days of tamoxifen injections beginning at birth, we then analyzed oIPC/OPC density in the SVZ at P8 (Fig. 5G), and found a significant increase in PDGFRα+ cell density in the iDAG cKO SVZ relative to the densities in control (cre−/−) littermates (25.5±1.9 × 105 mm3 vs. 18.4±1.4 × 105 mm3; Fig. 5H). These findings suggest that eliminating dystroglycan expression specifically in perinatal RGCs is sufficient to drive oIPC/OPC overproduction.
To determine whether changes in Notch activation occur concurrent to altered gliogenesis, we evaluated Notch ICD (NICD) levels in the iDAG cKO SVZ at P8 and observed increased NICD immunoreactivity in cells lining the lateral ventricle (Fig. 5I). Elevated levels of NICD following dystroglycan loss-of-function were confirmed in western blots of SVZ cultures treated with dystroglycan-blocking antibodies (Fig. S6). We also observed decreased γ-tubulin immunoreactivity along the ventricle wall (indicative of fewer maturing, multiciliated ependymal cells, confirming the reduction in multiciliated ependymal cells seen in the DAG cKO, Fig. 3E–K). Additionally, we observed increased coverage of GFAP+ immunoreactivity within the iDAG cKO SVZ (Fig. 5J), with cells exhibiting a radial glial phenotype (broad endfeet at the ventricle surface; Fig. 5I, lower panels), in keeping with the persistence of a RGC phenotype observed in DAG cKO (Fig. 3D). By evaluating changes in Notch target gene expression at P3 (Fig. 5L), we found elevated levels of Hes1 (1.91±0.28 fold control) and HeyL (2.05±0.29 fold control; Fig. 5M). We also observed increases in mRNA levels for Hes5 and Hey1 (2.19±0.49 and 1.90±0.44 fold control, respectively), as well as in pro-oligodendrogenic transcription factors stimulated by Notch activation, Olig2 and Sox9 (1.36±0.18 and 1.27±0.19 fold control, respectively), though these increases were variable and did not reach statistical significance.
As changes in Notch signaling in NSCs themselves might be dampened when evaluating the entire SVZ, we generated neurospheres from the P0 wild type SVZ, treated freshly-plated dissociated NSCs with dystroglycan-blocking or control antibodies for 6 hours, and evaluated mRNA levels of Notch target genes (Fig. 5K). We found that dystroglycan block in NSCs significantly increases mRNA levels for Hes1 (1.49±0.14 fold control), Hey1 (1.42±0.13 fold), Hes5 (1.72±0.22 fold), and HeyL (1.73±0.22 fold). Additionally, we found significantly elevated levels of Sox9 mRNA, a pro-oligodendrogenic transcription factor known to be stimulated by Notch signaling (Martini et al., 2013) (1.57±0.16 fold control). Of note, changes in Notch target gene expression in dissociated neurospheres are transient, and in many cases return to normal after longer-term treatment of neurosphere cultures with dystroglycan antibodies. Together, these data suggest that both in vitro and in vivo, dystroglycan loss-of-function acutely increases Notch-regulated gene expression. We tested the possibility that dystroglycan loss-of-function rapidly alters the expression of Notch ligands, but did not detect any changes in Notch ligands (or in Numb, a negative regulator of Notch activation; not shown).
To determine whether the increases in SVZ oIPC/OPC density in the constitutive and inducible DAG cKO mice arise from increased generation of these cells from RGCs, or from increased oIPC/OPC proliferation, we developed a strategy to acutely disrupt dystroglycan-ligand interactions in wild type rodents and track SVZ cell phenotypes shortly thereafter. Here, we injected either control or dystroglycan-blocking antibodies into the lateral ventricles of P2 rats, followed by analysis at either 6 or 24 hours post-injection (Fig. 5P). By visualizing laminin and β-catenin in SVZ wholemounts obtained 6 hours post-injection, we observed that ventricular surface laminin is found mainly in aggregates on the surface of immature ependymal cells in controls. These aggregates, however, are lost in rats injected with dystroglycan-blocking antibodies, with laminin immunoreactivity appearing more diffuse (Fig. 5N). At 24 hours post-injection of dystroglycan-blocking antibodies, however, laminin immunoreactivity recovers, likely due to antibody clearance from the lateral ventricles. We examined coronal sections from antibody-injected rodents for oIPC/OPCs (PDGFαR+), proliferating cells (PCNA+) and NSC/IPCs (Sox2+) within the SVZ. We found that dystroglycan blockade increases the oIPC/OPC population twofold by just 6 hours post-injection (Fig. 5O) (14.30±2.33 × 105 cells/mm3 vs. 6.96±1.94 × 105 in controls; Fig. 5Q), concomitant with approximately a 50% increase in Olig2+ cell densities (6.15±0.21 × 105 cells/mm3 vs. 4.33±0.30 × 105 cells/mm3 in controls; not shown), supporting PDGFαR immunoreactivity as a means to identify oIPC/OPCs. Importantly, this increase is not explained by greater oIPC/OPC proliferation as PCNA immunoreactivity within the oIPC/OPC population is similar between control and treated groups at this time (25.5±11.7% vs. 36.3±9.7% in controls; Fig. 5S). To confirm increased oIPC/OPC densities were indeed the result of de novo gliogenesis, we assessed PDGFαR expression within the Sox2+ NSC/IPC population. Six hours post-injection of dystroglycan-blocking antibodies, the proportion of Sox2+ cells co-expressing PDGFαR is nearly fourfold that of controls (38.2±8.3% vs. 9.6±4.4% in controls; Fig. 5T), a result consistent with an acute pro-gliogenic effect on neural stem- and/or uncommitted progenitor cells. PDGFαR+ cell populations within the SVZ returned to control levels by 24 hours post-injection, suggesting that the population of newly-formed oIPC/OPCs seen at 6 hours post-injection has exited the SVZ. In contrast, dystroglycan blocking antibodies have no effect on the density of cells expressing Pax6 (26.3±2.87 × 105 cells/mm3 vs. 25.2±2.44 × 105 in controls; Fig. 5R), a transcription factor that represses Olig2 to promote neuronal fate in postnatal SVZ progenitors (Heins et al., 2002; Jang and Goldman, 2011). Additionally, dystroglycan blockade leads to a twofold increase in Sox2+ cell proliferation (32.6±2.0% vs. 15.7±1.7% in controls; Fig. 5O,U), although this effect is not observed until 24 hours post-injection. These findings suggest that detachment from dystroglycan ECM ligands deregulates the proliferation and maturation of Sox2+ stem/progenitors and drives rapid oligodendroglial production without an intermediate transit-amplifying (i.e., proliferation) step. Overall, these results indicate that dystroglycan has multiple roles within the neonatal gliogenic SVZ, regulating the development and structural integrity of the niche (Fig. 2,Fig. 3,Fig. 4) as well as niche functional output (Fig. 5).
Dystroglycan loss delays the maturation of oligodendrocyte lineage cells
Newly-produced OPCs migrate dorsally and tangentially out of the SVZ into the overlying corpus callosum, where they differentiate into oligodendrocytes (OLs) that myelinate axons. To determine if dystroglycan loss affected OL lineage cells after they exited the SVZ, we examined PDGFαR, Sox2, and CC1 (a marker for mature OL cell bodies) in coronal sections from DAG cKO and WT mice (Fig. 6A). While WT and DAG cKO mice exhibit similar numbers of OPCs in the corpus callosum at P3 (Fig 6B), dystroglycan-deficient OPCs appeared less mature, with a higher proportion co-expressing the NSC/IPC marker Sox2 (36.0±3.9% vs. 22.1±5.4% in WT; Fig. 6C). By P8, DAG cKO callosa contain more OPCs and OLs than those of WT littermates (39.6±1.2 × 104 PDGFαR+ and 30.5±1.2 × 104 CC1+ cells/mm2 vs. 22.1±4.2 × 104 PDGFαR+ and 15.2±0.8 × 104 CC1+ in WT; Fig. 6D), though the OPC:OL ratio does not differ significantly (not shown). Furthermore, WT and DAG cKO OPC proliferation in the corpus callosum is similar (29.9±5.7% vs. 34.7±3.4% in WT; Fig. 5E), indicating that the OPC surplus in DAG cKO white matter is the result of SVZ overproduction, and not due to increased OPC proliferation within the white matter. These data also suggest that OPCs produced in the DAG cKO SVZ migrate effectively to the overlying corpus callosum. We then examined MBP immunoreactivity at P8 and P21 to assess oligodendrocyte myelination capacity in DAG cKO white matter. Despite there being higher densities of mature OLs (Fig. 6D), MBP immunoreactivity was surprisingly reduced in the DAG cKO corpus callosum at P8 relative to WT (Fig. 6F). Similarly, despite normal densities of mature OLs by P21 (not shown), MBP immunoreactivity remained lower in the DAG cKO corpus callosum than in WT littermates (Fig. 6F). Furthermore, western blots of cerebral cortices revealed lower levels of MBP protein in the DAG cKO cortex at P21 compared to that in WT littermates (Fig. 6G). By 3 months, however, MBP levels in DAG cKO cerebral cortices normalize (Fig. 6G), indicating that dystroglycan loss delays, but does not prevent, myelination by DAG cKO OLs.
Figure 6. Dystroglycan Promotes Timely Oligodendrocyte Lineage Progression.

(A) IHC to detect PDGFRα, Sox2, and CC1 in the corpus callosum in coronal sections from P3 and P8 WT and DAG cKO mice. (B) Quantification of PDGFRα+ OPC densities in the P3 corpus callosum (error bars, SEM; n=3). (C) Quantification of Sox2+ cells within PDGFRα+ cell population in the P3 corpus callosum (*p<0.05, Student’s t-test; error bars, SEM; n=3). (D) Quantification of PDGFRα+ and CC1+ cell densities in the P8 corpus callosum (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=3). (E) Quantification of OPC proliferation in the P8 corpus callosum (error bars, SEM; n=3). (F) IHC to detect myelin basic protein (MBP) in the corpus callosum in coronal sections from P8 and P21 WT and DAG cKO mice. (G) Western blot to detect MBP protein in cortical lysates obtained from WT and DAG cKO mice at P21 and 3 months. (H) WT and DAG cKO cells were dissociated from second passage neonatal SVZ neurospheres and differentiated on PDL. IHC was used to detect PDGFRα and Sox2 at differentiation day 3, or NG2, CNP, and MBP at differentiation day 7. (I) Quantification of Sox2+ cells within the PDGFRα+ cell population at day 3 in differentiation medium (*p<0.05, Student’s t-test; error bars, SEM; n=3). (J) Quantification of OPCs (NG2+) and OLs (CNP+) at day 3 in differentiation medium (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=7). (K) Quantification of OPCs (NG2+), OLs (CNP+) and myelin-competent OLs (MBP+) at day 7 in differentiation medium (*p<0.05, Student’s t-test; error bars, SEM; n=4). (L) Wild-type oligodendrocyte progenitor cells (OPCs) were plated on PDL in differentiation-permissive medium in the presence of dystroglycan-blocking (DG block) or control antibodies and evaluated 6 hours later for mRNA levels of Notch-regulated genes, Hes1 and Hey1 (*p<0.05, **p<0.01, Student’s t-test; error bars, SEM; n=6). (M) Treatment of OPCs as in L but analyzed at 24 hours (*p<0.05, , Student’s t-test; error bars, SEM; n=6). Scale bars: 50 µm (A,L), 100 µm (F).
To understand the effects of dystroglycan loss on oligodendrogenesis and OL lineage progression independent of niche alterations, we isolated SVZ cells from WT and DAG cKO mice at P0, cultured them as neurospheres, dissociated the cells, and allowed them to differentiate for 3 or 7 days (Fig. 6H). By detecting Sox2 (NSC/IPC), PDGFαR, NG2 (OPCs), CNPase (OLs) and MBP (myelinating OLs) we found that, in line with our in vivo observations, more PDGFRα+ oIPC/OPCs from DAG cKO neurospheres retain Sox2 expression than those derived from WT neurospheres (59.6±6.1% vs. 25.0±3.5% in WT; Fig. 6H,I), indicating a higher oIPC:OPC ratio. Dystroglycan-deficient cultures also contain higher percentages of NG2+ OPCs (56.1±4.5% vs. 39.7±2.7% in WT; Fig. 6J) and, although only a small number of CNP+ OLs had begun to differentiate from WT OPCs by day 3, almost no OLs were observed in DAG cKO cultures (1.1±0.3% vs. 4.5±1.4% in WT; Fig. 6J). After 7 days of differentiation, a significantly higher percentage of DAG cKO cells are OPCs (43.2±5.1% vs. 25.1±2.6% in WT; Fig. 6H,K), while the percentage of CNP+ OLs is significantly lower (5.2±1.8% vs. 12.3±3.2% in WT; Fig. 6K,L). These results suggest that dystroglycan loss perturbs the timely differentiation of OL lineage cells in a niche-independent manner. We did not, however, observe any significant proliferative changes in OPCs derived from dystroglycan-deficient neurospheres (not shown), indicating that increased oIPC/OPC proliferation in the DAG cKO SVZ is likely a result of disruption to the SVZ niche itself, e.g., loss of normal niche cellular architecture. Together these results suggest that from gliogenesis to myelination, dystroglycan is critical to the timely production of a competent OL pool. Furthermore, our in vitro results suggest that dystroglycan continues to regulate OL lineage progression outside of the SVZ stem cell niche, and thus is likely to play a role in OL maturation in white matter tracts (as is seen in the corpus callosum; Fig. 6F). In accordance with this hypothesis, we observed delayed myelination in the corpus callosum of CNP-cre+:DAGfI/fI mice (DG OL-cKO), which lack dystroglycan expression specifically in myelinating glia (Fig. S7). In DG OL-cKO mice, myelin basic protein levels are reduced in the corpus callosum at P21 (Fig. S7A,B) but reach normal levels by adulthood (not shown), suggesting that OL-specific loss of dystroglycan delays, but does not prevent myelination. In support of this, siRNA-mediated loss of dystroglycan expression in primary OPCs results in delayed OL maturation (Colognato et al., 2007; Eyermann et al., 2012; Galvin et al., 2010).
As Notch activation is also known to enhance OPC production, while delaying OL maturation (Kim et al., 2008; Park and Appel, 2003; Wang et al., 1998), mirroring the effects of dystroglycan loss-of-function we observed, we hypothesized that OPC Notch activity may be altered by dystroglycan loss-of-function. We plated OPCs from neonatal rat cerebral cortices in OL-differentiation-permissive medium (SATO with T3) in the presence of dystroglycan-blocking or control antibodies (Fig. 6L). At 6 hours, dystroglycan-blocking antibodies robustly increase Notch activation, as evidenced by elevated levels of Hes1 and Hey1 mRNA (2.27±0.12 and 4.24±0.76 fold control, respectively; Fig. 6L). However other Notch-regulated genes were unaffected, including HeyL, Olig2, Sox9, and Mash1 (Fig. 6L), in contrast to dissociated neurospheres and SVZ ependymal cell cultures, both of which show elevated HeyL mRNA at 6 hours following dystroglycan loss-of-function (Fig. 4B, Fig. 5K). By 24 hours both Hes1 and Hey1 mRNA levels return to near control levels (Fig. 6M), whereas HeyL mRNA levels are now significantly increased (2.02±0.20 fold control; Fig. 6O). Mash1 mRNA levels are also significantly elevated at 24 hours following dystroglycan loss-of-function (2.81±0.50 fold control; Fig. 6M), while Olig2 and Sox9 mRNA levels remained unchanged (Fig. 6M). These results are consistent with delayed oligodendroglial differentiation, as among Olig2, Sox9, and Mash1, only Mash1 is known to sharply decrease in expression as OPCs begin to differentiate into OLs (Nakatani et al., 2013).
Discussion
In the neonatal SVZ, embryonic radial glia transform into both ependymal cells and adult neural stem cells and reorganize into an adult niche configuration, while also generating the oligodendrogenic intermediate progenitor cells that will myelinate the majority of the forebrain. In this study we sought to identify the role of cell-ECM interactions in regulating SVZ niche maturation. We reveal that early postnatal neural development is accompanied by a profound dystroglycan-dependent reorganization of ependymal-associated ECM, with dystroglycan regulating niche organization, ependymal cell development, and gliogenesis. Finally, we demonstrate that dystroglycan mediates its regulatory effects, at least in part, by modulating Notch-dependent genes. Taken together, our results describe a dynamic dystroglycan-ECM axis responsible for tuning the development and function of the neonatal SVZ (Fig. 7).
Figure 7. Dystroglycan guides the development and function of the SVZ neural stem cell niche.

(i) Ependymal cell development and niche maturation: ystroglycan loss-of-function leads to decreased expression of MCI, myb, and FoxJ1, transcription factors known to be suppressed by Notch and required for multiciliogenesis, suggesting a model in which dystroglycan suppression of Notch leads to increased MCI, myb, and FoxJ1, thereby promoting the development of multiciliated ependymal cells. And, dystroglycan-mediated remodeling of ependymal cell-associated ECM helps to regulate the organization of ependymal cells into niche pinwheels. Suppression of Notch activation rescues several of these defects in niche maturation following dystroglycan loss-of-function, placing Notch downstream of dystroglycan in this process. However dystroglycan can also influence MCI expression in a Notch-independent fashion via an unknown mechanism, indicated by the dashed green arrow. (ii) Oligodendrogenesis: The dystroglycan-deficient SVZ generates additional oligodendrogenic progenitor cells (oIPC) and oligodendrocyte progenitor cells (OPCs), and dystroglycan loss-of-function rapidly (i.e, within 6 hours) leads to elevated Notch signaling as well as elevated expression of Sox9, a pro-oligodendrogenic transcription factor downstream of Notch. Thus, as Notch normally promotes the development of oIPCs in the SVZ, our data also supports a model in which dystroglycan suppression of Notch in turn inhibits, or delays, oligodendrogenesis. (iii) Oligodendrocyte development: Dystroglycan-deficient brains display delayed myelination, despite an overabundance of oligodendrocytes in developing white matter, and dystroglycan loss-of-function in differentiating OPCs leads to elevated Notch signaling as well as elevated expression of Mash1 (which normally decreases as OPCs transition into oligodendrocytes). These data support a model in which oligodendroglial dystroglycan suppresses Notch signaling to help regulate timely myelination.
V/SVZ ECM is extensively remodeled during postnatal development
The postnatal V/SVZ ECM undergoes a complex reorganization immediately after birth. During this period, a unique ventricular zone ECM begins to emerge, with both laminin and dystroglycan upregulated selectively on the surface of differentiating ependymal cells. By P8, ECM aggregates, or “hubs”, previously observed in association with GFAP+ apical processes in the adult niche (Shen et al, 2008), begin to emerge at the apical surface of ependymal cells, and by P21 are found at the interface between ependymal and adult neural stem cells in mature niche pinwheels. While laminin hubs in the adult SVZ were proposed to originate from the vascular basal lamina (Shen et al, 2008), the localization of early postnatal hubs on ependymal cell surfaces, abundant laminin deposition in ependymal cell cultures in vitro¸ and co-localization of dystroglycan with emerging hub structures indicate that these hubs may be produced, or at least assembled, by ependymal cells. Nonetheless, transient extra-vascular ECM structures appear during niche construction linking the VZ ependymal cell-associated laminin to the vascular basal lamina. Finally, fractones and fractone bulbs, extra-vascular ECM structures thought to have regulatory roles in the adult niche (Douet et al., 2012; Douet et al., 2013; Kerever et al., 2007; Mercier et al., 2002; Shen et al., 2008), are present in the neonatal SVZ. Thus while the early postnatal niche is “under construction”, it houses distinct ependymal and vascular niche ECM elements, with immature ependymal cells playing a key role in niche construction, helping establish their own regulatory environment via the expression of both ECM ligands and receptors.
Dystroglycan regulates stem and progenitor cell proliferation
As laminin-dystroglycan interactions contribute to RGC attachment to the pial basal lamina (Moore et al., 2002; Satz et al., 2010), we hypothesized that dystroglycan could also mediate cellular interactions with the laminin-rich ECM of the neonatal V/SVZ. RGCs associate with laminin-positive aggregates at the ventricular surface at birth, and deletion of neural dystroglycan virtually eliminates these aggregates (but does not precipitate RGC apical detachment). Nonetheless, RGCs exhibit proliferative abnormalities in the perinatal DAG cKO SVZ. For instance, RGC proliferative capacity at P0 is greatly reduced, but without a concomitant change in RGC density. This may indicate that dystroglycan supports the production of progenitors through asymmetric cell division and that these divisions are specifically reduced in DAG cKO mice. These effects are not likely a result of pial detachment, as disruption of RGC pial attachment in other mouse models (α6 integrin−/−, perlecan−/−, laminin γ1 nidogen-binding site mutation) does not alter RGC polarity, proliferation, or fate (Haubst et al., 2006). Supporting this conclusion, acute delivery of dystroglycan-blocking antibodies directly to the ventricle is sufficient to produce aberrant proliferation in Sox2+ V/SVZ progenitors. These results indicate that dystroglycan plays a role in the regulation of RGC division within the developing V/SVZ.
Dystroglycan promotes ependymal niche development
As cortical neurogenesis subsides in the immediate postnatal period, a subset of RGCs differentiate into ependymal cells and upregulate laminin and dystroglycan on their apical surfaces. In the DAG cKO SVZ, however, laminin deposition is profoundly dysregulated, and the accompanying RGC-to-ependymal cell transition stalls, resulting in delayed ependymal differentiation and maturation, with the resulting ependymal cells having smaller and more highly displaced basal body patches. Furthermore, in vitro blockade of dystroglycan solely during the ependymal cell maturation phase indicates that this delay in ependymal cell maturation is independent of any pre-existing RGC phenotype. Together these data suggest that dystroglycan in developing ependymal cells is required for appropriate ECM remodeling, differentiation, and maturation. Although the intricate cellular architecture of the adult V/SVZ has been extensively characterized, much less is known about how this architecture is attained. However, given that dystroglycan are required for the cellular reorganization necessary to produce organized adult niche pinwheel structures, it appears that cell-ECM interactions can drive this process.
Dystroglycan regulates the production and maturation of oligodendrocyte lineage cells
While multiple studies have implicated laminin-integrin interactions in adult SVZ niche maintenance and controlled neurogenesis (Kazanis et al.; Kokovay et al.; Shen et al., 2008), the regulation of ECM signaling and its significance in postnatal gliogenesis has remained unclear. Here we report a complex dysregulation of oligodendroglial dynamics in the DAG cKO brain. Although gliogenesis is initially delayed, by P3 a significant overproduction of oIPC/OPCs is observed in the SVZ, with a simultaneous increase in OPC proliferation. As a result, by P8, OPC density in the DAG cKO corpus callosum is nearly two-fold higher than that in WT. However, consistent with previous findings from our group implicating dystroglycan in regulated OPC maturation (Colognato et al., 2007; Eyermann et al., 2012; Galvin et al., 2010; Leiton et al., 2015), DAG cKO oligodendroglial differentiation is abnormally delayed and oligodendrocytes fail to myelinate callosal axons during the proper developmental window.
As with ependymal cells, dystroglycan’s ability to promote the production and differentiation of oligodendrocyte-lineage cells appears to be niche-independent, as P0 DAG cKO neurospheres overproduce OPCs whose development then stalls during differentiation. However, while oligodendrocyte densities are higher than normal in DAG cKO mice in vivo, the opposite is true in vitro. This may result from the lack of proliferative abnormalities in dystroglycan-deficient OPCs in vitro, and/or the absence of other in vivo factors promoting OPC maturation (discussed below). In antibody injection experiments, we found that the transient loss of dystroglycan binding is sufficient to induce rapid oligodendrogenesis in the absence of concomitant SVZ proliferation, suggesting that dystroglycan loss drives the direct acquisition of OPC identity by SVZ niche cells.
The complex nature of laminin dysregulation-initially deficient from DAG cKO ependymal cells, then overabundant, then markedly diminished- highlights the dynamic nature of the SVZ over the first postnatal week. This pattern is paralleled by cellular phenotypes in the DAG cKO SVZ: oligodendroglial fate specification and RGC proliferation are both dysregulated in a temporally-dependent manner. In contrast, ependymal cell maturation is uniformly delayed by dystroglycan loss-of-function and neurogenesis is unaffected. As laminin assembly, endocytosis, and degradation are all regulated by dystroglycan (Akhavan et al., 2012; Cohen et al., 1997; Colognato et al., 1999; Henry and Campbell, 1998; Henry et al., 2001; Leonoudakis et al., 2014; Leonoudakis et al., 2010; Montanaro et al., 1999; Weir et al., 2006), our findings suggest that the interpretation of signals from the dystroglycan-laminin axis heavily depends on the temporal and cell type-specific context in which they are received.
Dystroglycan regulates a Notch-dependent pathway important for SVZ development
Dystroglycan loss-of-function induces rapid changes in Notch signaling, increasing Hes1 expression within 6 hours, paralleling the rapid time course of induction of oligodendrogenic intermediate progenitors. Delayed maturation and decreased proliferation of DAG cKO RGCs are consistent with reports that in the late embryonic VZ, constitutive Notch signaling increases BLBP+ expression while inhibiting RGC proliferation (Gaiano et al., 2000). In the context of physiological gliogenesis, however, Notch promotes oligodendroglial fate specification (Kim et al., 2008; Taylor et al., 2007) with constitutive Notch activation leading to overproduction of OPCs (Park and Appel, 2003). These findings are in agreement with our data that OPC production is increased by dystroglycan loss during the first postnatal week - a period of robust gliogenesis. Importantly, however, while Notch signaling promotes gliogenesis, it inhibits oligodendrocyte terminal differentiation and myelination (Wang et al., 1998). Thus increased Notch activation (observed in our OPC cultures downstream of dystroglycan loss-of-function) may also explain the delayed myelination phenotype observed in the dystroglycan-deficient brain.
The cell-type context in which dystroglycan-mediated signals are received is clearly a critical factor for the signal-receiving cell. While Notch signaling is activated by dystroglycan loss-of-function in ependymal cells, neural stem cells, and oligodendrogenic lineage cells, the downstream effectors vary in each. For example, in dissociated neurospheres - which give rise to several lineages - Hes1, Hes5, Hey1, and HeyL are all suppressed by dystroglycan. However, in ependymal cell cultures, HeyL and Hes1 are likely the key immediate mediators of these signals, while in OPCs Hes1 and Hey1 respond first, followed by HeyL at 24 hours. One interpretation of our data is that, while dystroglycan broadly regulates Notch signaling, in a subset of NSCs or IPCs, perhaps those already primed towards a oligodendrogenic lineage, loss of dystroglycan “short-circuits” the normal brakes on differentiation, leading to rapid upregulation of Sox9, a direct activator of PDGFαR in these cells (Finzsch et al., 2008). However, bulk dissections of the SVZ did not reveal a significant increase in Sox9 upon loss of dystroglycan expression (despite an increased trend), perhaps because oligodendrogenic IPCs make up a small subset of the total SVZ population (and rapidly migrate out of the SVZ). In contrast, in neurosphere cultures from newborn mice, where oligodendrogenic IPCs are highly represented, we did observe a rapid increase in Sox9 levels upon dystroglycan loss-of-function. In OPCs themselves, dystroglycan continues to regulate Notch but this pathway no longer appears to be important for regulating Sox9 expression. Instead, loss of dystroglycan signaling increases levels of Mash1, impeding maturation.
In ependymal cell cultures, we also found that dystroglycan loss-of-function resulted in suppressed expression of Mcidas, c-Myb, and FoxJ1, genes for pro-multiciliogenic transcription factors known to be suppressed by Notch activation (Reviewed in (Brooks and Wallingford, 2014)), ultimately delaying the differentiation program of ependymal cells, a phenotype reversible by Notch inhibition. While Notch has been shown to act upstream of dystroglycan in Xenopus, (Sirour et al., 2011), these results are novel in demonstrating that in the SVZ, Notch signaling can act downstream of dystroglycan. Although Notch signaling engages in extensive feedback regulation, the concordance of increased NICD, suppression of FoxJ1, and delayed ependymal cell differentiation, as well as the rapid time-frame of increased Notch signaling (i.e., 6 hours), indicate that the changes seen in Notch are likely a primary effect of dystroglycan loss-of-function rather than a compensatory response. Despite this, several aspects of dystroglycan signaling remain unresolved. For instance, as dystroglycan blocking antibodies are able to suppress increases in MCI/Myb expression caused by γ-secretase inhibition, dystroglycan may also regulate the ependymal cell transcriptional program in a Notch-independent fashion by an as yet unknown mechanism. And, while dystroglycan loss-of-function leads to elevated Notch signaling, the precise nature by which dystroglycan interacts with the Notch signaling pathway remains to be determined. Finally, as dystroglycan loss leads to disruption of laminin organization, a role for integrin-laminin interactions in our phenotypes cannot be definitively ruled out. Several lines of evidence make this unlikely however, with ependymal cell differentiation being normal following both integrin blockade in ependymal cell cultures, and genetic loss of integrin expression in vivo (not shown). In addition, blockade of dystroglycan in SVZ ependymal cell cultures does not change FAK phosphorylation (at either tyrosine 397 or 576/7, not shown), a common readout of integrin activation.
Future directions and conclusions
It is becoming clear that the spatial and temporal regulation of ECM and ECM receptors is an important mechanism by which cells within stem cell niches modify their relationship to the extracellular environment. The dynamic and temporally distinct disruptions in the dystroglycan-deficient SVZ may reflect altered responses to dystroglycan signaling as well as other signals present in the extracellular environment. In the current study we show that dystroglycan regulates both the construction and output of a stem cell niche, and that dystroglycan does so, at least in part, by modulating Notch signaling. Further investigation of dystroglycan function has the potential to expand our understanding of stem cell regulation and reveal novel downstream targets, which could be harnessed to manipulate niche output to aid in tissue repair.
Experimental Procedures
Mice
Generation of conditional DAG1 mutant mice (nestin-Cre+:DAG1fl/fl) was achieved using DAG1fl/fl mice (Moore et al., 2002), nestin-Cre mice (Tronche et al., 1999). Generation of conditional DAG1 mutant mice (CNP-Cre+:DAG1fl/fl) was achieved using CNP-Cre mice (Lappe-Siefke et al., 2003). Generation of inducible conditional DAG1 mutant mice (nestin-CreERT2+;DAG1fl/fl) was achieved using nestin-CreERT2 mice (Lagace et al., 2007). For induction, tamoxifen was dissolved in corn oil at 1 µg/uL and delivered by intragastric injection for a final dose of 50 µg on three consecutive days beginning at birth. All rodent experiments were conducted with the approval by the Institutional Animal Care and Use Committee of Stony Brook University and conform to National Institutes of Health guidelines.
SVZ Wholemount Dissection and Immunohistochemistry
SVZ wholemounts were dissected as described (Doetsch et al., 1999). Briefly, the striatal wall of the lateral ventricle was dissected from mice at various postnatal ages as indicated, fixed with cold 4% paraformaldehyde in 0.1 M PBS for 12 hours at 4 °C and washed with PBS prior to staining. To visualize antigens at the ventricular surface, wholemounts were blocked in 10% donkey serum with 0.2% Triton-X100 and incubated with primary and secondary antibodies for 24 hours at 4°C. For deeper structures, wholemounts were blocked with 10% donkey serum with 2% Triton-X100 and incubated in primary and secondary antibodies for 48 hours. Details on primary antibodies used can be found in Supplemental Experimental Procedures.
Frozen Tissue Processing and Immunohistochemistry
Mice were intra-cardially perfused and brains post-fixed with 4% paraformaldehyde (PFA) in 0.1M PBS. Brains from mice younger than P14 were immersion-fixed in 4% PFA. Tissue was cryo-protected with 30% sucrose in PBS, embedded in OCT medium and frozen in dry ice cooled with isopentane. 18 µm sections were prepared on a cryostat, blocked in 10% donkey serum with 0.1% Triton X-100, incubated with primary antibodies diluted in blocking solution overnight at 4 °C and incubated with appropriate fluorophore-conjugated secondary antibodies (Jackson) in blocking solution at room temperature for 2 hours.
Cell Culture
For neurosphere and ependymal cell culture experiments, the lateral ventricular wall was dissected from P0/1 mice and mechanically dissociated in MEM with 25mM HEPES (Lonza) with 1% Pen/Strep (Mediatech). For neurosphere differentiation assays, cells isolated from Nestin-Cre/DAGfl/fl mice and their wild-type littermates were grown in suspension in DMEM/F12 (Thermo) with B27 (GIBCO) and 20 ng/mL EGF and FGF (Peprotech). Neurospheres were passaged at 5 and 10 days in vitro. Following the second passage, cells were resuspended in the same media without growth factors, plated at 15,000 cells/cm2 on PDL-coated chamber slides and differentiated for 3 or 10 days. Cells were fixed with 4% PFA in PBS for 15 minutes at room temperature and washed with PBS prior to staining. Ependymal cell cultures were performed as previously described (Paez-Gonzalez et al.). Briefly, cells isolated from wildtype or FoxJ1-GFP mice were resuspended in DMEM-High Glucose (Mediatech) with 10% FBS and 1% Pen/Strep and plated at 100,000 cells/cm2 on PDL-coated chamber slides. For ependymal cell differentiation experiments, cells were incubated under normal cell culture conditions until confluent (3–5 days), then media was switched to 2% FBS and cells were incubated for time periods as indicated, up to 7 days. Dystroglycan-blocking (IIH6C4, Millipore) or IgM control antibodies (Biolegend) were added to culture media at 10 µg/ml and refreshed every 3 days for long term experiments. DAPT and SAHM1 were used at 10 µM and 20 µM, respectively, in DMSO. Control cells were treated with equivalent volume of vehicle (DMSO). For all experiments, cells were fixed with 4% PFA in PBS. For oligodendrocyte progenitor cell (OPC) experiments, OPCs were isolated as previously described (Leiton et al., 2015). Briefly, cerebral cortices dissected from neonatal rats were dissociated using papain and trituration, then resuspended in DMEM supplemented with 10% FBS and seeded onto PDL-coated flasks. Medium was changed every 3 days, and after 10 days, OPCs were isolated by mechanical agitation and differential adhesion (McCarthy and de Vellis, 1980). Isolated OPCs were resuspended in SATO with T3 to induce differentiation and plated on PDL-coated dishes.
In Vivo Antibody Injections
Postnatal day 2 Sprague-Dawley rats were anaesthetized on ice, positioned in a stereotaxic device and given a single 2.5 µL injection of IIH6 antibody or mouse IgM control at 1 mg/mL into the lateral ventricle (1.4 lateral, 2.2 ventral to bregma).
qRT-PCR
Total RNA was isolated using with Qiazol and cleaned up with RNAeasy Kit (Qiagen), with DNase being used to eliminate DNA contamination. Synthesis of cDNA was carried out using the ProtoScript First Strand kit (New England Biolabs). qPCR was performed on a StepOnePlus system (Applied Biosystems) in a 20-µL reaction mixture using SYBR Green Fast PCR master mix. Cycle parameters were 3s at 95°C and 30s at 60°C, and data were normalized to GAPDH. Details on primers used can be found in Supplemental Experimental Procedures.
Image Acquisition and Analysis
Coronal sections and SVZ wholemounts were imaged with a Zeiss LSM 510 or a Leica SP5 confocal laser-scanning microscope. SVZ wholemount fields were random fields were selected from anterior dorsal areas. Images from cell culture preparations were acquired on a Zeiss Axiovert 200M epifluorescent microscope. All images were processed and quantified using ImageJ software. For in vivo ependymal cell pinwheel analysis and in vitro cluster analysis, pinwheels/clusters were defined as 4 or more ependymal cells immediately adjacent to each other and one or more monociliated cells. Clusters of basal bodies, visualized using γ-tubulin immunohistochemistry, in conjunction with apical cell area, were used to define multiciliated ependymal cells, whereas NSCs were defined by the presence of a single primary cilium and a smaller apical surface area. Where pinwheels overlapped, the larger of the two was measured.
Basal Body Patch Analysis
Cell outlines and basal body (BB) patches were manually outlined using ImageJ software. Absolute areas were directly calculated and reported while fractional areas were calculated by dividing the area of the BB patch by the apical cell surface area. The center of each area was calculated in ImageJ and the vector from the center of the cell and center of the BB patch was calculated. The angle of this vector was termed the BB patch angle, which was normalized to the average of all angles within a given field (set at 180 degrees). The percent distribution of patch angles was plotted in a histogram with a 15 degree bin. BB patch displacement was calculated by taking the magnitude of this vector. Fractional displacement was calculated by dividing the magnitude of the vector running from the center of the cell to the center of the BB patch by the magnitude of a manually drawn vector running from the center of the cell through the center of the BB and terminating at the cell border.
Statistics
All data are expressed as mean +/− SEM. Student’s t-tests were performed using SigmaPlot software. Patch angle distributions were analyzed using the Watson’s U2 test (MatLab). P values of <0.05 were defined as statistically significant.
Supplementary Material
Highlights.
As the SVZ niche emerges, dystroglycan reorganizes extracellular matrix into “hubs”
Dystroglycan regulates the development and organization of niche ependymal cells
Dystroglycan regulates SVZ stem cell proliferation and generation of glial progeny
Notch signaling in SVZ cells and progeny is regulated by dystroglycan
Acknowledgments
This study was supported by the Empire State Stem Cell Fund (NY State Department of Health contract CO26400). The authors wish to thank Ms. Haritha Desu and Mr. Azeez Aranmolate for technical assistance, Dr. Kevin Campbell (HHMI, Univ. of Iowa) for providing DAG1fl/fl mice, Dr. Ken-ichi Takemaru for providing FoxJ1-EGP mice, Dr. Shaoyu Ge for providing Nestin-CreER mice, Dr. Adan Aguirre and members of his lab for advice regarding Notch reagents, and members of the Colognato lab for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: The authors declare no conflict of interest.
Author Contributions
F.K.M., H.S., X.S., and C.E. conducted experiments, H.C., F.K.M., and H.S. designed the experiments, analyzed data, and wrote the paper.
References
- Ables JL, Breunig JJ, Eisch AJ, Rakic P. Not(ch) just development: Notch signalling in the adult brain. Nature reviews Neuroscience. 2011;12:269–283. doi: 10.1038/nrn3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhavan A, Griffith OL, Soroceanu L, Leonoudakis D, Luciani-Torres MG, Daemen A, Gray JW, Muschler JL. Loss of cell-surface laminin anchoring promotes tumor growth and is associated with poor clinical outcomes. Cancer research. 2012;72:2578–2588. doi: 10.1158/0008-5472.CAN-11-3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody SL, Yan XH, Wuerffel MK, Song SK, Shapiro SD. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. American journal of respiratory cell and molecular biology. 2000;23:45–51. doi: 10.1165/ajrcmb.23.1.4070. [DOI] [PubMed] [Google Scholar]
- Brooks ER, Wallingford JB. Multiciliated cells. Current biology : CB. 2014;24:R973–982. doi: 10.1016/j.cub.2014.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MW, Jacobson C, Yurchenco PD, Morris GE, Carbonetto S. Laminin-induced clustering of dystroglycan on embryonic muscle cells: comparison with agrin-induced clustering. The Journal of cell biology. 1997;136:1047–1058. doi: 10.1083/jcb.136.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colognato H, Galvin J, Wang Z, Relucio J, Nguyen T, Harrison D, Yurchenco PD, Ffrench-Constant C. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development. 2007;134:1723–1736. doi: 10.1242/dev.02819. [DOI] [PubMed] [Google Scholar]
- Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. The Journal of cell biology. 1999;145:619–631. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblandre GA, Wettstein DA, Koyano-Nakagawa N, Kintner C. A two-step mechanism generates the spacing pattern of the ciliated cells in the skin of Xenopus embryos. Development. 1999;126:4715–4728. doi: 10.1242/dev.126.21.4715. [DOI] [PubMed] [Google Scholar]
- Delgehyr N, Meunier A, Faucourt M, Bosch Grau M, Strehl L, Janke C, Spassky N. Ependymal cell differentiation, from monociliated to multiciliated cells. Methods in cell biology. 2015;127:19–35. doi: 10.1016/bs.mcb.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Douet V, Arikawa-Hirasawa E, Mercier F. Fractone-heparan sulfates mediate BMP-7 inhibition of cell proliferation in the adult subventricular zone. Neuroscience letters. 2012;528:120–125. doi: 10.1016/j.neulet.2012.08.077. [DOI] [PubMed] [Google Scholar]
- Douet V, Kerever A, Arikawa-Hirasawa E, Mercier F. Fractone-heparan sulphates mediate FGF-2 stimulation of cell proliferation in the adult subventricular zone. Cell proliferation. 2013;46:137–145. doi: 10.1111/cpr.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyermann C, Czaplinski K, Colognato H. Dystroglycan promotes filopodial formation and process branching in differentiating oligodendroglia. J Neurochem. 2012;120:928–947. doi: 10.1111/j.1471-4159.2011.07600.x. [DOI] [PubMed] [Google Scholar]
- Finzsch M, Stolt CC, Lommes P, Wegner M. Sox9 and Sox10 influence survival and migration of oligodendrocyte precursors in the spinal cord by regulating PDGF receptor alpha expression. Development. 2008;135:637–646. doi: 10.1242/dev.010454. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- Galvin J, Eyermann C, Colognato H. Dystroglycan modulates the ability of insulin-like growth factor-1 to promote oligodendrocyte differentiation. J Neurosci Res. 2010;88:3295–3307. doi: 10.1002/jnr.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubst N, Georges-Labouesse E, De Arcangelis A, Mayer U, Gotz M. Basement membrane attachment is dispensable for radial glial cell fate and for proliferation, but affects positioning of neuronal subtypes. Development. 2006;133:3245–3254. doi: 10.1242/dev.02486. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Gu YH, Izawa Y, Del Zoppo GJ. Disruption of dystroglycan-laminin interactions modulates water uptake by astrocytes. Brain research. 2013;1503:89–96. doi: 10.1016/j.brainres.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heins N, Malatesta P, Cecconi F, Nakafuku M, Tucker KL, Hack MA, Chapouton P, Barde YA, Gotz M. Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci. 2002;5:308–315. doi: 10.1038/nn828. [DOI] [PubMed] [Google Scholar]
- Henry MD, Campbell KP. A role for dystroglycan in basement membrane assembly. Cell. 1998;95:859–870. doi: 10.1016/s0092-8674(00)81708-0. [DOI] [PubMed] [Google Scholar]
- Henry MD, Satz JS, Brakebusch C, Costell M, Gustafsson E, Fassler R, Campbell KP. Distinct roles for dystroglycan, beta1 integrin and perlecan in cell surface laminin organization. Journal of cell science. 2001;114:1137–1144. doi: 10.1242/jcs.114.6.1137. [DOI] [PubMed] [Google Scholar]
- Hirrlinger PG, Pannicke T, Winkler U, Claudepierre T, Varshney S, Schulze C, Reichenbach A, Brunken WJ, Hirrlinger J. Genetic deletion of laminin isoforms beta2 and gamma3 induces a reduction in Kir4.1 and aquaporin-4 expression and function in the retina. PloS one. 2011;6:e16106. doi: 10.1371/journal.pone.0016106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet BV, Salinas-Mondragon R, Liang H, Therit B, Buie JD, Dykstra M, Campbell K, Ostrowski LE, Brody SL, Ghashghaei HT. FoxJ1-dependent gene expression is required for differentiation of radial glia into ependymal cells and a subset of astrocytes in the postnatal brain. Development. 2009;136:4021–4031. doi: 10.1242/dev.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang ES, Goldman JE. Pax6 expression is sufficient to induce a neurogenic fate in glial progenitors of the neonatal subventricular zone. PloS one. 2011;6:e20894. doi: 10.1371/journal.pone.0020894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanis I, Lathia JD, Vadakkan TJ, Raborn E, Wan R, Mughal MR, Eckley DM, Sasaki T, Patton B, Mattson MP, et al. Quiescence and activation of stem and precursor cell populations in the subependymal zone of the mammalian brain are associated with distinct cellular and extracellular matrix signals. J Neurosci. 2010;30:9771–9781. doi: 10.1523/JNEUROSCI.0700-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerever A, Mercier F, Nonaka R, de Vega S, Oda Y, Zalc B, Okada Y, Hattori N, Yamada Y, Arikawa-Hirasawa E. Perlecan is required for FGF-2 signaling in the neural stem cell niche. Stem cell research. 2014;12:492–505. doi: 10.1016/j.scr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerever A, Schnack J, Vellinga D, Ichikawa N, Moon C, Arikawa-Hirasawa E, Efird JT, Mercier F. Novel extracellular matrix structures in the neural stem cell niche capture the neurogenic factor fibroblast growth factor 2 from the extracellular milieu. Stem Cells. 2007;25:2146–2157. doi: 10.1634/stemcells.2007-0082. [DOI] [PubMed] [Google Scholar]
- Kim H, Shin J, Kim S, Poling J, Park HC, Appel B. Notch-regulated oligodendrocyte specification from radial glia in the spinal cord of zebrafish embryos. Dev Dyn. 2008;237:2081–2089. doi: 10.1002/dvdy.21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokovay E, Goderie S, Wang Y, Lotz S, Lin G, Sun Y, Roysam B, Shen Q, Temple S. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell. 2008;7:163–173. doi: 10.1016/j.stem.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrousi C, Arbi M, Pilz GA, Pefani DE, Lalioti ME, Ninkovic J, Gotz M, Lygerou Z, Taraviras S. Mcidas and GemC1 are key regulators for the generation of multiciliated ependymal cells in the adult neurogenic niche. Development. 2015;142:3661–3674. doi: 10.1242/dev.126342. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Whitman MC, Noonan MA, Ables JL, DeCarolis NA, Arguello AA, Donovan MH, Fischer SJ, Farnbauch LA, Beech RD, et al. Dynamic contribution of nestin-expressing stem cells to adult neurogenesis. J Neurosci. 2007;27:12623–12629. doi: 10.1523/JNEUROSCI.3812-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature genetics. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Leiton CV, Aranmolate A, Eyermann C, Menezes MJ, Escobar-Hoyos LF, Husain S, Winder SJ, Colognato H. Laminin promotes metalloproteinase-mediated dystroglycan processing to regulate oligodendrocyte progenitor cell proliferation. J Neurochem. 2015;135:522–538. doi: 10.1111/jnc.13241. [DOI] [PubMed] [Google Scholar]
- Leonoudakis D, Huang G, Akhavan A, Fata JE, Singh M, Gray JW, Muschler JL. Endocytic trafficking of laminin is controlled by dystroglycan and is disrupted in cancers. Journal of cell science. 2014;127:4894–4903. doi: 10.1242/jcs.152728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonoudakis D, Singh M, Mohajer R, Mohajer P, Fata JE, Campbell KP, Muschler JL. Dystroglycan controls signaling of multiple hormones through modulation of STAT5 activity. Journal of cell science. 2010;123:3683–3692. doi: 10.1242/jcs.070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–1122. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- Marcet B, Chevalier B, Luxardi G, Coraux C, Zaragosi LE, Cibois M, Robbe-Sermesant K, Jolly T, Cardinaud B, Moreilhon C, et al. Control of vertebrate multiciliogenesis by miR-449 through direct repression of the Delta/Notch pathway. Nat Cell Biol. 2011;13:693–699. doi: 10.1038/ncb2241. [DOI] [PubMed] [Google Scholar]
- Martini S, Bernoth K, Main H, Ortega GD, Lendahl U, Just U, Schwanbeck R. A critical role for Sox9 in notch-induced astrogliogenesis and stem cell maintenance. Stem Cells. 2013;31:741–751. doi: 10.1002/stem.1320. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. The Journal of cell biology. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood-brain barrier. J Neurosci. 2014;34:15260–15280. doi: 10.1523/JNEUROSCI.3678-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier F, Kitasako JT, Hatton GI. Anatomy of the brain neurogenic zones revisited: fractones and the fibroblast/macrophage network. J Comp Neurol. 2002;451:170–188. doi: 10.1002/cne.10342. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Radial glia give rise to adult neural stem cells in the subventricular zone. Proc Natl Acad Sci U S A. 2004;101:17528–17532. doi: 10.1073/pnas.0407893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh Z, Merkle FT, Soriano-Navarro M, Garcia-Verdugo JM, Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro F, Lindenbaum M, Carbonetto S. alpha-Dystroglycan is a laminin receptor involved in extracellular matrix assembly on myotubes and muscle cell viability. The Journal of cell biology. 1999;145:1325–1340. doi: 10.1083/jcb.145.6.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- Morimoto M, Liu Z, Cheng HT, Winters N, Bader D, Kopan R. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. Journal of cell science. 2010;123:213–224. doi: 10.1242/jcs.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myshrall TD, Moore SA, Ostendorf AP, Satz JS, Kowalczyk T, Nguyen H, Daza RA, Lau C, Campbell KP, Hevner RF. Dystroglycan on radial glia end feet is required for pial basement membrane integrity and columnar organization of the developing cerebral cortex. Journal of neuropathology and experimental neurology. 2012;71:1047–1063. doi: 10.1097/NEN.0b013e318274a128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani H, Martin E, Hassani H, Clavairoly A, Maire CL, Viadieu A, Kerninon C, Delmasure A, Frah M, Weber M, et al. Ascl1/Mash1 promotes brain oligodendrogenesis during myelination and remyelination. J Neurosci. 2013;33:9752–9768. doi: 10.1523/JNEUROSCI.0805-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nico B, Tamma R, Annese T, Mangieri D, De Luca A, Corsi P, Benagiano V, Longo V, Crivellato E, Salmaggi A, et al. Glial dystrophin-associated proteins, laminin and agrin, are downregulated in the brain of mdx mouse. Laboratory investigation; a journal of technical methods and pathology. 2010;90:1645–1660. doi: 10.1038/labinvest.2010.149. [DOI] [PubMed] [Google Scholar]
- Noell S, Wolburg-Buchholz K, Mack AF, Beedle AM, Satz JS, Campbell KP, Wolburg H, Fallier-Becker P. Evidence for a role of dystroglycan regulating the membrane architecture of astroglial endfeet. The European journal of neuroscience. 2011;33:2179–2186. doi: 10.1111/j.1460-9568.2011.07688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Gonzalez P, Abdi K, Luciano D, Liu Y, Soriano-Navarro M, Rawlins E, Bennett V, Garcia-Verdugo JM, Kuo CT. Ank3-dependent SVZ niche assembly is required for the continued production of new neurons. Neuron. 71:61–75. doi: 10.1016/j.neuron.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HC, Appel B. Delta-Notch signaling regulates oligodendrocyte specification. Development. 2003;130:3747–3755. doi: 10.1242/dev.00576. [DOI] [PubMed] [Google Scholar]
- Satz JS, Ostendorf AP, Hou S, Turner A, Kusano H, Lee JC, Turk R, Nguyen H, Ross-Barta SE, Westra S, et al. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci. 2010;30:14560–14572. doi: 10.1523/JNEUROSCI.3247-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang SM, Goderie SK, Roysam B, Temple S. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell. 2008;3:289–300. doi: 10.1016/j.stem.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirour C, Hidalgo M, Bello V, Buisson N, Darribere T, Moreau N. Dystroglycan is involved in skin morphogenesis downstream of the Notch signaling pathway. Molecular biology of the cell. 2011;22:2957–2969. doi: 10.1091/mbc.E11-01-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25:10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs JL, Vladar EK, Axelrod JD, Kintner C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol. 2012;14:140–147. doi: 10.1038/ncb2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FE, Vladar EK, Ma L, Fuentealba LC, Hoh R, Espinoza FH, Axelrod JD, Alvarez-Buylla A, Stearns T, Kintner C, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140:4277–4286. doi: 10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, Doetsch F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008;3:279–288. doi: 10.1016/j.stem.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MK, Yeager K, Morrison SJ. Physiological Notch signaling promotes gliogenesis in the developing peripheral and central nervous systems. Development. 2007;134:2435–2447. doi: 10.1242/dev.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature genetics. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 2009;136:2297–2307. doi: 10.1242/dev.034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron. 1998;21:63–75. doi: 10.1016/s0896-6273(00)80515-2. [DOI] [PubMed] [Google Scholar]
- Weir ML, Oppizzi ML, Henry MD, Onishi A, Campbell KP, Bissell MJ, Muschler JL. Dystroglycan loss disrupts polarity and beta-casein induction in mammary epithelial cells by perturbing laminin anchoring. Journal of cell science. 2006;119:4047–4058. doi: 10.1242/jcs.03103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.