

Abstract
Here we show that enzymatic reaction generates peptide assemblies to sequestrate proteins for selectively killing cancer cells. A phosphopeptide bearing the antagonistic motif (AVPI) to the inhibitors of apoptotic proteins (IAPs) enters cancer cells and normal cells via caveolin-dependent endocytosis and macropinocytosis, respectively. The AVPI-bearing peptide assemblies in the cytosol sequestrate IAPs and sensitize the cancer cells to bortezomib (BTZ), a proteasome inhibitor, but rescue the normal cells (i.e., HS-5) by trafficking the BTZ into lysosomes. Alkaline phosphatase (ALP) acts as a context-dependent signal for trafficking the peptide/BTZ assemblies and selectively inducing the death of the cancer cells. The assemblies of AVPI exhibit enhanced proteolytic resistance. This work, being the first example of utilizing the endocytic difference of enzymatically formed peptide assemblies for selective killing cancer cells, promises a new way to develop selective cancer therapeutics.
Keywords: Self-Assembly, Inhibitors of Apoptosis Proteins, Cell-Death, Cancer, Enzyme
Graphical Abstract
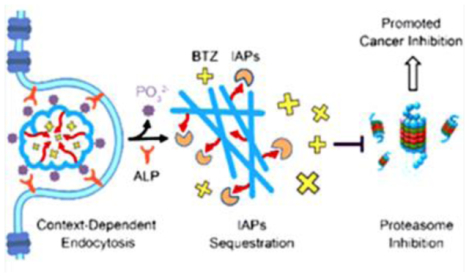
The enzymatic self-assembly of propapoptotic peptides sensitizes cancer cells to anticancer drugs while reduces the side effects on normal cell. This result illustrates the combination of enzymatic noncovalent synthesis with other therapeutic agents for treating diseases.
Cancer remains a major burden to public health[1] because multiple underlying cellular mechanisms complicate the cancer treatment.[2] Beside the problems of drug resistance in cancer therapy,[3] another challenge is how to increase treatment efficacy without increasing toxicity to normal tissues and organs.[4] Thus, it is necessary and beneficial to explore differences between cancer and normal cells for selectively killing cancer cells. One of well-known differences is the overexpression of alkaline phosphatase (ALP) by cancer cells.[5] In fact, ALP is the most tested cancer biomarker in clinical setting.[6] In the study of profiling of the ALP activities of live cells, we found that ALP presented on the surface of certain cancer cells (e.g., HeLa and Saos2), but not on bone marrow stromal cells (e.g., HS-5).[7] Thus, we decided to explore whether ALP on the cell surface could serve as a context-dependent signal for increasing the efficacy of bortezomib (BTZ), a clinically-used proteasome inhibitor, against cancer cells and reducing the known side effect of BTZ (toxicity to bone marrow stromal cells[8]). Considering that the upregulation of inhibitors of apoptotic proteins (IAPs) leads to the resistance to BTZ[9], we also introduced the binding motif (AVPI) of the IAPs to a phosphopeptide to generate intracellular assemblies of AVPI for antagonizing IAPs.
Our studies show that these ALP responsive peptide assemblies not only sequestrate IAPs in cancer cells to enhance the efficacy of BTZ, but also relocate BTZ into the lysosomes to reduce the toxicity to normal cells (HS-5)[10], as illustrated in Scheme 1. Specifically, conjugating AVPI to the ε-NH2 of lysine in a phosphotyrosine-bearing peptide generates a branched proapoptotic peptide (1), which self-assembles to form micelles that turn into nanofibers upon ALP-catalyzed dephosphorylation. Entering cancer cells via the caveolin-dependent endocytosis, the micelles of 1 mainly localize at the endoplasmic reticulum (ER) and form the assemblies of 2 to sequestrate the IAPs, thus significantly aggravating the BTZ-induced apoptosis of the cancer cells. Contrasting to the case of cancer cells, the micelles of 1 and BTZ, entering HS-5 cells via macropinocytosis, localize in the lysosome of HS-5, thus blocking BTZ to its target (proteasomes) and reducing the toxicity of BTZ. Differing from other ALP-instructed peptide assemblies that selectively gel and kill cancer cells as anticancer agents[11], the phosphopeptide reported in this work not only forms assemblies to deactivate the IAPs in cancer cells, but also rescues normal cells (e.g. HS-5) from the side effects of anticancer drugs (e.g., BTZ). Further study indicates that the micelles of 1 interact with the ALP enriched in lipid rafts[12] of the cancer cells for caveolin-mediated endocytosis,[13] confirming ALP as a context-dependent signal. Moreover, 1, as a heterochiral branched peptide, exhibits higher proteolytic resistance than AVPI peptide does. This work, utilizing the ALP difference between cancer and normal cells to control the endocytosis, the location of AVPI assemblies, and the location of proteasome inhibitor for selectively killing cancer cells and mitigating the side effect on normal cell, promises a new way to develop therapeutics with high selectivity.
Scheme 1.
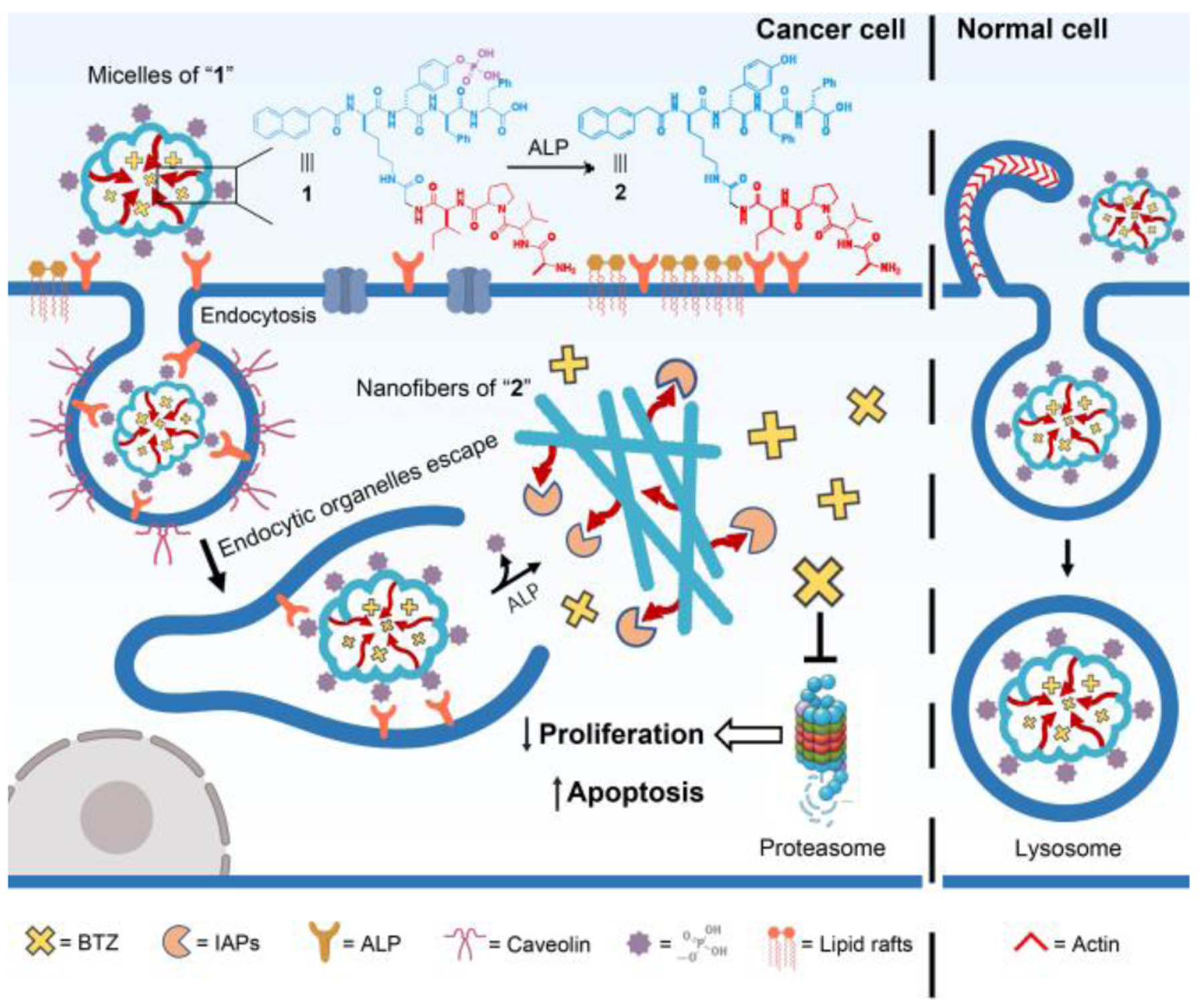
Illustration of enzymatically-forming peptide assemblies to sequestrate IAPs for selectively killing cancer cells.
As shown in Scheme 1, the precursor of the proapoptotic peptide consists of (i) the IAPs binding motif (NH2-Ala-Val-Pro-Ile, AVPI) from DIABLO[14] for sequestrating the IAPs in cells for apoptosis, (ii) a D-phenylalanine rich peptide containing phosphotyrosine (D1p, Scheme S1) for ALP-catalyzed self-assembly. Using Fmoc-based solid-phase peptide synthesis, we firstly produced the AVPI-Gly with protecting groups (Scheme S2). Subsequently, we connected the protected AVPI-Gly to D1p through C-terminal activation (Scheme S2). Finally, the removal of all protecting groups and the purification by high-pressure liquid chromatography (HPLC) produce the designed 1.
We first confirmed that 1 undergoes enzyme-instructed self-assembly (EISA).[15] Transmission electron microscopy (TEM) reveals that 1 forms micelles (27±6 nm in diameter) in solution (PBS, pH=7.4) at it critical micelle concentration (CMC, Figure S1), which turns into the nanofibers of 2 (23±4 nm in diameter) after the dephosphorylation catalyzed by ALP (Figure 1A and 1B). Circular dichroism (CD) spectrum shows that the secondary structure of the assemblies of 1 turns from random coil (featured by the trough around 195 nm) to β-sheet (featured by the peak around 200 nm and the trough around 215 nm) upon the addition of ALP (Figure 1C), which agrees with the TEM images (Figure 1A). Due to self-assembly and D-peptide backbone, 1 exhibits much higher stability than AVPI against the proteolytic hydrolysis[16] catalyzed by proteinase K or cell lysates (Figure 1D). The proteolytic stability of 1 confirms the emergent properties of the peptide assemblies and provides an enduring antagonistic effect against IAPs in cells.
Figure 1.
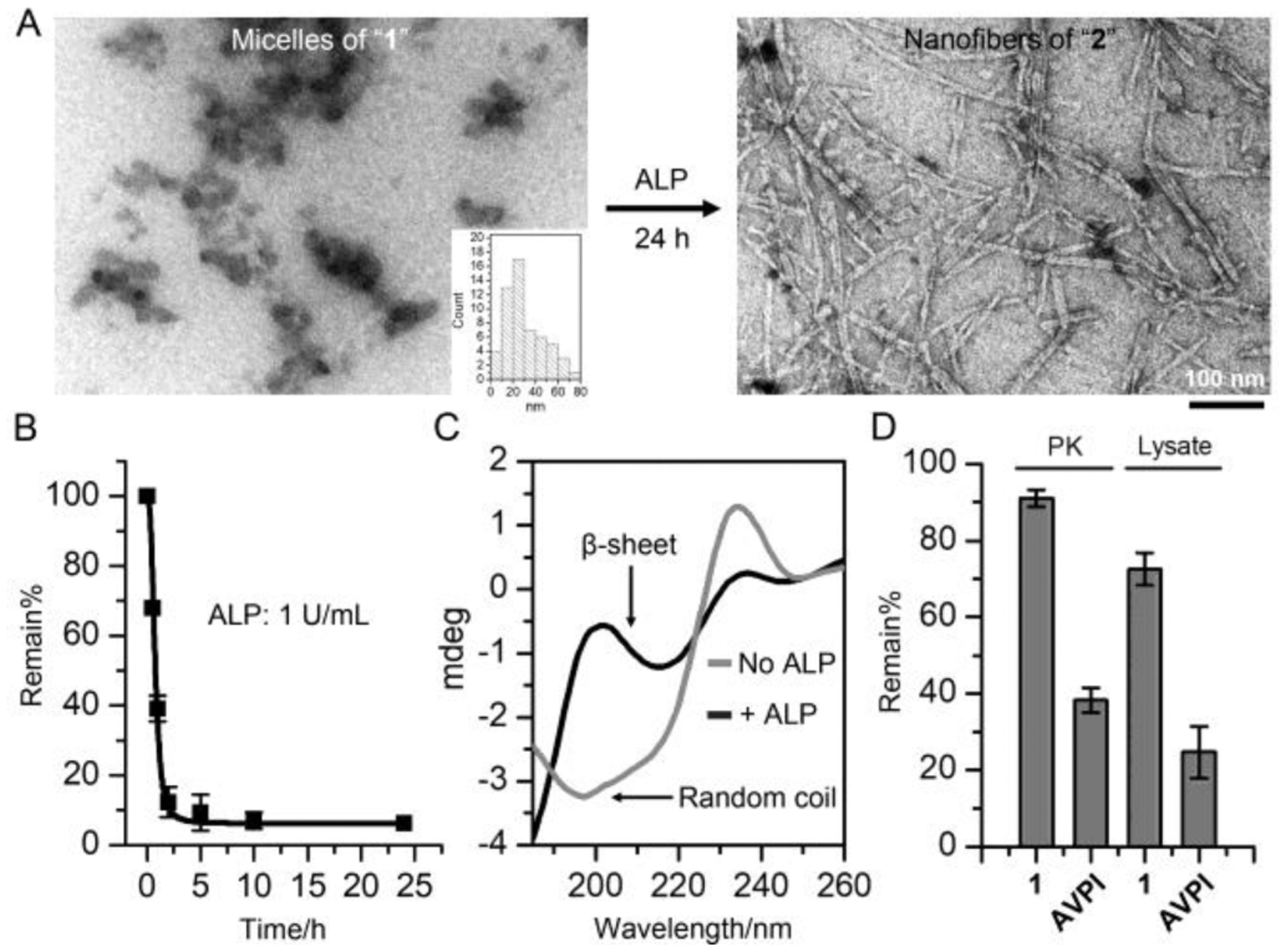
(A) TEM images of 1 (50 μM) before and after adding ALP (1 U/mL, 24 h). (B) Rapid dephosphorylation of 1 (200 μM) by ALP. (C) CD spectrum of 1 (200 μM) before and after adding ALP (1 U/mL, 24 h). (D) Remain% of 1 (200 μM) and AVPI peptide (200 μM) in the presence of proteinase K (PK, 2 U/mL, 24 h) or in Saos-2 cell lysate (24 h).
Although 1 shows little cytotoxicity to cells (Figure S2), the combination of BTZ and 1 exhibits significantly increased cytotoxicity against cancer cells (HeLa and Saos2), compared to BTZ only (Figure 2A, 2B and S3). The increase of the concentration of 1 in the combination (Figure 2C) results in more cell death, indicating that the proapoptotic peptide associates with the IAPs in the cancer cells, thus promoting cancer cell death. On the contrary, 1 efficiently rescues HS-5 cell, a normal cell line, from BTZ-induced apoptosis (Figure 2D and S3). To confirm that 1 decreases the cytotoxicity of BTZ against HS-5 cell, we determined the level of marker proteins of apoptotic signaling (e.g. DR5[17], DR4[18] and caspase-8[19]) in HS-5 cells by Western blot. HS-5 cells incubated with the mixture of 1 and BTZ express less DR5 and DR4, but more full-length caspase-8 than the cells treated by BTZ only (Figure 2E and 2F), confirming that 1 decreases the cytotoxicity of BTZ by helping reduce the apoptosis of the HS-5 cells.
Figure 2.

Cell viabilities of (A, B) HeLa and Saos-2 cells incubated with (A) 25 nM and (B) 200 nM of BTZ or the mixture of BTZ and 1 (50 μM), (C) HeLa cells incubated with 1 of different concentrations in the presence of BTZ (25 nM), and (D) HS-5 cells incubated with 25 nM and 200 nM of BTZ or the mixture of BTZ and 1 (50 μM). (E) Western blot of DR5, full-length caspase-8 and DR4 in the HS-5 cells treated by PBS, 1 (50 μM), BTZ (100 nM), and the mixture of BTZ (100 nM) and 1 (50 μM) for 2 days. (F) The relative protein levels of DR5, full-length caspase-8 and DR4 in (E). The protein level in the control group is defined to be 1.0.
To study the subcellular location of the proapoptotic peptide, we synthesized the nitrobenzofurazan (NBD) conjugated branched peptide, 3. As shown in Figure 3A, cancer cells incubated with 3 exhibit intensive fluorescence of NBD in ER, suggesting that the peptide escapes from endocytic organelles (Figure S4) and is further dephosphorylated in ER where ALP is produced.[20] Pretreating cancer cells with phospholipase C[21] (PLC) that removes the GPI-anchored ALP[22] from cell surface[21], M-β-CD[23] that disrupts the ALP-rich lipid rafts[12] and inhibits caveolin-dependent endocytosis, or replacing the phosphotyrosine in 3 by tyrosine (4, Scheme S1) significantly reduces the internalization of the peptide (Figure 3B and S5). Moreover, immunofluorescence staining reveals that the ALP density on cancer cell surface decreases after the incubation with 1 (Figure S6). The results suggest that in cancer cells, the phosphopeptides associate with the ALP in the lipid raft and initiate caveolin-dependent endocytosis[24]. Because the membranes of the phosphopeptide-carrying endocytic vesicles in cancer cells likely originate from the ALP-rich lipid rafts, the peptidic contents may undergo ALP-catalyzed self-assembly, producing nanofibers that cause mechanical stress against the endocytic organelles, leading to membrane leakage[25] followed by the escape of peptide assemblies into cytosol. The cytosolic peptides experience further dephosphorylation in ER where ALP is made[20], resulting in the ER-specific accumulation. This hypothesis is supported by the observation of punctate NBD fluorescence in the cancer cells incubated with 3 and ALP inhibitor (DQB)[26] (Figure 3B), indicating the endosomal retention of 3. This result suggests that ALP-catalyzed dephosphorylation of the peptide contributes to the peptides escaping from endocytic organelles via membrane leakage caused by the formation of nanofibers (Scheme 1). In contrast, 3 mostly locates in the lysosome of HS-5 cells, which presents low ALP level[7] (Figure 3C). EIPA[27], rather than M-β-CD, inhibits the endocytosis of 3 (Figure 3D) by HS-5 cells, indicating that HS-5 cells likely uptake the phosphopeptides via macropinocytosis[28] (Scheme 1). The results above suggest an ALP-dependent endocytosis and endosomal escaping in the cancer cells.
Figure 3.
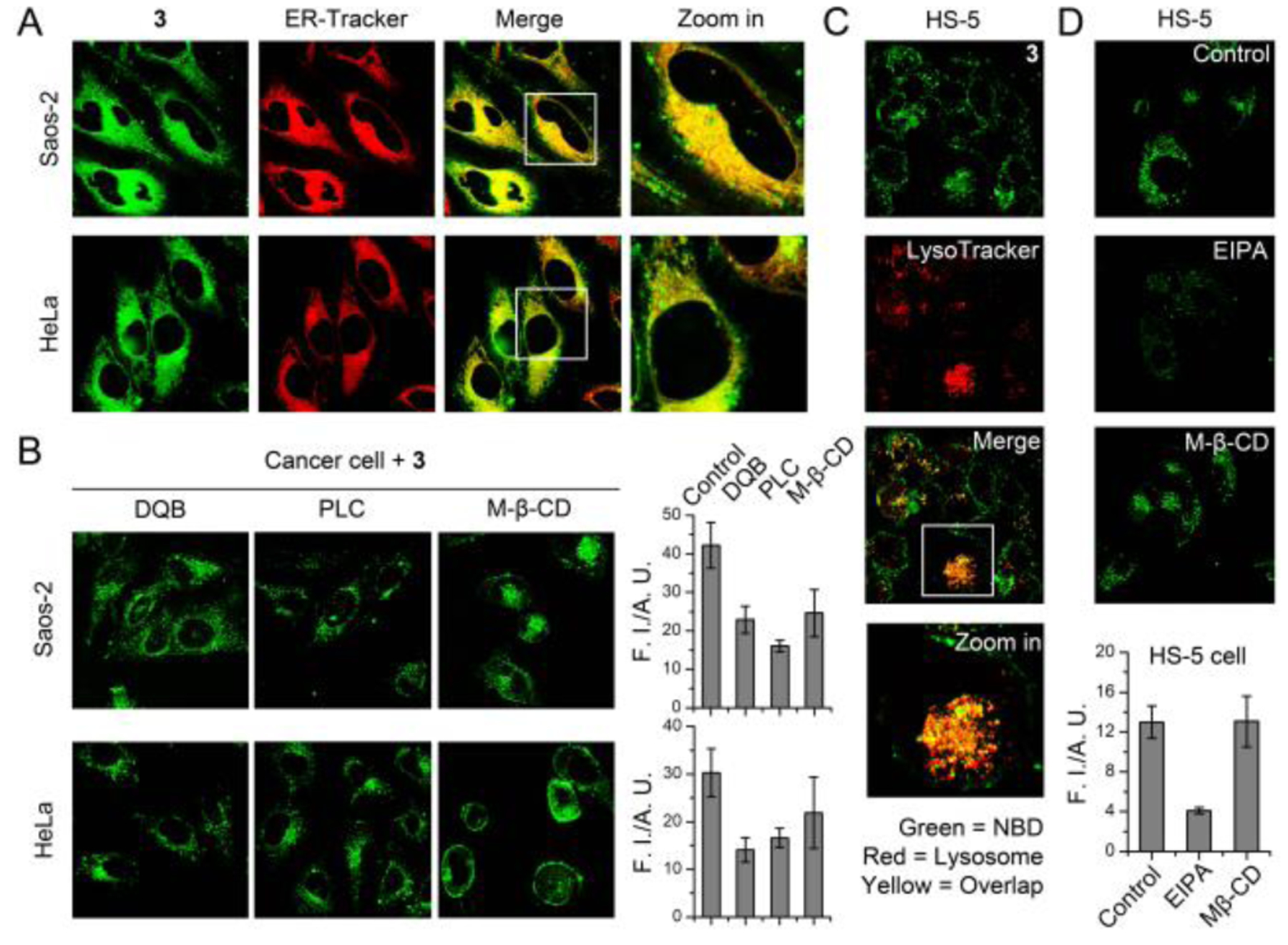
(A) Fluorescent images of HeLa and Saos-2 cells incubated with 3 (50 μM, 4 h). The ER was stained by ER-Tracker. (B) Intracellular 3 decreases after the treatment of DQB (25 μM), phospholipase C (0.2 U/mL, 24 h), and caveolin-dependent endocytosis inhibitors (M-β-CD). (C) Fluorescent images of HS-5 cells incubated with 3 (50 μM, 4 h). The lysosome was stained by LysoTracker. (D) The cellular uptake of 3 in HS-5 cell decreases in the presence of EIPA and M-β-CD.
To verify that the peptide assemblies sequestrate the IAPs in cancer cells to promote apoptosis, we examined the XIAP (member of IAPs) in cells using immunofluorescence staining (Figure 4 and S7). In the cancer cells incubated with 3, XIAP extensively colocalizes with the proapoptotic peptide assemblies (Figure 4A), indicating that the peptide assemblies sequestrate and antagonize the IAPs in cancer cells. Conversely, 3 hardly associates with the XIAP in HS-5 cells after the incubation (Figure 4B), which originates from the lysosomal encapsulation of the peptide in HS-5 cells (Figure 3C). These results agree with the cancer-specific apoptosis promoted by 1.
Figure 4.
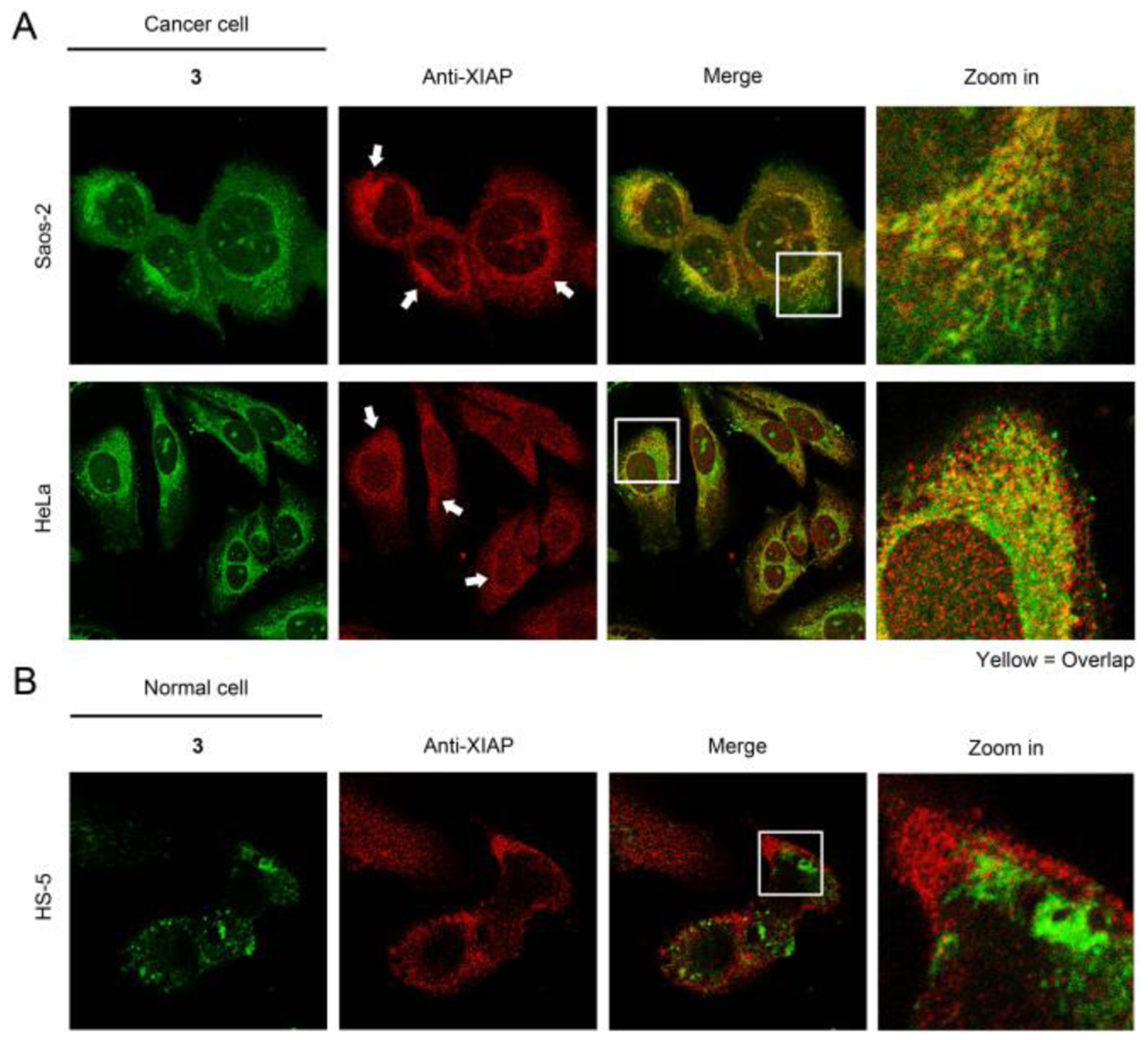
Immunofluorescence staining of XIAP for (A) cancer cells and (B) HS-5 cells pretreated by 3 (50 μM, 3h).
To investigate the mechanism that the phosphopeptide synergistically boosts the activity of BTZ against cancer cells but decreases the cytotoxicity of BTZ to HS-5 cells (antagonism, Table S1), we also made the NBD-labeled BTZ (5, Scheme S1) to study the localization of BTZ with 1. HeLa cells incubated with 5 and 1 exhibit higher cytosolic fluorescence than the cells treated by 5 only (Figure 5A), suggesting that the micelles formed by 1 likely encapsulate BTZ and facilitate the uptake of BTZ into cancer cells via endocytosis (Scheme 1), which contributes to cancer inhibition. Removing the phosphate group from 1 (2, Scheme S1) or adding ALP inhibitor (DQB), diminishes the efficiency of BTZ entering (facilitated by 1) the cancer cells (Figure 5A and 5B) and the inhibition of the cells (Figure 5C and S8). To validate that the IAPs sequestration is critical for the cancer cell death, we substituted the AVPI moiety in 1 by its D-enantiomer (6, Scheme S1), which would bind to IAPs poorly. While both 6 and 1 promote the cellular uptake of BTZ in a comparable extent (Figure 5A, 5B and S9), 6 is less effective than 1 for enhancing BTZ to inhibit the cancer cells (Figure 5D and S10). These results confirm that the IAPs sequestration by the proapoptotic peptide assemblies is essential for boosting the ability of BTZ to cause cancer cell death. However, HS-5 cells incubated with the mixture of 5 and 1 exhibit more lysosomal retention of 5 compared to the cells treated by 5 only (Figure 5E). This result indicates that the phosphopeptide primarily directs BTZ into lysosome where the peptide concentrates in HS-5 cell (Figure 3C), blocking the drug to proteasome, thus reducing the cytotoxicity of BTZ against HS-5 cells.
Figure 5.
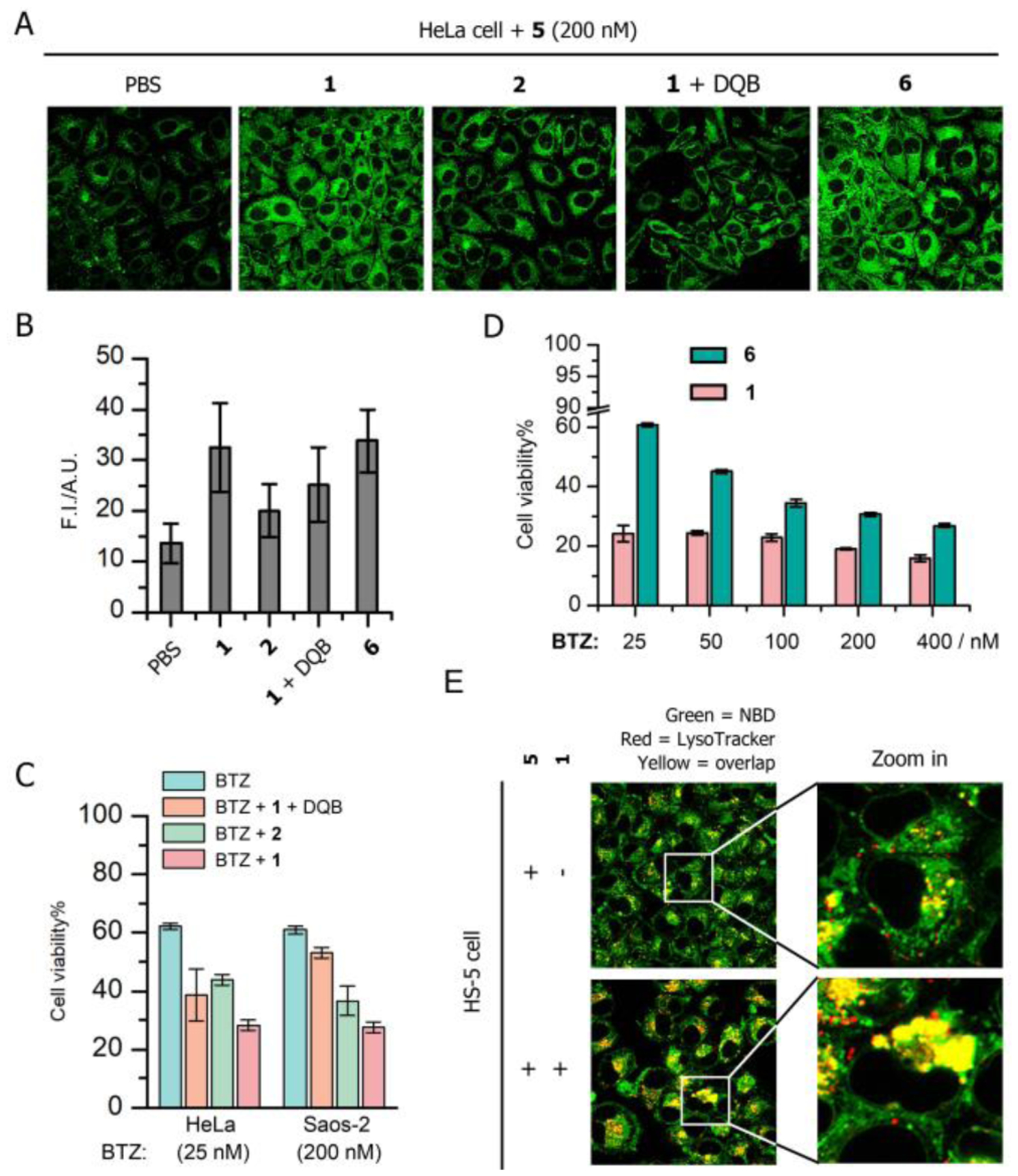
(A) Fluorescent images of HeLa cells incubated with 5 (200 nM, 4 h) in the presence of 50 μM 1, 2, the 1 mixed with DQB (25 μM) and 6 (50 μM). (B) Semi-quantification of the fluorescence of 5 in HeLa cells incubated with the condition of interest. (C) Cell viability of HeLa and Saos-2 cells incubated with BTZ (25 nM, 2nd day) in the presence of 50 μM 1, 2 and 1 mixed with DQB. (D) Cell viability of HeLa cells incubated with BTZ of different concentrations in the presence of 1 or 6. (E) Fluorescent images of HS-5 cells incubated with 5 (200 nM) and the mixture of 1 (50 μM) and 5 (200 nM).
In summary, we demonstrated that the EISA of IAPs-binding phosphopeptides sensitizes cancer cells to BTZ (proteasome inhibitor), while reduces the cytotoxicity of BTZ against normal cell (e.g. HS-5). The use of EISA of peptide assemblies to generate molecular condensates[29] as a context-dependent process[30] for controlling the location of BTZ and for antagonizing protein-protein interactions (PPI) of IAPs represents a new way for developing multispecific therapeutics.[31] Among numerous approaches for combating cancer, the use of supramolecular assemblies to combine surface enzymatic self-assembly[32] and endocytosis are particularly promising for cancer specificity.[33] The onset of cancer often associates with the ectopic elevation of ALP in serum[34], suggesting that the ALP level in tumors mostly prevails that in normal tissue. Thus, the reported proapoptotic phosphopeptide, which gains ALP-dependent selectivity, likely would act on tumor over normal tissue.[35] The concept demonstrated in this work may lead to the combination of self-assembly with other therapeutic agents[36] for increasing efficacy and reducing side effects in treating other diseases or enhancing the sensitivity and selective for molecular imaging.[37]
Supplementary Material
Acknowledgements
This work was partially supported by NIH (R01CA142746) and NSF (MRSEC-2011846).
References
- [1].Siegel RL, Miller KD, Jemal A, CA Cancer J Clin 2020, 70, 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Hanahan D, Weinberg RA, Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- [3].a) Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM, Nat. Rev. Cancer 2018, 18, 452–464; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Cell 2017, 168, 707–723; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW, Cortez MA, Pharmacol. Ther 2019, 195, 162–171. [DOI] [PubMed] [Google Scholar]
- [4].Sanmamed MF, Chen L, Cell 2018, 175, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Millán JL, Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology, John Wiley & Sons, 2006; [Google Scholar]; b) Bernhard A, Rosenbloom L, Science 1953, 118, 114–115; [DOI] [PubMed] [Google Scholar]; c) Fishman WH, Inglis NR, Green S, Anstiss CL, Gosh NK, Reif AE, Rustigia R, Krant MJ, Stolbach LL, Nature 1968, 219, 697–699. [DOI] [PubMed] [Google Scholar]
- [6].Lopez J, Indian J Clin. Biochem 2015, 30, 243. [Google Scholar]
- [7].Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B, Chem 2016, 1, 246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC, Cancer Res. 2001, 61, 3071–3076. [PubMed] [Google Scholar]
- [9].Nass J, Efferth T, Cancer Drug Resist. 2018, 1, 87–117. [Google Scholar]
- [10].Roecklein BA, Torok-Storb B, Blood 1995, 85, 997–1005. [PubMed] [Google Scholar]
- [11].a) Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q, Xu B, Angew. Chem. Int. Ed 2014, 53, 8104–8107; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang H, Feng Z, Xu B, Angew. Chem. Int. Ed 2019, 58, 5567–5571; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhou J, Du X, Xu B, Angew. Chem. Int. Ed 2016, 55, 5770–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Saslowsky DE, Lawrence J, Ren X, Brown DA, Henderson RM, Edwardson JM, J. Bio. Chem 2002, 277, 26966–26970; [DOI] [PubMed] [Google Scholar]; b) van der Luit AH, Budde M, Ruurs P, Verheij M, van Blitterswijk WJ, J. Bio. Chem 2002, 277, 39541–39547. [DOI] [PubMed] [Google Scholar]
- [13].Parton RG, Joggerst B, Simons K, J. Cell Bio 1994, 127, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wu G, Chai J, Suber TL, Wu J-W, Du C, Wang X, Shi Y, Nature 2000, 408, 1008–1012. [DOI] [PubMed] [Google Scholar]
- [15].Zhou J, Du X, Li J, Yamagata N, Xu B, J. Am. Chem. Soc 2015, 137, 10040–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ryu J, Park CB, Biotech. Bioeng 2010, 105, 221–230. [DOI] [PubMed] [Google Scholar]
- [17].Schneider P, Thome M, Burns K, Bodmer J-L, Hofmann K, Kataoka T, Holler N, Tschopp J, Immunity 1997, 7, 831–836. [DOI] [PubMed] [Google Scholar]
- [18].Kuang AA, Diehl GE, Zhang J, Winoto A, J. Bio. Chem 2000, 275, 25065–25068. [DOI] [PubMed] [Google Scholar]
- [19].Bodmer J-L, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J, Nat. Cell Biol 2000, 2, 241–243. [DOI] [PubMed] [Google Scholar]
- [20].Tokumitsu S, Fishman WH, J. Histochem. Cytochem 1983, 31, 647–655. [DOI] [PubMed] [Google Scholar]
- [21].Müller A, Klöppel C, Smith-Valentine M, Van Houten J, Simon M, Biochim. Biophys. Acta, Biomembr 2012, 1818, 117–124. [DOI] [PubMed] [Google Scholar]
- [22].Hooper NM, Clin. Chim. Acta 1997, 266, 3–12. [DOI] [PubMed] [Google Scholar]
- [23].Mahammad S, Parmryd I, in Methods in membrane lipids, Springer, 2015, pp. 91–102. [DOI] [PubMed] [Google Scholar]
- [24].Parton RG, Joggerst B, Simons K, J. Cell. Bio 1994, 127, 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Liang W, Lam JK, Mol. Regul. Endocytosis 2012, 429–456; [Google Scholar]; b) Mu Q, Broughton DL, Yan B, Nano Lett. 2009, 9, 4370–4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, J. Med. Chem 2009, 52, 6919–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fretz M, Jin J, Conibere R, Penning NA, Al-Taei S, Storm G, Futaki S, Takeuchi T, Nakase I, Jones AT, J. Controlled Release 2006, 116, 247–254. [DOI] [PubMed] [Google Scholar]
- [28].King JS, Kay RR, Philos. Trans. R. Soc. London, Ser. B 2019, 374, 20180158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Feng Z, Wang H, Xu B, J. Am. Chem. Soc 2018, 140, 16433–16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yao Q, Wang C, Fu M, Dai L, Li J, Gao Y, ACS Nano 2020. [DOI] [PubMed] [Google Scholar]
- [31].Deshaies RJ, Nature 2020, 580, 329–338. [DOI] [PubMed] [Google Scholar]
- [32].a) Vigier-Carrière C, Boulmedais F, Schaaf P, Jierry L, Angew. Chem. Int. Ed 2018, 57, 1448–1456; [DOI] [PubMed] [Google Scholar]; b) Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q, Xu B, Angew. Chem. Int. Ed 2014, 53, 8104–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tanaka A, Fukuoka Y, Morimoto Y, Honjo T, Koda D, Goto M, Maruyama T, J. Am. Chem. Soc 2015, 137, 770–775. [DOI] [PubMed] [Google Scholar]
- [34].a) Saif MW, Alexander D, Wicox CM, J. Appl. Res 2005, 5, 88; [PMC free article] [PubMed] [Google Scholar]; b) Stolbach LL, Krant MJ, Fishman WH, N. Engl. J. Med 1969, 281, 757–762. [DOI] [PubMed] [Google Scholar]
- [35].Feng Z, Han X, Wang H, Tang T, Xu B, Chem 2019, 5, 2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Wang H, Feng Z, Xu B, J. Am. Chem. Soc 2019, 141, 7271–7274; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H, Adv. Drug Delivery Rev 2017, 110, 112–126; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Alexopoulou O, Abrams P, Verhelst J, Poppe K, Velkeniers B, Abs R, Maiter D, Eur. J. Endocrinol 2004, 151, 317–324; [DOI] [PubMed] [Google Scholar]; d) Zheng M, Wang Y, Shi H, Hu Y, Feng L, Luo Z, Zhou M, He J, Zhou Z, Zhang Y, ACS Nano 2016, 10, 10075–10085; [DOI] [PubMed] [Google Scholar]; e) Yuan Y, Wang L, Du W, Ding Z, Zhang J, Han T, An L, Zhang H, Liang G, Angew. Chem. Int. Ed 2015, 54, 9700–9704. [DOI] [PubMed] [Google Scholar]
- [37].a) Hai Z, Li J, Wu J, Xu J, Liang G, J. Am. Chem. Soc 2017, 139, 1041–1044; [DOI] [PubMed] [Google Scholar]; b) Wang H, Feng Z, Del Signore SJ, Rodal AA, Xu B, J. Am. Chem. Soc 2018, 140, 3505–3509; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) ; d) Palner M, Pu K, Shao S, Rao J, Angew. Chem. Int. Ed 2015, 54, 11477–11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.