

Abstract
Background
Considering the boosting effect of glycolysis on tumor chemoresistance, this investigation aimed at exploring whether miR‐488/PFKFB3 axis might reduce drug resistance of colorectal cancer (CRC) by affecting glycolysis, proliferation, migration, and invasion of CRC cells.
Method
Totally, 288 CRC patients were divided into metastasis/recurrence group (n = 107) and non‐metastasis/recurrence group (n = 181) according to their prognosis about 1 year after the chemotherapy, and their 3‐year overall survival was also tracked. Besides, miR‐488 expression was determined in peripheral blood of CRC patients and also in CRC cell lines (ie, W620, HT‐29, Lovo, and HCT116). The targeted relationship between miR‐488 and PFKFB3 was predicted by Targetscan software and confirmed by dual‐luciferase reporter gene assay. Moreover, glycolysis and drug tolerance of CRC cells lines were assessed.
Results
MiR‐488 expression was significantly decreased in metastatic/recurrent CRC patients than those without metastasis/recurrence (P < .05), and lowly expressed miR‐488 was suggestive of unfavorable 3‐year survival, large tumor size, poor differentiation, in‐depth infiltration, and advanced Duke stage of CRC patients (P < .05). Besides, CRC cell lines transfected by miR‐488 mimic demonstrated decreases in glucose uptake and lactate secretion, increases in oxaliplatin/5‐Fu‐sensistivity, as well as diminished capability of proliferating, invading, and migratory (P < .05), which were reversible by extra transfection of pcDNA3.1‐PFKFB3 (ie, miR‐488 mimic + pcDNA3.1‐PFKFB3 group). Finally, the mRNA level of PFKFB3 was down‐regulated by miR‐488 mimic in CRC cell lines after being targeted by it (P < .05).
Conclusion
The miR‐488/PFKFB3 axis might clinically refine chemotherapeutic efficacy of CRC, given its modifying glycolysis and metastasis of CRC cells.
Keywords: 6‐phosphofructokinase‐2/fructose diphosphate‐2 isoenzyme 3, chemoresistance, colorectal cancer, glycolysis, miRNA‐488
MiR‐488 targeted PFKFB3 and suppressed its mRNA level, thereby inhibiting 5‐Fu/oxaliplatin‐resistance, glycolysis and malignant activity of colorectal cancer (CRC) cells.
![]()
1. INTRODUCTION
Colorectal cancer (CRC), a common malignancy arising from the digestive system of mankind, exhibited an obviously rising tendency in both prevalence and morbidity. Major triggers of CRC‐induced deaths included invasive growth and distant metastasis of tumor cells, which made it nearly impossible to radically excise tumor tissues and ultimately endangered people's lives. 1 As a matter of fact, rapid metastasis of malignant cells entailed sufficient nutrition and energy, a portion of which came from glycolysis, glutamate‐dependent anabolism, and lipogenesis. 2 Consequently, digging etiologies inherent in neoplastic metabolism, such as glycolysis, might help to improve prognosis of CRC patients.
Back to 1920s, Otto Warburg et al 3 proposed that tumor cells exposed to hypoxia or even aerobic circumstances primarily depended on glycolysis to acquire energy, which was then acknowledged as Warburg effect. There were solid evidences that malignant tumor cells usually possessed a strong capability in glucose uptake, that was, activity of glycolysis‐relevant enzymes, including hexokinase 2 (HK2), phosphofructokinase‐1 (PFK‐1), pyruvate kinase muscle isozyme (PKM2), lactate dehydrogenase‐A (LDH‐A), and pyruvate dehydrogenase‐1 (PDK‐1), were also intensified in tumor cells. 4 , 5 , 6 , 7 Besides, proliferation and metastasis of neoplasms (eg, CRC) could be abated in case of glycolytic insufficiency, 8 and chemoresistance of tumor cells, such as breast cancer and gastric cancer, was reversible when their glycolysis was held back. 9 , 10 To sum up, discovery of biomarkers that controlled glycolysis of tumors, including CRC, might efficaciously suppressed CRC progression and drug tolerance, which was conducive to improving chemotherapeutic prognosis of CRC patients.
MiR‐488 was suggested as a CRC‐resistant biomarker, given its undermining proliferative and migratory inclination of CRC cells by curbing function of plant homeodomain finger protein 8 (PHF8). 11 In vivo experiments also expounded the depression exerted by miR‐488 on epithelial‐mesenchymal transition (EMT) and lymph node metastasis (LNM) in CRC. 12 Actually, besides CRC, miR‐488 expression also was dampened in a myriad of other malignancies, such as non‐small‐cell lung cancer and osteosarcoma, while its high level biologically urges apoptosis of the cancer cells, 13 , 14 highlighting the anti‐oncogenic role of miR‐488. Despite these, the implication of miR‐488 in drug sensitivity of CRC remained unclear.
Interestingly, PFKFB3, involved with glycolysis, 15 was speculated as a target molecule of miR‐488, according to targetscan (http://www.targetscan.org/mamm_31/). It seemed that no other PFKFB isozyme was more productive than PFKFB3 in stimulating synthesis of fructose 2,6‐diphosphate and in encouraging glycolysis. Perhaps, it was this glycolysis‐promoting feature that rendered PFKFB3 as a contributor to angiogenesis and excessive growth of tumor cells. 16 Not only that, PFKFB3 was implicated in CRC pathogenesis by regulating glycolysis, 17 however, whether miR‐488 was also involved in CRC glycolysis by targeting PFKFB3 was still vague.
From the above, there existed a possibility that miR‐488 modulated CRC glycolysis and drug sensitivity by targeting PFKFB3, which has not been explored. Therefore, this investigation attempted to uncover this possible linkage, which might aid to improve CRC prognosis after chemotherapy.
2. MATERIALS AND METHODS
2.1. Gathering of CRC tissues
From May 2013 to June 2016, totally 288 CRC patients were retrospectively recruited from gastrointestinal surgical department of the First Affiliated Hospital of Bengbu Medical College, and they were grouped in line with the criterion set by International Alliance for Cancer Staging. 18 The recruited CRC patients should (a) be pathologically diagnosed as CRC and (b) conform to surgery and post‐chemotherapy indications. However, they would be excluded if: (a) they underwent radio‐/chemotherapy or molecular targeting treatment prior to surgery; or (b) they were complicated by other colon lesions, such as familial polyposis and inflammatory bowel disease. Finally, this program was approved by the First Affiliated Hospital of Bengbu Medical College and its ethics committee, and the study subjects have signed informed consents before conduction of this project. Peripheral venous blood (volume: 5 mL) was collected from each CRC patient before their chemotherapy, and the blood samples were reserved for examination of miR‐488 expression with RT‐PCR.
2.2. Drug treatment and follow‐up of CRC patients
The recruited CRC patients all underwent colorectal surgery, and 2 weeks later their chemotherapy (ie, FOLFOX4 regimen) was started. To be specific, they were treated by: (a) intravenous drip with 100 mg/m2 oxaliplatin on day 1; (b) intravenous drip with 200 mg/m2 calcium folinate from day 2 to day 6; and (c) intravenous drip with 350 mg/m2 5‐Fu from day 2 to day 6. The cycle of treatment was 21 days, and all CRC patients consecutively received 6 cycles of treatment.
Moreover, the subjects were followed up for 3 years after their chemotherapy. In the 1st year, their recurrent/metastatic condition was reviewed in clinic by computer tomography (CT) scanning, B‐ultrasound scanner, X‐ray film, and histopathological examination when necessary. 19 Patients whose tumor recurred or metastasized were categorized into the recurrent/metastatic group. In addition, 3‐year overall survival (OS) of CRC patients, defined as the time range from date of definite diagnosis to the date of cancer‐induced death or last contact, was also tracked by telephone or by visit.
2.3. Cell culture and transfection
The highly differentiated CRC cell line (ie, SW620), moderately differentiated CRC cell line (ie, HT‐29), and mildly differentiated CRC cell lines (ie, Lovo and HCT116) were all purchased from cell bank of Chinese Academy of Sciences (Shanghai, China), and the human‐origin normal intestinal epithelial cell line (ie, NCM‐460) was supplied by Jennio Biotech. The cell lines were cultivated in 5% CO2 at the constant temperature of 37°C, and their culture medium was designated as RPIM 1640 medium which was added by 10% fetal bovine serum (FBS) (Gibco). When the confluence of CRC cells rose to 80%, miR‐488 mimic (5′‐GGGTCTATTACCGTGAGAGTT‐3′) and pcDNA3.1‐PFKFB3 (Genepharma) were separately transfected into the cells as directed by Lipofectamine 3000 kit (Invitrogen). Cells transfected for 48 hours were separately prepared to extract mRNAs.
2.4. Real‐time PCR
Total RNAs were separated from blood samples and CRC cell lines with Trizol (Invitrogen), and they were then quantified on a spectrophotometer (model: SmartSpec Plus; Bio‐Rad). For determining the mRNA level of PFKFB3, precisely 100 ng total RNA was collected for reverse transcription, as per specifications of 1st Strand cDNA synthesis kit (model: Primerscript™; Takara). Then with sense (5′‐GACGCACCCTTCCTGTCCCTTTG‐3′) and anti‐sense primers (5′‐ACAAAGCCGCTGCACACACAA‐3′), cDNAs of PFKFB3 were amplified on the strength of GoTaq® Greeen Master Mix (Promega). When it came to miR‐488, the obtained miRNAs were reversely transcribed into cDNAs by feat of TaqMan®MicroRNA Reverse Transcription kit (ThermoFisher). The cDNAs were prepared to conduct real‐time PCR according to the instruction of TaqMan Universal Master Mix (ThermoFisher), assisted by its sense (5′‐GATGCTACCCAGATAATGGCACT‐3′) and anti‐sense primers (5′‐CAGTGCGTGTCGTGGAGT‐3′). Finally, miR‐488 level and mRNA level of PFKFB3 were drawn on the basis of 2−ΔΔ C t method. 20 U6 (sense: 5′‐TGACACGCAAATTCGTGAAGCGTTC‐3′, anti‐sense: 5′‐CCAGTCTCAGGGTCCGAGGTATTC‐3′) was set as the internal reference for miR‐488, whereas β‐actin (sense: 5′‐TGACGTGGACATCCGCAAAG‐3′, anti‐sense: 5′‐CTGGAAGGTGGACAGCGAGG‐3′) was arranged to standardize the mRNA level of PFKFB3.
2.5. Dual‐luciferase reporter gene assay
The 3′‐end untranslated region (UTR) of PFKFB3, which incorporated binding sites of miR‐488, was amplified on the strength of real‐time PCR as previously mentioned. In this manner, the reporter vectors called as Luc‐PFKFB3‐WT were constructed. Meanwhile, reporter vectors named as Luc‐PFKFB3‐MUT were created analogously, except that the vectors demanded mutating sites in PFKFB3 that bound to miR‐488. The pGL3‐PFKFB3‐WT and pGL3‐PFKFB3‐MUT were then transfected with miR‐488 mimic or miR‐NC into CRC cell lines for 72 hours, and Dual‐Luciferase Reporter Assay System (model: E1960; Promega) was then performed to determine the luciferase activity of the cell lines.
2.6. MTT assay for evaluating chemoresistance of CRC cells
Colorectal cancer cells, into 96‐well plates at the density of 2 × 103 per well, were treated by different concentrations of oxaliplatin and 5‐Fu. Forty‐eight hours later, each well was mixed by 10 μL MTT solution (Beyotime Biotechnology), whose concentration was 5 mg/mL. After 4‐hour treatment, absorbance value (A) of each well was determined at the wavelength of 490 nm. Inhibition rate (%) of the drugs on CRC cells was calculated, and corresponding IC50 values were drawn using an online tool (https://www.aatbio.com/tools/ic50-calculator).
2.7. Examination of sugar uptake and lactate secretion of CRC cells
Cell culture medium, separated from cells (concentration: 3 × 104 per well) via centrifugation, were managed by glucose detection kit (Biovision) and lactic acid detection kit (Biovision), respectively. The contents of glucose and lactic acid in CRC cells were monitored by colorimetric means.
2.8. CCK8 technique for measuring proliferation of CRC cells
The CCK8 reagent (Keygentec) was prepared to appraise the proliferative capacity of CRC cells, respectively, on the 1st, 2nd, and 3rd days. Two hours after supplementation of CCK‐8 reagent into CRC cells, the absorbance value (A) of each sample was detected at the wavelength of 450 nm, backed by enzyme‐linked immunosorbent assay (ELISA).
2.9. Transwell invasion and migration assay
The CRC cells cultivated in serum‐free RPMI 1640 medium for 24 hours were firstly digested and were then incubated into each upper Transwell chamber at a density of 5 × 104 per well. Additionally, 200 μL serum‐free RPMI 1640 medium that included 0.2% bovine serum albumin (BSA) was supplemented into the upper chamber, while 500 μL RPMI 1640 medium with 10% serum was added into the lower chamber. After 24 hours of incubation, cells left behind in the upper chamber were eliminated with a cotton bud, and the remaining cells were fixated by 4% methanol for 10 minutes and then dyed by 0.1% crystal violet. A total of eight views were randomly selected, and their cell number was counted to obtain the average value. With regard to migration assay, there was no need for Matrigel, and the incubation time was 16 hours.
2.10. Statistical analyses
The SPSS ver15.0 software was employed to analyze information derived from this investigation. The count data, expressed as number (N) or percentage (%), were compared by χ2 test, and the measurement data (mean ± standard deviation) were analyzed by student's test. Then survival curves were estimated adhering to Kaplan‐Meier method, and distinctions between overall survival (OS) of patients were assessed via log‐rank test or uni‐/multi‐variate Cox‐regression analyses. Ultimately, differences were deemed as statistically significant when P value was smaller than .05.
3. RESULTS
3.1. Clinical association of miR‐488 with post‐chemotherapeutic prognosis of CRC patients
All the CRC patients were effectively followed up, 107 (37.15%) of them developing tumor recurrence/metastasis (Table 1). Patients that assumed CRC metastasis or recurrence were associated with larger tumor size (>5 cm), poorer differentiation, deeper infiltration, and more advanced Duke stage than patients without CRC recurrence/metastasis (P < .05). Moreover, miR‐488 level was largely increased in recurrence/metastasis group as compared with non‐recurrence/metastasis group (P < .05) (Figure 1A). Beyond that, highly expressed miR‐488 was associated with more favorable 3‐year survival after chemotherapy than lowly expressed miR‐488 among the recruited CRC patients (P < .05) (Figure 1B).
Table 1.
Comparison of clinical features among colorectal cancer (CRC) patients with and without metastasis/recurrence
| Clinical features | Case number (N) | CRC patients (N = 288) | X2 | P value | OR | 95% CI | |
|---|---|---|---|---|---|---|---|
| Metastasis/recurrence (N = 107) | Non‐metastasis/recurrence (N = 181) | ||||||
| Gender (n) | |||||||
| Male | 143 | 50 (34.97%) | 93 (65.03%) | 0.5822 | .445 | 1.205 | 0.746‐1.945 |
| Female | 145 | 57 (39.31%) | 88 (60.69%) | ||||
| Age (years old) | |||||||
| <60 | 177 | 72 (40.68%) | 105 (59.32%) | 2.444 | .118 | 0.672 | 0.407‐1.108 |
| ≥60 | 111 | 35 (31.53%) | 76 (68.47%) | ||||
| Timor size (cm) | |||||||
| ≤5 | 147 | 41 (27.89%) | 106 (72.11%) | 11.03 | <.001* | 2.275 | 1.395‐3.711 |
| >5 | 141 | 66 (46.81%) | 75 (53.19%) | ||||
| Differentiation degree (n) | |||||||
| Moderate + high | 186 | 60 (32.26%) | 126 (67.74%) | 5.389 | .0203* | 1.795 | 1.093‐2.947 |
| Poor | 102 | 47 (46.08%) | 55 (53.92%) | ||||
| Infiltration into serosa (n) | |||||||
| No | 84 | 23 (27.38%) | 61 (72.62%) | 9.051 | .0026* | 2.345 | 1.336‐4.115 |
| Yes | 179 | 84 (46.93%) | 95 (53.07%) | ||||
| Dukes stage (n) | |||||||
| A + B | 107 | 21 (19.63%) | 86 (80.37%) | 22.4 | <.001* | 3.707 | 2.119‐6.486 |
| C + D | 181 | 86 (47.51%) | 95 (52.49%) | ||||
Abbreviations: CI, confidence interval; CRC, colorectal cancer; OR, odd ratio.
Statistical significance when P value was smaller than .05.
Figure 1.
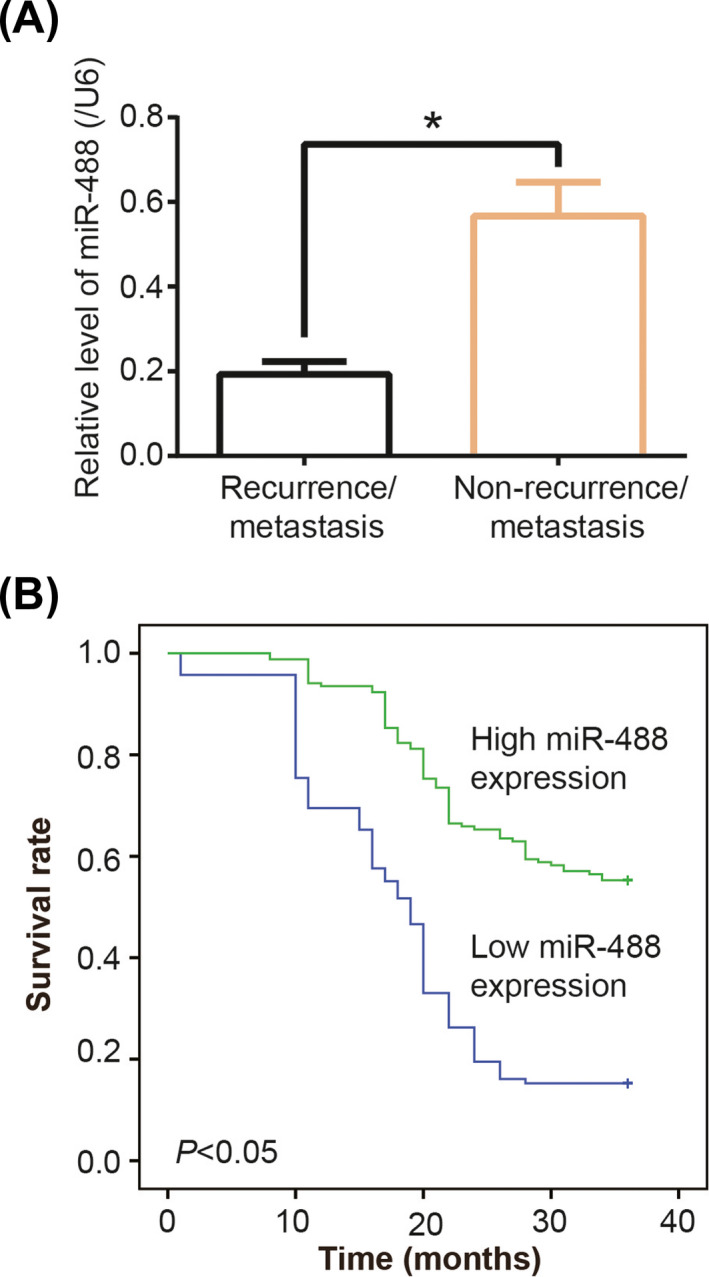
Low miR‐488 expression suggested poor prognosis of colorectal cancer (CRC) patients after chemotherapy. A, MiR‐488 expression was compared between CRC patients with recurrence/metastasis and patients without recurrence/metastasis. *: P < .05. B, High (>mean) miR‐488 expression was associated with more favorable 3‐y survival of CRC patients than lowly expressed miR‐488
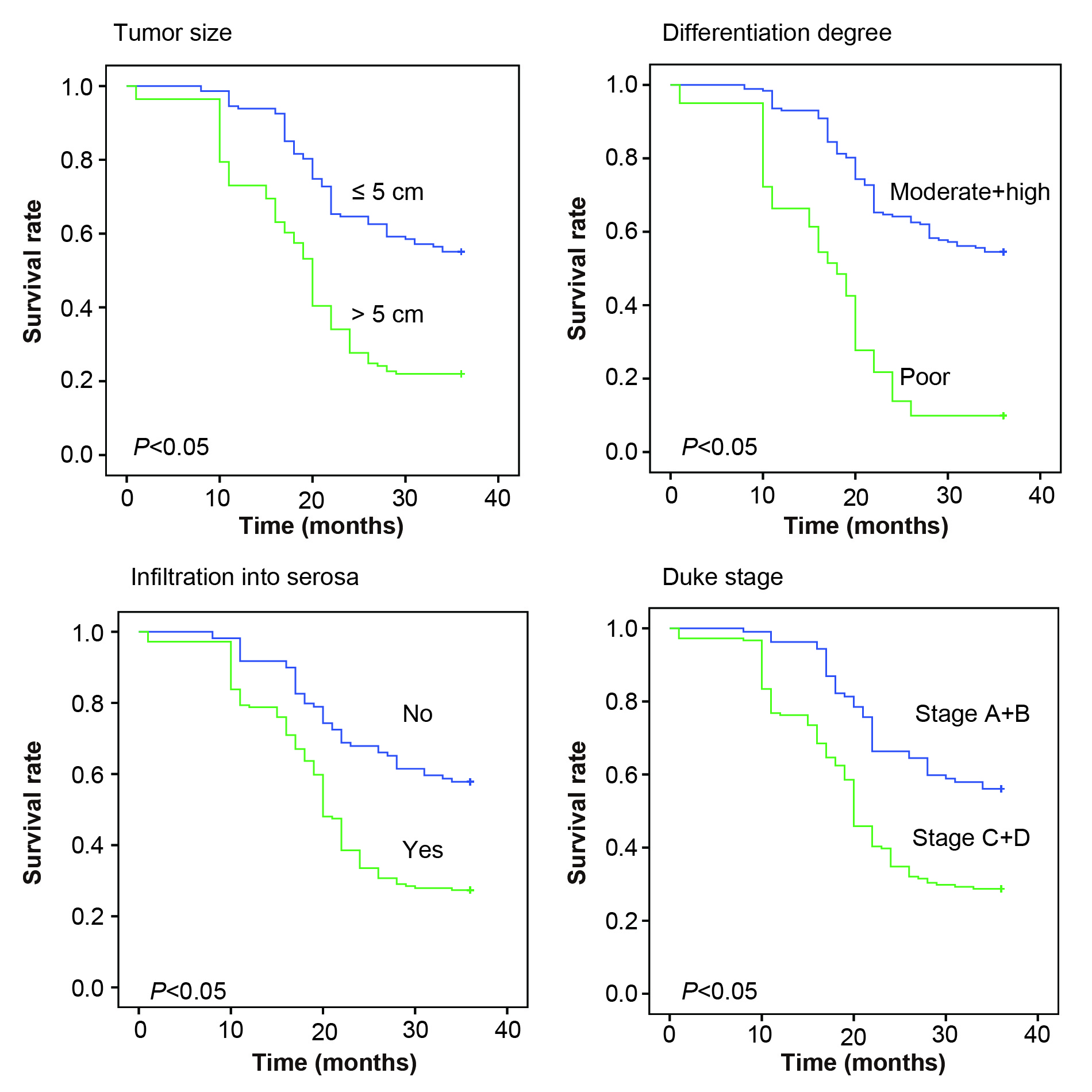
In addition, miR‐488 expression was significantly raised in CRC patients featured by large tumor size (>5 cm), poor differentiation, deep infiltration, and advanced Duke stage (all P < .05) (Figure 2A), which were pronounced indicators of poor 3‐year survival of CRC patients (all P < .05) (Figure S1). Besides, ROC curves also indicated that miR‐488 expression was able to discriminate CRC patients with large tumor size (>5 cm), poor differentiation, infiltration into serosa, and advanced Duke stage (A + D) from those with small tumor size (≤5 cm), high + moderate differentiation, non‐infiltration, and early Duke stage (A + B) (all P < .05) (Figure 2B).
Figure 2.
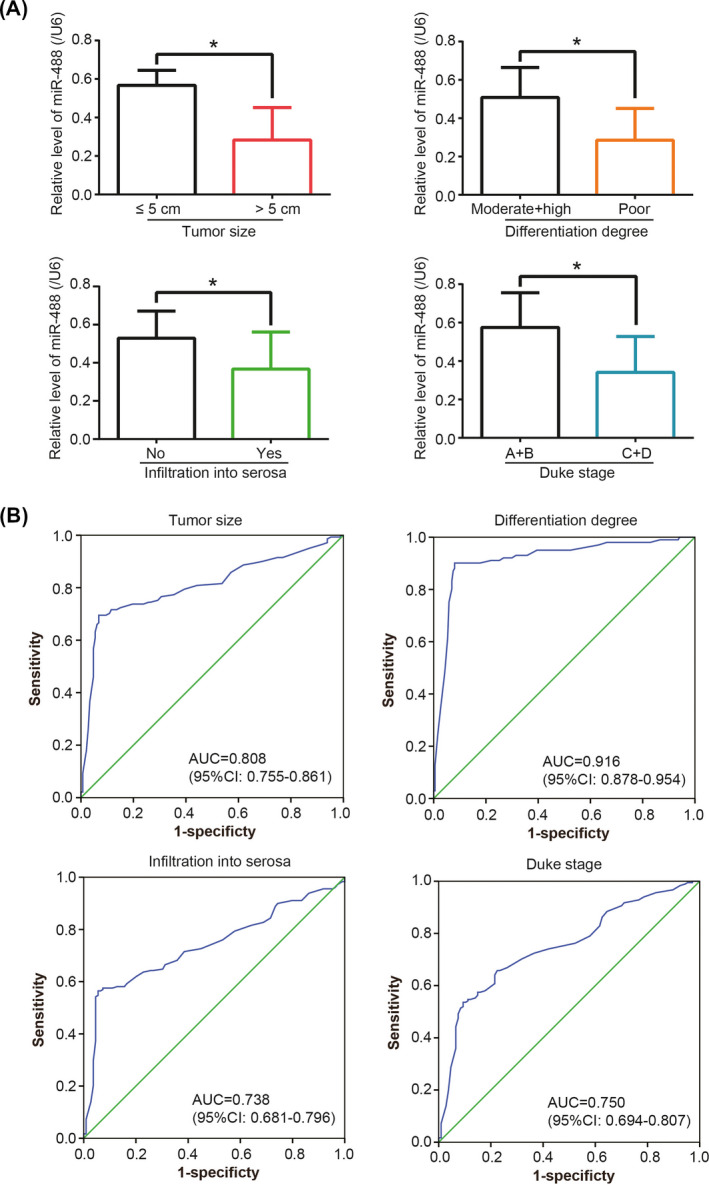
MiR‐488 expression was associated with malignant symptoms of colorectal cancer (CRC) patients. A, MiR‐488 expression was measured among CRC patients of different tumor sizes, differentiation degrees, infiltration depths, and Duke stages. *: P < .05. B, Receiver operating characteristic (ROC) curves were devised to evaluate the value of miR‐488 in discriminating CRC patients with different tumor sizes, differentiation degrees, infiltration depths, and Duke stages
3.2. MiR‐488 targeted PFKFB3 and lessened its mRNA level
Interestingly, both miR‐488 level and mRNA level of PFKFB3 were aberrantly displayed within CRC tissues (Figure 3A), as proposed by the microarray results summarized in the ENCORI website (http://starbase.sysu.edu.cn/). And the targetscan website (https://www.targetscan.org/) also suggested that there were targeting sites between miR‐488 and PFKFB3 (Figure 3B), a crucial participator in glycolysis of CRC. 17 There, hence, might be some hidden connection between miR‐488 and PFKFB3 underlying CRC pathogenesis.
Figure 3.
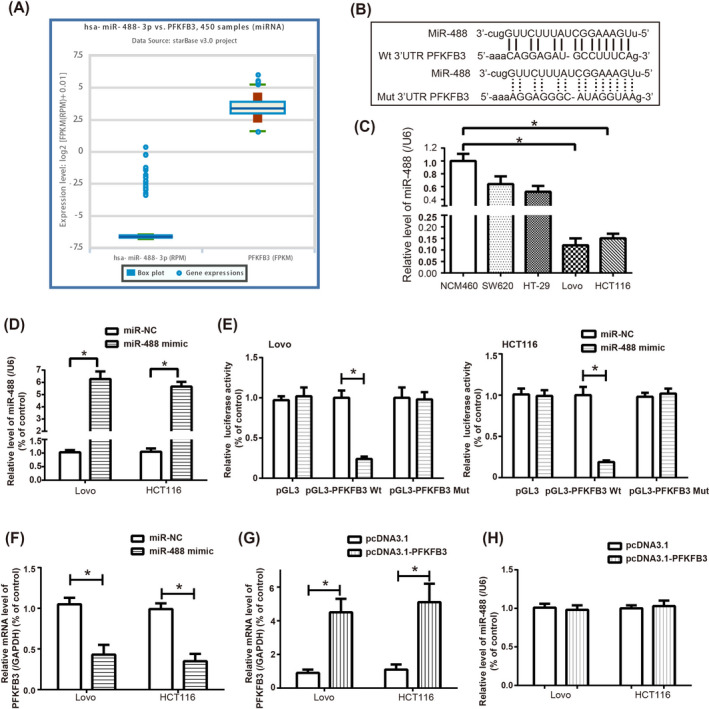
MiR‐488 targeted PFKFB3 in colorectal cancer (CRC) cell lines. A, The miR‐488 level and mRNA level of PFKFB3 were simultaneously abnormally expressed among 450 CRC samples, according to ENCORI software. B, The binding sites between PFKFB3 and miR‐488 were predicted by targetscan website. C, MiR‐488 expression was determined among NCM460, SW260, HT‐29, Lovo, and HCT116 cell lines. *: P < .05 when compared with NCM460 cell line. D, MiR‐488 level was heightened in CRC cell lines after transfection of miR‐488 mimic. *: P < .05 when compared with miR‐NC group. E, The luciferase activity of HCT116 and Lovo cells was weakened when PFKFB3 Wt and miR‐488 mimic were co‐transfected. *: P < .05 when compared with PFKFB3 Wt + miR‐NC group. F, The mRNA level of PFKFB3 was reduced by miR‐488 mimic. *: P < .05 when compared with miR‐NC group. G, The mRNA level of PFKFB3 was up‐regulated by transfection of pcDNA3.1‐PFKFB3. *: P < .05 when compared with pcDNA3.1 group. H, MiR‐488 level was determined when pcDNA3.1‐PFKFB3 was transfected. *: P < .05 when compared with pcDNA3.1 group
It was demonstrated that miR‐488 expression was higher in CRC cell lines (ie, SW260, HT‐29, Lovo and HCT‐116) than in NCM460 cell line (P < .05) (Figure 3C). And miR‐488 expression was dramatically raised within both HCT116 and Lovo cell lines, after transfection of miR‐488 mimic (P < .05) (Figure 3D). Furthermore, co‐transfection of miR‐488 mimic and pGL3‐PFKFB3 plasmid brought about weakening of luciferase activity in HCT116 and Lovo cells, as compared with miR‐NC + pcDNA‐PFKFB3 group and miR‐488 mimic + pcDNA group (P < .05) (Figure 3E). And the mRNA level of PFKFB3 was obviously lowered by miR‐488 mimic (P < .05) (Figure 3F). Besides, the mRNA level of PFKFB3 went up greatly under the force of pcDNA3.1‐PFKFB3 (P < .05) (Figure 3G), which, however, failed to generate any alteration of miR‐488 level in both HCT116 and Lovo cell lines (P > .05) (Figure 3H).
3.3. PFKFB3 blocked the impact of miR‐488 on chemoresistance and glycolysis of CRC cells
Oxaliplatin/5‐Fu‐resistance of HCT116 and Lovo cell lines was significantly relieved in the miR‐488 mimic in comparison with NC group (P < .05) (Figure 4A,B). Yet concurrent transfection of pcDNA3.1‐PFKFB3 and miR‐488 mimic remarkably enhanced oxaliplatin/5‐Fu‐resistance of CRC cells as relative to miR‐488 mimic group (P < .05). What is more, HCT116 and Lovo cell lines transfected by miR‐488 mimic were less capable of absorbing glucose and releasing lactate than those of NC group (P < .05) (Figure 4C,D). However, under co‐treatment of miR‐488 mimic and pcDNA3.1‐PFKFB3, the HCT116 and Lovo cells became more competent in assimilating glucose (Figure 4C) and releasing lactate (Figure 4D), as compared with miR‐488 mimic group (P < .05).
Figure 4.
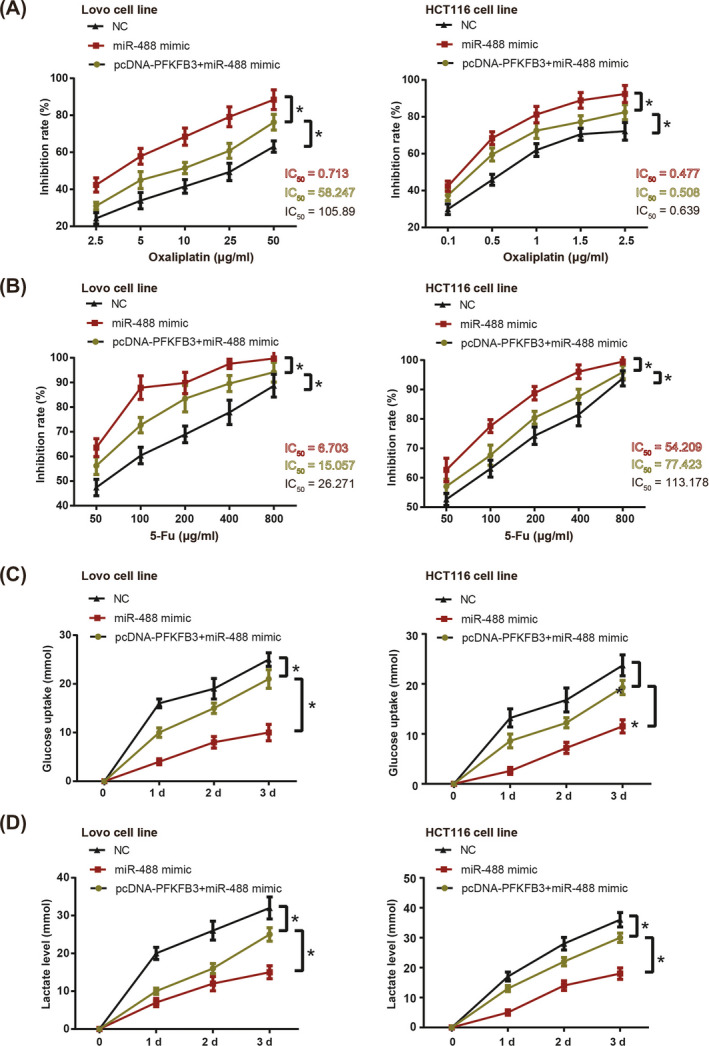
Oxaliplatin‐resistance (A), 5‐Fu‐tolerance (B), glucose uptake (C), and lactate production (D) of HCT116 and Lovo cell lines were compared among NC group, miR‐488 mimic group and miR‐488 + pcDNA3.1‐PFKFB3 group. *: P < .05
3.4. PFKFB3 interfered with the impact of MiR‐488 on proliferation, migration, invasion of CRC cells
The multiplicative capacity of HCT116 and Lovo cell lines was attenuated when miR‐488 mimic was transfected (P < .05) (Figure 5A). Besides that, HCT116 and Lovo cell lines were less capable of migrating (Figure 5B) and invading (Figure 5C) under treatment of miR‐488 mimic, with NC group as the reference (P < .05). Nevertheless, simultaneous treatment of pcDNA3.1‐PFKFB3 and miR‐488 mimic pronouncedly strengthened proliferation of HCT116 and Lovo cells, when compared with miR‐488 mimic group (P < .05) (Figure 5A). In addition, HCT116 and Lovo cell lines transfected by both miR‐488 mimic and pcDNA‐PFKFB3 displayed stronger ability to migrate than cells of the miR‐488 mimic group (P < .05) (Figure 5B). Not only that, HCT116 and Lovo cells of pcDNA3.1‐PFKFB + miR‐488 mimic group penetrated through the transwell membrane more aggressively than simply transfection of miR‐488 mimic (P < .05) (Figure 5C).
Figure 5.
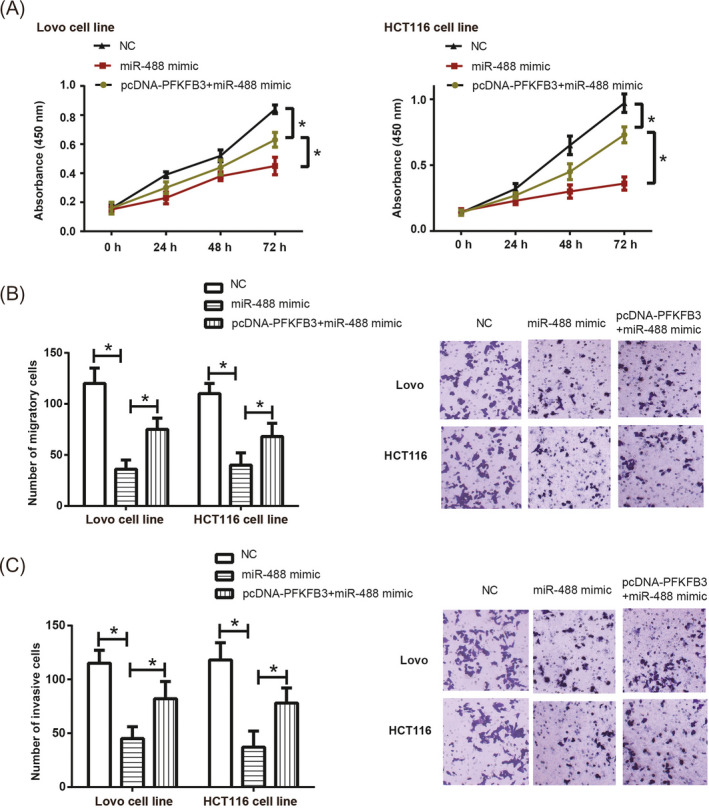
Proliferation (A), migration (B), and invasion (C) of HCT116 and Lovo cell lines were appraised among NC group, miR‐488 mimic group and miR‐488 + pcDNA3.1‐PFKFB3 group. *: P < .05
4. DISCUSSION
Tumor microenvironment, which was tightly connected with energetic metabolism, was principal to onset and progression of malignancies. The transformation from normal cells to tumor cells was often accompanied by the reshaping of metabolic pathways. 21 Whether oxygen supply was adequate or not, CRC cells preferred glycolysis to oxidative phosphorylation (CXPHOS) for energy. 22 Hence, valid suppression of glycolysis in CRC could be among one of effective CRC treatments.
MiR‐488, situated in 1q25.2 and comprised of 1 exon, was weakly expressed in prostate cancer and gastrointestinal stromal tumors, besides serving a protective role within cartilage tissue of osteoarthritis patients. 23 , 24 , 25 In addition to the clinical part, strongly expressed miR‐488 also impaired proliferative and invasive capability of gastric cancer cells. 26 With regard to CRC, miR‐488 was implied to retard progression of CRC, 12 and here we disclosed that lowly expressed miR‐488 was predictive of poor CRC prognosis after chemotherapy (Table 1, Figures 1 and 2), which was a bright spot of this investigation. Cellular experiments also corroborated the inhibitory effect of miR‐488 on CRC drug tolerance (Figure 4A), and we supposed that this might be relative to the negative role of miR‐488 in modulating glycolysis of CRC cells (Figure 4B), which was another novelty of this study. Apart from that, miR‐488 could also interrupt migration, invasion, and proliferation of CRC cells (Figure 5), which agreed with former studies, 11 and this might also explain the promoting effect of miR‐488 on CRC chemosensitivity. Altogether, miR‐488 was a potent driver of CRC chemosensitivity for its participation in diverse cell activities.
In addition, a broadly acknowledged hypothesis deemed that miRNAs modulated disease pathogenesis through silencing genes or combining with 3’‐UTR of mRNAs, so that functions of pathogenic genes were altered. This rule was also applicable for miR‐488, for instance, miR‐488 was documented to attenuate migration of prostate cancer cells by restraining androgen receptor expression. 27 And gastric cancer cells became disabled in proliferating and invading when miR‐488 bound to 3′‐untranslated region (UTR) of paired box 6 (PAX6). 26 Here, PFKFB3 was insinuated as the downstream molecule of miR‐488 (Figure 3B), and its mRNA level was negatively modulated by miR‐488 (Figure 3). What is more, it was intriguing to discover that elevating PFKFB3 mRNA level could reverse the inhibitory effect of miR‐488 on glycolysis, chemoresistance, proliferation, migration, and invasion of CRC cell lines (Figures 4 and 5), suggesting that miR‐488/PFKFB3 axis might be a critical involver in CRC etiology.
The PFKFB3 was a key enzyme responsible for controlling glycolysis, and it catalyzed transformation of fructose 6‐phosphate into fructose 2,6‐diphosphate (F2,6BP) and activated phosphofructokinase‐1 (PFK‐1) to postpone intracellular glycolysis. 28 Moreover, the kinase activity of PFKFB3 was nearly 700 folds stronger than biphosphatase, which dramatically accelerated glycolysis rate. 16 The interconnection of PFKFB3 and neoplastic progression was also intertwined. To be specific, PFKFB3 expression was heightened in a plethora of neoplasms, and the phosphorylation level of PFKFB3 presented an obvious elevation during both glycolysis and tumor progression. 29 Correspondingly, applying pharmacological inhibitor for PFKFB3, such as 3‐(3‐pyridinyl)‐1‐(4‐pyridinyl)‐2‐propen‐1‐one (3‐PO) and 7,8‐dihydroxy‐3‐(4‐hydroxyphenyl)‐chromen‐4‐one (YN1), could hinder glycolysis and suppress tumor growth. 30 , 31 Furthermore, repressing PFKFB3 expression seemed as a feasible approach to overcome drug tolerance in cancers, such as breast cancer and hepatocellular carcinoma. 32 , 33 This evidences could further state the tenability of applying miR‐488 for restraining CRC chemoresistance and progression, allowing for miR‐488‐induced changes of PFKFB3 mRNA level in CRC cells (Figure 3).
All in all, miR‐488/PFKFB3 axis might be clinically applicable for improving chemotherapeutic efficacy of CRC patients, owing to its controlling glycolysis, proliferation, migration, and invasion of CRC cells. Nonetheless, a few points in experimental design demanded improvement. Firstly, animal models were not established to simulate the in vivo effect of miR‐488/PFKFB3 axis on glycolysis. Secondly, molecular explanations for miR‐488 and glycolysis in CRC entailed more evidences, owing to that other molecules, more than PFKFB3, also functioned to regulate glycolytic rate. Finally, other glycolysis pathways were not explore here, such as hexokinase, zymohexase, and Sonic hedgehog. 34 , 35 These all entailed thorough explorations in the long run.
AUTHOR CONTRIBUTIONS
We declare that we have no conflict of interest, and all authors approved the final version of manuscript.
Supporting information
Figure S1
{kind=link}
ACKNOWLEDGMENT
This work was supported by Anhui Provincial Natural Science Foundation (No: 1808085MH240) and the Key Project of Natural Science Research of Universities of Anhui Province (No: KJ2019A0336, No: KJ2015A177).
Deng X, Li D, Ke X, et al. Mir‐488 alleviates chemoresistance and glycolysis of colorectal cancer by targeting PFKFB3. J. Clin. Lab. Anal. 2021;35:e23578 10.1002/jcla.23578
REFERENCES
- 1. Dekker E, Tanis PJ, Vleugels JLA, et al. Colorectal cancer. Lancet. 2019;394:1467‐1480. [DOI] [PubMed] [Google Scholar]
- 2. Branco C, Johnson RS. To PFKFB3 or not to PFKFB3, that is the question. Cancer Cell. 2016;30:831. [DOI] [PubMed] [Google Scholar]
- 3. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi X, You L, Luo RY. Glycolytic reprogramming in cancer cells: PKM2 dimer predominance induced by pulsatile PFK‐1 activity. Phys Biol. 2019;16:e066007. [DOI] [PubMed] [Google Scholar]
- 5. Garcia SN, Guedes RC, Marques MM. Unlocking the potential of HK2 in cancer metabolism and therapeutics. Curr Med Chem. 2019;26:7285‐7322. [DOI] [PubMed] [Google Scholar]
- 6. Sradhanjali S, Reddy MM. Inhibition of pyruvate dehydrogenase kinase as a therapeutic strategy against cancer. Curr Top Med Chem. 2018;18:444‐453. [DOI] [PubMed] [Google Scholar]
- 7. Ding J, Karp JE, Emadi A. Elevated lactate dehydrogenase (LDH) can be a marker of immune suppression in cancer: interplay between hematologic and solid neoplastic clones and their microenvironments. Cancer Biomark. 2017;19:353‐363. [DOI] [PubMed] [Google Scholar]
- 8. Wang G, Yu Y, Wang YZ, et al. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J Cell Physiol. 2019;234:17023‐17049. [DOI] [PubMed] [Google Scholar]
- 9. Chen F, Zhuang M, Zhong C, et al. Baicalein reverses hypoxia‐induced 5‐FU resistance in gastric cancer AGS cells through suppression of glycolysis and the PTEN/Akt/HIF‐1alpha signaling pathway. Oncol Rep. 2015;33:457‐463. [DOI] [PubMed] [Google Scholar]
- 10. Ruprecht B, Zaal EA, Zecha J, et al. Lapatinib resistance in breast cancer cells is accompanied by phosphorylation‐mediated reprogramming of glycolysis. Cancer Res. 2017;77:1842‐1853. [DOI] [PubMed] [Google Scholar]
- 11. Lv Y, Shi Y, Han Q, Dai G. Histone demethylase PHF8 accelerates the progression of colorectal cancer and can be regulated by miR‐488 in vitro. Mol Med Rep. 2017;16:4437‐4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang YB, Shi Q, Li G, et al. MicroRNA‐488 inhibits progression of colorectal cancer via inhibition of the mitogen‐activated protein kinase pathway by targeting claudin‐2. Am J Physiol Cell Physiol. 2019;316:C33‐C47. [DOI] [PubMed] [Google Scholar]
- 13. Fang C, Chen YX, Wu NY, et al. MiR‐488 inhibits proliferation and cisplatin sensibility in non‐small‐cell lung cancer (NSCLC) cells by activating the eIF3a‐mediated NER signaling pathway. Sci Rep. 2017;7:e40384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou C, Tan W, Lv H, et al. Hypoxia‐inducible microRNA‐488 regulates apoptosis by targeting Bim in osteosarcoma. Cell Oncol (Dordr). 2016;39:463‐471. [DOI] [PubMed] [Google Scholar]
- 15. Li FL, Liu JP, Bao RX, et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin‐induced apoptosis. Nat Commun. 2018;9:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi L, Pan H, Liu Z, et al. Roles of PFKFB3 in cancer. Signal Transduct Target Ther. 2017;2:17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Han J, Meng Q, Xi Q, et al. Interleukin‐6 stimulates aerobic glycolysis by regulating PFKFB3 at early stage of colorectal cancer. Int J Oncol. 2016;48:215‐224. [DOI] [PubMed] [Google Scholar]
- 18. Fielding LP, Arsenault PA, Chapuis PH, et al. Clinicopathological staging for colorectal cancer: an International Documentation System (IDS) and an International Comprehensive Anatomical Terminology (ICAT). J Gastroenterol Hepatol. 1991;6:325‐344. [DOI] [PubMed] [Google Scholar]
- 19. Sha D, Lee AM, Shi Q, et al. Association study of the let‐7 miRNA‐complementary site variant in the 3' untranslated region of the KRAS gene in stage III colon cancer (NCCTG N0147 Clinical Trial). Clin Cancer Res. 2014;20:3319‐3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods. 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 21. Martinez‐Outschoorn UE, Peiris‐Pages M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11‐31. [DOI] [PubMed] [Google Scholar]
- 22. Smith B, Schafer XL, Ambeskovic A, et al. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016;17:821‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J, Li X, Xiao Z, et al. MicroRNA‐488 inhibits proliferation and glycolysis in human prostate cancer cells by regulating PFKFB3. FEBS Open Bio. 2019;9:1798‐1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li Y, Li S, Luo Y, et al. LncRNA PVT1 regulates chondrocyte apoptosis in osteoarthritis by acting as a sponge for miR‐488‐3p. DNA Cell Biol. 2017;36:571‐580. [DOI] [PubMed] [Google Scholar]
- 25. Tong HX, Zhou YH, Hou YY, et al. Expression profile of microRNAs in gastrointestinal stromal tumors revealed by high throughput quantitative RT‐PCR microarray. World J Gastroenterol. 2015;21:5843‐5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao Y, Lu G, Ke X, et al. miR‐488 acts as a tumor suppressor gene in gastric cancer. Tumour Biol. 2016;37:8691‐8698. [DOI] [PubMed] [Google Scholar]
- 27. Sikand K, Slaibi JE, Singh R, et al. miR 488* inhibits androgen receptor expression in prostate carcinoma cells. Int J Cancer. 2011;129:810‐819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li X, Liu J, Qian L, et al. Expression of PFKFB3 and Ki67 in lung adenocarcinomas and targeting PFKFB3 as a therapeutic strategy. Mol Cell Biochem. 2018;445:123‐134. [DOI] [PubMed] [Google Scholar]
- 29. Minchenko OH, Tsuchihara K, Minchenko DO, et al. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. 2014;20:13705‐13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schoors S, De Bock K, Cantelmo AR, et al. Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 2014;19:37‐48. [DOI] [PubMed] [Google Scholar]
- 31. Seo M, Kim JD, Neau D, et al. Structure‐based development of small molecule PFKFB3 inhibitors: a framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS One. 2011;6:e24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ge X, Cao Z, Gu Y, et al. PFKFB3 potentially contributes to paclitaxel resistance in breast cancer cells through TLR4 activation by stimulating lactate production. Cell Mol Biol (Noisy‐le‐grand). 2016;62:119‐125. [PubMed] [Google Scholar]
- 33. Li S, Dai W, Mo W, et al. By inhibiting PFKFB3, aspirin overcomes sorafenib resistance in hepatocellular carcinoma. Int J Cancer. 2017;141:2571‐2584. [DOI] [PubMed] [Google Scholar]
- 34. Xintaropoulou C, Ward C, Wise A, et al. A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget. 2015;6:25677‐25695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ge X, Lyu P, Gu Y, et al. Sonic hedgehog stimulates glycolysis and proliferation of breast cancer cells: Modulation of PFKFB3 activation. Biochem Biophys Res Commun. 2015;464:862‐868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1