

Although combination antiretroviral (ARV) therapy is highly effective in controlling the progression of HIV disease, drug resistance can be a major obstacle. Recent findings suggest that resistance can develop without ARV target gene mutations.
KEYWORDS: antiretroviral drugs, envelope glycoprotein, virus transmission, gp41, human immunodeficiency virus
ABSTRACT
Despite the effectiveness of antiretroviral (ARV) therapy, virological failure can occur in some HIV-1-infected patients in the absence of mutations in drug target genes. We previously reported that, in vitro, the lab-adapted HIV-1 NL4-3 strain can acquire resistance to the integrase inhibitor dolutegravir (DTG) by acquiring mutations in the envelope glycoprotein (Env) that enhance viral cell-cell transmission. In this study, we investigated whether Env-mediated drug resistance extends to ARVs other than DTG and whether it occurs in other HIV-1 isolates. We demonstrate that Env mutations can reduce susceptibility to multiple classes of ARVs and also increase resistance to ARVs when coupled with target-gene mutations. We observe that the NL4-3 Env mutants display a more stable and closed Env conformation and lower rates of gp120 shedding than the WT virus. We also selected for Env mutations in clinically relevant HIV-1 isolates in the presence of ARVs. These Env mutants exhibit reduced susceptibility to DTG, with effects on replication and Env structure that are HIV-1 strain dependent. Finally, to examine a possible in vivo relevance of Env-mediated drug resistance, we performed single-genome sequencing of plasma-derived virus from five patients failing an integrase inhibitor-containing regimen. This analysis revealed the presence of several mutations in the highly conserved gp120-gp41 interface despite low frequency of resistance mutations in integrase. These results suggest that mutations in Env that enhance the ability of HIV-1 to spread via a cell-cell route may increase the opportunity for the virus to acquire high-level drug resistance mutations in ARV target genes.
INTRODUCTION
Antiretrovirals (ARVs) are categorized into five different classes based on their targets: nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), integrase (IN) strand transfer inhibitors (INSTIs), protease (PR) inhibitors (PIs), and entry inhibitors (Ent-Is) (1). The use of combinations of these ARVs (combination antiretroviral therapy [cART]) has proven remarkably effective in controlling HIV disease progression and prolonging survival. However, resistance to ARVs emerges in some patients because of poor adherence, use of a suboptimal drug regimen, and/or lack of viral load monitoring, particularly in resource-limited settings. Transmitted drug resistance is therefore becoming an increasingly serious problem in many parts of the world (2). In most cases, HIV drug resistance is the consequence of mutations that emerge in the viral genes targeted by the drugs (2). However, particularly in the case of PIs and INSTIs, virological failure can occur in the absence of target gene mutations (3–10), indicating that mutations outside the target gene can contribute to drug resistance. For example, in vitro selection studies have demonstrated that mutations in the 3′ polypurine tract (3′ PPT) or in the viral long terminal repeat (LTR) can confer resistance to INSTIs (11, 12), and mutations in Gag and the envelope glycoprotein (Env) have been implicated in PI resistance (13, 14).
HIV-1 Env is incorporated into virions as a noncovalently bound complex comprising three molecules each of gp120 and gp41 (15). The binding of gp120 to CD4 on the target cell surface triggers a conformational rearrangement in Env that exposes the coreceptor (CCR5 or CXCR4)-binding site. Interaction of gp120 with coreceptor promotes the refolding of gp41 heptad repeat 1 and 2 (HR1 and 2) to form an antiparallel six-helix bundle that mediates the fusion of viral and cellular membranes, allowing viral entry into the cytosol of the target cell (16).
HIV-1 Env is the only viral protein exposed on the surface of the infected cell or viral particle; it is therefore the target of neutralizing antibodies (NAbs) that can block viral entry and induce antibody-mediated effector functions. Single-molecule fluorescence resonance energy transfer (smFRET) analysis has revealed that Env trimers fluctuate between closed (state 1), intermediate (state 2), and open (state 3) conformations (17–19). The binding properties of NAbs are influenced by the conformational state of Env. Therefore, NAbs are useful as molecular probes to investigate the structure and conformation of Env (17, 19).
HIV-1 can spread either via a cell-free route or by cell-cell transmission at points of cell-cell contact known as virological synapses (VSs). The formation of a VS is initiated by the interaction of Env on the infected cell and CD4 on the target cell (20–22), although Gag can accumulate at the VS even in the absence of Env (23). The VS is stabilized by cellular adhesion proteins, such as LFA-1 and ICAM-1, and lipid raft microdomains and the actin cytoskeleton are also implicated in VS formation (24–26). At least in vitro, cell-cell transfer of HIV-1 is markedly more efficient than cell-free infection (27–29). HIV-1 transmission at a VS provides a higher multiplicity of infection (MOI) than cell-free infection, resulting in multiple copies of proviral DNA in the target cells (30–33). This high MOI allows HIV-1 to overcome multiple barriers to infection, such as those imposed by ARVs, NAbs, and inhibitory host factors (32–38). Although it is challenging to directly compare the relative contribution of cell-free infection and cell-cell transfer to HIV-1 dissemination and pathogenesis in vivo, several studies have suggested the importance of cell-associated virus in HIV-1 propagation in animal models of HIV-1 infection (30, 39–43). If cell-cell transfer, and the resulting higher MOI, is a common occurrence in vivo, one would expect to find large numbers of multiply infected cells in HIV-1-infected individuals. This has been observed in some (44–46) but not other (47) studies. Further analysis will be needed to understand the importance of cell-cell transfer in viral dissemination in vivo.
We previously reported that HIV-1 evades blocks to virus replication by acquiring Env mutations that enhance the capacity of the virus to spread via cell-cell transmission (48). By propagating HIV-1 mutants containing substitutions in the p6 domain of Gag that severely delay virus replication (49), we selected for second-site compensatory mutations Env-Y61H and P81S in gp120 and the Env-A556T mutation in gp41. These Env mutations decrease the sensitivity of the virus to the highly potent INSTI dolutegravir (DTG). Moreover, propagation of HIV-1 in the presence of DTG led to the selection of the DTG-escape mutant Env-A539V in the absence of any mutations in IN. These four Env mutations cluster within the C1 domain of gp120 and the HR1 domain of gp41 (Fig. 1), which have been shown through mutagenesis and structural studies to be critical for the stability of gp120-gp41 association (50–52). These results suggest that Env mutations that increase cell-cell spread may alter the stability of the gp120-gp41 interaction and demonstrate that mutations in Env can confer resistance to an ARV, at least in a subtype B, CXCR4-tropic laboratory-adapted HIV-1 strain. Whether Env-mediated drug resistance occurs in clinical HIV-1 isolates is unclear, because Env from laboratory-adapted and primary HIV-1 strains differs in significant functional and structural aspects. Laboratory-adapted isolates often sample the open Env conformation and are more sensitive to NAbs and soluble CD4 (sCD4) than primary isolates (17, 53, 54). sCD4 inhibits HIV-1 entry not only by competing with CD4 on the target cell but also by inducing the shedding of gp120 from the surfaces of viral particles. Env complexes from laboratory-adapted isolates are more prone to sCD4-induced gp120 shedding than primary isolates (55–57), indicating that Env stability differs between laboratory-adapted and primary isolates.
FIG 1.

The location of previously reported Env mutations that overcome blocks to virus replication (48). (A) Schematic of the HIV-1 Env-coding region with the position of mutations indicated using NL4-3 (and HXB2, in parentheses) numbering. Labeled domains are defined as follows: C1 to C5, constant regions 1 to 5; V1 to V5, variable regions 1 to 5; FP, fusion peptide; HR1/HR2, heptad repeat 1/2; DSL, disulfide loop; MPER, membrane proximal external region; TM, transmembrane; CT, cytoplasmic tail. (B) A Env structure of subtype B JR-FL SOSIP.664 (PDB accession number 5FYK [137]) with the position of the Env mutations highlighted. Most of gp120 is shown in white, with gp120 V1/V2 and V3 loops colored in light yellow and light green, respectively. gp41 is colored in tan. Y61, P81, A539, and A556 are highlighted in green, yellow, red, and blue, respectively. Structural model was generated using UCSF Chimera software (138).
In the present study, we examined whether our previously described Env mutations confer resistance not only to DTG but also to other classes of ARVs. To evaluate the impact of the Env mutations on the conformation and stability of the Env complex, we examined their effects on sensitivity to NAbs and on gp120 shedding. To further characterize the mechanism of Env-mediated drug resistance, we obtained Env mutants through in vitro selection experiments by propagating clinically relevant HIV-1 clones in the presence of ARVs and analyzed the phenotypes of the selected Env mutants. Finally, to address the possible in vivo relevance of Env-mediated drug resistance, we analyzed viral sequences obtained from the plasma of individuals failing a raltegravir (RAL)-containing regimen. The results indicate that Env mutations confer resistance across a broad range of ARVs in vitro and that the NL4-3 Env mutations decrease gp120 shedding and alter the sensitivity of Env to NAbs. Finally, we show that in some individuals failing a RAL-containing regimen in the absence of resistance mutations in IN, changes arise at the highly conserved gp120-gp41 interface, as observed during INSTI escape in vitro.
RESULTS
The A539V mutation in gp41 provides a replication advantage over WT NL4-3 in the presence of multiple classes of ARVs.
We previously reported that the Env-A539V mutation provides resistance to the INSTI DTG (48). To examine whether Env-A539V also provides a selective replication advantage in the presence of other ARVs, the SupT1 T-cell line was transfected with the WT NL4-3 molecular clone or the Env-A539V derivative in the absence or presence of various concentrations (0.1 to 3,000 nM) of a panel of ARVs. Replication kinetics were monitored by measuring the reverse transcriptase (RT) activity in the cell culture medium. Consistent with our previous report (48), Env-A539V exhibited faster replication kinetics than the wild type (WT) in the absence of drugs (Fig. 2). Whereas replication of WT NL4-3 was largely inhibited in the presence of 3.0 nM DTG, Env-A539V was able to replicate, albeit with delayed kinetics (Fig. 2A). By calculating the DTG 50% inhibitory concentration (IC50) based on the RT activity at the peak of replication in the absence of drugs, we found that Env-A539V showed 8.1-fold resistance to DTG relative to that of the WT (Fig. 2I). We also examined the sensitivity of Env-A539V to another INSTI, raltegravir (RAL). Env-A539V was able to replicate in the presence of 10 nM RAL and showed 5.4-fold resistance relative to that of WT NL4-3 (Fig. 2B and I). We also measured replication kinetics of Env-A539V in the presence of the NNRTI efavirenz (EFV) and the NRTI emtricitabine (FTC). Whereas replication of WT NL4-3 was strongly inhibited in the presence of 3.0 nM EFV or 30 nM FTC, Env-A539V still replicated at these concentrations (Fig. 2C and D). IC50 calculations showed that Env-A539V exhibited 28- and 5.4-fold resistance against EFV and FTC, respectively (Fig. 2I). Next, we examined the impact of PIs on the replication kinetics of Env-A539V. While replication of WT NL4-3 was strongly inhibited in the presence of 3.0 nM nelfinavir (NFV), the Env-A539V mutant replicated and exhibited 24-fold resistance (Fig. 2E and I). We also examined the sensitivity of Env-A539V to the allosteric integrase inhibitor (ALLINI) BI-224436, which induces aberrant IN multimerization and impairs the interaction between IN and the cellular cofactor LEDGF/p75 (58). As shown in Fig. 2F, Env-A539V caused a small but statistically significant reduction in the sensitivity to BI-224436 relative to that of WT NL4-3 (2.3-fold resistance) (Fig. 2I). These data demonstrate that Env mutations can overcome inhibition imposed by multiple classes of ARVs targeting not only postentry steps but also the maturation step of the HIV-1 replication cycle.
FIG 2.

Replication kinetics of Env-A539V in the presence of INSTIs, NRTI, NNRTI, PI, ALLINI, and Ent-Is. The SupT1 T-cell line was transfected with WT or Env-A539V proviral clones in the absence or in the presence of indicated concentrations of ARVs. (A and B) INSTIs; (C and D) NNRTI and NRTI; (E) PI; (F) ALLINI; (G and H) Ent-Is. Virus replication kinetics were monitored by measuring RT activity at the indicated time points. Data are representative of at least two independent experiments. (I) The SupT1 T-cell line was transfected with WT or Env-A539V proviral clones in the absence or in the presence of serial dilutions (0.01 to 3,000 nM) of ARVs. IC50 values were calculated based on RT levels at the peak of virus replication. Fold changes in IC50 were calculated compared to that for the WT. Data from at least two independent experiments are shown as means ± standard errors (SEs). ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired t test. The EFV and NFV data are shown on a separate bar graph to avoid compression of the y axis.
It has been suggested that entry inhibitors (Ent-Is) are effective in the context of cell-cell transmission (59, 60). In addition, several studies have shown that Env mutations selected in the presence of Ent-Is altered viral sensitivity to other anti-Env agents (61–64). These findings raise the possibility that Env mutations selected in the presence of ARVs might alter the viral sensitivity to Env-targeted inhibitors. To examine this question, we measured replication kinetics in the presence of the fusion inhibitor T-20 and BMS-378806, which blocks CD4-induced conformational changes in Env (19, 65–67). Interestingly, these Ent-Is efficiently suppressed replication of both WT NL4-3 and Env-A539V with no statistically significant differences in antiviral IC50s in a multicycle spreading infection (Fig. 2G to I). These observations suggest that Ent-Is can suppress the replication of Env mutants selected under the pressure of ARVs targeting the viral enzymes.
The Env-A539V mutation does not alter drug sensitivity in cell-free infection.
We previously proposed that Env mutations that confer resistance to ARVs do so by enhancing the efficiency of cell-cell transfer (48). Based on this hypothesis, the Env mutations would not be predicted to confer resistance in the context of cell-free infectivity. To test this hypothesis, we measured the single-round infectivity of WT NL4-3 and the Env-A539V mutant in the TZM-bl indicator cell line. As we reported previously (48), Env-A539V showed comparable cell-free infectivity relative to that of WT NL4-3 in the absence of inhibitor (Fig. 3A and Table 1). Consistent with the hypothesis that the reduced ARV sensitivity of the Env-A539V mutant is conferred at the level of cell-cell transmission, WT and Env-A539V infectivity was reduced to the same extent by the inhibitors (Fig. 3B to I and Table 1). Interestingly, Env-A539V did not alter susceptibility to either T-20 or BMS-378806, suggesting that Ent-Is efficiently suppress both cell-free infection and cell-cell transmission of HIV-1. We also measured cell-cell fusion activity of Env-A539V by coculture of TZM-bl cells with transfected 293T cells in the presence of a cocktail of EFV and DTG (200 nM each) to prevent infection of the TZM-bl cells (see Fig. S1 in the supplemental material). In contrast to the Y61H, P81S, and A556T mutations, which markedly suppressed cell-cell fusion activity (48), the Env-A539V mutation only slightly decreased cell-cell fusion compared to that of the WT.
FIG 3.
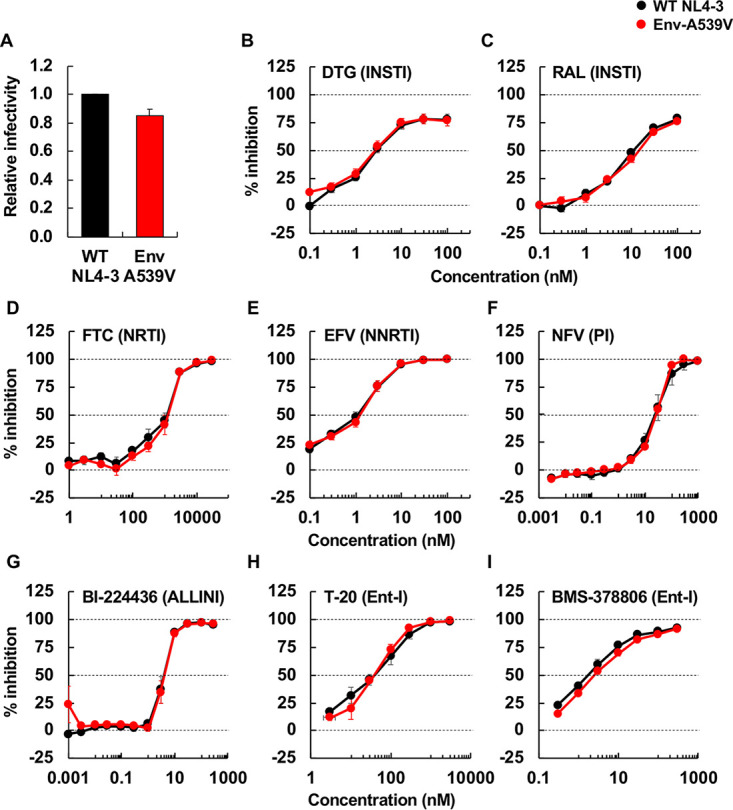
Cell-free infectivity of Env-A539V in the presence or absence of ARVs. (A) RT-normalized virus stocks produced from HeLa cells were used to infect TZM-bl cells. Luciferase activity was measured at 48 h postinfection. Infectivity of WT NL4-3 is normalized to 1.0. (B to I) TZM-bl cells were exposed to 100 TCID50 of WT or Env-A539V virus in the presence of various concentrations (from 0.003 to 3,000 nM) of ARVs and incubated for 48 h. For NFV and BI-224436, 293T cells were transfected with the indicated proviral clones in the presence of the inhibitors. At 48 h posttransfection, the supernatants were used to infect TZM-bl cells. Data from at least three independent experiments are shown as means ± SEs.
TABLE 1.
IC50 values in cell-free infection for WT NL4-3 and the A539V variant
| ARV class or NAb epitope | Drug or NAb | IC50 value (mean ± SE) |
Fold change | Significancea | |
|---|---|---|---|---|---|
| WT | Env-A539V | ||||
| INSTI | DTG (nM) | 3.3 ± 0.65 | 3.0 ± 0.72 | 0.91 | NS |
| RAL (nM) | 11 ± 0.94 | 15 ± 1.6 | 1.4 | NS | |
| NRTI | FTC (nM) | 558 ± 10 | 511 ± 30 | 0.92 | NS |
| NNRTI | EFV (nM) | 1.2 ± 0.31 | 1.4 ± 0.27 | 1.2 | NS |
| PI | NFV (nM) | 46 ± 21 | 28 ± 3.1 | 0.61 | NS |
| ALLINI | BI-224436 (nM) | 14 ± 2.0 | 14 ± 1.9 | 1.0 | NS |
| Ent-I | BMS-378806 (nM) | 2.3 ± 0.66 | 2.9 ± 0.54 | 1.3 | NS |
| T-20 (nM) | 42 ± 13 | 38 ± 2.9 | 0.90 | NS | |
| AMD3100 (nM) | 0.91 ± 0.070 | 0.77 ± 0.032 | 0.85 | NS | |
| sCD4 (0 h) (µg/ml) | 0.40 ± 0.031 | 0.97 ± 0.14 | 2.4 | * | |
| sCD4 (2 h) (µg/ml) | 0.30 ± 0.085 | 1.1 ± 0.21 | 3.7 | ** | |
| Anti-CD4 Ab | SIM.4 (dilution factor) | 88 ± 30 | 79 ± 24 | 0.90 | NS |
| Anti-V2 apex Ab | PG16 (µg/ml) | 13 ± 12 | 0.62 ± 0.30 | 0.048 | NS |
| PGT145 (µg/ml) | 1.3 ± 0.087 | 0.39 ± 0.22 | 0.30 | * | |
| Anti-V3 glycan Ab | 2G12 (µg/ml) | 1.3 ± 0.33 | 1.3 ± 0.54 | 1.0 | NS |
| PGT121 (µg/ml) | >10 | >10 | NDb | ND | |
| Anti-CD4bs Ab | VRC01 (µg/ml) | 0.11 ± 0.0070 | 0.11 ± 0.013 | 1.0 | NS |
| F105 (µg/ml) | 1.4 ± 0.51 | 4.3 ± 0.43 | 3.1 | ** | |
| Anti-CD4i Ab | 17b (µg/ml) | 0.81 ± 0.28 | 22 ± 12 | 27 | * |
| Anti-V3 loop Ab | 447-52D (µg/ml) | 0.39 ± 0.096 | 11 ± 2.6 | 28 | *** |
| Anti-gp41 MPER Ab | 10E8 (µg/ml) | 0.067 ± 0.020 | 0.12 ± 0.063 | 1.8 | * |
| Anti-gp120-gp41 interface Ab | 35O22 (µg/ml) | 0.062 ± 0.024 | 0.13 ± 0.026 | 2.1 | NS |
NS, not significant; ***, P < 0.001; **, P < 0.01; *, P < 0.05.
ND, not determined.
Cell-cell fusion activity of NL4-3 Env-A539V. HEK293T cells were transfected with the indicated proviral clones. At 24 h posttransfection, serial dilutions of the transfected HEK293T cells were cocultured with TZM-bl cells in the presence of a cocktail of EFV and DTG. Luciferase activity was measured at 24 h postinfection. Data from at least three independent experiments are shown as means ± SEs. ***, P < 0.001 by one-way ANOVA and Tukey’s multiple-comparison test. Download FIG S1, TIF file, 0.09 MB (95.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The Env-A539V mutation increases viral resistance to EFV when coupled with the RT-Y188L mutation.
To examine whether mutations in Env can increase the level of resistance conferred by drug target gene mutations, we tested EFV resistance of the RT-Y188L mutant (68) in the context of either WT or A539V Env. As shown in Fig. 4A (left), RT-Y188L showed a modest delay in replication compared to that of the WT in the absence of EFV, indicating that this RT mutation confers a small fitness defect. This replication defect was rescued by Env-A539V, consistent with our earlier finding that Env mutations can rescue the replication defect conferred by a mutation in IN (48). In the presence of 1.0 mM EFV, the Env-A539V mutation was able to rescue the replication kinetics of the RT-Y188L mutant at early time points (Fig. 4A, right), a result that was confirmed over a broad range of EFV concentrations (Fig. 4B and D). In contrast to the results for the multicycle spreading infection (Fig. 4B), the Env-A539V mutation did not increase EFV resistance of RT-Y188L in a single-cycle infectivity assay (Fig. 4C and D). Taken together, these results demonstrate that, in the context of a spreading infection in which virus transmission can take place via cell-cell transfer, an Env mutation can significantly increase the resistance conferred by a mutation in an ARV target gene.
FIG 4.
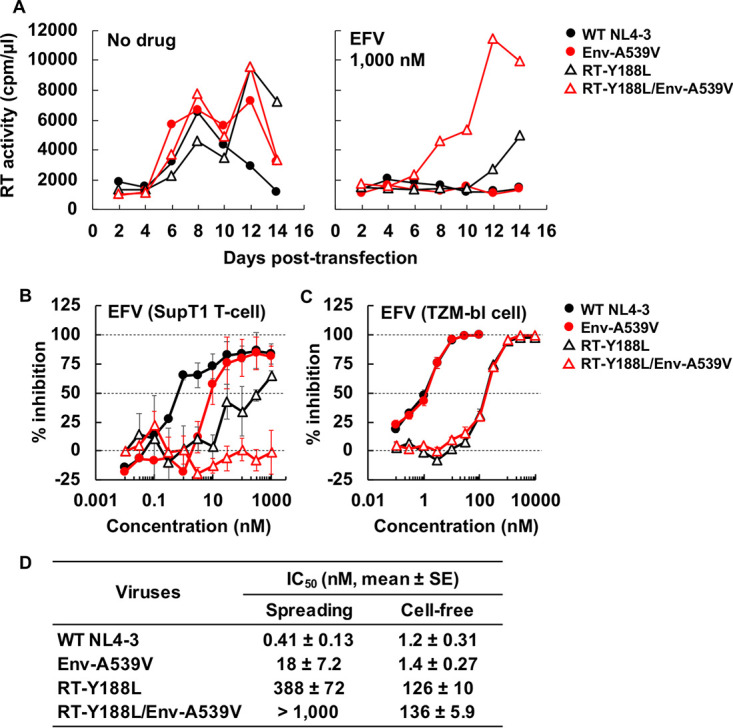
Effect of Env-A539V and RT-Y188L mutations on susceptibility to EFV. (A) The SupT1 T-cell line was transfected with the indicated proviral clones in the absence or presence of 1,000 nM EFV. Virus replication kinetics were monitored by measuring RT activity at the indicated time points. Data are representative of three independent experiments. (B) SupT1 T cells were transfected with the indicated proviral clones in the absence or presence of serial dilutions of EFV (0.03 to 1,000 nM). The dose-dependent inhibition curve was determined based on RT values at the peak of multicycle spreading virus replication. (C) TZM-bl cells were exposed to 100 TCID50 of the indicated viruses in the absence or presence of serial dilutions of EFV (0.1 to 10,000 nM). Luciferase activity was measured at 48 h postinfection. (D) IC50 values for EFV in multicycle spreading and cell-free infection based on the data in panels B and C. Data from at least three independent experiments are shown as means ± SEs. Statistical analysis by unpaired t test.
NL4-3 Env mutations that overcome blocks to HIV-1 replication in spreading infections stabilize the gp120-gp41 interaction.
Our previously reported Env mutations, which we selected for their ability to overcome blocks to virus replication in multicycle spreading infections, are located in the C1 domain of gp120 and the HR1 region of gp41 (Fig. 1B). In the crystal structure of the HIV-1 Env SOSIP trimer, which has recently been identified as adopting the state 2 conformation (69–72), these mutations are clustered at the gp120-gp41 interface. These domains of Env have been shown in mutagenesis and structural studies to be critical for the stability of the gp120-gp41 association (50–52, 62, 73). To provide clues regarding the mechanism by which Env mutations enhance the capacity of HIV-1 to spread via cell-cell transfer, we examined their impact on gp120-gp41 association. It is known that sCD4 induces gp120 shedding from virus particles (57); we therefore incubated purified virions with sCD4 at 37°C for 2 h and then measured the amount of particle-associated gp120 and p24 by Western blotting. As expected, levels of gp120 associated with WT NL4-3 particles were decreased in a dose-dependent fashion by sCD4 treatment (0.3 to 10 µg/ml). In contrast, the mutations in both gp120 (Env-Y61H and P81S) and gp41 (Env-A539V and A556T) showed significantly reduced sCD4-induced gp120 shedding (Fig. 5A). To quantify this effect, we calculated the 50% effective concentration (EC50) of sCD4-induced shedding. While the EC50 of sCD4-induced gp120 shedding for WT NL4-3 was 0.99 µg/ml sCD4, the EC50s for the Env-P81S and the other Env mutants were 3.5 and >10 µg/ml, respectively (Fig. 5B). To examine the interaction between sCD4 and a representative Env mutant, we measured the sensitivity of Env-A539V infectivity to sCD4 (Fig. 5C). Env-A539V showed a 3.7-fold resistance to sCD4 relative to that of the WT. Moreover, the IC50 of sCD4 against Env-A539V was 1.1 µg/ml (Table 1), which is ∼10-fold less than the EC50 of sCD4-induced gp120 shedding; this result suggests that sCD4 can bind to mutant Env at lower concentrations than needed for gp120 shedding, leading to inhibition of viral entry. To directly examine the interaction of Env with CD4, we measured the binding of a fusion protein (CD4-IgG2 [74]) comprising human IgG2 in which the Fv portions of both heavy and light chains have been replaced by the V1 and V2 domains of human CD4 (see Fig. S2). We observed comparable binding of WT and A539V Env to CD4-IgG2, suggesting that the Env-A539V mutation does not affect the binding affinity of Env for CD4. These results also suggest that sCD4 inhibits WT infection by inducing gp120 shedding and competing with gp120 binding to CD4 on target cells, whereas sCD4 inhibits infection of Env-A539V primarily by competing with CD4 binding on target cells. To further characterize the intrinsic stability of mutant Envs, we also performed a time-dependent gp120 shedding assay by incubating viruses at 37°C for up to 5 days (Fig. 5D). The Env mutations significantly reduced time-dependent gp120 shedding (Fig. 5E); whereas the half-life of WT NL4-3 gp120 shedding was 2.6 days, that of the Env mutants was >5 days. These data suggest that the NL4-3 Env mutations stabilize the gp120-gp41 interaction.
FIG 5.

Effect of Env mutations on sCD4-induced and time-dependent gp120 shedding. (A and B) Concentrated viruses were incubated with sCD4 at the indicated concentrations at 37°C for 0 or 2 h. Incubated viruses were subsequently purified through a 20% sucrose cushion, and viral proteins were detected by Western blotting. (A) A representative gel for the sCD4-induced gp120 shedding assay is shown. (B) The ratio of gp120 to p24 was quantified and plotted. Data from at least two independent experiments are shown as means ± SEs. (C) TZM-bl cells were exposed to 100 TCID50 of the indicated viruses incubated with sCD4 prior to infection for 0 or 2 h. Luciferase activity was measured at 48 h postinfection. Data from three independent experiments are shown as means ± SEs. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired t test, with asterisks for 0 h or 2 h incubation time points indicated in black or gray, respectively. (D and E) Concentrated viruses were incubated at 37°C for the indicated times. Incubated viruses were subsequently purified through a 20% sucrose cushion, and viral proteins were detected by Western blotting. (D) A representative gel for the time-dependent gp120 shedding assay is shown. (E) The ratio of gp120 to p24 was quantified and plotted. Data from at least three independent experiments are shown as means ± SEs. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired t test.
Binding of Env-A539V to CD4-IgG2. HEK293T cells transfected with the GFP-encoding reporter clone pBR43IeG expressing WT NL4-3 Env or the Env-A539V mutant were preincubated with 1.0 µg/ml of CD4-IgG2 at 4°C for 1 h. KFS is an Env-defective NL4-3 mutant included as a negative control. The cells were washed, and APC-conjugated anti-human IgG was used to detect bound CD4-IgG2. APC signals were normalized by GFP signal to calculate the CD4-IgG2 binding efficiency. Data are shown as means ± SEs from four independent experiments. *, P < 0.05 by one-way ANOVA and Tukey’s multiple-comparison test. Download FIG S2, TIF file, 0.09 MB (93.2KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The Env-A539V mutation reduces sensitivity to NAbs that recognize the CD4-induced Env conformation.
To probe the impact of the Env-A539V mutation on Env conformation, we examined the sensitivity of Env-A539V to a panel of NAbs that recognize different Env conformations (Fig. 6). We used three groups of NAbs. The first group (Fig. 6A to E), which included VRC01 (specific for the CD4 binding site [CD4bs]), PGT145, PG16 (specific for the V2 apex), 2G12, and PGT121 (V3-glycan specific), preferentially bind to the unliganded closed Env conformation (75–78). The second group (Fig. 6F) included 35O22, which is specific for the gp120-gp41 interface and shows no conformational preference (79). The third group (Fig. 6G to J) included F105 (CD4bs specific), 17b (specific for the CD4-induced epitope), 447-52D (V3 loop), and 10E8 (gp41 membrane-proximal external region [MPER]), which preferentially target the CD4-bound Env conformation (80–83). Although the Env-A539V mutation is not located within the epitopes of the NAbs used in this analysis, it altered the sensitivity to several NAbs, with reduced sensitivity to F105 (3.1-fold), 447-52D (28-fold), 17b (27-fold), and 10E8 (1.8-fold) and increased (33-fold) sensitivity to PGT145 (Table 1). To further probe the effect of the Env-A539V mutation on Env conformation, we compared NAb binding to Env on 293T cells by flow cytometry. As indicated in Fig. 6M and N, 17b and 10E8 bound less efficiently to Env-A539V than WT Env. In contrast, PGT145 and PG16 bound the mutant Env to a similar extent as WT NL4-3 Env (Fig. 6O and P). These data suggest that the Env-A539V mutation stabilizes the closed Env conformation. We also examined sensitivity to SIM.4 (an anti-CD4 Ab) (84) and AMD3100 (a CXCR4 antagonist) (85) to evaluate the impact of the Env-A539V mutation on Env function in HIV-1 entry. As shown in Fig. 6K and L, Env-A539V showed comparable sensitivity to SIM.4 and AMD3100 as WT NL4-3, suggesting that this mutation does not alter CD4 or CXCR4 dependency.
FIG 6.

Sensitivity or binding of Env-A539V to NAbs recognizing different Env conformations, anti-CD4 Ab, and coreceptor antagonist. TZM-bl cells were exposed to 100 TCID50 of WT NL4-3 or Env-A539V viruses in the presence of various concentrations of NAbs (A to J), anti-CD4 Ab (K), and CXCR4 antagonist (L); luciferase activity was measured at 48 h postinfection. Data from three independent experiments are shown as means ± SEs. 293T cells transfected with the green fluorescent protein (GFP)-encoding reporter clone pBR43IeG expressing WT NL4-3 Env or the Env-A539V mutant were preincubated with 17b (M), 10E8 (N), PG16 (O), or PGT145 (P) at 4°C for 1 h. KFS is an Env-defective mutant. The cells were washed, and APC-conjugated anti-human IgG was used to detect bound Ab. APC signals were normalized by GFP signal to calculate the Ab binding efficiency. Data are shown as means ± SEs from three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired t test or one-way ANOVA and Tukey’s multiple-comparison test.
Env mutations confer ARV resistance in the context of CCR5-tropic HIV-1.
To examine whether Env-mediated resistance to ARVs occurs in strains of HIV-1 other than the CXCR4-tropic lab-adapted strain NL4-3, we propagated the CCR5-tropic clone NL(AD8) (86) in the SupT1huR5 T-cell line, which expresses high levels of human CCR5, in the presence of DTG. Treatment of transfected SupT1huR5 cultures with 6.0 nM DTG markedly delayed NL(AD8) replication, but at 31 days posttransfection, we observed a peak of replication (Fig. 7A). Sequencing of the putative escape mutant indicated the absence of any mutations in IN but revealed the presence of an Env-N654K mutation in gp41 HR2 (Fig. 7B). To examine whether the Env-N654K mutation in NL(AD8) confers resistance to DTG, we introduced the mutation in WT NL(AD8) and examined replication kinetics in the presence of DTG (Fig. 7C). Interestingly, this mutant exhibited faster replication kinetics than WT and still replicated in the presence of 300 nM DTG (Fig. 7C, left). IC50 calculations indicate that the Env-N654K mutation confers 30-fold resistance to DTG relative to that of the WT NL(AD8) (Fig. 7D). As observed with several of the Env mutations selected in our previous study (48), the Env-N654K mutation impairs single-cycle infectivity (Fig. 7E) and does not confer DTG resistance in the context of cell-free virus (Fig. 7F and Table 2). These results are all consistent with the hypothesis that, as with the mutations selected in the context of NL4-3, the NL(AD8) Env-N654K mutation confers DTG resistance by enhancing the efficiency of cell-cell transfer.
FIG 7.
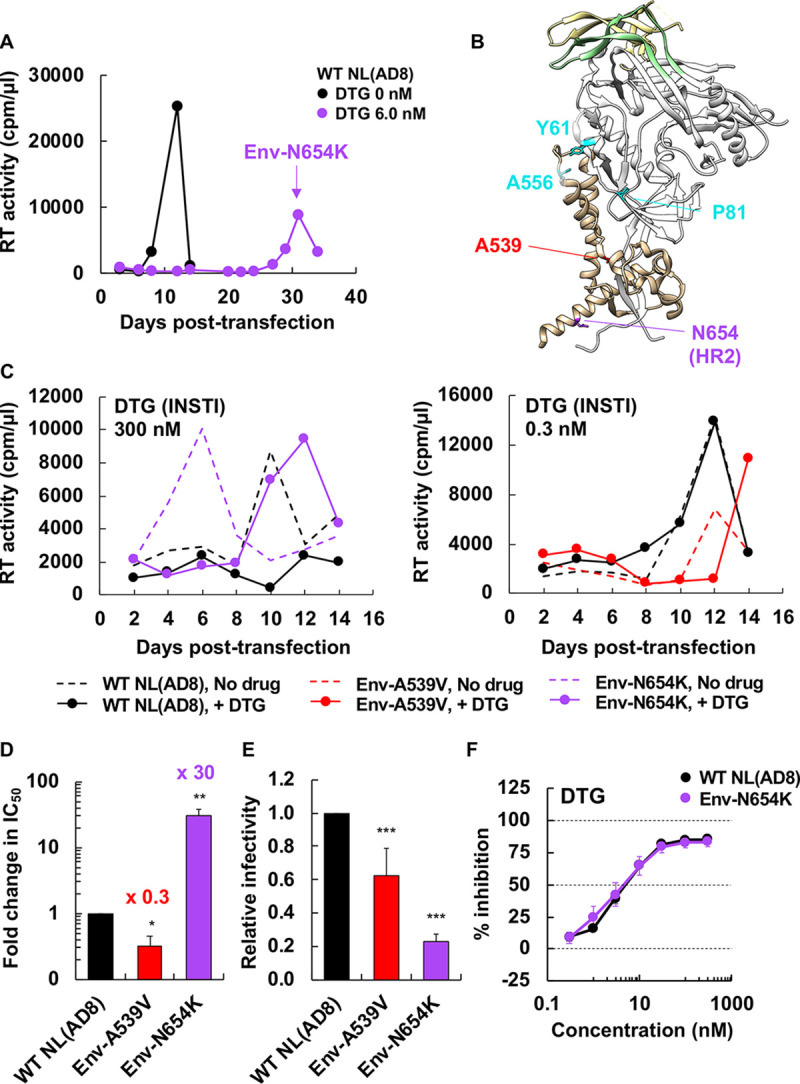
Selection for DTG resistance with the CCR5-tropic NL(AD8) strain. (A) The SupT1huR5 T-cell line was transfected with pNL(AD8) in the absence or presence of 6.0 nM DTG. At the time point indicated by the arrow, DNA was extracted from the DTG-treated culture and the IN- and Env-coding regions were sequenced, leading to the identification of the Env-N654K mutation. (B) An Env structure of subtype B JR-FL SOSIP.664 (PDB accession number 5FYK [137]), highlighting the location of Env mutations selected in the context of NL4-3 (48) and the Env-N654K mutation selected in NL(AD8). Env amino acid positions are indicated using the NL4-3 numbering. Most of gp120 is shown in white, with gp120 V1/V2 and V3 loops colored in light yellow and light green, respectively. Env-Y61, P81, and A556 are highlighted in cyan; Env-A539 and N654 are highlighted in red and purple, respectively. The structural model was generated using the UCSF Chimera software. (C) The SupT1huR5 T-cell line was transfected with WT or mutant pNL(AD8) in the absence or presence of 300 or 0.3 nM DTG. Replication kinetics were monitored by measuring RT activity at the indicated time points. Data are representative of at least three independent experiments. (D) The SupT1huR5 T-cell line was transfected with WT or mutant pNL(AD8) in the absence or presence of a serial dilution of DTG (1,000 nM to 0.03 nM). DTG IC50 values were calculated based on RT values at the peak of virus replication. Fold changes in IC50 relative to that for WT are indicated. Data from at least three independent experiments are shown as means ± SEs. **, P < 0.01; *, P < 0.05 by unpaired t test. (E) RT-normalized virus stocks produced from HeLa cells were used to infect TZM-bl cells. Luciferase activity was measured at 48 h postinfection. ***, P < 0.001 by unpaired t test. (F) TZM-bl cells were exposed to 100 TCID50 of the indicated viruses in the presence of various concentrations of DTG, and luciferase activity was measured at 48 h postinfection. Data are normalized to WT and are shown as means ± SEs from at least three independent experiments.
TABLE 2.
IC50 values in cell-free infection for NL(AD8) variants
| ARV class or NAb epitope | Drug or NAb | IC50 value (mean ± SE) |
Fold change | Significancea | |
|---|---|---|---|---|---|
| WT | Env-N654K | ||||
| INSTI | DTG (nM) | 4.9 ± 0.71 | 6.2 ± 2.7 | 1.3 | NS |
| Ent-I | BMS-378806 (nM) | 3.0 ± 0.24 | 2.8 ± 0.59 | 0.93 | NS |
| T-20 (nM) | 2,938 ± 724 | 797 ± 225 | 0.27 | ** | |
| MVC (nM) | 2.3 ± 0.36 | 1.1 ± 0.33 | 0.48 | * | |
| sCD4 (0 h) (µg/ml) | 4.1 ± 0.23 | 1.3 ± 0.42 | 0.32 | *** | |
| sCD4 (2 h) (µg/ml) | 3.6 ± 0.25 | 1.4 ± 0.31 | 0.39 | *** | |
| Anti-CD4 Ab | SIM.4 (dilution factor) | 113 ± 6.5 | 371 ± 44 | 3.3 | *** |
| Anti-V2 apex Ab | PG16 (µg/ml) | 0.040 ± 0.020 | 0.11 ± 0.053 | 2.8 | NS |
| PGT145 (µg/ml) | 0.068 ± 0.0060 | 0.062 ± 0.014 | 0.91 | NS | |
| Anti-V3 glycan Ab | 2G12 (µg/ml) | >30 | >30 | NDb | ND |
| PGT121 (µg/ml) | 0.043 ± 0.0026 | 0.036 ± 0.0073 | 0.84 | NS | |
| Anti-CD4bs Ab | VRC01 (µg/ml) | 0.40 ± 0.076 | 0.46 ± 0.14 | 1.2 | NS |
| F105 (µg/ml) | >10 | > 10 | ND | ND | |
| Anti-CD4i Ab | 17b (µg/ml) | >30 | > 30 | ND | ND |
| Anti-gp41 MPER Ab | 10E8 (µg/ml) | 0.47 ± 0.075 | 0.050 ± 0.012 | 0.11 | *** |
| Anti-gp120-gp41 interface Ab | 35O22 (µg/ml) | 4.1 ± 2.2 | 0.074 ± 0.037 | 0.018 | NS |
NS, not significant; ***, P < 0.001; **, P < 0.01; *, P < 0.05.
ND, not determined.
To examine whether the Env-A539V mutation, which was originally selected in the context of NL4-3 (48), would also confer ARV resistance in the context of another viral isolate, we introduced this mutation into the NL(AD8) molecular clone. In contrast to the phenotype of this mutant in NL4-3, the NL(AD8) Env-A539V mutant exhibited delayed replication in the absence of DTG and an increase (3.1-fold) in DTG sensitivity in multicycle spreading infections. Moreover, the cell-free infectivity of the NL(AD8) Env-A539V mutant was reduced relative to that of WT NL(AD8) (Fig. 7C to E). These data demonstrate that the impact of Env mutations on replication kinetics and drug sensitivity can be HIV-1 strain dependent.
We also propagated the subtype C, CCR5-tropic transmitted-founder isolate K3016 (87) in the presence of DTG, EFV, and rilpivirine (RPV) (Fig. 8A). We identified an Env-T541I mutation in gp41 HR1, which is located at the same position as NL4-3 Env-A539V (Fig. 8B), in the presence of DTG. We also identified an Env-E621V mutation in the gp41 disulfide loop (DSL) region in the presence of both FTC and RPV. To examine whether the Env-T541I and E621V mutations in K3016 confer resistance to ARVs, we introduced these mutations into WT K3016 and examined replication kinetics in the presence of DTG, EFV, NFV, and T-20 (Fig. 8C to J). These three Env mutants exhibited faster-than-WT replication kinetics in spreading infections but reduced cell-free infectivity relative to that of the WT. These mutants also showed reduced sensitivity to DTG, NFV, and EFV but not to T-20 in spreading infections (Fig. 8K and L). Again, these Env mutants did not alter sensitivity to DTG in the context of cell-free virus (Fig. 8M). These results indicate that Env-mediated drug resistance, which is linked to enhanced cell-cell transmission, can occur in clinically relevant HIV-1 strains independent of coreceptor usage, subtype, or ARV classes.
FIG 8.

Selection for DTG resistance with the subtype C CCR5-tropic isolate K3016. (A) The SupT1huR5 T-cell line was transfected with the K3016 molecular clone in the presence of 3.0 nM DTG, 2.0 nM RPV, or 100 nM FTC. DNA was extracted from the ARV-treated cultures at the peak of replication, and Env-coding regions were sequenced, leading to the identification of the Env-T541I and Env-E621V mutations. Amino acid numbers are based on HXB2 numbering. (B) gp41 HR1 sequences around the K3016 Env-T541I mutation are shown aligned with the NL4-3 sequence. (C to J) The SupT1huR5 T-cell line was transfected with WT K3016 or the indicated Env variants in the absence of DTG, EFV, NFV, and T-20. The supernatants were collected at the indicated time points and were assayed for replication kinetics by measuring RT activity. Data are representative of at three independent experiments. (K) The SupT1huR5 T-cell line was transfected with WT K3016 or the indicated Env variants in the absence or presence of a serial dilution of DTG, EFV, or NFV (0.01 to 300 nM) or T-20 (0.1 to 3,000 nM). IC50s were calculated based on RT values at the peak of virus replication. Fold changes in IC50 relative to that of WT were calculated. (L) RT-normalized virus stocks produced from 293T cells were used to infect TZM-bl cells. Luciferase activity was measured at 48 h postinfection. (M) TZM-bl cells were exposed to 100 TCID50 of the indicated viruses in the presence of a range of DTG concentrations, and luciferase activity was measured at 48 h postinfection. Data are shown as means ± SEs from three independent experiments. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by one-way ANOVA and Tukey’s multiple-comparison test.
Next, to examine the impact of the Env-N654K mutation on the conformation and neutralization properties of NL(AD8) Env, we examined the sensitivity of this mutant to a panel of NAbs (Fig. 9 and Table 2). Compared to NL4-3, NL(AD8) is more susceptible to NAbs, such as PGT121, PG16, and PGT145, that preferentially recognize the closed Env conformation (see Fig. S3 and Tables 1 and 2). Conversely, NL(AD8) is resistant to 17b (Fig. S3 and Tables 1 and 2) and F105 (Table 1). These results suggest that NL(AD8) tends to sample the closed Env conformation relative to NL4-3. NL(AD8) Env-N654K increased sensitivity to 35O22 (56-fold) and 10E8 (9.1-fold) (Fig. 9E and G and Table 2). These results suggest that the Env-N654K mutation in NL(AD8) has different impacts on Env conformation than the Env-A539V mutation in NL4-3. We also examined sensitivity to several entry inhibitors (Fig. 9H to L and Table 2). Env-N654K slightly increased sensitivity to SIM.4, maraviroc (MVC; a CCR5 antagonist), and T-20. Moreover, Env-N654K increased the sensitivity to sCD4. Unlike NL4-3 (Fig. S3J), incubation time did not affect the susceptibility of Env-N654K to sCD4 (Fig. 9H and Fig. S3J). This observation suggests that sCD4-induced gp120 shedding is minimal in the NL(AD8) strain, consistent with previous reports for non-lab-adapted isolates (55–57). Indeed, we did not observe sCD4-induced gp120 shedding from either WT or Env-mutant particles at sCD4 concentrations up to 10 µg/ml (see Fig. S4A, C, and E). We also examined time-dependent gp120 shedding (Fig. S4B and D). In contrast to that for NL4-3 (Fig. 5D and E, and Fig. S4F), only minimal gp120 shedding was observed for NL(AD8) Env over a 5-day incubation period at 37°C, and there was no significant difference in shedding between WT and Env-N654K NL(AD8) Env (Fig. S4B and D).
FIG 9.

The sensitivity of NL(AD8) Env-N654K to NAbs recognizing different Env conformations, anti-CD4 Ab, and entry inhibitors. TZM-bl cells were infected with 100 TCID50 of the indicated viruses in the presence of various concentrations of NAbs (A to G), sCD4 (H), anti-CD4 Ab SIM.4 (I), and entry inhibitors (J to L). Luciferase activity was measured at 48 h postinfection. Data from at least three independent experiments are shown as means ± SEs. ***, P < 0.001; **, P < 0.01; *, P < 0.05 by unpaired t test. The asterisks for 0 h or 2 h incubation with sCD4 (H) are indicated as black or gray, respectively.
Comparison of the sensitivity of NL4-3 and NL(AD8) to NAbs, DTG, T-20, and sCD4 in cell-free infection. TZM-bl cells were infected with 100 TCID50 of the indicated viruses in the presence of various concentrations of NAbs (A to F), ARVs (H and I), and sCD4 (J). sCD4 incubations were performed for 0 or 2 h. The data are related to Fig. 3 and 8. Data from at least three independent experiments are shown as means ± SEs. Download FIG S3, TIF file, 0.5 MB (468.7KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Analysis of sCD4-induced and time-dependent gp120 shedding of NL(AD8) Env mutants and comparison of NL4-3 versus NL(AD8) gp120 shedding. (A) Concentrated viruses were incubated with the indicated concentrations of sCD4 at 37°C for 2 h. Incubated viruses were subsequently purified through 20% sucrose cushions, and viral proteins were detected by Western blotting. A representative gel is shown. (B) Viruses were incubated at 37°C for the indicated times. Incubated viruses were subsequently purified through 20% sucrose cushions, and viral proteins were detected by Western blotting. A representative gel is shown. Quantification of sCD4-induced (C) and time-dependent (D) gp120 shedding from three independent experiments, calculated as the ratios of virion-associated gp120/p24 and shown as means ± SEs. Comparison of NL4-3 and NL(AD8) sCD4-induced (E) and time-dependent (F) gp120 shedding. The data are related to Fig. 5 andS3. Data from at least three independent experiments are shown as means ± SEs. Statistical analysis by unpaired t test. Download FIG S4, TIF file, 0.7 MB (719.9KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Sequence analysis of IN- and Env-coding regions of viruses from individuals failing a RAL-containing regimen.
Our results indicate that Env-mediated drug resistance can occur in clinically relevant HIV-1 strains in vitro. However, whether Env mutations contribute to drug resistance in vivo is unclear. To address this question, we used single-genome sequencing to compare viral sequences obtained from the plasma of participants failing a RAL-containing regimen in the SELECT study (5). The aim of the SELECT study was to examine whether RAL with boosted lopinavir (LPV) would be noninferior to boosted LPV with NRTIs. By 48 weeks of treatment, 10.3% of 258 participants treated with the RAL-containing regimen experienced virological failure, defined here as the inability to achieve a viral load <400 copies/ml at two consecutive time points or after 24 weeks of treatment. By the end of the follow-up in this study, 10 participants developed new resistance mutations in IN, including IN-N155H. In the present study, we compared changes in the sequences of IN- and Env-coding regions from five participants who experienced viral rebound (Table 3). These participants were infected with subtype C CCR5-tropic strains. Each participant had therapeutic plasma concentrations of RAL (>33 ng/ml) at the time of virological failure. No resistance mutations in IN were detected at day 0 (Table 3). While we identified the IN-N155H mutation in participant identifier (PID) A4 with high frequency (73%), PID A1, A3, and A5 had only a low frequency of resistance mutations in IN (Table 3), suggesting that virological failure may have occurred in these participants as a result of mutations elsewhere in the HIV-1 genome.
TABLE 3.
Information on study participants failing a RAL-containing regimen
| PIDa | Sample collectionb |
HIV-1 RNA (copies/ml) | RAL conc. (ng/ml) | IN accessory/resistance mutation(s) (%)c | |
|---|---|---|---|---|---|
| Time point | Day | ||||
| A1 | Pre-ART | 0 | 805,710 | No mutations detected | |
| Early RAL failure | 181 | 570 | 990 | Not determined | |
| Late RAL failure | 250 | 11,335 | 89 | T97A (33), N155H (5.6) | |
| A2 | Pre-ART | 0 | 17,450 | No mutations detected | |
| Late RAL failure | 195 | 1,772 | 93 | No mutations detected | |
| A3 | Pre-ART | 0 | 1,329,415 | No mutations detected | |
| Early RAL failure | 178 | 433 | 965 | Not determined | |
| Late RAL failure | 196 | 1,139 | 3,371 | F121Y (14) | |
| A4 | Pre-ART | 0 | 1,617,523 | No mutations detected | |
| Early RAL failure | 175 | 7,208 | 1,142 | E138K (2.7), N155H (73) | |
| Late RAL failure | 199 | 9,918 | 480 | Not determined | |
| A5 | Pre-ART | 0 | 30,206 | No mutations detected | |
| Late RAL failure | 194 | 18,806 | 2,729 | G163K (4.8) | |
PID, participant identifier.
Samples are derived from the SELECT study (5).
RAL resistance mutations are indicated in boldface font.
Evaluation of the Env-coding region before treatment and during virologic failure indicated the presence of many changes in gp120 and gp41 that emerged or were selected with failure. Most of the changes in gp120 were located in highly variable regions, such as the variable loops (V1/V2, V4, and V5 loops), and the C3 region (data not shown). Because these regions contain immunodominant epitopes of subtype C Env, mutations in these regions may have been driven by immune pressure (88, 89). Although we did not identify mutations at the same positions as those observed in our in vitro studies, we observed an accumulation of mutations in the gp120 C1 domain and gp41 ectodomain in PID A1, A3, A4, and A5 (Fig. 10A and B). The frequency of most of the mutations increased significantly with viral rebound. Although we identified a number of Env mutations in PID A2, the frequency of mutations before RAL therapy was similar to that after viral rebound, although the RAL-containing regimen decreased viral load before viral rebound (data not shown and Table 3). In many cases, amino acid residues were replaced with more conserved residues; however, we did identify changes to residues with very low prevalence, such as R577K (gp41 HR1) and L602R (gp41 HR2). Some mutated positions in the gp41 ectodomain were identified in several participants (Fig. 10A and B). The observation of changes arising in highly conserved positions at the gp120-gp41 interface—a region in which, based on our data, mutations that confer ARV resistance in vitro cluster—in the absence of drug resistance mutations in IN, suggests the possibility that these gp41 changes may have contributed to virological failure in a subset of participants in the study. Further analysis will be needed to explore this hypothesis in more detail.
FIG 10.


Frequency of observed mutations in gp120 C1 domain and gp41 ectodomain in patient-derived samples from the SELECT study. (A) PID A1, A3, and A5. (B) PID A4. Positions of mutations are indicated using HXB2 numbering. FP, fusion peptide; FPPR, fusion peptide proximal region; HR1/HR2, heptad repeat 1/2; DSL, disulfide loop. Mutations observed in multiple patients are shaded in yellow. Observed residues are indicated in boldface font. Mutations that changed conserved positions to less conserved residues are indicated with a superscript lowercase “a.” Frequencies were determined for subtype C sequences (n = 5,923) retrieved from the Los Alamos HIV databse. Fischer's exact test was performed to determine statistical significance. ***, P , 0.001; **, P , 0.01; *, P , 0.05; ns, not significant; N.D., not determined.
DISCUSSION
In this study, we demonstrate that the NL4-3 Env-A539V mutant, which was selected in the presence of DTG, confers broad resistance to multiple classes of ARVs in multicycle spreading infections. This Env mutation also increases resistance to ARVs when coupled with ARV target gene mutations, again in the context of spreading infections. However, this Env mutation does not alter ARV susceptibility in cell-free infection. The results demonstrate that Env mutations clustered at the gp120-gp41 interface can confer broad resistance by enhancing the ability of HIV-1 to spread via a cell-cell route of transmission. Interestingly, NL4-3 Env mutants exhibiting an enhanced ability to spread via cell-cell transmission have more stable and closed Env conformations than WT NL4-3. By propagating CCR5-tropic HIV-1 strains NL(AD8) and K3016 in the presence of DTG, we obtained gp41 ectodomain mutations Env-N654K and Env-T541I, respectively. We also identified Env-E621V by propagating K3016 in the presence of RT inhibitors. We demonstrate that these Env mutations reduce susceptibility to several ARVs in multicycle spreading infection, as observed for the NL4-3 Env mutants, indicating that Env-mediated drug resistance can occur in non-laboratory-adapted HIV-1 strains. However, the effects of the Env mutations on replication kinetics and Env structure are HIV-1 strain dependent. Finally, we performed single-genome sequencing analysis of the IN- and Env-coding regions of viruses from infected individuals failing a RAL-containing regimen. We observed that many mutations accumulated in the Env- but not IN-coding region in most study participants. While most of the gp120 changes that arose in vivo were located in highly variable regions, a number of changes in the gp41 ectodomain were observed at the highly conserved gp120-gp41 interface, as observed in vitro.
Several studies have demonstrated that cell-cell transfer is associated with a reduced susceptibility to ARVs relative to that with cell-free infection, but high concentrations of ARVs, or their use in combination, can block both cell-free and cell-cell infection (32–34). We previously proposed a model, consistent with other findings (33), whereby Env mutations that enhance cell-cell transmission increase the MOI following viral transfer across the VS; concentrations of DTG that are sufficient to inhibit cell-free infection by these mutants are therefore insufficient to block their cell-cell transmission (48). Based on this model, the selected Env mutations would be predicted to confer resistance to multiple ARVs independent of their mode of action and should confer resistance in the context of a spreading, but not cell-free, infection. Indeed, the NL4-3 Env-A539V mutant selected in the presence of DTG exhibits broad resistance to a number ARVs, including INSTIs, NRTIs, NNRTIs, and PIs, in spreading replication assays but not in cell-free infectivity assays. It is possible that additional mutations may accumulate during spreading replication of Env-A539V in the presence of ARVs; Env mutations may thus be the starting point on the path to acquisition of high-level drug resistance. It is also possible that mutations in Env could arise that confer drug resistance by greatly increasing the efficiency of cell-free infection. However, we have not observed such mutations in any of our selection experiments; rather, the mutations we have identified in this and our previous study (48) either greatly impair cell-free infectivity or have no major effect on this mode of transfer. Taken together, these observations suggest that the broad resistance conferred by the HIV-1 Env mutations will affect susceptibility not only to the currently approved ARVs but also potentially to next-generation drugs. It is noteworthy that other studies have also observed the selection of Env mutations upon propagation of HIV-1 in the presence of various inhibitors of virus replication (13, 90–92).
In contrast to the other classes of ARVs, the Env mutants we identified in NL4-3 and K3016 are sensitive to two types of Ent-Is: a fusion inhibitor (T-20) and a compound that blocks CD4-induced conformational changes in Env (BMS-378806) (19, 65–67). Previous studies have shown that Ent-Is targeting CD4 or a coreceptor are effective in blocking cell-cell transmission (34, 60, 93). While some CD4bs-targeting agents inefficiently inhibit cell-cell transmission, anti-gp41 MPER antibodies (Abs) and T-20 are effective (60, 94). These observations suggest that accessibility of epitopes or time of action may be important factors for the efficacy of Ent-Is in blocking cell-cell transfer. Interestingly, the Env-A539V mutation has been reported to emerge in the presence of low concentrations of fusion inhibitors in vitro; however, this mutation does not confer resistance to the fusion inhibitor and reverted back to WT at a high inhibitor concentration (95). Overall, our findings imply that including Env-targeted inhibitors in a cART regimen may help prevent the emergence of the type of broadly resistance-conferring Env mutations described here and previously (48).
Virological failure has been observed in patients on PI-containing regimens in the absence of PI-resistance mutations in PR or in the vicinity of Gag cleavage sites (7, 96–99). Likewise, recent studies have demonstrated that viral rebound can occur in individuals on INSTI-containing therapies without the emergence of INSTI resistance mutations in IN (5, 6). In some studies (100, 101), there are concerns about whether the infected individuals remained adherent (i.e., whether suppressive concentrations of the inhibitors were maintained), but in other studies (e.g., reference 5), the maintenance of adequate plasma drug concentrations was experimentally verified. In some cases in which target gene mutations were identified in the treated individuals experiencing virological failure, the observed resistance mutations conferred only low-level resistance (3–10). These findings suggest that mutations outside the pol-coding region (and not around Gag cleavage sites, in the case of PIs) also contribute to virological failure in association with the target gene mutations. Mutations in the cytoplasmic tail of gp41 were previously proposed to contribute to PI resistance in vivo (13), potentially reflecting the role of virion maturation in Env-mediated viral entry (102, 103). Mutations in the gp41 HR region have also been observed in association with PI failure, although their contribution to virological failure is unclear (9). In addition to mutations in IN, it has also been proposed that mutations in the 3′ PPT contribute to INSTI failure both in vitro and in vivo (12, 104). We demonstrate here that the Env-A539V mutation rescues the defect in virus replication imposed by the RT-Y188L mutation (68) at early time points and increases the resistance of this RT mutant to EFV in multicycle spreading but not cell-free infection. These observations indicate that Env mutations can increase resistance conferred by mutations in the ARV target gene by enhancing viral cell-cell transmission. Mathematical modeling suggests that cell-cell transmission increases the probability that NAb resistance mutations will emerge, relative to cell-free infection, as a result of reduced NAb potency in the context of cell-cell transmission (94). As shown in Fig. 2, Env-A539V provides a replication advantage over WT NL4-3 in the presence of ARVs, suggesting that mutations in Env such as those described here may facilitate the acquisition by the virus of resistance mutations in ARV target genes. According to this “stepping stone” model, by facilitating virus replication in the presence of ARVs, the Env mutations would facilitate the emergence of high-level resistance mutations in ARV target genes. Additional studies will be needed to investigate the role of HIV-1 Env in the development of high-level drug resistance.
We observed that Env mutations in the NL4-3 strain reduce sCD4-induced and time-dependent shedding of gp120 from viral particles. In addition, the Env-A539V mutation decreases sensitivity to NAbs preferring the CD4-bound conformation, whereas this mutant is more susceptible to PGT145, which recognizes the closed Env conformation. These observations suggest that the NL4-3 mutant Envs are more closed and stable on viral particles than WT NL4-3 Env. Flow cytometry analysis using Env-expressing cells indicates that the NAb-binding properties of the mutants largely parallel their neutralization properties, suggesting that mutant Envs on the surfaces of infected cells exhibit similar conformational dynamics to those on viral particles. To examine the impact of the Env-A539V mutation on the HIV-1 entry step, we also analyzed the sensitivity of this mutant to an anti-CD4 Ab (SIM.4) (84) and a CXCR4 antagonist (AMD3100) (85). Because the NL4-3 Env-A539V mutation does not alter sensitivity to either entry inhibitor, it is likely that the enhancement of cell-cell transfer is due to the increased probability of gp120-CD4 interactions rather than the increased affinity of gp120 for CD4 or CXCR4. Indeed, Env-A539V does not affect CD4-IgG2 binding. Our findings suggest that decreased gp120 shedding may contribute to enhanced Env-CD4 interactions at cell-cell contact sites. Further structural analysis will be needed to understand how the Env mutations stabilize the gp120-gp41 interaction in the unliganded Env conformation, because current structural models using HIV-1 Env SOSIP trimers represent the state-2 conformation (69–72).
We performed resistance analyses using the more clinically relevant, relative to NL4-3, HIV-1 strains NL(AD8) and K3016. We introduced the Env-A539V mutation into NL(AD8) and observed a different phenotype from that observed with this mutation in NL4-3. Whereas NL4-3 Env-A539V exhibited faster-than-WT replication kinetics, NL(AD8) Env-A539V exhibited impaired replication kinetics and was more susceptible to DTG than WT NL(AD8). These results imply that the Env-A539V mutation will not emerge in the context of the NL(AD8) strain. However, DTG selection experiments led to the emergence of an Env-T541I mutation, which is located at the same position as NL4-3 Env-A539V, in the context of the subtype C, CCR5-tropic transmitted founder isolate K3016. Whereas an Ala at Env-541 (539 in NL4-3) is highly conserved in subtypes B and C (98.00% and 98.65%, respectively), Thr, Ile, and Val are rare residues at this position in both subtypes. As observed with the NL4-3 Env mutants, K3016 Env-T541I exhibited faster-than-WT replication and conferred resistance to ARVs in spreading infections. These results highlight the context dependence of resistance-conferring Env mutations, consistent with the suggestion that residues in the gp41 ectodomain are functionally linked with gp120 and modulate conformational dynamics of Env in a strain-dependent manner (52, 62, 105, 106). As shown in Fig. S3 and S4 in the supplemental material, the stability of the gp120-gp41 interaction and the conformational dynamics of NL(AD8) Env are different from those of NL4-3 Env, suggesting that the differences in intra- and interprotomer interactions of Env are likely to affect the viral phenotypes of the Env mutants in terms of their fitness and ability to confer broad ARV resistance.
Propagation of NL(AD8) in the presence of DTG led to the selection of the Env-N654K mutation in gp41 HR2. Like several of the selected NL4-3 Env mutations (this report and in reference 48), the NL(AD8) Env-N654K mutation confers resistance to DTG and is highly fit in multicycle spreading infections but is also profoundly defective in cell-free infectivity. These observations suggest that this mutant spreads predominantly via a cell-cell route, as observed for the NL4-3 Env mutants. However, the effects of the NL(AD8) Env-N654K mutation on Env stability and conformation appear to be different from those of the NL4-3 Env mutants. This mutation increases the sensitivity to the anti-CD4 Ab, MVC, and to sCD4, T-20, and an anti-MPER Ab, suggesting that Env-N654K may be in a more open conformation than WT NL(AD8). As discussed above, WT AD8 is a primary isolate and prefers to adopt the closed Env conformation. It may have evolved in vivo while avoiding the N654 change to maintain the closed state. An Asn at Env-654 is highly conserved (99.76% in subtype B), and previous studies have shown that substitutions at this position decrease the affinity of HR2 for HR1, resulting in decreased cell-free infectivity and fusogenicity (107–109). Several lines of evidence suggest that syncytium formation is tightly regulated at the site of cell-cell contact by viral and cellular factors (102, 103, 110). Although the roles of syncytium formation in HIV-1 replication are still debated, the formation of large syncytia may negatively impact HIV-1 replication (110, 111). Several studies have reported that mutations in Env arose in infected SupT1 cells and improved replication in the absence of syncytium formation (112–115). As we reported previously, Env-Y61H, P81S, and A556T in the NL4-3 strain enhance cell-cell transmission while markedly impairing cell-free infectivity and fusogenicity, indicating that for these mutants, reduced fusogenicity is associated with enhanced cell-cell transmission capacity (48). In contrast, we show here that the Env-A539V mutant exhibits only a small impairment in fusogenicity in a cell-cell fusion assay. Additional studies will be needed to elucidate the precise mechanism of action of the Env-N654K mutation.
To explore the possibility that Env-mediated ARV resistance may occur in vivo, we performed single-genome sequence analysis using samples from the SELECT study (5), one of several studies that examined whether INSTI monotherapy would effectively suppress viral loads in animal models and human participants (6, 116, 117). In many cases, virological failure occurred in either the absence of mutations in IN or in association with low-frequency resistance mutations in IN (118). Consistent with these observations, our single-genome sequence analysis revealed that four of the five participants we analyzed had only low-frequency resistance mutations in the IN-coding region. These results suggest that virological failure occurred in these four participants as a result of mutations outside the Pol-coding region. As expected, we observed that many Env mutations accumulated in less-conserved regions of Env, such as gp120 C3 and variable loops, which are predominant targets for the Ab response (88, 89). More interestingly, we also found that several mutations in the gp41 ectodomain accumulated at the highly conserved gp120-gp41 interface, as observed in our in vitro selection experiments. As shown in our in vitro studies, it is unlikely that the identical mutations observed in vitro would emerge in vivo, because the effects of the Env mutations on viral fitness and drug resistance are virus strain dependent. It is interesting to note that viruses from PID A4, which had the IN-N155H mutation with high frequency, had many changes in the gp120 C1 domain and the gp41 ectodomain compared to viruses isolated from the other participants. A limitation of the current study is the relatively small number of INSTI failure samples analyzed. Larger numbers of samples will be needed to identify specific mutational patterns associated with virological failure. In addition, other regions outside IN, such as the 3′ PPT, may also be associated with virological failure (12, 104). Further analysis will be needed to understand the impacts of Env mutations on viral susceptibility to cART and their contribution to virological failure. Nevertheless, our findings are consistent with the possibility that HIV-1 Env simultaneously evolves to escape from both NAbs and ARVs in association with ARV target gene mutations in vivo.
In summary, the findings of this study demonstrate that mutations in Env can markedly reduce the susceptibility of HIV-1 to a broad panel of ARVs in vitro. These findings offer mechanistic insights into Env-mediated drug resistance in vitro and provide new clues to understand how HIV-1 develops high-level resistance to ARVs in vivo.
MATERIALS AND METHODS
Cell lines.
HeLa, HEK293T, and TZM-bl cells (119) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in 5% CO2. The SupT1 and SupT1huR5 T-cell lines (120) were cultured in RPMI 1640 medium supplemented with 10% FBS at 37°C in 5% CO2. The SupT1huR5 T-cell line was a kind gift from James Hoxie, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA.
Compounds and neutralizing antibodies.
DTG and BMS-378806 were purchased from MedChemExpress. sCD4 (four domains, consisting of amino acids 26 to 390 of the extracellular domain of human CD4) was purchased from R&D systems. RAL, FTC, EFV, RPV, NFV, T-20, AMD3100 (85), MVC, SIM.4 (84), PGT145 (76), PG16 (77), 2G12 (75), PGT121 (76), VRC01 (78), 447-52D (81), 17b (83), 10E8 (82), F105 (80), 35O22 (79), 16H3 (121), CD4-IgG2 (74), and HIV Ig were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. BI-224436 (58) was a kind gift from Alan Engelman, Dana Farber Cancer Institute, Boston, MA.
Cloning and plasmids.
The full-length HIV-1 molecular clones pNL4-3 (122) and pNL(AD8) (86) and the subtype C transmitted founder viral clone CH185 (here denoted K3016, a kind gift from Christina Ochsenbauer and John Kappes, University of Alabama) (87) were used in this study. pBR-NL43-IRES-eGFP-nef+ (pBR43IeG), a proviral clone that coexpresses Nef and eGFP from a single bicistronic RNA, was obtained from Frank Kirchhoff through the NIH AIDS Reagent Program (123, 124). pNL4-3 and pBR43IeG clones bearing Env mutations (Env-Y61H, P81S, A539V, and A556T) were described previously (48). The pBR43IeG/KFS clone, which does not express HIV-1 Env, was described previously (48, 125). pNL4-3 RT-Y188L (68) was constructed by an overlap PCR method using SpeI and AgeI restriction sites. pNL(AD8) Env mutants Env-A539V and N654K were constructed by an overlap PCR method using BamHI and EcoRI restriction sites. K3016 Env mutants Env-T541I and E621V were constructed by an overlap PCR method using XhoI and PacI restriction sites. Constructed plasmids were verified by Sanger DNA sequencing (Psomagen).
Preparation of virus stocks.
The HEK293T and HeLa cell lines were transfected with HIV-1 proviral DNA using Lipofectamine 2000 (Invitrogen). At 48 h posttransfection, virus-containing supernatants were filtered through a 0.45-µm membrane (Merck Millipore). The amount of virus in the supernatants was quantified by RT assay. RT assays were performed as described previously (126) with minor modification. Briefly, after incubation of the virus supernatants with RT reaction mixtures, which contained a template primer of poly(rA) (5 µg/ml) and oligo(dT)12-18 primers (1.57 µg/ml), in 50 mM Tris (pH 7.8), 75 mM KCl, 2 mM dithiothreitol, 5 mM MgCl2, 0.05% Nonidet P-40, and 0.25 µCi of 32P-dTTP at 37°C for 3 h, the mixtures were spotted onto Filtermat B (Perkin Elmer; catalog number [no.] 1450-521) soaked in 0.5% (vol/vol) branched-polyethylenimine (Merck Millipore; catalog no. 402727). After washing the spotted Filtermat with 2× SSC buffer (300 mM NaCl and 30 mM sodium citrate), levels of bound 32P were measured on a Wallac MicroBeta plate reader (PerkinElmer). The 50% tissue culture infective dose (TCID50) of the virus stocks was determined using TZM-bl cells.
Virus replication kinetics assays.
Virus replication was monitored in SupT1 cells as previously described with minor modifications (48). Briefly, SupT1 cells were incubated with pNL4-3 clones (1.0 µg/1.0 × 106 cells) in the presence of 700 µg/ml DEAE-dextran at 37°C for 15 min. Transfected cells (1.5 × 105 cells) were plated in 96-well flat-bottom plates and incubated at 37°C in the presence of various concentrations of ARVs. For CCR5-tropic viruses, we used SupT1huR5 cells, which express high levels of human CCR5. Aliquots of supernatants were collected to monitor RT activity, and cells were split 1:3 every other day with fresh drug and medium. As described previously (48), IC50 values were calculated based on RT activity at the peak of replication of each virus; specifically, the IC50 was defined as the amount of inhibitor required to reduce RT levels by 50% at the peak of virus replication. To identify the selected mutations in the Pol/Env-coding region, genomic DNA was extracted from infected cells by using a DNeasy Blood and Tissue minikit (Qiagen), and then the Pol- and Env-coding regions were amplified by PrimeSTAR GXL DNA polymerase (TaKaRa) and sequenced (Psomagen) using previously described primers (48).
Single-round infection assays.
For cell-free infectivity assays, TZM-bl cells (1.0 × 104 cells) in 96-well plates were exposed to RT-normalized virus stocks produced in HeLa cells. For drug sensitivity assays, 100 TCID50 of virus produced in 293T cells was exposed to TZM-bl cells (1.0 × 104 cells) in the presence of various concentrations of drugs or neutralizing antibodies. For the sCD4 sensitivity assay, prior to the infection, the viruses were incubated with various concentrations of sCD4 at 37°C for 0 or 2 h. For assays using PIs or ALLINIs, viruses were produced in the presence of serial dilutions of inhibitors as described previously (127). Briefly, 293T cells were transfected with WT or mutant molecular clones as indicated above. Six hours posttransfection, 1.5 × 105 transfected cells were incubated with a 3-fold serial dilution of the inhibitors at 37°C for 48 h. Virus-containing supernatants were harvested and used to infect TZM-bl cells. At 48 h postinfection, luciferase activity was measured using the Britelite plus reporter gene assay system and Wallac MicroBeta plate reader (PerkinElmer).
Cell-cell fusion assay.
The HEK293T cells were transfected with HIV-1 proviral DNA as described above. At 24 h posttransfection, serial dilutions of the transfected HEK293T cells were cocultured with 1.0 × 104 TZM-bl cells in the presence of a cocktail of EFV and DTG (200 nM each). At 24 h postcoculture, luciferase activity was measured as described above.
gp120 shedding assay.
To analyze time-dependent gp120 shedding, viruses produced from HeLa cells were incubated at 37°C for 0, 1, 3, or 5 days. For the sCD4-induced gp120 shedding assay, concentrated viruses were incubated with sCD4 (0, 0.3, 1.0. 3.0, and 10 µg/ml) at 37°C for 2 h. Following incubation, the viruses were purified by ultracentrifugation through 20% sucrose cushions (60,000 × g) for 45 min at 4°C. The levels of virion-associated gp160, gp120, and p24 were determined by Western blotting.
Western blotting.
Viral proteins were separated by SDS-PAGE and transferred to polyvinylidene disulfide (PVDF) membranes (Merck Millipore). After blocking the membranes with 5% skim milk, they were probed with primary antibodies for 1 h and then incubated for 1 h with species-specific horseradish peroxidase-conjugated secondary antibody. After the final washes, bands were detected by chemiluminescence with a Sapphire Biomolecular imager (Azure Biosystems). Quantification was performed using Image Studio Lite (LI-COR Biosciences) software. The p24 protein was detected with anti-HIV Ig at a final concentration of 5.0 µg/ml in 5% skim milk. gp160 and gp120 were detected with the 16H3 monoclonal antibody at a final concentration of 0.5 µg/ml in 5% skim milk.
Antibody binding assay.
HEK293T cells were transfected with pBR43IeG constructs as indicated above. At 24 h posttransfection, the cells were detached with 5 mM EDTA-phosphate-buffered saline (PBS) and then washed with PBS. Cells (2.0 × 105) were incubated with the specified antibodies at a final concentration of 1.0 µg/ml at 4°C. After 1 h of incubation, the cells were washed with PBS. The cells were then washed with PBS and incubated with allophycocyanin (APC)-conjugated F(ab′)2 fragment donkey anti-human IgG antibody (Jackson ImmunoResearch Laboratories; catalog no. 709-136-149) at a final concentration of 62.5 µg/ml. The cells were washed with PBS and fixed with 4% paraformaldehyde (PFA; Boston Biosciences). Fixed cells were analyzed with a BD LSR-II or FACSCalibur (BD Biosciences) flow cytometer. Data were analyzed by FCS Express Cytometry software 7 (De novo Software).
Ethics Statement.
Plasma samples were obtained prior to and after virologic failure from donors on combination ART of ritonavir (RTV)-boosted lopinavir (100 mg RTV, 400 mg LPV) plus 400 mg RAL twice a day. Samples were collected by the AIDS Clinical Trial Group (ACTG) study A5273, a randomized, open-label phase-3 noninferiority study at 15 research sites in nine resource-limited countries (three sites in India and South Africa, two in Malawi and Peru, and one each in Brazil, Kenya, Tanzania, Thailand, and Zimbabwe) (5). The primary endpoint was time to confirmed virologic failure (two measurements of HIV-1 RNA viral load >400 copies/ml). This trial is registered with ClinicalTrials.gov, NCT01352715. Entry and exclusion criteria are listed in the protocol (5). The study was approved by ethics committees at each site and written informed consent was obtained from each participant.
Plasma viral RNA extraction and cDNA synthesis.
Viral RNA was extracted from plasma containing 200 to 20,000 copies of HIV-1 RNA. Plasma was first centrifuged at 5,300 × g for 10 min at 4°C to remove cellular debris. The supernatant was then centrifuged at 21,000 × g for 1 h at 4°C. Viral RNA was extracted from the viral pellet as previously described (128) and resuspended in 40 µl of RNase-free 5 mM Tris-HCl (pH 8.0). Five microliters of each of 10 mM deoxynucleoside triphosphates (dNTPs) and 10 µM oligo(dT) were added prior to denaturing the RNA at 65°C for 10 min. cDNA was synthesized as previously described (129) except at 50°C for 1 h followed by 55°C for another hour. Two microliters RNase H (2 U/reaction) was added, and the RNA was digested at 37°C for 20 min.
Single-genome sequencing.
To obtain integrase PCR products from single cDNA molecules, cDNA was serially diluted to an endpoint (when approximately 30% of reactions yield PCR product). PCR master mix was prepared as previously described (129, 130), except primers targeting HIV integrase were used: Poli5 (OF) and Poli8 (OR) followed by Poli7 (IF) and Poli6 (IR) (131) in a nested PCR. PCR cycling conditions were modified by decreasing the annealing temperature to 50°C and increasing the elongation time to 1 min 30 s. To obtain env PCR products from single cDNA molecules, the same diluted cDNA was used but with the following primers, modified from refererence 132: HIVC.short.VIF1.F1 (5′-GTTTATTACAGGGACAGCAGA-3′) and HIV.short OFM.R1 (5′-CAAGGCAAGCTTTATTGAGGCTTA-3′). Alternatively, E0 forward primer (133) was used. PCR cycling was performed as follows: 95°C for 2 min and 44 cycles of 95°C for 30 s, 57°C for 30 s, and 68°C for 3 min to 3 min 30 s, followed by final elongation at 68°C for 5 min. Nested PCR was performed with the following primers, modified from reference 134: HIVC.short.ENVA.F2 (5′-GCATCTCCTATGGCAGGAAG-3′) and HIV.short.ENVN.R2 (5′-CAATCAGGGAAGTAGCCTTG-3′). Alternatively, E00 forward primer (133) was used. PCR cycling was performed as follows: 95°C for 2 min, 10 cycles 95°C for 15 s, 57°C for 30 s, and 68°C for 3 min, followed by 30 cycles of 95°C for 15 s, 57°C for 30 s, and 68°C for 2 min plus 5 s per cycle, and a final elongation of 68°C for 7 min. Positive wells were identified by detection using 1% agarose E-Gel 96-well gels (Thermo Fisher Scientific), and positive PCRs were sequenced by Sanger sequencing. For sequencing integrase, Poli7, Poli9D, and O-Poli2C forward and Poli6, Poli10B, and Poli3 reverse primers were used (131). For sequencing env, E00, For14, For15, For17, For18, and For19 forward and HIVCshort.ENVN.R2, Rev14, Rev16, Rev17, and Rev18 reverse primers were used (132, 133, 135). Additional primers used for env sequencing are as follows: For13, 5′-GAGAAAGAGCAGAAGACAGTGG-3′; For16, 5′-TTTAATTGTGGAGGAGAATTTTTCTA-3′; Rev19, 5′-ACTTTTTGACCACTTGCCACCCAT-3′; HIVC.REV20.SEQ, 5′-CACATGGAATTAAGCCAG-3′.
Sequence analysis.
Sanger sequencing data were analyzed using MEGA (136) to align and translate sequences. Sequences were analyzed for known INSTI resistance mutations using HIV Stanford HIV Drug Resistance database (https://hivdb.stanford.edu/).
Statistical analyses.
Two-tailed unpaired t tests and Fischer’s exact tests were performed using GraphPad Prism 8 (GraphPad). A P value of <0.05 was considered statistically significant. GraphPad Prism was also used to assess statistical significance by one-way analysis of variance (ANOVA) and Tukey’s multiple-comparison test.
ACKNOWLEDGMENTS
We thank members of the Freed laboratory for helpful discussion and critical review of the manuscript. We thank James Thomas (Leidos) for assistance with flow cytometry analysis. We also thank Michael Bale for assistance with sequence analysis and bioinformatics support. We thank James Hoxie for providing the SupT1 huCCR5 cell line and Alan Engelman for providing BI-224436. We are also grateful to Christina Ochsenbauer and John Kappes for providing pK3016. The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: efavirenz, emtricitabine, nelfinavir, maraviroc, AMD3100, T-20, raltegravir (from Merck & Company, Inc.), rilpivirine (from Tibotec Pharmaceuticals, Inc.), SIM.4 (from James Hildreth), PGT145, PGT121, and PG16 (from IAVI), 2G12 (from Polymun Scientific), VRC01 (from John Mascola), 447-52D (from Susan Zolla-Pazner), 17b (from James E. Robinson), 10E8 (from Mark Connors), F105 (from Marshall Posner and Lisa Cavacini), 35O22 (from Jinghe Huang and Mark Connors), 16H3 (from Barton Haynes and Hua-Xin Liao), HIV-Ig (from NABI and NHLBI), CD4-IgG2 (Progenics Pharmaceuticals), and pBR-NL43-IRES-eGFP-nef+ (from Frank Kirchhoff).
Research in the Freed lab is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Y.H. is supported by JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at the National Institutes of Health. This project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E to J.W.M. Research reported in this publication was also supported by the National Institute of Allergy and Infectious Diseases, of the National Institutes of Health, under award numbers UM1AI068636, UM1AI069494, and UM1AI106701 to the AIDS Clinical Trials Group, the Pitt-Ohio State Clinical Trials Unit, and the University of Pittsburgh VSL.
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
This article is a direct contribution from Eric O. Freed, a Fellow of the American Academy of Microbiology, who arranged for and secured reviews by Alon Herschhorn, University of Minnesota, and Andrés Finzi, CRCHUM, Université de Montréal.
Citation Hikichi Y, Van Duyne R, Pham P, Groebner JL, Wiegand A, Mellors JW, Kearney MF, Freed EO. 2021. Mechanistic analysis of the broad antiretroviral resistance conferred by HIV-1 envelope glycoprotein mutations. mBio 12:e03134-20. https://doi.org/10.1128/mBio.03134-20.
REFERENCES
- 1.Kemnic TR, Gulick PG. 2019. HIV antiretroviral therapy. StatPearls Publishing, Treasure Island, FL. [PubMed] [Google Scholar]
- 2.Collier DA, Monit C, Gupta RK. 2019. The impact of HIV-1 drug escape on the global treatment landscape. Cell Host Microbe 26:48–60. doi: 10.1016/j.chom.2019.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Perrier M, Castain L, Regad L, Todesco E, Landman R, Visseaux B, Yazdanpanah Y, Rodriguez C, Joly V, Calvez V, Marcelin AG, Descamps D, Charpentier C. 2019. HIV-1 protease, Gag and gp41 baseline substitutions associated with virological response to a PI-based regimen. J Antimicrob Chemother 74:1679–1692. doi: 10.1093/jac/dkz043. [DOI] [PubMed] [Google Scholar]
- 4.Manasa J, Varghese V, Pond SLK, Rhee SY, Tzou PL, Fessel WJ, Jang KS, White E, Rögnvaldsson T, Katzenstein DA, Shafer RW. 2017. Evolution of gag and gp41 in patients receiving ritonavir-boosted protease inhibitors. Sci Rep 7:11559. doi: 10.1038/s41598-017-11893-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Rosa AM, Harrison LJ, Taiwo B, Wallis CL, Zheng L, Kim P, Kumarasamy N, Hosseinipour MC, Jarocki B, Mellors JW, Collier AC. 2016. Raltegravir in second-line antiretroviral therapy in resource-limited settings (SELECT): a randomised, phase 3, non-inferiority study. Lancet HIV 3:e247–e258. doi: 10.1016/S2352-3018(16)30011-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hocqueloux L, Raffi F, Prazuck T, Bernard L, Sunder S, Esnault J-L, Rey D, Le Moal G, Roncato-Saberan M, André M, Billaud E, Valéry A, Avettand-Fènoël V, Parienti J-J, Allavena C, MONCAY study group. 2019. Dolutegravir monotherapy versus dolutegravir/abacavir/lamivudine for virologically suppressed people living with chronic human immunodeficiency virus infection: the Randomized Noninferiority MONotherapy of TiviCAY Trial. Clin Infect Dis 69:1498–1505. doi: 10.1093/cid/ciy1132. [DOI] [PubMed] [Google Scholar]
- 7.Havlir DV, Hellmann NS, Petropoulos CJ, Whitcomb JM, Collier AC, Hirsch MS, Tebas P, Sommadossi JP, Richman DD. 2000. Drug susceptibility in HIV infection after viral rebound in patients receiving indinavir-containing regimens. JAMA 283:229–234. doi: 10.1001/jama.283.2.229. [DOI] [PubMed] [Google Scholar]
- 8.Datir R, Kemp S, Bouzidi KE, Mlchocova P, Goldstein R, Breuer J, Towers GJ, Jolly C, Quiñones-Mateu ME, Dakum PS, Ndembi N, Gupta RK. 2020. In vivo emergence of a novel protease inhibitor resistance signature in HIV-1 matrix. mBio 11:e02036-20. doi: 10.1128/mBio.02036-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coetzer M, Ledingham L, Diero L, Kemboi E, Orido M, Kantor R. 2017. Gp41 and Gag amino acids linked to HIV-1 protease inhibitor-based second-line failure in HIV-1 subtype A from western Kenya. J Intern Aids Soc 20:e25024. doi: 10.1002/jia2.25024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castain L, Perrier M, Charpentier C, Palich R, Desire N, Wirden M, Descamps D, Sayon S, Landman R, Valantin MA, Joly V, Peytavin G, Yazdanpanah Y, Katlama C, Calvez V, Marcelin AG, Todesco E. 2019. New mechanisms of resistance in virological failure to protease inhibitors: selection of non-described protease, Gag and Gp41 mutations. J Antimicrob Chemother 74:2019–2023. doi: 10.1093/jac/dkz151. [DOI] [PubMed] [Google Scholar]
- 11.Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, Remillard RR, Walker MA, Langley DR, Krystal M. 2007. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J Biol Chem 282:31186–31196. doi: 10.1074/jbc.M704935200. [DOI] [PubMed] [Google Scholar]
- 12.Malet I, Subra F, Charpentier C, Collin G, Descamps D, Calvez V, Marcelin AG, Delelis O. 2017. Mutations located outside the integrase gene can confer resistance to HIV-1 integrase strand transfer inhibitors. mBio 8:e00922-17. doi: 10.1128/mBio.00922-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabi SA, Laird GM, Durand CM, Laskey S, Shan L, Bailey JR, Chioma S, Moore RD, Siliciano RF. 2013. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J Clin Invest 123:3848–3860. doi: 10.1172/JCI67399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fun A, Wensing AM, Verheyen J, Nijhuis M. 2012. Human immunodeficiency virus Gag and protease: partners in resistance. Retrovirology 9:63. doi: 10.1186/1742-4690-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Checkley MA, Luttge BG, Freed EO. 2011. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J Mol Biol 410:582–608. doi: 10.1016/j.jmb.2011.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2:a006866. doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, Kwong PD, Blanchard SC, Mothes W. 2014. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X, Lu M, Gorman J, Terry DS, Hong X, Zhou Z, Zhao H, Altman RB, Arthos J, Blanchard SC, Kwong PD, Munro JB, Mothes W. 2018. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 7:e34271. doi: 10.7554/eLife.34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herschhorn A, Ma X, Gu C, Ventura JD, Castillo-Menendez L, Melillo B, Terry DS, Smith AB, Blanchard SC, Munro JB, Mothes W, Finzi A, Sodroski J. 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. mBio 7:e01598-16. doi: 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hübner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FY, Li XD, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747. doi: 10.1126/science.1167525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudnicka D, Feldmann J, Porrot F, Wietgrefe S, Guadagnini S, Prévost MC, Estaquier J, Haase AT, Sol-Foulon N, Schwartz O. 2009. Simultaneous cell-to-cell transmission of human immunodeficiency virus to multiple targets through polysynapses. J Virol 83:6234–6246. doi: 10.1128/JVI.00282-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785–801. doi: 10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- 23.Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. 2008. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog 4:e1000015. doi: 10.1371/journal.ppat.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jolly C 2010. T cell polarization at the virological synapse. Viruses 2:1261–1278. doi: 10.3390/v2061261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent Synapse. J Exp Med 199:283–293. doi: 10.1084/jem.20030648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolly C, Mitar I, Sattentau QJ. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J Virol 81:13916–13921. doi: 10.1128/JVI.01585-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol 67:2182–2190. doi: 10.1128/JVI.67.4.2182-2190.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato H, Orenstein J, Dimitrov D, Martin M. 1992. Cell-to-cell spread of HIV-1 occurs within minutes and may not involve the participation of virus particles. Virology 186:712–724. doi: 10.1016/0042-6822(92)90038-Q. [DOI] [PubMed] [Google Scholar]
- 29.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. 2007. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol 81:1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Del Portillo A, Tripodi J, Najfeld V, Wodarz D, Levy DN, Chen BK. 2011. Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol 85:7169–7176. doi: 10.1128/JVI.00231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russell RA, Martin N, Mitar I, Jones E, Sattentau QJ. 2013. Multiple proviral integration events after virological synapse-mediated HIV-1 spread. Virology 443:143–149. doi: 10.1016/j.virol.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 32.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–99. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 33.Zhong P, Agosto LM, Ilinskaya A, Dorjbal B, Truong R, Derse D, Uchil PD, Heidecker G, Mothes W. 2013. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS One 8:e53138. doi: 10.1371/journal.pone.0053138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agosto LM, Zhong P, Munro J, Mothes W. 2014. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog 10:e1003982. doi: 10.1371/journal.ppat.1003982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bastarache SM, Mesplède T, Donahue DA, Sloan RD, Wainberg MA. 2014. Fitness impaired drug resistant HIV-1 is not compromised in cell-to-cell transmission or establishment of and reactivation from latency. Viruses 6:3487–3499. doi: 10.3390/v6093487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boullé M, Müller TG, Dähling S, Ganga Y, Jackson L, Mahamed D, Oom L, Lustig G, Neher RA, Sigal A. 2016. HIV cell-to-cell spread results in earlier onset of viral gene expression by multiple infections per cell. PLoS Pathog 12:e1005964. doi: 10.1371/journal.ppat.1005964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dufloo J, Bruel T, Schwartz O. 2018. HIV-1 cell-to-cell transmission and broadly neutralizing antibodies. Retrovirology 15:51. doi: 10.1186/s12977-018-0434-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Titanji BK, Aasa-Chapman M, Pillay D, Jolly C. 2013. Protease inhibitors effectively block cell-to-cell spread of HIV-1 between T cells. Retrovirology 10:161. doi: 10.1186/1742-4690-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. 2012. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490:283–287. doi: 10.1038/nature11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Law KM, Komarova NL, Yewdall AW, Lee RK, Herrera OL, Wodarz D, Chen BK. 2016. In vivo HIV-1 cell-to-cell transmission promotes multicopy micro-compartmentalized infection. Cell Rep 15:2771–2783. doi: 10.1016/j.celrep.2016.05.059. [DOI] [PubMed] [Google Scholar]
- 41.Ladinsky MS, Kieffer C, Olson G, Deruaz M, Vrbanac V, Tager AM, Kwon DS, Bjorkman PJ. 2014. Electron tomography of HIV-1 infection in gut-associated lymphoid tissue. PLoS Pathog 10:e1003899. doi: 10.1371/journal.ppat.1003899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ladinsky MS, Khamaikawin W, Jung Y, Lin S, Lam J, An DS, Bjorkman PJ, Kieffer C. 2019. Mechanisms of virus dissemination in bone marrow of HIV-1-infected humanized BLT mice. Elife 8:e46916. doi: 10.7554/eLife.46916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kieffer C, Ladinsky MS, Ninh A, Galimidi RP, Bjorkman PJ. 2017. Longitudinal imaging of HIV-1 spread in humanized mice with parallel 3D immunofluorescence and electron tomography. Elife 6:e23282. doi: 10.7554/eLife.23282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gratton S, Cheynier R, Dumaurier MJ, Oksenhendler E, Wain-Hobson S. 2000. Highly restricted spread of HIV-1 and multiply infected cells within splenic germinal centers. Proc Natl Acad Sci U S A 97:14566–14571. doi: 10.1073/pnas.97.26.14566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. 2002. Recombination: multiply infected spleen cells in HIV patients. Nature 418:144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- 46.Suspène R, Meyerhans A. 2012. Quantification of unintegrated HIV-1 DNA at the single cell level in vivo. PLoS One 7:e36246. doi: 10.1371/journal.pone.0036246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Josefsson L, Palmer S, Faria NR, Lemey P, Casazza J, Ambrozak D, Kearney M, Shao W, Kottilil S, Sneller M, Mellors J, Coffin JM, Maldarelli F. 2013. Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog 9:e1003432. doi: 10.1371/journal.ppat.1003432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Duyne R, Kuo LS, Pham P, Fujii K, Freed EO. 2019. Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc Natl Acad Sci U S A 116:9040–9049. doi: 10.1073/pnas.1820333116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. 2009. Functional role of Alix in HIV-1 replication. Virology 391:284–292. doi: 10.1016/j.virol.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korkut A, Hendrickson WA. 2012. Structural plasticity and conformational transitions of HIV envelope glycoprotein gp120. PLoS One 7:e52170. doi: 10.1371/journal.pone.0052170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pacheco B, Alsahafi N, Debbeche O, Prévost J, Ding S, Chapleau J-P, Herschhorn A, Madani N, Princiotto A, Melillo B, Gu C, Zeng X, Mao Y, Smith AB, Sodroski J, Finzi A. 2017. Residues in the gp41 ectodomain regulate HIV-1 envelope glycoprotein conformational transitions induced by gp120-directed inhibitors. J Virol 91:e02219-16. doi: 10.1128/JVI.02219-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wrin T, Loh TP, Vennari JC, Schuitemaker H, Nunberg JH. 1995. Adaptation to persistent growth in the H9 cell line renders a primary isolate of human immunodeficiency virus type 1 sensitive to neutralization by vaccine sera. J Virol 69:39–48. doi: 10.1128/JVI.69.1.39-48.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pugach P, Kuhmann SE, Taylor J, Marozsan AJ, Snyder A, Ketas T, Wolinsky SM, Korber BT, Moore JP. 2004. The prolonged culture of human immunodeficiency virus type 1 in primary lymphocytes increases its sensitivity to neutralization by soluble CD4. Virology 321:8–22. doi: 10.1016/j.virol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 55.Moore JP, McKeating JA, Huang YX, Ashkenazi A, Ho DD. 1992. Virions of primary human immunodeficiency virus type 1 isolates resistant to soluble CD4 (sCD4) neutralization differ in sCD4 binding and glycoprotein gp120 retention from sCD4-sensitive isolates. J Virol 66:235–243. doi: 10.1128/JVI.66.1.235-243.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore J, McKeating J, Weiss R, Sattentau Q. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139–1142. doi: 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 57.Hammonds J, Chen X, Ding L, Fouts T, De Vico A, Zur Megede J, Barnett S, Spearman P. 2003. Gp120 stability on HIV-1 virions and Gag-Env pseudovirions is enhanced by an uncleaved Gag core. Virology 314:636–649. doi: 10.1016/s0042-6822(03)00467-7. [DOI] [PubMed] [Google Scholar]
- 58.Engelman AN 2019. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J Biol Chem 294:15137–15157. doi: 10.1074/jbc.REV119.006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin N, Welsch S, Jolly C, Briggs JA, Vaux D, Sattentau QJ. 2010. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J Virol 84:3516–3527. doi: 10.1128/JVI.02651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abela IA, Berlinger L, Schanz M, Reynell L, Günthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshimura K, Harada S, Boonchawalit S, Kawanami Y, Matsushita S. 2014. Impact of maraviroc-resistant and low-CCR5-adapted mutations induced by in vitro passage on sensitivity to anti-envelope neutralizing antibodies. J Gen Virol 95:1816–1826. doi: 10.1099/vir.0.062885-0. [DOI] [PubMed] [Google Scholar]
- 62.Keller PW, Morrison O, Vassell R, Weiss CD. 2018. HIV-1 gp41 residues modulate CD4-induced conformational changes in the envelope glycoprotein and evolution of a relaxed conformation of gp120. J Virol 92:e00583-18. doi: 10.1128/JVI.00583-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hikichi Y, Yokoyama M, Takemura T, Fujino M, Kumakura S, Maeda Y, Yamamoto N, Sato H, Matano T, Murakami T. 2016. Increased HIV-1 sensitivity to neutralizing antibodies by mutations in the Env V3-coding region for resistance to CXCR4 antagonists. J Gen Virol 97:2427–2440. doi: 10.1099/jgv.0.000536. [DOI] [PubMed] [Google Scholar]
- 64.Berro R, Sanders RW, Lu M, Klasse PJ, Moore JP. 2009. Two HIV-1 variants resistant to small molecule CCR5 inhibitors differ in how they use CCR5 for entry. PLoS Pathog 5:e1000548. doi: 10.1371/journal.ppat.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zou S, Zhang S, Gaffney A, Ding H, Lu M, Grover JR, Farrell M, Nguyen HT, Zhao C, Anang S, Zhao M, Mohammadi M, Blanchard SC, Abrams C, Madani N, Mothes W, Kappes JC, Smith AB, III, Sodroski J. 2020. Long-acting BMS-378806 analogues stabilize the state-1 conformation of the human immunodeficiency virus type 1 envelope glycoproteins. J Virol 94:e00148-20. doi: 10.1128/JVI.00148-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schön A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci U S A 101:5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pancera M, Lai YT, Bylund T, Druz A, Narpala S, O'Dell S, Schon A, Bailer RT, Chuang GY, Geng H, Louder MK, Rawi R, Soumana DI, Finzi A, Herschhorn A, Madani N, Sodroski J, Freire E, Langley DR, Mascola JR, McDermott AB, Kwong PD. 2017. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat Chem Biol 13:1115–1122. doi: 10.1038/nchembio.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Melikian GL, Rhee SY, Varghese V, Porter D, White K, Taylor J, Towner W, Troia P, Burack J, Dejesus E, Robbins GK, Razzeca K, Kagan R, Liu TF, Fessel WJ, Israelski D, Shafer RW. 2014. Non-nucleoside reverse transcriptase inhibitor (NNRTI) cross-resistance: implications for preclinical evaluation of novel NNRTIs and clinical genotypic resistance testing. J Antimicrob Chemother 69:12–20. doi: 10.1093/jac/dkt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen HT, Alsahafi N, Finzi A, Sodroski JG. 2019. Effects of the SOS (A501C/T605C) and DS (I201C/A433C) disulfide bonds on HIV-1 membrane envelope glycoprotein conformation and function. J Virol 93:e00304-19. doi: 10.1128/JVI.00304-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu M, Ma X, Castillo-Menendez LR, Gorman J, Alsahafi N, Ermel U, Terry DS, Chambers M, Peng D, Zhang B, Zhou T, Reichard N, Wang K, Grover JR, Carman BP, Gardner MR, Nikic-Spiegel I, Sugawara A, Arthos J, Lemke EA, Smith AB, III, Farzan M, Abrams C, Munro JB, McDermott AB, Finzi A, Kwong PD, Blanchard SC, Sodroski JG, Mothes W. 2019. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 568:415–419. doi: 10.1038/s41586-019-1101-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alsahafi N, Debbeche O, Sodroski J, Finzi A. 2015. Effects of the I559P gp41 change on the conformation and function of the human immunodeficiency virus (HIV-1) membrane envelope glycoprotein trimer. PLoS One 10:e0122111. doi: 10.1371/journal.pone.0122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alsahafi N, Anand SP, Castillo-Menendez L, Verly MM, Medjahed H, Prevost J, Herschhorn A, Richard J, Schon A, Melillo B, Freire E, Smith AB, III, Sodroski J, Finzi A. 2018. SOSIP changes affect human immunodeficiency virus type 1 envelope glycoprotein conformation and CD4 engagement. J Virol 92:e01080-18. doi: 10.1128/JVI.01080-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Q, Liu L, Ren W, Gettie A, Wang H, Liang Q, Shi X, Montefiori DC, Zhou T, Zhang L. 2019. A single substitution in gp41 modulates the neutralization profile of SHIV during in vivo adaptation. Cell Rep 27:2593.e5–2607.e5. doi: 10.1016/j.celrep.2019.04.108. [DOI] [PubMed] [Google Scholar]
- 74.Allaway GP, Davis-Bruno KL, Beaudry GA, Garcia EB, Wong EL, Ryder AM, Hasel KW, Gauduin MC, Koup RA, McDougal JS. 1995. Expression and characterization of CD4-IgG2, a novel heterotetramer that neutralizes primary HIV type 1 isolates. AIDS Res Hum Retroviruses 11:533–539. doi: 10.1089/aid.1995.11.533. [DOI] [PubMed] [Google Scholar]
- 75.Buchacher A, Predl R, Strutzenberger K, Steinfellner W, Trkola A, Purtscher M, Gruber G, Tauer C, Steindl F, Jungbauer A. 1994. Generation of human monoclonal antibodies against HIV-1 proteins; electrofusion and Epstein-Barr virus transformation for peripheral blood lymphocyte immortalization. AIDS Res Hum Retroviruses 10:359–369. doi: 10.1089/aid.1994.10.359. [DOI] [PubMed] [Google Scholar]
- 76.Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P, Protocol G Principal Investigators. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR, Protocol G Principal Investigators. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang GY, Druz A, Doria-Rose NA, Laub L, Sliepen K, van Gils MJ, de la Peña AT, Derking R, Klasse PJ, Migueles SA, Bailer RT, Alam M, Pugach P, Haynes BF, Wyatt RT, Sanders RW, Binley JM, Ward AB, Mascola JR, Kwong PD, Connors M. 2014. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 515:138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cavacini LA, Emes CL, Power J, Buchbinder A, Zolla-Pazner S, Posner MR. 1993. Human monoclonal antibodies to the V3 loop of HIV-1 gp120 mediate variable and distinct effects on binding and viral neutralization by a human monoclonal antibody to the CD4 binding site. J Acquir Immune Defic Syndr (1988) 6:353–358. [PubMed] [Google Scholar]
- 81.Gorny MK, Conley AJ, Karwowska S, Buchbinder A, Xu JY, Emini EA, Koenig S, Zolla-Pazner S. 1992. Neutralization of diverse human immunodeficiency virus type 1 variants by an anti-V3 human monoclonal antibody. J Virol 66:7538–7542. doi: 10.1128/JVI.66.12.7538-7542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, Alam SM, Wang T, Yang Y, Zhang B, Migueles SA, Wyatt R, Haynes BF, Kwong PD, Mascola JR, Connors M. 2012. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491:406–412. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thali M, Moore JP, Furman C, Charles M, Ho DD, Robinson J, Sodroski J. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol 67:3978–3988. doi: 10.1128/JVI.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McCallus DE, Ugen KE, Sato AI, Williams WV, Weiner DB. 1992. Construction of a recombinant bacterial human CD4 expression system producing a bioactive CD4 molecule. Viral Immunol 5:163–172. doi: 10.1089/vim.1992.5.163. [DOI] [PubMed] [Google Scholar]
- 85.De Clercq E, Yamamoto N, Pauwels R, Balzarini J, Witvrouw M, De Vreese K, Debyser Z, Rosenwirth B, Peichl P, Datema R. 1994. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob Agents Chemother 38:668–674. doi: 10.1128/aac.38.4.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Freed EO, Englund G, Martin MA. 1995. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol 69:3949–3954. doi: 10.1128/JVI.69.6.3949-3954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP, Bjorkman PJ, Wilen CB, Doms RW, O'Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moore PL, Gray ES, Choge IA, Ranchobe N, Mlisana K, Abdool Karim SS, Williamson C, Morris L, CAPRISA 002 Study Team. 2008. The c3-v4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J Virol 82:1860–1869. doi: 10.1128/JVI.02187-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wibmer CK, Bhiman JN, Gray ES, Tumba N, Abdool Karim SS, Williamson C, Morris L, Moore PL. 2013. Viral escape from HIV-1 neutralizing antibodies drives increased plasma neutralization breadth through sequential recognition of multiple epitopes and immunotypes. PLoS Pathog 9:e1003738. doi: 10.1371/journal.ppat.1003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu J, Li M, Wilkins J, Ding S, Swartz TH, Esposito AM, Zheng YM, Freed EO, Liang C, Chen BK, Liu SL. 2015. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep 13:145–156. doi: 10.1016/j.celrep.2015.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 92.Ikeda T, Symeonides M, Albin JS, Li M, Thali M, Harris RS. 2018. HIV-1 adaptation studies reveal a novel Env-mediated homeostasis mechanism for evading lethal hypermutation by APOBEC3G. PLoS Pathog 14:e1007010. doi: 10.1371/journal.ppat.1007010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Durham ND, Yewdall AW, Chen P, Lee R, Zony C, Robinson JE, Chen BK. 2012. Neutralization resistance of virological synapse-mediated HIV-1 Infection is regulated by the gp41 cytoplasmic tail. J Virol 86:7484–7495. doi: 10.1128/JVI.00230-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Reh L, Magnus C, Schanz M, Weber J, Uhr T, Rusert P, Trkola A. 2015. Capacity of broadly neutralizing antibodies to inhibit HIV-1 cell-cell transmission is strain- and epitope-dependent. PLoS Pathog 11:e1004966. doi: 10.1371/journal.ppat.1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nameki D, Kodama E, Ikeuchi M, Mabuchi N, Otaka A, Tamamura H, Ohno M, Fujii N, Matsuoka M. 2005. Mutations conferring resistance to human immunodeficiency virus type 1 fusion inhibitors are restricted by gp41 and Rev-responsive element functions. J Virol 79:764–770. doi: 10.1128/JVI.79.2.764-770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Collier D, Iwuji C, Derache A, de Oliveira T, Okesola N, Calmy A, Dabis F, Pillay D, Gupta RK, French National Agency for AIDS and Viral Hepatitis Research (ANRS) 12249 Treatment as Prevention (TasP) Study Group. 2017. Virological outcomes of second-line protease inhibitor-based treatment for human immunodeficiency virus type 1 in a high-prevalence rural South African setting: a competing-risks prospective cohort analysis. Clinical Infectious Diseases 64:1006–1016. doi: 10.1093/cid/cix015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gallego O, de Mendoza C, Pérez-Elías MJ, Guardiola JM, Pedreira J, Dalmau D, Gónzalez J, Moreno A, Arribas JR, Rubio A, García-Arata I, Leal M, Domingo P, Soriano V. 2001. Drug resistance in patients experiencing early virological failure under a triple combination including indinavir. AIDS 15:1701–1706. doi: 10.1097/00002030-200109070-00014. [DOI] [PubMed] [Google Scholar]
- 98.Hosseinipour MC, Gupta RK, Van Zyl G, Eron JJ, Nachega JB. 2013. Emergence of HIV drug resistance during first- and second-line antiretroviral therapy in resource-limited settings. J Infect Dis 207 Suppl 2:S49–56. doi: 10.1093/infdis/jit107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pulido F, Arribas J, Hill A, Moecklinghoff C. 2012. No evidence for evolution of protease inhibitor resistance from standard genotyping, after three years of treatment with darunavir/ritonavir, with or without nucleoside analogues. AIDS Res Hum Retroviruses 28:1167–1169. doi: 10.1089/AID.2011.0256. [DOI] [PubMed] [Google Scholar]
- 100.Court R, Gordon M, Cohen K, Stewart A, Gosnell B, Wiesner L, Maartens G. 2016. Random lopinavir concentrations predict resistance on lopinavir-based antiretroviral therapy. Int J Antimicrobial Agents 48:158–162. doi: 10.1016/j.ijantimicag.2016.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Zyl GU, van Mens TE, McIlleron H, Zeier M, Nachega JB, Decloedt E, Malavazzi C, Smith P, Huang Y, van der Merwe L, Gandhi M, Maartens G. 2011. Low lopinavir plasma or hair concentrations explain second-line protease inhibitor failures in a resource-limited setting. J Acquir Immune Defic Syndr 56:333–339. doi: 10.1097/QAI.0b013e31820dc0cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murakami T, Ablan S, Freed EO, Tanaka Y. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78:1026–1031. doi: 10.1128/jvi.78.2.1026-1031.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, Aiken C. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78:3429–3435. doi: 10.1128/jvi.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wijting IEA, Lungu C, Rijnders BJA, van der Ende ME, Pham HT, Mesplede T, Pas SD, Voermans JJC, Schuurman R, van de Vijver DAMC, Boers PHM, Gruters RA, Boucher CAB, van Kampen JJA. 2018. HIV-1 resistance dynamics in patients with virologic failure to dolutegravir maintenance monotherapy. J Infect Dis 218:688–697. doi: 10.1093/infdis/jiy176. [DOI] [PubMed] [Google Scholar]
- 105.Drummer HE, Hill MK, Maerz AL, Wood S, Ramsland PA, Mak J, Poumbourios P. 2013. Allosteric modulation of the HIV-1 gp120-gp41 association site by adjacent gp120 variable region 1 (V1) N-glycans linked to neutralization sensitivity. PLoS Pathog 9:e1003218. doi: 10.1371/journal.ppat.1003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Narasimhulu VGS, Bellamy-McIntyre AK, Laumaea AE, Lay CS, Harrison DN, King HAD, Drummer HE, Poumbourios P. 2018. Distinct functions for the membrane-proximal ectodomain region (MPER) of HIV-1 gp41 in cell-free and cell-cell viral transmission and cell-cell fusion. J Biol Chem 293:6099–6120. doi: 10.1074/jbc.RA117.000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cao J, Bergeron L, Helseth E, Thali M, Repke H, Sodroski J. 1993. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J Virol 67:2747–2755. doi: 10.1128/JVI.67.5.2747-2755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Diaz-Aguilar B, Dewispelaere K, Yi HA, Jacobs A. 2013. Significant differences in cell-cell fusion and viral entry between strains revealed by scanning mutagenesis of the C-heptad repeat of HIV gp41. Biochemistry 52:3552–3563. doi: 10.1021/bi400201h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kahle KM, Steger HK, Root MJ. 2009. Asymmetric deactivation of HIV-1 gp41 following fusion inhibitor binding. PLoS Pathog 5:e1000674. doi: 10.1371/journal.ppat.1000674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roy NH, Lambelé M, Chan J, Symeonides M, Thali M. 2014. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. J Virol 88:7645–7658. doi: 10.1128/JVI.00550-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Symeonides M, Murooka TT, Bellfy LN, Roy NH, Mempel TR, Thali M. 2015. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 7:6590–6603. doi: 10.3390/v7122959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Compton AA, Schwartz O. 2017. They might be giants: does syncytium formation sink or spread HIV infection? PLoS Pathog 13:e1006099. doi: 10.1371/journal.ppat.1006099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Das AT, Land A, Braakman I, Klaver B, Berkhout B. 1999. HIV-1 evolves into a nonsyncytium-inducing virus upon prolonged culture in vitro. Virology 263:55–69. doi: 10.1006/viro.1999.9898. [DOI] [PubMed] [Google Scholar]
- 114.Das AT, van Dam AP, Klaver B, Berkhout B. 1998. Improved envelope function selected by long-term cultivation of a translation-impaired HIV-1 mutant. Virology 244:552–562. doi: 10.1006/viro.1998.9124. [DOI] [PubMed] [Google Scholar]
- 115.Sylwester A, Murphy S, Shutt D, Soll DR. 1997. HIV-induced T cell syncytia are self-perpetuating and the primary cause of T cell death in culture. J Immunol 158:3996–4007. [PubMed] [Google Scholar]
- 116.Van Rompay KKA, Hassounah S, Keele BF, Lifson JD, Ardeshir A, Watanabe J, Pham HT, Chertova E, Sowder R, Balzarini J, Mesplède T, Wainberg MA. 2018. Dolutegravir monotherapy of simian immunodeficiency virus-infected macaques selects for several patterns of resistance mutations with variable virological outcomes. J Virol 93:e01189-18. doi: 10.1128/JVI.01189-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wijting I, Rutsaert SL, Rokx C, Burger DM, Verbon A, van Kampen J, Boucher C, Rijnders B, Vandekerckhove L. 2019. Predictors of virological failure in HIV-1-infected patients switching to dolutegravir maintenance monotherapy. HIV Med 20:63–68. doi: 10.1111/hiv.12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Anstett K, Brenner B, Mesplede T, Wainberg MA. 2017. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology 14:36. doi: 10.1186/s12977-017-0360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D. 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol 72:2855–2864. doi: 10.1128/JVI.72.4.2855-2864.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Means RE, Matthews T, Hoxie JA, Malim MH, Kodama T, Desrosiers RC. 2001. Ability of the V3 loop of simian immunodeficiency virus to serve as a target for antibody-mediated neutralization: correlation of neutralization sensitivity, growth in macrophages, and decreased dependence on CD4. J Virol 75:3903–3915. doi: 10.1128/JVI.75.8.3903-3915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gao F, Scearce RM, Alam SM, Hora B, Xia S, Hohm JE, Parks RJ, Ogburn DF, Tomaras GD, Park E, Lomas WE, Maino VC, Fiscus SA, Cohen MS, Moody MA, Hahn BH, Korber BT, Liao HX, Haynes BF. 2009. Cross-reactive monoclonal antibodies to multiple HIV-1 subtype and SIVcpz envelope glycoproteins. Virology 394:91–98. doi: 10.1016/j.virol.2009.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. doi: 10.1128/JVI.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schindler M, Münch J, Kirchhoff F. 2005. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol 79:5489–5498. doi: 10.1128/JVI.79.9.5489-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schindler M, Würfl S, Benaroch P, Greenough TC, Daniels R, Easterbrook P, Brenner M, Münch J, Kirchhoff F. 2003. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J Virol 77:10548–10556. doi: 10.1128/jvi.77.19.10548-10556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Freed EO, Delwart EL, Buchschacher GL, Panganiban AT. 1992. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci U S A 89:70–74. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Willey RL, Smith DH, Lasky LA, Theodore TS, Earl PL, Moss B, Capon DJ, Martin MA. 1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J Virol 62:139–147. doi: 10.1128/JVI.62.1.139-147.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Urano E, Timilsina U, Kaplan JA, Ablan S, Ghimire D, Pham P, Kuruppu N, Mandt R, Durell SR, Nitz TJ, Martin DE, Wild CT, Gaur R, Freed EO. 2018. Resistance to second-generation HIV-1 maturation inhibitors. J Virol 93:e02017-18. doi: 10.1128/JVI.02017-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. 2005. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol 43:406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wiegand A, Spindler J, Hong FF, Shao W, Cyktor JC, Cillo AR, Halvas EK, Coffin JM, Mellors JW, Kearney MF. 2017. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc Natl Acad Sci U S A 114:E3659–E3668. doi: 10.1073/pnas.1617961114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kearney M, Palmer S, Maldarelli F, Shao W, Polis MA, Mican J, Rock-Kress D, Margolick JB, Coffin JM, Mellors JW. 2008. Frequent polymorphism at drug resistance sites in HIV-1 protease and reverse transcriptase. AIDS 22:497–501. doi: 10.1097/QAD.0b013e3282f29478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Swanson P, Devare SG, Hackett J. 2003. Molecular characterization of 39 HIV isolates representing group M (subtypes A-G) and group O: sequence analysis of gag p24, pol integrase, and env gp41. AIDS Res Hum Retroviruses 19:625–629. doi: 10.1089/088922203322231003. [DOI] [PubMed] [Google Scholar]
- 132.Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, Parrish NF, Shaw KS, Guffey MB, Bar KJ, Davis KL, Ochsenbauer-Jambor C, Kappes JC, Saag MS, Cohen MS, Mulenga J, Derdeyn CA, Allen S, Hunter E, Markowitz M, Hraber P, Perelson AS, Bhattacharya T, Haynes BF, Korber BT, Hahn BH, Shaw GM. 2009. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med 206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Myers GK, Hahn BH, Jeang K-T, Mellors JW, McCutchan FE, Henderson LE, Pavlakis GN. 1995. Human retroviruses and AIDS 1995: a compilation and analysis of nucleic acid and amino acid sequences. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM. [Google Scholar]
- 134.Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E, Allen S, Manigart O, Mulenga J, Anderson JA, Swanstrom R, Haynes BF, Athreya GS, Korber BT, Sharp PM, Shaw GM, Hahn BH. 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82:3952–3970. doi: 10.1128/JVI.02660-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Patro SC, Brandt LD, Bale MJ, Halvas EK, Joseph KW, Shao W, Wu X, Guo S, Murrell B, Wiegand A, Spindler J, Raley C, Hautman C, Sobolewski M, Fennessey CM, Hu WS, Luke B, Hasson JM, Niyongabo A, Capoferri AA, Keele BF, Milush J, Hoh R, Deeks SG, Maldarelli F, Hughes SH, Coffin JM, Rausch JW, Mellors JW, Kearney MF. 2019. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc Natl Acad Sci U S A 116:25891–25899. doi: 10.1073/pnas.1910334116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stewart-Jones GB, Soto C, Lemmin T, Chuang GY, Druz A, Kong R, Thomas PV, Wagh K, Zhou T, Behrens AJ, Bylund T, Choi CW, Davison JR, Georgiev IS, Joyce MG, Kwon YD, Pancera M, Taft J, Yang Y, Zhang B, Shivatare SS, Shivatare VS, Lee CC, Wu CY, Bewley CA, Burton DR, Koff WC, Connors M, Crispin M, Baxa U, Korber BT, Wong CH, Mascola JR, Kwong PD. 2016. Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell 165:813–826. doi: 10.1016/j.cell.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell-cell fusion activity of NL4-3 Env-A539V. HEK293T cells were transfected with the indicated proviral clones. At 24 h posttransfection, serial dilutions of the transfected HEK293T cells were cocultured with TZM-bl cells in the presence of a cocktail of EFV and DTG. Luciferase activity was measured at 24 h postinfection. Data from at least three independent experiments are shown as means ± SEs. ***, P < 0.001 by one-way ANOVA and Tukey’s multiple-comparison test. Download FIG S1, TIF file, 0.09 MB (95.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Binding of Env-A539V to CD4-IgG2. HEK293T cells transfected with the GFP-encoding reporter clone pBR43IeG expressing WT NL4-3 Env or the Env-A539V mutant were preincubated with 1.0 µg/ml of CD4-IgG2 at 4°C for 1 h. KFS is an Env-defective NL4-3 mutant included as a negative control. The cells were washed, and APC-conjugated anti-human IgG was used to detect bound CD4-IgG2. APC signals were normalized by GFP signal to calculate the CD4-IgG2 binding efficiency. Data are shown as means ± SEs from four independent experiments. *, P < 0.05 by one-way ANOVA and Tukey’s multiple-comparison test. Download FIG S2, TIF file, 0.09 MB (93.2KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Comparison of the sensitivity of NL4-3 and NL(AD8) to NAbs, DTG, T-20, and sCD4 in cell-free infection. TZM-bl cells were infected with 100 TCID50 of the indicated viruses in the presence of various concentrations of NAbs (A to F), ARVs (H and I), and sCD4 (J). sCD4 incubations were performed for 0 or 2 h. The data are related to Fig. 3 and 8. Data from at least three independent experiments are shown as means ± SEs. Download FIG S3, TIF file, 0.5 MB (468.7KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Analysis of sCD4-induced and time-dependent gp120 shedding of NL(AD8) Env mutants and comparison of NL4-3 versus NL(AD8) gp120 shedding. (A) Concentrated viruses were incubated with the indicated concentrations of sCD4 at 37°C for 2 h. Incubated viruses were subsequently purified through 20% sucrose cushions, and viral proteins were detected by Western blotting. A representative gel is shown. (B) Viruses were incubated at 37°C for the indicated times. Incubated viruses were subsequently purified through 20% sucrose cushions, and viral proteins were detected by Western blotting. A representative gel is shown. Quantification of sCD4-induced (C) and time-dependent (D) gp120 shedding from three independent experiments, calculated as the ratios of virion-associated gp120/p24 and shown as means ± SEs. Comparison of NL4-3 and NL(AD8) sCD4-induced (E) and time-dependent (F) gp120 shedding. The data are related to Fig. 5 andS3. Data from at least three independent experiments are shown as means ± SEs. Statistical analysis by unpaired t test. Download FIG S4, TIF file, 0.7 MB (719.9KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.