

Summary
Cortical microtubules (CMTs) play pivotal roles during plant cell growth and division. The organization of CMTs undergoes important changes during different cellular and developmental processes. Here, we describe two methods for the visualization of CMT organization in plant cells using confocal laser scanning microscopy. CMT networks in the outer tissue layers can be directly visualized by live imaging of a fluorescent reporter line, and a protocol combining sectioning and immunostaining is applied for visualization of CMTs throughout tissues.
For complete details on the use and execution of this protocol, please refer to Zhao et al. (2020).
Subject areas: Cell biology, Microscopy, Model organisms
Graphical Abstract
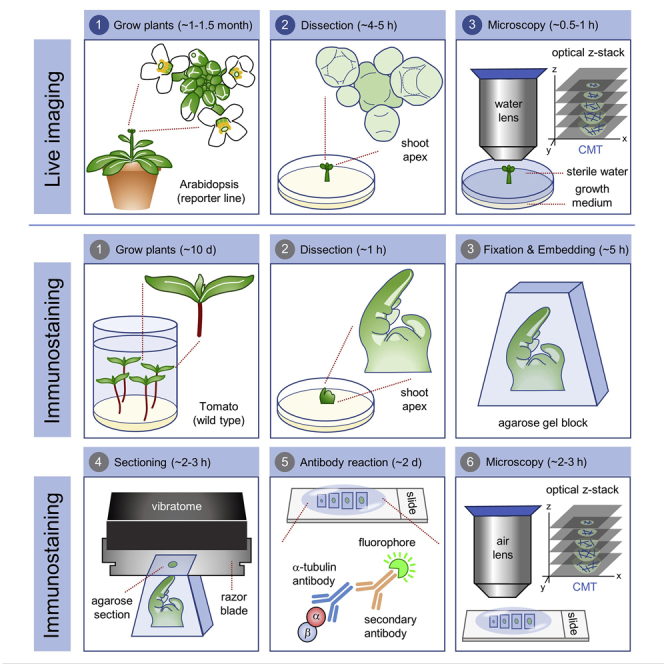
Highlights
-
•
Methods for live imaging of cortical microtubules in outer cell layers of plant tissues
-
•
Agarose-gel embedding and sectioning method for exposing inner plant cells
-
•
Methods for immunostaining of cortical microtubules in inner cells of plant tissues
Cortical microtubules (CMTs) play pivotal roles during plant cell growth and division. The organization of CMTs undergoes important changes during different cellular and developmental processes. Here, we describe two methods for the visualization of CMT organization in plant cells using confocal laser scanning microscopy. CMT networks in the outer tissue layers can be directly visualized by live imaging of a fluorescent reporter line, and a protocol combining sectioning and immunostaining is applied for visualization of CMTs throughout tissues.
Before you begin
Here, we give examples of the live imaging of CMTs in Arabidopsis sepal and leaf primordia, as well as the immunostaining of CMTs in tomato leaf primordia. It should be noted that both methods are applicable to inflorescence and vegetative meristems of both Arabidopsis and tomato and probably to other species as well.
Preparation for the live imaging of CMTs in Arabidopsis plants
Timing: ∼25–50 days
-
1.
Prepare Arabidopsis plants
Seeds of transgenic Arabidopsis plants that express fluorescent tags of microtubules (like p35S: GFP-MBD reporter line we used in this study) are sown on soil and kept under long day conditions (16 h light/8 h dark, LED 150 μE·m−2·s−1; 20°C–22°C day temperature; 60% humidity). The plants are used when they start bolting, 25–30 days after germination. To ease dissection of the inflorescence meristem, seeds can be sown under short-day conditions (8 h light/16 h dark) for 3–4 weeks. After that, plants are moved to long day conditions for an additional 2–3 weeks until bolting (Figures 1A and 1B). To prepare the vegetative shoot apical meristem for the visualization of CMT in leaf primordia, the plants cultured under short-day conditions for 33–37 days are appropriate for further dissection.
-
2.
Prepare MS medium
Figure 1.

Preparation of CMT live imaging in Arabidopsis flowers
(A) Arabidopsis plant cultured under short-day to long-day conditions with inflorescences of around 2 cm high.
(B) Detail of (A) showing the inflorescence shoot apex.
(C) Old flowers are dissected away and whole inflorescence shoot apices are inserted in ACM.
(D) Detail of (C) showing the pre-dissected shoot apices.
(E and F) Clean dissected shoot apices can be used for acquisition (E) or further cultured in ACM for at least 24 h (F).
(G) The shoot apices are submerged under water. A water immersion objective is used to image sepal primordia.
Scale bars: 10 mm in (A)–(C) and (G); 2 mm in (D); 100 μm in (E) and (F).
Dissolve 2.2 g Murashige & Skoog Basal Medium (without vitamins), 10 g sucrose in 800 mL ddH2O, adjust pH to 5.8 with 1 M KOH. Add 10 g agarose, fill up to 1 L with ddH2O, and autoclave. Store at room temperature (23°C–25°C).
-
3.Prepare vitamin stock (1,000×)
-
a.Dissolve 5 g Myo-inositol, 0.05 g Nicotinic acid, 0.05 g Pyridoxine hydrochloride, 0.5 g Thiamine hydrochloride, 0.1 g glycine, and fill up to 50 mL with ddH2O.
-
b.Sterilize using 0.22 μm filters in a laminar hood. Aliquot (500 μL for each) and store at −20°C.
-
a.
-
4.Prepare N6-Benzyladenine (BAP) stock
-
a.Dissolve 0.1 mg BAP in a small volume of 1 M NaOH, and then fill up to 5 mL with ddH2O. The final concentration of BAP stock is 100 μM.
-
b.Sterilize using 0.22 μm filters in a laminar hood. Aliquot (200 μL for each) and store at −20°C for up to half a year.
-
a.
-
5.Prepare staining dye solution
-
a.FM4-64 solution: prepare a 1 mg/mL stock solution in sterile water. Store aliquots (100 μL for each) at −20°C. The concentration of working solution is 0.33 mg/mL.
-
b.Propidium iodide solution: 1 mg/mL, ready to use stock, from Sigma-Aldrich. Store at 4°C.
-
a.
Preparation for immunostaining of CMTs in tomato plants
-
6.Prepare tomato germination medium
-
a.Dissolve 2.2 g Murashige & Skoog Basal Medium (with vitamins) and 30 g sucrose in 800 mL ddH2O, adjust pH to 5.8 with 1 M KOH. Add 3 g phytagel, and fill up to 1 L with ddH2O.
-
b.Sterilize by autoclaving for 15 min at 121°C. Aliquot 60–70 mL per sterilized Magenta box.
-
a.
-
7.Prepare tomato tissue culture medium
-
a.Dissolve 4.4 g Murashige & Skoog Basal Medium (with vitamins) and 20 g sucrose in 800 mL ddH2O, adjust pH to 6.0 with 1 M KOH. Add 3 g phytagel, and fill up to 1 L with ddH2O.
-
b.Sterilize by autoclaving for 15 min at 121°C. Add 1 mL trans-zeatin stock. Aliquot 30–40 mL per sterilized 100 mm × 20 mm Petri dish.
-
c.trans-Zeatin stock (1 mg/mL): Dissolve 50 mg trans-zeatin in 1 mL of 1 M NaOH. Fill up to 50 mL with ddH2O. Sterilize through a 0.22 μm filter. Store aliquots (1 mL for each) at −20°C.
-
a.
-
8.Grow tomato plants
-
a.Sterilize seeds of tomato (Solanum lycopersicum), e.g., cultivar M82 in this protocol, by shaking them in 95% ethanol for 5 min and 30% commercial bleach (10%–15% sodium hypochlorite) for 15 min. Wash the seeds five times with sterile ddH2O.
-
b.Sow sterilized seeds on germination medium in Magenta boxes (Figure 2A). Grow plants at 25°C under long day condition (16 h light/8 h dark) for 7–10 days.
- c.
-
a.
-
9.Prepare microtubule stabilizing buffer (MTSB).
-
a.Dissolve 7.56 g PIPES, 0.62 g MgSO4·7H2O and 0.95 g EGTA in 400 mL sterile ddH2O, adjust pH to 6.9 with KOH pellets. Fill up to 500 mL with sterile ddH2O. Store at 4°C for several weeks.
-
a.
-
10.Prepare poly-L-lysine coated glass slides
-
a.Soak glass slides in tap-water with drops of dish detergent for 30 min. Rinse with running tape-water for 2 h.
-
b.Soak in 10% HCl (v/v) overnight (12–16 h) in a glass beaker. The volume of 10% HCl should be sufficient to fully immerse all slides.
-
c.Rinse with running tape-water for 2 h. Air-dry slides.
-
d.Poly-L-lysine solution: add 2.5 mL poly-L-lysine solution stock (4 mg/mL) and 1 mL 1 M Tris-HCl (pH = 8.0) with 96.5 mL ddH2O. Normally, 100 mL poly-L-lysine solution is sufficient for treatment of 70–80 slides. To prepare 1 M Tris-HCl, add 121.1 g Tris-base in 800 mL ddH2O, adjust pH to 8.0 with HCl, and fill up to 1 L with ddH2O. Sterilize by autoclaving for 15 min at 121°C.
-
e.Arrange slides into a stainless steel slide rack. Soak the rack with slides in poly-L-lysine solution for 30 min in a glass container (e.g., glass staining jar). The volume of poly-L-lysine solution should be sufficient to fully immerse all slides. Air-dry in a laminar hood. Seal and store at −20°C.
-
f.Commercial poly-L-lysine coated glass slides (e.g., Sigma-Aldrich, P0425-72EA) can also be used.
-
a.
-
11.Prepare 1× TBS buffer
-
a.Firstly prepare 10× TBS buffer by mixing 88 g NaCl and 200 mL 1 M Tris-HCl (pH = 8.0), and fill up to 1 L with ddH2O. Sterilize by autoclaving for 15 min at 121°C. Store at room temperature (23°C–25°C).
-
b.Dilute 10× TBS buffer to 1× TBS buffer with sterilized ddH2O.
-
a.
-
12.Prepare 1× PBS buffer
-
a.Add 8 g NaCl, 0.2 g KCl, 1.42 g Na2HPO4 and 0.27 g KH2PO4 in 800 mL ddH2O. Adjust pH to 7.4 with HCl, and fill up to 1 L with ddH2O.
-
b.Sterilize by autoclaving for 15 min at 121°C. Store at room temperature (23°C–25°C).
-
a.
-
13.Prepare 3% agar plates
-
a.Dissolve 30 g agar in 1 L ddH2O.
-
b.Sterilize by autoclaving for 15 min at 121°C. Aliquot 30–40 mL per sterilized 100 mm × 20 mm Petri dish.
-
a.
-
14.Prepare DAPI solution
-
a.1 mg/mL DAPI stock solution: dissolve 1 mg DAPI in 1 mL ddH2O.
-
b.1 μg/mL DAPI working solution: 1:1000 dilution of DAPI stock solution with 1× PBS.
-
a.
Figure 2.

Preparation of agarose sections for CMT immunostaining in tomato leaves
(A) Tomato seedlings are grown on the germination medium in a Magenta box for 7–10 days.
(B) Seedlings with expanded cotyledons (c) and slightly visible true leaves are appropriate for further cultivation. Cotyledons are removed and the shoot apex is detached from the stem. Cutting positions are indicated by black lines.
(C) Detached shoot apex of tomato seedling in (B).
(D and E) Isolated shoot apices are cultivated in the tissue culture medium at day 1 (D) and day 3 (E).
(F) The shoot apex is dissected to remove old leaf primordia (P) before fixation.
(G) After fixation and rinsing, samples are embedded in 6%–7% low melting agarose in a plastic weighing dish. The agarose block with the plant tissue needs to be trimmed to the suitable size and shape before sectioning.
(H) The same shoot apex as in (F) embedded in a trimmed agarose block.
(I) The agarose block is arranged in a vibratome for sectioning.
(J) Ongoing sectioning of an agarose block with a razor blade.
(K) Sections are collected in MTSB in the PAP circle.
Scale bars: 5 mm in (A)–(E), (G), and (I)–(K); 1 mm in (H); 200 μm in (F).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-α-tubulin | Sigma-Aldrich | Cat# T5168, clone B-5-1-2 |
| Alexa Fluor 488 conjugated donkey anti-mouse IgG | Invitrogen | Cat# A-21202 |
| Chemicals, peptides, and recombinant proteins | ||
| Myoinositol | Sigma-Aldrich | Cat# I7508 |
| Nicotinic acid | Sigma-Aldrich | Cat# N4126 |
| Pyridoxine hydrochloride | Sigma-Aldrich | Cat# P8666 |
| Thiamine hydrochloride | Sigma-Aldrich | Cat# T1270 |
| Glycine | Sigma-Aldrich | Cat# G8898 |
| N6-Benzyladenine | Duchefa Biochemie | Cat# B0904 |
| FM4-64 | Thermo Fisher | Cat# T3166 |
| Propidium iodide | Sigma-Aldrich | Cat# P4170 |
| Agarose, low melting point | Promega | Cat# V2111 |
| Hemicellulase | Solarbio | Cat# H8110-15KU |
| Macerozyme R-10 | Yakult | Cat# L0021 |
| ProLong Gold antifade reagent | Thermo Fisher | Cat# P36930 |
| Poly-L-lysine | Sigma-Aldrich | Cat# P7890 |
| Murashige & Skoog Basal medium (with vitamins) | Phytotech | Cat# M519 |
| Phytagel | Sigma-Aldrich | Cat# P8169 |
| trans-Zeatin | Phytotech | Cat# Z125 |
| NaCl | Sigma-Aldrich | Cat# S5886 |
| KCl | Sigma-Aldrich | Cat# V900068 |
| KOH | Sigma-Aldrich | Cat# SA-P1767 |
| Na2HPO4 | Sigma-Aldrich | Cat# V900061 |
| KH2PO4 | Sigma-Aldrich | Cat# P5655 |
| Tris-base | AMRESCO | Cat# 0497 |
| Paraformaldehyde | Sigma-Aldrich | Cat# 158127 |
| 50% glutaraldehyde | Sigma-Aldrich | Cat# 340855 |
| Tween 20 | Sigma-Aldrich | Cat# V900548 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787 |
| PIPES | GENVIEW | Cat# BP248 |
| MgSO4·7H2O | Sigma-Aldrich | Cat# M1880 |
| EGTA | Sigma-Aldrich | Cat# E3889 |
| Bovine serum albumin (BSA), Fraction V | AMRESCO | Cat# 0332 |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| Agar | Sigma-Aldrich | Cat# V900500 |
| Sucrose | Sigma-Aldrich | Cat# V900116 |
| Experimental models: organisms/strains | ||
| Arabidopsis thaliana: p35S: GFP-MBD | (Hamant et al., 2008) | N/A |
| Solanum lycopersicum: cultivar M82 | Charles Rick Tomato Genetics Resource Center, UC Davis | N/A |
| Other | ||
| Stereo microscope | Nikon | SMZ1000 |
| Confocal microscope | Zeiss | LSM700 |
| Confocal microscope | Nikon | A1 |
| PAP pen | Daido Sangyo | Cat# 1-5902-01 |
| 1 mL insulin syringe | BD | Cat# 320310 |
| 15 mL conical tube | Corning | Cat# 430791 |
| Leica vibratome | Leica Biosystems | VT1000 S |
| 0.22 μm filter | Merck Millipore | Cat# SLGPR33RB |
| Cyanoacrylate adhesive (502 super glue) | Aibida | Cat# 20280 |
Materials and equipment
The Key resources table details all required materials and equipment.
Step-by-step method details
CMT live imaging: prepare apex culture medium (ACM)
-
1.
Melt MS medium using a microwave. If the medium is freshly autoclaved, it can be used directly.
-
2.
Add vitamins to the MS to a 1× final concentration.
-
3.
Add BAP stock to a final concentration of 200 nM.
-
4.
Mix gently and pour the medium into the Petri dishes (60 × 10 mm).
Note: The MS medium should be cooled down to ∼50°C before adding the vitamins and BAP. Steps 2–4 should be carried out in a laminar hood. We recommend using the ACM immediately. ACM plates could also be stored at 4°C for 2–3 weeks.
CMT live imaging: dissection
-
5.
Choose plants with inflorescences of about 2–3 cm high for further experiments (Figures 1A and 1B).
-
6.
Remove the outer mature flowers as much as possible using tweezers. Cut the stem at ∼1 cm length to the tip, and insert the stem into ACM.
-
7.
Dissect further using tweezers with extra fine tips, starting from the oldest flowers, removing the flowers one by one carefully until the floral stage you are interested in.
-
8.
Move the whole shoot apex to new ACM. Insert the stem into the ACM with an angle that makes the surface of the particular sepal primordium parallel to the medium surface (Figures 1C–1F).
-
9.
When finished, pour sterile water into the Petri dish and make sure the apex is under water. Rehydrate for around 10 min. Remove the water, seal the Petri dish, and move into a growth chamber for a recovery for 4 h or longer.
To dissect the vegetative shoot apex, keep the entire plant with as many roots as possible. If the plants are grown in soil, they should be washed in tap-water before dissection. Old leaves should be cut off as clean as possible using the needle from a 1 mL insulin syringe (BD). In order to expose the very young leaf primordia (e.g., P1-P3), a fine dissection is carried out to remove the leaves covering the shoot apical meristem, using fine tweezers under the stereo microscope at high magnification.
Note: When dissecting the flowers, the cutting should be at the basis of the pedicel as close to the stem as possible. To facilitate the cutting, the dissection can be performed under water by pouring sterile water into the Petri dish (from step 3 onward). The dissected shoot apex can be cultured for a long time in order to follow the growth and CMT dynamics (Figures 1E and 1F). For a video of the dissection process, refer to (Prunet, 2017).
CRITICAL: Fine tweezers and a stereo microscope with high magnifications are absolutely necessary for a successful dissection. The recovery period is important for the visualization of CMTs and should be at least 4 h.
CMT live imaging: staining the shoot apices (optional)
-
10.Staining cell membrane with FM4-64
-
a.Dilute the FM4-64 stock solution to a final concentration of 0.33 mg/mL.
-
b.Use a micropipette to apply 5 μL on the flower bud. Close the Petri dish and keep it in darkness for 5–10 min.
-
c.Rinse briefly with sterile water for two times.
-
a.
-
11.Staining cell wall with propidium iodide
-
a.Use a micropipette to apply 5 μL propidium iodide solution. Keep the Petri dish in darkness for 5–10 min.
-
b.Rinse briefly with sterile water for two times.
-
a.
Note: The cortical microtubule signal is very dense at both the anticlinal and periclinal cell membranes. This property makes it easy to distinguish the cell contour. However, if large scale segmentation and quantification is required, staining the membrane or cell wall will be very helpful.
CMT live imaging: acquisition
-
12.Prepare the microscope
-
a.We use upright confocal microscope LSM 700 (Zeiss) equipped with water immersion objectives (W Plan-Apochromat 40×/1.0 DIC and/or W Plan-Apochromat 63×/1.0 M27) to image sepal primordia.
-
b.Clean the objectives with 70% ethanol before starting the experiments using lens paper (swipe method) or high-purity cotton applicator (swirl method).
-
a.
-
13.Position the samples
-
a.Pour sterile water into the Petri dish and make sure the shoot apex is submerged.
-
b.Use the pipette to blow away any air bubble on the shoot apex.
-
c.Position the Petri dish on the stage center (Figure 1G). For the time-lapse imaging, it will be helpful to mark the front position on the Petri dish using a marker (optional).
-
a.
-
14.Acquisition
-
a.Use transmission light to find the flower bud.
-
b.Set up the pinhole size, laser power, gain, and offset. The detailed settings we used for CMT acquisition are listed below:
-
i.According to the strength of signal, use lower laser power as much as possible (e.g., 2%–10% of 10 mW laser) to avoid photo-bleaching and sample damage.
-
ii.Set the pinhole size to 1 AU (Airy unit).
-
iii.For optimal contrast and to avoid overexposure, we use the range indicator to set gain and offset. The ideal setting is that very few red points (overexposure noise) are in the preview image.
-
i.
-
c.We usually select the frame size as 1,024 × 1,024 which matches closely the diffraction-limited performance of the objectives. To precisely optimize the frame size, the user can also click the “optimal” button in the ZEN software for calculation of appropriate number of pixels. With these settings, our xy pixel dimensions are 114 μm.
-
d.High scanning speed under line scanning mode with 3× averaging will increase the signal-to-noise ratio.
- e.
-
f.After the acquisition, remove the water as much as possible, cover the Petri dish with lid, seal, and put the Petri dish back to the growth chamber.
-
a.
Note: Clean the objective before acquisition to make sure there is no contamination on the objective. 63× objective performs better in particular on small younger sepal primordia. During acquisition, avoid over-sampling in z steps to minimize the photo-bleaching.
Figure 3.

Morphology of cortical microtubules visualized by live imaging and immunostaining
(A–C) Live imaging of CMTs in Arabidopsis sepal primordium after staining with FM4-64. (A) 3D reconstruction of CMT array at the sepal surface (periclinal walls). (B) FM4-64 signals at the surface. (C) Tilted image of the area framed in (A) representing the CMTs in anticlinal walls (arrowheads).
(D and E) Immunostaining of CMTs in a cross-section of tomato leaf primordium. (D) Double labeling of CMTs (green color) and nucleus (red color) in each cell by tubulin antibodies and DAPI, respectively. (E) The organization of CMTs in (D). Scale bars: 20 μm.
CMT live imaging: image processing
-
15.3D reconstruction
-
a.To visualize the CMT network in 3D, we use the Zeiss software (ZEN, black edition), and choose the transparency rendering mode to visualize the signal at the sepal/leaf surface.
-
b.Surface renderings can also be obtained using MERRYPROJ (de Reuille et al., 2005) or MorphoGraphX software (Barbier de Reuille et al., 2015).
-
c.To visualize the CMT organization in anticlinal walls, choose subset of stack using ZEN software, make a 3D reconstruction, and tilt the 3D image properly to show the anticlinal CMT networks.
-
a.
-
16.
CMT anisotropy
We use Fiji plugin—FibrilTool to quantify CMT anisotropy. The detailed steps for the quantification can be obtained in (Boudaoud et al., 2014).
CMT immunostaining: tissue fixation
-
17.Prepare fixative
-
a.Add 4 g paraformaldehyde to 80 mL MTSB. Dissolve by adding a few NaOH pellets (around 0.1–0.15 g) and using a magnetic stir bar.
-
b.Add 1 mL 50% glutaraldehyde. Adjust pH to 7.0 with H2SO4.
-
c.Add 300 μL Tween 20 and 300 μL Triton X-100. Mix well and fill up to 100 mL with MTSB.
-
a.
-
18.Tissue fixation
-
a.Dissect tomato shoot apices under a dissecting microscope.
-
b.Dig a hole in a 3% agar plate. Vertically insert a tomato shoot apex from the tissue culture medium.
-
c.Remove leaves older than P3 using the needle from a 1 mL insulin syringe (Figure 2F).
-
d.Incubate the dissected shoot apices in a 15 mL conical tube with 10 mL fixative.
-
e.Vacuum infiltrate at −0.075 MPa (550 mm Hg) for 10 min at room temperature (23°C–25°C). Repeat for a total of 3 times (3 × 10 min). Renew the fixative each time.
-
f.Incubate in new fixative for an additional 3 h at room temperature (23°C–25°C) with the tube horizontally arranged.
-
g.Discard the fixative. Incubate samples in MTSB for 20 min with the tube horizontally arranged. Repeat for a total of 3 times (3 × 20 min). Renew MTSB each time.
-
a.
Note: Do not include too many samples for fixation in one 15 mL conical tube. The volume of fixative should be at least 20 times of the volume of samples. Release the vacuum pump slowly to avoid any damage to plant tissues.
CMT immunostaining: tissue embedding and sectioning
-
19.Prepare 6% low melting agarose
-
a.Add 1.8 g low melting agarose to 30 mL ddH2O. Heat to dissolve in a water bath at 96°C.
-
b.Start to prepare low melting agarose 1 h before the end of tissue fixation. Cool down the melted agarose to ∼40°C before tissue embedding.
-
a.
-
20.Tissue embedding
-
a.Pour fixed samples of tomato shoot apices into a 6 cm Petri dish together with MTSB.
-
b.Set a hot plate to 37°C.
-
c.Put a 44 × 44 × 7 mm (L × W × H) plastic weighing dish on the hot plate. Fill up with melted agarose (∼8–10 mL per plate).
-
d.Quickly put samples into the melted agarose with tweezers, and adjust to the desired orientation (Figure 2G).
-
e.When all samples are embedded, transfer the weighing dish to 4°C and leave for 15 min to let the agarose solidify.
-
a.
Note: Leave enough space among samples when you embed in agarose.
-
21.Tissue sectioning
-
a.Take a poly-L-lysine coated glass slide. Draw a circle on the slice using a PAP pen. Fill the circle with MTSB.
-
b.Cut a piece of solidified agarose containing an individual tomato shoot apex using a razor blade. Trim the agarose block to an appropriate size suitable for a vibratome (Figure 2H).
-
c.Fix the agarose block onto the specimen disc with cyanoacrylate adhesive (e.g., 502 super glue). Also evenly apply adhesive to the vertical faces of the block. Fill the cooling bath with ice, and fill the buffer tray with 1× PBS (Figure 2I).
-
d.Make agarose sections of 40–50 μm thickness with sectioning speed at 0.65–0.78 mm/s (set SPEED value as 7–7.5 for a VT1000 S vibratome) and the sectioning frequency as 40 Hz (set FREQ value at 4 for a VT1000 S vibratome) (Figure 2J).
-
e.Pick up sections using a brush and load them into the MTSB in the PAP circle (Figure 2K).
-
f.Put slides with sections in a humid box.
-
a.
Note: Wait a few seconds after drawing with the PAP pen to let the mark dry before adding MTSB. Otherwise, the ink of the PAP pen can mix with the buffer.
Note: The size of PAP circle depends on the number and size of agarose sections that are collected. To collect multiple sections (e.g., successive sections from one individual sample) on one slide, rather draw the PAP-rectangles which can cover a larger area of the slide.
Note: Sections are transparent, which makes them difficult to be seen in 1× PBS buffer. It is necessary to apply adhesive to the vertical faces of the block to make it visible. The cyanoacrylate adhesive is transparent when it is liquid, and becomes opaque after drying. It thus outlines the transparent section to make it visible in 1× PBS buffer.
CMT immunostaining: primary antibody incubation
-
22.Cell permeabilization
-
a.Prepare the enzymatic solution: 1% (w/v) hemicellulose with 1% (v/v) Triton X-100 in MTSB.
-
b.Replace MTSB by the enzymatic solution in the PAP circle using a pipettor. Incubate sections in the enzymatic solution for 15 min at room temperature (23°C–25°C).
-
a.
-
23.Incubation with primary antibody
-
a.Remove the enzymatic solution from the slices. Wash sections with 1× TBS three times for 5 min each by incubation without shaking.
-
b.Prepare the primary antibody solution: mouse anti-α-tubulin antibody is diluted in 1× TBS containing 1% (w/v) BSA at 1:800 dilution.
-
c.Remove 1× TBS from the slides and apply 200 μL primary antibody solution per slide. Incubate the slides in a humid box for overnight (12–16 h) at 4°C.
-
a.
Note: Hemicellulose digests the cell wall, while Triton X-100 increases the permeabilization of the plasma membrane by removing membrane remnants and soluble proteins. Combination of hemicellulose and Triton X-100 treatments increase the penetrance of antibodies. The enzymes used for cell wall digestion and the duration of Triton X-100 treatment should be optimized according to the plant materials. To label CMTs in tomato leaf primordia, we found it was sufficient to treat the samples with 1% hemicellulose and 1% Triton X-100 for 15 min. To label CMTs in Arabidopsis leaf primordia, samples should be treated with 1% hemicellulose, 0.1% Macerozyme R-10 and 1% Triton X-100 in MTSB for 15 min at room temperature (23°C–25°C), followed by 3 × 5 min wash in 1% Triton X-100 in MTSB. Including a step of cold-methanol treatment after enzymatic digestion can further increase the permeability of the cell wall and plasma membrane, and also facilitates the binding of antibodies (Shimamura, 2015).
CMT immunostaining: secondary antibody incubation
-
24.Incubation with secondary antibody
-
a.Transfer samples to room temperature (23°C–25°C). Remove primary antibody solution from the slides. Wash sections with 1× TBS three times for 10 min each by incubation without shaking.
-
b.Prepare secondary antibody solution: Alexa Fluor 488 conjugated donkey anti-mouse IgG is diluted in 1× TBS containing 1% (w/v) BSA at 1:500 dilution.
-
c.Remove 1× TBS from the slides and apply 200 μL secondary antibody solution per slide. Incubate the slides in a humid box for 2 h at 37°C in the dark.
-
a.
-
25.Nuclei staining (optional)
-
a.Remove secondary antibody solution from the slides. Wash sections with 1× TBS three times for 10 min each by incubation without shaking.
-
b.Apply 200 μL 1 μg/mL DAPI per slide. Incubate for 15 min at room temperature (23°C–25°C) in the dark.
-
a.
CMT immunostaining: CLSM
-
26.Mounting
-
a.Remove DAPI solution and incubate sections in 1× TBS for 10 min.
-
b.Remove 1× TBS from the slices. Mount the slide with ProLong Gold (1.47 RI) antifade reagent. This mounting media both preserves fluorescence and provides near aberration-free imaging on oil immersion lenses. Cover the slide with a coverslip.
-
a.
-
27.CLSM
-
a.CLSM is performed using a confocal laser scanning microscope with 60× oil objective. We used a Nikon A1 confocal microscope with a Nikon Plan Apo VC 60×/1.40 oil objective. Due to DAPI’s broad emission, sequential acquisition of DAPI an Alexa Fluor 488 stacks is important to minimize bleed-through.
-
b.To detect Alexa Fluor 488 signal, a 488 nm laser line is used for excitation and emission is collected at 500–530 nm. The laser power is 10 mW. The pinhole size is 1.2 AU (30.7 μm).
-
c.To detect the DAPI signal, a 405 nm laser line is used for excitation and emission is collected at 425–475 nm. The laser power is 10 mW. The pinhole size is 1.2 AU (30.7 μm).
-
d.Set the scanning step as 0.5 μm to collect more details of CMTs in each stack. Perform maxima z-projection of generated stacks to obtain the overall organization of CMTs in individual cells or the whole tissue section.
-
a.
Note: The amount of mounting medium (e.g., ProLong Gold antifade reagent) should be sufficient to fill the PAP circle. Care must be taken during loading the coverslip to avoid air bubbles which can interfere with the microscopy.
Note: The use of oil objectives gives higher resolution of CMT organization than air or long distance water objectives due to their higher numerical aperture. For visualizing CMTs in Arabidopsis cells, it is better to use a 100× oil objective.
Expected outcomes
Morphology of CMTs in Arabidopsis sepal primordium by live imaging
CMT bundles associated with GFP-MBD can be visualized in the epidermal cells of the abaxial side of Arabidopsis sepal primordium (Figures 3A and 3B). The orientation of CMTs depends on the developmental stages of sepal primordium (Zhao et al., 2020).
Morphology of CMTs in tomato leaf primordium by immunostaining
Labeling of CMTs in cells of inner tissues can be successfully achieved through immunostaining of agarose-gel sections (Kitaeva et al., 2016; Zhao et al., 2020). In the cross-sections of tomato leaf primordia, α-tubulin antibodies bind to the CMTs along the anticlinal cell walls. The overall organization of CMTs in majority of cells displays a highly anisotropic arrangement, in which the orientation of CMT bundles is parallel to the adaxial-abaxial axis (Figures 3C and 3D) (Zhao et al., 2020).
Limitations
-
1.
Due to several factors such as light scattering and light absorption by certain subcellular components, live imaging can only penetrate at limited tissue depth in plants. It is therefore more applicable to observing CMTs in the outer cell layers of the tissue.
-
2.
Immunostaining of agarose-gel sections enables the visualization of CMTs in cells within all tissues. However, two main limitations still exist. First, the signal can only be detected in 1 or 2 cell layers of tissue sections. This can be either due to the limited light penetration of fluorescent microscopy, or the limited antibody penetration. Second, since the relative position of each cell is different within a tissue, one cannot detect the overall CMT signals for every cell in a certain section. For example, if the cutting plane is away from the cortical region of a cell, the CMT signal may not be detected.
Troubleshooting
Problem 1
No or low signal during immunostaining.
Potential solution
There are multiple reasons for no or low signal. First, it can be largely due to an inadequate cell wall digestion and/or permeabilization. One can increase the duration of wall digestion and permeabilization. Second, it can be possible that the concentration of antibodies is too low or the duration of antibody incubation is not adequate. In this case, one can increase the concentration of primary and/or secondary antibody, or increase the antibody incubation time. The concentration and incubation time of antibodies should be adjusted for different plant tissues. In addition, it can also be due to loss of activity of antibodies. The antibodies should be aliquoted before storage to avoid repeated freezing and thawing. Moreover, samples that dried out also causes no or low signal. To avoid drying out, sections should be always submerged in buffers and the slides should be kept in a humid box.
Problem 2
No or few filamentous structures can be visualized.
Potential solution
Microtubules are highly dynamic structures, and many factors or stresses can result in depolymerization of microtubules. When depolymerization occurs, the percentage of filamentous structures of CMTs decreases, while the amount of tubulins and tubulin dimers increase. In that case, a diffuse signal can be observed in cells.
As to live imaging, shorten the time of dissection to avoid tissue drying, or perform the dissection of tissues in the water. It is better to dissect tissues a few hours before imaging to let tissues recover from the wounding stress (Hamant et al., 2019).
As to immunostaining, plant tissues must be fixed in the fixative for a sufficient long time. The fixation must be performed at room temperature (23°C–25°C) rather than at lower temperature such as 4°C, since chilling results in microtubule depolymerization.
Problem 3
Deformation of cells and tissues.
Potential solution
Carefully remove unnecessary tissues using a sharp tweezer or syringe. Do not destroy the desired tissues. The fixation time can be increased for samples undergoing immunostaining. When performing sectioning on the vibratome, always use a new razor blade each time and do not set the speed value too high.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jan Trass (jan.traas@ens-lyon.fr).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate unique datasets or code.
Acknowledgments
Y.J. acknowledges support from the K.C. Wong Education Foundation, the National Natural Science Foundation of China (NSFC) grants 31825002 and 31861143021, and CAS Key Research Project of the Frontier Science (grant ZDBS-LY-SM012). Y.J. is a Newton Advanced Fellow of the Royal Society. F.D. was supported by the Young Scientists Fund of NSFC (32000507). F.Z. and J.T. were supported by the ERC advanced grant MORPHODYNAMICS (grant number 294397) and by the ANR ERA CAPS grant Gene2Shape. J.T. was supported by the Inria Project Lab “Morphogenetics.”
Author contributions
Conceptualization, J.T. and Y.J.; Investigation, F.D. and F.Z.; Writing – Original Draft, F.D. and F.Z.; Writing – Review & Editing, J.T. and Y.J.; Funding Acquisition, J.T., Y.J., F.D., and F.Z.
Declaration of interests
J.T. is a member of the advisory board of Current Biology. The authors declare no other competing interests.
Contributor Information
Jan Traas, Email: jan.traas@ens-lyon.fr.
Yuling Jiao, Email: yljiao@genetics.ac.cn.
References
- Barbier de Reuille P., Routier-Kierzkowska A.L., Kierzkowski D., Bassel G.W., Schupbach T., Tauriello G., Bajpai N., Strauss S., Weber A., Kiss A. MorphoGraphX: a platform for quantifying morphogenesis in 4D. eLife. 2015;4:05864. doi: 10.7554/eLife.05864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaoud A., Burian A., Borowska-Wykret D., Uyttewaal M., Wrzalik R., Kwiatkowska D., Hamant O. FibrilTool, an ImageJ plug-in to quantify fibrillar structures in raw microscopy images. Nat. Protoc. 2014;9:457–463. doi: 10.1038/nprot.2014.024. [DOI] [PubMed] [Google Scholar]
- de Reuille P.B., Bohn-Courseau I., Godin C., Traas J. A protocol to analyse cellular dynamics during plant development. Plant J. 2005;44:1045–1053. doi: 10.1111/j.1365-313X.2005.02576.x. [DOI] [PubMed] [Google Scholar]
- Hamant O., Das P., Burian A. Time-lapse imaging of developing shoot meristems using a confocal laser scanning microscope. Methods Mol. Biol. 2019;1992:257–268. doi: 10.1007/978-1-4939-9469-4_17. [DOI] [PubMed] [Google Scholar]
- Hamant O., Heisler M.G., Jönsson H., Krupinski P., Uyttewaal M., Bokov P., Corson F., Sahlin P., Boudaoud A., Meyerowitz E.M. Developmental patterning by mechanical signals in Arabidopsis. Science. 2008;322:1650–1655. doi: 10.1126/science.1165594. [DOI] [PubMed] [Google Scholar]
- Kitaeva A.B., Demchenko K.N., Tikhonovich I.A., Timmers A.C., Tsyganov V.E. Comparative analysis of the tubulin cytoskeleton organization in nodules of Medicago truncatula and Pisum sativum: bacterial release and bacteroid positioning correlate with characteristic microtubule rearrangements. New Phytol. 2016;210:168–183. doi: 10.1111/nph.13792. [DOI] [PubMed] [Google Scholar]
- Prunet N. Live confocal imaging of developing Arabidopsis flowers. J. Vis. Exp. 2017;122:55156. doi: 10.3791/55156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura M. Whole-mount immunofluorescence staining of plant cells and tissues. Plant Microtechniques and Protocols. 2015:181–196. [Google Scholar]
- Zhao F., Du F., Oliveri H., Zhou L., Ali O., Chen W., Feng S., Wang Q., Lu S., Long M. Microtubule-mediated wall anisotropy contributes to leaf blade flattening. Curr. Biol. 2020;30:3972–3985. doi: 10.1016/j.cub.2020.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate unique datasets or code.