

Key Points
Question
Is there any phenotype–genotype correlation in patients harboring biallelic pathogenic variants in CLN3?
Findings
In a French cohort of 1533 patients with inherited retinal disorders (IRDs), 15 patients (1.0%) had retina-restricted CLN3-linked disease covering 2 phenotypes: mild (n = 6) and severe (n = 9) retinal degeneration with macular atrophy. The genetic background (variant/genotype) underlying the mild and the severe retina-restricted forms of CLN3-associated IRD was clearly distinct from neuronal ceroid lipofuscinosis.
Meaning
These results suggest that most patients harboring pathogenic variants in CLN3 might have a phenotype and a prognosis predicted from the genetic background.
This case series provides a detailed description of the retinal phenotype of autosomal recessive nonsyndromic inherited retinal disease harboring biallelic CLN3 pathogenic variants.
Abstract
Importance
Biallelic variants in CLN3 lead to a spectrum of diseases, ranging from severe neurodegeneration with retinal involvement (juvenile neuronal ceroid lipofuscinosis) to retina-restricted conditions.
Objective
To provide a detailed description of the retinal phenotype of patients with isolated retinal degeneration harboring biallelic CLN3 pathogenic variants and to attempt a phenotype–genotype correlation associated with this gene defect.
Design, Setting, and Participants
This retrospective cohort study included patients carrying biallelic CLN3 variants extracted from a cohort of patients with inherited retinal disorders (IRDs) investigated at the National Reference Center for Rare Ocular Diseases of the Centre Hospitalier National d’Ophtalmologie des Quinze-Vingts from December 2007 to August 2020. Data were analyzed from October 2019 to August 2020.
Main Outcome and Measures
Functional (best-corrected visual acuity, visual field, color vision, and full-field electroretinogram), morphological (multimodal retinal imaging), and clinical data from patients were collected and analyzed. Gene defect was identified by either next-generation sequencing or whole-exome sequencing and confirmed by Sanger sequencing, quantitative polymerase chain reaction, and cosegregation analysis.
Results
Of 1533 included patients, 843 (55.0%) were women and 690 (45.0%) were men. A total of 15 cases from 11 unrelated families harboring biallelic CLN3 variants were identified. All patients presented with nonsyndromic IRD. Two distinct patterns of retinal disease could be identified: a mild rod-cone degeneration of middle-age onset (n = 6; legal blindness threshold reached by 70s) and a severe retinal degeneration with early macular atrophic changes (n = 9; legal blindness threshold reached by 40s). Eleven distinct pathogenic variants were detected, of which 4 were novel. All but 1, p.(Arg405Trp), CLN3 point variants and their genotypic associations were clearly distinct between juvenile neuronal ceroid lipofuscinosis and retina-restricted disease. Mild and severe forms of retina-restricted CLN3-linked IRDs also had different genetic background.
Conclusions and Relevance
These findings suggest CLN3 should be included in next-generation sequencing panels when investigating patients with nonsyndromic rod-cone dystrophy. These results document phenotype–genotype correlations associated with specific variants in CLN3. However, caution seems warranted regarding the potential neurological outcome if a pathogenic variant in CLN3 is detected in a case of presumed isolated IRD for the onset of neurological symptoms could be delayed.
Introduction
Inherited retinal diseases (IRDs) are a clinically and genetically heterogeneous group of disorders, the most frequent of which being rod-cone dystrophy, also known as retinitis pigmentosa, with a prevalence of about 1:4000.1 The vast majority of cases are nonsyndromic, with the disease restricted to the retina. A lesser number of cases are syndromic and encompass a continuum of clinical manifestations, from retinopathy with subtle extraocular signs to multisystemic disorders.2,3 Both syndromic and nonsyndromic forms are characterized by a large genetic heterogeneity with more than 100 genes potentially implicated.4 Moreover, recent advances in high-throughput sequencing, including comprehensive targeted next-generation panels, have underlined genotypic overlaps—cases of restricted retinal disease being associated with variants in genes also implicated in syndromic IRD.1,5,6 For instance, pathogenic variants in a major ciliopathy-associated gene, USH2A, account for about 20% of cases of isolated autosomal-recessive retinitis pigmentosa.7,8,9
Another example is CLN3 (OMIM 607042), a ubiquitously expressed gene, variants of which have recently been associated with isolated retinal degeneration in 5 patients.10 Variants in CLN3 typically lead to the juvenile form of neuronal ceroid lipofuscinosis (JNCL), also known as Batten disease or Vogt-Spielmeyer disease (OMIM 204200),11 a severe neurometabolic disease with brain ceroid accumulation leading to neurodegeneration.12 Neuronal ceroid lipofuscinosis (NCL) is in fact a genetically heterogeneous group of metabolic disorders with lysosomal enlargement and ceroid material accumulation.12 Pathogenic variants in 13 genes, namely PPT1 (OMIM 600722), TPP1 (OMIM 607998), CLN3, CLN5 (OMIM 608102), CLN6 (OMIM 606725), MFSD8 (OMIM 611124), CLN8 (OMIM 607837), CTSD (OMIM 116840), DNAJC5 (OMIM 611203), CTSF (OMIM 603539), ATP13A2 (OMIM 610513), GRN (OMIM 138945), and KCTD7 (OMIM 611725), are known to cause NCL.12 NCL affects 1:100 000 live births worldwide.13 JNCL linked to CLN3 pathogenic variants accounts for approximately 13% of childhood-onset NCL.14 In its classic form, visual loss starts as early as age 5 to 6 years and precedes cognitive and motor regression.12,15 The earliest symptoms are poor vision, overlooking, bad color discrimination, and sometimes night blindness.16,17,18 Inner retinal dysfunction concomitant with the outer layer degeneration is a peculiar feature of the retinal dystrophy associated with JNCL.16,17,18
In this study, we provide further details on the retinal phenotype and novel pathogenic variants in a cohort of patients with IRD harboring biallelic CLN3 variants and evaluate phenotype–genotype correlations.
Methods
Research procedures adhered to the tenets of the Declaration of Helsinki and were approved by the local ethics committee (Comité de Protection des Personnes Ile de France 5). Prior to testing, written informed consent was obtained from each study participant. No compensation or incentives were offered to individuals to participate in the study.
Clinical Studies
The sporadic and familial cases affected with IRD were clinically investigated at the National Reference Center for Rare Ocular Diseases of the Centre Hospitalier National d’Ophtalmologie des Quinze-Vingts. Ophthalmic examination of affected individuals was performed as previously described.19 Patients’ data were collected from December 2007 to August 2020 and analyzed from October 2019 to August 2020. Information about genetic analyses can be found in the eMethods and eFigures 1 and 2 in the Supplement.
Statistical Analysis
Best-corrected visual acuity (BCVA) variables were expressed in logMAR, then mean BCVA between eyes (right eye and left eye) was calculated and then used for further analysis (no significant difference between both eyes was found with a 2-tailed Wilcoxon signed rank test). Given the small number of samples, we did not perform a regression analysis for BCVA decline but only plotted individual mean BCVA on a graph.
Two phenotypic groups of patients (group A, mild; group B, severe) were separated on the basis of the following criteria: age of onset of the first symptoms, progression of BCVA, and presence of macular atrophy. Kaplan-Meier survival curves were plotted for mean BCVA of 1.3 logMAR or greater (20/400 Snellen equivalent; the legal blindness threshold in France). The difference between the curves from each phenotypic group was determined using a 2-sided log-rank test. Significance was set at P < .05. Clinical variables were analyzed using SPSS Statistics software version 21.0 (IBM).
Results
Of 1533 patients with IR from a large French cohort, 843 (55.0%) were women and 690 (45.0%) were men. A total of 15 affected patients with nonsyndromic IRD, including 10 men and 5 women aged 19 to 65 years from 11 unrelated families, were identified with biallelic CLN3 variants. None of the 15 patients had neurological symptoms besides patient CIC00350, a woman in her 70s with extrapyramidal adverse effects from neuroleptic treatment given for depression, according to the neurologist.
Ocular findings are summarized in Figure 1 and eFigures 3 to 12 in the Supplement. Retinal findings from patients fall into 2 categories. In group A, 6 patients presented with mild retinal degeneration of middle-age onset (Figure 1A and B). Subjective symptoms appeared in individuals between their 20s and 40s. BCVA was relatively preserved (mean [range] BCVA, 20/32 [20/20 to 20/100]). Goldman visual field showed a general constriction of all isopters along with preservation of temporal crescent in 2 patients; no patient had absolute central scotoma. For the color vision defects, 4 patients exhibited a tritan axis, 1 patient had normal color vision, and 1 patient was not tested (Table 1). Fundus features included a waxy pallor of the optic disc, retinal vessel narrowing, midperipheral grayish discoloration, and sparse bone spicule–like pigmentation (eFigure 3 in the Supplement). Short-wavelength fundus autofluorescence showed a hyperautofluorescent ring in the posterior pole along with dark foveal dots in older patients (Figure 1A) (eFigure 3 in the Supplement). Spectral-domain optical coherence tomography (SD-OCT) revealed a variable extramacular outer retinal layer disruption. Foveal ellipsoid line was spared in 4 patients (Figure 1B) and disrupted or absent in 2 patients (eFigure 3 in the Supplement). Patients CIC08027, CIC08140 (eFigure 3 in the Supplement), and CIC09853 (Figure 1B) had cystoid macular changes in the progression of their disease. Electroretinogram (ERG) recordings were available for 5 patients in this group. Both rod and cone responses were undetectable in 3 patients (CIC00350, CIC08140, and CIC09152). It revealed a generalized rod-cone dysfunction with severely reduced or abolished dark-adapted responses and more preserved light-adapted responses in 2 patients (CIC02713 and CIC09853) (Table 1) (eFigure 5 in the Supplement).
Figure 1. Example of Milder (A and B) and Severe (C and D) Forms of CLN3-Related Isolated Retinal Degeneration.

A, Short-wavelength fundus autofluorescence perifoveal hyperautofluorescent annulus with macular edema in patient CIC09853 with genotype M2, M8, and M11. B, Spectral-domain optical coherence tomography (horizontal scan) of the same patient revealed cystoid maculopathy with a preserved ellipsoid zone. C, Short-wavelength fundus autofluorescence in patient CIC09088 with genotype M5 and M9. D, Spectral-domain optical coherence tomography widespread disruption of ellipsoid zone in the same patient.
Table 1. Clinical Characteristics of Patients With CLN3-Associated Retina-Restricted Diseases.
| Family | Patient | Sex | Age, y | Symptoms and comorbidities | BCVA, Snellen | Anterior segment | Fundus examination | SW-FAF | SD-OCT | Goldman visual fielda | Farnsworth 15 Hue | Full-field ERG | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| First examination | Last examination | ||||||||||||
| Mild retinal degeneration | |||||||||||||
| F234 | CIC00350 | F | 55 | Night blindness that developed in 40s; severe visual loss that developed in 50s; extrapyramidal syndrome that developed in 70s (medication associated: olanzapine, sertraline) | 20/100 OD; 20/200 OS | Age 76 y: LP OD; LP OS | Unilateral high myopia in the left eye (−7.50); left eye amblyopia from childhood | Optic disc pallor; vessel narrowing; posterior pole patchy chorioretinal atrophy; sparse bone spicule–like pigmentation in midperiphery | Global loss of autofluorescence; perifoveal hyperautofluorescent ring; round patches of hypoautofluorescence at the posterior pole | Widespread outer layer disappearance; indistinct retinal layering; thinning of the choriocapillaris | V4e constricted to 5° | Tritan | Undetectable at age 55 y |
| F976 | CIC02713 | F | 65 | Night blindness that developed in 40s; good vision | 20/25 OD; 20/25 OS | Age 69 y: 20/25 OD; 20/25 OS | Normal | Optic disc pallor; retinal vessel narrowing; macula seems normal; midperipheral retina grayish and atrophic; confluent bone spicule–like pigmentation | Perifoveal annulus; granularity and round hypoautofluorescent patches in the midperiphery | Outer retinal layers preserved in fovea, interrupted outside; choroidal thinning; epiretinal membrane | III4e constricted to 15° | Tritan | Dark-adapted ERG undetectable; light-adapted ERG severely reduced |
| F4480 | CIC08027 | M | 34 | Night blindness that developed in 20s; visual acuity loss and photophobia that developed in 30s | 20/50 OD; 20/20 OS | Age 39 y: 20/63 OD; 20/25 OS | Normal | Optic disc pallor; retinal vessel narrowing; cellophane light reflex from maculae; sparse bone spicule–like pigments and patchy chorioretinal atrophy in the midperiphery | Hypoautofluorescence from the fovea; perifoveal hyperautofluorescent ring; patchy hypoautofluorescence in the midperiphery | Outer retinal layers preserved at the fovea, interrupted outside; microcystic intraretinal changes in the right eye; epiretinal membrane | III4e constricted to 10°; large temporal crescents at III4e | NA | Dark-adapted ERG undetectable; light-adapted ERG severely reduced |
| F4545 | CIC08140 | M | 37 | Night blindness that developed when younger than 10 y; visual acuity loss that developed in 20s | 20/50 OD; 20/50 OS | Age 44 y: 20/25 OD; 20/500 OS | Posterior subcapsular cataract in both eyes; surgery at age 38 y | Optic disc pallor; retinal vessel narrowing; macular atrophy; dense bone spicule–like pigmentation and patchy chorioretinal atrophy in the midperiphery | Global hypoautofluorescence within the posterior pole; round patches of hypoautofluorescence in the midperiphery | Outer retinal layers preserved at the fovea, interrupted outside; microcystic intraretinal changes in both eyes | III4e constricted to 15° | Tritan | Undetectable at age 42 y |
| F5230 | CIC09152 | M | 37 | Night blindness that developed when younger than 10 y; congenital right esotropia | NA | Age 37 y: 20/40 OD; 20/32 OS | Keratoconus; with rigid contact lens carrier; clear lens | Optic disc pallor; retinal vessel narrowing; posterior pole patchy chorioretinal atrophy; fovea looks darkened; pigment clumping in the midperiphery | Central area of granular hypoautofluorescence (tiny black dots), involving the fovea and parafovea; large perimacular halo of hyperautofluorescence; patchy hypoautofluorescence in the midperiphery | Outer layer disappearance; hyperreflective subretinal dots | V4e constricted to 10°; temporal crescent | Tritan | Undetectable |
| F5662 | CIC09853 | M | 35 | Night blindness that developed in 20s; acetazolamide intake for microcystic retinal edema | 20/50 OD; 20/40 OS | Age 38 y: 20/40 OD; 20/50 OS | Normal | Disc and vessels seem normal; abnormal macular sheen; no obvious peripheral retinal changes | Perifoveal ring of hyperautofluorescence; granular hypoautofluorescence in the midperiphery | Outer retinal layers preserved in fovea, interrupted outside; microcystic retinal edema | III4e constricted to 10° | Normal | Dark-adapted ERG undetectable; light-adapted ERG severely reduced |
| Severe retinal degeneration | |||||||||||||
| F122 | CIC00166 | M | 34 | Night blindness and visual loss that developed in 20s | 20/100 OD; 20/125 OS | Age 37 y: 20/200 OD; 20/160 OS | Normal | Normal optic disc and retinal vessels; yellowish atrophic macular dots; sparse bone spicule–like pigmentation and perivascular pigmentary cuffs in midperiphy | Central area of granular hypoautofluorescence (tiny black dots), involving the fovea and parafovea | Widespread outer layers disappearance; hyperreflective subfoveal deposits | V4e constricted to 15°; temporal crescent | Tritan | Undetectable dark-adapted 0.01 and 3.0 ERG; electronegative dark-adapted 10.0 ERG (b/a ratio, 0.98); light-adapted ERG severely reduced |
| F699 | CIC01170 | F | 35 | Night blindness and visual acuity loss that developed in 20s | 20/400 OD; 20/400 OS | Age 38 y: 20/400 OD; 20/500 OS | Normal | Temporal optic disc pallor; yellowish macula with abnormal sheen and some pigmentary clumps; patchy chorioretinal atrophy and sparse bone spicule–like pigments in midperiphery | Hyperautofluorescence of the foveola; hypoautofluorescence and granularity of fovea; large perimacular halo of increased autofluorescence; patchy hypoautofluorescence of the midperiphery | Widespread outer layer disappearance; indistinct retinal layering; epimacular membrane | Absolute V4e central scotoma constricted to 5°; peripheral V4e constricted to 50° | Dyschromatopsia without axis | Undetectable at age 40 y |
| CIC01169 | M | 25 | Night blindness and visual acuity loss that developed when younger than 10 y | 20/200 OD; 20/200 OS | Age 32 y: CF OD; 20/400 OS | Normal | Temporal optic disc pallor; yellowish macula with atrophic changes; patchy chorioretinal atrophy and sparse bone spicule–like pigmentation in midperiphery | Hyperautofluorescence of the foveola; hypoautofluorescence and granularity of fovea; large perimacular halo of hyperautofluorescence; patchy hypoautofluorescence of the midperipheral retina | Widespread outer layer disappearance; indistinct retinal layering; epimacular membrane | V4e constricted to 15°; temporal crescent at V4e | NA | Undetectable at age 32 y | |
| CIC01168 | M | 30 | Night blindness and visual acuity loss that developed when younger than 10 y; neurological examination and brain MRI were normal at age 10 y; no vacuolated lymphocytes; death at 40 y from unspecified complications of long-term alcohol use | CF OD; CF OS | NA | Normal | Temporal optic disc pallor; yellowish macula with dark foveola and some pigmentary clumps; cellophane light reflex; patchy chorioretinal atrophy and sparse bone spicule–like pigment in the midperiphery | Hyperautofluorescence of the foveola; hypoautofluorescence and granularity of the fovea; large perifoveal hyperautofluorescent halo; patchy hypoautofluorescence of the midperiphery | Widespread outer layer disappearance; indistinct retinal layering | Small temporal crescent at V4e | NA | Undetectable at age 30 y | |
| F1517 | CIC03517 | F | 23 | Night blindness that developed when younger than 10 y; photophobia that developed in 20s | 20/160 OD; 20/160 OS | Age 32 y: HM OD; CF OS | High myopia (−18.0 OU); posterior subcapsular cataract in both eyes | Optic disc pallor; retinal vessel narrowing; atrophic maculae; sparse bone spicule–like pigments and patchy chorioretinal atrophy in the midperiphery | Hyperautofluorescence of the foveola; central area of granular hypoautofluorescence (tiny black dots), involving the fovea and parafovea; large perimacular halo of hyperautofluorescence | Widespread outer layer disappearance; indistinct retinal layering; developed a type 2 choroidal neovascular membrane (eFigure 6 in the Supplement) | V4e constricted to 5°; temporal crescent | Deutan | Undetectable dark-adapted 0.01 ERG, reduced; electronegative dark-adapted 3.0 and 10.0 ERG (b/a ratio, 0.75); light-adapted 3.0 and 30-Hz flicker ERG present, reduced, and delayed; ERG became undetectable at age 28 y |
| CIC05890 | M | 19 | Night blindness that developed when younger than 10 y; recent photophobia | NA | Age 19 y: CF OU | High myopia (−15.0 OU); posterior subcapsular cataract in both eyes | Optic disc pallor; retinal vessel narrowing; atrophic maculae; cellophane light reflex; sparse bone spicule–like pigmentation and patchy chorioretinal atrophy in the midperiphery | Central area of granular hypoautofluorescence (tiny black dots), involving the fovea and parafovea; large perimacular halo of hyperautofluorescence | Widespread outer layer disappearance; hyperreflective subfoveal deposit | NA | NA | NA | |
| F4639 | CIC08274 | M | 33 | Night blindness and photophobia that developed when younger than 10 y; visual acuity loss that developed in 20s | 20/400 OD; 20/800 OS | NA | Posterior subcapsular cataract in the right eye | Optic disc pallor and cupping; retinal vessels narrowing; pigmentary clumping in the macula and midperiphery | Round patches of atrophy (black holes) at the fovea; perifoveal hyperautofluorescent ring; round patches of hypoautofluorescence in the midperiphery | Outer layer disappearance; hyperreflective subretinal dots | V4e constricted to 20°; temporal crescents | NA | Undetectable at age 33 y |
| F5189 | CIC09088 | F | 38 | Night blindness that developed in the 20s; visual acuity loss that developed in the 30s | 20/250 OD; 20/250 OS | Age 42 y: 20/250 OD; 20/320 OS | Normal | Retinal vessels narrowing; macular depigmentation and atrophic changes; no pigmentary clumping | Central area of granular hypoautofluorescence (tiny black dots), involving the fovea and parafovea | Outer layer disappearance; hyperreflective subretinal dots | Absolute central scotoma of 5° at V4e; relative central scotoma of 30° at III1e; conserved peripheral isopters | Tritan | Dark-adapted ERG undetectable; light-adapted ERG severely reduced |
| 1037229 | M | 37 | Night blindness that developed in 20s; visual acuity loss that developed in 30s | 20/32 OD; 20/40 OS | Age 42 y: 20/63 OD; 20/125 OS | Normal | Retinal vessels narrowing; macular depigmentation and atrophic changes; no pigmentary clumping | Hyperautofluorescence of the foveola; central area of granular hypoautofluorescence (tiny black dots); involving the fovea and parafovea; irregular hypoautofluorescent patches in the midperiphery | Outer layer disappearance; hyperreflective subretinal dots | Relative central scotoma of 30° at III1e; conserved peripheral isopters | Tritan | Dark-adapted ERG undetectable; light-adapted ERG severely reduced | |
Abbreviations: BCVA, best-corrected visual acuity; CF, counting fingers; ERG, electroretinography; HM, hand motion; LP, light perception; MRI, magnetic resonance imaging; NA, not available; SD-OCT, spectral-domain optical coherence tomography; SW-FAF, short-wavelength fundus autofluorescence.
Isopter V4e: absolute visual field defect; isopter III4e: relative visual field defect (both requested in France for legal purposes).
In group B, 9 patients had a more severe retinal degeneration with macular atrophic changes (Figure 1C and D) (eFigure 4 in the Supplement). The onset of symptoms was earlier (age 10 to 20 years) than in group A, and BCVA was lower (mean [range] BCVA, 20/400 [20/160 to counting fingers]). For the Goldman visual field, 4 patients (CIC01168, CIC01170, CIC09088, and 1037229) exhibited central scotomata along with peripheral isopter constriction; 4 patients had an isopter constriction with residual temporal crescents (CIC00166, CIC01169, CIC08274, and CIC03517); and visual field was lacking for 1 patient (CIC05890) (Table 1). For color vision, 3 patients displayed a tritan axis, 1 patient had a deutan axis, and 1 patient had the color confusion errors without any axis; 4 patients could not be tested because they had a very low BCVA (Table 1). Fundus exhibited more widespread chorioretinal dystrophic changes, and all patients had atrophic changes with yellowish discoloration of the macula (particularly prominent in patients CIC01168, CIC01169, and CIC01170) along with grayish discoloration and subtle pigment migrations in the midperipheral retina (eFigure 4 in the Supplement). Optic disc pallor and retinal vessel narrowing were present in variable degrees but less obvious than in group A. SD-OCT revealed macular and extramacular outer retinal layer loss. Inner retinal layering was preserved; there was no visible alteration of the inner nuclear layer or the retinal nerve fiber layer thickness. A total of 7 patients had changes at the vitreoretinal interface; either an epiretinal membrane or retinal striae at the level of internal limiting membrane was also seen at fundoscopy (eFigure 4 in the Supplement). Short-wavelength fundus autofluorescence showed a macula of decreased autofluorescence in all patients surrounded by a large isoautofluorescent or hyperautofluorescent ring at the posterior pole with indistinct borders and patchy hypoautofluorescence outside the vascular arcades. In younger patients (Figure 1C) (eFigure 4 in the Supplement), macular hypoautofluorescence took a dotted, moth-eaten appearance while the extramacular retina looked isoautofluorescent. In more advanced cases (eFigure 4 in the Supplement), the fovea and extramacular retina were profoundly hypoautofluorescent and left between them an isoautofluorescent or slightly hyperautofluorescent ring. A total of 5 patients had undetectable ERG at the first assessment (Table 1). Patient CIC00166 had no response at the dark-adapted 0.01 ERG and dark-adapted 3.0 ERG but had a peculiar dark-adapted 10.0 ERG with reduced b/a ratio. Light-adapted ERG responses (light-adapted 3.0 ERG; light-adapted 30-Hz flicker ERG) were severely reduced (eFigure 5 in the Supplement). Patient CIC03517 had a clearly electronegative ERG (b wave less than a wave) with a reduced a wave; 5 years later, ERG became undetectable (eFigure 5 in the Supplement).
CLN3 variants are summarized in Table 2.10,11,20,21,22 Briefly, 11 variants in CLN3 were found, of which 4 were novel: the c.443_445del and c.906+15C>G variants identified in patient CIC09853 and variants c.938T>C p.(Leu313Pro) and c.1225A>G p.(Met409Val) in patients CIC00166 and CIC02713. All but 1 novel variant (c.906+15C>G, p.?) were classified as pathogenic or likely pathogenic in accordance with American College of Medical Genetics and Genomics standards.23
Table 2. CLN3 Variants in Study Cohort.
| Symbol | Location | Sequence variation | Protein change | ACMG interpretation | Comments | Source |
|---|---|---|---|---|---|---|
| M8 | Exon 7 | c.443_445dela | p.(Val148del)a | Likely pathogenic (IV: PM1, PM2, PM3, PM4, BP5) | rs752130042 gnomAD: MAF 0.00001592; never observed at homozygous state; allele count 4 | This cohort |
| M1 | Intron 7 | c.461-3C>G | p.? | Pathogenic (II: PS4, PP1-S, PM2, PM3, PP1) | rs181995380 gnomAD: MAF 0.000004311; never observed at homozygous state; allele count 1; reported only compound heterozygous; led to abnormal transcription; homozygous in patient CIC00350a | Ku et al,20 2017 |
| M2 | Intron 7_Intron 9 | Ex8_9del of 1.02 kilobase pairs (c.461-280_677+382del) | p.? | Pathogenic (Ia: PVS1, PS3, PS4, PP1-S, PM1, PM2, PM3, PP1-M, PP1) | Reported usually as Ex7_8del (exon 1 in 5′ untranslated region omitted); most frequent | Lerner et al,11 1995 |
| M7 | Exon 12 | c.868G>T | p.(Val290Leu) | Pathogenic (IIIa: PS4, PM1, PM2, PM3) | rs3690087702 gnomAD: MAF 0.000007953; never observed at homozygous state; allele count 2 | Wang et al,10 2014 |
| M10 | Exon12 | c.883G>A | p.(Glu295Lys) | Pathogenic (IIIa: PS4, PM1, PM2, PM3, PP2, PP3) | rs121434286 gnomAD: MAF 0.00002475; never observed at homozygous state; allele count 7 | Munroe et al,21 1997 |
| M11 | Intron 12 | c.906+15C>Ga | p.?a | Uncertain significance (PM2, PM3, PP2) | Unknown rs; new splicing donor site predicted: SpliceSiteFinder: 0→73 [0-100]; MaxEntScan: 0→4.7 [0-12] | This cohort |
| M5 | Exon 13 | c.938T>Ca | p.(Leu313Pro)a | Likely pathogenic (IV: PM1, PM2, PM3, PM6, PP2, PP3) | rs141816714 gnomAD: MAF 0.00003544; never observed at homozygous state; allele count 10; PolyPhen-2: probably damaging; SIFT: damaging; MutationTaster: disease causing | This cohort |
| M3 | Exon 14 | c.1000C>T | p.(Arg334Cys) | Pathogenic (II: PS4, PM1, PM2, PM3, PS1, PP2, PP3) | rs386833694 gnomAD: MAF 0.000004011; never observed at homozygous state; allele count 1 | Munroe et al,21 1997 |
| M9 | Intron 14 | c.1056+3A>C | p.? | Pathogenic (Ia: PVS1, PS3, PS4, PP1-S, PM1, PM2, PM3, PP1-M, PP1) | rs386833698; no frequency data | Lojewski et al,22 2014 |
| M4 | Exon 16 | c.1213C>T | p.(Arg405Trp) | Pathogenic (IIIa: PS4, PM1, PM2, PM3, PP2, PP3) | rs139842473 gnomAD: MAF 0.00006742; never observed at homozygous state; allele count 19 | Wang et al,10 2014 |
| M6 | Exon 16 | c.1225A>Ga | p.(Met409Val)a | Likely pathogenic (IV: PM1, PM2, PM3, PP3) | rs776443981 gnomAD: MAF 0.00003902; never observed at homozygous state; allele count 11; PolyPhen-2: probably damaging; SIFT: deleterious; MutationTaster: disease causing | This cohort |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; MAF, minor allele frequency; SIFT, Sorting Intolerant From Tolerant.
Novel variant.
Follow-up
All averaged BCVA measurements were plotted with age (eFigure 6 in the Supplement). All patients experienced a progressive BCVA reduction. Kaplan-Meier survival curves showing the survival distribution of BCVA of 1.3 logMAR or greater (20/400 Snellen equivalent; legal blindness threshold) were significantly different between the 2 phenotypic groups. A total of 3 of 6 patients from group A (50%) reached the legal blindness threshold by their 70s. A total of 11 patients from group B (70%) reached the legal blindness threshold by their 40s. Long-term follow-up data were available for patients CIC03517 (eFigure 7 in the Supplement), CIC00350 (eFigure 8 in the Supplement), and patients from families F699 (eFigures 9 to 11 in the Supplement) and F5189 (eFigures 12 and 13 in the Supplement).
Discussion
We report here 15 patients with isolated retinal degeneration presenting with biallelic variants in CLN3. A total of 11 distinct variants were found in this study, of which 4 were novel. To our knowledge, this is the largest series of patients with isolated CLN3-linked IRD reported so far. Biallelic CLN3 variants are usually associated with a severe neurometabolic disorder, JNCL.11 The classic form of the disease begins by visual symptoms between ages 4 and 6 years, months prior to the onset of neurological symptoms.12
Recent studies reported patients with isolated retinal degeneration and biallelic CLN3 variants.10,20,24,25 Our clinical data are mostly in line with these reports, with a further distinction of 2 phenotypic groups. Group A is characterized by a mild course of rod-cone degeneration, with an onset of symptoms between ages 20 and 40 years, a relatively preserved posterior pole and thus better visual acuity, and a slow progression, reaching legal blindness by the 70s. Group B shows more severe retinal degeneration with early macular atrophy, an onset of symptoms in the 10s or early 20s, rapid visual acuity loss, and legal blindness being reached by their 40s. Of note, there was no high prevalence of cystoid macular changes in our series (only 3 of 15 patients), in contrast with previous reports.20
Interestingly, cases from 2 families showed retinal findings reminiscent of JNCL with an additional inner retinal dysfunction to photoreceptor disease characteristic of JNCL-associated retinopathy.16 SD-OCT findings in JNCL-associated retinopathy may also include macular outer nuclear layer and ellipsoid zone disruption along with inner nuclear layer thinning.18 This latter change was not found in patients with isolated IRD in this study. The presence of striated macular pattern, found in 7 patients in this series, is another confounding feature. The radial macular striae at the vitreoretinal interface (either with or without evidence of epiretinal membrane on OCT) are a common finding in JNCL.17 However, unlike JNCL, the first symptoms of retinal degeneration in these patients occurred later, by the end of the second decade of life.
CLN3 is located in 16p12.1 (Figure 2) and encodes battenin. It is expressed in all retinal cell types, but the immunochemical staining is more intense in Müller cells, bipolar cells, and the inner segment of photoreceptors.26
Figure 2. Spectrum of Variants in CLN3.
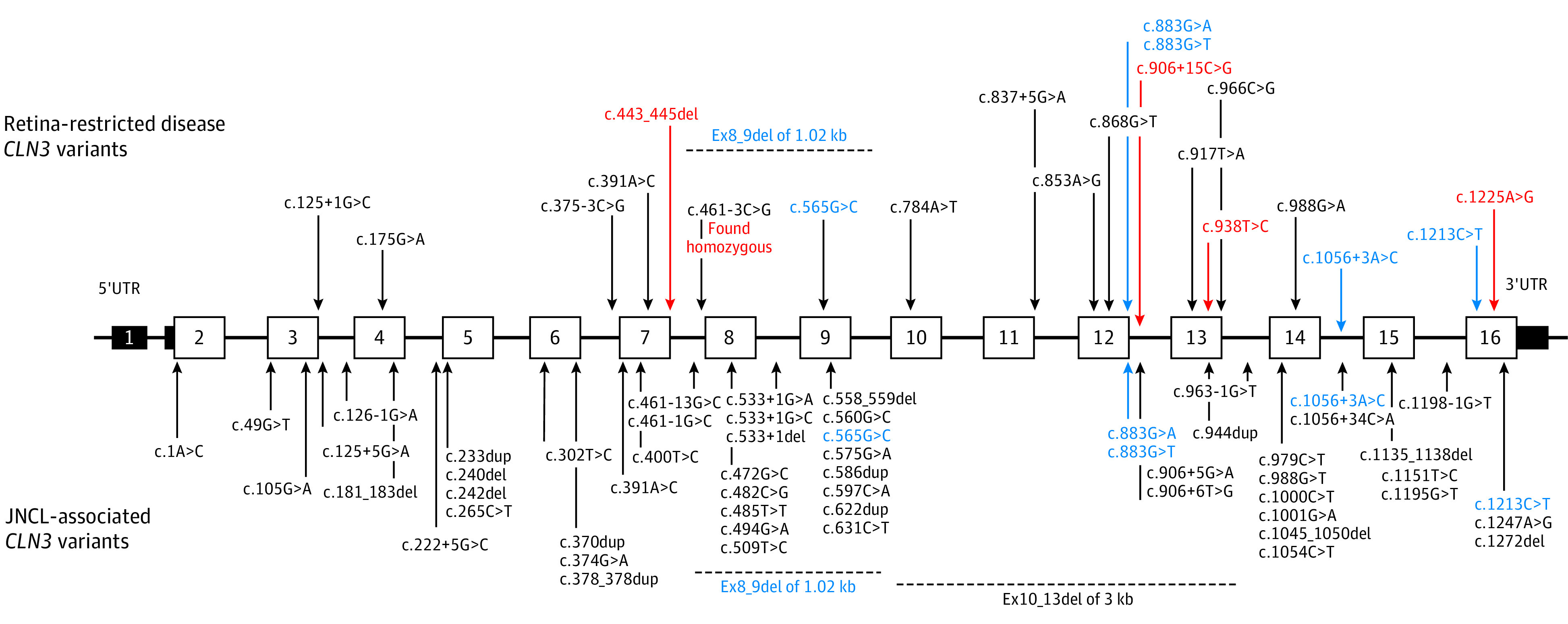
Variants reported in retina-restricted disease (top) and in juvenile neuronal ceroid lipofuscinosis (JNCL; bottom). Most variants are clearly distinct. Blue indicates variants reported in both retina-restricted disease and JNCL. Red indicates novel variants. UTR indicates untranslated region.
Battenin is a transmembrane protein containing 438 amino acids (eFigure 14 in the Supplement). The protein has not been crystallized yet; the topology is predicted in silico.27,28,29 Second cytoplasmic loop and C-terminal domain contain the lysosomal targeting motifs, responsible for correct addressing of battenin from the Golgi apparatus to lysosome.30,31 The exact cellular function of battenin is still unclear but appears to be linked with vesicular addressing and trafficking, posttranslational protein modification, autophagy, and overall lysosomal function.29,32,33 Variants involved in nonsyndromic retinal degeneration were reported to cluster near the fifth transmembrane domain and at the C-terminal end of the protein,20 but the same segment also harbors amino acid exchanges leading to JNCL. Based on the literature and the current study, an attempt of phenotype–genotype correlation can be drawn from confronting the clinical severity of CLN3-associated disease (both isolated retinopathy and JNCL) and the type of sequence variants (Figure 3).10,11,16,20,21,34,35,36,37,38,39
Figure 3. Phenotype–Genotype Correlations in Juvenile Neuronal Ceroid Lipofuscinosis (JNCL) and CLN3-Related Retina-Restricted Diseases.
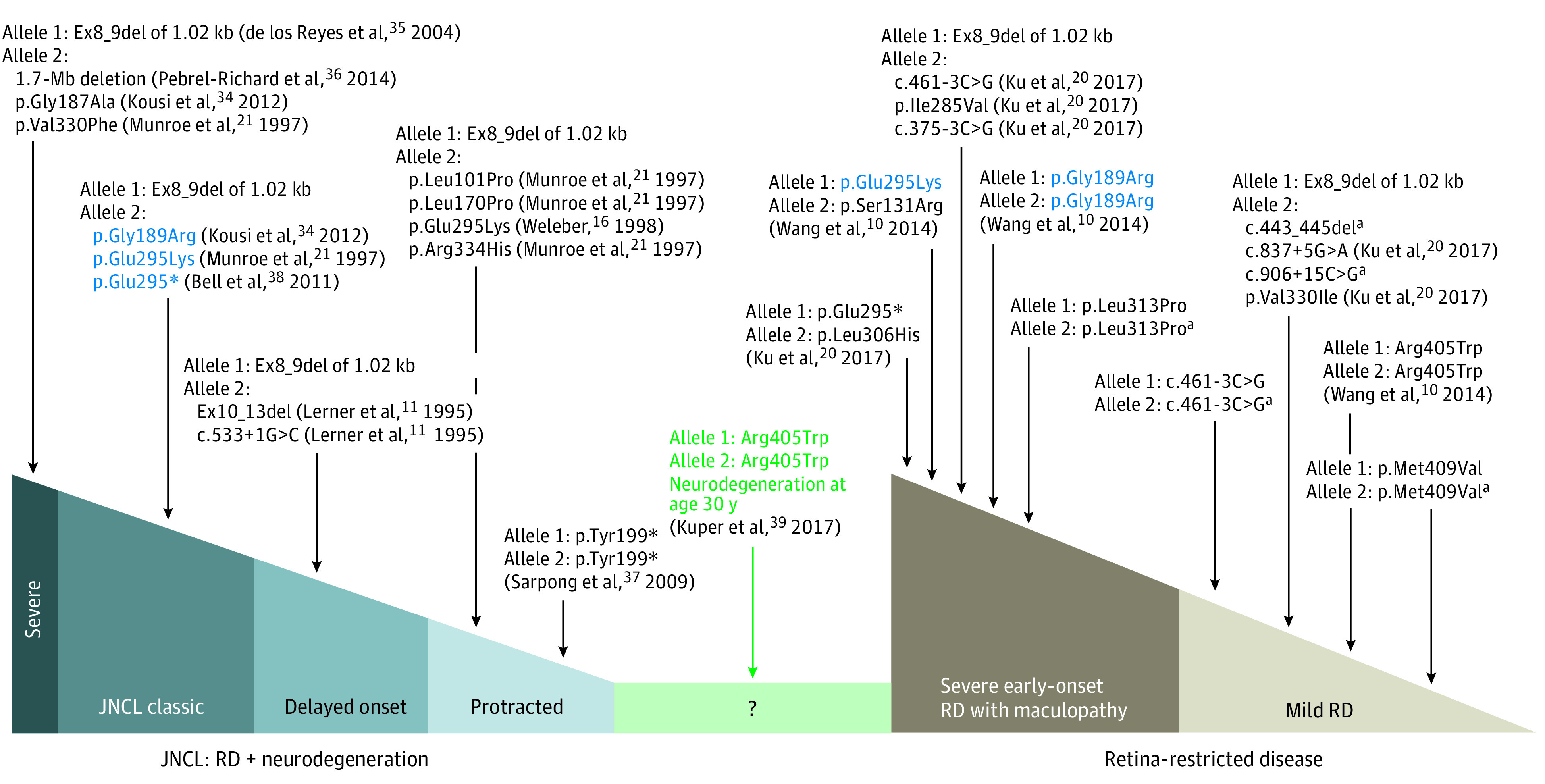
The patient harboring the p.(Arg405Trp) variant (in green) was recently reported to develop a late neurological involvement suggestive of JNCL. Blue indicates variants reported in both JNCL and retina-restricted disease. kb indicates kilobase pairs; Mb, megabase pairs; RD, retinal degeneration.
aNovel variant.
JNCL-Associated Retinopathy
A spectrum of severity is reported for JNCL associated with CLN3 variants with an early onset of disease around age 4 to 6 years. The most common pathogenic variant in CLN3, Ex8_9del of 1.02 kilobase pairs, is detected overall in 96% of patients with JNCL.21,34 Infantile-onset severe forms of JNCL were described in patients harboring Ex8_9del35 associated with the following variants in trans: 16p11.2 1.7–megabase pairs deletion including CLN3 and 2 point variants,36 c.560G>C (p.Gly187Ala)34 and c.988G>T (p.Val330Phe).21 The delayed-onset JNCL (with visual and neurological symptoms by age 8 to 10 years) was reported in a small number of patients with compound heterozygous variants in CLN3, including the common Ex8_9del and Ex10_13del or c.553+1G>C in trans.11 Protracted-onset JNCL (neurological symptoms occur years later after initial visual involvement) is also described with compound heterozygous variants, including the Ex8_9del as one allele and the following variations as the second allele: c.1A>C, c.302T>C p.(Leu101Pro), c.509T>C p.(Leu170Pro), c.883G>A p.(Glu295Lys), or c.1000C>T p.(Arg334His) or with c.597C>A p.(Tyr199*) in a homozygous state.37
CLN3-Associated Isolated Retinal Degeneration
Milder forms of retinal degeneration (group A) are associated with homozygous point variants affecting the lysosomal targeting motives of battenin, ie, c.1225A>G p.(Met409Val) and c.1213C>T p.(Arg405Trp),10 presumably precluding a normal trafficking of battenin to the lysosome. Similarly, the homozygous c.461-3C>G variant, which is probably a leaky variant with decreased but persistent expression, is associated with a milder phenotype.20 A mild phenotype is also found in genotypes associating the common Ex8_9del and the following point variants: c.837+5G>A, c.988G>A p.(Val330Ile),20 c.175G>A p.?,25 and c.906+15C>G p.?/c.443_445del p.(Val148del). In the latter case (patient CIC09853), one of the variants (c.443_445del) is predicted to be deleterious in silico and the second (c.906+15C>G) is of uncertain significance. More severe and earlier-onset forms of isolated retinal degeneration (group B) occur in homozygous and compound point biallelic variants of CLN3 coding sequence but also when an intronic variant (c.375-3C>G, c.461-3C>G) is associated with Ex8_9del in trans.
Three point variants—c.565G>C p.(Gly189Arg), c.883G>A p.(Glu295Lys), and c.883G>T p.(Glu295*)—are reported both in JNCL and isolated retinal disease.10,20,21,34,38 More specifically, compound heterozygous association of these variants with Ex8_9del give rise to JNCL, while isolated retinal phenotype is seen if any of these variants is in trans with another point variant, specific for only retinal involvement (eTable in the Supplement). Another intronic variant, c.1056+3A>C, was previously reported only in JNCL in association with the c.1247A>G variant34 or with Ex2_5del.17 We found this intronic variant in 2 affected members of the family F5189 along with c.938C>T p.(Leu313Pro). The latter variant, homozygous in patient CIC00166, also led to isolated retinal degeneration.
Of note, phenotype–genotype correlations in CLN3 variants are imperfect, and caution should be given to a falsely reassuring retina-restricted diagnosis. Although none of these patients manifested CLN3-related neurological symptoms (including a woman in her 70s who only manifested extrapyramidal iatrogenic symptoms according to the neurologist), a 2017 report mentioned 2 siblings with an initial isolated retinal disease carrying the homozygous c.1213C>T p.(Arg405Trp) variant (Figure 3) who developed neurological symptoms reminiscent of NCL in their 30s.39 The p.(Arg405Trp) variant was first reported in isolated retinal disease, both as homozygous and compound heterozygous with Ex8_9del in trans.10,20,40 In our series, 3 patients harbored the p.(Arg405Trp) variant, either with Ex8_9del or c.883G>A and c.1000C>T missense variants in trans. None of them have had extraocular involvement to date, but the patients are younger than 40 years. A very protracted course of ceroid lipofuscinosis, compatible with the diagnosis of Kufs disease (adult-onset ceroid lipofuscinosis),41,42,43 is in the spectrum of clinical manifestations of biallelic CLN3 variants. Thus, the long-term prognosis for development of the neurologic condition is very difficult if impossible to predict for these cases carrying CLN3 variants.
Limitations
Our study has some limitation. Biases from retrospective analyses of the collected data are obvious. Due to the limited number of cases, we could not realize a statistical analysis of phenotype–genotype correlations. Larger series with meta-analysis of previously published data should be performed to better understand the pathology and phenotype–genotype correlations. Functional studies of novel CLN3 variants could also be beneficial for deciphering the retinal degeneration pathophysiological mechanism.
To date, both JNCL and CLN3-associated isolated retinal degeneration are untreatable conditions.44 Gene replacement approaches in animals showed efficacy and safety both through systemic administration for neurological disease45 and through intraocular delivery for retinal degeneration.46 A phase 1/2 gene therapy study in human is under way to evaluate intrathecal injection of AAV9-CLN3 in pediatric patients with JNCL.47 Isolated retinal degeneration linked with CLN3 variants could also be a good target for intraocular gene delivery due to the simplicity of tissue access and functional assessment.
Conclusions
Biallelic CLN3 variants are responsible for a spectrum of diseases, varying from severe neurodegeneration to retina-restricted disease. These findings support adding CLN3 to NGS panels investigating presumed cases with IRD.48 Phenotype–genotype correlations can be drawn, but more data are needed to refine them. Caution should be taken regarding potential neurological outcome, and a long-term follow-up for patients with apparent retina-restricted disease linked with CLN3 variants is required. A better understanding of phenotype–genotype correlations would serve as a basis for the development of a CLN3 gene therapy.
eMethods.
eFigure 1. Pedigrees.
eFigure 2. qPCR for ex.8_ex.9 del.
eFigure 3. Mild forms of CLN3 isolated retinal degeneration.
eFigure 4. Severe forms of CLN3 isolated retinal degeneration.
eFigure 5. Full-field ERG.
eFigure 6. BCVA progression.
eFigure 7. CIC03517, progression.
eFigure 8. CIC00350, progression.
eFigure 9. CIC01170, progression.
eFigure 10. CIC01169, progression.
eFigure 11. CIC01168, progression.
eFigure 12. 1037229, progression.
eFigure 13. CIC09088, progression.
eFigure 14. Battenin.
eTable. CLN3 variants reported both in JNCL and isolated retinal degeneration.
eReferences.
References
- 1.Verbakel SK, van Huet RAC, Boon CJF, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157-186. doi: 10.1016/j.preteyeres.2018.03.005 [DOI] [PubMed] [Google Scholar]
- 2.Abu Diab A, AlTalbishi A, Rosin B, et al. The combination of whole-exome sequencing and clinical analysis allows better diagnosis of rare syndromic retinal dystrophies. Acta Ophthalmol. 2019;97(6):e877-e886. doi: 10.1111/aos.14095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X, Feng Y, Li J, et al. Retinal diseases caused by mutations in genes not specifically associated with the clinical diagnosis. PLoS One. 2016;11(10):e0165405. doi: 10.1371/journal.pone.0165405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daiger SP; The University of Texas Health Science Center . RetNet: Retinal Information Network. Accessed August 28, 2020. https://sph.uth.edu/retnet/
- 5.Karali M, Testa F, Brunetti-Pierri R, et al. Clinical and genetic analysis of a European cohort with pericentral retinitis pigmentosa. Int J Mol Sci. 2019;21(1):E86. doi: 10.3390/ijms21010086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estrada-Cuzcano A, Koenekoop RK, Senechal A, et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch Ophthalmol. 2012;130(11):1425-1432. doi: 10.1001/archophthalmol.2012.2434 [DOI] [PubMed] [Google Scholar]
- 7.Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res. 2004;79(2):167-173. doi: 10.1016/j.exer.2004.03.005 [DOI] [PubMed] [Google Scholar]
- 8.Ávila-Fernández A, Cantalapiedra D, Aller E, et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis. 2010;16:2550-2558. [PMC free article] [PubMed] [Google Scholar]
- 9.McGee TL, Seyedahmadi BJ, Sweeney MO, Dryja TP, Berson EL. Novel mutations in the long isoform of the USH2A gene in patients with Usher syndrome type II or non-syndromic retinitis pigmentosa. J Med Genet. 2010;47(7):499-506. doi: 10.1136/jmg.2009.075143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Wang H, Tuan H-F, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133(3):331-345. doi: 10.1007/s00439-013-1381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lerner TJ, Boustany R-MN, Anderson JW, et al. ; The International Batten Disease Consortium . Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82(6):949-957. doi: 10.1016/0092-8674(95)90274-0 [DOI] [PubMed] [Google Scholar]
- 12.Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews. University of Washington; 1993. Accessed November 24, 2019. https://www.ncbi.nlm.nih.gov/books/NBK1428/ [Google Scholar]
- 13.Geraets RD, Koh Sy, Hastings ML, Kielian T, Pearce DA, Weimer JM. Moving towards effective therapeutic strategies for neuronal ceroid lipofuscinosis. Orphanet J Rare Dis. 2016;11(1):40. doi: 10.1186/s13023-016-0414-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santorelli FM, Garavaglia B, Cardona F, et al. Molecular epidemiology of childhood neuronal ceroid-lipofuscinosis in Italy. Orphanet J Rare Dis. 2013;8:19. doi: 10.1186/1750-1172-8-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mink JW, Augustine EF, Adams HR, Marshall FJ, Kwon JM. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28(9):1101-1105. doi: 10.1177/0883073813494268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond). 1998;12(pt 3b):580-590. doi: 10.1038/eye.1998.148 [DOI] [PubMed] [Google Scholar]
- 17.Wright GA, Georgiou M, Robson AG, et al. Juvenile Batten disease (CLN3): detailed ocular phenotype, novel observations, delayed diagnosis, masquerades, and prospects for therapy. Ophthalmol Retina. 2020;4(4):433-445. doi: 10.1016/j.oret.2019.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Preising MN, Abura M, Jäger M, Wassill K-H, Lorenz B. Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life. Ophthalmic Genet. 2017;38(3):252-259. doi: 10.1080/13816810.2016.1210651 [DOI] [PubMed] [Google Scholar]
- 19.Audo I, Friedrich A, Mohand-Saïd S, et al. An unusual retinal phenotype associated with a novel mutation in RHO. Arch Ophthalmol. 2010;128(8):1036-1045. doi: 10.1001/archophthalmol.2010.162 [DOI] [PubMed] [Google Scholar]
- 20.Ku CA, Hull S, Arno G, et al. Detailed clinical phenotype and molecular genetic findings in CLN3-associated isolated retinal degeneration. JAMA Ophthalmol. 2017;135(7):749-760. doi: 10.1001/jamaophthalmol.2017.1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munroe PB, Mitchison HM, O’Rawe AM, et al. Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet. 1997;61(2):310-316. doi: 10.1086/514846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lojewski X, Staropoli JF, Biswas-Legrand S, et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. 2014;23(8):2005-2022. doi: 10.1093/hmg/ddt596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carss KJ, Arno G, Erwood M, et al. ; NIHR-BioResource Rare Diseases Consortium . Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100(1):75-90. doi: 10.1016/j.ajhg.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen FK, Zhang X, Eintracht J, et al. Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc Ophthalmol. 2019;138(1):55-70. doi: 10.1007/s10633-018-9665-7 [DOI] [PubMed] [Google Scholar]
- 26.Katz ML, Gao CL, Prabhakaram M, Shibuya H, Liu PC, Johnson GS. Immunochemical localization of the Batten disease (CLN3) protein in retina. Invest Ophthalmol Vis Sci. 1997;38(11):2375-2386. [PubMed] [Google Scholar]
- 27.Cotman SL, Staropoli JF. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin Lipidol. 2012;7(1):79-91. doi: 10.2217/clp.11.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nugent T, Mole SE, Jones DT. The transmembrane topology of Batten disease protein CLN3 determined by consensus computational prediction constrained by experimental data. FEBS Lett. 2008;582(7):1019-1024. doi: 10.1016/j.febslet.2008.02.049 [DOI] [PubMed] [Google Scholar]
- 29.Mirza M, Vainshtein A, DiRonza A, et al. The CLN3 gene and protein: what we know. Mol Genet Genomic Med. 2019;7(12):e859. doi: 10.1002/mgg3.859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kyttälä A, Ihrke G, Vesa J, Schell MJ, Luzio JP. Two motifs target Batten disease protein CLN3 to lysosomes in transfected nonneuronal and neuronal cells. Mol Biol Cell. 2004;15(3):1313-1323. doi: 10.1091/mbc.e03-02-0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyttälä A, Yliannala K, Schu P, Jalanko A, Luzio JP. AP-1 and AP-3 facilitate lysosomal targeting of Batten disease protein CLN3 via its dileucine motif. J Biol Chem. 2005;280(11):10277-10283. doi: 10.1074/jbc.M411862200 [DOI] [PubMed] [Google Scholar]
- 32.Yasa S, Modica G, Sauvageau E, Kaleem A, Hermey G, Lefrancois S. CLN3 regulates endosomal function by modulating Rab7A-effector interactions. J Cell Sci. 2020;133(6):jcs234047. doi: 10.1242/jcs.234047 [DOI] [PubMed] [Google Scholar]
- 33.Metcalf DJ, Calvi AA, Seaman MNj, Mitchison HM, Cutler DF. Loss of the Batten disease gene CLN3 prevents exit from the TGN of the mannose 6-phosphate receptor. Traffic. 2008;9(11):1905-1914. doi: 10.1111/j.1600-0854.2008.00807.x [DOI] [PubMed] [Google Scholar]
- 34.Kousi M, Lehesjoki A-E, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33(1):42-63. doi: 10.1002/humu.21624 [DOI] [PubMed] [Google Scholar]
- 35.de los Reyes E, Dyken PR, Phillips P, et al. Profound infantile neuroretinal dysfunction in a heterozygote for the CLN3 genetic defect. J Child Neurol. 2004;19(1):42-46. doi: 10.1177/08830738040190010703 [DOI] [PubMed] [Google Scholar]
- 36.Pebrel-Richard C, Debost-Legrand A, Eymard-Pierre E, et al. An unusual clinical severity of 16p11.2 deletion syndrome caused by unmasked recessive mutation of CLN3. Eur J Hum Genet. 2014;22(3):369-373. doi: 10.1038/ejhg.2013.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarpong A, Schottmann G, Rüther K, et al. Protracted course of juvenile ceroid lipofuscinosis associated with a novel CLN3 mutation (p.Y199X). Clin Genet. 2009;76(1):38-45. doi: 10.1111/j.1399-0004.2009.01179.x [DOI] [PubMed] [Google Scholar]
- 38.Bell CJ, Dinwiddie DL, Miller NA, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra4. doi: 10.1126/scitranslmed.3001756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuper WFE, van Alfen C, van Eck L, et al. A case of unexpected adult-onset neurologic decline in CLN3-associated retinal degeneration. JAMA Ophthalmol. 2017;135(12):1451-1453. doi: 10.1001/jamaophthalmol.2017.4353 [DOI] [PubMed] [Google Scholar]
- 40.Stone EM, Andorf JL, Whitmore SS, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124(9):1314-1331. doi: 10.1016/j.ophtha.2017.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berkovic SF, Carpenter S, Andermann F, Andermann E, Wolfe LS. Kufs’ disease: a critical reappraisal. Brain. 1988;111(pt 1):27-62. doi: 10.1093/brain/111.1.27 [DOI] [PubMed] [Google Scholar]
- 42.Tyynelä J, Lehesjoki A-E. Kufs or not Kufs: challenging diagnostics of a rare adult-onset neurodegenerative disease. Brain. 2019;142(1):2-5. doi: 10.1093/brain/awy312 [DOI] [PubMed] [Google Scholar]
- 43.Berkovic SF, Oliver KL, Canafoglia L, et al. Kufs disease due to mutation of CLN6: clinical, pathological and molecular genetic features. Brain. 2019;142(1):59-69. doi: 10.1093/brain/awy297 [DOI] [PubMed] [Google Scholar]
- 44.Mole SE, Anderson G, Band HA, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019;18(1):107-116. doi: 10.1016/S1474-4422(18)30368-5 [DOI] [PubMed] [Google Scholar]
- 45.Bosch ME, Aldrich A, Fallet R, et al. Self-complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis (CLN3). J Neurosci. 2016;36(37):9669-9682. doi: 10.1523/JNEUROSCI.1635-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wiley LA, Burnight ER, Drack AV, et al. Using patient-specific induced pluripotent stem cells and wild-type mice to develop a gene augmentation-based strategy to treat CLN3-associated retinal degeneration. Hum Gene Ther. 2016;27(10):835-846. doi: 10.1089/hum.2016.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gene therapy for children with CLN3 Batten disease. ClinicalTrials.gov identifier: NCT03770572. Updated March 25, 2020. Accessed August 28, 2020. https://clinicaltrials.gov/ct2/show/NCT03770572
- 48.Turriff AE, Cukras CA, Brooks BP, Huryn LA. Considerations in multi-gene panel testing in pediatric ophthalmology. J AAPOS. 2019;23(3):163-165.e1. doi: 10.1016/j.jaapos.2019.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eFigure 1. Pedigrees.
eFigure 2. qPCR for ex.8_ex.9 del.
eFigure 3. Mild forms of CLN3 isolated retinal degeneration.
eFigure 4. Severe forms of CLN3 isolated retinal degeneration.
eFigure 5. Full-field ERG.
eFigure 6. BCVA progression.
eFigure 7. CIC03517, progression.
eFigure 8. CIC00350, progression.
eFigure 9. CIC01170, progression.
eFigure 10. CIC01169, progression.
eFigure 11. CIC01168, progression.
eFigure 12. 1037229, progression.
eFigure 13. CIC09088, progression.
eFigure 14. Battenin.
eTable. CLN3 variants reported both in JNCL and isolated retinal degeneration.
eReferences.