

Abstract
About 10% of term neonates present with respiratory distress at birth. The most common aetiologies include transient tachypnoea of the newborn, pneumonia and meconium aspiration syndrome (MAS). Hyaline membrane disease (HMD) in a term infant occurs either as primary HMD, secondary surfactant deficiency or congenital surfactant dysfunction. A detailed history supported with appropriate radiological and laboratory investigations can help a clinician reach a diagnosis. We report a case of surfactant dysfunction disorder which presented as severe MAS and persistent pulmonary hypertension of the newborn. In the infant described, the significant history of a sibling death with severe neonatal respiratory disease led us to think of diffuse developmental lung diseases especially surfactant dysfunction syndromes. Exome sequencing detected a heterozygous missense variation in exon 21 of the ATP binding cassette protein member 3 (ABCA3) gene. Based on the clinical picture supported with the exome sequencing, a diagnosis of surfactant dysfunction disorder (ABCA3 deficiency) was confirmed.
Keywords: congenital disorders, neonatal health, neonatal and paediatric intensive care, materno-fetal medicine
Background
Hyaline membrane disease (HMD) is less likely in a term neonate but can be present in the rare scenario of secondary surfactant deficiency or congenital surfactant dysfunction due to genetic defects. A detailed history supported with appropriate radiological and laboratory investigations can help a clinician reach a diagnosis. Surfactant dysfunction disorders are associated with high morbidity and mortality.
We report a case of surfactant dysfunction disorder which presented as severe meconium aspiration syndrome (MAS) with severe persistent pulmonary hypertension of the newborn (PPHN).
Case presentation
A male infant from a non-consanguineous marriage with birth weight of 3100 g (25th centile) was delivered at 392/7 weeks of gestation to a 24-year-old gravida II, para I mother through a normal vaginal delivery. The mother’s previous delivery was a term male newborn who had MAS with features of severe PPHN refractory to inhaled nitric oxide (iNO) and had succumbed on day 23 of life. Autopsy was not carried out then.
Regarding the index case, prenatal check-ups and imaging were normal, amniotic fluid index was 13.2. The baby was born at a hospital with level II neonatal care. He did not require any resuscitation after birth. Apgar scores were 8 and 9 at 1 and 5 min of life, respectively. Amniotic fluid was meconium stained. He developed respiratory distress within 1 hour after birth and was initially started on continuous positive airway pressure with maximum positive end expiratory pressure of 8 cm H2O and maximum fraction of inspired oxygen (FiO2) of 80%. Initial diagnosis was MAS. Echocardiography was suggestive of severe PPHN. In view of progressively worsening respiratory status, he was referred to our centre.
At admission (9 hours of life), the baby had significant respiratory distress with severe retractions and grunting and the oxygen saturation was 85%–90% on FiO2 of 100%. The baby was intubated at admission. After a failed trial of conventional ventilation, the baby was started on high frequency oscillation (HFO) mode of ventilation with FiO2 of 100%. Arterial blood gas (ABG) revealed pH of 7.39, partial pressure of oxygen (PaO2) of 42 mm Hg, partial pressure of carbon dioxide (PaCO2) of 52 mm Hg, base deficit of 3 mmol/L with oxygenation index (OI) of 30. iNO was instituted at 12 hours of life in view of high OI. Repeat echocardiography was suggestive of severe pulmonary hypertension with a structurally normal heart (dilated right atrium and right ventricle with paradoxical movement of intraventricular septum, tricuspid regurgitation gradient 56 mm Hg with bidirectional shunting at the ductus arteriosus). Ventricular functions were within the normal range. Initial diagnosis was MAS with severe PPHN. There were no dysmorphism or chest deformity. Antibiotics were initiated at admission suspecting severe sepsis. Sildenafil was added in view of persistently high pulmonary arterial pressures.1 Chest radiograph revealed bilateral reticulogranular shadows with diffuse air bronchograms (figure 1) without any evidence of pleural effusion. The high ventilator settings and the X-ray picture were suggestive of secondary surfactant deficiency. Surfactant was administered to the baby (Survanta, AbbVie, USA) at a dose of 150 mg/kg (6 mL/kg) at 14 hours of life and repeated once again after 6 hours. There was clinical response to the above interventions (iNO, sildenafil and surfactant) and OI decreased from 30 to 5. There was improvement in the respiratory parameters over the next 36 hours and the baby was extubated to non-invasive ventilation. However, this improvement was short lived and the baby had to be reintubated after 48 hours in view of recurrence of respiratory distress. ABG revealed pH of 7.45, PaO2 of 53 mm Hg, PaCO2 of 59 mm Hg and base deficit of 2 mmol/L with OI of 19.3. Repeat chest radiograph was similar to the first one with persistent bilateral reticulogranular shadows and air bronchograms. A repeat dose of surfactant resulted in a brief period of improvement in the FiO2 requirement, while the baby was continued on HFO mode of ventilation. Antibiotics were stopped once blood culture was sterile. After discussion with the paediatric pulmonologist, a trial of azithromycin and steroids was instituted in view of suspected interstitial lung disease. Fibreoptic bronchoscopy was performed and bronchoalveolar lavage (BAL) was done with surfactant. Despite the varied interventions, the baby continued to deteriorate.
Figure 1.
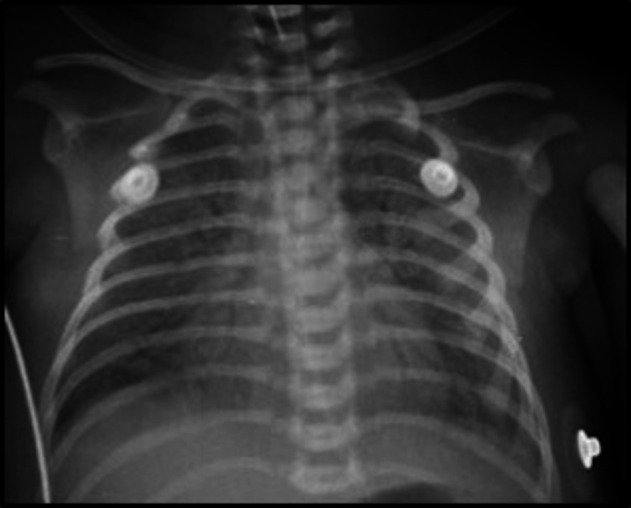
Chest radiograph depicting bilateral reticulogranular shadows with diffuse air bronchograms.
Investigations
Other causes of severe neonatal respiratory disease including congenital heart disease, sepsis and cystic fibrosis were ruled out with appropriate investigations. Sepsis screen was negative and blood culture was sterile. High-resolution CT (HRCT) of the chest exhibited ground glass opacities and extensive areas of septal thickening in bilateral lung fields. CT pulmonary angiogram ruled out total anomalous pulmonary venous circulation (TAPVC) as well as malaligned pulmonary veins (MPV). The significant history of a sibling death with severe neonatal respiratory disease led us to think of diffuse developmental lung diseases especially congenital surfactant deficiency syndromes.
In view of suspected interstitial lung disease, BAL obtained by bronchoscopy was analysed for hemosiderin laden macrophages and periodic acid–Schiff staining, which were negative. On day 15 of life, after consultation with a geneticist, exome sequencing was carried out which detected a heterozygous missense variation (c.2816T>C) in exon 21 of the ATP binding cassette protein member 3 (ABCA3) gene. This variation which is of uncertain significance may be associated with surfactant metabolism dysfunction-3 (OMIM# 610921). Lung biopsy was refused by the parents.
Based on the clinical and radiological picture supported with the exome sequencing, a diagnosis of surfactant dysfunction disorder (ABCA3 deficiency) was considered.
Outcome and follow-up
The baby continued to deteriorate with progressively increasing ventilatory requirement. Once the exome sequencing results were known, the geneticist was involved again, and poor prognosis was discussed with the parents. Extracorporeal membrane oxygenation (ECMO) and lung transplantation were not feasible. In the absence of lung transplantation, the chances of survival were grim.2 Despite optimised medical therapy for pulmonary hypertension, the baby had a deteriorating clinical course and he succumbed on day 52 of life. Autopsy was refused by the parents. In view of the high risk of similar illness in future progenies, the importance of appropriate parental testing and pre-implantation genetic analysis in the next pregnancy were discussed with the family.
Discussion
Respiratory distress is a common morbidity in neonates with a high incidence in preterm infants. About 10% of term neonates present with respiratory distress at birth.3 4 The most common aetiologies leading to respiratory distress in term neonates are transient tachypnea of the newborn and congenital pneumonia. Another common aetiology for respiratory distress in term neonates is MAS (index case). The pathogenesis is multifactorial in this condition with aspiration pneumonitis, obstruction of airways, pneumothorax and pulmonary hypertension.5 PPHN implies ‘persistence’ of supra-systemic pulmonary pressures as is generally seen in the fetus and can be secondary to meconium aspiration, pneumonia or can be idiopathic. Congenital lung malformations like congenital pulmonary airway malformations, congenital lobar emphysema, bronchogenic cyst, pulmonary sequestration, alveolar capillary dysplasia and tracheobronchomalacia are rare causes of respiratory distress at birth. Congenital cyanotic heart disease can present as respiratory distress at birth with poor response to respiratory support and oxygen. HMD is less likely in a term neonate but can be present in the rare scenario of congenital surfactant deficiency due to genetic defect in surfactant production.
A detailed history supported with appropriate radiological and laboratory investigations can help a clinician reach a diagnosis. Presence of bilateral reticulogranular shadows and air bronchograms along with a low volume lung, as seen in our case, is suggestive of surfactant deficiency. Exogenous surfactant has been used successfully in severe MAS, decreasing air leak and need for ECMO.6 Other entity which can present with a similar radiological picture include TAPVC. Congenital pneumonia can present with a similar X-ray findings but generally respond to antibiotics which was not seen in our case. The other diagnostic possibilities with similar radiological picture are diffuse developmental disorders of lung comprising of acinar dysplasia (AD), congenital alveolar dysplasia and alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV). A HRCT can aid in the diagnosis of structural malformations and developmental disorders of the lung.
In view of the severe hypoxaemic respiratory failure refractory to conventional therapy along with a background of a sibling death due to a similar illness, a genetic basis of the disease pathology was possible. Chest X-ray and HRCT picture of diffuse lung parenchymal involvement along with transient improvement with exogenous surfactant administration with recurrence once the exogenous pool of surfactant depleted pointed towards the diagnosis of surfactant dysfunction disorder.
Surfactant dysfunction disorders encompass different disorders characterised by mutations in genes encoding various proteins required for normal surfactant function. Surfactant proteins B and C are hydrophobic, their genes encoded on chromosomes 2 and 8, respectively, and are essential for surfactant function. Surfactant is synthesised by alveolar type 2 epithelial cells and stored in special organelles called lamellar bodies. ABCA3, a member of the ATP binding cassette transporter, is located on the membrane of lamellar bodies and is involved in the transport of phospholipid into the lamellar bodies essential for surfactant production.7 ABCA3 is a 1704-amino acid protein and is localised to chromosome 16. Genetic defects in surfactant protein B or C or in ABCA3 protein can present with severely abnormal pulmonary function in the neonatal period.7 8 The clinical presentation is that of a term neonate with diffuse lung disease mimicking HMD. The lung disease progressively worsens and is refractory to medical therapy. Shulenin et al have reported a case series of 21 neonates with fatal surfactant deficiency, out of which 16 neonates had ABCA3 gene mutation and died within 3 months after birth.8 Similarly, Somaschini et al have described the genetic analysis of 17 neonates who died due to unexplained respiratory failure, out of which 12 neonates had ABCA3 mutation.9 In this case series, similar to our index case, seven neonates had significant family history of neonatal death due to respiratory failure. Unlike previous cases reported, the infant described here had clinical features of severe MAS and PPHN, which can mimic severe surfactant deficiency and thus can delay suspicion of genetic basis for respiratory distress. Rare presentation in a child or adult as interstitial lung disease has also been reported.10 Lung transplantation is the only known salvage therapy, although mortality and morbidities remain considerable for such infants even after lung transplantation. The 5-year survival rate after lung transplantation is nearly 50% with infections (including cytomegalovirus infection), graft-related complications, bronchiolitis obliterans and lymphoproliferative disorders contributing to the mortality.11 Prolonged ventilation, significant growth impairment, motor and speech delays contribute to their poor quality of life.11
Chest radiography features include diffuse alveolar and interstitial infiltrates throughout the lung fields. Typical findings on HRCT include presence of ground glass opacities along with thickened interlobular and intralobular septa. Bronchoscopy with BAL analysis can help in refuting certain aetiologies with similar clinical presentation like pulmonary alveolar proteinosis (PAP), eosinophilic lung disease and infections. The availability of genetic testing has changed the paradigm towards non-invasive testing. In case of surfactant dysfunction disorders, testing for mutations in the common genes, namely, SFTPB, SFTPC, ABCA3 and NKX 2.1, can aid in the diagnosis without resorting to invasive diagnostic modalities like lung biopsy.12 Presence of a specific mutation can guide therapy and help in prognostication in the index case. ABCA3 deficiency is an autosomal recessive disorder with nearly 150 reported mutations.8 Our patient was found to be heterozygous for mutation of the ABCA3 gene. Case reports have identified patients with a clinical and histopathological picture consistent with surfactant dysfunction disorders but exhibiting a single heterozygous gene mutation.8–10 13 14 This could be attributed to functionally significant mutation in the untranslated region or a novel mutation which is not detected by the usual approaches. The lacunae with genetic analysis is the long turnover time which generally precludes an early diagnosis in a rapidly deteriorating baby.
Lung biopsy is generally warranted only in specific circumstances where the genetic analysis is negative or an early diagnosis is desirable. The usual histopathological findings include PAP and desquamative interstitial pneumonitis.15 Electron microscopy reveals presence of abnormal lamellar bodies in the alveolar epithelial cells (AEC2) giving a ‘fried egg appearance’.
In conclusion, a term neonate presenting with (1) severe respiratory distress with diffuse parenchymal lung disease not responding to conventional management or (2) a family history of a neonatal death from respiratory disease should undergo a detailed diagnostic workup including advanced imaging studies. Genetic analysis should be performed at the earliest as this may guide therapy and also assist in prognostication. Surfactant dysfunction disorders, especially ABCA3 deficiency, are associated with high morbidity and mortality. Once a specific genetic mutation is detected, it is imperative to discuss the short-term and long-term outcomes of the index case with the parents along with the risk to future offspring (25% recurrence risk) and the role of pre-implantation genetic analysis.
Learning points.
Hyaline membrane disease (HMD) is less likely in a term neonate but can be present in the rare scenario of secondary surfactant deficiency or congenital surfactant deficiency due to genetic defect in surfactant production.
A term neonate presenting with respiratory distress and a family history of a neonatal death from respiratory disease should undergo a detailed diagnostic workup including advanced imaging studies.
Genetic analysis should be performed at the earliest as this may guide therapy and also assist in prognostication.
ABCA3 deficiency is associated with high morbidity and mortality risk (especially in the absence of lung transplantation) and has significant recurrence risk in future pregnancies.
Acknowledgments
We are grateful to Dr Krishan Chugh, Consultant Pediatric Pulmonologist, Department of Pediatrics, Fortis Memorial Research Institute, Gurugram, Haryana, India, for his valuable inputs.
Footnotes
Contributors: SG, GA, MB: conceived and wrote the manuscript. SW: critically reviewed and finalised the manuscript. All authors (SG, GA, MB, SW): involved in clinical care of the infant, read and approved the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Parental/guardian consent obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Steinhorn RH, Kinsella JP, Pierce C, et al. Intravenous sildenafil in the treatment of neonates with persistent pulmonary hypertension. J Pediatr 2009;155:841–7. 10.1016/j.jpeds.2009.06.012 [DOI] [PubMed] [Google Scholar]
- 2.Hamvas A. Evaluation and management of inherited disorders of surfactant metabolism. Chin Med J 2010;123:2943–7. [PubMed] [Google Scholar]
- 3.Kumar A, Bhat BV. Epidemiology of respiratory distress of newborns. Indian J Pediatr 1996;63:93–8. 10.1007/BF02823875 [DOI] [PubMed] [Google Scholar]
- 4.Clark RH. The epidemiology of respiratory failure in neonates born at an estimated gestational age of 34 weeks or more. J Perinatol 2005;25:251–7. 10.1038/sj.jp.7211242 [DOI] [PubMed] [Google Scholar]
- 5.Meritano J, Abraham S, Di Pietro S. Respiratory distress syndrome associated with meconium stained amniotic fluid in term and post-term newborns: incidence, risk factors, morbidity, and mortality. Pediatric Research Conference: 47th Annual Meeting of the Latin American Society for Paediatric Research, LASPR Asuncion Paraguay Conference Start, 2010:446. [Google Scholar]
- 6.El Shahed AI, Dargaville PA, Ohlsson A. Surfactant for meconium aspiration syndrome in full term infants. Cochrane Database Syst Rev 2007;3:CD002054 10.1002/14651858.CD002054 [DOI] [PubMed] [Google Scholar]
- 7.Bullard JE, Wert SE, Nogee LM. Abca3 deficiency: neonatal respiratory failure and interstitial lung disease. Semin Perinatol 2006;30:327–34. 10.1053/j.semperi.2005.12.001 [DOI] [PubMed] [Google Scholar]
- 8.Shulenin S, Nogee LM, Annilo T. ABCA3 gene is frequently mutated in human newborns with fatal surfactant deficiency. N Engl J Med 2004;350:1296–303. 10.1056/NEJMoa032178 [DOI] [PubMed] [Google Scholar]
- 9.Somaschini M, Nogee LM, Sassi I, et al. Unexplained neonatal respiratory distress due to congenital surfactant deficiency. J Pediatr 2007;150:649–53. 10.1016/j.jpeds.2007.03.008 [DOI] [PubMed] [Google Scholar]
- 10.Bullard JE, Wert SE, Whitsett JA, et al. Abca3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med 2005;172:1026–31. 10.1164/rccm.200503-504OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eldridge WB, Zhang Q, Faro A, et al. Outcomes of lung transplantation for infants and children with genetic disorders of surfactant metabolism. J Pediatr 2017;184:157–64. 10.1016/j.jpeds.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatr Dev Pathol 2009;12:253–74. 10.2350/09-01-0586.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wambach JA, Casey AM, Fishman MP, et al. Genotype-Phenotype correlations for infants and children with ABCA3 deficiency. Am J Respir Crit Care Med 2014;189:1538–43. 10.1164/rccm.201402-0342OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kunig AM, Parker TA, Nogee LM, et al. Abca3 deficiency presenting as persistent pulmonary hypertension of the newborn. J Pediatr 2007;151:322–4. 10.1016/j.jpeds.2007.05.054 [DOI] [PubMed] [Google Scholar]
- 15.Trapnell BC, Carey BC, Uchida K, et al. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr Opin Immunol 2009;21:514–21. 10.1016/j.coi.2009.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]