

Abstract
Cereblon (CRBN) is an E3 ligase adapter protein that can be reprogrammed by imide-class compounds such as thalidomide, lenalidomide, and pomalidomide to induce the degradation of neo-substrate proteins. In order to identify additional small molecule CRBN modulators, we implemented a focused combinatorial library approach where we fused an imide-based CRBN binding pharmacophore to a heterocyclic scaffold which could be further elaborated. We screened the library for CRBN-dependent antiproliferative activity in the multiple myeloma cell line MM1.S and identified five hit compounds. Quantitative chemical proteomics of hit compounds revealed that they induced selective degradation of GSPT1, a translation termination factor that is currently being explored as a therapeutic target for the treatment of acute myeloid leukemia. Molecular docking studies with CRBN and GSPT1 followed by analog synthesis identified a possible hydrogen bond interaction with the central pyrimidine ring as a molecular determinant of hit compounds’ selectivity. This study demonstrates that focused combinatorial library design, phenotypic screening, and chemical proteomics can provide a suitable workflow to efficiently identify novel CRBN modulators.
Graphical Abstract
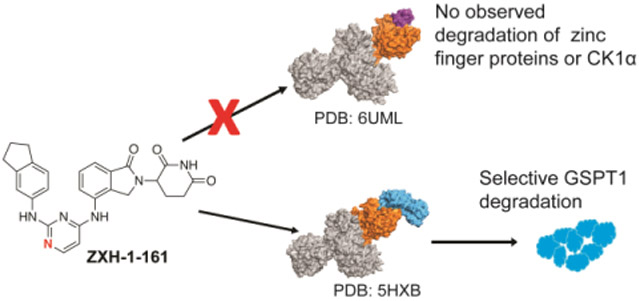
INTRODUCTION
During the late 1950s and early 1960s thalidomide was sold as a sedative that was frequently prescribed to pregnant women. Infamously, thalidomide use during pregnancy had teratogenic activity, leading to birth defects such as limb, ear, cardiac, and gastrointestinal malformations.1 In the following decades thalidomide was demonstrated to have immunomodulatory, anti-inflammatory, and anti-angiogenic properties, which reignited interest in its therapeutic potential.2 This led to thalidomide being granted its first FDA approval in 1998 for the treatment of erythema nodosum leprosum (ENL), a life-threatening inflammatory complication of lepromatous leprosy.2 Shortly afterwards thalidomide was shown to be an effective treatment for multiple myeloma, resulting in the development of more potent immunomodulatory drugs (IMiDs) with lower toxicities, including lenalidomide and pomalidomide as novel therapeutics to treat multiple myeloma.2-4
Thalidomide’s molecular mechanism of action was found to depend on its ability to bind to the adapter protein cereblon (CRBN), a substrate receptor component of the large E3 ubiquitin ligase complex CUL4-RBX1-DDB1-CRBN (CRL4CRBN).1,5-7 Thalidomide directs the activity of CRBN towards neo-substrates, leading to their ubiquitination and subsequent proteasomal degradation. IMiDs and other compounds with similar mechanisms of action are frequently referred to as “molecular glues” due to their ability to mediate interactions between two proteins that have not evolved to interact and have no measurable binding affinity in the absence of a molecular glue compound. The antiproliferative effects seen in multiple myeloma cells depend primarily on the ability of thalidomide to induce the degradation of the zinc finger transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) which are regulators of hematopoietic lineage, while the teratogenic activity appears to depend on its ability to degrade SALL4, a zinc finger protein which is essential for limb development.8-10 Structural biology studies have revealed that all known IMiD-dependent CRL4CRBN neo-substrates contain a conserved beta-hairpin motif which acts as a structural degron that is recruited to a pocket in CRBN by imide-based compounds.11,12 In addition to zinc finger proteins, some imide-based compounds can also induce the degradation of other proteins, such as casein kinase 1A1 (CK1α), which is degraded by lenalidomide, and translation termination factor G1 to S phase transition protein 1 (GSPT1), degraded by CC-885.12,13
These findings have stimulated tremendous interest in discovering new small molecules that can direct CRBN to recruit substrates for degradation. The most popular strategy currently involves the development of heterobifunctional small molecules frequently called PROTACs (PROteolysis Targeting Chimeras) where a CRBN binding motif is tethered via a linker to a recruiting element for a new target of interest.14-16 A second approach includes thalidomide-analogs as CRBN modulators (molecular glues) that can recruit neo-substrates to the CRBN E3 ubiquitin ligase complex.17,18 Both PROTACs and molecular glues have the ability to exhibit differentiated pharmacology relative to traditional occupancy-based inhibitors due to their ability to degrade proteins.19 An advantage of PROTACs is that they can be targeted towards distinct proteins by basing their design on known binding ligands, whereas glues and their targets have been discovered serendipitously or through phenotypic screening.18 Although clinical stage PROTACs have been developed,19,20 it is challenging to obtain desirable drug-like properties due to their typically higher molecular weight relative to glue-type molecules.
Here we develop a structured workflow for the identification, optimization, and validation of CRBN modulators. Our approach uses a focused combinatorial analog library, designed based on the fusion of heterocyclic scaffolds to an imide-based CRBN binding pharmacophore. We screened this library for antiproliferative activity in the multiple myeloma cell line MM1.S (wild-type (WT) and CRBN knockout). Compounds with CRBN-dependent antiproliferative activity were selected for expression proteomics in order to identify degraded proteins (Figure 1). Through this workflow we identified five hits from a library of 51 compounds. These five new CRBN modulators were all shown to induce selective degradation of GSPT1. We used molecular docking to rationalize the selectivity profile of hit compounds. We identified and validated a specific hydrogen bond as a possible determinant of selectivity.
Figure 1.

Workflow for identifying new CRBN modulators.
RESULTS AND DISCUSSION
IMiDs have shown the ability to drug targets traditionally considered to be “undruggable” due to not possessing a small molecule binding pocket, such as IKZF1/3 and GSPT1. A literature survey revealed that introducing small motifs (like −NH2, urea, or aniline) into the IMiD scaffold can have a major impact on protein-protein complementarity between CRBN and the target neo-substrate, resulting in differentiated neo-substrate selectivity. Inspired by these insights, we designed a focused combinatorial library by replacing one chloro group in dichloroheterocyclic scaffolds (including substituted pyrimidine and purine) with a lenalidomide moiety. The remaining chloro group is then derivatized with a nucleophilic SNAr or palladium-mediated coupling reaction with different anilines, alkyl amines and boronic acids/esters, which introduce various interactions with potential target proteins. We explored two different pyrimidine scaffolds (scaffolds A and B), where one is directly attached to lenalidomide (scaffold B) and the other has a short linkage in the middle which covers more chemical and conformational space (scaffold A) (Table 1). Additionally, we explored a purine scaffold directly attached to lenalidomide (scaffold C).
Table 1.
IC50 (μM) values of thalidomide analogs.a
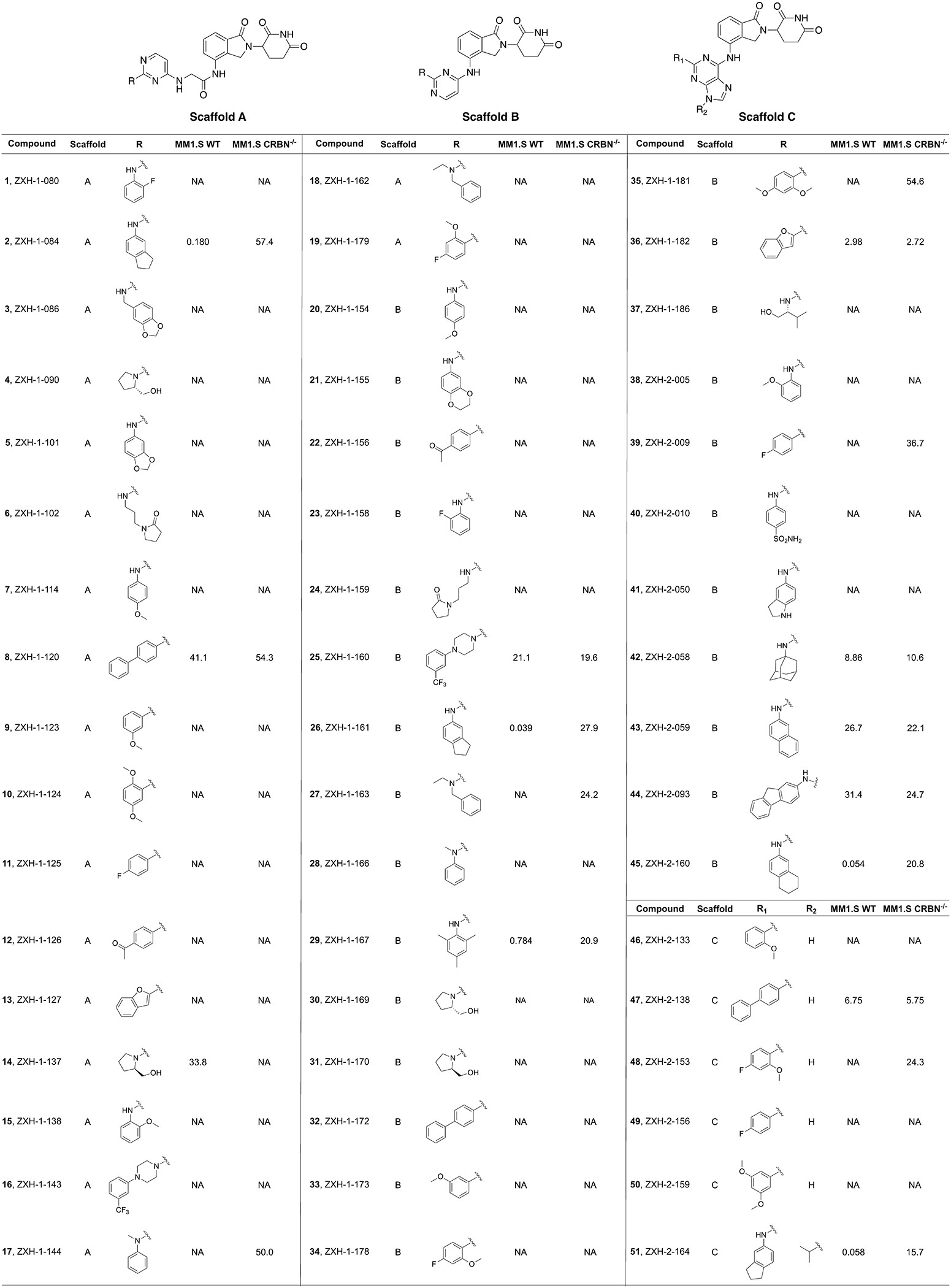 |
Cell viability in MM1.S WT and CRBN−/− cells after 48 h treatment (3 biological replicates). NA = no activity. IC50s of 2, 26, and 29 shown as average of 2 separate runs (3 biological replicates each).
The majority of the 51 compounds in the library exhibited no antiproliferative activity in either MM1.S WT or CRBN−/− up to a concentration of 20 μM. Seven compounds (8, 25, 36, 42, 43, 44, and 47) had notable to slight antiproliferative activity in both cell lines (Table 1, Supplementary Figure S1). This indicated that these compounds have CRBN-independent cytotoxicity. We identified five hit compounds with strong CRBN-dependent antiproliferative activity: 2, 26, 29, 45, and 51. The five hit compounds all share a similar hydrophobic moiety even though they do not belong to a single scaffold: compounds 2, 26, and 51 share the exact same 2,3-dihydro-1H-inden-5-amine; 45 contains a similar 5,6,7,8- tetrahydronaphthalen-2-amine moiety, and 29 has a trimethylaniline. The similarity of hydrophobic features suggested that these compounds could recruit the same neo-substrate(s).
To validate hit compounds further, we conducted an in vitro competitive CRBN binding assay with a TR-FRET readout. The in vitro assay demonstrated that all five hit compounds bound CRBN to a similar degree as lenalidomide (Figures 2B, Supplementary Figure S2). In addition, we performed a cellular CRBN engagement assay. In brief, compounds are co-treated with the BRD4 degrader dBET6 and cellular CRBN engagement is measured as the relative abundance of BRD4BD2-GFP, as described previously.21 In the cellular assay, the hit compounds were also able to engage CRBN to a similar extent as lenalidomide with the exception of 2 (Figures 2C, Supplementary Figure S3); this may indicate that 2 has some difficulties with cell permeability since the compound was able to bind CRBN in vitro.
Figure 2.

Hit compounds from CRBN-dependent antiproliferative activity screening and their degradative activity of known CRBN modulator targets. (A) Structures of hit compounds. (B) in vitro TR-FRET CRBN binding assay. (C) Cellular CRBN engagement assay. (D) Immunoblots after 4 h treatments in MM1.S cells. Vinculin representative of 4 blots. Quantification shown as percentage of DMSO control normalized to vinculin. CC-885, a CRBN modulator that induces GSPT1 degradation, is shown for comparison.12
We then examined via immunoblot whether these hit compounds induced the degradation of previously identified targets of CRBN modulators. All five compounds induced some degradation of GSPT1 in MM1.S cells after 4 h of treatment, with little to no detectable activity against IKZF1, IKZF3, and CK1α (Figure 2D). Co-treatment with the proteasome inhibitor carfilzomib demonstrated that the observed GSPT1 degradation for all five compounds was proteasome mediated, while co-treatment with the neddylation inhibitor MLN4924 confirmed that the GSPT1 degradation was occurring via recruitment of CRL4CRBN (Supplementary Figure S4).
In order to characterize the selectivity profile of the hit compounds further and examine whether they induce degradation of any additional proteins, we used quantitative proteomics with compounds 29, 2, and 51; these compounds were selected due to their low degradative activity of known CRBN modulator targets in order to identify unexpected degraded proteins, if any. In brief, protein abundance was measured in multiplexed mass spectrometry-based proteomics using tandem mass tag (TMT) isobaric labels as described previously10,22 after 6 h treatment in MM1.S cells with 0.1 μM of CC-885, 1 μM of 29, 1 μM of 2, 1 μM of 51, or vehicle control. This demonstrated that while CC-885 induced the downregulation of many proteins in MM1.S cells, 29 selectively induced the degradation of GSPT1, with no other statistically significant downregulated targets after triplicate analysis (Figures 3A and 3B). Singlicate analysis of expression proteomics with 2 (Figure 3C) and 51 (Figure 3D) showed similarly selective degradation of GSPT1, with GSPT2, a close homolog of GSPT1,23 being the only other downregulated target observed after treatment with 51. A single unique peptide of GSPT2 was quantified and measured as downregulated for 2 and 29, but the resulting data was filtered to only include proteins that had a minimum of three unique peptides in order to reduce false positives.
Figure 3.

Expression proteomics in MM1.S cells after 6 h treatment with compound or DMSO. (A) Duplicate analysis of CC-885 (0.1 μM). (B) Triplicate analysis of 29 (1 μM). (C) Singlicate analysis of 2 (1 μM). (D) Singlicate analysis of 51 (1 μM).
To rationalize how our hit compounds might induce the binding of CRBN and GSPT1, all five compounds were docked into the ternary complex model of CC-885 with CRBN and GSPT1 (PDB: 5HXB) using InducedFit protocol (Schrodinger suite release 2019-2). The binding mode of CC-885 is shown in gray in Figure 4A and docking of 26 is shown in yellow in Figure 4B; all five hit compounds showed similar binding modes as 26 (data not shown). Overall, 26 occupies the same binding cavity formed at the interface between GSPT1 (cyan) and CRBN (green), and has a very similar shape compared to CC-885. Besides forming the same interactions via the IMiD portion of the molecule, both CC-885 and 26 form a hydrogen bond with His353 of CRBN. However, we observed that 26 forms a hydrogen bond with Lys628 of GSPT1, which is not seen with CC-885. In addition, the central pyrimidine ring of 26 was observed to retain a minimal twist in the tail phenyl group when compared to the CC-885 phenyl group. Thus, the nitrogen of the pyrimidine ring appears to play a critical role in GSPT1 binding. Therefore, we hypothesized that if we replaced the N-1 with a carbon, it would disrupt the hydrogen bond with Lys628 and force the terminal phenyl group to adopt a twisted conformation. Both effects together should have detrimental consequences on the recruitment of GSPT1. To test this hypothesis, we synthesized 52 (Figure 4C).
Figure 4.

Molecular modeling of 26 identifies that the nitrogens in the pyrimidine ring are critical for GSPT1 degradation. (A) CC-885 (gray) bound to CRBN (green) and GSPT1 (cyan) in a ternary complex (PDB: 5HXB). (B) Docking of 26 (yellow) into PDB: 5HXB. (C) Structures of 26 and its analog 52, with the N-1 replaced with carbon. (D) Antiproliferative IC50 curves ± SD after 48 h treatments (three biological replicates; Graphpad Prism 8 software). (E) Immunoblot after 4 h treatments in MM1.S cells. Vinculin representative of 4 blots. Quantification shown as percentage of DMSO control normalized to vinculin.
Direct comparison of 26 and 52 demonstrated that the replacement of N-1 with carbon resulted in a loss of CRBN-dependent antiproliferative activity in MM1.S cells, despite 52 retaining its ability to bind CRBN (Figures 4D, Supplementary Figure S5). Additionally, examination by immunoblot showed that 52 does not induce degradation of GSPT1 to the same extent as 26 (Figure 4E). Together this indicates that the nitrogens in the pyrimidine ring of our hit compound are essential for GSPT1 degradation, and likely essential for the recruitment of GSPT1 to the CRBN E3 ubiquitin ligase complex. Furthermore, it is possible that an equivalent hydrogen bond interaction cannot be made with zinc finger proteins, accounting for the observed selectivity profile.
CONCLUSION
We have developed a prototype focused combinatorial library strategy that would be suitable for building collections of imide-based compounds. Screening for CRBN-dependent antiproliferative effects led to the identification of CRBN modulators that can induce the selective degradation of GSPT1. In contrast to CC-885, which induces degradation of GSPT1, IKZF1/3 and a large number of other proteins,12,24,25 our tool compounds are highly selective GSPT1/2 degraders with little to no activity on IKZF1/3 and no observable off-target activity in global proteomics. This is confirmed by our selective tool compounds exhibiting antiproliferative activity that is strictly dependent on the expression of CRBN, while CC-885 retains antiproliferative activity with unspecific toxicity in a CRBN−/− cell line (Figure 4D). The use of MM1.S cells likely biased our results towards the identification of compounds that degrade factors such as IKZF1 and GSPT1, which are known dependencies in these cells. Further exploration of our library with additional cell lines may therefore reveal new molecular glues and their targets.
Taken together, we have successfully identified novel CRBN modulators with selective activity towards GSPT1 and have identified a potential structural determinant for this selectivity. Due to its potency and selectivity for GSPT1, compound 26 is a superior tool to CC-885 for exploring the therapeutic potential of GSPT1 degradation and may provide additional opportunities for therapeutically targeting GSPT1, where a degradation-based strategy has already shown potential in the treatment of acute myeloid leukemia (AML).26-29
METHODS
Cell Culture.
MM1.S WT and MM1.S CRBN−/− cells were generously provided by James Bradner (DFCI, Boston, MA). MM1.S cell lines were cultured in RPMI-1640 media containing L-glutamine, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HEK293T cells were cultured om DMEM media containing L-glutamine, supplemented with 10% FBS and 1% penicillin/streptomycin. Mycoplasma testing was performed on a monthly basis and all lines were negative.
Cell Viability Assays.
Cell viability was evaluated using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) following the manufacturer’s standards.
Immunoblotting.
Cells were washed with PBS before being lysed with Cell Lysis Buffer (Cell Signaling) supplemented with protease and phosphatase inhibitor cocktails (Roche) at 4°C for 15 minutes. The cell lysate vortexed before being centrifuged at 14,000 x g for 20 min at 4°C. Protein in cell lysate was quantified by BCA assay (Pierce). Primary antibodies used in this study include β-Actin (Cell Signaling Technology, 3700s), CK1α (Abcam, ab206652), CRBN (Novus Biologicals, NBP1-91810), eRF3/GSPT1 (Abcam, ab49878), IKZF1 (Cell Signaling Technology, 5443S), IKZF3 (Cell Signaling Technology, 15103S), and vinculin (Abcam, ab130007). Blot quantification was performed using Image Studio 4.0 software, normalizing to loading controls.
Purification of biotinylated DDB1ΔB-CRBN.
Human DDB1ΔB and human CRBN were cloned in pAC-derived vectors30 and recombinant proteins were expressed as N-terminal His6 (DDB1ΔB) or StrepII-Avi (CRBN) fusions in Trichoplusia ni High-Five insect cells using the baculovirus expression system (Invitrogen). For purification, cells were resuspended in buffer containing 50 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) pH 8.0, 200 mM NaCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1x protease inhibitor cocktail (2 μg/ml Aprotinin, 10μM Bestatin, 2μM E-64, 10μM Leupeptin, 1μM Pepstatin A and 10μM 1,10 - Phenanthrolin and 1μM Phosphoramidon) and lysed by sonication. Following ultracentrifugation, the soluble fraction was passed over Strep-Tactin XT (IBA, 2-4030-025) resin and eluted with wash buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 1 mM TCEP) supplemented with 50 mM biotin (MedChemExpress, HY-B0511). The affinity-purified protein was subjected to ion exchange chromatography (Poros 50HQ) followed by size exclusion chromatography (16/60 S200, GE) in 50 mM HEPES pH 7.4, 200 mM NaCl and 1 mM TCEP. Biotinylation of was performed as previously described.31 The protein-containing fractions were concentrated using ultrafiltration (Millipore) and flash frozen in liquid nitrogen at 40-120 μM concentration and stored at −80°C.
In vitro CRBN Binding Assay.
Competitive binding to DDB1ΔB-CRBN1 was measured in vitro by titrating compounds into 10 nM Lenalidomide-BodipyFL, 100 nM biotinylated DDB1ΔB-CRBN, 2 nMTerbium-Streptavidin in a buffer containing 50 mM Tris pH 7.5, 200 mM NaCl, 0.1% Pluronic F-68 solution (Sigma). Titrations of compounds were performed using d300 dispenser (HP) and final DMSO concentration normalized to 1%. All biochemical assays were performed in 384-well low volume plates (Corning, 4514) using 15 μL assay volume. Substrate recruitment was measured using Pherastar (BMG) plate reader with excitation at 337 nm using 520/490 nm filter for signal detection. The TR-FRET signal was calculated as ratio of 520/490 emission. The IC50 values were calculated using a nonlinear fit variable slope model (GraphPad Prism Software). Data are presented as means normalized to DMSO ± standard deviation of n = 3 technical replicates.
Cellular CRBN engagement assay.
HEK293T cells stably expressing BRD4BD2-eGFP protein fusion with mCherry reporter were seeded at a density of 1000-4000 cells/well in a 384-well plate (Corning, 3764). BRD4BD2-GFP reporter cells were treated with increasing concentrations of lenalidomide or indicated compounds for 5 hrs in the presence of 100 nM dBET6. Relative abundance of BRD4BD2-GFP was measured by Laser Scanning Cytometry (Acumen). Green fluorescent signal (excitation laser: 488 nm; filter: 500-530 nm) and red florescent signal (excitation laser: 561 nm; filter: 575-640 nm) were individually measured and exported for analysis. Data analysis was performed using Cell Profiler,32 as previously described.21 The compound concentrations which resulted in 50% degradation of BRD4BD2-GFP (DC50) were calculated using a nonlinear fit variable slope model (GraphPad Prism Software). Data are represented as means ± s.d of three replicates (n = 3).
Sample preparation TMT LC-MS3 mass spectrometry.
MM1.S cells were treated with DMSO or 1 μM of compound ZXH-1-167 (29) in biological triplicates, 0.1 μM of DGY-4-189 (CC-885) in biological duplicates, and 1 μM of ZXH-1-084 (2) and 1 μM of ZXH-1-164 (51) in biological singlicate for 6 h. Cells were harvested by centrifugation and prepared for mass spectrometry as described previously.33,34 Data was collected as reported.33,34
LC-MS data analysis.
Data was analyzed using Proteome Discoverer 2.2 (Thermo Fisher Scientific) as described.33,34 Reporter ion intensities were normalized and scaled using in-house scripts in the R framework.35 Statistical analysis was carried out using the limma package within the R framework.36
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank M. Kostic for helpful feedback on the manuscript.
Funding Sources
Funding for this work was received from the National Institutes of Health (NIH): Grant R01 CA214608 (E.S.F.) and 5 F31 CA210619-02 (C.E.P.).
ABBREVIATIONS
- AML
acute myeloid leukemia
- CK1α
casein kinase 1A1
- CRBN
cereblon
- GSPT1
G1 to S phase transition protein 1
- IKZF1
Ikaros
- IKZF3
Aiolos
- IMiD
immunomodulatory drug
Footnotes
Supporting Information
The Supporting Information (PDF) is available free of charge on the ACS Publications website.
Antiproliferative IC50 curves in MM1.S cells.
In vitro and cellular CRBN binding assay IC50s.
Immunoblots of proteasome inhibitor co-treatments.
Chemical synthesis experimental methods.
The authors declare no competing financial interest.
REFERENCES
- (1).Ito T; Ando H; Suzuki T; Ogura T; Hotta K; Imamura Y; Yamaguchi Y; Handa H Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327 (5971), 1345–1350. 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- (2).Bartlett JB; Dredge K; Dalgleish AG The Evolution of Thalidomide and Its IMiD Derivatives as Anticancer Agents. Nat Rev Cancer 2004, 4 (4), nrc1323 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- (3).Rajkumar SV; Hayman SR; Lacy MQ; Dispenzieri A; Geyer SM; Kabat B; Zeldenrust SR; Kumar S; Greipp PR; Fonseca R; Lust JA; Russell SJ; Kyle RA; Witzig TE; Gertz MA Combination Therapy with Lenalidomide plus Dexamethasone (Rev/Dex) for Newly Diagnosed Myeloma. Blood 2005, 106 (13), 4050 10.1182/blood-2005-07-2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lacy MQ; Hayman SR; Gertz MA; Dispenzieri A; Buadi F; Kumar S; Greipp PR; Lust JA; Russell SJ; Dingli D; Kyle RA; Fonseca R; Bergsagel PL; Roy V; Mikhael JR; Stewart AK; Laumann K; Allred JB; Mandrekar SJ; Rajkumar SV Pomalidomide (CC4047) Plus Low-Dose Dexamethasone As Therapy for Relapsed Multiple Myeloma. J Clin Oncol 2009, 27 (30), 5008–5014. 10.1200/jco.2009.23.6802. [DOI] [PubMed] [Google Scholar]
- (5).Fischer ES; Böhm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith REJ; Harper JW; Jenkins JL; Thomä NH Structure of the DDB1-CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512 (7512), 49 53. 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chamberlain PP; Lopez-Girona A; Miller K; Carmel G; Pagarigan B; Chie-Leon B; Rychak E; Corral LG; Ren YJ; Wang M; Riley M; Delker SL; Ito T; Ando H; Mori T; Hirano Y; Handa H; Hakoshima T; Daniel TO; Cathers BE Structure of the Human Cereblon–DDB1–Lenalidomide Complex Reveals Basis for Responsiveness to Thalidomide Analogs. Nat Struct Mol Biol 2014, 21 (9), 803–809. 10.1038/nsmb.2874. [DOI] [PubMed] [Google Scholar]
- (7).Lopez-Girona A; Mendy D; Ito T; Miller K; Gandhi AK; Kang J; Karasawa S; Carmel G; Jackson P; Abbasian M; Mahmoudi A; Cathers B; Rychak E; Gaidarova S; Chen R; Schafer PH; Handa H; Daniel TO; Evans JF; Chopra R Cereblon Is a Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26 (11), 2326 2335. 10.1038/leu.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lu G; Middleton RE; Sun H; Naniong M; Ott CJ; Mitsiades CS; Wong K-K; Bradner JE; Kaelin WG The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343 (6168), 305–309. 10.1126/science.1244917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kronke J; Udeshi ND; Narla A; Grauman P; Hurst SN; McConkey M; Svinkina T; heckl D; Comer E; Li X; Ciarlo C; Hartman E; Munshi N; Schenone M; Schreiber SL; Carr SA; Ebert BL Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343 (6168), 301 305. 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Donovan KA; An J; Nowak RP; Yuan JC; Fink EC; Berry BC; Ebert BL; Fischer ES Thalidomide Promotes Degradation of SALL4, a Transcription Factor Implicated in Duane Radial Ray Syndrome. Elife 2018, 7, e38430 10.7554/elife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Petzold G; Fischer ES; Thomä NH Structural Basis of Lenalidomide-Induced CK1α Degradation by the CRL4CRBN Ubiquitin Ligase. Nature 2016, 532 (7597), 127 130. 10.1038/nature16979. [DOI] [PubMed] [Google Scholar]
- (12).Matyskiela ME; Lu G; Ito T; Pagarigan B; Lu C-C; Miller K; Fang W; Wang N-Y; Nguyen D; Houston J; Carmel G; Tran T; Riley M; Nosaka L; Lander GC; Gaidarova S; Xu S; Ruchelman AL; Handa H; Carmichael J; Daniel TO; Cathers BE; Lopez-Girona A; Chamberlain PP A Novel Cereblon Modulator Recruits GSPT1 to the CRL4CRBN Ubiquitin Ligase. Nature 2016, 535 (7611), 252–257. 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- (13).Krönke J; Fink EC; Hollenbach PW; MacBeth KJ; Hurst SN; Udeshi ND; Chamberlain PP; Mani DR; Man H-W; Gandhi AK; Svinkina T; Schneider RK; McConkey M; Järås M; Griffiths E; Wetzler M; Bullinger L; Cathers BE; Carr SA; Chopra R; Ebert BL Lenalidomide Induces Ubiquitination and Degradation of CK1α in Del(5q) MDS. Cell 2015, 523 (7559), 183 188. 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as a Strategy for in Vivo Target Protein Degradation. Science 2015, 348 (6241), 1376 1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; Besten W. den ; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in Vivo Protein Knockdown by Small-Molecule PROTACs. Behav Brain Res 2015, 11 (8), 611 617. 10.1016/s0166-4328(01)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Paiva S-L; Crews CM Targeted Protein Degradation: Elements of PROTAC Design. Curr Opin Chem Biol 2019, 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Matyskiela ME; Zhang W; Man H-W; Muller G; Khambatta G; Baculi F; Hickman M; LeBrun L; Pagarigan B; Carmel G; Lu C-C; Lu G; Riley M; Satoh Y; Schafer P; Daniel TO; Carmichael J; Cathers BE; Chamberlain PP A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem 2017, 61 (2). 10.1021/acs.jmedchem.6b01921. [DOI] [PubMed] [Google Scholar]
- (18).Chamberlain PP; Cathers BE Cereblon Modulators: Low Molecular Weight Inducers of Protein Degradation. Drug Discov Today Technologies 2019, 31 (Curr Med Chem 25 31 2018). 10.1016/j.ddtec.2019.02.004. [DOI] [PubMed] [Google Scholar]
- (19).Chamberlain PP; Hamann LG Development of Targeted Protein Degradation Therapeutics. Nat Chem Biol 2019, 15 (10), 937–944. 10.1038/s41589-019-0362-y. [DOI] [PubMed] [Google Scholar]
- (20).Mullard A First Targeted Protein Degrader Hits the Clinic. Nat Rev Drug Discov 2019. 10.1038/d41573-019-00043-6. [DOI] [PubMed] [Google Scholar]
- (21).Gasic I; Groendyke BJ; Nowak RP; Yuan JC; Kalabathula J; Fischer ES; Gray NS; Mitchison TJ Tubulin Resists Degradation by Cereblon-Recruiting PROTACs. Cells 2020, 9 (5), 1083 10.3390/cells9051083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McAlister GC; Nusinow DP; Jedrychowski MP; Wühr M; Huttlin EL; Erickson BK; Rad R; Haas W; Gygi SP MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal Chem 2014, 86 (14), 7150–7158. 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Goff CL; Zemlyanko O; Moskalenko S; Berkova N; Inge-Vechtomov S; Philippe M; Zhouravleva G Mouse GSPT2, but Not GSPT1, Can Substitute for Yeast ERF3 in Vivo. Genes Cells 2002, 7 (10), 1043–1057. 10.1046/j.1365-2443.2002.00585.x. [DOI] [PubMed] [Google Scholar]
- (24).Hansen JD; Condroski KR; Correa M; Muller GW; Man H-W; Ruchelman AL; Zhang W; Vocanson F; Crea T; Liu W; Lu G; Baculi F; LeBrun L; Mahmoudi A; Carmel G; Hickman MG; Lu C-C Protein Degradation via CRL4CRBN Ubiquitin Ligase: Discovery and Structure-Activity Relationships of Novel Glutarimide Analogs That Promote Degradation of Aiolos and/or GSPT1. J Med Chem 2017, 61 (2). 10.1021/acs.jmedchem.6b01911. [DOI] [PubMed] [Google Scholar]
- (25).Hao B; Li X; Jia X; Wang Y; Zhai L; Li D; Liu J; Zhang D; Chen Y; Xu Y; Lee S; Xu G; Chen X; Dang Y; Liu B; Tan M The Novel Cereblon Modulator CC-885 Inhibits Mitophagy via Selective Degradation of BNIP3L. Acta Pharmacol Sin 2020, 1–9 10.1038/s41401-020-0367-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jin L; Mbong N; Ng SWK; Wang JCY; Minden MD; Fan J; Pierce DW; Pourdehnad M; Dick JE A Novel Cereblon E3 Ligase Modulator Eradicates Acute Myeloid Leukemia Stem Cells through Degradation of Translation Termination Factor GSPT1. Blood 2019, 134 (Supplement_1), 3940–3940. 10.1182/blood-2019-128450. [DOI] [Google Scholar]
- (27).Lopez-Girona A; Lu G; Rychak E; Mendy D; Lu C-C; Rappley I; Fontanillo C; Cathers BE; Daniel TO; Hansen J CC-90009, a Novel Cereblon E3 Ligase Modulator, Targets GSPT1 for Degradation to Induce Potent Tumoricidal Activity Against Acute Myeloid Leukemia (AML). Blood 2019, 134 (Supplement_1), 2703–2703. 10.1182/blood-2019-127892. [DOI] [Google Scholar]
- (28).Lu G; Surka C; Lu C-C; Jang IS; Wang K; Rolfe M Elucidating the Mechanism of Action of CC-90009, a Novel Cereblon E3 Ligase Modulator, in AML Via Genome-Wide CRISPR Screen. Blood 2019, 134 (Supplement_1), 405–405. 10.1182/blood-2019-125492. [DOI] [Google Scholar]
- (29).Uy GL; Minden MD; Montesinos P; DeAngelo DJ; Altman JK; Koprivnikar J; Vyas P; Fløisand Y; Vidriales MB; Gjertsen BT; Esteve J; Buchholz TJ; Couto S; Fan J; Hanna B; Li L; Pierce DW; Hege K; Pourdehnad M; Zeidan AM Clinical Activity of CC-90009, a Cereblon E3 Ligase Modulator and First-in-Class GSPT1 Degrader, As a Single Agent in Patients with Relapsed or Refractory Acute Myeloid Leukemia (R/R AML): First Results from a Phase I Dose-Finding Study. Blood 2019, 134 (Supplement_1), 232–232. 10.1182/blood-2019-123966. [DOI] [Google Scholar]
- (30).Abdulrahman W; Uhring M; Kolb-Cheynel I; Garnier J-M; Moras D; Rochel N; Busso D; Poterszman A A Set of Baculovirus Transfer Vectors for Screening of Affinity Tags and Parallel Expression Strategies. Anal Biochem 2009, 385 (2), 383–385. 10.1016/j.ab.2008.10.044. [DOI] [PubMed] [Google Scholar]
- (31).Cavadini S; Fischer ES; Bunker RD; Potenza A; Lingaraju GM; Goldie KN; Mohamed WI; Faty M; Petzold G; Beckwith REJ; Tichkule RB; Hassiepen U; Abdulrahman W; Pantelic RS; Matsumoto S; Sugasawa K; Stahlberg H; Thomä NH Cullin–RING Ubiquitin E3 Ligase Regulation by the COP9 Signalosome. Nature 2016, 531 (7596), 598–603. 10.1038/nature17416. [DOI] [PubMed] [Google Scholar]
- (32).Carpenter AE; Jones TR; Lamprecht MR; Clarke C; Kang IH; Friman O; Guertin DA; Chang JH; Lindquist RA; Moffat J; Golland P; Sabatini DM CellProfiler: Image Analysis Software for Identifying and Quantifying Cell Phenotypes. Genome Biol 2006, 7 (10), R100 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li Z; Pinch BJ; Olson CM; Donovan KA; Nowak RP; Mills CE; Scott DA; Doctor ZM; Eleuteri NA; Chung M; Sorger PK; Fischer ES; Gray NS Development and Characterization of a Wee1 Kinase Degrader. Cell Chem Biol 2019. 10.1016/j.chembiol.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brand M; Jiang B; Bauer S; Donovan KA; Liang Y; Wang ES; Nowak RP; Yuan JC; Zhang T; Kwiatkowski N; Müller AC; Fischer ES; Gray NS; Winter GE Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol 2019, 26 (2), 300–306.e9. 10.1016/j.chembiol.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Team, R. D. C. R: A Language and Environment for Statistical Computing. http://www.R-project.org/.
- (36).Ritchie ME; Phipson B; Wu D; Hu Y; Law CW; Shi W; Smyth GK Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res 2015, 43 (7), e47 e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.