

Abstract
The fact that Fat-1 transgenic mice producing n-3 polyunsaturated fatty acids via overexpressed 3-desaturase significantly mitigated Helicobacter pylori (H. pylori)-associated gastric tumorigenesis through rejuvenation of chronic atrophic gastritis (CAG) led us to study whether dietary intake of walnut plentiful of n-3 PUFAs can be nutritional intervention to prevent H. pylori-associated gastric cancer. In our model that H. pylori-initiated, high salt diet-promoted gastric carcinogenesis, pellet diet containing 100 mg/kg and 200 mg/kg walnut was administered up to 36 weeks. As results, control mice (24 weeks) developed significant chronic CAG, in which dietary walnuts significantly ameliorated chronic atrophic gastritis. Expressions of COX-2/PGE2/NF-κB/c-Jun, elevated in 24 weeks control group, were all significantly decreased with walnut (p<0.01). Tumor suppressive enzyme, 15-PGDH, was significantly preserved with walnut. Control mice (36 weeks) all developed significant tumors accompanied with severe CAG. However, significantly decreased tumorigenesis was noted in group treated with walnuts, in which expressions of COX-2/PGE2/NF-κB/IL-6/STAT3, all elevated in 36 weeks control group, were significantly decreased with walnut. Defensive proteins including HO-1, Nrf2, and SOCS-1 were significantly increased in walnut group. Proliferative index as marked with Ki-67 and PCNA was significantly regulated with walnut relevant to 15-PGDH preservation. Conclusively, walnut can be an anticipating nutritional intervention against H. pylori.
Keywords: H. pylori, walnut, rejuvenation, cancer prevention, 15-PGDH
Introduction
Though Helicobacter pylori (H. pylori) infection had been defined as class I, definite, carcinogen for gastric cancer and has been associated with diverse gastric diseases as well as several extra-gastric diseases, the bacterium frequently persists in the human host without inducing disease and plays a beneficial role in health in some as commensals.(1) Furthermore, as efforts to decrease gastric cancer mortality, general eradication strategy had been performed in Japan from 2013, but we should wait for long-term outcome whether simple eradication strategy was right for cancer prevention or not. Therefore, as another intervention for similar purpose of gastric cancer prevention, non-microbial or dietary intervention had been tried. However, among these, in gastric carcinogenesis, dietary factors themselves are dual-edged swords, that is, implicated in carcinogenesis, but possibly preventive on others. For example, salty food and red/processed meat intakes were proven to be associated with an increased risk of gastric non-cardia cancer, whereas vegetable and fruits are protective factors especially in H. pylori antibody-positive subjects.(2) Although the cohort-based evidence is still lacking, a few dietary approaches with antioxidants or nutraceuticals are available to prevent H. pylori-associated gastric diseases.
Among dietary intervention as non-microbial or dietary approach targeted for H. pylori infection, quite several agents had been studied, including blueberry plants,(3) phytochemicals from fruits,(4) genistein from soy rich diet,(5) dietary antioxidant β-carotene or astaxanthin,(6,7) and n-3 polyunsaturated fatty acids (n-3 PUFAs)-rich walnut.(8) Stimulated with significant cancer preventive outcome of walnut that n-PUFAs synthesizing Fat-1 transgenic mice synthesizing n-3 PUFAs such as DHA or EPA significantly resisted from chronic H. pylori-associated gastric carcinogenesis and walnut polyphenol extracts significantly inhibited interleukin-6 (IL-6)/Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signaling via peroxisome proliferator-activated receptor γ (PPAR-γ)/suppressors of cytokine secretion (SOCS) 1 stimulation in gastric cells,(8,9) we put hypothesis that dietary intake of walnut (Juglans regia L.) can reduce H. pylori-associated gastric carcinogenesis. In this study, we have explored the efficacy of dietary intake of walnut against H. pylori-associated chronic atrophic gastritis (CAG) or gastric cancer model and extended documentation the molecular pathologic mechanisms.
Material and Methods
H. pylori culture
H. pylori strain ATCC43504 (American Type Culture Collection, cagA+ and vacA s1-m1 type’s strain) was used for in vitro cell model (Supplemental Fig. 1*) and Sydney strain (SS1, a cagA+, vacA s2-m2 strain adapted for mice infection) for in vivo model. H. pylori (Fig. 1A and 4A) were cultured at 37°C in BBL Trypticase soy (TS) agar plate with 5% sheep blood (TSAII; BD Biosciences, Franklin Lakes, NJ) under microaerophilic condition (BD GasPaK EZ Gas Generating Systems, BD Biosciences) for 3 days. The bacteria were harvested in clean TS broth, centrifuged at 3,000 × g for 5 min, and resuspended in the broth at a final concentration of 109 colony-forming units (CFUs)/ml. In all experiments, cultures grown for 72 h on TS agar plates were used.
Fig. 1.
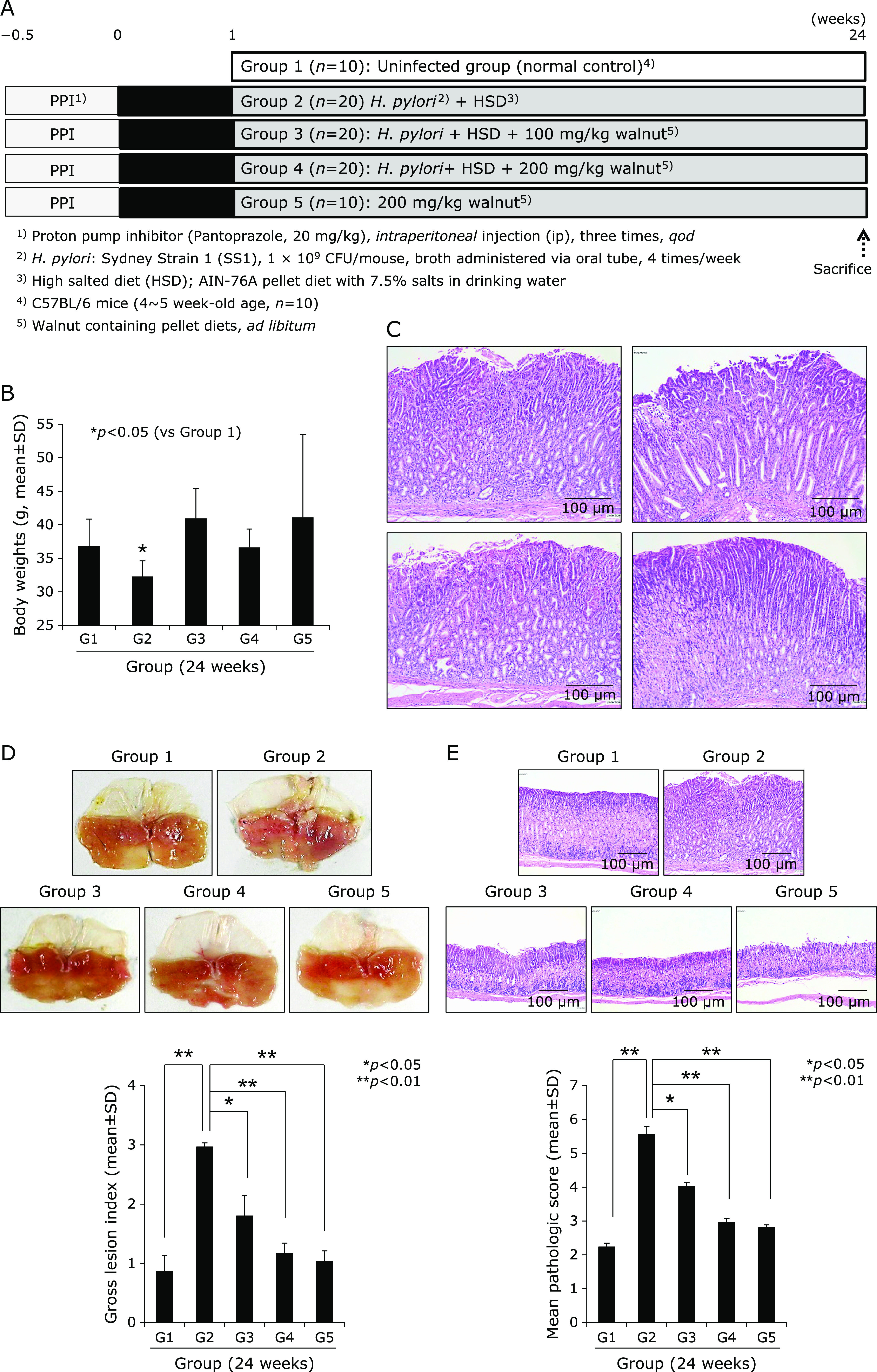
Influence of dietary intake of walnuts on H. pylori-initiated, high salt diet-promoted CAG, (24 weeks). (A) Scheme for group, Group 1; normal control, Group 2; H. pylori-associated CAG disease control, Group 3; disease control treated with 100 mg/kg walnut diet, and Group 4; disease control treated with 200 mg/kg walnut diet. (B) Body weight changes according to group. (C) Representational photo showing gastric diseases after 24 weeks of H. pylori infection. (D) Representative photo of resected stomach according to group and mean gross lesion scores according to group. (E) Representational pathology and mean pathological scores according to group.
H. pylori-infected mice model
Experimental protocol
Five-week-old male C57BL/6 mice (WT mice) were purchased from Orient (Seoul, Korea) and they were housed in a cage maintained at 23°C in a 12 h/12 h light/dark cycle under specific pathogen-free conditions (n = 140). We divided mice into four groups; Group 1 (n = 20); WT mice as vehicle control group, Group 2 (n = 40); WT mice as H. pylori-infected disease group, Group 3 (n = 40); WT mice as H. pylori-infected disease group administered with 100 mg/kg walnut containing pellet diet, and Group 4 (n = 40); WT mice as H. pylori-infected disease group administered with 200 mg/kg walnut containing pellet diet. However, we determined two times of sacrifice, half at 24 weeks, and the other remaining half at 36 weeks, respectively. All groups were given intraperitoneal (i.p.) injections of pantoprazole, 20 mg/kg; Amore-Pacific Pharma) as proton pump inhibitor, three times per week, to increase successful H. pylori colonization through lowered gastric acidity, then, each mouse was intragastrically inoculated with a suspension of H. pylori containing 108 CFUs/ml or with an equal volume (0.1 ml) of clean TS broth using gastric intubation needles. The H. pylori-infected mice were fed a special pellet diet based on AIN-46A containing 7.5% NaCl (high salt diet, Biogenomics, Seongnam, Korea) for total 36 weeks (Fig. 1A and 4A) to promote H. pylori-induced carcinogenic process in all infected animals.(10) Randomized groups of mice (n = 20) sacrificed at 24 or 36 weeks of post H. pylori infection, respectively based on our previous experience that CAG was generated at 24 weeks and gastric tumorigenesis was generated after 36 weeks. The body weight was checked in all mice every 3 days up to observational periods. The stomachs of mice were opened along the greater curvature and washed with ice cold PBS. The numbers of either erosions/ulcers or protruded nodule/mass were determined under the magnified photographs (Fig. 1B and 4B). Stomachs were isolated and subjected to a histologic examination, ELISA, Western blotting, and RT-PCR. All animal studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of CHA University CHA Cancer Institute after institutional review board of IACUC approval (IRB #17-0901).
Preparation of walnut containing pellet diet and preparation of walnut polyphenol extract (WPE)
Whole, fresh walnut was provided from California walnut commission (CA). Ina fresh condition, pellet diets were generated containing dose of 100 mg/kg or 200 mg/kg according to pellet diet manufacture manual. Walnut polyphenol extract (WPE) from English walnuts (J. regia L., California Walnut Commission, CA) was prepared according to a previously described methanolic extraction method.(11) Briefly, after the walnuts were frozen for 24 h, the shelled kernels were finely ground and immersed in a solution of 75% acetone containing 526 µm/L sodium metabisulfite. The solution was subsequently purged with N2 to prevent oxidation and was incubated at 4°C. After 24 h, the solution was decanted, thereby resulting in a cold extract that was centrifuged at 8,000 × g for 10 min. The resulting supernatant was filtered using Whatman filter paper No. 2. To remove lipids from the sample, the acetone was removed under reduced pressure and methanol (50% aqueous, v/v) was added. After three consecutive hexane extractions, the extracts were lyophilized to a dry powder after removing the methanol to prevent oxidation. All of the prepared samples were stored at 80°C until needed.
Gross lesion index
After sacrificing the mice, the isolated stomachs were open along the greater curvature and washed in ice-cold saline. To investigate the degree of gross mucosal pathology, the mucosal sides of the stomachs were photographed using a digital camera (Fig. 1D and 4B) and part of the mucosa was immediately fixed with 10% formalin solution. The gross damage of the gastric mucosa was assessed by three gastroenterologists, who were blinded to the treatments, using a gross ulcer index.(12)
Index of histopathologic injury
For histopathological analysis, the stomach were fixed in 10% neutralized buffered formalin, processing using the standard method and embedded in paraffin. Sections of 4 µm thickness were then stained with hematoxylin and eosin. The glandular mucosae of corpus and antrum were examined histologically. The pathological changes of H. pylori-infection, such as inflammatory cells infiltration, erosive lesions, ulceration, dysplasia, adenoma formation (precancerous lesion), were graded by three gastroenterologists, who were blinded to the group, using an index of histologic injury defined.(13) In this study, inflammation was defined as grade the infiltration of inflammatory cells, 0: none, 1: under the lamina propria, 2: half of mucosa 3: until the epithelial gland layer (all mucosa). The erosion was defined as proportion of erosive lesion, 0: none, 1: loss of epithelial gland layer (1/3 proportion), 2: two-three portion of mucosa (2/3 proportion) 3: all mucosa (3/3 proportion).
Immunohistochemical staining
Immunohistochemistry was performed on replicate sections of mouse gastric tissues. After deparaffinization were dewaxed and rehydrated with graded alcohol, and boiled three times in 100 mM Tris buffered saline (pH 6) with 5% urea in an 850 W microwave oven for 5 min each. And then cooling in water for 15 min and washed in PBS, and slides were incubated overnight with the primary antibody at 4°C. Antibodies:F4/80 (1:500; eBioscience, San Diego, CA) or CD31 (1:300; Dako, Santa Clara, CA) or Ki-67 (1:300; Santa Cruz Biotechnology, Santa Cruz, CA) in the presence of 1.0% bovine serum albumin respectively. Slides incubated with secondary antibody (1:300) for 1 h at room temperature, and then with 40-6-diamidino-2-phenylindole (DAPI, 100 ng/ml) for 1 min at room temperature. And finally the slides were counterstained with hematoxylin (Sigma-Aldrich).
RT-PCR
Total RNA was isolated using the Trizol (Invitrogen, Carlsbad, CA). Trizol was added to 1.5 ml tube, which were then incubated 10 min at 4°C and gently mixed with 100 µl chloroform (Merck, Rahway, NJ). After incubation for 10 min in ice, samples were centrifuged at 10,000 × g for 30 min. Supernatants were extracted and mixed with 200 µl isopropanol (Merck), and mixtures were incubated at 4°C for 1 h. After centrifuging at 13,000 × g for 30 min, pellets were washed with 70% (v/v) ethanol. After allowing the ethanol to evaporate completely, pellets were dissolved in 40 µl diethylene pyrocarbonate-treated water (Invitrogen Life Technologies). cDNA was prepared using reverse transcriptase originating from Murine-Moloney leukemia virus (Promega, Madison, WI), according to the manufacturer’s instructions. The polymerase chain reaction (PCR) was performed over 25 cycles of: 94°C for 20 s, 58.5 for 30 s, and 72°C for 45 s. Oligonucleotide primers were purchased from Bioneer (Daejeon, Korea). Oligonucleotide primers were as follows; for cyclooxygenase (COX)-2, sense 5'-GAA ATG GCT GCA GAG TTG AA-3' and antisense 5'-TCA TCT AGT CTG GAG TGG GA-3', for IL-1β, sense 5'-CAG GCT CCG AGA TGA ACA ACA AAA-3' and antisense 5'-TGG GGA ACT CTG CAG ACT CAA ACT-3', for IL-8, sense 5'-GGG GCT TTG CCG TGC AAT AA-3' and antisense 5'-GCA CAG GGT TGA GCC AAA A-3', for IL-6, sense 5'-AAG AGA CTT CCA GCC AGT TG-3' and antisense 5'-TGG ATG GTC TTG GTC CTT AG-3', and for GAPDH, sense 5'-GGT GCT GAG TAT GTC GTG GA-3' and antisense 5'-TTC AGC TCT GGG ATG ACC TT-3'.
Western blotting
Resected gastric tissues were harvested and lysed in lysis buffer (Cell Signaling Technology) containing 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma Aldrich, St. Louis, MO). After 30 min of incubation, samples were centrifuged at 12,000 × g for 15 min at 4°C. The supernatants were then collected and protein quantification was carried out with a Bio-Rad protein assay. Equal amounts soluble protein (30 µg) were denaturated by heating at 100°C for 3 min. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% BSA in PBST for 30 min. And then, the membranes probed initially with specific primary antibody, washed, incubated with peroxidase-conjugated secondary antibodies, and rewashed. The protein bands were detected by chemiluminescence (Supersignal, Pierce) exposure on chemiluminescence system (GE Healthcare, Buckinghamshire, UK). The general procedure for Western blot analysis of cultured mouse gastric mucosal cells was similar to the procedures described above. Antibodies used in the current study were COX-2, purchased from Thermo, β-actinpurchased from Santa Cruz, 15-hydroxyprostaglandin dehydrogenase (15-PGDH), purchased from Cayman. RPMI-1640 medium, fetal bovine serum, penicillin (FBS), streptomycin were products of GIBCO BRL (Grand Island, NY) and materials for culturing H. pylori were sheep blood agar, Gaspak and anaerobic jars (BD Biosciences, Sparks, MD). Primary antibody against actin was purchased from Sigma-Aldrich Co., antibodies for lamin B from Santa Cruz Biotechnology, other antibodies for p-STAT3Tyr705, total STAT3 from Cell Signaling Technology (Beverly, MA), horseradish peroxidase (HRP)-conjugated secondary antibody from Pierce Biotechnology (Rockford, IL). dl-dithiothreitol (DTT), TRIzol, 4',6-diamidino-2-phenylindole (DAPI) from Invitrogen, and polyvinylidene difluoride (PVDF) membranes were supplied from Gelman laboratory (Ann Arbor, MI). The ECL chemiluminescent detection kit was purchased from LPS solution (Daejon, South Korea) and protein assay dye (Bradford) reagent was supplied by Bio-Rad Laboratories (Hercules, CA), bicinchonic acid (BCA) protein assay reagent was obtained from Pierce Biotechnology.
Preparation of cytosolic and nuclear extracts
After H. pylori infection, resected stomach tissues were washed twice with ice-cold PBS and scraped in 1 ml of PBS, followed by centrifugation at 1,700 × g for 5 min at 4°C. Pellets were resuspended in hypotonic buffer A [10 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT and 0.2 mM phenylmethylsulfonylfluoride (PMSF)] for 15 min on ice. Ten % Nonidet P-40 was then added to final concentration of 0.1% for less 3 than 5 min. The mixture was then centrifuged at 6,000 × g for 5 min at 4°C. Supernatant was collected as the cytosolic extract and stored at −80°C. The pellets were washed twice with hypotonic buffer A and resuspended again in hypertonic buffer C [20 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (pH 7.9), 20% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid, 0.5 mM DTT, and 0.2 mM PMSF] for 1 h on ice and centrifuged at 18,000 × g for 15 min at 4°C. The supernatant containing nuclear proteins was collected and stored at −80°C. The protein concentrations of both fractions were determined by using the BCA protein assay reagent.
Statistical analysis
Results are expressed as the mean SD. Statistical analyses were conducted with GraphPad Prism (GraphPad Software, La Jolla, CA) and SPSS software (ver. 12.0; SPSS Inc., Chicago, IL). Statistical significance between groups was determined by Mann-Whitney U test. Statistical significance was accepted at p<0.05.
Results
Twenty-four weeks dietary administration of walnut significantly mitigated H. pylori-associated chronic atrophic gastritis
We used our model that H. pylori-initiated, high salt condition-promoted chronic atrophic gastritis (CAG) model to address the efficacy of dietary intake of walnut on mitigation of CAG, for which we have injected pantoprazole as proton pump inhibitor before infecting H. pylori (to facilitate colonization of H. pylori), 20 mg/kg intraperitoneally three time every other day, 1 × 108 CFU/mouse adapted H. pylori SS1 strain was administered via zoned for intragastric administration, four times/week. This is based on our previous protocol that hypoacidity before H. pylori infection significantly increased colonization (Fig. 1A). Then, in all group, we provided 7.5% salt containing drinking water to facilitate the pathogenic condition of colonized H. pylori. The mice were sacrificed 24 weeks after H. pylori infection, by which we could confirm 100% colonization of SS1. As result of these animal model, we found the mean body weight was significantly decreased in Group 2, H. pylori-infected group (p<0.05, Fig. 1B). However, Group 3 and Group 4, group administered with diet intake of 100 mg/kg or 200 mg/kg walnut in pellet significantly maintained body weight in spite of same H. pylori infection. On gross morphology of resected stomach according to group, as shown in Fig. 1D, stomach of Group 2 was swollen, edematous, and erythematous compared to Group 1 normal control, whereas similar looking in Group 3 (100 mg/kg walnut diet under H. pylori), Group 4 (200 mg/kg walnut diet under H. pylori), and Group 5 (only walnut diet in the absence of H. pylori infection). Using score system (edema, 0–2; erythema, 0–2; erosions, 0–2; ulcer, 0–2; atrophy, 0–2), mean gross lesion scores were significantly increased in Group 2 compared to Group 1, whereas scores were significantly decreased in Group 3 and Group 4 (p<0.01, Fig. 1D). As seen in Fig. 1C, H. pylori-control group, Group 2, showed significant pathological changes of chronic atrophic gastritis as well as erosions. These pathologic findings were evaluated with microscopic pathologic score (inflammation, 0–3; ulcer, 0–3; atrophy, 0–3; atrophy, 03), which was quite similar changes with gross lesion score. As seen in Fig. 1C, our control model showed significant changes including hypertrophic gastritis on junction of fore stomach, atrophic changes with loss of parietal cells on corpus and antral area with significant infiltrations of inflammatory mononuclear cells, and focal changes of erosions. As seen in Fig. 1E, the mean pathological scores of control group were significantly increased in Group 2, but significantly mitigated in group treated with diet pellet containing walnut (p<0.05). Using in vitro cell model, as seen in Supplemental Fig. 1*, WPE significantly attenuated genes promoted with H. pylori infection. Supported with these gross and pathological changes according to group, COX-2 expressions were checked according to group. As seen in Fig. 2A, Western blot for COX-1 and COX-2 noted significantly increased COX-2/significantly decreased COX-1 in Group 2, whereas these changes were reverse, signifying COX-2 relevant to H. pylori was significantly attenuated with walnut diet administration. When we measured serum levels of PGE2 according group, the mean levels of PGE2 were significantly increased in Group 2 (p<0.05), but the mean levels of PGE2 were significantly decreased in Group 3 and Group 4 (p<0.05, Fig. 2B). Since increased COX-2 produced inflammatory PGE2, the 15-PGDH can regulate PGE2. As seen in Fig. 2C, 24 weeks H. pylori infection significantly decreased 15-PGDH (p<0.001), but group administered with walnut significantly maintained 15-PGDH in spite of chronic H. pylori infection. These results of 15-PGDH were further validated with immunostaining of 15-PGDH, as seen in Fig. 2D, the expressions of 15-PGDH were significantly decreased in Group 2, but significantly preserved in Group 3 or Group 4. These findings stimulated us to search for associated mediators and signaling genes implicated in H. pylori-associated chronic atrophic gastritis including nuclear factor κB (NF-κB), c-Fos/c-Jun, c-Myc, SOCS1, STAT, and p-STAT molecules. Cytoplasmic levels of phosphorylated NF-κB p65, control group showed significantly increased levels, but repressed levels in group administered with walnut (Fig. 3A). Using nuclear homogenates, significantly increased nuclear translocation and binding of NF-κB was noted in Group 2, but not in Group 3 and Group 4. Acute phase response is also one of response after H. pylori infection. As seen in Fig. 3B, c-Fos/c-Jun was significantly activated in H. pylori infected control group, while walnut administration significantly attenuated these c-Jun/c-Fos responses. SOCS-1 was identified as one of defense response against H. pylori infection, in contrast c-Myc is reverse response. As seen in Fig. 3C, SOCS-1 was significantly decreased in control group, while c-Myc was significantly increased in H. pylori control group (p<0.001). However, walnut administration for 24 weeks significantly increased SOCS-1 defensive response (p<0.001). STAT3 gene is very critically involved in either precancerous condition of H. pylori-associated atrophic gastritis or mutagenesis. As seen in Fig. 3D, H. pylori infection led to significant increased expression in gastric cells or significantly increased nuclear translocation, spurting mutagenesis (p<0.01). However, 24 weeks administration of diet pellet containing walnut significantly ameliorated H. pylori-associated STAT3 activation in the stomach.
Fig. 2.
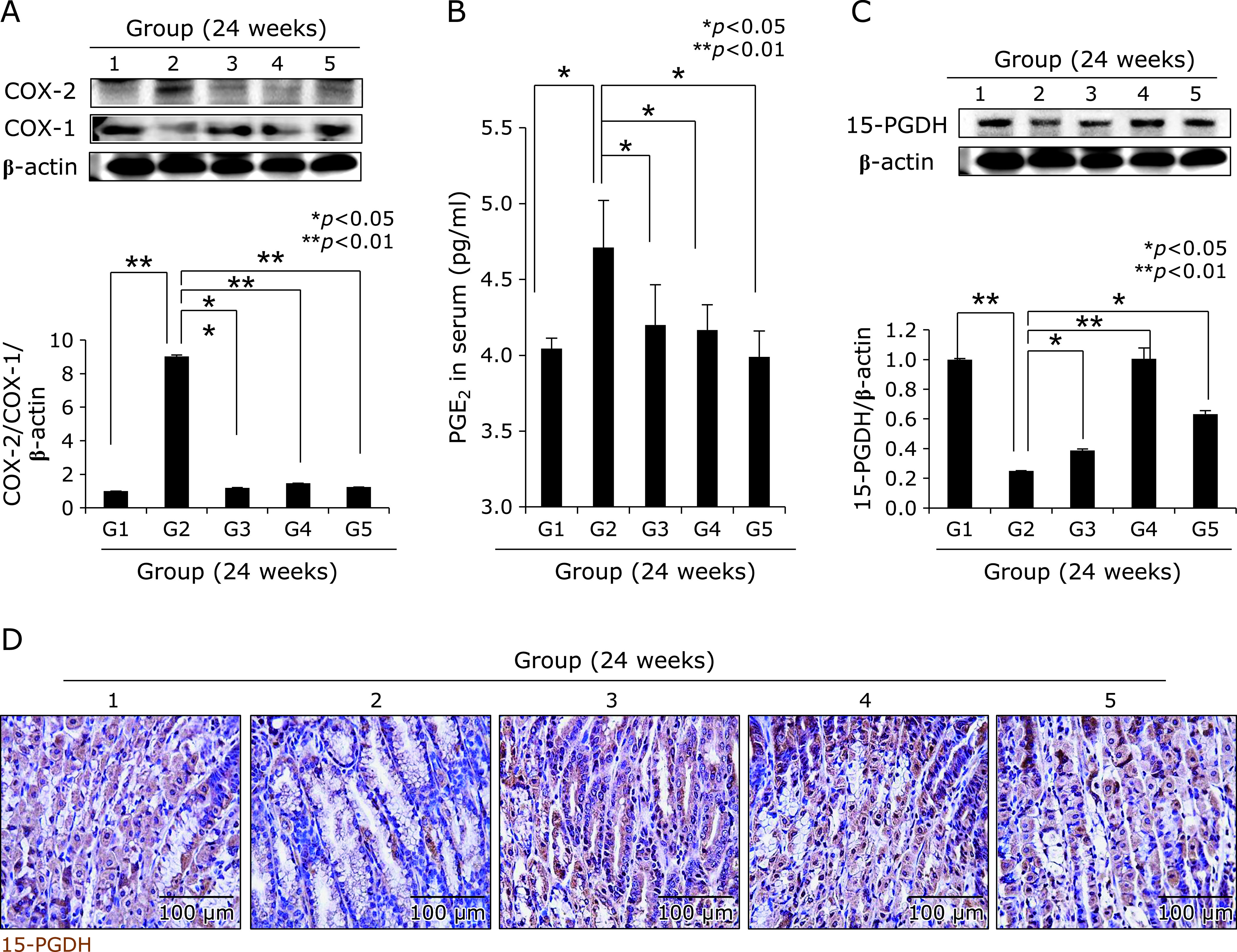
Changes of COX-1, COX-2, PGE2, and 15-PGDH according to group. (A) Western blot for COX-1 and COX-2. (B) Mean PGE2 in sera of each group. (C) Western blot for 15-PGDH. (D) Immunohistochemical staining of 15-PGDH, ×100 magnification。
Fig. 3.
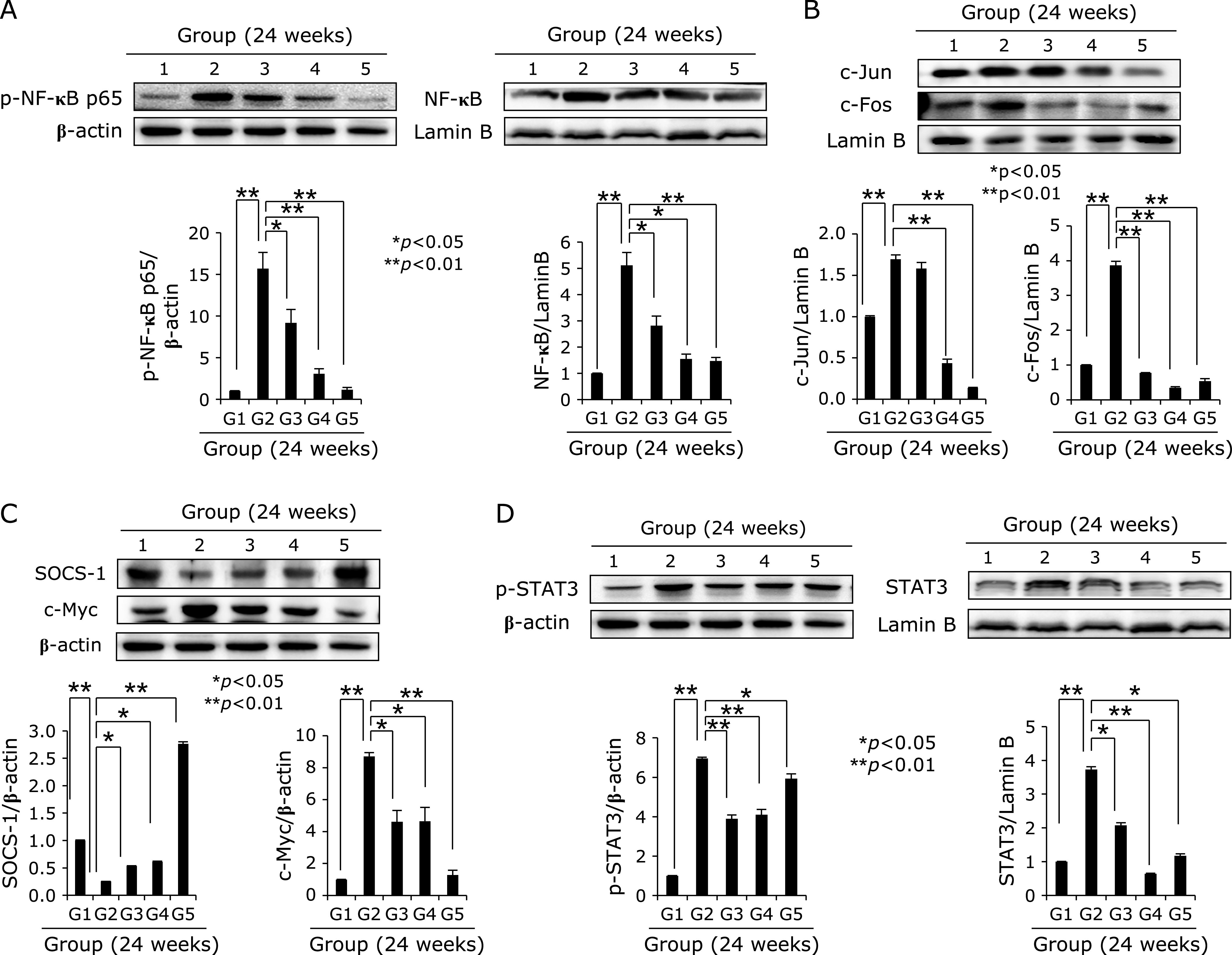
Comparison of NF-κB, acute phase protein like c-Jun and c-Fos, SOCS-1, c-Myc, and STAT3. (A) Western blot for NF-κB p65 in cytoplasmic homogenates and NF-κB in nuclear fraction according to group. (B) Western blot for c-Jun and c-Fos in nuclear fraction of gastric homogenates according to group. (C) Western blot for SOCS-1 and c-Myc. (D) Western blot for p-STAT3 in cytoplasmic fraction and STAT3 in nuclear fraction according to group.
Thirty-six weeks dietary administration of walnut significantly prevented H. pylori-associated gastric tumorigenesis gastritis
We extended the above H. pylori infection up to 36 weeks because the current model, H. pylori-initiated, salt-promoted model, developed gastric tumorigenesis around 36–45 weeks, in which condition we want to explore the efficacy of diet walnut administration on the preventive action of H. pylori-associated gastric tumorigenesis. Our model that H. pylori-initiated, high salt condition-promoted significant gastric carcinogenesis around 36 weeks in detail, the hypoacidity before H. pylori infection significantly increased colonization (Fig. 4A). Then, in all group, we provided 7.5% salt containing drinking water to facilitate the carcinogenesis of colonized H. pylori. The mice were sacrificed 36 weeks after H. pylori infection, by which we could confirm 100% colonization of mouse adapted strain of SS1. As result of these animal model, we found the mean body weight was significantly decreased in Group 2, H. pylori-infected group (p<0.05, data not shown). As seen in Fig. 4B, our model developed significant mucosal changes such as multiple tumorigenesis, pale and thin gastric corpus mucosa, and several shallow ulcerations (Fig. 4B). Scoring according to criteria (gastric tumor, 0–2; gastric ulcer, 0–2; pale and thin mucosa, 0–2; gastric mucosa edema, 0–2), the mean pathological scores were significantly increased in Group 2 (p<0.01), but pathological scores of Group 3 and Group 4 were significantly decreased (p<0.05, Fig. 4B). Control group showed significant development of gastric adenoma and focal carcinoma in all numbers of Group 2, while these changes of tumorigenesis as well as mucosal erosions/ulcers were significantly decreased in Group 3 and Group 4 (p<0.001, Fig. 4C). All of these findings significantly suggested dietary walnut administration significantly ameliorated CAG or prevented gastric tumorigenesis. In order to explore molecular mechanisms, first, we have measured COX related mechanisms, COX-1, COX-2, and 15-PGDH as noted in 24 weeks model. As seen in Fig. 5A, no significant changes of COX-1 according to group, but the levels of COX-2 were significant. Group 4 showed significant decreases of COX-2. When checked the expression of tumor suppressive 15-PGDH, as seen in Fig. 5B and C, the expressions of 15-PGDH were significantly decreased in H. pylori control group, but significantly preserved with walnut administration. Looking at Ki-67, the expressions were remarkable, significantly increased in Group 2, but reversed state in Group 3 and Group 4. In accordant with COX changes, IL-6 has been known to be implicated in H. pylori-associated gastric carcinogenesis, IL-6 mRNA and serum levels of IL-6 were measured respectively. As seen in Fig. 6A, IL-6 mRNA was significantly increased in either 24 weeks or 36 weeks, but not in group administered with walnut pellet diet (p<0.01, Fig. 6A and B). Also, when measured NF-κB and acute phase response genes, c-Fos and c-Jun, walnut administration significantly decreased chronic H. pylori infection-associated NF-κB or acute phase response genes (p<0.01, Fig. 6C and D). We repeated the investigation of genes implicated in 24 weeks H. pylori infection relevant to gastric carcinogenesis including c-Myc, p-STAT3, STAT3 according to group. As seen in Fig. 7A, B, and C, c-Myc and STAT3 activation was significantly increased in Group 2, but not in group administered with walnut, signifying that long-term intake of walnut significantly mitigated chronic H. pylori-associated gastric carcinogenesis pathway. Since results from 24 weeks and general facts that phytonutrient can provide significant phase 2 enzyme response like hemeoxygenase-1 (HO-1) and nuclear factor erythroid 2-related factor 2 (Nrf2), we have measured the levels of HO-1 and Nrf2 according to group. As seen in Fig. 7C and D, walnut administration significantly increased HO-1 mediated through increased Nrf-2 nuclear translocation (Fig. 7D) and significant inactivation of Kelch-like ECH-associated protein 1 (Keap1).
Fig. 4.
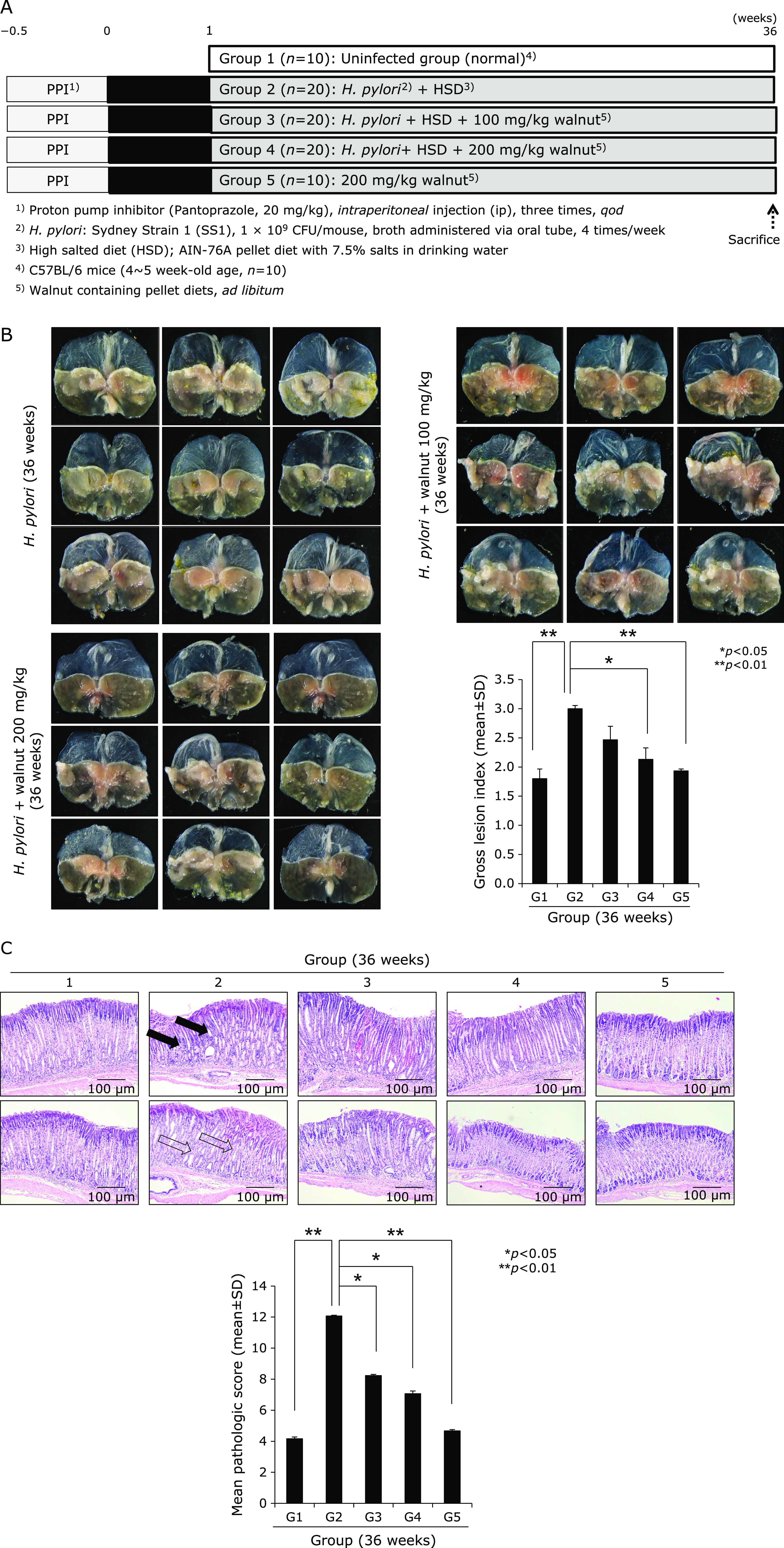
Influence of dietary intake of walnuts on H. pylori-initiated, high salt diet-promoted gastric tumorigenesis (36 weeks). (A) Scheme for group, Group 1; normal control, Group 2; H. pylori-associated CAG disease control, Group 3; disease control treated with 100 mg/kg walnut diet, and Group 4; disease control treated with 200 mg/kg walnut diet, all sacrificed at 36 weeks after H. pylori infection. (B) Representative photo of resected stomach according to group and mean gross lesion scores according to group. (C) Representational pathology and mean pathological scores according to group.
Fig. 5.
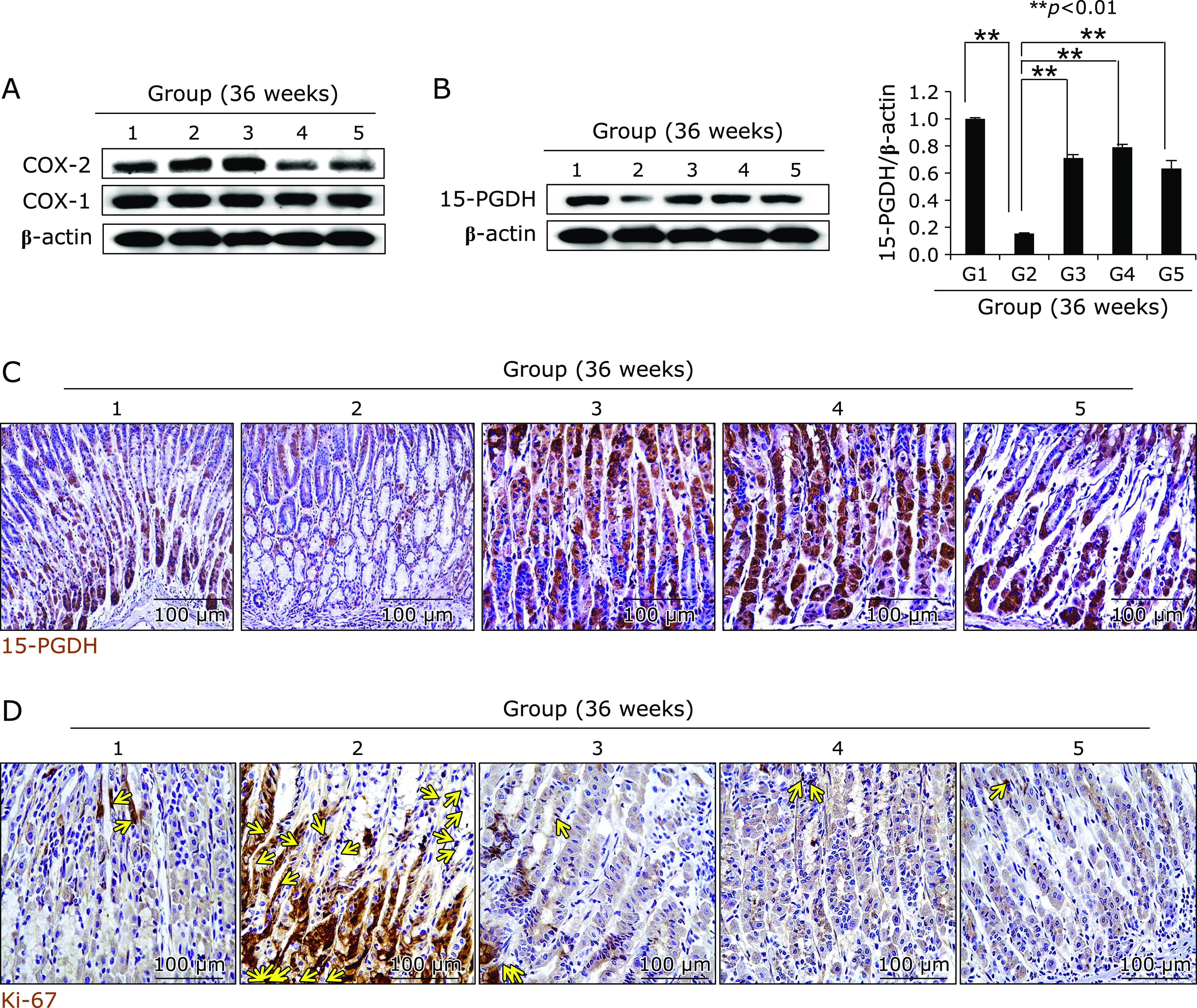
Changes of COX-1, COX-2, 15-PGDH, and Ki-67 according to group. (A) Western blot for COX-1 and COX-2. (B) Western blot for 15-PGDH. (C) Immunihistochemical staining of 15-PGDH, ×100 magnification. (D) Western blot for Ki-67, ×100 magnification.
Fig. 6.
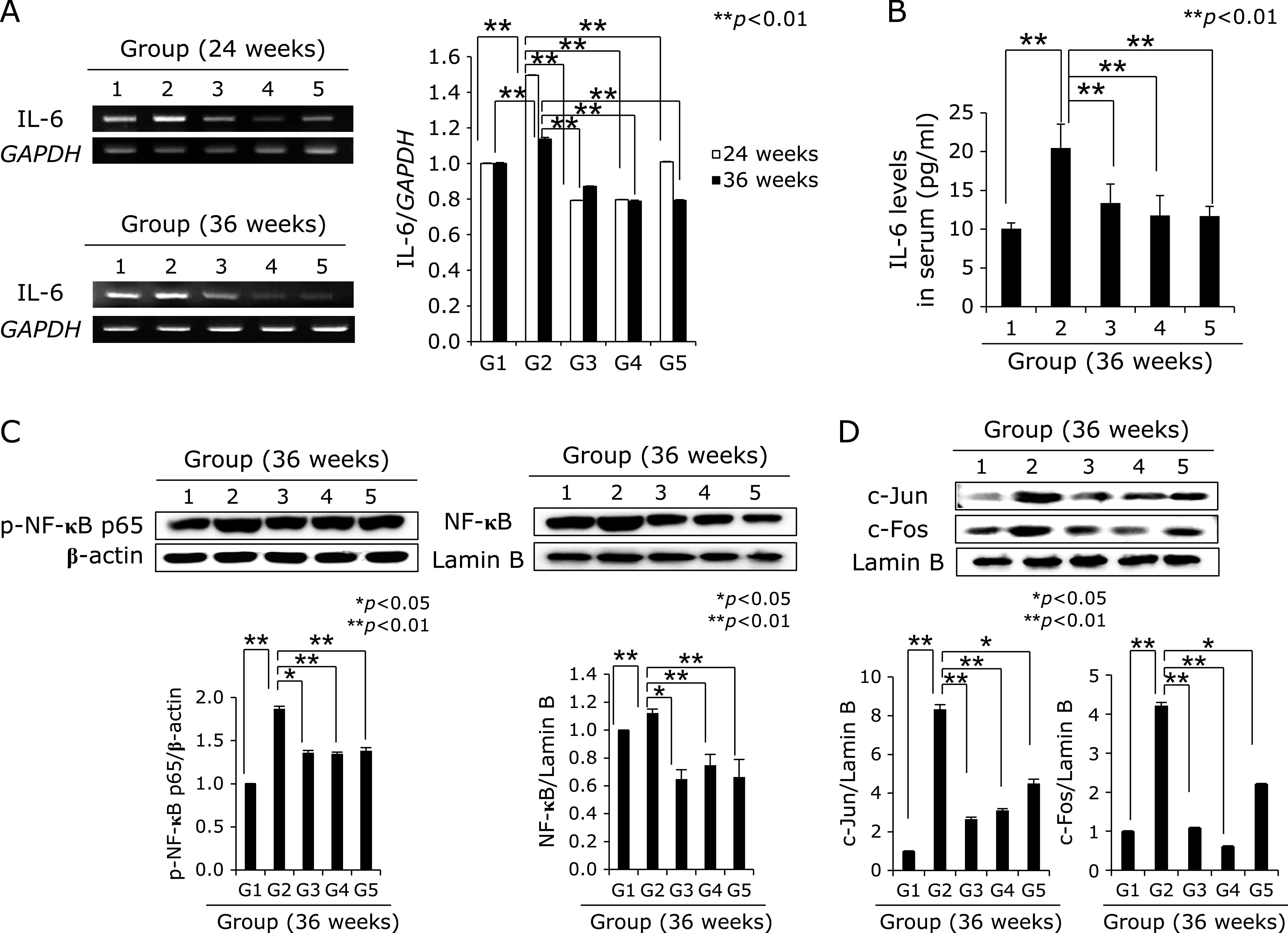
IL-6, NF-κB, and c-Fos/c-Jun according to group. (A) RT-PCR for IL-6 at 24 week and 36 weeks. (B) Serum levels of IL-6 according to group. (C) Western blot for cytoplasmic expressions of p-NF-κB p65 and nuclear expressions of NF-κB according to group. Mean expressions levels after triplicate measurement according to group. (D) Western blot for c-Jun and c-Fos.
Fig. 7.
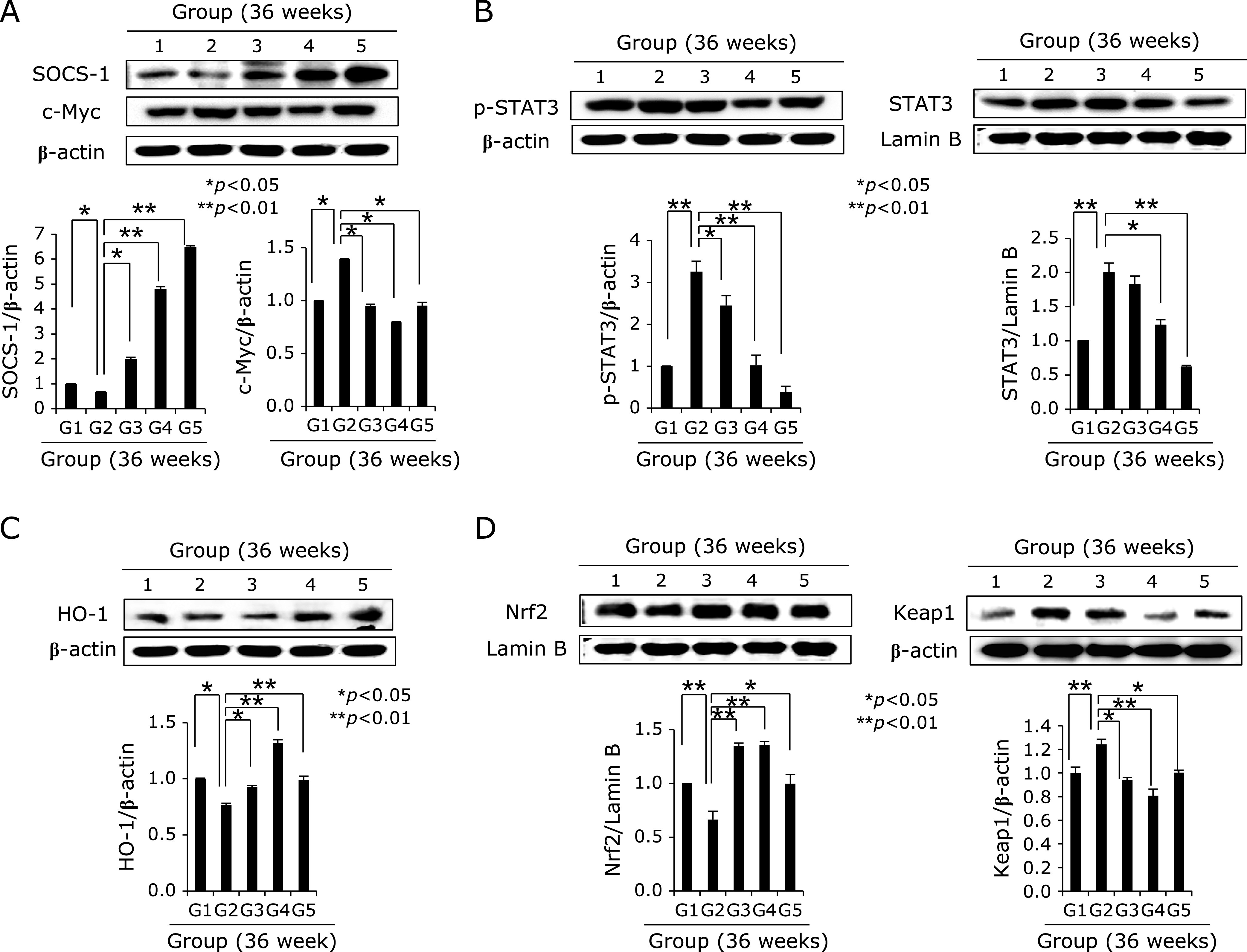
Expression of SOCS-1, c-Myc, STAT3, HO-1, Nrf2, and keap2. (A) Western blot for SOCS-1 and c-Myc according to group. (B) Western blot for cytoplasmic p-STAT3 and nuclear STAT3 according to group. (C) HO-1 expression according to group. (D) Western blot for Nrf2 and keap1 according to group.
Discussion
In the current study, we confirmed, for the first time, that long-term dietary intake of walnut plentiful of n-3 PUFAs, monounsaturated fatty acids, and vitamins rejuvenated H. pylori-associated CAG and prevented gastric carcinogenesis. This is an extended study of previous publication that walnut polyphenol extract (WPE) significantly ameliorated H. pylori-associated inflammation through sequential actions of PPAR-γ activation, SOCS-1 induction, IL-6/gp130 inhibition, and STAT3 inactivation.(8) Translating these in vitro actions of WPE, in the current study, we documented that dietary intake of walnut could significantly ameliorate gastric inflammation, significantly rejuvenate CAG, and mitigate gastric carcinogenesis via concerted actions of attenuated inflammatory mediators, increased preservation of 15-PGDH, and enhanced gastric defense mechanisms (Fig. 8).
Fig. 8.

Schematic presentation showing dietary intake of walnut mitigated H. pylori-associated gastric pathologies such as chronic atrophic gastritis, gastric erosions, and gastric ulcers, leading to prevention of H. pylori-associated tumorigenesis, for which we have documented that walnut intake significantly inhibited mutagenic COX-2 and PGE2 via 15-PGDH induction, redox-sensitive NF-κB repression, IL-6 inhibition, STA3 inactivation, and Nrf2 activation.
As the representational example of “inflammation-carcinogenesis”, chronic H. pylori infection developed gastric cancer via chronic gastritis, CAG, intestinal metaplasia, and gastric adenoma according to Prof. Correa P hypothesis.(14,15) Therefore, according to International Agency for Research on Cancer (IARC) definition that H. pylori as class I carcinogen, either eradication or attenuation of mutagenic inflammation can be a way of cancer prevention. Recent investigation by Choi et al.,(16) the eradication of H. pylori in patients with endoscopic resection of early gastric cancer significantly prevented metachronous gastric cancer and ensuing publication by same group that primary relative of gastric cancers developed less gastric cancer in whom eradication was done.(17) These results suggested the removal of bug in high risk population for gastric cancer. However, this is not for general population. Though general population eradication had been done in Japan, it is not generally acknowledged in the world. Then, the other option might be attenuation of mutagenic gastric inflammation. In addition, though H. pylori infection had been defined as definite carcinogen for gastric cancer and has been associated with extra-gastric diseases, the bacterium frequently persists in the human host without inducing disease and plays a beneficial role in health in some, commensals. Therefore, non-microbial approach could be considered to prevent H. pylori-associated diseases.(18–20)
Among several trials for regulating H. pylori, dietary or non-microbial approach can be a feasible and ideal step because of no risk of microbial resistance, safety, feasibility, and long-term application.(21–23) Furthermore, since no availability of vaccine and possible attenuation of mutagenic gastric inflammations is presented, dietary intervention is preferred in regulating H. pylori.(24–28) The concept of “pharmanutrient” required the following three aspects, one is continuity, the second is hormesis, and lastly mode of actions. In this concept, authors produced the proof of concept studies that fat-1 transgenic mice producing n-3 PUFAs by way of n-3 desaturase over-expression that the transgenic mice can produce n-3 PUFAs in the stomach irrespective of diet. n-3 PUFAs significantly attenuated H. pylori-associated CAG or prevention of H. pylori-induced gastric cancer.(9,21,25) Supported with these faithful backgrounds, the authors proceed current experiment under the hope that dietary intake of whole walnut can rescue from H. pylori-associated gastric carcinogenesis pathway via CAG.
H. pylori, a gram-negative bacterial pathogen living in stomach, has been implicated in development of gastric cancer through the development of hypertrophic and atrophic gastritis, is well-known as initiating and progressing mechanism of gastric carcinogenesis.(29–31) Among mechanisms in H. pylori-carcinogenesis, including NF-κB based oxidative stress, cytokines mediated mutagenesis, apoptosis induced atrophic changes through loss of parietal cells and some gastric stem cells, and more carcinogenic actions like CagA oncogenic proteins,(32,33) it has been well documented that H. pylori infection causes activation of STAT3 activation, forms a dimer, trans-locates to nucleus, and functions as transcription factor to regulate the target genes implicated in H. pylori-associated inflammation and carcinogenesis subsequent to IL-6.(27,34–36) Furthermore, CagA oncogenic cytotoxin from H. pylori led to significant cytokine signaling pathways via MAPK activation and is responsible for gastric inflammation and carcinogenes is including increased cell proliferation, angiogenesis, inflammation, inhibition of immunocytes, and epithelial cell apoptosis.(37) Among these cytokines, IL-6, IL-6R, and gp130 with subsequent JAKs/STAT3 activation are representationally dysregulated pathways in H. pylori infection.(38) In this point, agents or intervention to regulate IL-6 signaling seem to be of potential significance to solve unmet medical needs of H. pylori-associated gastric pathologies. As seen in the current study, walnut administration efficiently inhibited either IL-6/IL-6R/gp130 or JAK/STAT3 activation.
Though H. pylori infection led to development of gastritis and consequent gastric cancer, more than 90% of infected patients do not develop gastric cancer, suggesting the existence of host defense systems.(39) Nrf2 is a transcription factor that plays a key role in cellular defense against H. pylori infection, in which the Nrf2-HO-1 axis is considered to play a role in cellular adaptive survival response to H. pylori. On the other aspect, H. pylori incites a futile inflammatory response as the key feature of its immunopathogenesis enable to survive H. pylori in hostile environment of stomach and recruitment of myeloid cells to the gastric mucosa during H. pylori infection, during which robust host defense against the bacterium and gastric inflammation occurs, a mechanism by which H. pylori impairs the immune response and favors its own survival via activation of mononuclear cell HO-1.(40) Therefore, the fact that H. pylori cytotoxic gene, CagA, favors its own enhanced inflammation by inhibiting HO-1 induction,(41) whereas inhibition of phosphorylation of CagA by HO-1 in gastric epithelial cells highlighted a mechanism that host HO-1 response can limit the pathogenesis of H. pylori infection.(42) In the current investigation, walnut intake consistently increased host defense system against H. pylori infection via HO-1, which was transcribed with Nrf2 and keap1 inactivation.
In our investigation, walnut achieved significant rejuvenation of H. pylori-induced CAG and cancer prevention under the significant preservation of tumor suppressive 15-PGDH (Fig. 2B and 5B). 15-PGDH as a key PG catabolic enzyme catalyzing the oxidation and inactivation of PGE2 synthesized from the COX pathway is known to be a novel candidate for early detection of field defects in carcinogenesis and strong tumor suppressive enzyme since early inactivation of 15-PGDH,(43) followed by activation of COX-2 contributes to PGE2 production, leading to colon carcinogenesisand loss of 15-PGDH increases PGE2 in GI cancers.(44,45) In addition, 15-PGDH is associated with GI inflammation and immune response,(46,47) concluding that PG catabolic enzymes as tumor suppressor.(48) Consistently, walnut antagonized H. pylori-associated COX-2 oncogene via preservation of 15-PGDH.
As molecular mechanisms of preventive strategies achieved with walnuts, first, we have focused on IL-6/JAK/STAT3 pathway and COX-2/PGE2/NF-κB pathway because they are responsible for transcribing inflammatory and mutagenic mediators such as COX-2, IFN-γ, iNOS, IL-1β, and c-Myc relevant to H. pylori infection. STAT3 is a transcription factor activated by various external stimuli including cytokines and growth factors. JAK1/STAT3 is known to be an upstream signaling of NF-κB activation producing IL-8/IL-1β/IL-2 and generating ROS after H. pylori infection.(36,49,50) Conclusively, H. pylori-induced STAT3 activation is mediated through ROS-induced upregulation of IL-6 expression, necessitating inhibiting STAT3 to mitigate gastric damages after H. pylori infection. Consequent to IL-6 generation after H. pylori infection, augmented gp130-mediated cytokine signaling should be blocked in order to prevent H. pylori-associated inflammation and carcinogenesis.(51,52) In addition to these epithelial components, though not studied in the current investigation, STAT3 imparted a profound influence on immune response to H. pylori infection,(53) for instance, inhibition of dendritic cell maturation via IL-10 mediated STAT3 activation to facilitate H. pylori infection,(54) triggering the expression of the bactericidal lectin REG3γ to allow H. pylori to manipulate host immunity in order to favor bacterial survivalin the gastric mucosal niche.(55,56) Especially among H. pylori, also used in our study, H. pylori CagA activates STAT3 pathway in this propagation of gastric inflammation.(38) Since H. pylori-induced STAT3 activation is considered as significant signaling network in gastric cancer,(57–59) from the current study, walnut intake can contribute to significant inhibitory action of STAT3 via IL-6/gp130 inhibition.
Walnut (Juglans regia L.) contains a complex array of natural compounds and phytochemicals, for instance, in the metabolite-profiling analysis, walnut caused a significant increase in several PUFAs, including docosahexaenoic acid (DHA) and 9-oxo-10(E),12(E)-octadecadienoic acid (9-oxoODA), as well as kynurenic acid, that exhibits a wide range of health benefits, including anti-inflammatory, antioxidative, and regenerating actions, by which dietary walnut supplementation (14% walnut) showed significant effects in recovery from dextran sulfate sodium-induced colitis,(60) significant protection against fenitrothion- or malathion-mediated immunotoxicity,(61,62) enriching intestinal microbiome for improving health condition,(63) reducing telomere length,(64) ameliorating colitis and colitis-associated cancer,(65) suppressing colon cancer cell growth,(66) and regulating anti-cancer stem cells.(67) Though not touched in the current investigation, cancer stem cell markers including CD133, CD44, DLK1, and Notch1 as well as the β-catenin/p-GSK3β signaling pathway were significantly down-regulated and the self-renewal capacity of cancer stem cells was suppressed. Taken together with our investigation, dietary intake of walnut can impose significant cancer preventive action as well as anti-cancer effects in H. pylori infection (Fig. 8). In clinic, the importance of either H. pylori eradication or their regulation impose significant impact on either gastric cancer prevention or gastric diseases amelioration, for which several kinds of intervention like natural products, probiotics, and more effective anti-H. pylori therapy are under active investigation.(68–71) In conclusion, walnut seems to be very effective and attractive because of well-acknowledgement as “pharmanutrient” against H. pylori infection. However, well-designed clinical trial should be followed.
Author Contributions
Study concept and design: JM Park and KB Hahm; acquisition of data: YM Han and JM Park; analysis and statistical analysis: KB Hahm; interpretation of data: YM Han, JM Park and YJ Park; drafting of manuscript: YM Han and KB Hahm. All authors approved the final version of this manuscript to be published.
Acknowledgments
This work was supported by California Walnut Commission and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (No. NRF-2017R1C1B2009057 to YM Han).
Abbreviations
- CAG
chronic atrophic gastritis
- COX
cyclooxygenase
- H. pylori
Helicobacter pylori
- HO-1
hemeoxygenase-1
- IARC
International Agency for Research on Cancer
- JAK
Janus kinase
- Keap1
Kelch-like ECH-associated protein 1
- n-3 PUFA
n-3 polyunsaturated fatty acid
- NF-κB
nuclear factor-κ-light chain-enhancer of activated B cells
- Nrf2
nuclear factor erythroid 2-related factor 2
- 15-PGDH
15-hydroxyprostaglandin dehydrogenase
- PPAR-γ
peroxisome proliferator-activated receptor gamma
- SOCS
suppressor of cytokine secretion
- STAT3
signal transducer and activator of transcription 3
- TNF-α
tumor necrosis factor-α
- WPE
walnut polyphenol extract
Conflict of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
References
- 1.Bravo D, Hoare A, Soto C, Valenzuela MA, Quest AF. Helicobacter pylori in human health and disease: mechanisms for local gastric and systemic effects. World J Gastroenterol 2018; 24: 3071–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gonzalez CA, Riboli E. Diet and cancer prevention: where we are, where we are going. Nutr Cancer 2006; 56: 225–231. [DOI] [PubMed] [Google Scholar]
- 3.Ribera-Fonseca A, Jiménez D, Leal P, et al. The anti-proliferative and anti-invasive effect of leaf extracts of blueberry plants treated with methyl jasmonate on human gastric cancer in vitro is related to their antioxidant properties. Antioxidants (Basel) 2020; 9: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Ishaq RK, Overy AJ, Büsselberg D. Phytochemicals and gastrointestinal cancer: cellular mechanisms and effects to change cancer progression. Biomolecules 2020; 10: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spagnuolo C, Russo GL, Orhan IE, et al. Genistein and cancer: current status, challenges, and future directions. Adv Nutr 2015; 6: 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park Y, Lee H, Lim JW, Kim H. Inhibitory effect of β-carotene on Helicobacter pylori-induced TRAF expression and hyper-proliferation in gastric epithelial cells. Antioxidants (Basel) 2019; 8: 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SH, Lim JW, Kim H. Astaxanthin inhibits mitochondrial dysfunction and interleukin-8 expression in Helicobacter pylori-infected gastric epithelial cells. Nutrients 2018; 10: 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park JM, An JM, Han YM, et al. Walnut polyphenol extracts inhibit Helicobacter pylori-induced STAT3Tyr705 phosphorylation through activation of PPAR-γ and SOCS1 induction. J Clin Biochem Nutr 2020; 67: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han YM, Kim KJ, Jeong M, et al. Suppressed Helicobacter pylori-associated gastric tumorigenesis in Fat-1 transgenic mice producing endogenous omega-3 polyunsaturated fatty acids. Oncotarget 2016; 7: 66606–66622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaddy JA, Radin JN, Loh JT, et al. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect Immun 2013; 81: 2258–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson KJ, Teuber SS, Gobeille A, Cremin P, Waterhouse AL, Steinberg FM. Walnut polyphenolics inhibit in vitro human plasma and LDL oxidation. J Nutr 2001; 131: 2837–2842. [DOI] [PubMed] [Google Scholar]
- 12.Nam SY, Kim N, Lee CS, et al. Gastric mucosal protection via enhancement of MUC5AC and MUC6 by geranylgeranylacetone. Dig Dis Sci 2005; 50: 2110–2120. [DOI] [PubMed] [Google Scholar]
- 13.Lacy ER, Ito S. Microscopic analysis of ethanol damage to rat gastric mucosa after treatment with a prostaglandin. Gastroenterology 1982; 83: 619–625. [PubMed] [Google Scholar]
- 14.Mera RM, Bravo LE, Camargo MC, et al. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut 2018; 67: 1239–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piazuelo MB, Correa P. Gastric cáncer: overview. Colomb Med (Cali) 2013; 44: 192–201. [PMC free article] [PubMed] [Google Scholar]
- 16.Choi IJ, Kook MC, Kim YI, et al. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N Engl J Med 2018; 378: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 17.Choi IJ, Kim CG, Lee JY, et al. Family history of gastric cancer and Helicobacter pylori treatment. N Engl J Med 2020; 382: 427–436. [DOI] [PubMed] [Google Scholar]
- 18.Nagashima H, Yamaoka Y. Importance of toll-like receptors in pro-inflammatory and anti-inflammatory responses by Helicobacter pylori infection. Curr Top Microbiol Immunol 2019; 421: 139–158. [DOI] [PubMed] [Google Scholar]
- 19.Han HS, Lee SY, Oh SY, Moon HW, Cho H, Kim JH. Correlations of the gastric and duodenal microbiota with histological, endoscopic, and symptomatic gastritis. J Clin Med 2019; 8: 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu YL, Pang W, Huang Y, Zhang Y, Zhang CJ. The gastric microbiome is perturbed in advanced gastric adenocarcinoma identified through shotgun metagenomics. Front Cell Infect Microbiol 2018; 8: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han YM, Park JM, Jeong M, et al. Dietary, non-microbial intervention to prevent Helicobacter pylori-associated gastric diseases. Ann Transl Med 2015; 3: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SH, Kangwan N, Park JM, Kim EH, Hahm KB. Non-microbial approach for Helicobacter pylori as faster track to prevent gastric cancer than simple eradication. World J Gastroenterol 2013; 19: 8986–8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JS, Paek NS, Kwon OS, Hahm KB. Anti-inflammatory actions of probiotics through activating suppressor of cytokine signaling (SOCS) expression and signaling in Helicobacter pylori infection: a novel mechanism. J Gastroenterol Hepatol 2010; 25: 194–202. [DOI] [PubMed] [Google Scholar]
- 24.An JM, Kang EA, Han YM, et al. Dietary threonine prevented stress-related mucosal diseases in rats. J Physiol Pharmacol 2019; 70. DOI: 10.26402/jpp.2019.3.14 [DOI] [PubMed] [Google Scholar]
- 25.Lee HJ, Han YM, An JM, et al. Role of omega-3 polyunsaturated fatty acids in preventing gastrointestinal cancers: current status and future perspectives. Expert Rev Anticancer Ther 2018; 18: 1189–1203. [DOI] [PubMed] [Google Scholar]
- 26.Park JM, Jeong M, Kim EH, Han YM, Kwon SH, Hahm KB. Omega-3 polyunsaturated fatty acids intake to regulate Helicobacter pylori-associated gastric diseases as nonantimicrobial dietary approach. Biomed Res Int 2015; 2015: 712363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong M, Park JM, Han YM, et al. Dietary prevention of Helicobacter pylori-associated gastric cancer with kimchi. Oncotarget 2015; 6: 29513–29526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeong M, Park JM, Han YM, et al. Dietary intervention of artemisia and green tea extracts to rejuvenate Helicobacter pylori-associated chronic atrophic gastritis and to prevent tumorigenesis. Helicobacter 2016; 21: 40–59. [DOI] [PubMed] [Google Scholar]
- 29.Stewart OA, Wu F, Chen Y. The role of gastric microbiota in gastric cancer. Gut Microbes 2020; 11: 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Zhang J, Cheng ASL, Yu J, To KF, Kang W. Gastric cancer: genome damaged by bugs. Oncogene 2020; 39: 3427–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petryszyn P, Chapelle N, Matysiak-Budnik T. Gastric cancer: Where are we heading? Dig Dis 2020; 38: 280–285. [DOI] [PubMed] [Google Scholar]
- 32.Eid R, Moss SF. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2002; 346: 65–67. [PubMed] [Google Scholar]
- 33.Zullo A, Hassan C, Morini S. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2002; 346: 65–67. [PubMed] [Google Scholar]
- 34.Ishii Y, Shibata W, Sugimori M, et al. Activation of signal transduction and activator of transcription 3 signaling contributes to Helicobacter-associated gastric epithelial proliferation and inflammation. Gastroenterol Res Pract 2018; 2018: 9050715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao S, Zhu C, Feng J, et al. Helicobacter hepaticus infection induces chronic hepatitis and fibrosis in male BALB/c mice via the activation of NF-kappaB, Stat3, and MAPK signaling pathways. Helicobacter 2020; 25: e12677. [DOI] [PubMed] [Google Scholar]
- 36.Piao JY, Lee HG, Kim SJ, et al. Helicobacter pylori activates IL-6-STAT3 signaling in human gastric cancer cells: potential roles for reactive oxygen species. Helicobacter 2016; 21: 405–416. [DOI] [PubMed] [Google Scholar]
- 37.Howlett M, Menheniott TR, Judd LM, Giraud AS. Cytokine signalling via gp130 in gastric cancer. Biochim Biophys Acta 2009; 1793: 1623–1633. [DOI] [PubMed] [Google Scholar]
- 38.Bronte-Tinkew DM, Terebiznik M, Franco A, et al. Helicobacter pylori cytotoxin-associated gene A activates the signal transducer and activator of transcription 3 pathway in vitro and in vivo. Cancer Res 2009; 69: 632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paik JY, Lee HG, Piao JY, et al. Helicobacter pylori infection promotes autophagy through Nrf2-mediated heme oxygenase upregulation in human gastric cancer cells. Biochem Pharmacol 2019; 162: 89–97. [DOI] [PubMed] [Google Scholar]
- 40.Gobert AP, Verriere T, Asim M, et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to Helicobacter pylori. J Immunol 2014; 193: 3013–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gobert AP, Asim M, Piazuelo MB, et al. Disruption of nitric oxide signaling by Helicobacter pylori results in enhanced inflammation by inhibition of heme oxygenase-1. J Immunol 2011; 187: 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gobert AP, Verriere T, de Sablet T, Peek RM, Jr, Chaturvedi R, Wilson KT. Haem oxygenase-1 inhibits phosphorylation of the Helicobacter pylori oncoprotein CagA in gastric epithelial cells. Cell Microbiol 2013; 15: 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki Y, Nakatani Y, Hara S. Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis. Prostaglandins Other Lipid Mediat 2015; 121 (Pt A): 42–45. [DOI] [PubMed] [Google Scholar]
- 44.Yang DH, Ryu YM, Lee SM, et al. 15-Hydroxyprostaglandin dehydrogenase as a marker in colon carcinogenesis: analysis of the prostaglandin pathway in human colonic tissue. Intest Res 2017; 15: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pham H, Chen M, Li A, et al. Loss of 15-hydroxyprostaglandin dehydrogenase increases prostaglandin E2 in pancreatic tumors. Pancreas 2010; 39: 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai L, King DW, Perera DS, Lubowski DZ, Burcher E, Liu L. Inverse expression of prostaglandin E2-related enzymes highlights differences between diverticulitis and inflammatory bowel disease. Dig Dis Sci 2015; 60: 1236–1246. [DOI] [PubMed] [Google Scholar]
- 47.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol 2012; 188: 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tai HH. Prostaglandin catabolic enzymes as tumor suppressors. Cancer Metastasis Rev 2011; 30: 409–417. [DOI] [PubMed] [Google Scholar]
- 49.Cha B, Lim JW, Kim H. Jak1/Stat3 is an upstream signaling of NF-κB activation in Helicobacter pylori-induced IL-8 production in gastric epithelial AGS cells. Yonsei Med J 2015; 56: 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gobert AP, Wilson KT. Polyamine- and NADPH-dependent generation of ROS during Helicobacter pylori infection: a blessing in disguise. Free Radic Biol Med 2017; 105: 16–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson CB, Judd LM, Menheniott TR, et al. Augmented gp130-mediated cytokine signalling accompanies human gastric cancer progression. J Pathol 2007; 213: 140–151. [DOI] [PubMed] [Google Scholar]
- 52.Xiong H, Du W, Sun TT, et al. A positive feedback loop between STAT3 and cyclooxygenase-2 gene may contribute to Helicobacter pylori-associated human gastric tumorigenesis. Int J Cancer 2014; 134: 2030–2040. [DOI] [PubMed] [Google Scholar]
- 53.Menheniott TR, Judd LM, Giraud AS. STAT3: a critical component in the response to Helicobacter pylori infection. Cell Microbiol 2015; 17: 1570–1582. [DOI] [PubMed] [Google Scholar]
- 54.Rizzuti D, Ang M, Sokollik C, et al. Helicobacter pylori inhibits dendritic cell maturation via interleukin-10-mediated activation of the signal transducer and activator of transcription 3 pathway. J Innate Immun 2015; 7: 199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee KS, Kalantzis A, Jackson CB, et al. Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3γ via gastric STAT3 activation. PLoS One 2012; 7: e30786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaebisch R, Mejías-Luque R, Prinz C, Gerhard M. Helicobacter pylori cytotoxin-associated gene A impairs human dendritic cell maturation and function through IL-10-mediated activation of STAT3. J Immunol 2014; 192: 316–323. [DOI] [PubMed] [Google Scholar]
- 57.Zhao J, Dong Y, Kang W, et al. Helicobacter pylori-induced STAT3 activation and signalling network in gastric cancer. Oncoscience 2014; 1: 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun TT, Tang JY, Du W, et al. Bidirectional regulation between TMEFF2 and STAT3 may contribute to Helicobacter pylori-associated gastric carcinogenesis. Int J Cancer 2015; 136: 1053–1064. [DOI] [PubMed] [Google Scholar]
- 59.Giraud AS, Menheniott TR, Judd LM. Targeting STAT3 in gastric cancer. Expert Opin Ther Targets 2012; 16: 889–901. [DOI] [PubMed] [Google Scholar]
- 60.Nakanishi M, Matz A, Klemashevich C, Rosenberg DW. Dietary walnut supplementation alters mucosal metabolite profiles during DSS-induced colonic ulceration. Nutrients 2019; 11: 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu H, Wan Y, Wang Y, et al. Walnut polyphenol extract protects against fenitrothion-induced immunotoxicity in murine splenic lymphocytes. Nutrients 2018; 10: 1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao Y, Fan C, Zhang A, et al. Walnut polyphenol extract protects against malathion- and chlorpyrifos-induced immunotoxicity by modulating TLRx-NOX-ROS. Nutrients 2020; 12: 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bamberger C, Rossmeier A, Lechner K, et al. A walnut-enriched diet affects gut microbiome in healthy caucasian subjects: a randomized, controlled trial. Nutrients 2018; 10: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shin PK, Zoh Y, Choi J, Kim MS, Kim Y, Choi SW. Walnut phenolic extracts reduce telomere length and telomerase activity in a colon cancer stem cell model. Nutr Res Pract 2019; 13: 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koh SJ, Choi YI, Kim Y, et al. Walnut phenolic extract inhibits nuclear factor kappaB signaling in intestinal epithelial cells, and ameliorates experimental colitis and colitis-associated colon cancer in mice. Eur J Nutr 2019; 58: 1603–1613. [DOI] [PubMed] [Google Scholar]
- 66.Lee J, Kim YS, Lee J, et al. Walnut phenolic extract and its bioactive compounds suppress colon cancer cell growth by regulating colon cancer stemness. Nutrients 2016; 8: 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chung J, Kim YS, Lee J, Lee JH, Choi SW, Kim Y. Compositional analysis of walnut lipid extracts and properties as an anti-cancer stem cell regulator via suppression of the self-renewal capacity. Food Sci Biotechnol 2016; 25: 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.An JM, Kim E, Lee HJ, Park MH, Son DJ, Hahm KB. Dolichos lablab L. extracts as pharmanutrient for stress-related mucosal disease in rat stomach. J Clin Biochem Nutr 2020; 67: 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mukai R, Handa O, Suyama Y, Majima A, Naito Y. Effectiveness of including probiotics to Helicobacter pylori eradication therapies. J Clin Biochem Nutr 2020; 67: 102–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Handa O, Naito Y, Osawa M, et al. Nutirents and probiotics: current trends in their use to eradicate Helicobacter pylori. J Clin Biochem Nutr 2020; 67: 26–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Horie R, Handa O, Ando T, et al. Helicobacter pylori eradication therapy outcome according to clarithromycin susceptibility testing in Japan. Helicobacter 2020; 25: e12698. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.