

Abstract
PREMISE
New sequencing technologies facilitate the generation of large‐scale molecular data sets for constructing the plant tree of life. We describe a new probe set for target enrichment sequencing to generate nuclear sequence data to build phylogenetic trees with any flagellate land plants, including hornworts, liverworts, mosses, lycophytes, ferns, and all gymnosperms.
METHODS
We leveraged existing transcriptome and genome sequence data to design the GoFlag 451 probes, a set of 56,989 probes for target enrichment sequencing of 451 exons that are found in 248 single‐copy or low‐copy nuclear genes across flagellate plant lineages.
RESULTS
Our results indicate that target enrichment using the GoFlag451 probe set can provide large nuclear data sets that can be used to resolve relationships among both distantly and closely related taxa across the flagellate land plants. We also describe the GoFlag 408 probes, an optimized probe set covering 408 of the 451 exons from the GoFlag 451 probe set that is commercialized by RAPiD Genomics.
CONCLUSIONS
A target enrichment approach using the new probe set provides a relatively low‐cost solution to obtain large‐scale nuclear sequence data for inferring phylogenetic relationships across flagellate land plants.
Keywords: flagellate plants, next‐generation sequencing, nuclear loci, phylogenomics, target enrichment
For the first ~300 million years after the movement of plants to land, Earth’s terrestrial flora consisted of flagellate plants, or plants with mobile flagellate male gametes (i.e., spermatozoids). The modern descendants of these lineages that have retained flagellate sperm include the hornworts, liverworts, mosses, lycophytes, ferns, and some gymnosperms, which together comprise approximately 30,000 extant species. During the evolution of these groups, numerous anatomical innovations arose, including stomata, vascular tissue, roots and leaves, lignified stems with secondary growth, and seeds. Collectively, these plants hold the keys to understanding the early evolution of these and other critical features of modern land plant diversity, which is overwhelmingly represented by non‐flagellate angiosperms. The phylogenetic relationships among many flagellate land plant taxa remain poorly understood, and the lack of a consistent molecular toolkit makes resolving these relationships difficult.
Analyses of large numbers of nuclear loci can provide the power to resolve difficult phylogenetic relationships and the ability to address patterns of lineage sorting and reticulate evolution. Recent analyses of single‐copy nuclear genes from transcriptome data have provided insights into backbone relationships among flagellate plants (Wickett et al., 2014; Qi et al., 2018; Shen et al., 2018; Leebens‐Mack et al., 2019a). However, transcriptome sequencing requires access to freshly collected tissue, is often expensive and impractical, and many loci are either not useful for phylogenetics or only expressed in specific tissues or stages of development. Therefore, although transcriptomic sequencing can provide much information about candidate loci for phylogenetics, it may be difficult to scale up for systematic studies with extensive species‐level sampling (see McKain et al., 2018). Target enrichment methods use short RNA (or occasionally DNA) probes, corresponding to selected loci, to bind to DNA (or RNA) from sequencing libraries. The bound DNA is then sequenced, while much of the unbound DNA is discarded (Gnirke et al., 2009; Cronn et al., 2012; Weitemier et al., 2014). Target enrichment approaches can be used to obtain data from hundreds of phylogenetically informative nuclear loci at relatively low cost. These approaches also appear to work well even with low‐quantity, potentially degraded DNA samples, like those extracted from herbarium specimens (see Brewer et al., 2019; Forrest et al., 2019). Target enrichment approaches have been used to generate nuclear data sets to resolve relationships within several flagellate land plant clades, including mosses (Liu et al., 2019; Medina et al., 2019), ferns (Wolf et al., 2018), and pines (Gernandt et al., 2018; Montes et al., 2019). However, no probe sets exist that can generate nuclear data sets for all flagellate land plant taxa.
In this study, we leveraged recent transcriptome data from the One Thousand Plant Transcriptomes Initiative (1KP; see Leebens‐Mack et al., 2019a) and whole genome sequence data, mostly available in Phytozome (https://phytozome.jgi.doe.gov/), to design a “universal” probe set that enables target enrichment sequencing across all flagellate land plant lineages, as well as the non‐flagellate gymnosperms. The probes target 451 relatively conserved exons found within 248 single‐ or low‐copy nuclear loci. Furthermore, the target enrichment protocol typically also yields sequence data from the more variable flanking regions that may be useful to resolve relationships among closely related taxa. We demonstrate the target enrichment protocol using representatives from all major flagellate land plant lineages and provide an analytical pipeline to process the resulting data. We also describe an optimized probe set covering 408 of the 451 original exons.
METHODS
Probe design
We designed target enrichment probes to cover all flagellate land plant groups, including mosses, liverworts, hornworts, lycophytes, ferns, and all gymnosperms (including non‐flagellate gnetophytes and conifers), using existing genomic and transcriptomic data. We designed probes to cover conserved exons among a set of 410 single‐copy (or low‐copy) nuclear genes identified by the 1KP initiative (Carpenter et al., 2019; Leebens‐Mack et al., 2019b), which consists of assembled transcriptomes from 1173 green plant species, including 241 flagellate land plant samples (Appendix S1). We examined 23 available genome sequences from land plant taxa to identify exons that belong to the single‐copy loci identified by 1KP that were at least 120 bp in length (Appendix S2), and used a pairwise BLAST search of selected exons to find those shared across multiple genomes that had at least 65% average pairwise identity. Only regions represented across at least six of the 23 genomes from at least three selected taxonomic groups (i.e., bryophytes, lycophytes, gymnosperms, “basal” angiosperms, monocots, “basal” eudicots, Caryophyllales, asterids, and rosids; Appendix S2), indicating conservation of exon content and splice sites across a large diversity of land plants, were used to design probes. For the probe kit, we identified the best 451 loci (i.e., exons) that met our criteria. In some cases, multiple exons used in the probe set are found within the same gene; in total, the 451 exons we used are found in 248 genes (see Appendix S3). Data regarding the number of 1KP samples found for each locus, the pairwise identity and length of loci, and the genomes that contained each locus are available in Dryad (Breinholt et al., 2020).
We aligned and then cut the 451 exonic target loci out of the 1KP alignments that included only the flagellate plant taxa. These alignments include data from 244 flagellate land plant samples (241 from transcriptomes and three from genomes), including eight hornworts, 22 liverworts, 42 mosses, 22 lycophytes, 69 ferns, and 81 gymnosperms (Appendix S1). We clustered the cut sequences for each locus at 90% similarity and took the centroid sequence of each cluster using UCLUST (Edgar, 2010). We designed the probe set from these sequences with a 2× tiling density. The resulting GoFlag 451 probe set consists of 56,989 probes covering 451 loci and is available on Dryad (Breinholt et al., 2020). The term GoFlag refers to the Genealogy of Flagellate plants project, which was funded through the U.S. National Science Foundation Genealogy of Life (GoLife) program. The average length of reference sequences was 186.5 bp, for a total average length of 84,107.9 bp.
These exonic target loci generally have little overlap with target loci from previous flagellate plant targeted enrichment probe sets. Using a BLAST search of the 1KP reference loci, we determined that five of the 451 target exons are found within three of the 25 genes covered by the Wolf et al. (2018) fern probe set, and 17 of the 451 target exons are found within eight of the 105 nuclear genes covered by the Liu et al. (2019) moss probe set, which also includes probes for plastid and mitochondrial loci (Appendix S3). However, there is much overlap between the 451 target exons and the 353 genes covered by the angiosperm probe set of Johnson et al. (2019); 422 of the 451 target exons are found within 228 of the 353 genes in the Johnson et al. (2019) probe set (Appendix S3). This overlap is perhaps not surprising because both probe sets were designed from the 1KP single‐copy gene data set.
Although the GoFlag 451 probe set only includes probes for flagellate land plants, it was designed from conserved alignments across all land plants. Therefore, to test whether data from the 451 exons can resolve relationships across land plants, we extracted these exons from the 1KP translated nucleotide alignments and removed sequences from non‐land plants from the alignments. We concatenated the exon alignments into a supermatrix and ran a maximum likelihood (ML) search with 100 nonparametric bootstrap (BS) replicates using RAxML 8.2.10 with the GTR CAT model (Stamatakis, 2014). Alignments for this analysis also are available on Dryad (Breinholt et al., 2020).
Taxon selection
We assembled a collection of 188 samples of flagellate land plants for our pilot study (Appendix S3). These include representatives of major clades within hornworts (14), liverworts (46), mosses (48), lycophytes (16), ferns (48), and gymnosperms (16). Within these groups we also included some sets of closely related taxa (e.g., congeners) to test the probe set’s ability to resolve close relationships (see Appendix S4 for voucher information). Some of these samples came from herbarium specimens, whereas others were from recently collected silica‐dried tissue. We extracted DNA using a cetyltrimethylammonium bromide (CTAB) extraction as described in Doyle and Doyle (1987), modified for 2‐mL extractions, using a Geno/Grinder 2010 mill (SPEX CertiPrep, Metuchen, New Jersey, USA), and with 2.5% polyvinylpyrrolidone and 0.4% beta‐mercaptoethanol, and two rounds of chloroform washes followed by an isopropanol precipitation and an ethanol wash. To remove RNA contamination, between chloroform washes we added 2 μL of 10 mg/mL RNase A (QIAGEN, Valencia, California, USA) to each sample.
Sequence capture and sequencing
The library construction, target enrichment, and sequencing were done by RAPiD Genomics (Gainesville, Florida, USA). After a bead‐based DNA cleanup step, DNA was normalized to 250 ng and mechanically sheared to an average size of 300 bp. We constructed Illumina‐comparable libraries by repairing the ends of the sheared fragments followed by the addition of an adenine residue to the 3′‐end of the blunt‐end fragments (Bentley et al., 2008). Next, we ligated barcoded adapters suited for the Illumina sequencing platform to the libraries. Ligated fragments were PCR‐amplified for 9–11 cycles. We pooled 16 barcoded libraries equimolarly to a total of 500 ng for hybridization. Target enrichment was performed using the custom‐designed probes and protocols modified from Gnirke et al. (2009). After enrichment, samples were re‐amplified for an additional 6–12 cycles. All enriched samples were sequenced using an Illumina HiSeq 3000 (Illumina, San Diego, California, USA) with paired‐end 100‐bp reads. The sequence reads were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA; BioProject PRJNA630729).
Bioinformatic and phylogenetic analyses
Targeted nuclear exon loci were recovered from enriched Illumina data using a modified version of the iterative baited assembly pipeline described by Breinholt et al. (2018). This six‐step pipeline, with all scripts and necessary input files, is available in Dryad (Breinholt et al., 2020). In step 1 (trim reads), adapters and bases with Phred scores less than 20 were trimmed from paired‐end reads with Trim Galore! version 0.4.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Only pairs of reads in which both the forward and reverse read were at least 30 bp long were retained for assembly. In step 2 (assembly), the targeted loci were assembled using iterative baited assembly (IBA) implemented in a previously published Python script (IBA.py; Breinholt et al., 2017, 2018). For each locus, the script first finds raw reads with significant homology to the exonic target region based on the reference transcriptome sequences from 1KP data and whole genome sequences (Appendix S1) using USEARCH version 7.0 (Edgar, 2010) and then performs an iterative de novo assembly with the subset of reads for each locus with BRIDGER version 2014‐12‐01 (Chang et al., 2015). In the IBA script, we set the BRIDGER k‐mer size parameter to 25 and the minimum depth of coverage for the k‐mers to be included in the assembly to 10. We set the number of IBA iterations to 3 in order to extend the assembly beyond the exonic target regions. In step 3 (probe trimming), we separate the exonic target region sequences to be used in the next step to assess orthology and format the output.
Although the probes were designed from exons in single‐ or low‐copy genes across land plants, it is possible that paralogous or other non‐targeted sequences were assembled from the enriched data. Thus, in step 4 (orthology to reference) we assessed orthology based on the best tBLASTx (Camacho et al., 2009) hit of the exonic target region of each assembled sequence to the coordinates of 10 plant genomes representing hornworts, liverworts, mosses, lycophytes, ferns, and gymnosperms (see Dryad; Breinholt et al., 2020). We called an assembled sequence an ortholog of the probe region if it had no additional tBLASTx hits with >95% of the best bit score, outside of a 1000‐bp flanking window around the genomic coordinates of the exonic target locus in a reference genome. We only required that a sequence have evidence of orthology in any one of the reference genomes. At this point in the pipeline, a taxon may retain more than one orthologous sequence for a single locus, potentially representing allelic variation, homeolog, or recent duplication.
In the fifth step (contamination filter), in order to filter out likely contaminants, for each assembled sequence we performed a tBLASTx search against the respective reference 1KP and genomic sequences for that locus. If a sequence’s best hit was not from the taxonomic group (i.e., hornwort, liverwort, moss, lycophyte, fern, or gymnosperm) from which the sequence came, that sequence was removed as a potential contaminant. Finally, in the sixth step (alignment and merge isoforms), we aligned the resulting sequences using MAFFT version 7.425 (Katoh and Standley, 2013). Alignments can be done for either only the sequences representing the exonic target loci or the full sequences, including the exonic target loci and flanking intron sequences. Aligning the conserved exonic target regions across all flagellate land plants is usually straightforward, but it can be difficult to confidently align the more variable intronic flanking regions from distantly related taxa. After the alignments, sequences from the same taxon with mismatches due to heterozygous sites were merged with a Perl script, using International Union of Pure and Applied Chemistry codes to represent heterozygous sites.
We performed a linear regression using R version 3.3.3 (R Core Team, 2017) to examine the relationship between the amount of library DNA in each sample and the number of total reads resulting from each sample to the number of loci recovered. In addition, in order to evaluate how well the exonic target loci could resolve relationships among flagellate land plants, we ran a ML analysis on a supermatrix of the targeted exonic locus alignments. To build the supermatrix, we removed sequences from all samples from which we recovered fewer than 10% (i.e., 45) of the loci and then pruned the alignments so that they only included sites (i.e., columns) that had data from at least four samples. After completing the pipeline, it is possible that a sample would still have multiple sequences in an exon alignment where BRIDGER determined that reads represented more than simple allelic diversity. In our samples, the pipeline retained multiple copies for 6.1% (3790/62,200) of the exonic target region sequences across all samples. In order to ameliorate possible paralogy issues, when a sample had multiple sequences for a locus, we excluded all copies of that sequence from that sample. We then concatenated all loci into a single supermatrix and ran a ML search and 100 nonparametric BS replicates using RAxML 8.2.10 with the GTR CAT model (Stamatakis, 2014). The scripts used to process the data for phylogenetic analysis and the supermatrix alignment with locus boundaries are available on Dryad (Breinholt et al., 2020).
Optimizing the GoFlag 451 probe set
Based on the results of this pilot study, we refined the original GoFlag 451 probe set to optimize the performance of the target enrichment across flagellate land plants. The resulting GoFlag 408 probe set is a subset of the original GoFlag 451 probe set, containing 52,306 probes covering 408 of the original 451 loci. These 408 loci are found in 229 genes. For the GoFlag 408 probe set, we removed probes for all but two of the loci that produced sequences from fewer than 104 samples in this study, along with other probes that were either underperforming or exhibited strong taxonomic biases (see Appendix S3). The GoFlag 408 probe set is also available on Dryad (Breinholt et al., 2020) and commercialized by RAPiD Genomics (http://rapid‐genomics.com). Although we did not run a separate target enrichment experiment to assess the performance of the GoFlag 408 set, we inferred a tree using data from the 408 selected loci that were generated using the GoFlag 451 probe set. Specifically, we made a concatenated matrix of just the target regions corresponding to the GoFlag 408 probe set for the same taxa as the GoFlag 451 supermatrix, and we ran a ML phylogenetic analysis on that supermatrix as described above (data available on Dryad; Breinholt et al., 2020).
RESULTS
The supermatrix of the 451 exons used for the design of the probe set from the 1KP data had 943 taxa and was 150,369 bp in length, with 85,112 variable sites (i.e., columns in the alignment that have at least two different nucleotides) and 58.2% missing data. The ML phylogenetic analysis of the supermatrix produced a land plant tree with relationships that are generally consistent with those from formal 1KP analyses (Appendix S5; Leebens‐Mack et al., 2019a). Throughout the tree, 81.5% (767/941) of the internal branches had 100% BS support, 89.3% of the branches had at least 90% BS support, and 94.6% of the branches had at least 70% BS support (Appendix S5). This suggests that the 451 relatively conserved loci covered by the GoFlag 451 probe set provide sufficient data to resolve many relationships throughout land plants (Appendix S5; Leebens‐Mack et al., 2019a).
One measure of the performance of the probe set is the proportion of sequences from each library that mapped to the probe loci. The target enrichment ranged from 0.1% (Aneura pinguis (L.) Dumort., a liverwort) to 89.9% (Rhynchostegium murale (Hedw.) Schimp., a moss) of the reads, with an average across samples of 42.5% and a median of 40.9% (Appendix S4). The number of loci recovered (out of a possible 451) ranged from three (Mesoptychia badensis (Gottsche ex Rabenh.) L. Söderstr. & Váňa, a liverwort) to 436 (Podocarpus smithii de Laub., a gymnosperm), with an average of 332.4 and a median of 394.0 (Fig. 1, Appendix S4). Although we recovered fewer than 10% of the possible loci in 16 samples, in 82 of the 188 samples we recovered at least 90% of the possible loci (Fig. 1, Appendix S4). The samples with fewer than 10% of the samples, including ferns and liverworts, do not appear to be phylogenetically clustered (Fig. 1, Appendix S4). Overall, the probes worked well across flagellate plant lineages, with the fewest average number of loci in the gymnosperm samples, and the highest average number of loci in the mosses (Table 1). There were 17 species in our target enrichment experiment that also had transcriptome data generated by 1KP (Leebens‐Mack et al., 2019a). In 13 of the 17 common species, our target enrichment study generated data from more of the 451 loci than 1KP (Appendix S6), suggesting either that these loci were not amplified in the transcriptome sequencing or our experiment amplified divergent copies that were excluded from the 1KP alignments. It is difficult to compare the results of a targeted enrichment experiment with transcriptome sequencing, but this indicates that the targeted enrichment is not missing many loci that we know exist from transcriptome data.
FIGURE 1.
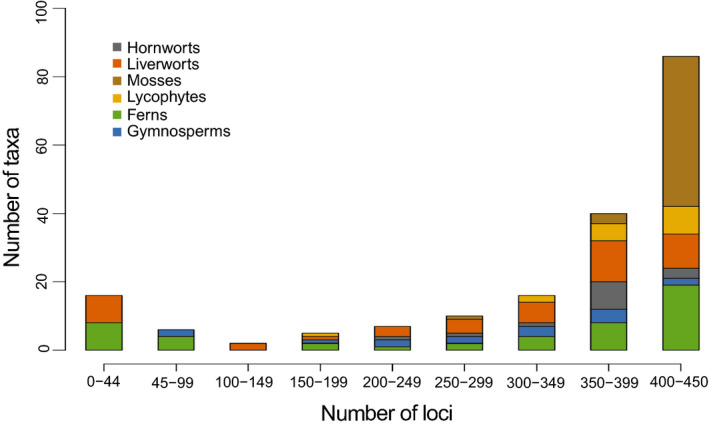
Distribution of the number of successfully sequenced loci (out of a possible 451) per taxon sample. Each locus is a relatively conserved exon from a single‐copy or low‐copy nuclear gene.
TABLE 1.
Distribution of loci with sequence data (out of a possible 451) across major flagellate plant lineages.
| Lineage | No. of samples | Average loci recovered | Median loci recovered |
|---|---|---|---|
| Ferns | 48 | 291.9 | 369.0 |
| Gymnosperms | 16 | 291.3 | 314.5 |
| Hornworts | 14 | 365.9 | 387.5 |
| Liverworts | 46 | 281.7 | 346.5 |
| Lycophytes | 16 | 379.8 | 401.0 |
| Mosses | 48 | 409.8 | 415.0 |
The samples for which we recovered few loci could have had highly diverged sequences from the probe sites or had poor quality DNA. However, in a few cases species from which we recovered few loci are closely related to species from which we recovered many loci (e.g., Dryopteris pentheri (Krasser) C. Chr. [21 loci] vs. D. patula (Sw.) Underw. [431 loci], or Elaphoglossum yatesii (Sodiro) Christ [27 loci] vs. E. bellermannianum (Klotzsch) T. Moore [418 loci]; Appendix S4), suggesting that probe site evolution is unlikely to explain at least some of the failed captures. Similarly, the relationship between the amount of DNA from a given specimen and the number of recovered loci is weak at best (Fig. 2A). A linear regression in which we counted all samples with >250 ng of library DNA as having 250 ng of DNA (the maximum used in any library) had an R 2 = 0.07 (P = 0.0002). However, some samples with very little DNA were successful, and some samples with abundant DNA were not (Fig. 2A), suggesting that DNA quality rather than quantity may have a greater effect on these libraries. The relationship between the number of reads and the number of loci recovered was much stronger (R 2 = 0.23; P < 0.0001), and samples from which we recovered few loci all had relatively few reads (Fig. 2B). We obtained sequence data from an average of 138.6 (median = 147.0) out of 188 total samples across the 451 loci (Appendix S3), but there was also variation in the number of samples that recovered each locus, and some loci had a taxonomic bias (Fig. 3, Appendix S3).
FIGURE 2.
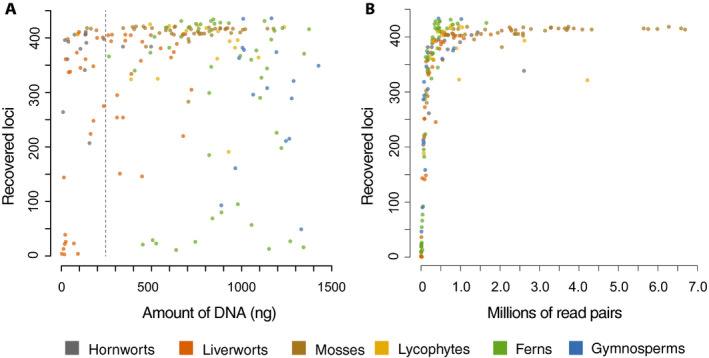
Associations of the amount of DNA and number of reads with the number of resulting loci from a sample. (A) Amount of library DNA in each sample vs. the number of resulting loci obtained in the targeted enrichment analysis. The dashed line at 250 ng represents the amount of DNA at which samples were normalized for the library preparation. (B) Number of reads obtained from each sample vs. the number of loci obtained from the targeted enrichment analysis.
FIGURE 3.
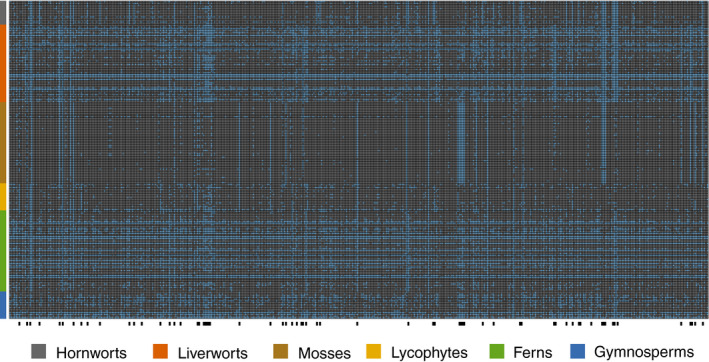
Heat map showing the distribution of data in the flagellate plant samples across the 451 probe regions (i.e., exons). Loci that were missing for an individual are colored blue in the heatmap while sampled loci are grey. Black bars along the bottom of the heatmap indicate loci with biases among major plant groups, where less than 25% of one group had the locus and over 75% of another group had the locus.
Alignments for the exonic target regions were on average 214 bp, with 9.4% missing data, 154 variable sites, and 138 parsimony informative sites (Appendix S3). To evaluate how well the target loci could resolve flagellate land plant relationships, we constructed a 172‐taxon phylogenetic supermatrix of the 451 loci (i.e., exonic probe regions) that was 89,973 nucleotides in length, with 67,634 variable sites and 29.2% missing data. Again, the resulting phylogenetic tree is generally consistent with recent phylogenetic studies (e.g., Leebens‐Mack et al., 2019a). Of the 170 clades in the ML tree, 134 (79%) had 100% BS support, 86% of the clades had at least 90% BS support, and only 16 clades had less than 70% BS support (Fig. 4). One unexpected result is the non‐monophyly of the two Targionia hypophylla L. (liverwort) samples (Fig. 4). This could be the result of misidentification of the specimens. However, many of the bryophytes were sampled from mixed herbarium samples that contained tissue from multiple taxa. This may also explain the relatively large number of contaminant sequences identified in many of the bryophyte samples (Appendix S4).
FIGURE 4.
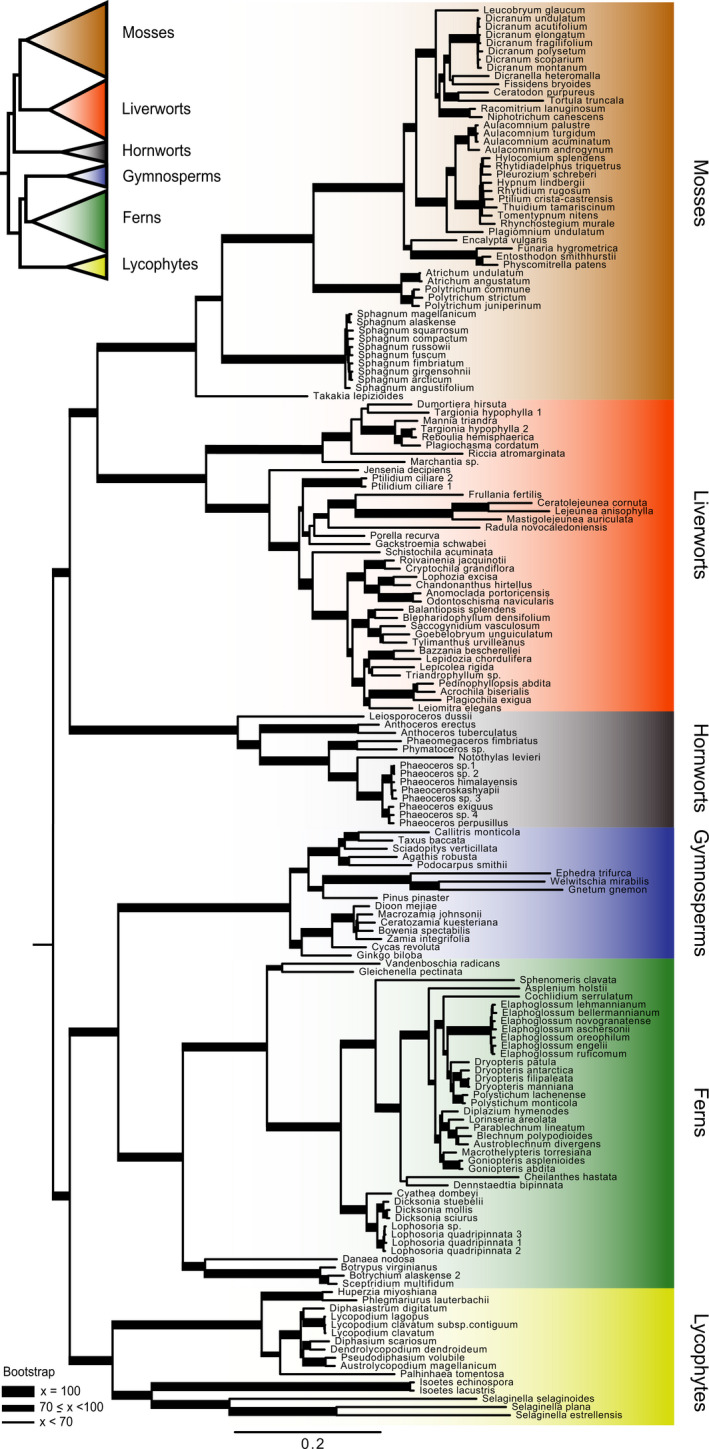
Phylogram from a maximum likelihood analysis of the supermatrix made by concatenating the alignments from the GoFlag 451 probe regions (i.e., exons) for the samples with at least 45 loci. The tree was rooted between the bryophytes and vascular plants.
To explore the potential for the probe set to resolve relationships among closely related taxa, we assembled supermatrices including both the exonic target regions and the more variable flanking regions for samples from the seven genera from which we had at least four samples. In these supermatrices we only included loci with data from at least four taxa, and within each locus alignment we only included columns with at least four nucleotides. By including the flanking regions, the length of the alignments was between 1.8 and 5.8 times longer than the target region–only alignments, with between 2.7 and 11.7 times more variable sites (Table 2). In contrast to the exonic target regions, which can be easily aligned across land plants, it can be difficult to align the variable flanking regions across distantly related taxa. Nevertheless, the flanking regions potentially can provide a tremendous amount of additional data to infer phylogenies among more closely related taxa.
TABLE 2.
Comparison of phylogenetic data in concatenated supermatrices from exonic target regions (i.e., exons) and the exonic target and flanking regions for the seven genera with at least four samples. In the locus alignments prior to concatenation, if a sample had more than one sequence, all copies for that sample were removed, and all columns in the alignment with fewer than four nucleotides were removed.
| Probe region only | Probe and flanking regions | ||||||
|---|---|---|---|---|---|---|---|
| Genus | Lineage | Samples | Loci | Alignment (bp) | Variable characters | Alignment (bp) | Variable characters |
| Aulacomnium | Moss | 4 | 369 | 67,739 | 2190 | 377,479 | 25,649 |
| Dicranum | Moss | 7 | 382 | 70,000 | 1214 | 388,915 | 11,965 |
| Dryopteris | Fern | 5 | 203 | 44,303 | 2619 | 79,832 | 6979 |
| Elaphoglossum | Fern | 8 | 373 | 71,759 | 1695 | 176,667 | 6379 |
| Lophosoria | Fern | 4 | 365 | 69,239 | 566 | 193,704 | 2423 |
| Phaeoceros | Hornwort | 8 | 379 | 71,806 | 3128 | 237,677 | 18,795 |
| Sphagnum | Moss | 10 | 391 | 71,186 | 3124 | 415,123 | 34,349 |
Finally, the 172‐taxon supermatrix for the loci in the optimized GoFlag 408 probe set from the samples with data from at least 45 loci was 80,582 bp long, with 60,873 variable sites and 25.0% missing data. Although this supermatrix alignment was 9391 bp shorter than the supermatrix made from the GoFlag 451 loci, the topology and levels of bootstrap support from the resulting trees were virtually identical (Appendix S7).
CONCLUSIONS
Here, we have described a probe set targeting nuclear loci across flagellate plants that likely diverged more than 450 million years ago (Morris et al., 2018). Data from the exonic target region sequences can help resolve phylogenetic relationships across the flagellate land plants (Fig. 4; Appendices S5, S7). Furthermore, the more variable flanking regions may provide much more data for resolving relationships among closely related species, or potentially even populations within a species (Table 2). Although the GoFlag 451 probe set recovered a large number of loci in samples from all major extant flagellate land plant lineages (Table 1; Figs. 1, 3), they may not work as well in some flagellate land plant taxa. Our strategy was to develop a “universal” probe set that covers the majority of these groups, and the GoFlag 451 probe set and the analysis pipeline provide a core set of validated tools accessible to all scientists. However, some evolutionary questions in the flagellate land plants may require more loci or a more specific probe set (e.g., Jantzen et al., 2020). While the GoFlag 451 probe set facilitates target enrichment projects in any flagellate land plant group, a probe set designed for a particular lineage could easily have more specific probes that cover either more loci, loci of special interest (e.g., Medina et al., 2019; Montes et al., 2019), or loci with higher substitution rates (de La Harpe et al., 2019). Resolving some of the more contentious flagellate plant relationships may likewise require a larger, more specific probe set. In those cases, the GoFlag 451 probes define a core set of loci that can be built upon. Nuclear gene evolution within land plants is often extremely complex, with, for example, frequent gene and whole genome duplications. Although nuclear loci have the potential to resolve complicated evolutionary relationships, their own complex histories can mislead phylogenetic inference. Our test for orthology in the analytical pipeline is simplistic, and in this study, we did not carefully examine potential issues of paralogy or homoeology in the 451 loci within flagellate plants. However, the resulting sequence data potentially can be used to examine gene or genome duplication, or even allelic variation and heterozygosity.
In subsequent sequencing runs, the GoFlag project has used the GoFlag 408 probe set, and results have been similarly successful (J.G.B., unpublished data). Due to the large number of probes needed to cover the diversity of flagellate land plants, we did not include the angiosperms when designing the GoFlag probe sets. However, the probe set was designed to cover exons that were conserved across all land plants, including angiosperms, and the target regions appear to provide sufficient data to resolve many angiosperm relationships (Appendix S5). Thus, the loci in this probe set may provide a foundation for constructing large‐scale nuclear phylogenies across land plants.
Supporting information
APPENDIX S1. Samples from the 1KP transcriptome data (Leebens‐Mack et al., 2019a) used to make the reference alignments from which the GoFlag 451 probe set was designed.
APPENDIX S2. Genomes used to design the GoFlag 451 probe set.
APPENDIX S3. Description of the loci from the GoFlag 451 probe set.
APPENDIX S4. Samples used in the GoFlag 451 targeted enrichment experiment.
APPENDIX S5. Phylogram from a maximum likelihood analysis of a supermatrix made by concatenating 1KP transcriptome sequences from the gene regions covered by the GoFlag 451 probe set. Due to the extremely long branch leading to the closest outgroups, the tree was rooted between the bryophytes and vascular plants.
APPENDIX S6. Comparison of the number of loci covered by the GoFlag 451 probe set that were recovered by targeted enrichment (“GoFlag loci”) and those recovered by the 1KP transcriptome sequencing (Leebens‐Mack et al., 2019a).
APPENDIX S7. Phylogram from a maximum likelihood analysis of the supermatrix made by concatenating the alignments from the GoFlag 408 probe regions (i.e., exons) for the samples with at least 45 loci. The tree was rooted between the bryophytes and vascular plants.
ACKNOWLEDGMENTS
This work was funded by the U.S. National Science Foundation (DEB‐1541506). The authors thank Jim Leebens‐Mack (University of Georgia) and Gane Wong (University of Alberta and BGI‐Shenzhen) for early access to 1KP transcriptome data, Matt Johnson (Texas Tech University) for discussions and advice about probe design, and Adam Payton (University of Florida and RAPiD Genomics) for lab help, especially with scaling up the DNA extraction capacity.
Breinholt, J. W. , Carey S. B., Tiley G. P., Davis E. C., Endara L., McDaniel S. F., Neves L. G., Sessa E. B., et al. 2021. A target enrichment probe set for resolving the flagellate land plant tree of life. Applications in Plant Sciences 9(1): e11406.
DATA AVAILABILITY
Sequence reads have been deposited to the National Center for Biotechnology (NCBI) Sequence Read Archive (PRJNA630729). The GoFlag 451 and GoFlag 408 probe sets are available on Dryad (https://doi.org/10.5061/dryad.7pvmcvdqg; Breinholt et al., 2020) with the pipeline scripts and reference sequences, the post‐processing scripts, and all phylogenetic matrices and trees from this study. Accessions and voucher information are provided in Appendix S4.
LITERATURE CITED
- Bentley, D. R. , Balasubramanian S., Swerdlow H. P., Smith G. P., Milton J., Brown C. G., Hall K. P., et al. 2008. Accurate whole genome sequencing using reversible terminator chemistry. Nature 456: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breinholt, J. W. , Earl C., Lemmon A. R., Lemmon E. M., Xiao L., and Kawahara A. Y.. 2017. Data from: Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for anchored phylogenomics. Dryad Dataset. 10.5061/dryad.rf7g5. [DOI] [PubMed] [Google Scholar]
- Breinholt, J. W. , Earl C., Lemmon A. R., Moriarty Lemmon E., Xiao L., and Kawahara A. Y.. 2018. Resolving relationships among the megadiverse butterflies and moths with a novel pipeline for anchored phylogenomics. Systematic Biology 67: 78–93. [DOI] [PubMed] [Google Scholar]
- Breinholt, J. W. , Carey S. B., Tiley G. P., Davis E. C., Endara L., McDaniel S. F., Neves L. G., et al. 2020. Target enrichment probe set for resolving the flagellate land plant tree of life. Dryad Dataset. 10.5061/dryad.7pvmcvdqg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, G. E. , Clarkson J. J., Maurin O., Zuntini A. R., Barber V., Bellot S., Biggs N., et al. 2019. Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of angiosperms. Frontiers in Plant Science 10: 1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C. , Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., and Madden T. L.. 2009. BLAST+: Architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter, E. J. , Matasci N., Ayyampalayam S., Wu S., Sun J., Yu J., Vieira F. R. J., et al. 2019. Access to RNA‐sequencing data from 1,173 plant species: The 1000 Plant transcriptomes initiative (1KP). GigaScience 8: giz126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, Z. , Li G., Liu J., Zhang Y., Ashby C., Liu D., Cramer C. L., and Huang X.. 2015. Bridger: A new framework for de novo transcriptome assembly using RNA‐seq data. Genome Biology 16: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronn, R. , Knaus B. J., Liston A., Maughan P. J., Parks M., Syring J. V., and Udall J.. 2012. Targeted enrichment strategies for next‐generation plant biology. American Journal of Botany 99: 291–311. [DOI] [PubMed] [Google Scholar]
- de La Harpe, M. , Hess J., Loiseau O., Salamin N., Lexer C., and Paris M.. 2019. A dedicated target capture approach reveals variable genetic markers across micro‐ and macro‐evolutionary time scales in palms. Molecular Ecology Resources 19: 221–234. [DOI] [PubMed] [Google Scholar]
- Doyle, J. J. , and Doyle J. L.. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Edgar, R. C. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. [DOI] [PubMed] [Google Scholar]
- Forrest, L. L. , Hart M. L., Hughes M., Wilson H. P., Chung K.‐F., Tseng Y.‐H., and Kidner C. A.. 2019. The limits of Hyb‐Seq for herbarium specimens: Impact of preservation techniques. Frontiers in Ecology and Evolution 7: 439. [Google Scholar]
- Gernandt, D. S. , Aguirre‐Dugua X., Vázquez‐Lobo A., Willyard A., Moreno Letelier A., Pérez de la Rosa J. A., Piñero D., and Liston A.. 2018. Multi‐locus phylogenetics, lineage sorting, and reticulation in Pinus subsection Australes . American Journal of Botany 105: 711–725. [DOI] [PubMed] [Google Scholar]
- Gnirke, A. , Melnikov A., Maguire J., Rogov P., LeProust E. M., Brockman W., Fennell T., et al. 2009. Solution hybrid selection with ultra‐long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology 27: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzen, J. R. , Amarasinghe P., Folk R. A., Reginato M., Michelangeli F. A., Soltis D. E., Cellinese N., and Soltis P. S.. 2020. A two‐tier bioinformatic pipeline to develop probes for target capture of nuclear loci with applications in Melastomataceae. Applications in Plant Sciences 8: e11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M. G. , Pokorny L., Dodsworth S., Botigué L. R., Cowan R. S., Devault A., Esienhardt W. L., et al. 2019. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k‐medoids clustering. Systematic Biology 68: 594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens‐Mack, J. H. , Barker M. S., Carpenter E. J., Deyholos M. K., Gitzendanner M. A., Graham S. W., Grosse I., et al. 2019a. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574: 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens‐Mack, J. H. , Wong G. K. S., and the One Thousand Plant Transcriptomes Initiative . 2019b. Data packages for One Thousand Plant transcriptomes and phylogenomics of green plants. CyVerse Data Commons 10.25739/8m7t-4e85. Website https://datacommons.cyverse.org/browse/iplant/home/shared/commons_repo/curated/oneKP_capstone_2019 [accessed 18 November 2020]. [DOI]
- Liu, Y. , Johnson M. G., Cox C. J., Medina R., Devos N., Vanderpoorten A., Hedenäs L., et al. 2019. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nature Communications 10: 1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKain, M. R. , Johnson M. G., Uribe‐Convers S., Eaton D., and Yang Y.. 2018. Practical considerations for plant phylogenomics. Applications in Plant Sciences 6: e1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, R. , Johnson M. G., Liu Y., Wickett N. J., Shaw A. J., and Goffinet B.. 2019. Phylogenomic delineation of Physcomitrium (Bryophyta: Funariaceae) based on targeted sequencing of nuclear exons and their flanking regions rejects the retention of Physcomitrella, Physcomitridium and Aphanorrhegma . Journal of Systematics and Evolution 57: 404–417. [Google Scholar]
- Montes, J. R. , Peláez P., Willyard A., Moreno‐Letelier A., Piñero D., and Gernandt D. S.. 2019. Phylogenetics of Pinus subsection Cembroides Engelm. (Pinaceae) inferred from low‐copy nuclear sequences. Systematic Botany 44: 501–518. [Google Scholar]
- Morris, J. L. , Puttick M. N., Clark J. W., Edwards D., Kenrick P., Pressel S., Wellman C. H., et al. 2018. The timescale of early land plant evolution. Proceedings of the National Academy of Sciences, USA 115: E2274–E2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, X. , Kuo L.‐Y., Guo C., Li H., Li Z., Qi J., Wang L., et al. 2018. A well‐resolved fern nuclear phylogeny reveals the evolutionary history of numerous transcription factor families. Molecular Phylogenetics and Evolution 127: 961–977. [DOI] [PubMed] [Google Scholar]
- R Core Team . 2017. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: Website http://www.R‐project.org/ [accessed 19 November 2020]. [Google Scholar]
- Shen, H. , Jin D., Shu J.‐P., Zhou X.‐L., Lei M., Wei R., Shang H., et al. 2018. Large‐scale phylogenomic analysis resolves a backbone phylogeny in ferns. GigaScience 7: gix116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis, A. 2014. RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitemier, K. , Straub S. C. K., Cronn R. C., Fishbein M., Schmickl R., McDonnell A., and Liston A.. 2014. Hyb‐Seq: Combining target enrichment and genome skimming for plant phylogenetics. Applications in Plant Sciences 2: 1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickett, N. J. , Mirarab S., Nguyen N.‐P., Warnow T., Carpenter E., Matasci N., Ayyampalayam S., et al. 2014. A phylotranscriptomics analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences, USA 111: E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf, P. , Robison T. A., Johnson M. G., Sundue M. A., Testo W. L., and Rothfels C. J.. 2018. Targeted sequence capture of nuclear‐encoded genes for phylogenetic analysis of ferns. Applications in Plant Sciences 6: e1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Samples from the 1KP transcriptome data (Leebens‐Mack et al., 2019a) used to make the reference alignments from which the GoFlag 451 probe set was designed.
APPENDIX S2. Genomes used to design the GoFlag 451 probe set.
APPENDIX S3. Description of the loci from the GoFlag 451 probe set.
APPENDIX S4. Samples used in the GoFlag 451 targeted enrichment experiment.
APPENDIX S5. Phylogram from a maximum likelihood analysis of a supermatrix made by concatenating 1KP transcriptome sequences from the gene regions covered by the GoFlag 451 probe set. Due to the extremely long branch leading to the closest outgroups, the tree was rooted between the bryophytes and vascular plants.
APPENDIX S6. Comparison of the number of loci covered by the GoFlag 451 probe set that were recovered by targeted enrichment (“GoFlag loci”) and those recovered by the 1KP transcriptome sequencing (Leebens‐Mack et al., 2019a).
APPENDIX S7. Phylogram from a maximum likelihood analysis of the supermatrix made by concatenating the alignments from the GoFlag 408 probe regions (i.e., exons) for the samples with at least 45 loci. The tree was rooted between the bryophytes and vascular plants.
Data Availability Statement
Sequence reads have been deposited to the National Center for Biotechnology (NCBI) Sequence Read Archive (PRJNA630729). The GoFlag 451 and GoFlag 408 probe sets are available on Dryad (https://doi.org/10.5061/dryad.7pvmcvdqg; Breinholt et al., 2020) with the pipeline scripts and reference sequences, the post‐processing scripts, and all phylogenetic matrices and trees from this study. Accessions and voucher information are provided in Appendix S4.