

Abstract
Background:
Chamber-specific and temporally regulated perinatal cardiac growth and maturation is critical for functional adaptation of the heart and may be altered significantly in response to perinatal stress, such as systemic hypoxia (hypoxemia), leading to significant pathology, even mortality. Understanding transcriptome regulation of neonatal heart chambers in response to hypoxemia is necessary to develop chamber-specific therapies for infants with cyanotic congenital heart defects (CHDs).
Methods and Results:
We sought to determine chamber-specific transcriptome programming during hypoxemic perinatal circulatory transition. We performed transcriptome-wide analysis on right ventricle (RV) and left ventricle (LV) of postnatal day 3 (P3) mouse hearts exposed to perinatal hypoxemia. Hypoxemia decreased baseline differences between RV and LV leading to significant attenuation of ventricular patterning (AVP), which involved several molecular pathways, including Wnt signaling suppression and cell cycle induction. Notably, robust changes in RV transcriptome in hypoxemic condition contributed significantly to the AVP. Remarkably, suppression of epithelial mesenchymal transitions (EMT) and dysregulation of the TP53 signaling were prominent hallmarks of the AVP genes in neonatal mouse heart. Furthermore, members of the TP53-related gene family were dysregulated in the hypoxemic RVs of neonatal mouse and cyanotic TOF hearts.
Conclusion(s):
Integrated analysis of chamber-specific transcriptome revealed hypoxemia-specific changes that were more robust in RVs compared to LVs, leading to previously uncharacterized AVP induced by perinatal hypoxemia. Remarkably, reprogramming of EMT process and dysregulation of the TP53 network contributed to transcriptome remodeling of neonatal heart during hypoxemic circulatory transition. These insights may enhance our understanding of hypoxemia-induced pathogenesis in newborn infants with cyanotic CHD phenotypes.
Keywords: Neonatal Heart Maturation, Congenital Heart Defects, Transcriptome, Tetralogy of Fallot, Hypoxia
INTRODUCTION
Early development of postnatal heart in mammals is a tightly regulated process that involves not only sequential transition in chamber-specific morphology and function, but also dramatic changes in chamber-specific cellular and transcriptional architecture that ultimately lead to the mature postnatal heart [1–7]. This tightly regulated process may alter significantly during pathological perinatal transition in response to systemic hypoxia (hypoxemia) leading to significant morbidity and mortality.
Soon after birth, the mammalian heart encounters rapid changes in hemodynamic load and external environment [2–6]. During the critical transition window, structural and hemodynamic changes ultimately lead to functional maturation of the left ventricle (LV) and the right ventricle (RV) as they functionally synchronize to support the systemic and the pulmonary circuits, respectively. This tightly regulated process may become rapidly disrupted in response to perinatal stress, such as systemic hypoxia (hypoxemia). Transcriptome programming is a major driving force of cardiac chamber development and pathological remodeling in heart. However, chamber-specific transcriptome changes during hypoxemic perinatal transition have not been fully revealed [8–11]. At present, a gap of knowledge exists regarding the mechanisms that control the molecular signaling of the RV vs LV. Consequently, our management of the diseased RV is largely shaped by our approaches to the diseased LV. Characterizing the discernible genetic programs and molecular signaling of the LV vs RV transcriptomes in response to perinatal hypoxemia is a very important task with potential implications in CHDs.
Hypoxemia is a common stress factor for newborn infants with cyanotic CHDs [8–11]. In previous studies on neonatal mice [1, 2], we demonstrated that perinatal hypoxemia induces myocyte proliferation more robustly in RVs and suppresses Wnt11 expression in a chamber-specific manner, eliminating the differences in myocyte proliferation and Wnt11 signaling between RV and LV, observed under normal conditions [2]. Importantly, we discovered, for the first time, differential activation of Wnt pathways in infants with cyanotic (hypoxemic) vs noncyanotic (non-hypoxemic) Tetralogy of Fallot (TOF) [1, 2]. However, the observed changes in transcriptome regulation can potentially be contributed by other factors including abnormal RVOT development and pressure overload. Therefore, further studies are needed to confirm the chamber-specific molecular signature, induced specifically by hypoxemia.
In order to determine the specific effects attributable to hypoxemia in left and right ventricular chambers, we performed systematic analysis on chamber-specific transcriptomes of postnatal day 3 (P3) male newborn mouse hearts during perinatal circulatory transition under normal and hypoxia conditions. The data provide a comprehensive spatial-temporal landscape of major molecular signaling pathways and transcription regulators in P3 neonatal heart chambers during hypoxemic vs normal perinatal circulatory transition. Importantly, hypoxemia induced attenuation of ventricular patterning (AVP), which involved several signaling pathways and transcriptional regulators. Moreover, we identified a previously unrecognized dysregulation of the TP53-regulated network in the hypoxemic neonatal mouse heart, suggesting a potential contribution of the TP53 pathway to hypoxia–induced pathology during hypoxemic perinatal circulatory transition and in cyanotic CHDs.
RESULTS
Transcriptome Landscape of Neonatal Mouse Heart during Hypoxemic Perinatal Circulatory Transition
To determine the impact of perinatal hypoxemia on chamber-specific transcriptome regulation, we induced systemic hypoxia at FIO2 (fraction of inspired oxygen)=10% in neonatal mice as we described previously [2] [Figure 1. A]. According to previous studies [8], this level of hypoxia corresponds to an oxygen tension ~14 mmHG, which results in tissue hypoxia. Consistently, histological examination of heart sections from hypoxia-treated neonatal mice confirmed the status of significant tissue hypoxia as demonstrated by nuclear deposits of stabilized Hif1a protein [Figure 1. B].
Figure 1. Perinatal Induction of Systemic Hypoxia (Hypoxemia) in Neonatal Mouse Heart.
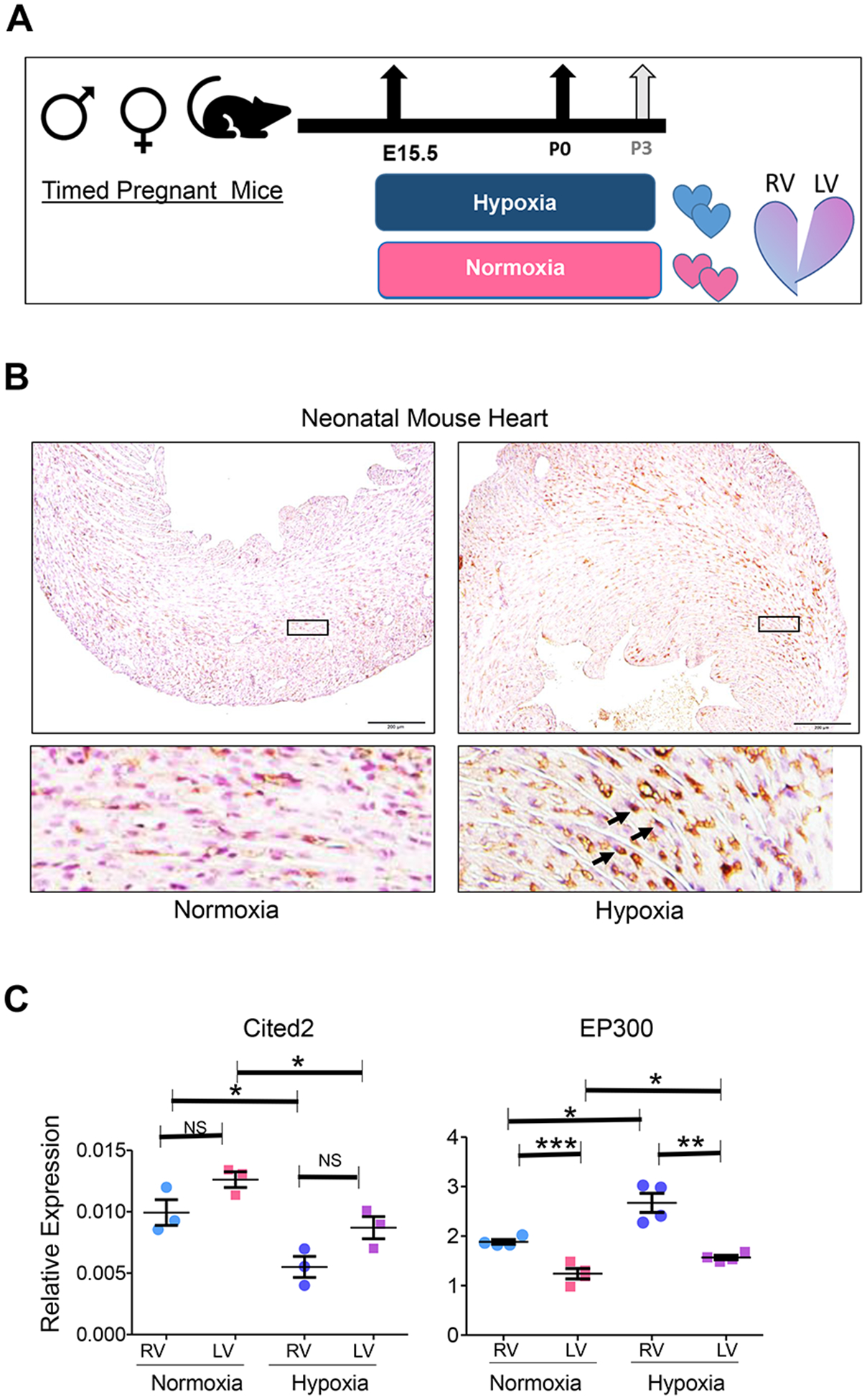
A. Schematic illustration of experimental design for perinatal systemic hypoxia exposure. The dams carrying the experimental group (hypoxia) were acclimatized in the hypoxia chamber by decreasing FIO2 by 2% daily for at least 5 days preceding the experiment starting at E15.5 to reach 10% at E20.5. Neonatal pups were reared with their dams in either normoxia or hypoxia (10% FIO2) and maintained until postnatal day 3 (P3). B. Hif1a immunohistochemistry (IHC) staining of normoxia-treated or hypoxia-treated P3 neonatal mouse hearts demonstrates nuclear Hif1a stabilization in response to perinatal hypoxia. C. Quantitative expression analysis (qRT-PCR) of Hif1a homeostasis genes, Cited2 and EP300, in RV and LV of P3 neonatal mouse hearts in normoxia and hypoxia conditions. Error bars represent standard error of means (SEM). *P ≤0.05, **P≤0.01, ***P≤0.005; two tailed Student’s t test; n=3 (Cited2), n=4 (Ep300).
To evaluate whether transcriptome reprogramming reflects Hif1a-dependent regulation, we specifically examined the expression of known modulators of Hif1a activity [12–14]. Indeed, EP300, a known cofactor that promotes Hif1a interaction with Hif2a (Arnt) in association with Creb, was upregulated in heart tissue from hypoxemic neonatal heart, while Cited2, the inhibitor to Hif1a interaction with EP300, was downregulated [Figure 1. C]. Together, these results indicate successful induction of hypoxia at the tissue level. Transcriptome analysis using expression array data of total RNAs derived from neonatal heart RVs and LVs revealed significant transcriptome changes in both ventricles in response to hypoxemia. Unsupervised hierarchical clustering at all transcript levels, and heat map of the top DEGs, revealed diminished chamber-specific signature in response to hypoxemia. Remarkably, hypoxemic RV samples clustered closer to the normoxic LV samples in both datasets. Consistently, the number of significantly differentially expressed genes decreased in response to hypoxemia [Figure 2. A–C]. Pair-wise differential gene expression (DGE) analysis revealed 55 DGE in RV and 61 DGE in LV under hypoxic conditions compared to baseline normoxia [adjusted P value ≤ 0.05]. Among them 17 overlapped DGE between RV and LV. Consistently, pathway heat map revealed distinct patterns that were associated with ventricular chamber and/or hypoxemia condition [Figure 2. D, E]. Remarkably, chamber-specific signature was more distinct in normoxia compared to hypoxia-treated neonatal mouse hearts.
Figure 2. Hypoxia-Induced Attenuation of Ventricular Patterning in Neonatal Mouse Heart.
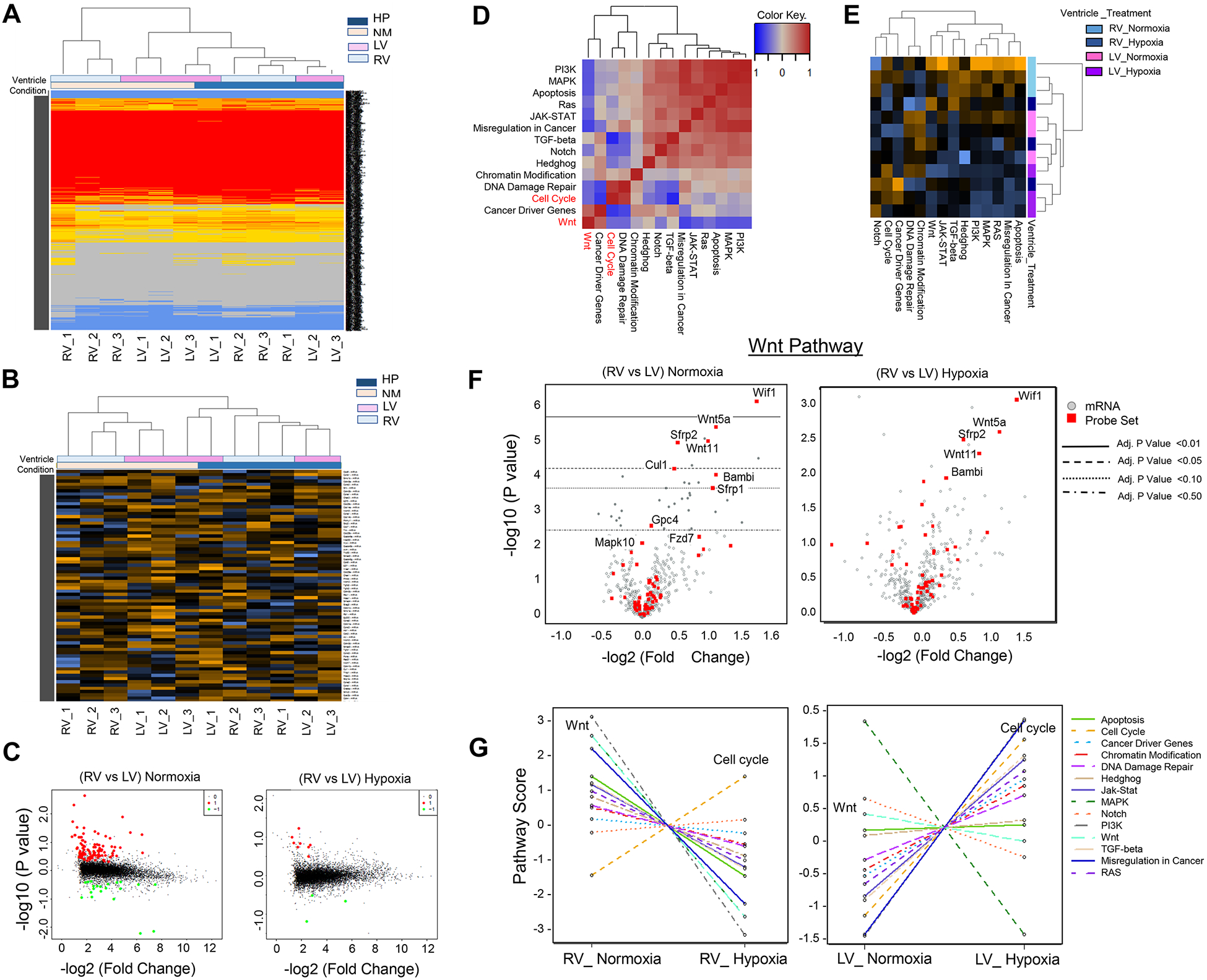
A. Unsupervised Hierarchical clustering of all transcripts from RV and LV of normoxia-treated and hypoxia-treated neonatal mouse hearts. B. Expression heat map of significant differentially expressed genes (DEGs). C. Volcano Plots display pair-wise comparison analysis (RV vs LV) in normoxia and hypoxia conditions. D. Pathway correlation plot of normoxia-treated RV (X Axis) compared to hypoxia-treated RV (Y Axis) depicts significant inverse correlation between cell cycle processes and Wnt signaling. E. Heat map depicts pathway enrichment signature in RV compared to LV in normoxia and hypoxia conditions. The chamber-specific signature is lost in hypoxia-treated hearts more significantly in RV. Blue: negative score. Orange: positive score. F. Volcano blots of Wnt signaling genes display reduced DGE in RV vs LV in neonatal mouse heart in hypoxia-treated neonatal mouse hearts compared to normoxia condition. G. Pathway enrichment analysis demonstrates trend changes of pathway scores (Bonferroni-adjusted P value) in RV (normoxia vs hypoxia) and in LV (normoxia vs hypoxia).
Perinatal Hypoxemia Attenuates Ventricular Patterning
Consistent with our previous transcriptomic studies [1, 2], we observed a significant reciprocal correlation between Wnt signaling enrichment in RV and cell cycle activation in LV under normoxia conditions [Figure 2. D–G]. These data are consistent with Wnt11/Rb1 suppression in RVs of cyanotic TOF compared to noncyanotic TOF cases, as we have previously reported [1, 2]. Furthermore, several pathways exhibited chamber-specific enrichment in RV compared to LV at the baseline (during normal perinatal transition), including apoptosis pathway, JAK-STAT, TGF-β, MAPK, and PIK signaling [Figure 2. G]. Remarkably, chamber-specific patterns observed under normoxia condition were significantly attenuated following hypoxemia induction [Figure 2. D–G]. While several of these pathways exhibited significant co-repression in hypoxemic RVs, some pathways exhibited co-activation in LV, demonstrating inversed patterns of regulation in RV compared to LV under hypoxic condition [Figure 2. G, and Figure 3]. Consistently, the statistical significance and number of the differentially expressed genes between RV and LV under hypoxia decreased compared to baseline normoxia condition [Figure 4. A and B], indicating hypoxemia-induced attenuation of ventricular patterning (AVP). Other than Wnt signaling and E2F1-regulated cell cycle program, epithelial mesenchymal transition (EMT) [15], extracellular matrix (ECM) remodeling process and TP53 pathway were significantly enriched biological terms in the hypoxia-regulated AVP gene set [Figure 4. C], defined as the genes that lost their statistically significant differences between RV and LV in hypoxemic condition [Supplemental Table 2A], with a subset confirmed by quantitative real time PCR (qRT-PCR) [Figure 4. D].
Figure 3. Perinatal Hypoxemia Regulates Major Signaling Pathways in Neonatal Mouse Heart.
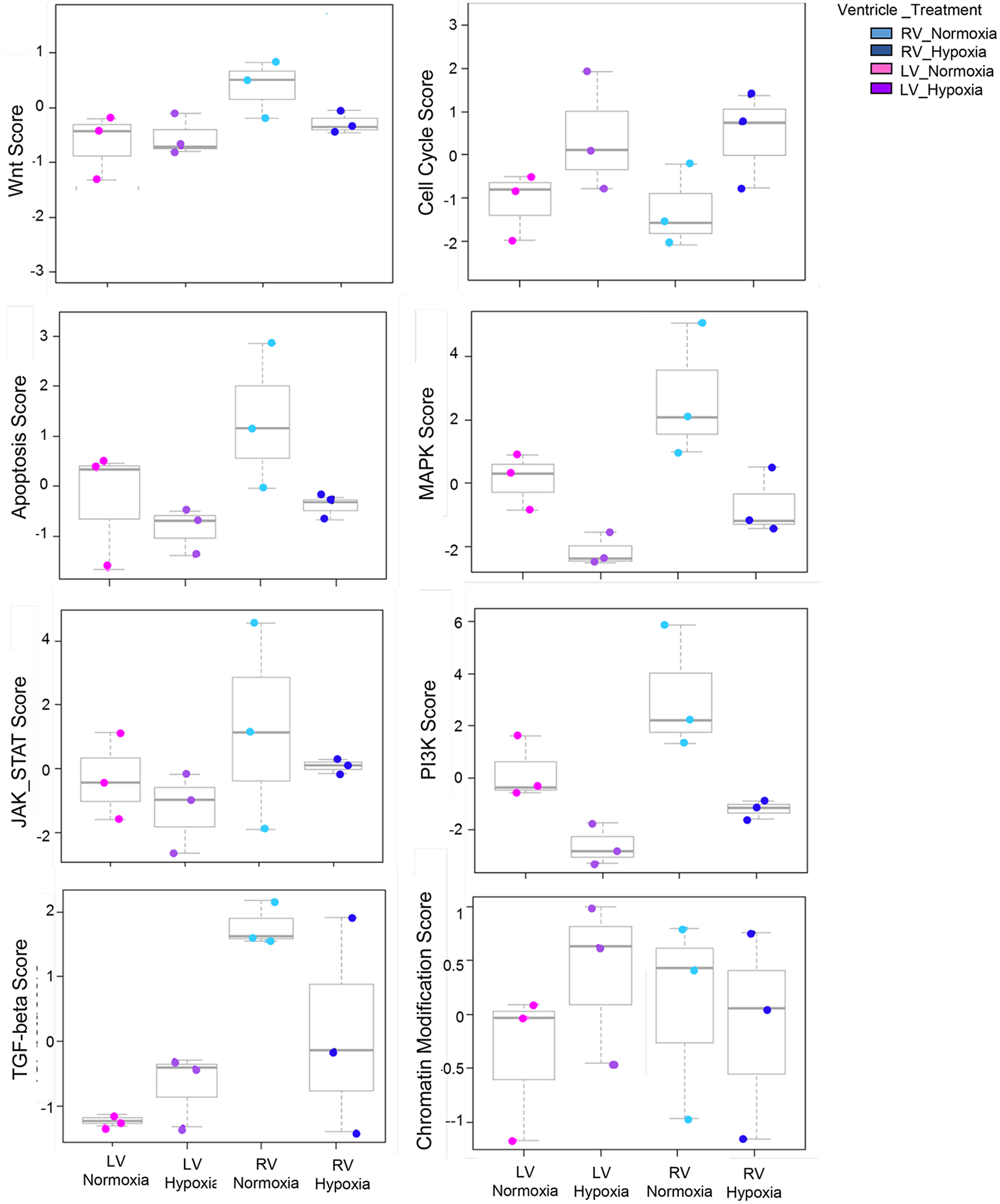
Box plots depict global changes of major signaling pathways scores in RV (normoxia vs hypoxia) and in LV (normoxia vs hypoxia).
Figure 4. Hypoxemia-Induced Attenuation of Ventricular Patterning (AVP).
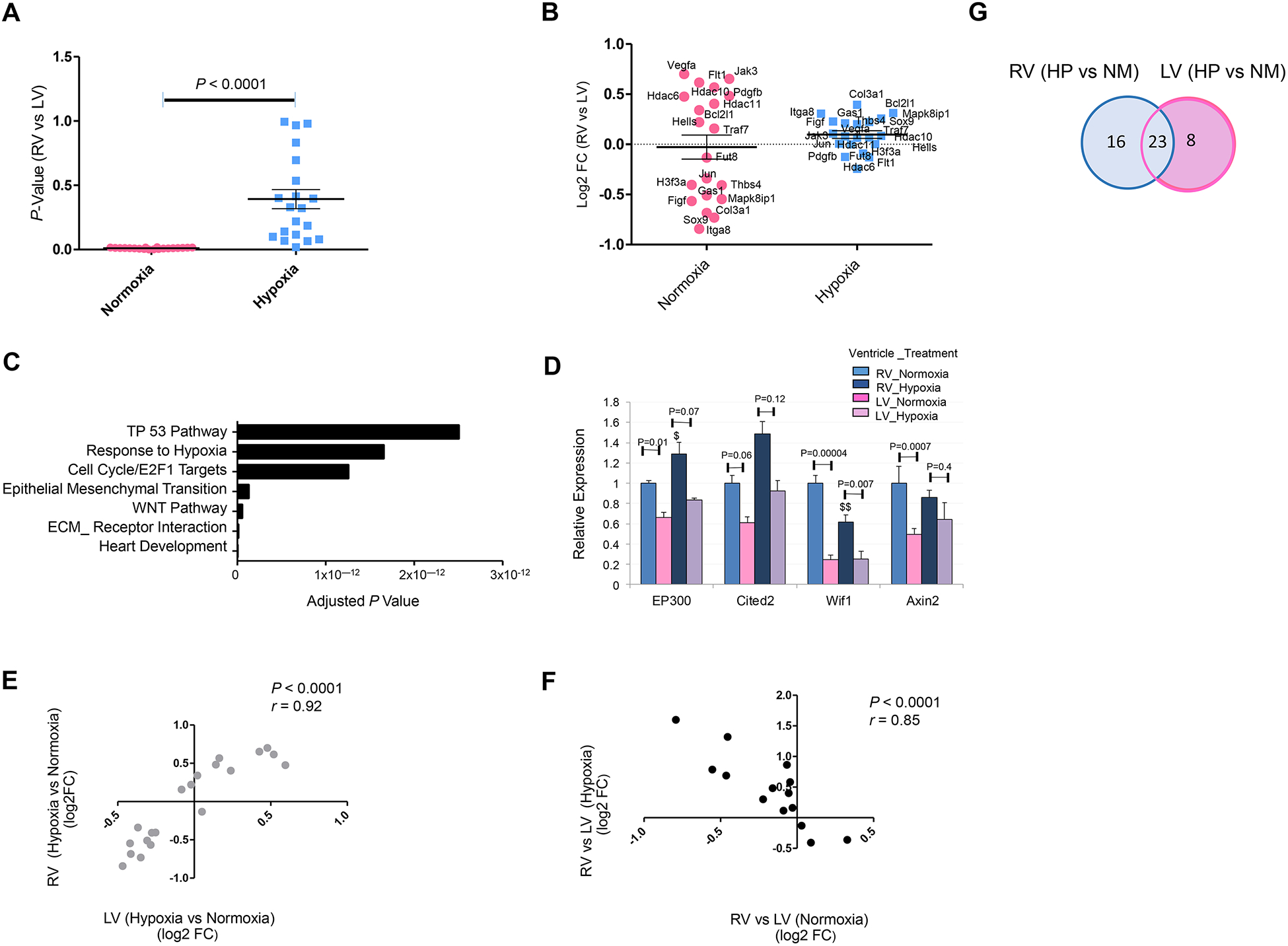
A and B. P value distribution (A) and magnitude of expression differences [Log2 Fold Change (FC)] (B) of top 20 differentially expressed genes (RV vs LV) that exhibited attenuation of ventricular patterning (AVP) in P3 neonatal mouse heart in response to systemic hypoxia (hypoxemia) treatment compared to normoxia condition (FDR P Value ≤ 0.05). C. Top GO terms enriched in hypoxia-induced AVP genes in neonatal mouse (Bonferroni-adjusted p value). D. Quantitative expression analysis (qRT-PCR) of hypoxia homeostasis genes (EP300 and Cited2) and Wnt related genes (Wif1 and Axin2) in LV and RV of WT P3 mouse demonstrates significant attenuation of ventricular patterning (fold change and P Values) in hypoxia-treated hearts compared to normoxia condition. $P ≤ 0.05, $$P ≤ 0.01 (hypoxia-treated RV compared to RV normoxia); two-tailed Student’s t test; n=4 per group per condition. E. Correlation analysis of top twenty AVP genes reveals significant concordance in mouse RV and LV. F. Correlation analysis of top genes with inversed attenuation of ventricular patterning (IVP) in hypoxia compared to baseline normoxia. G. Venn diagram depicting the distribution of DEGs in RV vs LV in hypoxia (HP) vs Normoxia (NM) conditions.
We next examined whether the observed changes in gene regulation are preserved in RV and LV. Interestingly, the hypoxia-induced changes in AVP genes exhibited concordant patterns in RV and LV [Figure 4. E], but were more robust in magnitude and significance in RV compared to those observed in LV in neonatal mouse hearts [Supplemental Table 2A, and Figure 4.G]. Moreover, some of these genes exhibited reciprocal patterns of regulation in response to hypoxia treatment, compared to baseline normoxia, leading to inversed ventricular patterning (IVP) as shown in Figure 4. F, and Supplemental Table 2B.
Perinatal Hypoxemia Dysregulates TP53-Signaling
Among the pathways enriched in the AVP genes, the TP53 pathway [16] was prominent [Figure 4. C], with significant dysregulation of TP53 downstream signaling. Importantly, TP53-regulated genes constituted approximately 1/3 of all AVP genes [Figure 5. A and B and Supplemental Table 3A]. Furthermore, IPA-based upstream analysis predicted TP53 among the top regulators of the DGE in response to hypoxia, including genes involved in EMT and ECM [Figure 5. C]. Consistently, transcription factor binding site (TFBS) enrichment analysis for hypoxemia-associated genes using Gene Set Enrichment Analysis (GSEA) suite [17], identified 59 genes with putative TP53 DNA binding motifs overrepresented in their promoters, including known coactivators of Hif1a (EP300/Crebbp) [13]. Moreover, among these TP53 target genes, 14 genes exhibited significant concordant regulation in the hypoxemic RVs from neonatal mice and cyanotic TOF data previously reported by us [1], including important mediators of EMT and ECM remodeling as well as cell cycle regulators [Figure 5. D, and Supplemental Table 3B.].
Figure 5. TP53 is a Potential Key Regulator of Hypoxia-Induced Transcriptome Reprogramming.
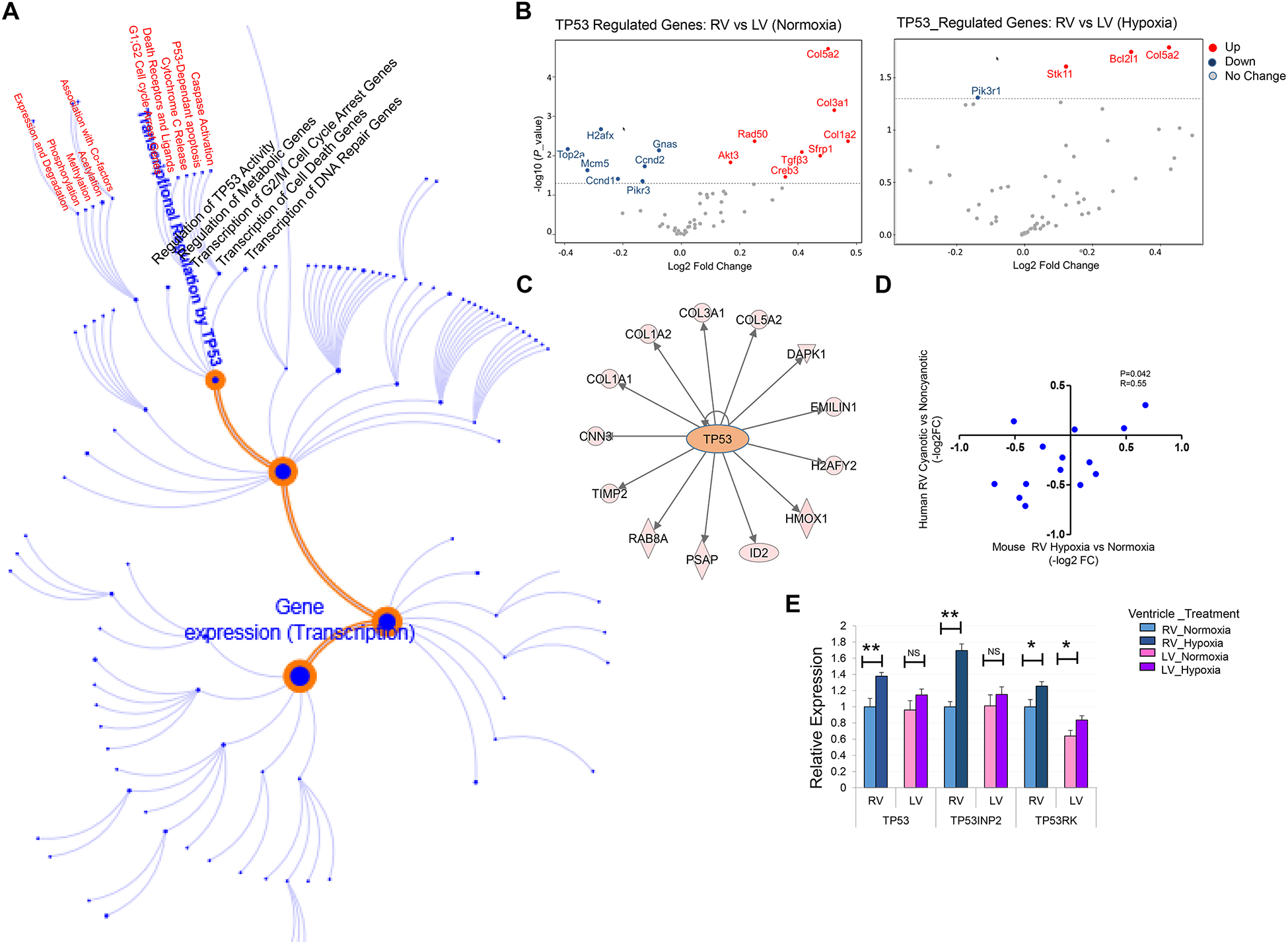
A. Schematic representation of P53 transcriptional network. B. Volcano blots of TP53_regulated genes display reduced DGE in RV vs LV in neonatal mouse heart in hypoxia-treated neonatal mouse hearts compared to normoxia condition. C. IPA-based upstream analysis reveals TP53 as a key regulator of the DGE genes involved in epithelial-mesenchymal transition and extracellular matrix remodeling. D. Correlation plot depicts significant concordance of TP53 -target gene expression in hypoxemic RVs from mouse and human TOF cases. E. Quantitative expression analysis (qRT-PCR) of TP53, TP53INP2, TP53RK in RV and LV myocardium of P3 neonatal mouse depicts robust induction in hypoxia-treated RVs compared to normoxia condition; n=3 per group per condition. *P ≤ 0.05, **P ≤ 0.01, (hypoxia compared to normoxia condition).
To investigate the biological relevance of this putative TP53 loop, we examined the impact of hypoxemia on expression of selected members of the TP53 gene family [18–21] using qRT-PCR. We found TP53, TP53 regulatory kinase (TP53rk) and TP53-inducible nuclear protein (TP53inp2), a known mediator of autophagy, were significantly induced in hypoxemic mouse hearts, more robustly in RV compared to LV [Figure 5. E]. Together, these analyses suggested potential hypoxemia-induced dysregulation of the TP53-regulated network contributing to the expression pattern of AVP genes. Together, these findings suggest TP53 is a potential key modulator of hypoxemia-induced transcriptome regulation in neonatal heart.
DISCUSSION
In this study, we performed comprehensive analysis on chamber-specific transcriptome derived from P3 neonatal male mouse hearts during normal and hypoxemic perinatal circulatory transition. Significant attenuation of chamber-specific (RV vs LV) transcriptome signature was observed in hypoxemic hearts compared to control. The observed attenuation of ventricular patterning, denoted as AVP, was mostly contributed by robust changes in hypoxemic RV and found to be significantly concordant with the changes observed in LV in response to hypoxemia. Moreover, a subset of genes exhibited inversed ventricular patterning (IVP) in RV compared to LV in response to hypoxia. Therefore, our animal model of perinatal hypoxemia induction allowed us to determine transcriptome changes that are chamber-specific and those that are conserved in RV and LV under hypoxia condition. Furthermore, our findings suggest that neonatal RV is more vulnerable to significant transcriptome alteration in hypoxemic conditions.
Although the exact mechanisms that regulate RV-specific responses to hypoxia remain to be fully investigated, it could be contributed in part by the suppression of chamber-specific expression of important signaling molecules, including Wnt11, as demonstrated in our previous publications [1, 2]. Consistent with our previous reports, chamber-specific differences in Wnt signaling and cellular proliferations were reciprocally affected by perinatal hypoxemia conditions, leading to significant reduction in the baseline differences between RV and LV. However, although these patterns were anticipated based on our previous observation, it is quite interesting to observe that significant chamber-specific changes affecting several other signaling pathways that are involved in stress response, cellular viability and tissue homeostasis (P38, apoptosis and Jak-stat signaling) were also attenuated by hypoxemia exposure. Together, these findings suggest that systemic hypoxia induces complex and pluripotent responses in neonatal heart chambers, requiring well-designed mechanistic studies to elucidate the regulatory cascade and the underlying mechanisms.
Our findings introduce a new regulatory loop that is primarily dominated by dysregulated TP53 network in response to hypoxemia. The TP53-dependent pathway removes DNA-damaged cells through either apoptosis or cell-cycle arrest [16, 21]. Hypoxemia is known to modulate the TP53 pathway, dependent or independent on HIF1A. The interaction between these two major pathways has been broadly investigated in cancer [22, 23]. Synergetic and antagonistic effects of TP53 and HIF1A have both been reported in regulating many cellular processes, including apoptosis, cell cycle arrest, glycolysis, ROS production and EMT through direct and indirect interactions [15, 22, 24]. Additionally, the severity and duration of hypoxemia can influence TP53 level and activity very differently [22]. Consistent with these reports, our findings illustrate that the role of TP53 in cardiac tissue homeostasis is complex under hypoxemic conditions acting in concert with other transcriptional components that are necessary for the regulation of cardiac response to hypoxic stress. This model is consistent with the known ability of TP53 to physically interact with key transcription factors and co-factors, such as Hif1a, Crebbbp, and EP300, or transcription repressors, such as Rb1. Indeed, despite TP53 induction by hypoxia [Figure 5. C], we observed repression of TP53 downstream targets in hypoxemic neonatal heart. Remarkably, among the repressed genes are known mediators of EMT and fibrosis (Col3a1, col5a2). In contrast, cell cycle regulator (MDC1) and DNA damage response (DDIT4) were induced [Supplemental Table 3A]. This selective pattern of TP53 target gene expression indicates that, in hypoxemic conditions TP53 may function as both a transcription activator and repressor in a gene-specific manner. Furthermore, we observed concordant regulation of some TP53 target genes in hypoxemic RVs from mouse and human TOF. Together, our results suggest a potentially complex trans-regulation between Hif1a and TP53 contributing to hypoxemia-induced transcriptome changes. Further in vivo studies focused on TP53-Hif1a interactions are necessary and can potentially pave the way for future therapeutic applications for infants with cyanotic CHDs.
Among the different biological processes involved in hypoxemia-induced transcriptome reprogramming, the enrichment with EMT-related terms in DGE and AVP genes is particularly prominent. In heart, different cells arise from one or more EMT event (s) and errors of EMT are known contributors to CHDs [15, 24]. EMT regulation involves some common cellular features and transcriptional re-programming mechanisms [15, 24–28]. However, the exact roles of EMT in neonatal heart maturation are unknown. Furthermore, the potential contribution of Hif1a-TP53 interplay in EMT regulation of hypoxemic hearts requires specific attention. In cancer, a reciprocal regulation between these two families of genes has been shown [29, 30]. Antagonistic effects of TP53 and Hif1a induce EMT in colorectal cancer cells, where Hif1a increases, and TP53 represses EMT. Along with these lines of evidence, our findings suggest that the mechanisms of EMT regulation in hypoxemic neonatal heart possibly involve a TP53-mediated regulatory loop that can potentially be modified and modulated in a gene-specific and a context-dependent manner in response to hypoxemia.
Finally, we acknowledge important limitations of this study: 1) Hypoxemia level, duration, and other associated variables may influence the molecular signature of chamber-specific transcriptome. Therefore, the insights revealed from this study provide only a foundation for further investigation focused on stratifying these environmental variables. 2) The functional data did not reveal underlying mechanisms involved in transcriptome regulation. 3) The impact of anatomical defects and pressure overload vs hypoxemia condition should be evaluated in further mechanistic studies using mouse models of TOF and other congenital heart defects.
METHODS
Animal Studies:
All animal-related experimental protocols were approved by the University of California Los Angeles Animal Care and Use Committee (ACUC). Gene expression data will be deposited within the Gene Expression Omnibus repository (www.ncbi.nlm.nih.gov/geo) under Neonatal Heart Maturation SuperSeries GSE85728 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85728) [31].
Experimental Animals and Perinatal Hypoxemia Induction:
The experiments were conducted under an active animal protocol approved by the Institutional ACUC at UCLA. Pathogen-free male and female C57BL/6J mice were obtained from Charles River Laboratories. BioSpherix OxyCycler normobaric hypoxia chamber was used to induce systemic hypoxia [FIO2=10%] as we previously described [1]. Timed pregnant dams were allowed to deliver normally. The dams carrying the experimental group (hypoxia) were maintained with their offspring in the hypoxia chamber until P3, whereas the dams carrying the control neonates were maintained in ambient air.
Quantitative Expression Array (NanoString Gene Expression Profiling):
Total RNA was isolated from LV and RV of wild type P3 neonatal mice reared in hypoxia or normoxia condition (n=3 per group per condition). Mus musculus (Mm) PanCancer: Gene expression CodeSet profiling 750 mouse genes that belong to 14 different pathways and 20 housekeeping genes were used as internal reference control. The NanoString ‘nSolver advanced’ software was used for the analysis of the gene expression files. To avoid spurious conclusions based on analysis of background rather than signal genes, we omitted low count data. The ‘mean+/−2SD’ of the negative controls was used to estimate each sample’s background count level. Then, the experiment’s background count was estimated by calculating the average of all samples’ mean+/−2SD. For each probe per sample, the signal to noise ratio was estimated by dividing each probe’s count by the experiment’s background count. The probe threshold for estimating the frequency level of samples below background was set at ≤0.5 (at least half of the samples must have a signal to noise ratio <1). To balance the counts between samples, and therefore make meaningful biological comparisons, normalization was performed implementing the geNorm algorithm [32]. To investigate differential gene expression (DGE), nSolver implements multivariate linear regression. This analysis was run twice utilizing all 12 samples, one adjusted for ventricle-type (left, right), and the other without this confounder. Similar DGE analyses were run using only the left or right ventricle data. Within each condition, analyses were run to examine DGE between left and right ventricles. The DGE results were overlaid on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways by implementing the Pathview R package [33]. For each variable in the DGE analysis and for each pathway with a KEGG graph, a Pathview plot was generated. On the plot, the coloring of the nodes was performed according to the DGE of their genes, measured by fold change. A p-value=0.05 threshold was selected so that genes above this threshold had their fold change set to 0 before Pathview was run. DGE was also examined at the pathway level by running the Gene Set Analysis (GSA) [17], which calculates the global significance scores by summarizing the overall level of statistical significance of each gene in each pathway. Pathway scores (or dysregulation scores) were also generated from principal component analysis. Pearson’s correlation, linear regression module, was implemented for correlation studies and a P value less than or equal to 0.05 was considered significant.
Differential Gene Expression (DGE) Analysis.
DGE analysis was performed with expression levels normalized for gene length, library size and GC contents. An LME module framework was used to assess differential expression in log2 [normalized RPKM] values for each gene. The age, sex, diagnosis and hypoxemia status were treated as fixed parameters. Significant results were reported at Benjamini_Hochberg FDR ≤ 0.05.
Correlation Studies.
Throughout the study, we assessed replication between datasets by evaluating the concordance between independent sample sets by comparing the correlation of fold changes in each sample set at a defined statistical cut-off. We set the statistical cut-off in one sample set (the y-axis in the scatterplots) and computed the r (correlation coefficient) with fold changes in these genes in the compared sample set (the x-axis in the scatterplots).
Other Bioinformatics and Computational Methods.
The statistical significance for differential gene expression was assessed by Fisher’s exact test. Pearson’s correlation coefficients (r) for gene expression were calculated in R. Principal Component Analysis (PCA) was conducted using R function prcomp. The top 500 varied mRNAs based on Tophat alignment results were used to generate PCAs. The heat map function of R, which employs a hierarchical cluster algorithm, was used to draw heat map figures. The log2-transformed data were preprocessed by median centering of the data for each set and then hierarchically clustered using centered correlation as the similarity metric and average linkage as the clustering method.
Quantitative Real-Time PCR.
Total RNA was isolated from pooled male pups’ LV free walls and RV free walls separately, using an RNeasy Mini Kit (QIAGEN). For reverse transcription, one microgram of total RNA was used to generate first-strand cDNA with oligo-dT primers. Real-time PCR was performed using the SYBR Green Mix (Bio-Rad) on a CFX96 Real-time System (Bio-Rad). Primer sequences are listed in Supplemental Table 4.
Immunohistochemistry (IHC).
Heart tissue was fixed in 4% (v/v) formaldehyde, embedded into paraffin, and cut into 5-μm-thick tissue sections. After deparaffinization, slides were subjected to antigen retrieval. After blocking in PBS containing 10% bovine serum albumin, tissue sections were incubated with primary antibodies overnight and then appropriate AlexaFluor-conjugated secondary antibodies.
Statistical Analysis.
Quantified results are presented as mean ± SEM. Student’s t-test (unpaired, 2-tailed) and ANOVA with post-hoc Kruskal-Wallis were used for comparing 2 groups and more than 2 groups, respectively; P value less than or equal to 0.05 was considered significant, unless specified otherwise. The correlation of gene expression for each mRNA/trait pair was calculated using Pearson’s correlation and Benjamini-Hochberg correction methods. A Benjami-ni-Hochberg–adjusted correlation P value less than or equal to 0.05 was considered significant.
Supplementary Material
Supplemental Table 1 (Attached Excel File. Sheet 1). Top 20 differential gene expression (DGE) in RV and LV of P3 neonatal mouse heart in response to hypoxia treatment.
Supplemental Table 2 (Attached Excel File. Sheet 2). 2A. Attenuation of Ventricular Patterning (AVP) Genes. Top 20 AVP genes revealed by pair-wise differential gene expression (DGE) analysis (RV vs LV) of P3 neonatal mouse heart in hypoxia vs normoxia conditions. 2B. Inversion of Ventricular Patterning (IVP) Genes. IVP gene subset revealed by pair-wise differential gene expression (DGE) analysis (RV vs LV) of P3 neonatal mouse heart in hypoxia vs normoxia conditions.
Supplemental Table 3 (Attached Excel File. Sheet 3). 3A. TP53 Network. Representative list of dysregulated genes of TP53 network that overlapped with the AVP genes. 3B. Representative TP53-target genes that exhibited concordant regulation in hypoxemic RVs of mouse and human.
Supplemental Table 4 (Attached Excel File. Sheet 4). List of primers used for quantitative RT-PCR.
KEY MESSAGES.
During perinatal circulatory transition, transcriptome programming is a major driving force of cardiac chamber-specific maturation and adaptation to hemodynamic load and external environment.
During hypoxemic perinatal transition, transcriptome reprogramming may affect chamber-specific growth and development, particularly in newborns with congenital heart defects (CHDs).
Chamber-specific transcriptome changes during hypoxemic perinatal transition are yet to be fully elucidated.
Systems-based analysis of hypoxemic neonatal hearts at postnatal day 3 reveals chamber-specific transcriptome signatures during hypoxemic perinatal transition, which involve attenuation of ventricular patterning (AVP) and repression of epithelial mesenchymal transition (EMT).
Key regulatory circuits involved in hypoxemia response were identified including suppression of Wnt signaling, induction of cellular proliferation and dysregulation of TP53-network.
ACKNOWLEDGMENT
This work was supported by grants from the American Heart Association Career Development Award [18CDA34110414]; the Department of Defense-Congressionally Directed Medical Research Programs [W81XWH-18-1-0164]; the NIH/NHLBI [1R56HL146738-01], and the UCLA David Geffen School of Medicine Research Innovation Seed Grant to M. Touma.
We acknowledge the support from UCLA Clinical Genomics Center, Animal Physiology Core, and the Congenital Heart Defects-BioCore at UCLA.
NON-STANDARD ABBREVIATIONS
- CHD
Congenital Heart Defect
- RVOT
Right Ventricle Outflow Tract
- TOF
Tetralogy of Fallot
- EMT
Epithelial Mesenchymal Transition
- AVP
Attenuation of Ventricular Patterning
Footnotes
CONFLICT OF INTEREST:
None.
DISCLOUSURES
None.
SUPPLEMENTAL TABLES
Please refer to [Supplemental Tables (1–5)] provided in the SUPPLEMETAL DATA file and accompanied EXCEL sheets.
ETHICAL STANDARD.
All animal-related experimental protocols were approved by the UCLA Institutional Animal Care and Use Committee (IACUC). Therefore, all studies have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
REFERENCES
- 1.Zhao Y, Kang X, Gao F, Guzman A, Lau RP, Biniwale R, Wadehra M, Reemtsen B, Garg M, Halnon N, Quintero-Rivera F, Arsdell GV, Giovanni C, Nelson SF, Touma M & the UCLA Congenital Heart Defects BioCore Faculty. Gene-Environment Regulatory Circuits of Right Ventricular Pathology in Tetralogy of Fallot JMolMed Ms. No. JMME-D-19–00563R1. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Touma M, Kang X, Gao F, Zhao Y, Cass AA, Biniwale R, Xiao X, Eghbali M, Coppola G, Reemtsen B, Wang Y. Wnt11 regulates cardiac chamber development and disease during perinatal maturation. JCI Insight. 2017;2(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finnemore A, Groves A. Physiology of the fetal and transitional circulation. Semin Fetal Neonatal Med. 2015;20(4):210–216. [DOI] [PubMed] [Google Scholar]
- 4.Rudolph AM. Myocardial growth before and after birth: clinical implications. Acta Paediatr. 2000; 89(2):129–133. [DOI] [PubMed] [Google Scholar]
- 5.Sinha SK, Donn SM. Fetal-to-neonatal maladaptation. Semin Fetal Neonatal Med. 2006;11(3):166–173. [DOI] [PubMed] [Google Scholar]
- 6.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Touma M, Reemtsen B, Halnon N, Alejos J, Finn JP, Nelson SF, Wang Y. A Path to Implement Precision Child Health Cardiovascular Medicine. Front Cardiovasc Med. 2017;4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pattersin AJ, Zhang L. Hypoxia and fetal Heart Development. Curr Mol Med. 2010;10(7):653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohlicek CV, Matsuka T, Saiki C. Cardiovascular response to acute hypoxemia in adult rats hypoxemic neonatally. Cardiovasc Res. 2002;53(1):263–270. [DOI] [PubMed] [Google Scholar]
- 10.Kimura W, Sadek HA. The cardiac hypoxic niche: emerging role of hypoxic microenvironment in cardiac progenitors. Cardiovasc Diagn Ther. 2012;2(4):278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541(7636): 222–227. [DOI] [PubMed] [Google Scholar]
- 12.Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007;80(2):51–60. [PMC free article] [PubMed] [Google Scholar]
- 13.Dyson HJ, Wright PE. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J Biol Chem. 2016;291(13):6714–6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoon H, Lim JH, Cho CH, Huang LE, Park JW. CITED2 controls the hypoxic signaling by snatching p300 from the two distinct activation domains of HIF-1α. Biochim Biophys Acta. 2011;1813(12):2008–2016. [DOI] [PubMed] [Google Scholar]
- 15.Von Gise A, Pu WT. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ Res. 2012;110(12):1628–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan KD, Galbraith MD, Andrysik Z, Espinosa JM. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018;25(1):133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roder J, Linstid B, Oliveira C. Improving the power of gene set enrichment analyses. BMC Bioinformatics. 2019;20(1):257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peterson D, et al. A chemosensitization screen identifies TP53RK, a kinase that restrains apoptosis after mitotic stress. Cancer Res. 2010;70(15):6325–6335. [DOI] [PubMed] [Google Scholar]
- 19.Okamura S, et al. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell. 2001;8(1):85–94. [DOI] [PubMed] [Google Scholar]
- 20.Sancho A, et al. DOR/Tp53inp2 and Tp53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS One. 2012;7(3):e34034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer M, Steiner L, Engeland K. The transcription factor p53: not a repressor, solely an activator. Cell Cycle. 2014;13(19):3037–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011;2:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amelio I, Mancini M, Petrova V, Cairns RA, Vikhreva P, Nicolai S, Marini A, Antonov AA, Le Quesne J, Baena Acevedo JD, Dudek K, Sozzi G, Pastorino U, Knight RA, Mak TW, Melino G. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc Natl Acad Sci U S A. 2018;115(46):E10869–E10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139(5):871–890. [DOI] [PubMed] [Google Scholar]
- 25.Von Gise A, Zhou B, Honor LB, Ma Q, Petryk A, Pu WT. WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev Biol. 2011;356(2):421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai X, Zhang W, Hu J, Zhang L, Sultana N, Wu B, Cai W, Zhou B, Cai CL. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development. 2013;140(15):3176–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jesse S, Koenig A, Ellenrieder V, Menke A. Lef-1 isoforms regulate different target genes and reduce cellular adhesion. Int J Cancer. 2010;126(5):1109–1120. [DOI] [PubMed] [Google Scholar]
- 28.Blom JN, Feng Q. Cardiac repair by epicardial EMT: Current targets and a potential role for the primary cilium. Pharmacol Ther. 2018;186:114–129. [DOI] [PubMed] [Google Scholar]
- 29.Tsai YP, Wu KJ. Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci. 2012;19:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Rokavec M, Jiang L, Horst D, Hermeking H. Antagonistic Effects of p53 and HIF1A on microRNA-34a Regulation of PPP1R11 and STAT3 and Hypoxia-induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells. Gastroenterology. 2017;153(2):505–520. [DOI] [PubMed] [Google Scholar]
- 31.Touma M, Kang X, Zhao Y, Cass AA, Gao F, Biniwale R, Coppola G, Xiao X, Reemtsen B, Wang Y. Decoding the Long Noncoding RNA During Cardiac Maturation: A Roadmap for Functional Discovery. Circ Cardiovasc Genet. 2016;9(5):395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 (Attached Excel File. Sheet 1). Top 20 differential gene expression (DGE) in RV and LV of P3 neonatal mouse heart in response to hypoxia treatment.
Supplemental Table 2 (Attached Excel File. Sheet 2). 2A. Attenuation of Ventricular Patterning (AVP) Genes. Top 20 AVP genes revealed by pair-wise differential gene expression (DGE) analysis (RV vs LV) of P3 neonatal mouse heart in hypoxia vs normoxia conditions. 2B. Inversion of Ventricular Patterning (IVP) Genes. IVP gene subset revealed by pair-wise differential gene expression (DGE) analysis (RV vs LV) of P3 neonatal mouse heart in hypoxia vs normoxia conditions.
Supplemental Table 3 (Attached Excel File. Sheet 3). 3A. TP53 Network. Representative list of dysregulated genes of TP53 network that overlapped with the AVP genes. 3B. Representative TP53-target genes that exhibited concordant regulation in hypoxemic RVs of mouse and human.
Supplemental Table 4 (Attached Excel File. Sheet 4). List of primers used for quantitative RT-PCR.