

Abstract
Background
Treacher Collins syndrome (TCS) is a rare autosomal dominant or recessive disorder, that involves unique bilateral craniofacial malformations. The phenotypes of TCS are extremely diverse. Interventional surgery can improve hearing loss and facial deformity in TCS patients.
Method
We recruited seven TCS families. Variant screening in probands was performed by targeted next‐generation sequencing (NGS). The variants identified were confirmed by Sanger sequencing. The pathogenicity of all the mutations was evaluated using the guidelines of the American College of Medical Genetics and Genomics (ACMG) and InterVar software.
Results
Three frameshift variants, two nonsense variants, one missense variant, and one splicing variant of TCOF1 were identified in the seven TCS probands. Five variants including c.1393C > T, c.4111 + 5G>C, c.1142delC, c.2285_2286delCT, and c.1719delG had not been previously reported. Furthermore, we report the c.149A > G variant for the first time in a Chinese TCS patient. We provided prenatal diagnosis for family 4. Proband 7 chose interventional surgery.
Conclusion
We identified five novel variants in TCOF1 in Chinese patients with TCS, which expands the mutation spectrum of TCOF1 in TCS. Bone conduction hearing rehabilitation can improve hearing for TCS patients and prenatal diagnosis can provide fertility guidance for TCS families.
Keywords: next‐generation sequencing, Sanger sequencing, TCOF1, Treacher Collins syndrome
Figure 1. The facial features of these patients are characteristic of TCS. Figure 2. Sanger sequencing of TCOF1: P is proband, F is father, M is mother, AF is amniotic fluid. None of the parents of these seven families detected the same variation as the proband had. The Sanger sequencing of fetus for family 4 was normal

1. INTRODUCTION
Treacher Collins syndrome (TCS) is a disorder of craniofacial development characterized by hypoplasia of the facial bones, coloboma of the lid, downward slanting palpebral fissures, prominent nose, cleft palate, malformation of the external ears, atresia of the external auditory canals, and conductive hearing loss. 1 , 2 The occurrence of TCS is about 1/50 000 live births. 3 , 4
Treacher Collins syndrome has three clinical subtypes. Treacher Collins syndrome‐1 (Online Mendelian Inheritance in Man [OMIM], 154500) is an autosomal dominant disorder caused by heterozygous mutation of the TCOF1 gene (OMIM, 606847). 1 Treacher Collins syndrome‐2 (TCS2, OMIM, 613717) is an autosomal dominant or autosomal recessive disorder caused by mutations in the POLR1D gene (OMIM, 613715). 2 , 5 Treacher Collins syndrome‐3 (TCS3, OMIM, 248390) is an autosomal recessive disorder caused by mutations in the POLR1C gene (OMIM, 610060). 2 Mutations of TCOF1 account for approximately 63%‐80% of TCS cases. 6 , 7 However, mutations or intragenic deletions of the POLR1D and POLR1C genes have been observed only in a few TCS patients. 6 Furthermore, the TCS phenotype exhibits extreme variability. Consequently, correlations between phenotypic variability and TCOF1 mutations have not been established. 6 , 8 , 9
In this study, we recruited seven Chinese TCS patients from unrelated families in this study. All of them were sporadic cases. We established accurate molecular diagnoses by using next‐generation sequencing (NGS) and Sanger sequencing for both the probands and their families. Five novel TCOF1 variants and two previously reported TCOF1 variants were identified in these families. We assessed the pathogenicity of the five novel variants and performed genetic counseling for all these seven families.
2. MATERIALS AND METHODS
2.1. Patients
We recruited seven patients with sporadic TCS that had been treated at Gansu Provincial Maternal and Child Health Care Hospital. Based on physical examinations, all seven probands were diagnosed with the clinical features of TCS, including bilateral craniofacial malformations, 7 atresia of the external auditory canals, and conductive hearing loss. We obtained photographs from five probands; however, two families did not consent to providing photographs. The clinical features of the probands are shown in Table 1 and Figure 1. This study was carried out according to the tenets of the Declaration of Helsinki. This study was approved by the Ethics Committee of the Gansu Provincial Maternal and Child Health Care Hospital (No. 4 of hospital ethics review, 2016). Written informed consent was obtained from all participants in this study.
Table 1.
Clinical characteristics in this study
| Case No. | Age | Sex | Downward slanting palpebral fissures | Hypoplasia of the facial bones | Auricle deformity | Stenosis/atresia of external ear canal | Conductive deafness |
|---|---|---|---|---|---|---|---|
| 1 | 2 mo | Male | + | + | + | + | + |
| 2 | 20 d | Male | + | + | − | + | + |
| 3 | 2 y | Female | + | + | + | + | + |
| 4 | 2 d | Female | + | + | + | + | + |
| 5 | 9 d | Female | + | + | + | + | + |
| 6 | 1 d | Male | + | + | + | + | + |
| 7 | 2 mo | Female | + | + | + | + | + |
Figure 1.
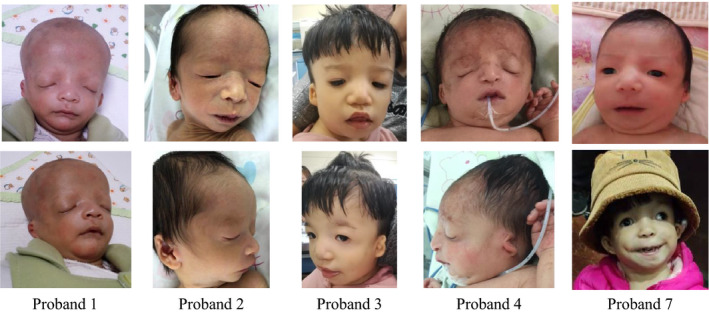
The facial features of these patients are characteristic of Treacher Collins syndrome (TCS)
2.2. Methods
2.2.1. Sample collection and genomic DNA preparation
2‐3 mL blood samples were collected from the probands and their parents. Genomic DNA was extracted using a Tiangen Biotech DNA extraction kit.
2.2.2. Targeted NGS and Sanger sequencing
TCOF1, POLR1D, and POLR1C were screened in the probands by targeted gene capture and sequencing of the 121 deafness‐related genes by MyGenostics Corporation (MyGenostics GenCap Enrichment technologies). The data analysis and bioinformatics analysis procedures were performed as previously described. 10
The candidate gene variants were observed in the probands in each family by Sanger sequencing. Polymerase chain reaction (PCR) primers were prepared and the amplification conditions for Sanger sequencing were as described previously. 9 DNA sequencing was performed on an ABI 3500DX Genetic Analyzer (Applied Biosystems).
2.2.3. Bioinformatics analysis
Genetic variants are described according to the nomenclature recommended by the Human Genomic Variation Society. Novel variants were checked in the Human Gene Variant Database (HGMD, http://www.hgmd.cf.ac.uk/), ClinVar database (https:// www.ncbi.nlm.nih.gov/clinvar/), EXAC database (http://exac.broadinstitute.org/), and in 100 healthy controls. Human Splicing Finderv3.1 (HSF, http://www.umd.be/HSF3/) 11 was used to predict the functional effects of the splicing site variants. InterVar software (http://wintervar.wglab.org/) was used to evaluate the pathogenicity of all variants by reference to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG). 12
3. RESULTS
3.1. Clinical features and variant analysis of the probands
All seven probands have the typical clinical features shown in Table 1 and Figure 1, proband 1‐5 and 7 have auricle and ear canal malformations, however proband 6 does not have an auricle deformity. Using targeted NGS and Sanger sequencing, seven TCOF1 variants were identified in these seven unrelated families with TCS (Table 2). The parents of the probands were tested for these variants; however, none were found to have same variants as the respective probands, implying de novo origin. Two nonsense, one missense, one splicing, and three frameshift variants were identified (Table 2, Figure 2). In our seven probands, no variants were identified in POLR1D or POLR1C.
Table 2.
Classification of TCOF1 mutations in this study according to ACMG guideline
| Case No. | Variation | Classification | Evidences | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|
| cDNA change | p.change | Exon | Status | Type | SNP | ||||
| 1 | c.1393C > T | p.Gln465Ter | 10 | Hetero | Missense | — | Pathogenic | PVS1, PS2, PM2, and PM4 | novel |
| 2 | c.4131_4135delAAAAG | p.Lys1380Glufs*12 | intron24 | Hetero | Frameshift | — | Pathogenic | PVS1, PS2, and PM4 | #1 |
| 3 | c.2285_2286delCT | p.Ser762Ter | 14 | Hetero | Frameshift | — | Pathogenic | PVS1, PS2, PM2, and PM4 | novel |
| 4 | c.1142delC | p.Arg383Glyfs*109 | 9 | Hetero | Frameshift | — | Pathogenic | PVS1, PS2, and PM2 | novel |
| 5 | c.4111 + 5G>C | — | 23 | Hetero | Splice | — | Pathogenic | PVS1, PS2, and PM2 | novel |
| 6 | c.149A > G | p.Tyr50Cys | 2 | Hetero | Missense | rs28941769 | Likely pathogenic | PS1 and PM2 | #2 |
| 7 | c.1719delG | p.Asn574Thrfs*22 | 12 | Hetero | Frameshift | — | Pathogenic | PVS1, PS2, and PM2 | novel |
The type of evidence refers to ACMG/AMP 2015 guideline (http://wintervar.wglab.org/). #1: Edwards et al 1997, Li et al 2019; #2: Splendore et al 2002.
Abbreviation: Hetero, heterozygosity,
Figure 2.
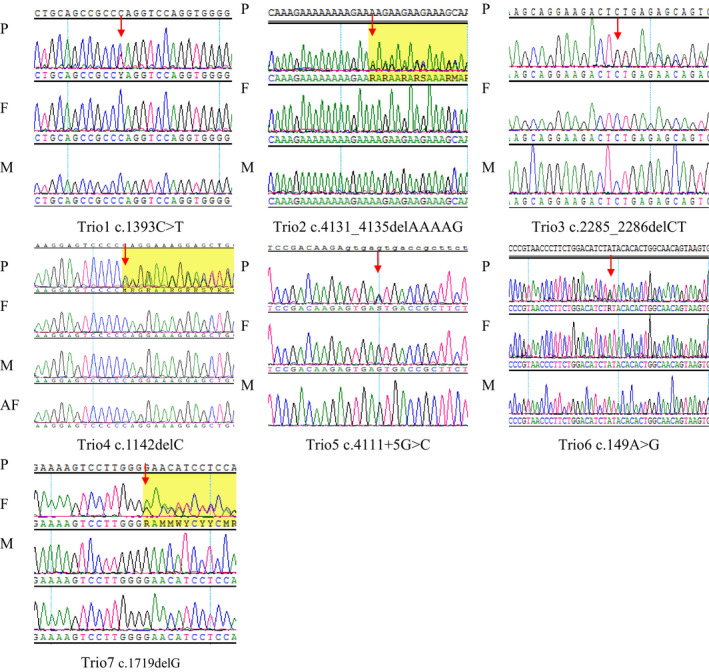
Sanger sequencing of TCOF1: P is proband, F is father, M is mother, AF is amniotic fluid. None of the parents of these seven families detected the same variation as the proband had. The Sanger sequencing of fetus for family 4 was normal
3.2. Bioinformatics analysis
Sequencing fragments were mapped with the hg19 reference genome using BWA software (version 0.7.9a). PCR duplications were removed using Picard software (version 1.115). Variant calling was done using The Genome Analysis Tool Kit, version 3.2; the processing included base quality score recalibration, insertion/deletion (indel) realignment, duplicate removal, indel calling, and single‐nucleotide variant detection. Variants were annotated with Variant Effect Predictor, Ensembl 73 software. Mutations in the functional coding region and splice site, which mainly involved loss‐of‐function mutations, missense mutations, and non‐frameshift indel mutations, were included in the next step of the analysis. Loss‐of‐function mutations include nonsense, frameshift, and splicing site variants. Three major databases that contain pathogenic or likely pathogenic variants, ClinVar, HGMD, and OMIM, were used for further screening. Pathogenicity analysis of the variants was done using InterVar software based on the criteria recommended by the ACMG/Association for Molecular Pathology (AMP) guidelines. 12 The process was automatically performed as described in previously published studies. 13 , 14 According to the ACMG/AMP guidelines and InterVar software, six of the proband TCOF1 variants were categorized as “pathogenic” and one as “likely pathogenic” (Table 2).
3.3. Treatment and pregnancy outcome
Following molecular analysis, we provided genetic counseling to all seven TCS families included in this study. We provided prenatal diagnosis for family 4 by using Sanger sequencing. During their pregnancy, they underwent regular prenatal screenings that did not detect the presence of the c.1142delC variant in the fetus (Figure 2). The baby was born in January 2020, with normal facial features; both ears passed the hearing screening test. Proband 7 chose bone conduction hearing rehabilitation and general surgery to improve hearing and correct facial deformity. The patient reported good outcomes. The unaided frequency‐specialized hearing thresholds were approximately 65‐70 dB SPL initially, but improved to 20‐30 dB SPL after hearing intervention with bone conduction aids.
4. DISCUSSION
In this study, we performed mutation screening in seven Chinese sporadic TCS cases using targeted NGS and Sanger sequencing. In these seven probands, the following TCOF1 mutations were identified: (c.1142delC [p.Arg383Glyfs*109], c.4131_4135delAAAAG [p.Lys1380Glufs*12], and c.1719delG [p.Asn574ThrfsTer22]), two nonsense variants (c.2285_2286delCT [p.Ser762Ter], c.1393C > T [p.Gln465Ter]), one missense variant (c.149A > G [p.Tyr50Cys]), and one splicing variant (c.4111 + 5G>C) (Table 2, Figure 2). Among these variants, c.1393C > T, c.4111 + 5G>C, c.1142delC, c.2285_2286delCT, and c.1719delG had not previously been submitted to the HGMD, ClinVar, or EXAC databases, whereas c.149A > G has been identified in previous TCS reports. 8 , 15 However, this variant has not been reported in Chinese TCS cases. According to the ACMG/AMP guidelines and InterVar software, six of the above variants were categorized as “pathogenic” and one was categorized as “likely pathogenic” (Table 2).
Treacher Collins syndrome was first defined by the English ophthalmologist Edward Treacher Collins. During the early fetal development (5th‐8th weeks), the abnormal formation of the first and second branchial arches causes TCS, which results in profound craniofacial malformation. 16 TCS has typical bilateral facial features, such as hypoplasia of the facial bones, coloboma of the lid, downward slanting palpebral fissures, and a prominent nose. These features constitute the typical bilateral craniofacial malformations of TCS. 7 However, TCS is both genetically and phenotypically heterogeneous.
Treacher Collins syndrome can be caused by mutations in three genes that are associated with ribosome biogenesis and synthesis, that is, TCOF1, POLR1D, and POLR1C. Most TCS cases (63%‐80%) are caused by variants in TCOF1 and include frameshift, nonsense, splice, missense, and intragenic microdeletions. 6 , 7 Bowman et al 3 reported that 57% of the causal variants for TCS were frameshift variants, 23% were nonsense variants, 16% were splicing variants, and only 4% were missense variants. In our seven TCS patients, we only identified variants in TCOF1, and the predominant variant type (57.1%) was frameshift.
Currently, no genotype–phenotype correlations have been established for TCS. However, the severity of TCS phenotypes does not significantly differ between different variant types. 6 , 9 In our study, six TCS probands had typical phenotypes and one proband had an atypical phenotype that was caused by c.4131_4135delAAAAG in intron 24 of TCOF1. Vincent, et al 6 used a scoring system to evaluate the severity of the phenotype and found that few patients with severe scores had variants in exon 24 of TCOF1. However, whether the TCS caused by variants in exon 24 or the last two exons would have a lower potential for gene inactivation remains to be elucidated.
Treacher Collins syndrome presents with complex mandibulofacial dysplasia and requires lifelong multidisciplinary interventions. 16 TCS phenotypes are extremely diverse, and the needs of TCS patients for aesthetic, functional, and the psychosocial interventions also vary. Some TCS patients choose general surgery to improve the presentation of mandibulofacial dysplasia. However, the development of individualized treatment options is needed. 17 , 18 , 19 , 20 Fan et al 16 reported that bone conduction hearing rehabilitation was a very helpful improvement measure for TCS patients with bilateral conductive hearing loss. In our study, proband 7 chose interventional surgery to improve hearing loss and correct facial deformity and had a good outcome. Prenatal diagnosis is important for the birth of healthy fetuses in families. We provided prenatal diagnosis for family 4 by Sanger sequencing. The fetus tested negative for the variant (Figure 2). The facial features of the baby were normal and both ears passed the hearing screening test.
Treacher Collins syndrome, Goldenhar syndrome, Miller syndrome, and Nager syndrome can overlap in terms of their variable phenotypic expression, asymmetric involvement of facial structures, and familial occurrence of microtia or related anomalies. This complicates the diagnosis of such diseases based on the patient's clinical manifestation. However, we can use molecular diagnosis, especially NGS, to classify patients clinically.
In conclusion, we performed a mutation screening for seven TCS patients from seven unrelated families by using targeted NGS and Sanger sequencing. Currently, about 345 pathogenic/likely pathogenic variants causing TCS are listed in HGMD. We identified seven different TCOF1 variants in these seven TCS patients, and five of these variants had not previously been reported. Our results expand the mutational spectrum of TCS and could be useful for genetic counseling, treatment, and prenatal diagnosis of TCS in China. Interventional surgery can improve hearing loss and correct facial deformity in TCS patients and prenatal diagnosis can provide fertility guidance for TCS families.
ACKNOWLEDGMENTS
We would like to thank all the participants in this study.
Zhang C, An L, Xue H, et al. Mutation analysis of TCOF1 gene in Chinese Treacher Collins syndrome patients. J Clin Lab Anal.2021;35:e23567 10.1002/jcla.23567
Funding information
This work was supported by the Gansu Natural Science Foundation (grant: No.18JR3RA036; 1606RJZA151; 1606RJZA159);The National Key Research and Development Program of China (grant No.:2016YFC1000307); Non‐profit Central Research Institute Fund of National Research Institute For Family Planning (grant No.:2019GJZ07); the National Population and Reproductive Health Science Data Center (grant No.:2005DKA32408), People's Republic of China.
Contributor Information
Zongfu Cao, Email: zongfu_cao@163.com.
Xu Ma, Email: maxubioinfo@163.com.
REFERENCES
- 1. Dixon MJ. Treacher Collins syndrome. Hum Mol Genet. 1996;5:1391‐1396. [DOI] [PubMed] [Google Scholar]
- 2. Dauwerse JG, Dixon J, Seland S, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43:20‐22. [DOI] [PubMed] [Google Scholar]
- 3. Bowman M, Oldridge M, Archer C, et al. Gross deletions in TCOF1 are a cause of Treacher‐Collins‐Franceschetti syndrome. Eur J Hum Genet. 2012;20:769‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosas MG, Lorenzatti A, Porcel de Peralta MS, et al. Proteasomal inhibition attenuates craniofacial malformations in a zebrafish model of Treacher Collins Syndrome. Biochem Pharmacol. 2019;163:362‐370. [DOI] [PubMed] [Google Scholar]
- 5. Schaefer E, Collet C, Genevieve D, et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet Med. 2014;16:720‐724. [DOI] [PubMed] [Google Scholar]
- 6. Vincent M, Geneviève D, Ostertag A, et al. Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genet Med. 2016;18:49‐56. [DOI] [PubMed] [Google Scholar]
- 7. Li X, Su Y, Huang S, et al. Genotype‐phenotype variability in Chinese cases of Treacher Collins syndrome. Acta Oto laryngol. 2019;139:567‐575. [DOI] [PubMed] [Google Scholar]
- 8. Edwards SJ, Gladwin AJ, Dixon MJ. The mutational spectrum in Treacher Collins syndrome reveals a predominance of mutations that create a premature termination codon. Am J Hum Genet. 1997;60:515‐524. [PMC free article] [PubMed] [Google Scholar]
- 9. Teber OA, Gillessen‐Kaesbach G, Fischer S, et al. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004;12:879‐890. [DOI] [PubMed] [Google Scholar]
- 10. Zhang C, Hao S, Liu Y, et al. A novel LOXHD1 variant in a Chinese couple with hearing loss. J Int Med Res. 2019;47:6082‐6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Desmet FO, Hamroun D, Lalande M, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. Am J Hum Genet. 2017;100:267‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhuang J, Cao Z, Zhu Y, et al. Mutation screening of crystalline genes in Chinese families with congenital cataracts. Mol Vis. 2019;25:427‐437. [PMC free article] [PubMed] [Google Scholar]
- 14. Jin X, An L, Hao S, et al. Compound heterozygous variants of the FBXO7 gene resulting in infantile‐onset Parkinsonian‐pyramidal syndrome in siblings of a Chinese family [published online ahead of print, 2020 Apr 10]. J Clin Lab Anal. 2020;34:e23324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Splendore A, Jabs EW, Passos‐Bueno MR. Screening of TCOF1 in patients from different populations: confirmation of mutational hot spots and identification of a novel missense mutation that suggests an important functional domain in the protein treacle. J Med Genet. 2002;39:493‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fan X, Wang Y, Fan Y, et al. TCOF1 pathogenic variants identified by Whole‐exome sequencing in Chinese Treacher Collins syndrome families and hearing rehabilitation effect. Orphanet J Rare Dis. 2019;14:178‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Posnick JC, Tiwana PS, Costello BJ. Treacher Collins syndrome: comprehensive evaluation and treatment. Oral Maxillofac Surg Clin NorthAm. 2004;16:503‐523. [DOI] [PubMed] [Google Scholar]
- 18. Cobb AR, Green B, Gill D, et al. The surgical management of Treacher Collins syndrome. Br J Oral MaxillofacSurg. 2014;52:581‐589. [DOI] [PubMed] [Google Scholar]
- 19. Plomp RG, van Lieshout MJ, et al. Treacher Collins syndrome: a systematic review of evidence‐based treatment and recommendations. PlastReconstr Surg. 2016;137:191‐204. [DOI] [PubMed] [Google Scholar]
- 20. Aljerian A, Gilardino MS. Treacher Collins syndrome. Clin Plast Surg. 2019;46:197‐205. [DOI] [PubMed] [Google Scholar]