

Abstract
The structural and regulatory elements in therapeutically relevant RNAs offer many opportunities for targeting by small molecules, yet fundamental understanding of what drives selectivity in small molecule:RNA recognition has been a recurrent challenge. In particular, RNAs tend to be more dynamic and offer less chemical functionality than proteins, and biologically active ligands must compete with the highly abundant and highly structured RNA of the ribosome. Indeed, the only small molecule drug targeting RNA other than the ribosome was just approved in August 2020, and our recent survey of the literature revealed fewer than 150 reported chemical probes that target non-ribosomal RNA in biological systems. This Feature outlines our efforts to improve small molecule targeting strategies and gain fundamental insights into small molecule:RNA recognition by analyzing patterns in both RNA-biased small molecule chemical space and RNA topological space privileged for differentiation. First, we synthesized libraries based on RNA binding scaffolds that allowed us to reveal general principles in small molecule:recognition and to ask precise chemical questions about drivers of affinity and selectivity. Elaboration of these scaffolds has led to recognition of medicinally relevant RNA targets, including viral and long noncoding RNA structures. More globally, we identified physicochemical, structural, and spatial properties of biologically active RNA ligands that are distinct from those of protein-targeted ligands, and we have provided the dataset and associated analytical tools as part of a publicly available online platform to facilitate RNA ligand discovery. At the same time, we used pattern recognition protocols to identify RNA topologies that can be differentially recognized by small molecules and have elaborated this technique to visualize conformational changes in RNA secondary structure. These fundamental insights into the drivers of RNA recognition in vitro have led to functional targeting of RNA structures in biological systems. We hope that these initial guiding principles, as well as the approaches and assays developed in their pursuit, will enable rapid progress toward the development of RNA-targeted chemical probes and ultimately new therapeutic approaches to a wide range of deadly human diseases.
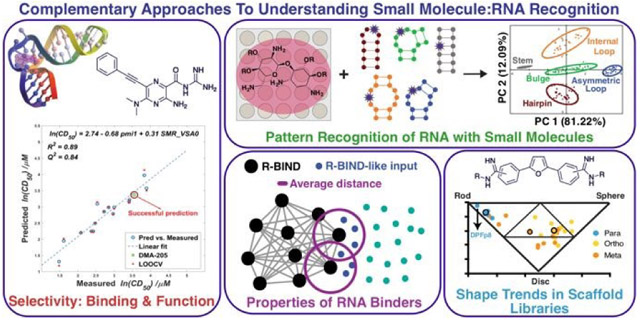
Graphical Abstract
Complementary approaches such as scaffold-based synthesis and screening, cheminformatics analysis, assay development, and pattern recognition have progressed fundamental understanding of small molecule:RNA recognition and led to the development of bioactive RNA-targeted ligands.
1. Introduction
RNA molecules are increasingly recognized both for their regulatory roles and as potential therapeutic targets in a range of human diseases.1, 2 If developed, drugs that target these RNAs would offer novel treatment strategies toward multiple deadly illnesses, including multi-drug-resistant bacterial, fungal, and viral infections as well as metastatic cancer.3-9 Despite this potential, the development of drugs targeted to RNA other than bacterial ribosomes has been slow, leading many to term RNA “undruggable.” Indeed, the first small molecule drug targeting RNA other than the ribosome was just approved by the US FDA in August of 2020.10 While antisense oligonucleotides offer high RNA specificity via base pair complementarity and are beginning to be FDA approved, in vivo delivery outside of the liver or central nervous remains a significant barrier.11-13 Small molecules offer several potential benefits, including extensive tunability in terms of delivery, uptake, immunogenicity, and other medicinal parameters as well as the ability to access a broad range of size, shape, and chemical functionality through organic synthesis. However, selective targeting of RNA with small molecules has been elusive.14-16 Fundamental challenges of RNA targeting include the limited chemical functionality of RNA relative to protein, the generally dynamic structure of RNA, and the difficulty of specific RNA target engagement in a cellular environment where 85% of the total RNA is ribosomal (rRNA) and chemically similar genomic DNA abounds. These challenges are exacerbated by the protein-centric nature of currently available screening libraries and methodologies. Furthermore, many RNA-targeted screening campaigns yield nonspecific hits that rely on electrostatic interactions with the phosphate backbone and/or stacking interactions with the RNA bases. In addition to the lack of exploration into RNA-targeted small molecule chemical space, the limited number of distinct RNA targets pursued to date has hindered our understanding of the RNA structures that can be recognized by small molecules.17
Another barrier to understanding small molecule:RNA recognition is the inherent difficulty of RNA structural characterization,18-20 which often prevents atomic-level interpretation of these interactions and limits both structure-based design toward specific targets and our ability to discern patterns in the small molecule recognition of RNA structures. For 2D RNA structures, computational predictions augmented by chemical probing data, which report on the likelihood of base-pairing at a given position, have seen great utility and are broadly implemented.21-23 At the same time, these methods are limited by the algorithms employed, which can yield different solutions for the same data set and overlook complex structures such as triple helices, thus requiring input from other experimental and phylogenetic analyses.24, 25 For 3D RNA structures, de novo computational prediction is largely limited to short sequences26, 27 while high-resolution experimental characterization of RNA can be difficult and time-consuming, particularly for large sequences, using traditional methods such as NMR and X-ray diffraction. Cryo-electron microscopy (cryo-EM) of RNA is becoming increasingly effective, though it has been used primarily for large RNA:protein complexes to date,28-31 with fewer examples of RNA alone.32, 33 Continuous improvements on traditional methods, as well as ongoing work that combines 2D probing and 3D predictions34 or evaluates RNA dynamics and functional ensembles20 promises to further our understanding of 3D RNA structure and thus RNA molecular recognition.
Despite these challenges, a number of emerging examples have confirmed that non-ribosomal RNA can be targeted by small molecules (Figure 1) and that a number of strategies, both RNA-centric and more general, may be successful.3, 5, 6, 9, 16, 17, 35-39 For example, the Disney laboratory has used selection-based strategies to match small molecules with specific RNA sequences,40 which ultimately led to small molecules with activity against RNA repeat-associated diseases41 and against microRNAs in triple negative breast cancer,42 both with efficacy in mouse models. Modular assembly of RNA binding units into large and often multivalent ligands can increase affinity and specificity and has also proven effective in biological systems. Examples include work by: the Zimmerman laboratory in which a multivalent ligand targeting repeat RNA of myotonic dystrophy reverses phenotype in fly and mouse models43; the Miller laboratory in which dynamic combinatorial chemistry produced ligands targeting the HIV-1 frameshift sequence with antiviral activity;44, 45 and the Disney laboratory using the above mentioned sequence-based approach.46, 47 In another RNA-centric example, the Al-Hashimi laboratory identified novel ligands for HIV-1-TAR RNA by docking to an experimentally-informed ensemble of several RNA conformations, enabling a structure-based approach to be applied to this highly dynamic system.48, 49
Figure 1.

Example structures of small molecule:RNA complexes. RNA structures rendered in ICM and small molecules highlighted in purple. A) SMN-C5 bound to the RNA duplex of the 5’-end splice site of Survival Of Motor Neuron 2 (SMN2) exon 7 (PDB 6HMO, Ref. 132). B) Ribocil-C bound to the flavin mononucleotide (FMN) riboswitch (PDB 5C45, Ref. 55). C) DMA-135 bound to stem loop II of the internal ribosomal entry site (IRES) of enterovirus 71 (EV71) (PDB 6XB7, Ref. 91). D) Benzimidazole 2 bound to subdomain IIa of the hepatitis C virus (HCV) IRES (PDB 3TZR, Ref. 61).
More traditional screening methods, using scaffold-based or general libraries, have also been successful. Well-studied scaffold-based libraries have included oxazolidinones,50-55 diphenylfurans,56-60 benzimidazoles,61-64 and aminoglycosides,65-69 with derivatives showing a range of RNA binding properties and biological activity. In a very recent example, Dutta and co-workers synthesized a library of quinoxaline derivatives that target the HCV IRES and inhibit viral translation and replication.70 Successful higher-throughput screens have included work by Schneekloth and co-workers, who leveraged microarray screening of roughly 20,000 molecules to identify selective ligands for HIV-1-TAR,71 miRNA-21,72 the Pre-Q1 riboswitch,73 and the MALAT1 3’-triple helix,74 many of which show efficacy in cell-based systems, including evidence of anti-HIV and anti-cancer activity. The Pyle lab developed an activity-based screen for inhibition of the group II intron ribozyme and identified several active antifungal molecules from a library of 10,000 compounds that were further optimized using traditional medicinal chemistry methods.75 Several examples from industrial laboratories have also come to the forefront. For example, a Merck group screened ~57,000 molecules with antibacterial activity to identify ribocil, a ligand for the flavin mononucleotide (FMN) riboswitch that was optimized (ribocil-C) to demonstrate efficacy in a murine model of sepsis (Figure 1 B).76 Finally, both Novartis77, 78 and PTC / Roche 79, 80 have developed RNA-targeted small molecules that lead to exon 7 inclusion in the SMN2 gene in patients with spinal muscular atrophy (Figure 1A). Both small molecules appear well-tolerated and are proceeding through clinical trials, with risdiplam (Evrysdi) recently approved by the US FDA.10 These examples, in addition to other success stories of RNA targeting in vivo, have led to a surge in RNA-targeted small molecule programs in the pharmaceutical industry, both within larger companies and in startups, with a corresponding increase in venture capital investment.81-83
Inspired by the potential of RNA targeting in drug discovery, the Hargrove Lab takes a very fundamental approach: to elucidate guiding principles for achieving selectivity in small molecule:RNA recognition and to apply these principles to develop RNA-targeted chemical probes that modulate RNA functions in cell culture. As has been seen with protein-targeted chemical probes, RNA-targeted chemical probes would be expected to both elucidate fundamental RNA biology and provide insight into how disease pathways might be modulated with RNA-targeted drugs. To begin, we have generated RNA-biased small molecule libraries and screening methodologies that are expected to allow rational targeting of a wide range of disease-related RNAs. Concurrently, we are utilizing the power of differential sensing and pattern recognition to elucidate the shape-based drivers of small molecule:RNA recognition and to classify functional RNAs. We hypothesize that this framework will facilitate characterization and targeting of regulatory RNAs with the resulting potential to transform our understanding of molecular biology. This Feature provides an overview of our work toward a fundamental understanding of selective small molecule:RNA interactions as well as an outlook on the future of the small molecule:RNA targeting field.
2. Understanding and engineering selectivity in small molecule: RNA recognition
To help overcome the selectivity barrier in small molecule:RNA targeting, we asked if specific chemical scaffolds and/or chemical properties of small molecules may bias them toward selective RNA interactions. In these efforts, we have investigated synthetically tractable scaffold-based libraries that allow atom-level tuning of individual molecules, globally analyzed chemical properties of known biologically active RNA ligands, and developed screening procedures that allow us to rapidly assess selectivity.
2.1. Scaffold-based libraries
Scaffold-based libraries offer several advantages in the pursuit of guiding principles for selective small molecule:RNA targeting, including the ability to ask precise chemical questions, to build into a desired chemical space, and to rapidly generate structure-activity relationships that are not readily available with commercial libraries. We herein discuss how this strategy has been applied to the amiloride and diphenylfuran scaffolds and has revealed both preliminary guidelines for small molecule design as well as lead molecules for chemical probe development against viral and oncogenic noncoding RNAs.
Amiloride:
In our first efforts to explore the amiloride scaffold, we collaborated with the Al-Hashimi laboratory to target the HIV-1 Trans-Activation Response RNA (TAR) element, a conserved structure known to be critical for HIV replication.84 Historically, amiloride has been used as an FDA-approved diuretic that functions by blocking sodium channels, and amiloride derivatives have also been tuned to target urokinase plasminogen activator (uPA) in cancer cells and a range of GPCRs.85-87 One such derivative, dimethyl amiloride (DMA-001), also appeared as a hit in a docking-based screen against HIV-1-TAR led by the Al-Hashimi laboratory.48 We then evolved DMA-001 from a weak ligand to a strong, selective TAR ligand (DMA-169) through a combination of synthetic and analytical methods (Figure 2).88 Specifically, iterative modifications at the C(5)- and C(6)- positions (DMA-101 and DMA-132, respectively) yielded DMA-169, which has a 100-fold increase in displacement activity over the parent DMA-001 (Figure 2A). Screening was performed using a displacement assay in which a peptide fragment from the native protein binding partner, Tat, was labeled with a FAM-TAMRA FRET pair, and selectivity was evaluated by the addition of 100-fold excess tRNA or DNA to the assay. The impact of these amilorides on TAR conformations was assessed by NMR chemical shift mapping using the 2D SOFAST-[1H-13C] HMQC NMR method. Tighter and more selective binders were found to perturb chemical shifts corresponding to the bulge region of TAR while others displayed broad chemical shift perturbations (Figure 2B,C). In addition to the identification of lead molecule DMA-169, we found that we could predict the selectivity of the amiloride derivatives, though not their affinity, based on cheminformatic properties using linear discriminate analysis (LDA)(Figure 2D).
Figure 2. Dimethylamiloride (DMA) as a tunable RNA-binding scaffold.

A) Stepwise modification at the C(5) and C(6) positions of amiloride scaffold to give lead DMA-169. Competitive displacement dose (CD50) for Tat peptide assays shown below each ligand B) Heat maps of 1H-13C [HMQC] SOFAST NMR experiments with amiloride and HIV-1-TAR RNA. C) Docked pose of DMA-169 with HIV-1-TAR, which shows interactions near the trinucleotide bulge (shown in orange). D) Linear discriminate analysis based on 20 cheminformatic parameters clusters selective amiloride ligands from non-selective ligands. Sample parameters are shown to the right, with trend for selectivity indicated by the arrow. Panels A-D reproduced from Ref. 88 with permission from the Royal Society of Chemistry. E) QSAR study on ESSV ligands generated a robust model and predicted binding affinity of a new ligand (DMA-205). LOOCV = leave-one-out cross validation. Panel E reproduced from Ref. 90 with permission from the Royal Society of Chemistry.
In the course of developing general screening assays utilizing the Tat peptide,89 we demonstrated that some amilorides showed selectivity for HIV-1 TAR over other regulatory RNAs known to bind small molecules, including HIV RRE-IIB and the bacterial ribosomal A-site. However, a subset of amilorides that were selective against tRNA in the original study88 also bound the HIV RRE-IIB and A-site controls. These findings inspired us to perform a broader structure-activity relationship (SAR) study on amiloride derivatives, specifically focused on regulatory RNAs in HIV that may also represent promising therapeutic targets. We incorporated a range of modifications at the C(5) and C(6) positions based on previous work and expanded synthetic routes to study the structure- activity/selectivity relationships with a collection of HIV related RNA structures, namely HIV-1 TAR, HIV-2 TAR, HIV-1-RRE-IIB, HIV-1-FSS, and HIV-1-ESSV.90 Our profiling analysis revealed a number of interesting trends. For example, the C(6) phenyl group significantly improves the activity and selectivity of the amiloride derivatives for HIV-1-TAR, but other aryl subunits at C(6) such as biphenyl, naphthyl, or heteroaryl groups proved to be detrimental to activity. In contrast, large subunits at the C(6) position increased binding for ESSV. Reducing the length of the linker between the pyrazine core and indole ring at the C(5) position of DMA-169 increased both affinity and selectivity for HIV-1-TAR, suggesting that flexible ligands may pay an entropic cost in binding. Cheminformatic analysis further suggested that weakly binding ligands tended to have an increase in oxygen count and flexibility while promiscuous ligands had very high nitrogen counts. Quantitative structure-activity relationships correlated chemical properties to CD50 values of amilorides binding to both HIV-1-TAR and ESSV, with distinct driving properties identified for each target (Figure 2E).
Leveraging the tunability of the amiloride scaffold, we then collaborated with the Tolbert and Brewer/Li laboratories to identify ligands for stem loop II (SLII) of the internal ribosomal entry site (IRES) of human enterovirus 71 (EV71),91 which had been previously shown to drive viral translation.92 We first screened our amiloride library by adapting the Tat peptide assay to identify a number of SLII ligands.91 Screening in a dual luciferase assay revealed that DMA-135 inhibited translation dependent on the EV71 IRES without impacting normal translation. Similar activity was observed in viral titer assays where viral replication was eliminated at concentrations with no observed toxicity. Mechanistic studies found that DMA-135 increased binding of human AUF1 repressive protein to SLII both in vitro via ITC and in cell culture via pull-down assays. Finally, DMA-135 induced a dramatic (~77º) conformational change in the linear SLII structure by NMR (Figure 1C). We hypothesized that this conformational change exposes an AUF1 binding site on SLII, leading to stabilization of a ternary DMA-135:SLII:AUF1 complex that prevents translation and ultimately viral replication. This example demonstrated the potential of modulating the conformational landscape of a dynamic RNA to impact function.
Finally, the amiloride scaffold provided the opportunity to establish a robust method for the selection of high affinity RNA ligands from a dynamic combinatorial pool using imine-based chemistry.93 This method allowed the identification of ligands for three RNA targets without a priori synthesis of discrete library members and is being expanded to additional, multifunctional scaffolds.
Diphenylfuran:
The diphenylfuran (DPF) scaffold was explored in the context of targeting the 3’-triple helix of the long noncoding RNA MALAT1 (Figure 3).94, 95 MALAT1 is thought to play a number of important roles in healthy cells, particularly in splicing, but accumulates at high levels in many cancer types.96 The formation of a stable triple helix at the 3’-end has been shown to prevent degradation of the MALAT1 transcript while destabilizing mutants led to increased degradation.97, 98 As a result, the MALAT1 triple helix has been considered a putative drug target in metastatic cancers. The DPF scaffold is promising as it has been shown to be tunable for a range of RNA and DNA duplex and stem loop targets, as well as T-A-T DNA triple helices, and has demonstrated biological activity.59, 99, 100 In addition, the scaffold’s inherent fluorescence allows for direct measurement of binding changes via emission intensity.101 The simplest core, furamidine, was found to have modest affinity toward the MALAT1 triple helix in our preliminary studies. We thus developed an efficient route to DPF di-amidines and synthesized 33 library members based on ortho-, meta-, and para-substituted scaffolds with eleven side chains (Figure 3A).94 By monitoring changes in fluorescence, we identified a ligand selective for the MALAT1 triple helix over control sequences, namely DPF-p8. Investigation of putative structure activity relationships revealed a general trend between the computationally predicted shape of DPFs and triple helix selectivity and binding strength (Figure 3B). Shape was largely dictated by subunit composition and positioning, along with predicted intramolecular interactions. For example, para-substituted derivatives were found to be the most rod-like in shape, with few predicted intramolecular interactions, and to be the most effective ligands.
Figure 3. Diphenylfuran scaffold reveals shape-dependent RNA recognition of MALAT1 triplex.

A. Schematic of diphenylfuran (DPF) scaffold library with symmetric regio-substitution. B. Principle moments of inertia (PMI) analysis of DPF library (blue-para, orange-meta, yellow-ortho substituted) revealed a correlation between triple helix binding strengths and small molecule 3D shape, with the highest affinity triplex binder (DPFp8) as the most rod-like. Reproduced from Ref. 94 with permission from John Wiley and Sons. C. Docking model of MALAT1 triple helix structure (PDB: 4PLX, Ref. 98) with DPFp8 (blue) illustrating the importance of rod-like shape and preorganization. D) Left: Correlation of DPF-induced changes in triplex melting temperature with amount of RNA remaining in an enzyme (RNaseR) degradation assay. Right: Structures of DPFp20, the most stabilizing DPF (purple) and DPFp8, the highest affinity DPF (blue). Shown EC50 values are based on RNA titration of fluorescent DPF scaffold. E. Gel image showing stabilization of MALAT1 triplex by DPF20 to RNaseR degradation. Panels D,E reproduced from Ref. 95 with permission from Oxford University Press.
In follow up work, we generated additional para-substituted DPF derivatives that tested different aspects of the DPF-p8 subunit in a range of assays.95 We first performed docking against the triple helix (Figure 3C), and the most favorable docking energies were found for small molecule structures that underwent minimal predicted conformational change between the starting minimized free energy structure and the bound structure, and these docking energies generally correlated with binding affinities. At the same time, we were able to observe selectivity trends between MALAT1 and other triple helices, including the mammalian NEAT1 / MENβ. Finally, we evaluated the impact of these DPFs on MALAT1 triple helix stability and identified correlations between increased melting temperature and protection against ribonuclease R degradation (Figure 3D). Interestingly, DPF-p20, a modest binder and the only molecule derived from an aniline subunit rather than an alkyl amine, led to the most dramatic protection from exonucleolytic degradation (Figure 3E). This work reinforced the importance of pre-organization and shape-based recognition in selective triple helix binding but also the potential for discrepancies between affinity- and function-based assays.
Our studies with amiloride and the diphenylfuran scaffolds revealed design principles for these scaffolds against viral and lncRNA targets, respectively, and supported our hypothesis that chemical properties may bias small molecules toward selective RNA interactions. The initial amiloride studies provided one of the tightest selective ligands for HIV-1-TAR to date and the first reported ligand for ESSV while demonstrating that the scaffold is tunable to a range of RNA secondary structures and that QSAR can be used for rational RNA ligand design. In addition, both the HIV-1-TAR and EV71 studies highlighted the importance of conformational dynamics in small molecule:RNA targeting, with the EV71 ligand representing one of few examples where modulation of RNA conformation is shown to directly influence biological function. This data supports small molecule regulation of RNA dynamics as an emerging mode of action for functional targeting, shifting the view of RNA dynamics from an obstacle to an opportunity. With the diphenylfuran scaffold, we identified not only the first reported ligands for the MALAT1 3’-triple helix but also the first experimental support for the impact of molecular shape in RNA-ligand design. Observed discrepancies between binding affinity and function with this scaffold underscored considerations of binding mode and conformational landscapes in SM:RNA targeting. We are moving these scaffolds forward to test and refine these guiding principles in biological systems and to identify and optimize potential leads for targeting disease-related RNA. At the same time, we are expanding our repertoire of scaffolds and methods, including dynamic combinatorial chemistry, in the pursuit of fundamental discoveries.
2.2. Computational, Screening and Selection Methods to Evaluate RNA Privileged Small Molecule Space
Given the fundamental differences between RNA and proteins, including their chemical properties, it has been hypothesized that the small molecule space needed to selectively target RNA might be distinct. We have demonstrated preliminary support for this hypothesis by comparing bioactive non-ribosomal RNA-targeted ligands to bioactive protein-targeted ligands represented as FDA-approved small molecule drugs. This work also made it possible to compare discovery methods and techniques that have been successful in identifying these bioactive RNA-targeted ligands, curate a searchable database, and optimize an online platform to facilitate progression of the field.
RNA-Biased Physicochemical, Structural and Spatial Properties of Small Molecules:
Our initial work revealed physicochemical, structural, and spatial properties that differentiate bioactive RNA ligands from bioactive protein ligands, represented as a subset of FDA-approved drugs (Figure 4A,B). In this work, bioactive RNA ligands were collected from the literature based on demonstrated activity in vitro and in vivo (cell or animal) against a non-ribosomal RNA target.35 This collection was termed the “RNA-targeted Bioactive Ligand Database” or RBIND. At the end of 2016, 104 small molecules were identified, including both traditionally-defined small molecules (< ~500 Da) and multivalent ligands that link multiple binding cores for increased affinity and selectivity. When the traditional small molecules were compared to FDA-approved drugs in the same molecular weight range, several trends emerged for bioactive RNA ligands, including: 1) compliance with medicinal chemistry rules, 2) distinctive structural features, and 3) enrichment in rod-like shape over others. Importantly, we found that bioactive RNA-targeted ligands can be found in existing drug-like chemical space, though in a specific subset of that space that may warrant further expansion. In addition, while the number of R-BIND ligands increased by 50% between the initial analysis in 2016 and the latest in 2018, no change has been observed in the chemical space occupied by these ligands.36 These properties were further supported by analysis of small molecule:RNA high resolution structures.102 A significant increase in hydrogen bonding and stacking, along with a decrease in hydrophobic effects, was observed for small molecule:RNA interactions relative to interactions in small molecule: protein structures.
Figure 4. RNA-targeted BIoactive LigaNd Database (R-BIND) reveals distinct properties of RNA-targeted small molecules.

A. Principle component analysis of R-BIND small molecules (blue), nucleic acid ligand database (NALDB) RNA-binding small molecules (orange), FDA-approved drugs (gray), R-BIND multivalent ligands (green), NALDB multivalent ligands (yellow) based on 20 cheminformatic parameters showing that R-BIND small molecules represent a subset of traditional medicinal chemistry (FDA) space. B. Box-whisker plots of representative parameters showing structural differences between RNA-targeted bioactives (R-BIND SM), protein-targeted bioactivites (FDA), and general RNA ligands (NALDB). Panel A,B reproduced from Ref. 35 with permission from John Wiley and Sons. C. Schematic of nearest neighbor analysis where the distance between R-BIND small molecules (black) and input ligands is measured using cheminformatic parameters. The distance between each R-BIND small molecule and its nearest neighbor is averaged (purple) and “R-BIND like ligands” (blue) are defined as those within the average distance to at least one R-BIND ligand. Reprinted with permission from Ref. 36. Copyright 2019 American Chemical Society.
The R-BIND collection also allowed for comparison of RNA targets, design and discovery strategies, and chemical probe characterization techniques.17 While a diverse range of discovery and development strategies were found to be successful, conclusions were limited by the relative paucity of distinct RNA targets explored and a lack of standardization in chemical probe characterization. Both will need to be addressed as the field moves forward.
To make this work more accessible, we developed an online platform that provides a user-friendly interface to search the available collection of R-BIND ligands along with tools to analyze existing and user-input molecules for similarity to the current set (https://rbind.chem.duke.edu).36 For example, users can search for and evaluate R-BIND ligands based on physicochemical, structural, and spatial properties as well functional groups and user-input substructures. Additional search features include RNA target, ligands with PDB-deposited structures, and types of in vitro or biological assays. Finally, a similarity search based on a nearest-neighbor algorithm allows researchers to identify RBIND ligands that are similar in the available parameter space, either to ligands within R-BIND or of user uploaded ligands (Figure 4C). This analysis can be used to design “R-BIND-like” small molecule libraries, optimize lead ligands into RNA-biased chemical space, or select targets, probes, assays, and control experiments based on similarity to a known R-BIND ligand. We expect that this platform will provide the scientific community valuable insight into past successes in small molecule:RNA targeting along with tools for future discovery, ultimately reducing barriers in RNA-targeted chemical probe discovery.
Screening Assays for Profiling Small Molecule:RNA Interactions:
Generalizable and simple screening assays are critical to carefully assessing selectivity among RNA targets but can be challenging to develop, in part due to the inapplicability of enzyme activity-based approaches and antibody-based methods (e.g., ELISA) to RNA. Fluorescent indicator displacement assays are particularly promising as these methods are often sensitive, high throughput, and do not require small molecule or RNA modification.103 One example from our lab takes advantage of the highly basic Tat peptide, which had been previously used in screening against HIV-1-TAR by appending the ends with a FRET pair that is sensitive to RNA binding.104 We tested the utility of this method against other RNAs as a way to rapidly screen for both binding and selectivity.89 First, four similarly sized RNA targets with varied secondary structure motifs were evaluated for binding to the Tat peptide and found to have low nanomolar dissociation constants, allowing the use of minimal RNA material. From a library of 30 RNA-targeted small molecules, the screening assay identified ligands for all four RNA structures, with a range of selectivity observed. This assay further revealed ligands that bound multiple RNAs with simple secondary structures but were not impacted by tRNA and DNA controls in previous work,88 confirming the value of using multiple targetable RNAs to evaluate specificity. Screening against multiple targets allowed statistical analyses to be used to assess small molecule binding patterns and begin to elucidate the relationship between small molecule structures and RNA binding affinity and selectivity. The broad applicability, low material cost, and rapid assessment of small molecule:RNA binding patterns available with Tat peptide displacement confirm the potential utility of generalizable binding assays for evaluating small molecule:RNA interactions.
Assays that directly relate small molecule impact on RNA function are less common but particularly valuable when available.75, 77, 79, 105, 106. For example, we recently tested differential scanning fluorimetry (DSF), in which a fluorescent dye (RiboGreen) reports on RNA melting temperatures via qPCR machine. We demonstrated the utility of this method for the 3’-MALAT1 triple helix and found that melting temperature changes observed in traditional UV-melts matched results from DSF.95 Importantly, changes in melting temperature correlated to stability in an RNase R enzyme degradation assay, suggesting that the results from this high-throughput screen may directly indicate the stabilizing or destabilizing function of the small molecule. We anticipate that this approach will allow for high-throughput stability-based screens for several RNA structures, including complex triple helices.
Moving forward, we are applying these and other screening methods to larger libraries. We hope to not only identify potential leads for chemical probe design but also to refine our cheminformatic analyses of selective RNA ligands and examine the influence of different screening methods, if any, on the outcome of these analyses. We expect the combination of binding and functional assays to yield additional and powerful insight into rational development of small molecule probes for RNA.
In summary, our combined small molecule-based efforts have led to the identification of small molecule characteristics that distinguish selective RNA-ligands from non-selective RNA and/or protein-targeted ligands as well as the identification of novel small molecule leads for viral and oncogenic ncRNAs. Importantly, this work enables the rational generation of RNA-biased libraries and/or rational lead optimization that, along with our identification of efficient screening procedures, will significantly increase the productivity of RNA-targeted screening campaigns and chemical probe development.
3. Differentiation and Characterization of RNA Structures: Pattern Recognition of RNA with Small Molecules (PRRSM)
As a complement to our understanding of small molecule properties that facilitate selective interactions, we also explored the properties of RNA structures that allow differentiation by small molecules, in this case using pattern recognition. This work has the potential to reveal selectively targetable RNA structures along with driving factors in small molecule:RNA recognition.
Molecular-scale, pattern-based sensing relies on the use of receptors, in this case small molecules, that interact differentially with the analyte of interest, in this case RNA structures, to elucidate underlying patterns or classifications in the analytes without the need for highly specific receptor:analyte pairings.19, 107, 108 We have recently developed a method termed Pattern-Recognition of RNA using Small Molecules (PRRSM, Figure 5)109, 110 and published proof-of-concept studies demonstrating that small molecules can classify RNA secondary structure and that RNA and small molecule shape plays a critical role in RNA recognition.109 Follow up studies have revealed the importance of conformational dynamics in this recognition111 and the ability of this method to predict RNA secondary structures at specific nucleotide positions.112
Figure 5: Pattern recognition of RNA by small molecules (PRRSM).

An array of small molecule receptors is titrated with RNA secondary structure analytes. Utilizing the small molecule differential binding and an unbiased statistical method allows for clustering based on the RNA structural motifs. Reprinted with permission from Ref. 19. Copyright 2019 American Chemical Society.
Development of the PRRSM Technique and Preliminary Insights
We first evaluated the ability of aminoglycosides, arguably the best characterized RNA ligands, to differentiate canonical RNA secondary structure motifs such as bulges, internal and apical loops.109 Eleven aminoglycosides, nine commercial and two synthetically modified, were evaluated for binding against a training set of 16 RNAs with well-predicted structures that varied in the size and sequence of the motifs (Figure 6A). To measure site-specific binding, we incorporated the solvatochromic chemosensor benzofuranyluridine (BFU) 113, 114 via solid-phase synthesis. The BFU-labeled RNA training set was incubated with the aminoglycosides at varying concentrations in a 384-well plate and the emission data was used as input for principal component analysis (PCA). PCA revealed an unbiased clustering based on secondary structure class and leave-one-out cross validation (LOOCV) confirmed that PRRSM was able to predict these secondary structure motif classes with 100% accuracy.
Figure 6. PRRSM classifies individual RNA secondary structures under dynamic conditions.

A. Schematic of secondary structures (unpaired regions) in PRRSM RNA training set with BFU-labeled sites noted with a blue star. Reprinted with permission from Ref. 109. Copyright 2017 American Chemical Society. B. PCA plot of training set under conditions including polyethylene glycol (PEG) and increased temperature. Open ovals indicate 95% confidence intervals. The predictive power for the sequences was 92%. Buffer: 10 mM NaH2PO4, 25 mM NaCl, 4 mM MgCl2, 0.5 mM EDTA, 8 mM PEG 12 000, pH 7.4 at 37 °C. Reproduced from Ref. 111 with permission from the Royal Society of Chemistry.
These trends, including the modest differentiation of individual sequences, allowed preliminary insights into aminoglycoside:RNA molecular recognition. The largest amount of variance, i.e. differentiation, within the data was found to correlate with the motif size of the RNA secondary structures followed by sequence of the motif. This qualitative analysis aligned with previously published work showing that RNA recognition is heavily dependent on the topology of the RNA structure,115 which is driven by motif size and then sequence. To evaluate small molecule-based trends, we first compared Tanimoto coefficients among the aminoglycosides. While some trends were observed, globally consistent correlations could not be identified based on these fingerprints or through further analysis of simple physicochemical properties (total charge, molecular weight, etc.). The lack of correlation with physicochemical properties and fingerprint analysis, along with the influence of topology, are in line with other evidence that aminoglycoside recognition may be driven largely by three-dimensional properties or shape.116, 117 While the local flexibility of aminoglycosides renders the in-solution structures difficult to predict computationally, this work suggests that different aminoglycosides access distinct conformations that ultimately allow differentiation of RNA structures.
PRRSM Reveals Impact of RNA Dynamics
The initial success of the PRRSM method inspired a range of both fundamental and applied investigations, including further probing of the influence of RNA topology on small molecule:RNA recognition. One way to purposefully modulate RNA topology is through alteration of the RNA environment via changing buffer conditions. For example, the modulation of mono- and divalent cation concentrations, presence or absence of molecular crowders, and changes in pH and temperature are known to alter the stability of RNA secondary and tertiary structures.118, 119 Utilizing predictive power (LOOCV) to assess RNA differentiation, we first evaluated the impact of several buffer conditions often used in small molecule:RNA assays.111 High sodium (140 mM) and low pH (5.0) were found to significantly reduce differentiation, likely due to a decrease in binding affinity as a result of interfering with the electrostatic nature of aminoglycoside:RNA interactions. Removal of magnesium, near neutral pH (6-8), and different buffer composition (phosphate versus Tris) had minimal impact relative to the original conditions. The addition of polyethylene glycol (PEG) and increased temperature (25°C to 37°C), however, significantly improved differentiation despite the reported destabilization of secondary structure motifs under these conditions (Figure 6B).119 The opposite was observed for increased magnesium concentration, which would be expected to stabilize secondary structure, though the competition between the magnesium ions and the aminoglycosides for RNA binding may also play a role. These combined results suggested that specific RNA secondary structures are best recognized under conditions that favor dynamic motion, i.e. access to multiple conformations, which is consistent with previously published work demonstrating that RNA structures sample a set of defined but distinct conformations, thus facilitating differentiation.20 Such work, along with the examples above,88, 91 is shifting the view of RNA dynamics from a hindrance to a property that can be leveraged for specific recognition by small molecules.
PRRSM Structural Classification and Prediction
Along with these fundamental insights, we investigated the power of the PRRSM method to gather site-specific structural information for RNA. We began with biologically relevant RNA constructs with multiple identified secondary structure motifs and/or inducible conformational changes.109, 112 For each nucleotide position of interest, we synthesized the corresponding BFU-labeled construct. Samples included a truncated version of HIV-1 TAR RNA labeled at the 3 nucleotide (nt) bulge or 6 nt hairpin loop109 along with labeled constructs of the prequeuosine-1 riboswitch (PreQ1) and fluoride riboswitch (FR) (three each),112 which both undergo analyte-induced conformational changes (Figure 7A).120-124 Of the eight RNA constructs analyzed via PRRSM, six of the constructs were classified as expected based on the experimentally determined or predicted structures (Figure 7B). Control experiments, along with literature precedence,124 suggested that the two poorly-predicted constructs, the U6- and U11-sites of the fluoride riboswitch, were modified at positions that inhibit proper RNA folding. All PRRSM-based observations of unfolded and folded riboswitch states were confirmed via NMR. PRRSM was thus able to classify structures specific to RNA conformations, including folded and unfolded states of the same RNA, and provide insight into modification-induced changes in the structure.
Figure 7. PRRSM classifies apo and bound secondary structures in prequeuosine-1 (PreQ1) riboswitch.

A. Left: PreQ riboswitch secondary structure in the unbound and bound state. Right: Bound structure of PreQ1 (PDB 2L1V) with highlighted sites of BFU fluorophore insertion (U9-red, U11-blue, U14-green) and PreQ1 ligand (orange). B. PCA plot of the U9, U11, and U14 modified RNA in the absence (w/o Lig) and presence (w Lig) of PreQ1 ligand. All constructs were predicted to be the correct structure in both the unbound and bound states under standard conditions (10 mM NaH2PO4, 25 mM NaCl, 4 mM MgCl2, 0.5 mM EDTA, pH 7.3 at 25 °C. Reprinted with permission from Ref. 112. Copyright 2019 American Chemical Society.
In summary, this method has allowed us to: 1) elucidate small molecule properties that impact selective small molecule:RNA interactions; 2) identify the structural and topological determinants of RNA recognition along with the impact of environment; and 3) classify and predict functionally-relevant changes in RNA structure. Ongoing work includes using computational methods to understand the contribution of conformations/dynamics to differentiation, the development of more general (i.e. label free) methods to assess binding, and expansion in terms of both small molecule ligands and diversity of RNA structures evaluated. We are also applying this method to larger RNAs, both to gain site-specific structural information in complex structures and to pursue classification of molecules such as long noncoding RNAs (lncRNAs).
Summary and Outlook
Our work has leveraged a range of approaches to elucidate the drivers of selective small molecule:RNA recognition. Scaffold-driven synthetic efforts have led to discoveries such as amiloride as an RNA-privileged scaffold and to design principles to tune amilorides to bind viral RNAs and to tune diphenylfurans for selective binding of the triple helix of oncogenic lncRNA MALAT1. In a complementary approach, we identified features of published RNA ligands with biological activity that distinguish them from protein-targeted drugs. We worked to generalize screening methods for assessing RNA binding and stability, including FRET-based peptide displacement, fluorescent indicator displacement, and differential scanning fluorimetry. To investigate targetable properties of RNA, we developed a pattern recognition method (PRRSM) and first elucidated distinguishing features of RNA secondary structures, which included not only the size and shape but also the conformational dynamics. Indeed, recurring themes in this work that are being further explored include the importance of shape / shape complementarity in small molecule:RNA interactions as well as opportunities in recognizing and modulating the RNA conformational landscape.
Importantly, these approaches and insights complement and are bolstered by the growing body of research in the field. First, on the question of what properties biologically active RNA-targeting small molecules might require, it has become clear that RNA bioactives can be “drug-like” and can be identified in large chemical libraries.36 At the same time, RNA ligands tend to have distinct structural and chemical properties compared to protein-binding ligands, as seen in our work35, 36 and supported by others.125, 126 These results suggest that specifically curated or focused libraries may be even more effective for RNA ligand discovery and that such principles can be used to guide lead optimization. As the number of RNA bioactive ligands grows, it will be worthwhile to evaluate whether distinct classes of RNA select for specific properties similar to the way protein classes have distinct ligands. In addition, the fact that RNA bioactives have these distinguishing properties suggests that there might be historically underexplored chemical space for this class of chemical probes. Furthermore, larger molecules such as multivalent ligands, peptides, and aminoglycoside-conjugates are also showing promise. As broad, guiding principles for small molecules that target RNA continue to be refined, high-throughput screens that readily assess selectivity will be critical and screens that incorporate function especially valuable. Detailed studies of thermodynamic and kinetic drivers of small molecule:RNA interactions along with structural studies will provide the needed rationale for these distinguishing features. Our understanding of what makes an ideal RNA target also continues to expand. The importance of carefully considering, and possibly leveraging, conformational dynamics observed via PRSSM was supported by previous and continuing work of the Al-Hashimi lab, who has shown that structure-based approaches are more successful for RNAs such as HIV-1-TAR and RRE when docking against multiple experimentally informed conformations.49, 127 Along these lines, allosteric mechanisms in RNA-targeting were illustrated by Hermann and co-workers when targeting the HCV IRES sequence62 and in our work targeting the EV71 IRES.91 The Disney lab has targeted several functionally important secondary structures in pre-microRNAs that inhibit processing by the Dicer enzyme.128 Given that the conformation of these sites may impact protein binding129 and/or be altered upon binding to the enzyme,130 it would not be surprising if stabilization of alternative conformations inhibits processing. At the same time, Weeks and co-workers used lessons from protein-based targeting to propose that complex RNAs, such as ribosomal RNA, offer the greatest chance of success for selective RNA targeting due to the formation of “high-quality pockets.”16 The targeting of an RNA:protein complex by the SMN2 small molecule splicing modifiers supports this hypothesis.77, 131, 132 It indeed makes sense that these RNAs would follow protein-type rules given that they target similar functions as those targeted in traditional protein campaigns, i.e. sites of chemical reactivity (translation, splicing) or small molecule binding (riboswitches). In a complementary approach, Schneekloth and co-workers analyzed “ligandable” pockets in RNA PDB structures using similar approaches to those used for proteins and found significant overlap in the pocket characteristics between RNAs and proteins, particularly for more complex RNAs.133 Such analyses, and RNA-targeting in general, will be significantly bolstered by additional elucidation of RNA structures, particularly in ligand-bound states, and continued progress in RNA 3D structure prediction based on experimental inputs such as chemical probing.134
As we look to understand how in vitro knowledge transfers to biological systems, other important considerations will include the level of expression of a given RNA,42, 135 which is influenced by multiple factors, including healthy versus disease states, the lifetime of the RNA, and the cell cycle. Highly expressed RNAs may be easier to target as the criteria for affinity and selectivity will be less stringent, and some have made the case that expression level may be one reason that the ribosome has been so successfully targeted.16 Additional work is needed to elucidate the criterion for the relative affinity and selectivity needed for a given small molecule:RNA interaction and whether this is truly influenced by the expression level of the RNA target. Methods to assess target engagement, such as the Chem-CLIP136, 137 and Ribo-SNAP138, 139 used by Disney or the use of sequencing in splicing targets, will be critical to answering this question and to progressing drug development. Methods that assess RNA structure and dynamics in cells, such as in-cell chemical probing methods,140 should also facilitate evaluation of target engagement.
While challenges remain, the progress being made in understanding small molecule:RNA recognition and the promise this holds for both elucidation of RNA function and therapeutic targeting is inspiring. As a field, we will need to both deepen and expand upon existing knowledge by pursuing a wide range of approaches to small molecule discovery and assessment. Given historic frustrations in RNA targeting, it is critically important that we maintain rigor and transparency in our analyses of chemical probes while also recognizing that many “rules” of affinity, selectivity, and assessment of target engagement may be different for RNA than proteins and many protein-targeting small molecules also break these rules. Without a doubt, it is an incredibly exciting and dynamic time to be in the small molecule:RNA targeting field, where so-called barriers are continuously overcome and each new success story re-shapes our view of RNA recognition. The potential for revealing new biology and for relieving suffering from incurable human diseases is overwhelming – won’t you join us?
Acknowledgements
The work described here was made possible by a brilliant and incredibly dedicated team of undergraduate, graduate, postdoctoral and staff researchers hailing from seven different countries over seven years. I am proud and grateful to have worked with each of them. This manuscript specifically relied on insight and edits from lab members Aline Umuhire Juru, Christopher Laudeman, Martina Zafferani, Giacomo Padroni, Emily McFadden, Anita Donlic, Sarah Wicks, Zhengguo Cai, Kamillah Kassam, James Falese, and Emily Swanson along with figure-making assistance from Martina Zafferani and Kamillah Kassam.
Funding for this writing was provided by Duke University, the National Science Foundation (CAREER 1750375), and the U.S. National Institutes of Health (R35 GM124785; U54 AI150470).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Morris KV and Mattick JS, Nat. Rev. Genet, 2014, 15, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cech TR and Steitz JA, Cell, 2014, 157, 77–94. [DOI] [PubMed] [Google Scholar]
- 3.Deigan KE and Ferre-D'Amare AR, Acc. Chem. Res, 2011, 44, 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson DN, Nat Rev Microbiol, 2014, 12, 35–48. [DOI] [PubMed] [Google Scholar]
- 5.Hermann T, Wiley Interdiscip Rev RNA, 2016, 7, 726–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donlic A and Hargrove AE, Wiley Inter. Rev. RNA, 2018, 9, e1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt AM and Chang HY, Cancer Cell, 2016, 29, 452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling H, Fabbri M and Calin GA, Nat. Rev. Drug Discov, 2013, 12, 847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sztuba-Solinska J, Chavez-Calvillo G and Cline SE, Bioorg. Med. Chem, 2019, 27, 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.FDA Approves Oral Treatment for Spinal Muscular Atrophy, https://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy (accessed August 7, 2020).
- 11.Juliano RL, Nucleic Acids Res., 2016, 44, 6518–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein CA and Castanotto D, Molecular Therapy, 2017, 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin W and Rogge M, Clin. Transl. Sci, 2019, 12, 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas J and Hergenrother P, Chem. Rev, 2008, 108, 1172–1224. [DOI] [PubMed] [Google Scholar]
- 15.Xie Y, Tam VK and Tor Y, in The Chemical Biology of Nucleic Acids, John Wiley & Sons, Ltd, 2010, DOI: 10.1002/9780470664001.ch6, pp. 115–140. [DOI] [Google Scholar]
- 16.Warner KD, Hajdin CE and Weeks KM, Nat. Rev. Drug Discovery, 2018, 17, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan BS, Forte JE and Hargrove AE, Nucleic Acids Res., 2018, 46, 8025–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strobel EJ, Yu AM and Lucks JB, Nat. Rev. Genet, 2018, 19, 615–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eubanks CS and Hargrove AE, Biochemistry, 2019, 58, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganser LR, Kelly ML, Herschlag D and Al-Hashimi HM, Nature Reviews Molecular Cell Biology, 2019, 20, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seetin MG and Mathews DH, Methods Mol. Biol. (N. Y., NY, U. S.), 2012, 905, 99–122. [DOI] [PubMed] [Google Scholar]
- 22.Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH and Weeks KM, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, 5498–5503, S5498/5491-S5498/5434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegfried NA, Busan S, Rice GM, Nelson JAE and Weeks KM, Nature Methods, 2014, 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kladwang W, VanLang CC, Cordero P and Das R, Biochemistry, 2011, 50, 8049–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keane SC, Heng X, Lu K, Kharytonchyk S, Ramakrishnan V, Carter G, Barton S, Hosic A, Florwick A, Santos J, Bolden NC, McCowin S, Case DA, Johnson BA, Salemi M, Telesnitsky A and Summers MF, Science, 2015, 348, 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das R, Karanicolas J and Baker D, Nat. Methods, 2010, 7, 291–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao ZC, Adamiak RW, Blanchet MF, Boniecki M, Bujnicki JM, Chen SJ, Cheng C, Chojnowski G, Chou FC, Cordero P, Cruz JA, Ferre-D'amare AR, Das R, Ding F, Dokholyan NV, Dunin-Horkawicz S, Kladwang W, Krokhotin A, Lach G Magnus M, Major F, Mann TH, Masquida B, Matelska D, Meyer M, Peselis A, Popenda M, Purzycka KJ, Serganov A, Stasiewicz J, Szachniuk M, Tandon A, Tian S, Wang J, Xia Y, Xu XJ, Zhang JW, Zha PN, Zok T and Westhof E, RNA, 2015, 21, 1066–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu G, Kaushal PS, Wang J, Shigematsu H, Piazza CL, Agrawal RK, Belfort M and Wang HW, Nat Struct Mol Biol, 2016, 23, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agafonov DE, Kastner B, Dybkov O, Hofele RV, Liu WT, Urlaub H, Luhrmann R and Stark H, Science, 2016, 351, 1416–1420. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen TH, Galej WP, Bai XC, Oubridge C, Newman AJ, Scheres SH and Nagai K, Nature., 2016, 530, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Leiro R and Scheres SHW, Nature., 2016, 537, 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang K, Keane SC, Su Z, Irobalieva RN, Chen M, Van V, Sciandra CA, Marchant J, Heng X, Schmid MF, Case DA, Ludtke SJ, Summers MF and Chiu W, Structure, 2018, 26, 490–498.e493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang K, Li S, Kappel K, Pintilie G, Su Z, Mou T-C, Schmid MF, Das R and Chiu W, Nat. Commun, 2019, 10, 5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng CY, Chou FC, Kladwang W, Tian S, Cordero P and Das R, Elife, 2015, 4, e07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan BS, Forte JE, Culver RN, Zhang Y and Hargrove AE, Angew. Chem., Int. Ed, 2017, 56, 13498–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan BS, Sanaba BG, Donlic A, Karloff DB, Forte JE, Zhang Y and Hargrove AE, ACS Chem. Biol, 2019, 14, 2691–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Connelly CM, Moon MH and Schneekloth JS, Cell Chem. Biol, 2016, 23, 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ursu A, Vezina-Dawod S and Disney MD, Drug Discov Today, 2019, 24, 2002–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Giorgio A and Duca M, MedChemComm, 2019, 10, 1242–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Disney MD, Velagapudi SP, Li Y, Costales MG and Childs-Disney JL, in Methods Enzymol., ed. Hargrove AE, Academic Press, 2019, vol. 623, pp. 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angelbello AJ, Rzuczek SG, Mckee KK, Chen JL, Olafson H, Cameron MD, Moss WN, Wang ET and Disney MD, Proc. Natl. Acad. Sci. U. S. A, 2019, 116, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costales MG, Haga CL, Velagapudi SP, Childs-Disney JL, Phinney DG and Disney MD, J. Am. Chem. Soc, 2017, 139, 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J, Bai Y, Chembazhi UV, Peng S, Yum K, Luu LM, Hagler LD, Serrano JF, Chan HYE, Kalsotra A and Zimmerman SC, Proc. Natl. Acad. Sci. U. S. A, 2019, 116, 8709–8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hilimire TA, Chamberlain JM, Anokhina V, Bennett RP, Swart O, Myers JR, Ashton JM, Stewart RA, Featherston AL, Gates K, Helms ED, Smith HC, Dewhurst S and Miller BL, ACS Chem. Biol, 2017, 12, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anokhina VS, McAnany JD, Ciesla JH, Hilimire TA, Santoso N, Miao H and Miller BL, Bioorg. Med. Chem, 2019, 27, 2972–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rzuczek SG, Gao Y, Tang ZZ, Thornton CA, Kodadek T and Disney MD, ACS Chem. Biol, 2013, 8, 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Velagapudi SP, Cameron MD, Haga CL, Rosenberg LH, Lafitte M, Duckett DR, Phinney DG and Disney MD, Proc. Natl. Acad. Sci. U. S. A, 2016, 113, 5898–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM and Al-Hashimi HM, Nat. Chem. Biol, 2011, 7, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ganser LR, Lee J, Rangadurai A, Merriman DK, Kelly ML, Kansal AD, Sathyamoorthy B and Al-Hashimi HM, Nat. Struct. Mol. Biol, 2018, 25, 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Means J, Katz S, Nayek A, Anupam R, Hines JV and Bergmeier SC, Bioorg. Med. Chem. Lett, 2006, 16, 3600–3604. [DOI] [PubMed] [Google Scholar]
- 51.Anupam R, Nayek A, Green NJ, Grundy FJ, Henkin TM, Means JA, Bergmeier SC and Hines JV, Bioorg. Med. Chem. Lett, 2008, 18, 3541–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maciagiewicz I, Zhou S, Bergmeier SC and Hines JV, Bioorg. Med. Chem. Lett, 2011, 21, 4524–4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Orac CM, Zhou S, Means JA, Boehm D, Bergmeier SC and Hines JV, J. Med. Chem, 2011, 54, 6786–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbachyn MR and Ford CW, Angew. Chem., Int. Ed, 2003, 42, 2010–2023. [DOI] [PubMed] [Google Scholar]
- 55.Ippolito JA, Kanyo ZF, Wang D, Franceschi FJ, Moore PB, Steitz TA and Duffy EM, J. Med. Chem, 2008, 51, 3353–3356. [DOI] [PubMed] [Google Scholar]
- 56.Zhao M, Ratmeyer L, Peloquin RG, Yao S, Kumar A, Spychala J, Boykin DW and Wilson WD, Bioorg. Med. Chem, 1995, 3, 785–794. [DOI] [PubMed] [Google Scholar]
- 57.Gelus N, Bailly C, Hamy F, Klimkait T, Wilson WD and Boykin DW, Bioorg. Med. Chem, 1999, 7, 1089–1096. [DOI] [PubMed] [Google Scholar]
- 58.Xiao G, Kumar A, Li K, Rigl CT, Bajic M, Davis TM, Boykin DW and Wilson WD, Bioorg. Med. Chem, 2001, 9, 1097–1113. [DOI] [PubMed] [Google Scholar]
- 59.Chaires JB, Ren J, Hamelberg D, Kumar A, Pandya V, Boykin DW and Wilson WD, J. Med. Chem, 2004, 47, 5729–5742. [DOI] [PubMed] [Google Scholar]
- 60.Yang WY, Gao R, Southern M, Sarkar PS and Disney MD, Nat Commun, 2016, 7, 11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dibrov SM, Ding K, Brunn ND, Parker MA, Bergdahl BM, Wyles DL and Hermann T, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 5223–5228, S5223/5221-S5223/5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dibrov SM, Parsons J, Carnevali M, Zhou S, Rynearson KD, Ding K, Garcia Sega E, Brunn ND, Boerneke MA, Castaldi MP and Hermann T, J. Med. Chem, 2014, 57, 1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Velagapudi SP, Gallo SM and Disney MD, Nat. Chem. Biol, 2014, 10, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA and Disney MD, J. Am. Chem. Soc, 2012, 134, 4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhao F, Zhao Q, Blount KF, Han Q, Tor Y and Hermann T, Angew. Chem., Int. Ed, 2005, 44, 5329–5334. [DOI] [PubMed] [Google Scholar]
- 66.Staple DW, Venditti V, Niccolai N, Elson-Schwab L, Tor Y and Butcher SE, ChemBioChem., 2008, 9, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar S, Ranjan N, Kellish P, Gong C, Watkins D and Arya DP, Org. Biomol. Chem, 2016, 14, 2052–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watkins D, Maiti K and Arya DP, Methods Mol Biol, 2019, 1973, 147–162. [DOI] [PubMed] [Google Scholar]
- 69.Waduge P, Sati GC, Crich D and Chow CS, Bioorg. Med. Chem, 2019, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chakraborty J, Kanungo A, Mahata T, Kumar K, Sharma G, Pal R, Ahammed KS, Patra D, Majhi B, Chakrabarti S, Das S and Dutta S, Chem. Commun, 2019, 55, 14027–14030. [DOI] [PubMed] [Google Scholar]
- 71.Sztuba-Solinska J, Shenoy SR, Gareiss P, Krumpe LR, Le Grice SF, O'Keefe BR and Schneekloth JS Jr., J. Am. Chem. Soc, 2014, 136, 8402–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Connelly CM, Boer RE, Moon MH, Gareiss P and Schneekloth JS Jr., ACS Chem. Biol, 2017, 12, 435–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Connelly CM, Numata T, Boer RE, Moon MH, Sinniah RS, Barchi JJ, Ferré-D’Amaré AR and Schneekloth JS, Nat. Commun, 2019, 10, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abulwerdi FA, Xu W, Ageeli AA, Yonkunas MJ, Arun G, Nam H, Schneekloth JS, Dayie TK, Spector D, Baird N and Le Grice SFJ, ACS Chem. Biol, 2019, 14, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fedorova O, Jagdmann GE, Adams RL, Yuan L, Van Zandt MC and Pyle AM, Nature Chemical Biology, 2018, 14, 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinverni JC, Mayhood T, Villafania A, Nahvi A, Murgolo N, Barbieri CM, Mann PA, Carr D, Xia E, Zuck P, Riley D, Painter RE, Walker SS, Sherborne B, de Jesus R, Pan WD, Plotkin MA, Wu J, Rindgen D, Cummings J, Garlisi CG, Zhang RM, Sheth PR, Gill CJ, Tang HF and Roemer T, Nature., 2015, 526, 672–677. [DOI] [PubMed] [Google Scholar]
- 77.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang XL, Van Hoosear M, Shin Y, Chin DN, Keller CG, Beibel M, Renaud NA, Smith TM, Salcius M, Shi XY, Hild M, Servais R, Jain M, Deng L, Bullock C, McLellan M, Schuierer S, Murphy L, Blommers MJJ, Blaustein C, Berenshteyn F, Lacoste A, Thomas JR, Roma G, Michaud GA, Tseng BS, Porter JA, Myer VE, Tallarico JA, Hamann LG, Curtis D, Fishman MC, Dietrich WF, Dales NA and Sivasankaran R, Nature Chemical Biology, 2015, 11, 511–517. [DOI] [PubMed] [Google Scholar]
- 78.Cheung AK, Hurley B, Kerrigan R, Shu L, Chin DN, Shen YP, O'Brien G, Sung MJ, Hou Y, Axford J, Cody E, Sun R, Fazal A, Fridrich C, Sanchez CC, Tomlinson RC, Jain M, Deng L, Hoffmaster K, Song C, Van Hoosear M, Shin Y, Servais R, Towler C, Hild M, Curtis D, Dietrich WF, Hamann LG, Briner K, Chen KS, Kobayashi D, Sivasankaran R and Dales NA, J. Med. Chem, 2018, 61, 11021–11036. [DOI] [PubMed] [Google Scholar]
- 79.Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KKY, Karp GM, Qi H, Woll MG, Chen G, Zhang N, Gabbeta V, Vazirani P, Bhattacharyya A, Furia B, Risher N, Sheedy J, Kong R, Ma J, Turpoff A, Lee C-S, Zhang X, Moon Y-C, Trifillis P, Welch EM, Colacino JM, Babiak J, Almstead NG, Peltz SW, Eng LA, Chen KS, Mull JL, Lynes MS, Rubin LL, Fontoura P, Santarelli L, Haehnke D, McCarthy KD, Schmucki R, Ebeling M, Sivaramakrishnan M, Ko C-P, Paushkin SV, Ratni H, Gerlach I, Ghosh A and Metzger F, Science, 2014, 345, 688. [DOI] [PubMed] [Google Scholar]
- 80.Ratni H, Ebeling M, Baird J, Bendels S, Bylund J, Chen KS, Denk N, Feng Z, Green L, Guerard M, Jablonski P, Jacobsen B, Khwaja O, Kletzl H, Ko C-P, Kustermann S, Marquet A, Metzger F, Mueller B, Naryshkin NA, Paushkin SV, Pinard E, Poirier A, Reutlinger M, Weetall M, Zeller A, Zhao X and Mueller L, J. Med. Chem, 2018, 61, 6501–6517. [DOI] [PubMed] [Google Scholar]
- 81.Mullard A, Nat. Rev. Drug Discovery, 2017, 16, 813–815. [DOI] [PubMed] [Google Scholar]
- 82.Chakradhar S, Nature Medicine, 2017, 23, 532–534. [DOI] [PubMed] [Google Scholar]
- 83.Petrone J and DeFrancesco L, Nat. Biotechnol, 2018, 36, 787–790. [DOI] [PubMed] [Google Scholar]
- 84.Stevens M, De Clercq E and Balzarini J, Med. Res. Rev, 2006, 26, 595–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Massey AP, Harley WR, Pasupuleti N, Gorin FA and Nantz MH, Bioorg. Med. Chem. Lett, 2012, 22, 2635–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Buckley BJ, Aboelela A, Minaei E, Jiang LX, Xu Z, Ali U, Fildes K, Cheung C-Y, Cook SM, Johnson DC, Bachovchin DA, Cook GM, Apte M, Huang M, Ranson M and Kelso MJ, J. Med. Chem, 2018, 61, 8299–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Massink A, Amelia T, Karamychev A and Ijzerman AP, Med. Res. Rev, 2020, 40, 683–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patwardhan NN, Ganser LR, Kapral GJ, Eubanks CS, Lee J, Sathyamoorthy B, Al-Hashimi H and Hargrove AE, MedChemComm, 2017, 8, 1022–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patwardhan NN, Cai Z, Newson CN and Hargrove AE, Org. Biomol. Chem, 2019, 17, 1778–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patwardhan NN, Cai Z, Umuhire Juru A and Hargrove AE, Org. Biomol. Chem, 2019, 17, 9313–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davila-Calderon J, Patwardhan NN, Chiu L-Y, Sugarman A, Cai Z, Penutmutchu SR, Li M-L, Brewer G, Hargrove AE and Tolbert BS, Nat. Commun, 2020, 11, 4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tolbert M, Morgan CE, Pollum M, Crespo-Hernández CE, Li ML, Brewer G and Tolbert BS.
- 93.Umuhire Juru A, Cai Z, Jan A and Hargrove AE, Chem. Commun, 2020, 56, 3555–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr. and Hargrove AE, Angew. Chem., Int. Ed, 2018, 57, 13242–13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Donlic A, Zafferani M, Padroni G, Puri M and Amanda E Hargrove, Nucleic Acids Res., 2020, 48, 7653–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arun G, Aggarwal D and Spector DL, Non-Coding RNA, 2020, 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA and Steitz JA, Nat Struct Mol Biol, 2014, 21, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilusz JE, Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms, 2016, 1859, 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao M, Ratmeyer L, Peloquin RG, Yao SJ, Kumar A, Spychala J, Boykin DW and Wilson WD, Bioorg. Med. Chem, 1995, 3, 785–794. [DOI] [PubMed] [Google Scholar]
- 100.Yang W-Y, Gao R, Southern M, Sarkar PS and Disney MD, Nat. Commun, 2016, 7, 11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ratmeyer L, Zapp M, Green M, Vinayak R, Kumar A, Boykin DW and Wilson WD, Biochemistry, 1996, 35, 13689–13696. [DOI] [PubMed] [Google Scholar]
- 102.Padroni G, Patwardhan NN, Schapira M and Hargrove AE, RSC Medicinal Chemistry, 2020, 11, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wicks SL and Hargrove AE, Methods, 2019, 167, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Matsumoto C, Hamasaki K, Mihara H and Ueno A, Bioorg. Med. Chem. Lett, 2000, 10, 1857–1861. [DOI] [PubMed] [Google Scholar]
- 105.Hung M, Patel P, Davis S and Green SR, J. Virol, 1998, 72, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lorenz DA and Garner AL, Springer Berlin Heidelberg, Berlin, Heidelberg, DOI: 10.1007/7355_2017_3, pp. 1–32. [DOI] [Google Scholar]
- 107.Anzenbacher P, Lubal P, Bucek P, Palacios MA and Kozelkova ME, Chem. Soc. Rev, 2010, 39, 3954–3979. [DOI] [PubMed] [Google Scholar]
- 108.Umali A and Anslyn E, Curr. Opin. Chem. Biol, 2010, 14, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eubanks CS, Forte JE, Kapral GJ and Hargrove AE, J. Am. Chem. Soc, 2017, 139, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Padroni G, Eubanks CS and Hargrove AE, in Methods Enzymol., ed. Hargrove AE, Academic Press, 2019, vol. 623, pp. 101–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eubanks CS and Hargrove AE, Chem. Commun. (Cambridge, U. K.), 2017, 53, 13363–13366. [DOI] [PubMed] [Google Scholar]
- 112.Eubanks CS, Zhao B, Patwardhan NN, Thompson RD, Zhang Q and Hargrove AE, J. Am. Chem. Soc, 2019, 141, 5692–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tanpure AA and Srivatsan SG, ChemBioChem, 2012, 13, 2392–2399. [DOI] [PubMed] [Google Scholar]
- 114.Tanpure AA and Srivatsan SG, Nucleic Acids Res., 2015, 43, e149/141–e149/110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bailor MH, Sun X and Al-Hashimi HM, Science, 2010, 327, 202–206. [DOI] [PubMed] [Google Scholar]
- 116.Vicens Q and Westhof E, Chembiochem, 2003, 4, 1018–1023. [DOI] [PubMed] [Google Scholar]
- 117.Kondo J, Hainrichson M, Nudelman I, Shallom-Shezifi D, Barbieri CM, Pilch DS, Westhof E and Baasov T, Chembiochem, 2007, 8, 1700–1709. [DOI] [PubMed] [Google Scholar]
- 118.Draper DE, RNA, 2004, 10, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tyrrell J, Weeks KM and Pielak GJ, Biochemistry, 2015, 54, 6447–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roth A, Winkler WC, Regulski EE, Lee BW, Lim J, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, Iwata-Reuyl D and Breaker RR, Nat Struct Mol Biol, 2007, 14, 308–317. [DOI] [PubMed] [Google Scholar]
- 121.Kang M, Peterson R and Feigon J, Mol. Cell, 2010, 39, 653–655. [Google Scholar]
- 122.Zhang Q, Kang M, Peterson RD and Feigon J, J. Am. Chem. Soc, 2011, 133, 5190–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ren A, Rajashankar KR and Patel DJ, Nature., 2012, 486, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhao B, Guffy SL, Williams B and Zhang Q, Nat. Chem. Biol, 2017, 13, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rizvi NF, Santa Maria JP, Nahvi A, Klappenbach J, Klein DJ, Curran PJ, Richards MP, Chamberlin C, Saradjian P, Burchard J, Aguilar R, Lee JT, Dandliker PJ, Smith GF, Kutchukian P and Nickbarg EB, Slas Discovery, 2020, 25, 384–396. [DOI] [PubMed] [Google Scholar]
- 126.Haniff HS, Knerr L, Liu XH, Crynen G, Bostrom J, Abegg D, Adibekian A, Lekah E, Wang KW, Cameron MD, Yildirim I, Lemurell M and Disney MD, Nature Chemistry, 2020, 12, 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chu CC, Plangger R, Kreutz C and Al-Hashimi HM, Nucleic Acids Res., 2019, 47, 7105–7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Velagapudi SP, Vummidi BR and Disney MD, Curr. Opin. Chem. Biol, 2015, 24, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nam Y, Chen C, Gregory Richard I., Chou James J. and Sliz P, Cell, 2011, 147, 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu Z, Wang J, Cheng H, Ke X, Sun L, Zhang QC and Wang H-W, Cell, 2018, 173, 1191–1203.e1112. [DOI] [PubMed] [Google Scholar]
- 131.Sivaramakrishnan M, McCarthy KD, Campagne S, Huber S, Meier S, Augustin A, Heckel T, Meistermann H, Hug MN, Birrer P, Moursy A, Khawaja S, Schmucki R, Berntenis N, Giroud N, Golling S, Tzouros M, Banfai B, Duran-Pacheco G, Lamerz J, Hsiu Liu Y, Luebbers T, Ratni H, Ebeling M, Cléry A, Paushkin S, Krainer AR, Allain FHT and Metzger F, Nat. Commun, 2017, 8, 1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Campagne S, Boigner S, Rüdisser S, Moursy A, Gillioz L, Knörlein A, Hall J, Ratni H, Cléry A and Allain FHT, Nature Chemical Biology, 2019, 15, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hewitt WM, Calabrese DR and Schneekloth JS, Bioorg. Med. Chem, 2019, 27, 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kappel K, Zhang KM, Su ZM, Watkins AM, Kladwang W, Li SS, Pintilie G, Topkar VV, Rangan R, Zheludev IN, Yesselman JD, Chiu W and Das R, Nat. Methods, 2020, 17, 698-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu XH, Haniff HS, Childs-Disney JL, Shuster A, Aikawa H, Adibekian A and Disney MD, J. Am. Chem. Soc, 2020, 142, 6970–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guan L and Disney MD, Angew. Chem., Int. Ed, 2013, 52, 10010–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yang WY, Wilson HD, Velagapudi SP and Disney MD, J. Am. Chem. Soc, 2015, 137, 5336–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rzuczek SG, Colgan LA, Nakai Y, Cameron MD, Furling D, Yasuda R and Disney MD, Nat. Chem. Biol, 2017, 13, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li Y and Disney MD, ACS Chem. Biol, 2018, 13, 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mitchell D, Assmann SM and Bevilacqua PC, Curr. Opin. Struct. Biol, 2019, 59, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]