

A 3D PNS disease–on–a–chip enables the monitoring of pathophysiological processes, including demyelination and remyelination.
Abstract
Demyelinating diseases involve loss of myelin sheaths and eventually lead to neurological problems. Unfortunately, the precise mechanisms remain unknown, and there are no effective therapies. To overcome these limitations, a reliable and physiologically relevant in vitro model is required. Here, we present a three-dimensional peripheral nervous system (PNS) microfluidic platform that recapitulates the full spectrum of myelination, demyelination, and remyelination using primary Schwann cells (SCs) and motor neurons (MNs). The platform enables reproducible hydrogel patterning and long-term stable coculture of MNs and SCs over 40 days in vitro based on three distinct design factors. Furthermore, the on-demand detachable substrate allows in-depth biological analysis. We demonstrated the possibility of mimicking segmental demyelination by lysophosphatidylcholine, and recovery of myelin structure by application of two drugs: benzatropine or methylcobalamin. This 3D PNS disease–on–a–chip may serve as a potential platform for understanding the pathophysiology of demyelination and screening drugs for remyelination.
INTRODUCTION
Myelin is a multilayered membrane produced by glial cells, particularly Schwann cells (SCs) in the peripheral nervous system (PNS) and oligodendrocytes in the central nervous system (CNS) (1, 2). These glial cells stretch and form myelin sheaths along axons through numerous processes called myelination. Myelin structures are important for the propagation of action potentials, enhancing the speed and efficiency of electrical communication between neurons via electrical shielding.
Demyelinating diseases involve the loss of myelin, which is caused by multiple factors: genetic mutations, inflammation, and other chemical or environmental factors (3, 4). Among demyelinating diseases, the types of PNS neuropathy are classified as segmental demyelination, Wallerian degeneration, and distal axonopathy (5). In particular, segmental demyelination leads to the loss of myelin sheaths without axonal damage, while the other two types affect both axons and SCs (6). SCs are unwrapped from axons, and myelin sheaths are destroyed. Moreover, repetitive segmental demyelination and remyelination processes cause onion bulb–like structures with concentric layers of SCs around axons (7). Unfortunately, the mechanisms underlying these diseases (e.g., Charcot-Marie-Tooth and Guillain-Barre syndromes) remain unclear (8, 9). Furthermore, there are few treatments for regenerating lost myelin (10, 11).
To more fully understand the relevant mechanisms and develop therapeutic drugs, organ-on-a-chip technology has recently emerged to recapitulate physiological and pathological in vivo conditions (12, 13). Some studies have presented neural development models (14, 15) and disease models such as Alzheimer’s disease (16), Parkinson’s disease (17), and amyotrophic lateral sclerosis (ALS) (18). Furthermore, other research groups introduced new approaches such as photolithography and three-dimensional (3D) printing and demonstrated the relationship between artificial axon fibers and oligodendrocytes to understand the underlying cellular mechanisms and achieve efficient drug screening (19, 20). In contrast to the in vitro model of the CNS, there have been few studies regarding the reconstruction of PNS motor nerves (18, 21, 22). Human spheroid models have been widely adapted to reconstruct the microphysiology of the PNS (18, 21). With these models, development or disease stage with spheroid motor neuron (MN) units could be reliably established, which allowed the confirmation of the nerve function through electrophysiological recording. In addition, optogenetic simulation promotes the maturation of myelination during the developmental process (22). Among these various models, to the best of our knowledge, no 3D PNS disease models have mimicked both the demyelination and remyelination processes on a single platform while focusing on SCs and MNs.
Here, we present an in vitro 3D PNS disease model, consisting of primary SCs and MNs cocultured in a 3D hydrogel microenvironment. This platform is designed to recapitulate a broad range of myelination procedures, such as development of myelin, acute demyelination upon biochemical stimulation, and subsequent recovery with the aid of pharmaceutical agents. This 3D organ-on-a-chip platform enables long-term coculture of essential cells for PNS modeling such as SCs and MNs over 40 days in vitro (DIV). It also allows in-depth biological analysis [e.g., Western blot analysis and transmission electron microscopy (TEM)] with the aid of on-demand detachable substrates. In terms of pathology, we established a robust demyelination model by the introduction of lysophosphatidylcholine (LPC) and a remyelination model by the application of benzatropine (Benz) or methylcobalamin (MeCbl). We confirmed our model at the cellular and molecular levels, as well as at the functional and morphological levels, by measuring electrophysiological signals and the thickness of the myelin sheath. Hence, our platform can help clarify the underlying mechanisms of normal and diseased myelination. Moreover, the platform is expected to aid in the discovery of new therapeutic drugs for remyelination.
RESULTS
Design of the 3D PNS disease microfluidic platform
Our 3D PNS disease microfluidic platform provides a method to reconstruct a full myelination process of normal, disease, and regeneration (Fig. 1A). This platform is fabricated by bonding the micromolded polydimethylsiloxane (PDMS) compartment to a pressure-sensitive adhesive (PSA)–coated polycarbonate (PC) film (fig. S1A). The platform consists of one hydrogel channel (cell culture area filled with Matrigel located in the middle, shown in sky blue), two cell-loading channels for SCs (left channel shown in green), and MNs (right channel shown in red) (Fig. 1B). A micropost array (Fig. 1B, shown in white) separates the two cell-loading channels, thus enabling only sprouted cells to make physical contact in the hydrogel channel. The platform is composed of three distinct elements: a tail, an N-gap, and a bump (Fig. 1, B to D). We designated the narrow gap between microposts as the N-gap (pink arrowheads in Fig. 1, B and D, and pink circle in Fig. 1C) and the semicircle as the bump (blue arrowheads in Fig. 1, B and D, and pink circle in Fig. 1C). All microstructures have a height of 100 μm.
Fig. 1. Design of 3D PNS disease platform.
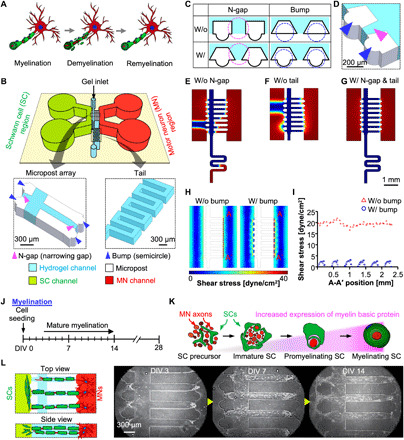
(A) Schematic illustrations displaying the full process of myelination, demyelination, and remyelination. (B) Schematics of the design of the platform: SC channel (green), MN channel (red), micropost array (white), and hydrogel channel (sky blue). (C) Enlarged 2D view of the microstructure of the chip with or without an N-gap (a pink circle) and bump structure (a blue circle). (D) (bottom of B) Enlarged 3D view of the microstructure of the chip with the micropost array consisting of the N-gap (magenta arrowhead) and bump (blue arrowheads) and tail. COMSOL finite-element simulation results for liquid patterning in the platform without an N-gap (E) or without a tail (F) and with an N-gap and a tail (G). The volume fractions of air (red) and liquid (blue) were simulated. (H) Simulation results of fluidic shear stress depending on the bump structure and (I) distribution of shear stress on hydrogel surface to which cells were attached (A-A′). Schematic diagram (J) and illustration (K) showing the time course of mature myelination in the SC-MN coculture model. (L) In the hydrogel channel, migrated SCs interacted with axons of MNs, leading to the wrapping process. Representative microscopic images showing the process of mature myelination at DIV 3, 7, and 14.
The importance of those design parameters was confirmed using a COMSOL multiphysics simulation. N-gap and tail structures help with successful 3D liquid patterning, which is defined as preservation of the 3D liquid interface integrity only in the hydrogel channel. Considering the Young-Laplace equation, we designed the N-gap, which acts as a capillary valve that resists high capillary pressure against the penetration of liquid into SC and MN channels (23, 24). The N-gap structure allows liquid pinning at the end of the micropost. Without the N-gap, the liquid interface cannot withstand pressure, which leads to bursting (Fig. 1E and fig. S1C). The tail structure allows higher fluidic resistance due to its serpentine structure of microfluidic channel. This extended fluidic channel structure helps the efficient filling of hydrogels within the hydrogel channel by inhibiting flowing out of hydrogels during the gel-filling process (Fig. 1F and fig. S1B). Therefore, considering the aforementioned factors, we successfully patterned Matrigel within the hydrogel channel for 3D cell culture (Fig. 1G and fig. S1D).
In addition, a bump structure was incorporated in this platform to establish a stable SC-MN microenvironment. When seeding cells or changing culture medium, flow inevitably occurs in the MN and SC channels, which subsequently generates shear stress (fig. S1E). Because shear stress affects the cell viability and MNs are very sensitive cells (25, 26), this mechanical stress on MNs should be minimized as much as possible. The simulation results revealed that the bump structure reduced the shear stress on the side surface of the hydrogel channel to which cells were attached (Fig. 1, H and I, A-A′ position). Furthermore, as the low number of MNs can be acquired from one mouse (27), efficient use of cells is important to reconstruct the SC-MN model. We observed that more cells were captured on the hydrogel surface with the bump than without the bump during cell seeding (fig. S1F).
Moreover, we investigated the diffusion of chemicals from the cell channels to the hydrogel channel, because various chemicals are introduced to induce demyelination and remyelination (fig. S1G). In this platform, the concentration of exogenously injected biochemical becomes uniform within 240 min, suggesting a minimized region-to-region variability.
Together, we verified that the platform design was suitable for 3D fluid patterning, stable coculture environment, and diffusion of chemicals into the cell culture area. Considering microscale design factors in the microfluidic platform, we fabricated a reliable microfluidic platform that guarantees reproducible gel filling, reduced fluidic shear stress, and uniform distribution of exogenously added reagents.
Modeling of 3D MN demyelination by LPC treatment
We reconstructed a normal 3D PNS myelination model on the platform to be used in demyelination experiments. (Fig. 1, J and K). In the PNS motor system, the nuclei of MNs are located in the spinal cord and extend their axons, while SCs have direct contact with the axons and form the myelin sheath. To mimic these structures, SCs and MNs were seeded in each cell channel and incubated to induce myelination in the gel channel prefilled with Matrigel at DIV 0 (Fig. 1L). SCs rapidly proliferated and grew toward the MN channel, whereas axons of MNs extended longitudinally into the hydrogel channel within DIV 3. SCs wrapped around the axons and interacted directly with the axons of MNs within DIV 7. Abundantly proliferating SCs and axons filled in the hydrogel channel, forming a myelin sheath at DIV 14.
To model the demyelination in vitro, we used LPC, a chemical compound that causes dedifferentiation of readily differentiated SCs (28, 29). We assessed the viability of SCs or MNs upon exposure to LPC. Because of the low MN survival rates in the absence of SCs (30, 31), the viability of each cell type was confirmed in a coculture environment. To this end, we used a Transwell system, in which two cellular components are physically separated for individual cell imaging, while paracrine-mediated cell-cell interactions are promoted in a culture medium. Each SC or MN was plated on coverslips at the bottom compartment of a Transwell, with the other cell type cultured on the Transwell insert (fig. S2A). Two different concentrations of LPC (0.35 or 0.7 mM) were added to the Transwell at DIV 3. For SCs, the viability was greater than 93% under all conditions [control (ctrl): 94.7 ± 2.4%; DMSO: 93.5 ± 4.3%; 0.35 mM LPC: 95.2 ± 2.3%; 0.7 mM LPC: 96.3 ± 0.1%] (fig. S2, B and C). Similarly, MNs also exhibited greater than 80% viability under all conditions (ctrl: 90.5 ± 3%; N.C.: 86.1 ± 5.4%; 0.35 mM LPC: 85.8 ± 3.3%; 0.7 mM LPC: 80.3 ± 4.5). These results suggested that 0.35 and 0.7 mM LPC did not affect the viability of SCs and MNs.
After mature myelination at DIV 14 (22, 32), LPC was added to the SC-MN myelination model at either of the two concentrations (0.35 or 0.7 mM). We analyzed the effect of LPC at three incubation times (DIV 17, 20, and 23) (Fig. 2A). At DIV 20, we observed partially broken myelinated forms, and some axons appeared to be unmyelinated (Fig. 2, B and C, yellow arrowheads). To determine the dose- and time-dependent effects of LPC on the completed demyelination in the 3D microfluidic platform, we investigated the effects of LPC on the expression of myelin-related proteins at the cellular level. Under demyelination conditions, dedifferentiation of myelinated glial cells affects the expression levels of proteins such as c-Jun and myelin basic protein (MBP) (33, 34). Specifically, c-Jun is an important negative regulator of the myelination process, which is typically seen in injured nerves and demyelination, while MBP is highly expressed in myelinating glial cells. These indicators exhibit an inverse relationship with each other.
Fig. 2. Reconstruction of 3D demyelination in SC-MN coculture by LPC treatment.
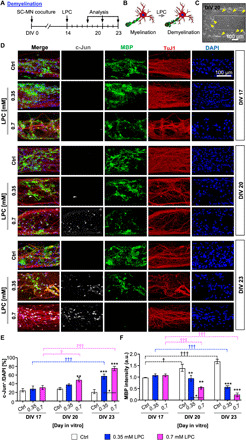
(A) Schematic diagram displaying the time course of LPC introduction and demyelination analysis in the SC-MN myelination model. To induce demyelination, cocultures were treated with 0.35 or 0.7 mM LPC, and the demyelination of SC-MN cocultures was measured by immunocytochemistry at DIV 17, 20, and 23. Fixed samples were immunostained with antibodies against c-Jun (gray), myelin basic protein (MBP; green), tubulin beta III (TuJ1; red), and 4′,6-diamidino-2-phenylindole (DAPI) (blue). (B) Schematic illustrations displaying the SC-MN coculture from myelination to demyelination. (C) Representative bright-field image for establishment of a demyelinating coculture system. Demyelination was rapidly induced by LPC treatment, and uneven nerve fiber (yellow arrowheads) was observed in the gel channel at DIV 20. (D) Representative confocal images of cocultures and quantification of c-Jun+ cells (E) and MBP intensity (F) with or without 0.35 or 0.7 mM LPC treatment, respectively. The graph shows mean ± SEM values from at least three independent experiments using different mice. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control. †P < 0.05 and †††P < 0.001 compared with each value at DIV 17 [one-way analysis of variance (ANOVA), Tukey’s multiple comparison tests]. a.u., arbitrary units.
The number of c-Jun+ cells was very low and remained consistent in the control over time, while it increased gradually in both 0.35 and 0.7 mM LPC treatments (Fig. 2, D and E). In particular, at DIV 17, the number of c-Jun+ cells did not vary among cocultures according to LPC treatment. After 0.7 mM LPC treatment, approximately half of all cells were c-Jun+ at DIV 20, while 74.7% of cells were c-Jun+ at DIV 23. Similar results were obtained under the 0.35 mM LPC condition, although the percentage of c-Jun+ cells was 1.3-fold lower than that in the 0.7 mM LPC condition.
In contrast to c-Jun, MBP expression decreased progressively after LPC treatment (0.35 or 0.7 mM), while it increased gradually in the control (Fig. 2, D and F). Specifically, 0.7 mM LPC treatment caused the MBP expression to decrease significantly at DIV 20 and 23. Similar results were obtained under the 0.35 mM LPC condition. The difference between the MBP expression under the two concentrations of LPC was significant at DIV 20, while it became barely detectable at DIV 23. Notably, the MBP expression level decreased significantly compared with the control, while the MBP levels under the 0.35 or 0.7 mM LPC conditions were 1.4- and 3.2-fold or 2.5- and 5.6-fold lower than the control at DIV 20 or 23, respectively. Together, these results demonstrate that LPC induces dedifferentiation of SCs and the demyelination process, showing an inverse relationship between increased expression of c-Jun+ and decreased MBP expression.
Protein-level validation of SC dedifferentiation in the 3D demyelinating MN model
The cultured cells were easily accessible for the in-depth biological analysis, because we used a removable substrate that could be easily detached on demand (Fig. 3A). After detachment of PSA film from a PDMS piece, we tested the molecular and structural changes on the myelination model. To more precisely confirm demyelination by the LPC, we analyzed the expression of c-Jun and MBP at the molecular level using Western blotting (Fig. 3B).
Fig. 3. Confirmation of 3D demyelination with effective biochemical analysis on a 3D PNS disease platform.
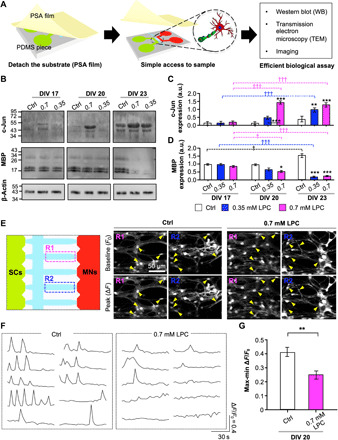
(A) Schematic descriptions of our easy-to-use platform with the PSA-coated PC film for efficient biological analysis [Western blot (WB), TEM, and imaging]. (B) Representative immunoblots and quantification of c-Jun (C) and MBP (D). *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control. †P < 0.05 and †††P < 0.001 are compared with each value at DIV 17 (one-way ANOVA, Tukey’s multiple comparison tests). Intracellular Ca2+ levels of coculture with or without LPC were analyzed at DIV 20 using the Oregon Green 488 BAPTA-1 AM staining kit. (E) Representative inverted confocal images of BAPTA-1 AM signals in two random gel channel regions—R1 and R2 (left)—showing the time points of the baseline (F0) and peak (∆F) levels (middle and right), (F) cell recording, and (G) quantification of the relative change of intracellular Ca2+ on coculture with or without LPC. Arrowheads indicate cocultured cells at the F0 and ∆F levels. The graph shows mean ± SEM values from three [in (B)] and at least seven independent experiments [in (G)] using different mice. In (G), an average of 14 cells per independent experiment was calculated. **P < 0.01 compared with control (unpaired, two-tailed t test with Welch’s correction).
Consistent with our immunocytochemistry results, c-Jun expression increased gradually over time following treatment with LPC (both 0.35 and 0.7 mM), which was almost consistent with the results from the control (Fig. 3C). The c-Jun levels were increased by 14- and 12-fold, compared with the control at DIV 20 and 23, respectively. Notably, under the 0.7 mM LPC condition, the c-Jun expression at DIV 20 increased significantly. Under the 0.35 mM LPC condition, c-Jun expression was slightly enhanced at DIV 20 and increased further at DIV 23.
The MBP expression was inversely correlated with expression of c-Jun under all conditions over time (Fig. 3D). In the control, we observed a gradual increase in MBP expression over time. At DIV 20, the MBP expression level was slightly lower in the 0.7 mM LPC condition than the control, while there were no significant differences between the 0.35 mM LPC condition and the control. Notably, we detected a remarkable decrease in MBP expression at DIV 23 under both LPC concentrations, and the MBP level decreased by 7.1- and 6.5-fold under the 0.35 and 0.7 mM LPC conditions, respectively, compared with the control.
Together with the confirmation at the cellular and molecular levels, we observed that demyelination of MN occurred at DIV 20 under the 0.7 mM LPC condition. These findings support the notion that we established a demyelination model with SCs-MNs by the introduction of chemicals. Next, we verified the disease model in terms of its electrophysiological signals.
Reduced intracellular calcium level in the 3D demyelinating MN model
Ca2+ signals play a pivotal role in our understanding of both physiological and pharmacological functions in neuronal networks (35). To investigate how neuronal networks change in both normal and diseased MN models, we measured the real-time intracellular Ca2+ in the SC-MN coculture model with or without 0.7 mM LPC treatment at DIV 20 using live-cell imaging (movie S1). Cultured SC-MN cocultures were treated with or without LPC at DIV 14, and the concentration of LPC was maintained throughout the culture period until calcium imaging analysis. For the analysis, we removed extracellular Ca2+ by washing with a standard buffer (see Materials and Methods) and recorded the neural activity upon loading a Ca2+ indicator (Oregon Green BAPTA-1 AM). When we measured intracellular Ca2+ signals in two random fields in the hydrogel channel (Fig. 3E, left), Ca2+ signals from the SC-MN coculture with 0.7 mM LPC were considerably reduced compared with the control, in terms of both frequency and amplitude (Fig. 3E, middle and right panels). Under the 0.7 mM LPC condition, there was a very small intensity difference between the resulting peak and the baseline (Fig. 3, E and F, yellow arrowheads). Furthermore, cell recordings revealed that intracellular Ca2+ signals were reduced upon LPC treatment compared with the control (Fig. 3F). The intensity difference (maximum ΔF/F0 minus minimum ΔF/F0) was 1.99-fold lower than that of the control (control: 0.41 ± 0.032; 0.7 mM LPC: 0.25 ± 0.034) (Fig. 3G). Therefore, these results revealed that LPC decreased the intracellular Ca2+ concentration, indicating similarity to the pathophysiological environment.
On the basis of validation at the cellular, molecular, and functional levels, we confirmed that demyelination of MN occurred under 0.7 mM of LPC treatment at DIV 20. Accordingly, we created stable and robust 3D myelinating and demyelinating MNs in our microfluidic platform.
Modeling of 3D MN remyelination by Benz or MeCbl treatment
Many therapeutic candidates have been investigated to promote remyelination. Benz and MeCbl are known as potent differentiating chemicals for oligodendrocytes or SCs, accelerating remyelination after injury (29, 36). Thus, to induce a remyelination process in our LPC-induced demyelination model, we introduced Benz or MeCbl at DIV 20 after demyelination with 0.7 mM LPC treatment and then analyzed MBP expression 8 days later (DIV 28) (Fig. 4A). Untreated cocultures without chemicals were used as controls. With the aid of Benz or MeCbl, dedifferentiated SCs were processed to redifferentiate their structures, and many axons exhibited remyelinated morphologies (Fig. 4, B and C, yellow arrowheads).
Fig. 4. Reconstruction of 3D remyelination by either Benz or MeCbl treatment on demyelinated SC-MN coculture.
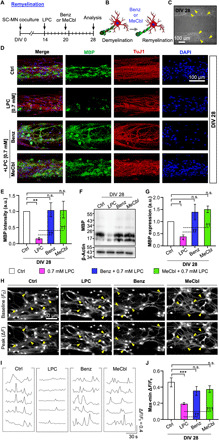
(A) To verify remyelination, cocultures were cotreated with Benz (1.5 μM) or MeCbl (10 μM) in the presence of LPC at DIV 20 and analyzed at DIV 28. (B) Schematic illustrations displaying the coculture from demyelination to remyelination. (C) Representative bright-field image for establishment of a remyelinating coculture system. (D) Representative confocal images of cocultures with MBP (green), TuJ1 (red), and DAPI (blue); (E) quantification of MBP intensity and (F) immunoblots; and (G) quantification of MBP on cocultures with Benz or MeCbl in the presence and absence of LPC are shown. (H) Representative inverted confocal images of BAPTA-1 AM signals showing the time points of the baseline (F0) and peak (∆F) levels, (I) cell recording, and (J) quantification of the relative changes in intracellular Ca2+ in cocultures with LPC only and with LPC plus Benz or MeCbl treatment. In (F) and (J), the graph shows mean ± SEM values from three independent experiments using different mice. In (J), an average of 13 cells per independent experiment was calculated. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with the control; ††P < 0.01 and †††P < 0.001 compared with the LPC treatment (one-way ANOVA, Tukey’s multiple comparison tests). n.s., no statistically significant difference.
After treatment with the two chemicals, the 3D MN demyelinating model exhibited increased MBP expression (Fig. 4D). Compared with LPC treatment only, the addition of Benz and MeCbl significantly accelerated MBP expression, increasing the level by 6.6- or 6.7-fold, respectively (Fig. 4E). Notably, after Benz or MeCbl treatment, MBP expression in the demyelination model recovered, reaching approximately the same level as the control. The two drugs had similar effects, with no significant differences in the recovery rate of the MBP expression level.
Furthermore, our Western blotting data revealed that MBP expression was substantially increased compared with the LPC-induced demyelinated model, which is consistent with the MBP intensity data obtained from MBP immunostaining (Fig. 4, F and G). Similar levels of MBP expression were observed in the three groups (control, Benz, and MeCbl), in contrast to the LPC-treated SC-MN model. These remarkable effects were consistent with previous in vivo remyelination tests of demyelinated nerve mice (29, 36).
We also observed intracellular Ca2+ signals in the LPC-induced demyelination model upon application of Benz or MeCbl to demonstrate functional regeneration of disease model (movie S2). The results showed that the initially reduced intracellular Ca2+ signals were considerably enhanced upon the addition of Benz or MeCbl (Fig. 4, H to J). Cell recording data revealed that Ca2+ signals in Benz or MeCbl treatment were markedly activated, compared with the LPC treatment only (Fig. 4I). In addition, the signal intensity differences of Ca2+ level increased 1.9- and 2.0-fold, respectively, compared with the LPC treatment only (Fig. 4J). Notably, the signal intensity differences did not differ significantly between the control and the Benz or MeCbl treatment, indicating that these remyelination reagents can aid in the functional recovery of the PNS neural network. These results support our conclusion that we induced remyelination in our 3D PNS disease platform. In addition, we were able to screen the efficacy of two therapeutic candidates that promote SC-MN remyelination.
Ultrastructural analysis of demyelination and remyelination
To investigate ultrastructural changes in myelin sheaths under Benz or MeCbl treatment, we analyzed the thickness of nerve fibers using TEM at DIV 40. By using TEM imaging, we frequently found “onion bulb” formation and thin myelin structures (Fig. 5, A and B). These results implied that our model has characteristics of segmental demyelination (6, 7). These observations support the usefulness of our PNS demyelination/remyelination platform for studying the segmental demyelination.
Fig. 5. Confirmation of 3D remyelination based on myelin thickness of demyelinated SC-MN cocultures.
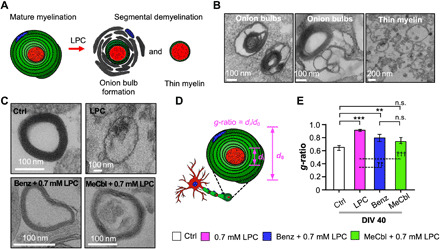
The extent of myelin sheath formation of LPC-induced SC-MN demyelination by Benz or MeCbl was analyzed by TEM at DIV 40. (A) Schematic diagram of structural alterations of mature myelin sheaths by LPC. (B) Representative TEM images of nerve cross sections of SC-MN cocultures with LPC, demonstrating segmental demyelination, including onion bulb formation (left and middle) and thin myelinated axons (right). (C) Representative TEM images of transected nerve fibers in the control, Benz, or MeCbl conditions in the presence and absence of LPC. (D) Schematic illustration of the g-ratio, defined as the ratio of the axonal diameter to the total nerve fiber diameter. A g-ratio close to 0.6 indicated a more mature myelinated axon. (E) Quantification of g-ratios of transected nerve fibers in the control, Benz, or MeCbl samples in the presence and absence of LPC. The graph shows mean ± SEM values from at least seven independent experiments using different mice. **P < 0.01 and ***P < 0.001 compared with the control; ††P < 0.01 and †††P < 0.001 compared with LPC (one-way ANOVA, Tukey’s multiple comparison tests).
In terms of demyelination, the LPC treatment almost completely destroyed the myelin structure (Fig. 5C, top right). Notably, even under LPC treatment, only SCs were damaged, whereas axons exhibited a normal ultrastructure, suggesting the features of segmental demyelination. By contrast, the treatment with Benz or MeCbl led to a considerably more compact structure, although these conditions required additional time to reach a level similar to that of control (Fig. 5C). Therefore, we confirmed that Benz or MeCbl treatments promote remyelination and significantly thicker ultrastructure, despite considerable demyelination by the LPC treatment.
Next, we measured the g-ratio of the transected nerve fiber. The g-ratio is defined as the ratio of the axonal diameter to the total nerve fiber diameter, which is used as a criterion to classify remyelination (Fig. 5D) (32, 37). In particular, a g-ratio of 0.6 serves widely as the theoretical estimated value for optimization. The g-ratio of the control was almost 0.6 (0.6 ± 0.02). Unexpectedly, the g-ratio of the nerve fibers under Benz (0.7 ± 0.05) or MeCbl (0.7 ± 0.04) treatment was significantly smaller than LPC only (0.9 ± 0.008) and was thus closer to the control (Fig. 5E). No significant differences were observed between the control and MeCbl conditions, while differences were detected between the control and Benz conditions. This result was presumably due to the accelerated redifferentiation of dedifferentiated SCs, which was induced more robustly by MeCbl than by Benz. Therefore, we established a remyelination model in LPC-induced demyelinated MNs by Benz or MeCbl treatment, validated at the cellular, molecular, and morphological levels. We confirmed that both Benz and MeCbl play critical roles in the remyelination of SC-MNs and can be used as therapeutic agents.
DISCUSSION
Here, we have presented a 3D PNS disease model that recapitulates a sequential demyelination and remyelination process in PNS MNs in a 3D microfluidic platform. After establishment of a stable long-term environment for SC-MN coculture (40 days), we introduced several chemical that induced dysfunction in myelinated MNs and subsequent recovery of myelin. The disease and regeneration models were demonstrated at the cellular, molecular, functional, and morphological levels. Therefore, our reliable and reproducible model is expected to aid our understanding of disease mechanisms and facilitate drug screening for PNS disease.
We developed a full-spectrum 3D PNS demyelination and remyelination model process, with particular emphasis on tight SC-MN interaction. For demyelination, we verified the dedifferentiation of SCs and decrease in intracellular calcium levels. Notably, only SCs were destroyed by LPC, while MNs were not damaged. Thin myelinated axons and onion bulb–like structures were also observed. These results suggest that we recapitulated important features of segmental demyelination (6, 7). Furthermore, we expanded our disease model to a regeneration model using Benz and MeCbl. To the best of our knowledge, no in vitro PNS models have been reported that reconstruct normal to disease and disease to regeneration states in a single platform. Therefore, our platform serves as a powerful tool for both basic and applied PNS research, which would be difficult to undertake using conventional in vivo or in vitro models.
Our model is expected to elucidate the underlying mechanisms and novel biological pathways of myelination. Recent evidence suggests that Benz and MeCbl are effective for remyelination in multiple sclerosis or peripheral neuropathies (29, 36). Benz is currently approved by the U.S. Food and Drug Administration for treatment of Parkinson’s disease and acts as a regulator of oligodendrocytes, which are precursors for cell differentiation and remyelination after cuprizone-induced toxic demyelination (38). MeCbl is a vitamin B12 analog, which promotes remyelination through down-regulation of the Efk1/2 pathway in the rat sciatic nerve model (39). LPC activates protein kinase C, MAP (mitogen-activated protein) kinase, and the JNK (c-Jun N-terminal kinase) pathway, which stimulate the transcription of c-Jun (40). Together, we speculate that the activity of Benz or MeCbl may be correlated with these pathway cascades, based on the remyelination results in our demyelination model. Further research will be of interest to investigate the key pathways governing remyelination.
Furthermore, our platform serves as a drug screening model. For an organ-on-a-chip to be of practical use in drug screening, it must be easy to use. We used PSA film to develop a user-friendly platform in terms of fabrication and analysis. PSA film has been adapted as a substrate in many organ-on-a-chip studies, because it is attachable and detachable, as well as biocompatible and transparent similar to glass (14, 41). Compared with conventional PDMS-based platforms, the fabrication process is simpler because no plasma treatment is required for bonding. Furthermore, the on-demand removal of substrates could facilitate in-depth biological assays such as Western blotting and TEM, serving as a new tool for fundamental study. Therefore, we straightforwardly demonstrated the effects of two drugs by carrying out various analyses in a high-throughput microfluidic platform.
Emerging evidence suggests the importance of SCs for both developmental and pathological processes in the PNS (1, 11). SCs are essential for the growth of axons, regulation of myelination, and recovery of function at nerve lesions. Furthermore, the viability of MNs is very low without SCs in vitro (30, 31). Because our model showed pathophysiological changes in SCs during demyelination and remyelination, glial biology will be of interest to establish the mechanism of demyelination, the contributions of proteins to myelin damage, and the factors that drive regeneration of damaged myelin, in terms of SCs.
To improve our model, several modifications could be considered. First, introduction of muscle cells may broaden our model to include PNS motor units, especially neuromuscular units (42, 43). Many PNS diseases (e.g., ALS and myasthenia gravis) are related to muscle cells, as myelinated MNs propagate axon potentials to muscle fiber, causing muscle contractions (18). Therefore, if we establish a triculture environment of muscle cells with SC-MNs, our model could be applied to various PNS disease models, beyond segmental demyelination. Second, induced pluripotent stem cell (iPSC)–derived cells from patients provide more reliable pathological results, similar to in vivo conditions. Recent in vitro models have attempted to introduce patient samples or human iPSC cells (44, 45). If we use iPSC cells from patients, our model could be improved to mimic both demyelination and remyelination more accurately.
MATERIALS AND METHODS
Fabrication of the 3D disease microfluidic platform
The microfluidic platform fabrication process is described in detail previously (13, 24). Briefly, the 3D microfluidic platform was fabricated using photolithography and soft lithography. An SU-8 master mold (MicroChem) on a 101.6 mm silicon wafer was manipulated by photolithography. A mixture of PDMS (Sylgard 184, Dow Corning) and curing agent was prepared at 10:1 ratio. The mixture was poured onto the master mold, degassed in a vacuum chamber, and thermally cured to obtain a replica PDMS piece. The medium reservoirs and hydrogel injection ports were punched out with a biopsy punch (6 mm) and a syringe needle (0.5 mm). Instead of a glass coverslip for the substrate, a PSA-coated PC film was used, which is easily detachable (14, 41). The complete chip was fabricated by bonding the PDMS piece to the PSA-coated PC film. Before the experiment, the chip was sterilized under ultraviolet irradiation.
Simulation analysis
COMSOL multiphysics software (COMSOL, Burlington, MA) was used to perform simulations on the 3D PNS disease platform. For finite-element analysis of hydrogel filling, computer-aided design of the platform was imported, and laminar flow and phase field modules were applied (46, 47). Under the laminar flow condition, a pressure gradient of 500 Pa between the inlet and outlet pressure was set. Under the phase field condition, the volume fractions of air (red) and liquid (blue) were simulated. The inlet was set as water, and the other part was set as air to represent the air/liquid interface. For the analysis of shear stress, a laminar flow module was used to simulate 50 μl of medium changes. The pressure gradient of 20 Pa was set as medium height difference of 2 mm. To analyze the biochemical diffusion effect that induces demyelination and remyelination, a diluted species transport module was used (48, 49). A porous media module was applied to simulate Matrigel with a porosity of 0.99. The chemical inlet was set to the side of the hydrogel channel where cells were attached. The diffusion coefficient of chemicals was set to 5 × 10−11 m2 s−1, which is the diffusion coefficient of 5-kDa fluorescein isothiocyanate (FITC)–dextran in Matrigel (50). As LPC and MeCbl have respective molecular weights of 299.26 and 1344.4 Da, the diffusion coefficient of these chemicals was presumed to be greater than the coefficient of 5-kDa FITC-dextran and diffusion, which would result in more rapid diffusion. Dynamic viscosity of culture medium at 37°C of 0.748 cP and density of 1000 kg m−3 were used in the simulation (51, 52).
Preparation of the SC-MN coculture
The preparation of SC-MN was performed as described previously (22, 32). SCs isolated from sciatic nerves and MNs isolated from the lumbar regions of spinal cords were cocultured. For the SC culture, the sciatic nerves isolated from CD-1 mice (postnatal 4) were treated with 2.5% trypsin (Thermo Fisher Scientific) for 30 min, followed by inactivation of trypsinization with 10% horse serum (Gibco). For the MN culture, the lumbar regions of spinal cords isolated from embryonic day 12 mice were treated with 1% TRL3 trysin solution (Trypsin chromatographically purified, diafiltered, Worthington) for 15 min, and then 1% trypsin inactivation solution was added (Sigma-Aldrich). MNs were purified from spinal cord cells using a p75NTR (Abcam)–coated immunopanning dish and placed into coculture medium containing neurobasal/B27 medium (Gibco) supplemented with 2% horse serum, 0.5 mM l-glutamine (Thermo Fisher Scientific), 0.5 μM forskolin (Sigma-Aldrich), 1% penicillin/streptomycin (Thermo Fisher Scientific), 1× B27 supplement (Gibco), bovine pituitary extract (1 mg/ml; Gibco), brain-derived neurotrophic factor (10 μg/ml; Gibco), and 1 mM β-mercaptoethanol (Sigma-Aldrich). The final concentration of cell suspension was 2 million/ml of MNs and 1.5 million/ml of SCs. All procedures and experiments were approved by the Institutional Animal Care and Use Committee of Seoul National University. All cells were cultured in a 5% CO2 incubator at 37°C.
Cell seeding and SC-MN coculture
The SC-MN coculture on the 3D PNS disease platform was performed as described previously (22, 32). The growth factor–reduced Matrigel (Corning, Corning, NY, USA) was injected into the hydrogel channel and incubated for 20 min at room temperature. After gel polymerization, each 50 μl of cell suspension was introduced into the cell channel, respectively, and cells were attached to each side surface of the hydrogel channel by tilting the chip at 90° for 30 to 60 min. The seeded cells were settled and trapped very close to the N-gap between bumps. To induce myelination, SCs and MNs were incubated in coculture medium with ascorbic acid (50 μg/ml; Sigma-Aldrich) after DIV 7. For medium changes, 25 μl of culture medium was removed from all cell chambers, and 50 μl of fresh coculture medium was added to one side of each cell chamber at 3-day intervals (fig. S1F).
Transwell coculture
To evaluate the viability of SCs growing in Transwells, isolated MNs were seeded on Transwell inserts that had been precoated with poly-ornithine hydrobromide (0.5 mg/ml; Sigma-Aldrich) and laminin (2.5 μg/ml; Invitrogen). In addition, SCs plated on coverslips precoated with poly-l-lysine (10 μg/ml; Sigma-Aldrich) were placed in the bottom compartment of the Transwell coculture. To assess the viability of MNs, SCs were cultured on inserts precoated with poly-l-lysine (10 μg/ml), and coverslips seeded with MNs were placed in the bottom compartments of the Transwell coculture. Coverslips were coated with poly-ornithine hydrobromide (0.5 mg/ml; Sigma-Aldrich) and laminin (2.5 μg/ml; Invitrogen). To assess cell viability, SCs and MNs were treated with different concentrations of LPC at DIV 3, and the LPC concentration was maintained during the culture period.
Chemical treatments
All chemicals including LPC (Sigma-Aldrich), Benz (Thermo Fisher Scientific), and MeCbl (Sigma-Aldrich) were purchased. LPC was dissolved in a mixture of chloroform:methanol (1:1, v/v) to a concentration of 25 mg/ml and then diluted in cell culture medium to final concentrations of 0.7 and 0.35 mM. Benz and MeCbl were, respectively, dissolved in dimethyl sulfoxide (DMSO) to 1.5 μM and ethanol to 100 μM. To maintain the concentrations of the chemicals during the culture period, each chemical was mixed with culture medium when the culture medium was changed.
Live and dead cell assays
The cell viability of SCs and MNs was assessed using a Live-Dead cell staining kit (Abcam, Cambridge, UK), in accordance with the manufacturer’s instructions. At DIV 7, SCs and MNs were washed once with phosphate-buffered saline (PBS), and a mixture of live (calcein-AM, green) and dead solution (propidium iodide, red) was added to the cells, which were then incubated for 30 min at room temperature. Each cell was washed twice with PBS and then examined using confocal microscopy (IX81; Olympus). The number of stained cells was counted in five random fields of view under a microscope.
Immunocytochemistry
Cocultures were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) for 20 min at room temperature and then treated with 0.2% Triton X-100 for 15 min. Subsequently, coculture samples were incubated in 4% bovine serum albumin (BSA; Millipore, Burlington, MA, USA) at 4°C overnight. Primary antibodies diluted in 1% BSA were added to cells, and the mixture was incubated at 4°C overnight. Cells were then incubated with secondary antibodies at room temperature for 2 hours. The primary antibodies used were anti–c-Jun (1:500; Abcam), anti-MBP (1:500; Abcam), and anti-TuJ1 (1:1000; Abcam). The secondary antibodies used were goat anti-rabbit immunoglobulin G (IgG) heavy and light chains (H&L) (1:500 to 1:1000; Abcam), goat anti-rat IgG H&L (1:500; Abcam), and goat anti-chicken IgY H&L (1:500; Abcam). Last, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Carlsbad, CA, USA) for 15 min. All images were acquired using an inverted confocal laser scanning microscope (LSM 700) equipped with solid-state lasers (405, 488, 555, and 649 nm).
Measurement of florescence signals
To assess c-Jun and MBP fluorescence signals, stained coculture samples were processed sequentially, and confocal microscopy images were acquired in a single session using identical exposure and gain settings. A threshold was set for both c-Jun (for Fig. 2C) and MBP intensities (for Figs. 2D and 5C), which were analyzed in five random fields of view. In Fig. 2F, each bar represents the average expression level of a protein of interest normalized to that of the control at DIV 17. Five regions in each condition were randomly chosen and analyzed.
Western blotting analysis
Expression levels of c-Jun and MBP were quantitatively analyzed using Western blot analysis. After PC film had been detached from the 3D PNS disease platform, all samples were lysed in radioimmunoprecipitation assay buffer (T&I, Chuncheon, Korea) containing protease inhibitors (aprotinin, leupeptin, pepstatin A, and phenylmethylsulfonyl fluoride). The protein concentrations of the coculture lysates were measured using the Bradford assay (Sigma-Aldrich). Twelve to 15 μg of protein from each coculture sample were loaded in SDS–polyacrylamide gel electrophoresis gel and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with anti-rabbit c-Jun (1:500; Abcam) and anti-rat MBP (1:500; Abcam) at 4°C overnight. The membranes were washed three times in tris-buffered saline solution with Tween 20 and then incubated with goat anti-rabbit IgG or anti-rat IgG conjugated with horseradish peroxidase (1:1000; Sigma-Aldrich) for 2 hours. Bands were visualized using an ECL system, and the intensities of the bands were quantified using ImageJ software. In Figs. 3 (C and D) and 4, each bar represents the average expression level of a protein of interest normalized to that of the control at DIV 17 and 28, respectively. Each protein level was normalized against β-actin, which was used as a loading control.
Calcium imaging
To measure the intracellular Ca2+ levels in cocultures in the demyelination and remyelination model, Oregon Green 488 BAPTA-1 AM was used in accordance with the manufacturer’s instructions. Cocultures were rinsed three times with standard buffer (139 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM Hepes, and 10 mM glucose), followed by treatment with a mixture of 3 mM Oregon Green 488 BAPTA-1 AM and 10% (w/v) pluronic F-127 (at a 1:1 ratio) for 30 min in an incubator. Cocultures were washed three times with standard buffer. Time-lapse images were taken for 5 min, and cocultures were excited at the same intensity using a 488-nm laser line. The intensity of intracellular Ca2+ in each coculture was measured using Zen software (LSM 700), and the intracellular Ca2+ signals were determined by calculating the ratios between changes in fluorescence signal intensity (∆F) and baseline fluorescence (F). In Figs. 3 (E to G) and 4 (H to J), the time intervals between F0 and ∆F were adjusted to be consistent among the groups.
Transmission electron microscopy
To assess myelin sheath thickness, cocultures were fixed with 2% glutaraldehyde overnight at 4°C, followed by postfixation with 1% osmium tetroxide for 30 min at room temperature. After the samples had been washed three times with ultrafiltered water, they were dehydrated in an ethanol series (50, 70, 80, 85, 90, 95, and 100%). Using an ultramicrotome (Ultra Cut C; Leica, Wetzlar, Germany), samples were sectioned and mounted on copper grids. Grids were contrast stained with uranyl acetate and lead citrate. Images were captured by cryo-TEM (Tecnai F20 Cryo; FEI, Hillsboro, OR, USA). To quantitatively determine the relative thickness of individual remyelinated nerves, the g-ratio was analyzed with a g-ratio calculator plug-in for ImageJ software (available at https://imagej.nih.gov/ij/), which allowed semiautomated analysis of randomly selected nerve fibers (53, 54). At least seven remyelinated axons per group were randomly selected for measurement, and the mean g-ratio was calculated for each group by averaging across all individual g-ratios.
Statistical analysis
All statistical analyses were performed using Prism (GraphPad Software, USA). All statistical data are presented as the means ± SEM. We performed the Shapiro-Wilk test to assess normality. Normal distributions were analyzed using parametric tests [analysis of variance (ANOVA) with Tukey’s multiple comparison test for comparing values among three or more groups in Figs. 2 (E and F), 3 (C and D), 4 (E, G, and J), and 5E, and unpaired, two-tailed t tests with Welch’s correction in Fig. 3G]. *, **, and *** or †, ††, and ††† in the figures denote P < 0.05, 0.01, and 0.001, respectively. n.s. indicates no statistically significant difference. Calculated P values are specified in table S1.
Acknowledgments
Funding: This work was supported by the National Research Foundation of Korea (NRF; grant nos. NRF-2018R1A2A1A05019550 to N.L.J. and 2018R1A2A3075013 to H.N.K.). Author contributions: S.H. and S.-R.L. performed all the experiments and drafted the manuscript. J.K., Y.K., and S.K. performed design and fabrication of the platform under the supervision of N.L.J. H.N.K. participated in the TEM experiment and wrote relevant sections in the manuscript. S.H., S.-R.L., and N.L.J. designed the research, and N.L.J. supervised the work. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd9749/DC1
REFERENCES AND NOTES
- 1.Mirsky R., Jessen K. R., Schwann cell development, differentiation and myelination. Curr. Opin. Neurobiol. 6, 89–96 (1996). [DOI] [PubMed] [Google Scholar]
- 2.Jessen K. R., Mirsky R., The repair Schwann cell and its function in regenerating nerves. J. Physiol. 594, 3521–3531 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamil K., Yazid M. D., Idrus R. B. H., Das S., Kumar J., Peripheral demyelinating diseases: From biology to translational medicine. Front. Neurol. 10, 87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin R., Mcfarland H. F., Mcfarlin D. E., Immunological aspects of demyelinating diseases. Annu. Rev. Immunol. 10, 153–187 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Pardo I. D., Rao D. B., Butt M. T., Jortner B. S., Valentine W. M., Arezzo J., Sharma A. K., Bolon B., Toxicologic pathology of the peripheral nervous system (PNS): Overview, challenges, and current practices. Toxicol. Pathol. 46, 1028–1036 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Moss T. H., Segmental demyelination in the peripheral-nerves of mice affected by a hereditary neuropathy (Dystonia Musculorum). Acta Neuropathol. 53, 51–56 (1981). [DOI] [PubMed] [Google Scholar]
- 7.Schoene W. C., Carpenter S., Behan P. O., Geschwind N., ’Onion bulb’ formations in the central and peripheral nervous system in association with multiple sclerosis and hypertrophic polyneuropathy. Brain 100, 755–773 (1977). [DOI] [PubMed] [Google Scholar]
- 8.Young P., Suter U., Disease mechanisms and potential therapeutic strategies in Charcot-Marie-Tooth disease. Brain Res. Brain Res. Rev. 36, 213–221 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Park H. T., Kim J. K., Tricaud N., The conceptual introduction of the "demyelinating Schwann cell" in peripheral demyelinating neuropathies. Glia 67, 571–581 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Manganas L. N., Maletic-Savatic M., Stem cell therapy for central nervous system demyelinating disease. Curr. Neurol. Neurosci. Rep. 5, 225–231 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y., Notterpek L., Promoting peripheral myelin repair. Exp. Neurol. 283, 573–580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huh D., Matthews B. D., Mammoto A., Montoya-Zavala M., Hsin H. Y., Ingber D. E., Reconstituting organ-level lung functions on a chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor A. M., Blurton-Jones M., Rhee S. W., Cribbs D. H., Cotman C. W., Jeon N. L., A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat. Methods 2, 599–605 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S. R., Hyung S., Bang S., Lee Y., Ko J., Lee S., Kim H. J., Jeon N. L., Modeling neural circuit, blood-brain barrier, and myelination on a microfluidic 96 well plate. Biofabrication 11, 035013 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Osaki T., Shin Y., Sivathanu V., Campisi M., Kamm R. D., In vitro microfluidic models for neurodegenerative disorders. Adv. Healthc. Mater. 7, 10.1002/adhm.201700489, (2017). [DOI] [PubMed] [Google Scholar]
- 16.Park J., Wetzel I., Marriott I., Dréau D., D’Avanzo C., Kim D. Y., Tanzi R. E., Cho H., A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolognin S., Fossépré M., Qing X., Jarazo J., Ščančar J., Moreno E. L., Nickels S. L., Wasner K., Ouzren N., Walter J., Grünewald A., Glaab E., Salamanca L., Fleming R. M. T., Antony P. M. A., Schwamborn J. C., 3D cultures of Parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv. Sci. 6, 1800927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osaki T., Uzel S. G. M., Kamm R. D., Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4, eaat5847 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espinosa-Hoyos D., Jagielska A., Homan K. A., Du H., Busbee T., Anderson D. G., Fang N. X., Lewis J. A., Van Vliet K. J., Engineered 3D-printed artificial axons. Sci. Rep. 8, 478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mei F., Fancy S. P. J., Shen Y. A. A., Niu J., Zhao C., Presley B., Miao E., Lee S., Mayoral S. R., Redmond S. A., Etxeberria A., Xiao L., Franklin R. J. M., Green A., Hauser S. L., Chan J. R., Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nat. Med. 20, 954–960 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma A. D., Coy L. M., Jacobs E., Willey H., Behn J. Q., Nguyen H., Bolon B., Curley J. L., Moore M. J., Engineering a 3D functional human peripheral nerve in vitro using the Nerve-on-a-Chip platform. Sci. Rep. 9, 8921 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hyung S., Lee S. R., Kim Y. J., Bang S., Tahk D., Park J. C., Suh J. K. F., Jeon N. L., Optogenetic neuronal stimulation promotes axon outgrowth and myelination of motor neurons in a three-dimensional motor neuron-Schwann cell coculture model on a microfluidic biochip. Biotechnol. Bioeng. 116, 2425–2438 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Cho H., Kim H. Y., Kang J. Y., Kim T. S., How the capillary burst microvalve works. J. Colloid Interf. Sci. 306, 379–385 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Kim S., Lee H., Chunga M., Jeon N. L., Engineering of functional, perfusable 3D microvascular networks on a chip. Lab Chip 13, 1489–1500 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Bucchia M., Merwin S. J., Re D. B., Kariya S., Limitations and challenges in modeling diseases involving spinal motor neuron degeneration in vitro. Front. Cell. Neurosci. 12, 61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu M., Song W., Li P., Huang Y., Gong X., Zhou G., Jia X., Zheng L., Fan Y., Galanin protects against nerve injury after shear stress in primary cultured rat cortical neurons. PLOS ONE 8, e63473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiese S., Herrmann T., Drepper C., Jablonka S., Funk N., Klausmeyer A., Rogers M. L., Rush R., Sendtner M., Isolation and enrichment of embryonic mouse motoneurons from the lumbar spinal cord of individual mouse embryos. Nat. Protoc. 5, 31–38 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Zhang H., Jarjour A. A., Boyd A., Williams A., Central nervous system remyelination in culture—A tool for multiple sclerosis research. Exp. Neurol. 230, 138–148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishimoto S., Tanaka H., Okamoto M., Okada K., Murase T., Yoshikawa H., Methylcobalamin promotes the differentiation of Schwann cells and remyelination in lysophosphatidylcholine-induced demyelination of the rat sciatic nerve. Front. Cell. Neurosci. 9, 298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haastert K., Grosskreutz J., Jaeckel M., Laderer C., Bufler J., Grothe C., Claus P., Rat embryonic motoneurons in long-term co-culture with Schwann cells—A system to investigate motoneuron diseases on a cellular level in vitro. J. Neurosci. Methods 142, 275–284 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Gingras M., Beaulieu M. M., Gagnon V., Durham H. D., Berthod F., In vitro study of axonal migration and myelination of motor neurons in a three-dimensional tissue-engineered model. Glia 56, 354–364 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Hyung S., Yoon Lee B., Park J. C., Kim J., Hur E. M., Francis Suh J. K., Coculture of primary motor neurons and schwann cells as a model for in vitro myelination. Sci. Rep. 5, 15122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parkinson D. B., Bhaskaran A., Arthur-Farraj P., Noon L. A., Woodhoo A., Lloyd A. C., Feltri M. L., Wrabetz L., Behrens A., Mirsky R., Jessen K. R., c-Jun is a negative regulator of myelination. J. Cell Biol. 181, 625–637 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sternberger N. H., Itoyama Y., Kies M. W., Webster H. D., Myelin basic protein demonstrated immunocytochemically in oligodendroglia prior to myelin sheath formation. Proc. Natl. Acad. Sci. U.S.A. 75, 2521–2524 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berridge M. J., Neuronal calcium signaling. Neuron 21, 13–26 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Deshmukh V. A., Tardif V., Lyssiotis C. A., Green C. C., Kerman B., Kim H. J., Padmanabhan K., Swoboda J. G., Ahmad I., Kondo T., Gage F. H., Theofilopoulos A. N., Lawson B. R., Schultz P. G., Lairson L. L., A regenerative approach to the treatment of multiple sclerosis. Nature 502, 327–332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chomiak T., Hu B., What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PLOS ONE 4, e7754 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeuner K. L., Schaffer E., Hopfner F., Bruggemann N., Berg D., Progress of pharmacological approaches in Parkinson’s disease. Clin. Pharmacol. Ther. 105, 1106–1120 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Okada K., Tanaka H., Temporin K., Okamoto M., Kuroda Y., Moritomo H., Murase T., Yoshikawa H., Methylcobalamin increases Erk1/2 and Akt activities through the methylation cycle and promotes nerve regeneration in a rat sciatic nerve injury model. Exp. Neurol. 222, 191–203 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Ozaki H., Ishii K., Arai H., Kume N., Kita T., Lysophosphatidylcholine activates mitogen-activated protein kinases by a tyrosine kinase-dependent pathway in bovine aortic endothelial cells. Atherosclerosis 143, 261–266 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Lee Y., Choi J. W., Yu J., Park D., Ha J., Son K., Lee S., Chung M., Kim H. Y., Jeon N. L., Microfluidics within a well: An injection-molded plastic array 3D culture platform. Lab Chip 18, 2433–2440 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Sanes J. R., Lichtman J. W., Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22, 389–442 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Li L., Xiong W. C., Mei L., Neuromuscular junction formation, aging, and disorders. Annu. Rev. Physiol. 80, 159–188 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Vatine G. D., Barrile R., Workman M. J., Sances S., Barriga B. K., Rahnama M., Barthakur S., Kasendra M., Lucchesi C., Kerns J., Wen N., Spivia W. R., Chen Z., Van Eyk J., Svendsen C. N., Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 24, 995–1005.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Liu C., Oikonomopoulos A., Sayed N., Wu J. C., Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 145, dev156166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee H., Kim S., Chung M., Kim J. H., Jeon N. L., A bioengineered array of 3D microvessels for vascular permeability assay. Microvasc. Res. 91, 90–98 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Huang C. P., Lu J., Seon H., Lee A. P., Flanagan L. A., Kim H. Y., Putnam A. J., Jeon N. L., Engineering microscale cellular niches for three-dimensional multicellular co-cultures. Lab Chip 9, 1740–1748 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shirure V. S., Bi Y., Curtis M. B., Lezia A., Goedegebuure M. M., Goedegebuure S. P., Aft R., Fields R. C., George S. C., Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 18, 3687–3702 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao Y., Kim D., Dura B., Zhang K., Yan R., Li H., Han E., Ip J., Zou P., Liu J., Chen A. T., Vortmeyer A. O., Zhou J., Fan R., Ex vivo dynamics of human glioblastoma cells in a microvasculature-on-a-chip system correlates with tumor heterogeneity and subtypes. Adv. Sci. 6, 1801531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galgoczy R., Pastor I., Colom A., Giménez A., Mas F., Alcaraz J., A spectrophotometer-based diffusivity assay reveals that diffusion hindrance of small molecules in extracellular matrix gels used in 3D cultures is dominated by viscous effects. Colloids Surf. B Biointerfaces 120, 200–207 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Hsu Y. H., Moya M. L., Abiri P., Hughes C. C. W., George S. C., Lee A. P., Full range physiological mass transport control in 3D tissue cultures. Lab Chip 13, 81–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helm C. L. E., Fleury M. E., Zisch A. H., Boschetti F., Swartz M. A., Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 15779–15784 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malheiro A., Morgan F., Baker M., Moroni L., Wieringa P., A three-dimensional biomimetic peripheral nerve model for drug testing and disease modelling. Biomaterials 257, 120230 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Babetto E., Wong K. M., Beirowski B., A glycolytic shift in Schwann cells supports injured axons. Nat. Neurosci. 23, 1215–1228 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd9749/DC1