

Historically well-known tumor-promoting gene family plays a key role in cell identity regulation in small cell lung cancer.
Abstract
Comprehensive genomic analyses of small cell lung cancer (SCLC) have revealed frequent mutually exclusive genomic amplification of MYC family members. Hence, it has been long suggested that they are functionally equivalent; however, more recently, their expression has been associated with specific neuroendocrine markers and distinct histopathology. Here, we explored a previously undescribed role of L-Myc and c-Myc as lineage-determining factors contributing to SCLC molecular subtypes and histology. Integrated transcriptomic and epigenomic analyses showed that L-Myc and c-Myc impart neuronal and non-neuroendocrine–associated transcriptional programs, respectively, both associated with distinct SCLC lineage. Genetic replacement of c-Myc with L-Myc in c-Myc–SCLC induced a neuronal state but was insufficient to induce ASCL1-SCLC. In contrast, c-Myc induced transition from ASCL1-SCLC to NEUROD1–SCLC characterized by distinct large-cell neuroendocrine carcinoma–like histopathology. Collectively, we characterize a role of historically defined general oncogenes, c-Myc and L-Myc, for regulating lineage plasticity across molecular and histological subtypes.
INTRODUCTION
Small cell lung cancer (SCLC) represents about 15% of all lung cancers with a median survival time of approximately 10 months and 5-year overall survival at 6% (1). SCLCs are characterized by neuroendocrine differentiation, rapid growth, early metastatic spread, and poor prognosis (2). The standard of care has remained cytotoxic chemotherapy for decades, mainly etoposide combined with a platinum agent (3), which are only temporarily effective for the vast majority of patients (4). Even with recent developments in immune checkpoint inhibitors, activity against SCLCs when combined with chemotherapy has been marginal with a modest improvement in the median survival (10.3 months versus 12.3 months) for extensive-stage SCLCs treated with immunotherapy (5). These data reflect the urgent need for more effective therapeutics for patients with SCLC. The lack of effective therapeutics for SCLC stands in stark contrast to the breadth of targeted therapies for non-small cell lung cancer (NSCLC), particularly lung adenocarcinoma (6). The progress in drug development for NSCLCs is largely attributable to more comprehensive understanding of molecular subtypes and to identification of targetable driver oncogenes (7). Therefore, better characterization of the molecular subtypes of SCLC should aid future drug development and permit patient stratification for targeted therapies, a strategy that has been remarkably effective for specific subsets of patients with advanced lung adenocarcinoma.
Characterization of SCLC subtypes was first noted by morphological differences over three decades ago when human SCLC cell lines were implanted as xenografts and distinguished as two primary subtypes: classic and variant SCLC (8, 9). The classic subtype featured relatively small cells with high nuclear:cytoplasm ratio, while the variant subtype exhibited relatively larger cells and moderate amounts of cytoplasm. However, the World Health Organization (WHO) classification, updated in 2015, histologically recognizes SCLC as a homogeneous disease with neuroendocrine features defined by small cells, scant cytoplasm, nuclear morphology with fine granular chromatin, and lacking prominent nucleoli, reminiscent of the features of the “classic” SCLC. The originally described “variant” subtype may represent combined SCLC with large-cell neuroendocrine carcinoma (LCNEC) in the current classification (10).
More recent efforts to distinguish SCLC molecular subtypes include profiling gene expression and genome-wide methylation in primary human tumors and patient-derived xenografts. These profiles revealed three clusters, with a dichotomy between ASCL1 and NEUROD1 expression, in addition to a cluster with low expression of both (11). The expression of ASCL1 and NeuroD1 has been implicated to confer SCLC heterogeneity by imparting distinct transcriptional profiles (12). The third neuroendocrine-low cluster led to further classification into two subtypes characterized by transcriptional driver YAP1 or POU2F3 (13, 14). SCLC cell line xenograft histology has been correlated with these contrasting factors, where variant SCLC was positively correlated with a higher NeuroD1:ASCL1 ratio and classic SCLC was positively correlated with a higher ASCL1:NeuroD1 ratio in SCLC cell lines (11).
Observations on Myc family characteristics in SCLC genetically engineered mouse models (GEMMs) have provided insight in their contribution to histopathological characteristics (15, 16). The SCLC GEMM with conditional loss of Rb1 and Trp53 mouse (RP) harbored stochastic MYCL amplifications or overexpression associated with classic SCLC histopathology (17). On the contrary, consistent with the original report on the variant subtype to harbor frequent MYC amplification (9), a more recent study showed that additional c-Myc overexpression, in the RP SCLC GEMM, drives the progression of murine SCLC with variant histopathology and reduced neuroendocrine gene expression including ASCL1 but higher NeuroD1 expression (18). However, the molecular mechanisms underlying the distinction between L-Myc– and c-Myc–driven subsets of SCLC remain unexplored.
c-Myc (MYC) and L-Myc (MYCL) belong to the MYC family of basic helix-loop-helix (bHLH) transcription factors. The paralogs contain functionally relevant highly conserved amino acid sequences and are structurally homologous (19, 20). c-Myc is a well-characterized oncogene; L-Myc, although understudied, is implicated to have a similar oncogenic role. Amplification of Myc family members is mutually exclusive and overall accounts for ~20% of SCLC and overexpression for ~50% of SCLC in primary human tumors (21). In contrast to the fact that MYC is commonly amplified across all three major lung cancer subtypes—lung adenocarcinomas, squamous cell lung carcinomas, and SCLC (21–23)—MYCL and MYCN are uniquely amplified in SCLC, in a manner suggestive of their role as lineage-amplified genes.
In this study, we investigated a previously undescribed of c-Myc and L-Myc as lineage-specific factors to associate SCLC molecular subtypes with histological classes. We investigated the potential of L-Myc and c-Myc to regulate lineage state and identified transcriptional programs unique to each Myc family member, wherein L-Myc regulates neuronal developmental pathways and c-Myc regulates epithelial-to-mesenchymal transition and Notch signaling, biological pathways that are associated with distinct molecular subsets. We showed that c-Myc expression is required to maintain lineage state marker NeuroD1 in NeuroD1-positive SCLC. In addition, c-Myc is incompatible with ASCL1-positive SCLC that ultimately leads to transdifferentiation to NeuroD1-SCLC, consistent with previous findings (24). Furthermore, this transdifferentiation is characterized by variant histopathology mediated by a transcriptional repressor, Rest.
RESULTS
SCLC network reveals unique and distinct subnetworks for c-Myc and L-Myc
To understand biological processes that may be unique to c-Myc and L-Myc in SCLC, we sought to investigate potential causal regulations among genes. To this end, we first built a molecular causal network using primary SCLC datasets and then focused on networks that involve c-Myc and L-Myc. Combining two independent primary SCLC transcriptomic datasets (21, 25), we built a Bayesian network using RIMBANet (Reconstructing Integrative Molecular Bayesian Network), a software package we previously developed (see Methods). The Bayesian network comprised 8451 unique genes (nodes) and 9301 regulations (edges) among these genes. The regulations inferred in this Bayesian network provide insights into biological functions associated with molecular features such as pathways or gene signatures (26, 27). This enables us to discern transcriptional subnetworks associated with c-Myc and L-Myc that may reflect their unique biological roles. We first aimed to generate a signature associated with each factor and then project the signature to the SCLC Bayesian network to identify specific subnetworks (Fig. 1A). To generate these signatures, we sought to select an independent dataset. Hence, we examined copy number alteration and expression for each Myc family member in 49 SCLC cell lines from the Cancer Cell Line Encyclopedia (CCLE) and classified them into four groups representing each Myc family member and low Myc (fig. S1A). Then, we selected cell lines that belong to MYC and MYCL groups and examined mRNA expression MYC and MYCL to select cell lines for c-Myc with high expression of MYC and low expression of MYCL and vice versa (fig. S1B). We identified 457 differentially expressed genes (t test, P < 0.01; fold change, >1.5), 147 and 310 genes overexpressed in MYC and MYCL SCLC cell lines, respectively, and defined them as their introductory gene signatures (fig. S1C and table S1).
Fig. 1. Bayesian network analysis reveals unique L-Myc and c-Myc networks associated with distinct biological processes.
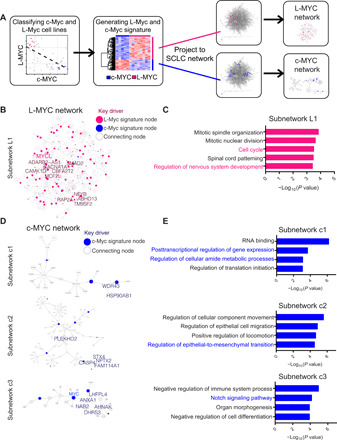
(A) Schematic of workflow to use SCLC Bayesian causal gene regulatory network to identify networks involving c-Myc and L-Myc. (B) L-Myc subnetwork showing directionality and association of genes when L-Myc gene signature (fig. S1C and table S1) is projected to SCLC Bayesian network. Circles colored in pink represent nodes from L-Myc gene signature. Size of pink circles is directly proportional to the number of outgoing nodes. Nodes indicated in larger text are key drivers of the subnetwork (table S2). (C) Gene ontology (GO) analysis for L-Myc neighbor subnetwork. Enriched functions for these genes are identified on the basis of hypergeometric test against GO terms. (D) Three c-Myc subnetworks showing directionality and association of genes when c-Myc–associated gene signature (fig. S1C and table S1) is projected to SCLC Bayesian network. Circles colored in blue represent nodes from c-Myc gene signature. Size of blue circles is directly proportional to the number of outgoing nodes. Nodes indicated in larger text are key drivers of the subnetwork (table S3). (E) GO analysis for corresponding c-Myc neighbor subnetwork. Enriched functions for these genes are identified on the basis of hypergeometric test against GO terms.
To explore the subnetworks associated with L-Myc, we projected the genes up-regulated in the L-Myc–expressing subset onto the network and collected all nodes within two layers from them (see Methods). We identified one large closed subnetwork (L1; Fig. 1B) that comprises 959 gene nodes that included 120 of 310 genes from the L-Myc signature. To identify master regulators of the L-Myc subnetwork, we performed key driver analysis (see Methods) that revealed 13 statistically significant genes (table S2). Examining protein expression of Smad2, a node in the L-Myc subnetwork, revealed higher expression in L-Myc–classified cell lines compared to c-Myc–classified cell lines (fig. S1D). Gene ontology (GO) analysis of this L-Myc subnetwork revealed enrichments of two biological processes: cell cycle progression and neuronal development (Fig. 1C). These processes have been previously implicated as core descriptors of classic SCLC (2). On the other hand, when we projected the c-Myc signature onto the network, the c-Myc network was organized into three unique subnetworks, c1, c2, and c3, and consists of 25, 95, and 29 gene nodes, respectively (Fig. 1D). Key driver analysis of all three subnetworks revealed 15 statistically significant regulators (table S3). Subnetwork c1 was enriched for canonical c-Myc functions in transcriptional, translational, and metabolic regulation (Fig. 1E) (20). c-Myc subnetwork c2 was enriched for pathways in epithelial-to-mesenchymal transition (Fig. 1E) that has been shown to be relevant in the neuroendocrine-low subtype (28). Last, c-Myc subnetwork c3 was enriched for Notch signaling pathway (Fig. 1E), which has been implicated to mediate a transition from neuroendocrine to non-neuroendocrine fate of tumor cells in a murine SCLC model (29). These functional pathways enriched in subnetworks c2 and c3 describe a relationship between c-Myc and lineage state determination pathways in SCLC.
Together, these findings suggest that c-Myc and L-Myc are associated with different molecular mechanisms that have been implicated in SCLC biology. Specifically, it implicates that c-Myc is associated with neuroendocrine-low differentiation state in addition to its canonical oncogenic functions in SCLC and, by contrast, L-Myc is associated with classic neuroendocrine state of SCLC.
c-Myc– and L-Myc–driven SCLCs exhibit distinct chromatin states to exert differential transcriptional programs
Next, we sought to determine whether the distinct c-Myc and L-Myc networks are associated with their distinct cistromes. To examine the role of each Myc family member, we selected representative cell lines for c-Myc (NCI-H82, NCI-H524, NCI-H2171, and NCI-H2081) and L-Myc (CORL-88, NCI-H1963, and NCI-H209) based on the classification described in fig. S1A and confirmed protein expression for these transcription factors (Fig. 2A). Notably, NCI-H1963 expressed MYCL-RLF fused protein and the wild-type protein. The fused protein that lacks the first 27 amino acids of L-Myc and is fused to 79 amino acids of RLF protein has been previously shown to be functional in SCLCs (30).
Fig. 2. c-Myc– and L-Myc–driven SCLCs exhibit distinct chromatin states to regulate distinct transcriptional programs.
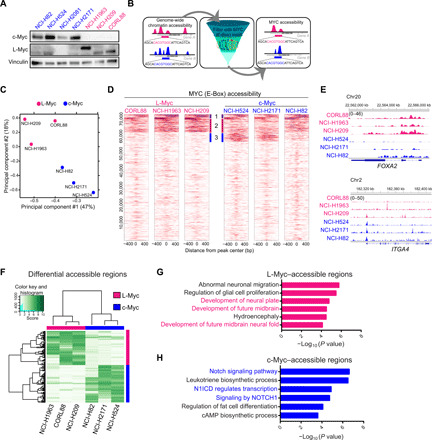
(A) Protein expression of c-Myc and L-Myc as well as vinculin as a loading control in a panel of representative SCLC cell lines (blue, c-MYC–classified lines; pink, L-MYC–classified lines). (B) Schematic showing the analytic approach to defining MYC-accessible regions. (C) PCA of open chromatin regions with E-box motif in six SCLC cell lines. Each dot represents a SCLC cell line that is colored on the basis of Myc status (blue, c-Myc–classified cell lines; pink, L-Myc–classified cell lines). (D) Each heatmap depicting global MYC (E-box) accessibility in individual SCLC cell lines at 72,833 combined peaks. ATAC-seq signal intensity is shown by color shading. bp, base pair. (E) Genome browser view tracks of ATAC-seq signals at the FOXA2 and ITGA4 loci (pink, L-Myc–classified cell lines; blue, c-Myc–classified cell lines). (F) Heatmap showing 2808 differentially accessible regions [fold change, ≥5; false discovery rate (FDR), ≤0.05] between three L-Myc cell lines shown in pink and three c-Myc cell lines shown in blue. (G) Enriched ontology by GREAT (Genomic Regions Enrichment of Annotations Tool) analyses for regions differentially accessible in L-Myc–classified cells. (H) Enriched ontology by GREAT analyses for regions differentially accessible in c-Myc–classified cells. cAMP, cyclic adenosine 3′,5′-monophosphate.
To define open regulatory elements potentially regulated by c-Myc and L-Myc, we first performed the assay for transposase-accessible chromatin sequencing (ATAC-seq) on three representative cell lines for each Myc (selected from Fig. 2A) and identified accessible chromatin regions, and then we filtered these regions with motif matrices for Myc binding (E-box) referring to as Myc-accessible regions hereafter (Fig. 2B and see Methods). Notably, inferred Myc-accessible regions reasonably recapitulated c-Myc binding sites previously determined by c-Myc chromatin immunoprecipitation (ChIP) in NCI-H2171 cells (fig. S2, A and B) (31). This approach eliminates confounding factors from antibody-dependent sensitivity and specificity that are intrinsic to ChIP-based assays to compare cistromes of two distinct proteins.
Principal components analysis (PCA) of E-box–containing peaks in all six cell lines separated these cell lines according to Myc status (Fig. 2C). In addition, a correlation matrix of pairwise Pearson correlation coefficient of Myc accessibility signal captured distinct clusters for the c-Myc– and L-Myc–classified cell lines (fig. S2C). Examining genome-wide Myc-accessible regions in each individual cell line, we observed a fraction of peaks that overlapped between c-Myc– and L-Myc–classified cells (cluster 1; Fig. 2D), suggestive of common functional binding that the Myc family members share. We also found distinct groups of peaks with higher signal intensity unique to L-Myc–classified cells (cluster 2; Fig. 2D) and in c-Myc–classified cell lines (cluster 3; Fig. 2D), suggesting distinctive DNA binding profiles for L-Myc and c-Myc. We observed preferential accessibility at distinct genetic loci in L-Myc cell lines when compared to c-Myc cell lines (Fig. 2E). This included augmented accessibility in L-Myc cell lines at lung and neuron development factor FOXA2 (32, 33) (Fig. 2E) and POU3F2 (Brn2), a neuronal lineage factor of lung squamous cancer (fig. S2D) (34), while in c-Myc cell lines at cell surface adhesion factor ITGA4 (Fig. 2E) and neuronal repressor, REST (fig. S2E). Consistent with the epigenetic results, we found that protein expression of Brn2 was significantly higher in L-Myc–classified cell lines and Rest expression was highest in two c-Myc–classified cell lines with the highest Myc accessibility at its locus while there was no statistically significant difference between c-Myc– and L-Myc–classified cell lines (fig. S2F). Notably, the chromatin landscape of NCI-H82 appeared to be intermediate to the L-Myc and c-Myc profiles. Together, these findings reveal the commonality and distinction between c-Myc and L-Myc binding profiles.
To gain insights into the unique biological processes that c-Myc and L-Myc may impart, we analyzed differentially enriched Myc-accessible sites comparing c-Myc cell lines and L-Myc cell lines. We identified 2808 differentially accessible regions; 1235 peaks enriched in L-Myc–classified cell lines and 1573 peaks enriched in c-Myc–classified cell lines (Fig. 2F). Next, we performed GREAT (Genomic Regions Enrichment of Annotations Tool) analysis (35) on the differentially accessible peaks. Unique L-Myc–accessible sites were enriched for neuronal pathways such as glial cell proliferation, development of the neural plate, neural fold, and future midbrain (Fig. 2G). By contrast, unique c-Myc–accessible sites were enriched for pathways involved in Notch signaling (Fig. 2H). While specific Notch signaling activity in MYC-amplified SCLCs has not been previously noted, there have been previous reports showing Rest as a target of the Notch signaling pathway to suppress the expression of neuronal genes (21, 24, 29). These findings are consistent with the observations from the Bayesian network analysis, providing additional evidence to suggest distinct transcriptional programs in c-Myc– and L-Myc–driven SCLCs.
Next, to explore the relationship between c-Myc and L-Myc transcriptomic and epigenomic signatures, we examined average Myc accessibility signal at L-Myc and c-Myc subnetwork genes in c-Myc– and L-Myc–classified cell lines, respectively. This analysis revealed that L-Myc subnetwork genes were preferentially accessible in L-Myc–classified cell lines, while c-Myc subnetwork genes were preferentially accessible in c-Myc lines when compared to all other genes (fig. S3, A and B). Gene set enrichment analysis of Myc-accessible regions revealed that c-Myc–specific accessible regions were significantly enriched for the introductory c-Myc transcriptomic signature (defined in fig. S1C), while L-Myc–specific accessible regions were significantly enriched for the introductory L-Myc transcriptomic signature (fig. S3C), confirming the consistency between two datasets. Together, these independent datasets reveal concordant L-Myc and c-Myc signatures to distinguish the two factors.
c-Myc and L-Myc expression is associated with distinct lineage state markers
A recent review synthesized SCLC profiling studies to identify four molecular subtypes, where two of these subtypes are classified as neuroendocrine SCLC that are driven by either ASCL1 or NeuroD1 and the other two are classified as non-neuroendocrine driven by YAP1 or POU2F3 (6). Given our findings from the Bayesian network and Myc accessibility profiling that c-Myc and L-Myc regulate transcriptional programs associated with distinct lineage-determining programs, we sought to investigate the relationship between Myc family members and the proposed SCLC master regulator classifiers.
Myc accessibility profile at the individual locus level of our c-Myc– and L-Myc–classified cell lines for these four factors revealed augmented accessibility in the c-Myc–classified cell lines at the NEUROD1 locus in contrast to L-Myc–classified cell lines that had augmented accessibility at the ASCL1 locus (fig. S4, A and B). We observed no differential or preferential accessibility signal in either c-Myc– or L-Myc–classified cell lines at POU2F3 and YAP1 loci, likely reflecting that these cell lines do not represent these subtypes (fig. S4, C and D). Protein expression of these lineage state markers showed that our L-Myc–classified cell lines had exclusive expression of ASCL1, while the c-Myc–classified cell lines exclusively expressed NeuroD1, consistent with the chromatin profile. (fig. S4E).
To understand how c-Myc and L-Myc expression correlated with the four molecular subtypes in CCLE cell lines, we examined the expression of c-Myc and L-Myc in each of the molecular subtypes as classified in (6). We observed that the non-neuroendocrine, POU2F3, and YAP1, as well as NeuroD1, subtypes had higher expression of c-Myc (fig. S4F). On the other hand, L-Myc expression was enriched in the neuroendocrine-high ASCL1 subtype (fig. S4G).
We further sought to reproduce the classification with data from primary tumors (21) and CCLE cell lines with additional data from 77 primary tumors (Fig. 3A and table S4) (25). Notably, while our unsupervised clustering was in general agreement with the published classification (6), the ASCL1 subtype cluster revealed heterogeneity particularly of primary tumors with the dual expression of ASCL1 and other lineage factors that may indicate lineage transition or intratumor heterogeneity (Fig. 3A). In this classification, we found that L-Myc expression was higher in samples classified as ASCL1 or NeuroD1 subtype when compared to the non-neuroendocrine subtype, thus indicating unique expression of L-Myc in neuroendocrine SCLC (Fig. 3B). On the other hand, the expression of c-Myc was higher in tumors and cell lines clustered in the NeuroD1 subtype and non-neuroendocrine low: POU2F3 and YAP1, as compared to those classified as ASCL1 (Fig. 3C), suggesting a negative relationship between ASCL1 and c-Myc. We found that the expression of c-Myc is anticorrelated with ASCL1 [Pearson’s correlation coefficient (r) = −0.53] across the combined primary tumor and cell line datasets (Fig. 3D). Together, these data imply the dichotomy of c-Myc expression and L-Myc expression in SCLC lineages.
Fig. 3. c-Myc and L-Myc expression is associated with distinct molecular subtype markers.
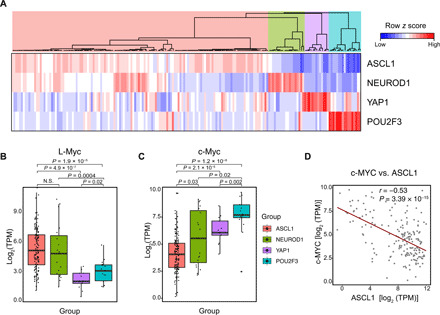
(A) Unsupervised clustering of 137 primary tumors and 51 CCLE cell lines using the expression of ASCL1, NeuroD1, POU2F3, and YAP1 distinguishing four molecular subtypes (table S4). (B) Box plot showing mRNA expression of L-Myc across four molecular subtypes for combined primary tumor and cell line classification described in (A). (C) Box plot showing mRNA expression of c-Myc across four molecular subtypes for combined primary tumor and cell line classification described in (A). (D) Scatterplot with best-fit line showing negative correlation between c-Myc and ASCL1 expression for combined primary tumor and cell lines. Pearson’s correlation coefficient (r) = −0.53, P < 0.01, t test. N.S., not significant; TPM, transcripts per million.
L-Myc induces a neuronal state but fails to transition to ASCL1-expressing SCLC
Given that L-Myc expression and c-Myc expression are enriched in distinct subtype(s) and they regulate programs associated with its respective subtype, we hypothesized that part of the role Myc family members play is to serve as a lineage factor. To test the potential of L-Myc to establish the neuroendocrine lineage, we modified two c-Myc–expressing NeuroD1-classified cell lines NCI-H82 and NCI-H524 by exogenous overexpression of L-Myc and CRISPR-Cas9–mediated deletion of c-Myc (Fig. 4A). We found that c-Myc–expressing cell lines NCI-H82 and NCI-H524 were able to tolerate the overexpression of L-Myc (Fig. 4B and fig. S5, A and B). Cells engineered to express both L-Myc and c-Myc exhibited comparable proliferation rates compared to parental cells expressing only c-Myc (Fig. 4C and fig. S5C). We further demonstrated that replacement of c-Myc with the expression of L-Myc allowed the cells to retain proliferative potential in the absence of c-Myc in NCI-H82 and NCI-H524 (Fig. 4C and fig. S5C). This indicates redundant roles among the two Myc family members in regard to maintaining cell viability.
Fig. 4. L-Myc induces neuronal state not defined by ASCL1 expression.
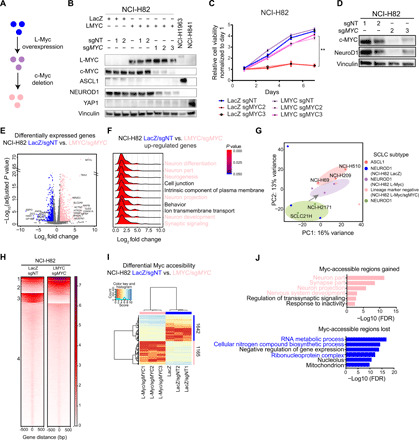
(A) Workflow schematic to genetically engineer replacement of c-Myc with L-Myc. (B) Protein expression of L-Myc, c-Myc, ASCL1, NeuroD1, and YAP1, with vinculin as a loading control in replaced NCI-H82 cells. Quantified in fig. S5A. (C) Cell growth curve for genetically engineered NCI-H82 cells. **P < 0.01 versus Lac-Z/sgNT NCI-H82 cells, t test. (D) Protein expression of c-Myc and NeuroD1 with vinculin as a loading control in NCI-H82 cells with c-Myc ablation. Quantified in fig. S5F. (E) Volcano plot showing 1213 differentially expressed genes (713 up-regulated in pink and 500 down-regulated in blue). NCI-H82 (LacZ and LacZ/sgNT) versus NCI-H82 (LMYC/sgMYC). (F) Ridge plot showing distribution of fold change for genes in each enriched GO term for up-regulated genes (E). (G) PCA of 14 SCLC cell lines; including 3 ASCL1, 2 NEUROD1, and 9 genetically engineered NCI-H82 cells; 3 control (LacZ, LacZ/sgNT1, or LacZ/sgNT2); 3 L-Myc overexpressed (L-Myc, L-Myc/sgNT1, and L-Myc/sgNT2); and 3 c-Myc replaced with L-Myc (L-Myc/sgMYC1, L-Myc/sgMYC2, and L-Myc/sgMYC3). PC, principal component. (H) Each heatmap depicting k-means (k = 4) clustering of global Myc accessibility in NCI-H82 LacZ/sgNT (left) and NCI-H82 L-MYC/sgMYC (right) at 170,711 combined peaks. ATAC-seq signal intensity is shown by color shading. (I) Heatmap showing 2807 differentially accessible regions (fold change, >16; FDR, <0.01) between three NCI-H82 control cells (LacZ, LacZ/sgNT1, and LacZ/sgNT2) and three c-Myc replaced by L-Myc (L-Myc/sgMYC1, L-Myc/sgMYC2, and L-Myc/sgMYC3). (J) Enriched ontology by Cistrome-GO analyses for Myc-accessible regions gained (top) and lost (bottom) after replacement.
To inquire their contribution to lineage determination, we evaluated the expression of lineage state markers on day 5. Addition of L-Myc neither induced expression of neuroendocrine marker ASCL1 nor altered the expression of NeuroD1 (Fig. 4B and fig. S5B). By contrast, the replacement of c-Myc with L-Myc led to the down-regulation of NeuroD1 (Fig. 4B and fig. S5B). The lack of expression of both neuroendocrine lineage marker (ASCL1, NeuroD1 or ATOH1) and neuroendocrine-low lineage markers (YAP1 and POU2F3) in c-Myc replaced NCI-H82 and NCI-H524 cells indicates a unique lineage-negative state (fig. S5, D and E).
Without L-Myc replacement, consistent with previous findings (36), c-Myc was essential for survival in NCI-H82 (Fig. 4C), thus precluding us from evaluating converted lineage state. However, during the short window of survival, c-Myc ablation led to significant down-regulation of NeuroD1, suggesting a role of c-Myc on maintaining expression of lineage state marker (Fig. 4D and fig. S5F).
Even with the loss in lineage marker expression, we observed no notable morphological changes in vitro (fig. S5G). However, morphological characteristics in suspension cells can be but are not always reflective of a neuroendocrine or non-neuroendocrine state. Previous literature showed that deletion of Mycl (L-Myc) in the RP GEMM resulted in tumors with mixed and NSCLC morphology (16) in contrast to the c-Myc–expressing SCLC with variant morphology (9, 18). Therefore, to investigate L-Myc’s contribution to SCLC histology, we implanted genetically engineered NCI-H82 cells into immunodeficient mice. Histological analysis of NCI-H82 expressing L-Myc and NCI-H82 L-Myc ablated with c-Myc xenografts revealed morphology similar to the control NCI-H82–LacZ/sgNT cells with polygonal cells, large nucleoli, and moderate amounts of cytoplasm representative of variant SCLC consistent with previous reports (fig. S5H) (9). The insufficiency of L-Myc to induce histological transdifferentiation may partially explain the low frequency of transition from variant to classic SCLC (29).
To investigate the state of these lineage marker–negative cells, we performed transcriptomic profiling. GO analysis of 235 significantly up-regulated genes by L-Myc in the presence of c-Myc in NCI-H82 revealed enrichment for neurogenesis and neuronal-associated pathways (fig. S6, A and B), suggesting that L-Myc imparts neuronal pathways while not fully inducing ASCL1 lineage state. Ablation of c-Myc further up-regulated 170 genes compared to the previous condition, which are enriched for neuronal structure, suggesting that the cells are further committed to a neuronal state (fig. S6, C and D). A direct comparison of the control NCI-H82 cells with lineage-negative NCI-H82 cells revealed 713 significantly up-regulated genes enriched for neuronal-associated pathways and 500 down-regulated genes enriched for canonical Myc function (Fig. 4, E and F, and fig. S6E). Collectively, it indicates the significance of L-Myc in inducing a more neural state. Next, to investigate which state these lineage marker–negative cells most closely resemble, we compared the transcriptomic profile of these genetically engineered cells to cell lines classified as ASCL1, NEUROD1, YAP1, and POU2F3. PCA of these cell lines revealed with the replacement of c-Myc with L-Myc in NCI-H82 cells clustered away from NEUROD1, YAP1, and POU2F3 cells and shifted toward ASCL1-classified cell lines (fig. S6F). To more clearly see the shift, we compared the state of these cells only to ASCL1 and NEUROD1 SCLC that revealed replacement of c-Myc with L-Myc in NCI-H82 cells transitioned the cells closer to ASCL1 (Fig. 4G). Together, these data suggest that replacement of c-Myc with L-Myc, although induces a more neural state, does not fully transdifferentiate to ASCL1-positive state and remains to be negative for lineage markers used in the current consensus molecular classification (6).
To investigate how L-Myc rewires the Myc-accessible landscape, we profiled Myc-accessible regions of three NCI-H82 control cells (LacZ, LacZ/sgNT1, and LacZ/sgNT2) and three NCI-H82 cells with c-Myc replaced by L-Myc (L-Myc/sgMYC1, L-Myc/sgMYC2, and L-Myc/sgMYC3). Analysis of genome-wide Myc accessibility in control cells and replacement cells identified a common cluster of Myc-accessible regions, indicating an aspect of functional redundancy between c-Myc and L-Myc (cluster 1; Fig. 4H). In addition, we identified unique clusters gained and lost with L-Myc overexpression and c-Myc deletion (clusters 2 and 3; Fig. 4H). To investigate what these unique clusters represent, we identified 1165 regions uniquely enriched in replaced NCI-H82 cells compared to the controls (Fig. 4I). Integrative ontology analysis of these regions with the transcriptomic profile (from Fig. 4E) revealed enrichment for neuronal-associated pathways, suggesting direct activation of these genes (Fig. 4J, top). On the other hand, Myc-accessible regions lost with this genetic engineering were enriched for functions to maintain high metabolic state that have been reported to be regulated by Myc (Fig. 4J, bottom). These data suggest that L-Myc has the potential to up-regulate neuronal lineage–associated pathways, suggestive of its lineage-determining function in SCLC.
Aurora kinase A inhibition sensitivity is altered with change in Myc status
Previous findings suggest that MYC (c-Myc)–driven SCLC is more responsive to Aurora kinase A inhibition (18, 37). We confirmed previous findings that MYC-amplified cell lines exhibit increased sensitivity and that MYCL-amplified cell lines are resistant to alisertib (fig. S7A). Alisertib treatment of the NCI-H82 cells expressing both c-Myc and L-Myc revealed no changes in sensitivity to the drug nor did changes in expression of the variant state marker. However, the cells that replaced c-Myc with L-Myc became resistance to aurora kinase inhibition (fig. S7B). On the other hand, overexpression of c-Myc increased sensitivity at lower concentrations of the drug (fig. S7C). This suggests that alisertib sensitivity is attributable to either c-Myc–driven lineage state of the cells or difference in molecular interaction of Aurora kinase A with each Myc protein.
c-Myc causes loss of classic neuroendocrine SCLC features
The dependency of lineage state marker NeuroD1 on c-Myc led us to hypothesize that c-Myc exerts a role in addition to its oncogenic role to regulate and establish a variant differentiation state. To this end, we genetically engineered neuroendocrine-high L-Myc–expressing NCI-H1963 and NCI-H187 with exogenous overexpression of c-Myc (Fig. 5, A and B). Notably, exogenous expression of c-Myc led to down-regulation of L-Myc in NCI-H1963 and NCI-H187 (Fig. 5, A and B, and fig. S8, A and B).
Fig. 5. c-Myc expression in ASCL1 subtype SCLC induces transdifferentiation to histologically distinct NeuroD1-positive state.
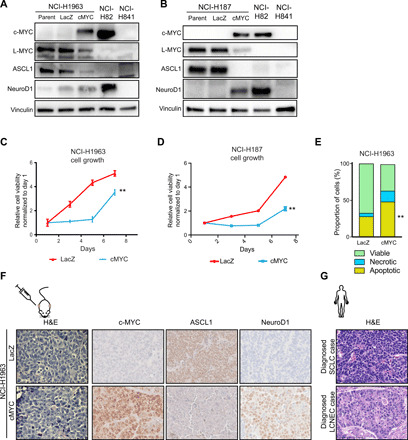
(A and B) Protein expression of c-Myc, L-Myc, ASCL1, and NeuroD1, as well as vinculin, as a loading control in genetically engineered NCI-H1963 (A) and NCI-H187 (B) cells. Quantification in fig. S8A (NCI-H1963) and fig. S8B (NCI-H187). (C and D) Cell growth curve for NCI-H1963 (C) and NCI-H187 (D) cells with overexpression of c-Myc or control Lac-Z. Means ± SD of five technical replicates. **P < 0.01 versus Lac-Z–overexpressed control cells, t test. (E) Bar chart shows proportions of viable, necrotic, and apoptotic cells in NCI-H1963 c-Myc–overexpressed and control cells measured by annexin V staining. Means ± SD of three biological replicates. **P < 0.01, t test. Scatterplots shown in fig. S8E. (F) Hematoxylin and eosin (H&E) staining and immunohistochemical staining of c-Myc, ASCL1, and NeuroD1 in LacZ-overexpressed (top) or c-Myc–overexpressed (bottom) NCI-H1963 xenograft. H&E original images, ×600; immunohistochemistry original images, ×20. (G) H&E staining of typical SCLC and LCNEC primary human tumors. Original images, ×400.
When c-Myc expression is introduced in NCI-H1963 or NCI-H187, we observed an initial phase of growth suppression (Fig. 5, C and D). This observation stands in contrast to the established role of c-Myc to promote cell cycle and cell proliferation (38). The growth suppression is unlikely to be due to oncogene-induced senescence as these cells stained negative for β-galactosidase (fig. S8C) and are deficient for Rb1 and p53. We investigated the cell cycle dynamics in their growth-suppressive phase and found that initial c-Myc expression (day 2) modestly increased the proportion of cells in S and G2-M phases but did not significantly alter the distribution of cells or induce cell cycle arrest to explain slower cell growth (fig. S8D). Alternatively, we investigated the proportion of annexin V–positive cells on day 2 and found that c-Myc overexpression increased the proportion of necrotic and apoptotic cells as compared to control cells (Fig. 5E and fig. S8E) in NCI-H1963.
We hypothesized that cell death was a consequence of incompatibility of the neuroendocrine differentiation state with c-Myc expression and that the persisting cells eventually grown out were cells that tolerated the expression of c-Myc, potentially by transdifferentiating to a state compatible with c-Myc expression (Fig. 5, C and D). Note that we define transdifferentiation as change in cancer lineage state by the expression of lineage markers and histological features, not a conversion to a different lineage hierarchy. Therefore, we investigated the expression of lineage state markers and found that the overexpression of c-Myc down-regulated ASCL1 and up-regulated NeuroD1 in NCI-H1963 and NCI-H187 cells (Fig. 5, A and B), indicating the transdifferentiation to NeuroD1 subtype of SCLC and that c-Myc exerts a role in dictating a distinct NeuroD1-positive neuroendocrine differentiation state. We observed no notable morphological changes in these cells in vitro (fig. S8F).
To investigate whether the transdifferentiation between molecular subtypes was accompanied by histological switch in vivo, we injected NCI-H1963 cells genetically engineered with stable c-MYC expression. Tumor growth dynamics recapitulated the slower growth of c-Myc–overexpressed NCI-H1963 observed in vitro (fig. S9A). This was associated with comparable levels of proliferation measured by Ki67 and increased levels of apoptosis measured by cleaved caspase-3 (fig. S9B). We observed that NCI-H1963 overexpressed with LacZ (control cells) had oval and elongated nuclei with a high nuclear:cytoplasm ratio, smooth granular chromatin, and no nucleoli consistent with classic SCLC histology (Fig. 5F and fig. S9C), similar to the histology that would be typically diagnosed with SCLC in primary human tumors (Fig. 5G). Histological analysis of xenografts of NCI-H1963 overexpressed with c-Myc showed presence of nucleoli and polygonal-shaped larger cells, reminiscent of variant SCLC histology as defined in (9) (Fig. 5F and fig. S9C). The histopathology observed here, according to the current WHO classification, would likely be recognized as either purely LCNEC or “combined SCLC” that includes a component of LCNECs, potentially reflective of the discrepancies between clinical practice and experimental findings (Fig. 5G) (10, 39). Together, these results suggest that c-Myc expression drives the transition from ASCL1-positive state characterized by classic SCLC histology to NeuroD1-positive state SCLC characterized by variant SCLC or LCNEC histology.
To examine the relevance of c-Myc expression in LCNEC, we compared distribution of its mRNA expression levels in tumors diagnosed as LCNEC (40) and those diagnosed as SCLC (21). After batch correction, we found that a greater proportion of LCNEC tumors express c-Myc at a level comparable to c-Myc–expressing SCLC (fig. S9D). These data suggest that c-Myc expression is likely as relevant in human LCNEC as in SCLC and are consistent with the evidence where c-Myc overexpression can induce histological characteristics reminiscent of LCNEC in our model, while it does not necessarily indicate that LCNEC histology is defined by the expression of c-Myc.
c-Myc mediates transdifferentiation through REST
We sought to investigate the mechanism through which c-Myc mediates transdifferentiation to the ASCL1-negative/NeuroD1-positive state. To this end, we profiled the accessible chromatin profile of transdifferentiated NCI-H1963. Filtering the regions to reflect regions with Myc (E-box) motif matrices reflected a common pattern (clusters 1 to 4). c-Myc overexpression resulted in the gain of additional regulatory regions (cluster 5) and a decrease in accessibility at a fraction of sites in cluster 6 (Fig. 6A), revealing an alteration of the Myc (E-box) cistrome and further suggesting nonredundancy of this family of transcription factors. Next, we sought to investigate how the rewiring of these regulatory regions compared to c-Myc– and L-Myc–expressing SCLC. PCA of Myc accessibility in these genetically engineered cells and c-Myc– and L-Myc–classified cell lines revealed NCI-H1963 expressing c-Myc clustered closer to c-Myc–classified cell lines along PC1, mapping NCI-H1963 transitioning from L-Myc cistrome toward a c-Myc cistrome (Fig. 6B). These data reiterate c-Myc–mediated alteration in regulatory regions that dictate cellular identity.
Fig. 6. c-Myc rewires Myc-accessible chromatin regulatory elements to regulate distinct lineage-associated pathways.
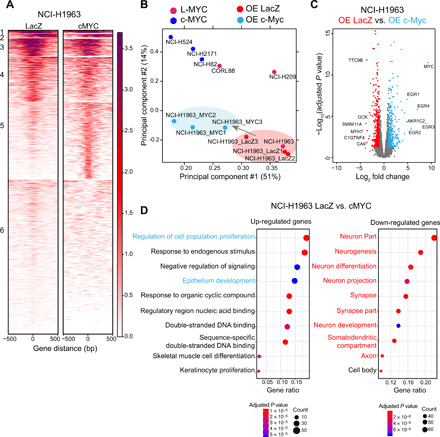
(A) Each heatmap depicting k-means (k = 6) clustering of global Myc (E-box) accessibility in NCI-H1963 overexpressing (OE) LacZ (left) and NCI-H1963 overexpressing cMYC (right) at 44,722 combined peaks. ATAC-seq signal intensity is shown by color shading. (B) PCA of open chromatin regions with E-box motif in six SCLC cell lines in addition to NCI-H1963–LacZ (control) and NCI-H1963–c-Myc in triplicate. Each dot represents a SCLC cell line that is colored on the basis of Myc status (pink, L-MYC; blue, c-MYC; red, NCI-H1963 LacZ; light blue, NCI-H1963 cMYC). (C) Volcano plot showing 775 differentially expressed genes (375 up-regulated in light blue and 400 down-regulated in red) between control NCI-H1963 cells with LacZ overexpression cells and NCI-H1963 cells with c-Myc overexpression. (D) Dot plot showing GO analysis for differentially up-regulated (right) and down-regulated (left) genes.
To identify the transcriptional programs associated with the changes in regulatory regions of the Myc cistrome, we performed RNA sequencing (RNA-seq) on transdifferentiated NCI-H1963. We identified 775 differentially expressed genes (375 up-regulated genes and 400 down-regulated genes) (Fig. 6C). GO analysis for up-regulated genes revealed an enrichment for epithelium development in addition to canonical c-Myc functions including regulation of cell proliferation and regulatory region nucleic acid binding (Fig. 6D, left), while GO analysis for down-regulated of genes revealed an enrichment for neuronal-associated genes (Fig. 6D, right). These findings suggest that c-Myc expression in ASCL1-expressing SCLC induces a transition to a more epithelial state and are consistent with the loss in neuroendocrine lineage state marker (Fig. 5A) but did not reveal further specific mechanistic insights.
Previous works have shown the negative regulation of ASCL1 by Notch signaling during lung development (41), and activation of Notch signaling in SCLC suppresses ASCL1 expression (21, 42, 43). In addition, our SCLC transcriptional network analysis and Myc accessibility data revealed Notch pathway activation preferentially in c-Myc–expressing tumors and cell lines (Figs. 1E and 3C). Although we did not observe an enrichment for an active Notch signaling signature among the up-regulated genes when we investigated the expression of known Notch signaling pathway targets in transdifferentiated NCI-H1963 (24), we identified that Notch target REST was induced upon c-Myc expression (Fig. 7, A and B). Prior works have shown that Rest acts as a neuronal differentiation repressor (44), and REST inhibition removes neural reprogramming block that allows for the conversion to neuronal fate in human adult lung and dermal fibroblasts (45). Therefore, we overexpressed Rest expression in NCI-H1963, inquired the status of ASCL1, and found that Rest in this context can also mediate the suppression of ASCL1 (Fig. 7C).
Fig. 7. c-Myc induces neuronal repressor REST to mediate lineage conversion.
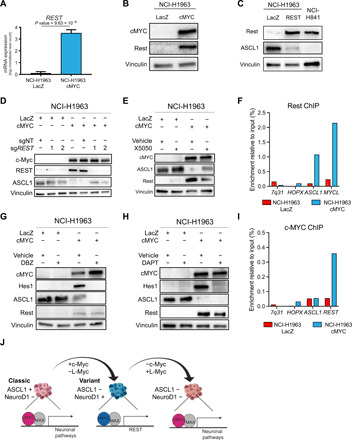
(A) Expression of REST in NCI-H1963 cells with LacZ overexpression and NCI-H1963 with c-Myc overexpression (n = 3). (B) Protein expression of c-Myc and Rest, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells. (C) Protein expression of Rest and ASCL1, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells to overexpress Rest. (D) Protein expression of c-Myc, Rest, and ASCL1, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells genetically deleted for REST and overexpressed with either LacZ as control or c-Myc. (E) Protein expression of c-Myc, L-Myc, ASCL1, and Rest, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells treated with vehicle [dimethyl sulfoxide (DMSO)] or 10 μM Rest inhibitor (X5050) for 48 hours. (F) Rest ChIP-qPCR in NCI-H1963 c-Myc–overexpressed cells and LacZ-overexpressed cells (negative control) at the ASCL1 and MYCL loci with 7q31 and HOPX as negative control loci. One representative of three biological replicates is shown. (G) Protein expression of c-Myc, L-Myc, ASCL1, and Rest, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells treated with vehicle (DMSO) or 0.5 μM Notch inhibitor (DBZ) for 48 hours. (H) Protein expression of c-Myc, L-Myc, ASCL1, and Rest, as well as vinculin, as a loading control in genetically engineered NCI-H1963 cells treated with vehicle (DMSO) or 10 μM Notch inhibitor (DAPT) for 48 hours. (I) c-Myc ChIP-qPCR in NCI-H1963 c-Myc–overexpressed cells and LacZ-overexpressed cells (negative control) at the ASCL1 and REST loci with 7q31 and HOPX as negative control loci. One representative of three biological replicates is shown. (J) Graphical schematic representing role of c-Myc and L-Myc as lineage-determining factors in SCLC.
To further investigate whether Rest is required for c-Myc–mediated suppression of ASCL1 in SCLC, we deleted REST mediated by CRISPR-Cas9 before c-Myc overexpression in NCI-H1963 cells. Suppression of Rest reversed c-Myc overexpression–induced decrease in the level of ASCL1 (Fig. 7D). Simultaneous pharmacological inhibition of Rest (46) and c-Myc overexpression on NCI-H1963 cells recapitulated these findings (Fig. 7E). Further, ChIP–quantitative polymerase chain reaction (qPCR) for Rest in c-Myc–overexpressed NCI-H1963 indicated that Rest associates more significantly with the ASCL1 and MYCL loci compared to control LacZ-overexpressed NCI-H1963, suggesting that Rest directly modulates the expression of these neuroendocrine markers (Fig. 7F).
In murine SCLC cells, gain of Rest expression has been associated with active Notch signaling that down-regulate the expression of Ascl1 (29). Therefore, we sought to investigate the c-Myc–Notch–Rest axis in mediating transdifferentiation. Pharmacological inhibition of Notch using a γ-secretase inhibitor, DBZ (47), and DAPT in combination with c-Myc expression in NCI-H1963 cells reduced the level of Hes1, confirming a suppression of the pathway, but did not alter expression of Rest and ASCL1 (Fig. 7, G and H). Therefore, we investigate how c-Myc activates the expression of Rest, and we performed c-Myc ChIP-qPCR on c-Myc–overexpressed NCI-H1963 cells and found that c-Myc binding is more enriched at the REST locus but not at the ASCL1 locus compared to NCI-H1963 LacZ control cells, suggesting that c-Myc directly, at least partially, modulates the expression of Rest (Fig. 7I). Together, these data suggest a novel Notch-independent, c-Myc–mediated activation of Rest and Rest-mediated suppression of ASCL1.
DISCUSSION
In this study, we showed a novel functional distinction between c-Myc and L-Myc in SCLC. Moreover, our data from functional replacement of c-Myc with L-Myc in c-Myc-SCLC suggest that c-Myc is responsible for the maintenance of NeuroD1 expression and that further replacement with L-Myc leads to transition into a neuronal state more similar to ASCL1-SCLC; however, it does not shift into full ASCL1-SCLC state but instead to neuronal lineage–negative state. On the other hand, c-Myc overexpression in ASCL1-SCLC with classic SCLC histology resulted in transdifferentiation to NeuroD1-SCLC accompanied by LCNEC-like/variant SCLC histology partially mediated through the direct up-regulation of Rest (Fig. 7J).
We describe that c-Myc is associated not only with canonical roles including transcriptional and translational regulation but also with lineage-associated pathways such as Notch signaling and epithelial-to-mesenchymal transitions. Our findings are supported by a previous report that described active Notch signaling negatively regulating neuroendocrine lineage by suppressing the expression of ASCL1 (29) and a study that showed mesenchymal-like features in neuroendocrine-low subtype of SCLC (28). On the other hand, we found that L-Myc is associated with neuronal pathways, implicating their relevancy in determining the classic neuroendocrine state of SCLC. These findings are consistent with previous observation showing a correlation of expression between L-Myc and neuronal proteins (28, 48). Together, contrary to the notion that Myc family members, c-Myc in particular, exclusively act as oncogenes and general amplifiers of expression and metabolism, our data suggest that they additionally exert lineage-defining transcriptional programs (24). We propose that the Myc family members regulate a defined set of transcriptional programs that are essential in SCLC lineage subtype determination. Notably, these biological pathways have been implicated for lineage maintenance and determination in distinct subtypes of SCLC and correlated to different Myc family members (28); here, we show that Myc family members indeed regulate these SCLC lineage–associated pathways.
We found that L-Myc failed to induce the expression of a neuroendocrine lineage state marker ASCL1, suggesting its inability to fully control transdifferentiation from neuroendocrine-low/variant state to neuroendocrine-high/classic state. However, on the basis of previous work suggesting requirement of L-Myc to drive tumors of neuroendocrine origin in SCLC GEMM (16) and our findings that revealed L-Myc regulating and inducing neuronal development pathways, L-Myc likely plays roles in lineage state maintenance and/or is more compatible with the neuroendocrine lineage.
Here, we report the functional importance of c-Myc in regulating cellular states (18, 24). We also report a previously undescribed function for c-Myc in lineage maintenance in NeuroD1-positive SCLC. c-Myc expression was incompatible with ASCL1-positive SCLC and induces transdifferentiation to NeuroD1-positive state, revealing the role for c-Myc in regulating lineage plasticity (24). We show that this transdifferentiation between molecular subtypes of SCLC is accompanied by alterations in the transition of histopathology in SCLC from classic SCLC features to variant histopathology. These data reflect that the variant histopathology that would typically be classified as combined SCLC or LCNEC per the current guidelines of WHO can indeed be derived from SCLC. This itself has substantial clinical implication. This phenotype is reflective of biology distinct from classic SCLC consistent with prior reports (14, 18, 37, 49) and, therefore, warrants distinct therapeutic strategies.
We identified that the transdifferentiation is mediated independent of Notch signaling but through the direct activation of a Notch signaling target, Rest, a transcriptional repressor. These findings may be clinically important when evaluating the rationale for targeting SCLC by activating Notch signaling with drugs including LSD1 and KDM5A inibitors (42, 43). Our data suggest that downstream actors of Notch signaling induce c-Myc–mediated transdifferentiation. This may not be a favorable outcome since the variant subtype has been reported to be more frequent in tumors after initial response to chemotherapy (9, 18). We found that the altered differentiation state with the gain of L-Myc and loss of c-Myc cells conferred resistance to Aurora kinase A inhibition and, in contrast, the transdifferentiation as a result of c-Myc gain and loss of L-Myc cells conferred sensitivity to Aurora kinase A inhibition (37). The differential sensitivity associated with distinct lineage state with Myc family member status as a biomarker could translate to valuable clinical implications in stratification of patients for targeted therapy (50).
SCLC is a recalcitrant disease typically characterized by neuroendocrine differentiation; nonetheless, approximately 10 to 20% of SCLCs may lack expression of diagnostic neuroendocrine markers. Here, we report that the plasticity between these histological subtypes and molecular subtypes is regulated by c-Myc and L-Myc. The role in lineage determination and maintenance for this family of transcription factors is notable since the Myc family has historically been grouped as general oncogenes. Our data suggest that the role of Myc family in SCLC tumorigenesis could be redefined. This will enable us to categorize these subtypes to develop effective therapies to combat this highly lethal disease.
METHODS
CCLE RNA-seq analysis
RNA-seq gene expression data and SNP array 6.0 copy number data for MYC, MYCL, and MYCN were downloaded from the CCLE website. Log2[RNA-seq Expectation Maximization (RSEM) (transcripts per million) + 1] values for mRNA expression and log2(inferred copy number / 2) values for copy numbers were used to depict a heatmap for 49 SCLC cell lines. MYC-classified cell lines were further refined with expression cutoff for MYC at a log2 RSEM of >8 and expression of MYCL at a log2 RSEM of <2. For MYCL-classified cell lines, expression cutoff was set at an MYCL log2 RSEM of >8 and expression of an MYC log2 RSEM of <2.
Bayesian network analysis
A comprehensive regulatory network was built using the software suite RIMBANet (26) by combining RNA-seq data from two independent SCLC primary cohorts (21, 25). To identify subnetworks enriched for L-Myc and c-Myc signatures, the gene signature was overlaid on the network, and neighboring nodes within two layers for each signature were selected. Then, closed subnetworks with more than 25 nodes were collected for L-MYC and c-MYC subnetworks. Then, key driver analysis (51) identified master regulators associated with L-Myc and c-Myc signatures in two steps. First, for each node in the network, we compared neighboring nodes within two layers for each signature to obtain a list of potential master regulators whose overlap is statistically significant [Fisher’s exact test (FET), P < 0.05 / (number of nodes)]. After sorting candidate genes by corresponding FET P values, the gene with the strongest P value was determined as a key regulator. Any candidate regulators in its two-layered neighbors were excluded from the candidate list. The process was iterated throughout the sorted candidate list. Enriched functions were identified using GO terms in Molecular Signatures Database (MSigDB). The significance of the enrichment was determined on the basis of FET P value considering multiple testing [FET, P < 0.05 / (number of dataset tested)] in each category.
Cell lines
All cells were cultured at 37°C in a humidified incubator at 5% CO2. SCLC cell lines NCI-H2171, NCI-H82, NCI-H524, NCI-H209, NCI-H1963, PC-9, CORL88, HCC33, NCI-H69, and NCI-H526 were maintained in RPMI 1640 (Gibco) and supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich) and 1 mM penicillin/streptomycin (P/S; Gibco). Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco’s modified Eagle’s medium and supplemented with 10% FBS (Sigma-Aldrich) and 1 mM P/S. HU-EV cultured cells were regularly tested for mycoplasma using the MycoAlert Detection Kit (Lonza).
ATAQ-seq
ATAC-seq was performed as previously described (52). Five thousand cells were harvested and pretreated with deoxyribonuclease I (DNase I) (Thermo Fisher Scientific) at 37°C for 20 min to eliminate DNA contamination from dead cells. The cells were washed in cold phosphate-buffered saline (PBS) to eliminate traces of DNase. Nuclei were isolated with nuclear lysis buffer (10 mM tris, 10 mM NaCl, 3 mM MgCl2, and 0.1% IGEPAL-630) and centrifuged at low speeds. The nuclei were resuspended in transposase reaction mixture [10 mM tris (pH 8.0), 5 mM MgCl2, 10% dimethlyformamide, and Tn5 transposase complex (0.2 mg/ml)]. Transposition was carried out at 37°C for 30 min, followed by DNA purification with DNA Clean and Concentrator-25 (Zymo Research) according to the manufacturer’s recommendation. Following purification, library fragments were PCR-amplified with Nextera adapter primers. Sequencing was performed at the Tisch Cancer Institute sequencing core on the NextSeq 500 (Illumina) for 38 nucleotides (nt) each from paired ends according to the manufacturer’s instructions.
ATAC-seq data analysis
Illumina sequencing adapter was removed using Cutadapt from raw sequence files in fastq format. The reads were aligned to the hg19 reference genome using Bowtie2 with -k 1 and default parameters. The aligned reads were used after removing PCR duplicates using SAMtools and filtered off an ATAC blacklist (52) for mitochondrial DNA and homologous sequences. Both fragment ends were shifted +4 nt for positive strand and −5 nt for negative strand to account for the distance from Tn5 binding and helicase activity to identify cut sites. Extended Tn5 cut sites were used for peak calling with MACS2 with parameters --nomodel --extsize 100 --shift 50 --nolambda --keep-dup all. Fraction of reads in peak score was calculated for each sample and was used as a normalization factor for pileup rescaled to a total number of uniquely alignable sequences by WigMath function of Java-Genomic Toolkit. Peaks were filtered by MYC motif using findMotifsGenome function in Homer using four motif matrices MA0058.2 (MAX), MA0059.1 (MAX::MYC), MA0104.4 (MYCN), and MA0147.3 (MYC). The peaks filtered with MYC motifs were visualized in heatmaps by k-means clustering (k = 8) using Cistrome (10, 11, 35) or using k-means clustering (k = 6 for genetically engineered NCI-H1963 and k = 4 for genetically engineered NCI-H82 cells) using plotHeatmap function of deepTools (53) with wiggle files. Normalized ATAC-seq signals for each sample were visualized on Integrative Genome Viewer genome browser (54).
Differentially bound sites comparing three c-Myc–classified cell lines versus three L-Myc–classified cell lines were identified using R package DiffBind with cutoffs at a fold change of ≥5 and a false discovery rate (FDR) of ≤0.05. Functional analysis of the differentially bound regions was performed using GREAT (35). For NCI-H82, differentially bound sites were identified using R package DiffBind between three NCI-H82 control cells (LacZ, LacZ/sgNT1, and LacZ/sgNT2) and three c-Myc replaced by L-Myc (L-Myc/sgMYC1, L-Myc/sgMYC2, and L-Myc/sgMYC3) with cutoffs at a fold change of >16 and an FDR of <0.01. Functional analysis of the differentially bound regions integrated with differentially expressed genes from transcriptomic data of the same samples was performed using Cistrome-GO (55). Aggregate Myc accessible signal at TSS around c-Myc or L-Myc signature genes was computed using deeptools (53).
Western blot and antibodies
Protein lysates were prepared by washing cells with 1 ml of cold PBS and resuspended in lysis buffer [150 mM NaCl, 50 mM tris-HCl (pH 8.0), 1% NP-40, 0.5% Na deoxycholate, 0.1% SDS, and protease inhibitors] for 30 min at 4°C. Lysates were centrifuged at 5°C for 15 min at 13,000 rpm to remove insoluble debris. Protein concentrations were quantified using Pierce BCA (Thermo Fisher Scientific). Proteins were separated by electrophoresis on an SDS–polyacrylamide gel electrophoresis gel (Bio-Rad), transferred to a polyvinylidene difluoride membrane (Thermo Fisher Scientific), and blocked with 5% milk in tris-buffered saline (TBS) with Tween 20. The membranes were immunoblotted with anti–c-Myc (Santa Cruz Biotechnology, sc-40), anti–L-Myc (University of Iowa, PCRP-MYCL1-1A3), anti-vinculin (Sigma-Aldrich), anti-ASCL1 (BD Biosciences, #556604), anti-NeuroD1(Proteintech, 12081-1-AP), anti-Smad2 (Cell Signaling Technology, #5339), anti-Rest (Millipore Sigma, 07-579), anti-Hes1 (Santa Cruz Biotechnology, sc-166410), anti-Brn2 (Cell Signaling Technology, 12137), anti-YAP1 (Proteintech, 13584-1-AP). Immunoblots were quantified using ImageJ.
Lentiviral introduction of genes
c-Myc, L-Myc, or LacZ open reading frame was cloned into pLEX_307 (a gift from D. Root; Addgene, #41392) using the Gateway cloning methods according to the manufacturer’s recommendations (Thermo Fisher Scientific). HEK293T cells were seeded in a 10-cm tissue culture dish and incubated at 37°C and 5% CO2. At 80% confluency, the cells were cotransfected with 10 μg of plasmid constructs, 7.5 μg of psPAX2 (a gift from D. Trono; Addgene, #12260), and 2.5 μg of pMD2.G (a gift from D. Trono; Addgene, #12259) vectors using TransIT-Lenti (Mirus) following the manufacturer’s recommendations. At 48 hours after transfection, virus-containing supernatants were collected, filtered (0.45 μm), and stored at −80°C. Cells were infected with lentiviral supernatant supplemented with polybrene at a final concentration of 8 μg/ml or LentiBlast (OZ Biosciences) at a ratio of 1:1000. Cells were selected with puromycin (1 to 2 μg/ml for 4 to 6 days).
CRISPR-Cas9 genome editing
Cells with stable human codon-optimized Streptococcus pyogenes Cas9 expression were generated by infection with the lentiCas9-Blast plasmid (a gift from F. Zhang; Addgene, #52962). Single-guide RNAs (sgRNAs) targeting MYC and MYCL were selected from the Brunello library (56). Nontarget sgRNAs from the Gecko library v2 (57) were used as nontarget sgRNAs. sgRNA target sequences are listed in table S5. sgRNAs were cloned using Bbs I site downstream of the human U6 promoter in a lentiviral vector containing enhanced green fluorescent protein (EGFP) downstream of the human Phosphoglycerate kinase (PGK) promoter [a gift of Brown laboratory, Icahn School of Medicine at Mount Sinai (ISMMS)]. Lentivirus was produced as described above. Cas9-expressing cells were then infected with pLenti-GFP-sgRNA.
Cell proliferation assay
Cells were plated at a density of 5000 cells per well with five replicates in a 96-well plate; four identical plates were prepared. Cell viability was assayed at 0, 2, 4, and 6 days after plating with alamarBlue Cell Viability Reagent (Thermo Fisher Scientific), and fluorescence at 585 nm was measured on a SpectraMax3 plate reader (Molecular Devices) according to the manufacturer’s protocol at an excitation of 555 nm. Cell viability at 2, 4, and 6 days were corrected for the ratio to control cells from the day 0 reading to account for plating unevenness.
RNA sequencing
Total RNAs from engineered NCI-H82 and NCI-H1963 cell lines were extracted using an RNeasy kit (QIAGEN). Polyadenylated RNA was enriched from 1 μg of RNA for each sample with the NEBNext PolyA mRNA Magnetic Isolation Module [New England Biolabs (NEB)], incubated at 94°C for 15 min, and double-stranded complementary DNA (cDNA) was synthesized using SuperScript III reverse transcriptase (Thermo Fisher Scientific) and NEBNext Ultra II Directional RNA Second Strand Synthesis Module (NEB). Up to 10 ng of cDNA was used for the Illumina sequencing library construction using the NEBNext Ultra DNA Library Prep Kit (NEB). Paired-end sequencing was performed on NextSeq 500 (Illumina) for 38 nt from each end according to the manufacturer’s instructions.
RNA-seq analyses
Sequencing reads were pseudo-aligned to the human reference transcriptome GRCh38.95 from Ensembl, and transcript abundance was estimated using kallisto (v0.45.0) (58). Transcript abundance was aggregated to gene level abundance using biomaRt annotation. DEseq2 (59) was used to identify differentially expressed genes between control NCI-H82 cells (LacZ, LacZ/sgNT1, and LacZ/sgNT2) and L-Myc–overexpressed NCI-H82 cells (L-Myc, L-Myc/sgNT1, and L-Myc/sgNT2). The same strategy was used for L-Myc–overexpressed NCI-H82 cells (L-Myc, L-Myc/sgNT1, and L-Myc/sgNT2) and c-Myc replaced with L-Myc NCI-H82 cells (L-Myc/sgMYC1, L-Myc/sgMYC2, and L-Myc/sgMYC3), as well as for LacZ-overexpressed NCI-H1963 cells and c-Myc–overexpressed NCI-H1963 cells.
Differentially expressed genes were determined on the basis of cutoffs of a fold change of >2 and an FDR of <0.05. To identify potentially enriched functions of selected gene sets of interest, we compared these gene sets with the genes annotated by GO terms curated in the MSigDB. Each of 5917 GO terms included in “C5” collection (version 7.0) was compared with query gene sets using FET. The significance of the overlap was determined on the basis of P value adjusted for multiple comparisons (FDR, <0.05). Any GO terms consisting of more than 2000 genes were considered nonspecific and removed from the analysis.
β-Galactosidase staining
Cells were stained with the β-galactosidase FluoReporter Galactosidase Quantification Kit (Thermo Fisher Scientific, F2905) according to the manufacturer’s recommendations in technical triplicates.
Cell cycle analysis
Cells were harvested at day 2 after selection and fixed with 70% ethanol overnight at 4°C, washed with PBS, and then incubated in PBS containing ribonuclease (RNase; 100 μg/ml) and propidium iodide (50 μg/ml) at room temperature for 1 hour. DNA content was analyzed by FACSCanto II (BD Biosciences), and quantitative analyses for the proportions of cells in cell cycle were performed using FlowJo software (BD Biosciences).
Annexin V staining
Cells were harvested at day 2 after selection and washed with PBS. Cells were stained with annexin V and propidium iodide using an Alexa Fluor 488 annexin V/dead cell apoptosis kit (Thermo Fisher Scientific) according to the manufacturers’ recommendations. Fluorescence was measured by FACSCanto II (BD Biosciences). Quantitative analyses for the cell viability proportions were performed using FlowJo software (BD Biosciences).
Xenograft model
All animal procedures and studies were approved by the Mount Sinai Institutional Animal Care Use Committee (protocol number, IACUC-2018-0021). H1963 cells that overexpressed LacZ or c-Myc (5 × 106 cells) were injected with a 1:1 mixture of 50-μl cell suspension and 50-μl Matrigel (Corning) subcutaneously into both flank regions of 4- to 6-week-old male nonobese diabetic–severe combined immunodeficient gamma mice (the Jackson laboratory). Tumor volume (length × width2 / 2) was measured twice a week. When tumor size reached at 1000 mm3, mice were euthanized, and tumors were immersed in formalin for histological analysis. Histopathology was reviewed by a board-certified lung cancer pathologist (M.B.B.).
Immunohistochemistry
Xenograft tumor specimen formalin-fixed, paraffin-embedded tissue slides (5 μm in thickness) were deparaffinized and rehydrated. For antigen retrieval, slides were heated at 95°C in 10 mM citrate buffer (pH 6.0) for 30 min. The sections were incubated with 0.3% H2O2 in TBS for 15 min to block endogenous peroxidase activity and were incubated with 10% normal horse serum (Jackson ImmunoResearch) in TBS for 30 min to block nonspecific staining. The sections were rinsed with TBS and 0.025% Triton X-100 and then incubated with anti–c-Myc antibody (1:150; Abcam, #ab32072), anti-NeuroD1 (1:1000; Abcam, #ab60704), or anti-ASCL1 (1:100; BD Biosciences, #556604) at 4°C overnight. This was followed by incubation with biotin-conjugated horse anti-mouse secondary antibody (Vector Laboratories) at room temperature for 1 hour. Then, the sections were incubated with the ABC reagent (Vector Laboratories, CA) and visualized with ImmPACT-DAB Peroxidase Substrate (Vector Laboratories). All slides were counterstained with hematoxylin before mounting.
Aurora kinase inhibition assay
Cells were seeded in a 96-well plate at a density of 5000 cells per well with five replicates. Cells were treated with alisertib at concentrations ranging from 1 nM to 10 μM for 96 hours. Cell viability was measured using alamarBlue Cell Viability Reagent (Thermo Fisher Scientific), and fluorescence at 585 nm was measured on a SpectraMax3 plate reader (Molecular Devices, CA) according to the manufacturer’s protocol at an excitation of 555 nm.
ChIP followed by qPCR
ChIP was performed as previously described (34). A total of 1 × 107 cells were cross-linked with 1% formaldehyde in PBS for 10 min at room temperature, washed in bovine serum albumin (5 mg/ml) in PBS and then in just cold PBS, resuspended in lysis buffer [50 mM tris-HCl (pH 8.1), 10 mM EDTA, 1% SDS, and 1× protease inhibitor cocktail (Thermo Fisher Scientific)], and sonicated with the Covaris M220 sonicator to obtain chromatin fragment lengths of 100 to 1000 base pairs judged by the Bioanalyzer DNA High Sensitivity Kit (Agilent). Fragmented chromatin was diluted in immunoprecipitation buffer [20 mM tris-HCl (pH 8.1), 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100] and incubated overnight at 4°C with protein G magnetic beads (Dynabeads, Thermo Fisher Scientific) that had been preincubated with anti-MYC (Cell Signaling Technology, #9402) or anti-REST (Millipore Sigma, 07-579) antibodies. Immunoprecipitates were washed six times with the wash buffer [50 mM Hepes (pH 7.6), 0.5 M LiCl, 1 mM EDTA, 0.7% sodium deoxycholate, and 1% IGEPAL CA-630] and twice with tris-EDTA buffer. Immunoprecipitated (or no-immunoprecipitation input) DNA was treated with RNase A and proteinase K on the beads, recovered in 1% SDS and 0.1 M NaHCO3 over a period of 7 hours at 65°C, and purified with DNA Clean and Concentrator-25 (Zymo Research).
Real-time qPCRs were performed in duplicate (Applied Biosystems) on a 7500 Real-Time PCR System (Applied Biosystems) with results normalized to input. Primers are listed in table S6.
Quantitative reverse transcription PCR
For expression analysis, RNA was extracted from cells using the QIAGEN RNeasy Mini Kit, and cDNA was prepared with the QIAGEN QuantiTect Reverse Transcription Kit. Real-time qPCRs were performed as described above with results normalized to GAPDH expression. Primers are listed in table S6.
Acknowledgments
We thank D. Yang, F. Jiang, Y. Tian, B. Shewale, N. Stokes, D. Dominique-Sola, and Y. Sun for helpful discussions and technical assistance; C. Nguyen and E. Bernstein for providing the Rest antibody; P. Guo for providing HU-EV cells; A. Wroblewska and B. D. Brown for providing a lentiviral vector for cloning sgRNAs; and NextSeq Sequencing Facility of the Department of Oncological Sciences at ISMMS, S. Hekmaty, G. Panda, and R. Sachidanandam for sequencing assistance. We also thank the Flow Cytometry Core facility and the Biorepository and Pathology Core Facility at ISMMS. Funding: This work was supported in part through Tisch Cancer Institute at ISMMS and the computational resources and staff expertise provided by the Scientific Computing at ISMMS. H.W. has been supported by the ATS Foundation Unrestricted Grant, the American Lung Association of the Northeast Lung Cancer Discovery Award, Department of Defense (W81XWH-19-1-0613), and NIH (R01CA240342). T.S. is supported by the Japanese Respiratory Society the 6th Lilly Oncology Fellowship Program and the Uehara Memorial Foundation. R.K. is supported by Shaanxi Provincial Natural Science Foundation, China (2017JM8046). C.A.P. and H.W. are supported by NIH (R01CA163772). Author contributions: Conceptualization and methodology: A.S.P. and H.W. Data collection: A.S.P., S.Y., R.K., T.S., A.S., S.K., L.B., M.F., K.E., and H.W. Resources: M.B.B., C.A.P., and H.W. Data analysis: A.S.P., S.Y., G.N., M.B.B., and H.W. Writing—original draft: A.S.P. and H.W. Writing—review and editing: A.S.P., S.Y., C.A.P., M.B.B., J.Z., and H.W. Supervision: C.A.P., J.Z., and H.W. Project administration and funding acquisition: H.W. Competing interests: J.Z. is the head of data science, and S.Y. is an employee at Sema4 Genomics, a Mount Sinai venture, a for-profit organization that promotes genomic sequencing for patient-centered health care. C.A.P. serves as a consultant to Daiichi Sankyo Inc. and AstraZeneca PLC for which C.A.P. has received consulting fees. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The sequencing data have been deposited into GEO with the accession number GSE156334. The vectors pLEX_307 (Addgene, #41392), psPAX2 (Addgene, #12260), pMD2.G (Addgene, #12259), and lentiCas9-Blast (Addgene, #52962) used in this manuscript can be provided via Addgene pending scientific review and a completed material transfer agreement. Requests for the vectors should be submitted to Addgene.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc2578/DC1
REFERENCES AND NOTES
- 1.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Gazdar A. F., Bunn P. A., Minna J. D., Small-cell lung cancer: What we know, what we need to know and the path forward. Nat. Rev. Cancer 17, 725–737 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Alvarado-Luna G., Morales-Espinosa D., Treatment for small cell lung cancer, where are we now?-a review. Transl. Lung Cancer Res. 5, 26–38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietanza M. C., Byers L. A., Minna J. D., Rudin C. M., Small cell lung cancer: Will recent progress lead to improved outcomes? Clin. Cancer Res. 21, 2244–2255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horn L., Mansfield A. S., Szczęsna A., Havel L., Krzakowski M., Hochmair M. J., Huemer F., Losonczy G., Johnson M. L., Nishio M., Reck M., Mok T., Lam S., Shames D. S., Liu J., Ding B., Lopez-Chavez A., Kabbinavar F., Lin W., Sandler A., Liu S. V.; IMpower133 Study Group , First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 379, 2220–2229 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Rudin C. M., Poirier J. T., Byers L. A., Dive C., Dowlati A., George J., Heymach J. V., Johnson J. E., Lehman J. M., Pherson D. M., Massion P. P., Minna J. D., Oliver T. G., Quaranta V., Sage J., Thomas R. K., Vakoc C. R., Gazdar A. F., Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 19, 289–297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zappa C., Mousa S. A., Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 5, 288–300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carney D. N., Gazdar A. F., Bepler G., Guccion J. G., Marangos P. J., Moody T. W., Zweig M. H., Minna J. D., Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 45, 2913–2923 (1985). [PubMed] [Google Scholar]
- 9.Gazdar A. F., Carney D. N., Nau M. M., Minna J. D., Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res. 45, 2924–2930 (1985). [PubMed] [Google Scholar]
- 10.Travis W. D., Brambilla E., Nicholson A. G., Yatabe Y., Austin J. H. M., Beasley M. B., Chirieac L. R., Dacic S., Duhig E., Flieder D. B., Geisinger K., Hirsch F. R., Ishikawa Y., Kerr K. M., Noguchi M., Pelosi G., Powell C. A., Tsao M. S., Wistuba I.; WHO Panel , The 2015 world health organization classification of lung tumors: Impact of genetic, clinical and radiologic advances since the 2004 classification. J. Thorac. Oncol. 10, 1243–1260 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Poirier J. T., Dobromilskaya I., Moriarty W. F., Peacock C. D., Hann C. L., Rudin C. M., Selective tropism of seneca valley virus for variant subtype small cell lung cancer. J. Natl. Cancer Inst. 105, 1059–1065 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borromeo M. D., Savage T. K., Kollipara R. K., He M., Augustyn A., Osborne J. K., Girard L., Minna J. D., Gazdar A. F., Cobb M. H., Johnson J. E., ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 16, 1259–1272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McColl K., Wildey G., Sakre N., Lipka M. B., Behtaj M., Kresak A., Chen Y., Yang M., Velcheti V., Fu P., Dowlati A., Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 8, 73745–73756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y.-H., Klingbeil O., He X.-Y., Wu X. S., Arun G., Lu B., Somerville T. D. D., Milazzo J. P., Wilkinson J. E., Demerdash O. E., Spector D. L., Egeblad M., Shi J., Vakoc C. R., POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 32, 915–928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brägelmann J., Böhm S., Guthrie M. R., Mollaoglu G., Oliver T. G., Sos M. L., Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 16, 1489–1498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim D.-W., Wu N., Kim Y.-C., Cheng P. F., Basom R., Kim D., Dunn C. T., Lee A. Y., Kim K., Lee C. S., Singh A., Gazdar A. F., Harris C. R., Eisenman R. N., Park K.-S., Pherson D. M., Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 30, 1289–1299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meuwissen R., Linn S. C., Linnoila R. I., Zevenhoven J., Mooi W. J., Berns A., Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Mollaoglu G., Guthrie M. R., Böhm S., Brägelmann J., Can I., Ballieu P. M., Marx A., George J., Heinen C., Chalishazar M. D., Cheng H., Ireland A. S., Denning K. E., Mukhopadhyay A., Vahrenkamp J. M., Berrett K. C., Mosbruger T. L., Wang J., Kohan J. L., Salama M. E., Witt B. L., Peifer M., Thomas R. K., Gertz J., Johnson J. E., Gazdar A. F., Wechsler-Reya R. J., Sos M. L., Oliver T. G., MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31, 270–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DePinho R., Mitsock L., Hatton K., Ferrier P., Zimmerman K., Legouy E., Tesfaye A., Collum R., Yancopoulos G., Nisen P., Kriz R., Alt F., Myc family of cellular oncogenes. J. Cell. Biochem. 33, 257–266 (1987). [DOI] [PubMed] [Google Scholar]
- 20.Conacci-Sorrell M., McFerrin L., Eisenman R. N., An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 4, a014357 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.George J., Lim J. S., Jang S. J., Cun Y., Ozretić L., Kong G., Leenders F., Lu X., Fernández-Cuesta L., Bosco G., Müller C., Dahmen I., Jahchan N. S., Park K.-S., Yang D., Karnezis A. N., Vaka D., Torres A., Wang M. S., Korbel J. O., Menon R., Chun S.-M., Kim D., Wilkerson M., Hayes N., Engelmann D., Pützer B., Bos M., Michels S., Vlasic I., Seidel D., Pinther B., Schaub P., Becker C., Altmüller J., Yokota J., Kohno T., Iwakawa R., Tsuta K., Noguchi M., Muley T., Hoffmann H., Schnabel P. A., Petersen I., Chen Y., Soltermann A., Tischler V., Choi C.-m., Kim Y.-H., Massion P. P., Zou Y., Jovanovic D., Kontic M., Wright G. M., Russell P. A., Solomon B., Koch I., Lindner M., Muscarella L. A., la Torre A., Field J. K., Jakopovic M., Knezevic J., Castaños-Vélez E., Roz L., Pastorino U., Brustugun O.-T., Lund-Iversen M., Thunnissen E., Köhler J., Schuler M., Botling J., Sandelin M., Sanchez-Cespedes M., Salvesen H. B., Achter V., Lang U., Bogus M., Schneider P. M., Zander T., Ansén S., Hallek M., Wolf J., Vingron M., Yatabe Y., Travis W. D., Nürnberg P., Reinhardt C., Perner S., Heukamp L., Büttner R., Haas S. A., Brambilla E., Peifer M., Sage J., Thomas R. K., Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weir B. A., Woo M. S., Getz G., Perner S., Ding L., Beroukhim R., Lin W. M., Province M. A., Kraja A., Johnson L. A., Shah K., Sato M., Thomas R. K., Barletta J. A., Borecki I. B., Broderick S., Chang A. C., Chiang D. Y., Chirieac L. R., Cho J., Fujii Y., Gazdar A. F., Giordano T., Greulich H., Hanna M., Johnson B. E., Kris M. G., Lash A., Lin L., Lindeman N., Mardis E. R., Mc Pherson J. D., Minna J. D., Morgan M. B., Nadel M., Orringer M. B., Osborne J. R., Ozenberger B., Ramos A. H., Robinson J., Roth J. A., Rusch V., Sasaki H., Shepherd F., Sougnez C., Spitz M. R., Tsao M.-S., Twomey D., Verhaak R. G. W., Weinstock G. M., Wheeler D. A., Winckler W., Yoshizawa A., Yu S., Zakowski M. F., Zhang Q., Beer D. G., Wistuba I. I., Watson M. A., Garraway L. A., Ladanyi M., Travis W. D., Pao W., Rubin M. A., Gabriel S. B., Gibbs R. A., Varmus H. E., Wilson R. K., Lander E. S., Meyerson M., Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The Cancer Genome Atlas Research Network , Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ireland A. S., Micinski A. M., Kastner D. W., Guo B., Wait S. J., Spainhower K. B., Conley C. C., Chen O. S., Guthrie M. R., Soltero D., Qiao Y., Huang X., Tarapcsák S., Devarakonda S., Chalishazar M. D., Gertz J., Moser J. C., Marth G., Puri S., Witt B. L., Spike B. T., Oliver T. G., MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 38, 60–78.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang L., Huang J., Higgs B. W., Hu Z., Xiao Z., Yao X., Conley S., Zhong H., Liu Z., Brohawn P., Shen D., Wu S., Ge X., Jiang Y., Zhao Y., Lou Y., Morehouse C., Zhu W., Sebastian Y., Czapiga M., Oganesyan V., Fu H., Niu Y., Zhang W., Streicher K., Tice D., Zhao H., Zhu M., Xu L., Herbst R., Su X., Gu Y., Li S., Huang L., Gu J., Han B., Jallal B., Shen H., Yao Y., Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLOS Genet. 12, e1005895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu J., Lum P. Y., Lamb J., Thakurta D. G., Edwards S. W., Thieringer R., Berger J. P., Wu M. S., Thompson J., Sachs A. B., Schadt E. E., An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet. Genome Res. 105, 363–374 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Zhu J., Zhang B., Smith E. N., Drees B., Brem R. B., Kruglyak L., Bumgarner R. E., Schadt E. E., Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 40, 854–861 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W., Girard L., Zhang Y.-A., Haruki T., Papari-Zareei M., Stastny V., Ghayee H. K., Pacak K., Oliver T. G., Minna J. D., Gazdar A. F., Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl. Lung Cancer Res. 7, 32–49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim J. S., Ibaseta A., Fischer M. M., Cancilla B., O’Young G., Cristea S., Luca V. C., Yang D., Jahchan N. S., Hamard C., Antoine M., Wislez M., Kong C., Cain J., Liu Y.-W., Kapoun A. M., Garcia K. C., Hoey T., Murriel C. L., Sage J., Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 545, 360–364 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudin C. M., Durinck S., Stawiski E. W., Poirier J. T., Modrusan Z., Shames D. S., Bergbower E. A., Guan Y., Shin J., Guillory J., Rivers C. S., Foo C. K., Bhatt D., Stinson J., Gnad F., Haverty P. M., Gentleman R., Chaudhuri S., Janakiraman V., Jaiswal B. S., Parikh C., Yuan W., Zhang Z., Koeppen H., Wu T. D., Stern H. M., Yauch R. L., Huffman K. E., Paskulin D. D., Illei P. B., Varella-Garcia M., Gazdar A. F., de Sauvage F. J., Bourgon R., Minna J. D., Brock M. V., Seshagiri S., Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 44, 1111–1116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin C. Y., Lovén J., Rahl P. B., Paranal R. M., Burge C. B., Bradner J. E., Lee T. I., Young R. A., Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan H., Kaestner K. H., Ang S.-L., Ikegami M., Finkelman F. D., Stahlman M. T., Fulkerson P. C., Rothenberg M. E., Whitsett J. A., Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131, 953–964 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Ferri A. L. M., Lin W., Mavromatakis Y. E., Wang J. C., Sasaki H., Whitsett J. A., Ang S.-L., Foxa1 and Foxa2 regulate multiple phases of midbrain dopaminergic neuron development in a dosage-dependent manner. Development 134, 2761–2769 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Sato T., Yoo S., Kong R., Sinha A., Chandramani-Shivalingappa P., Patel A., Fridrikh M., Nagano O., Masuko T., Beasley M. B., Powell C. A., Zhu J., Watanabe H., Epigenomic profiling discovers trans-lineage SOX2 partnerships driving tumor heterogeneity in lung squamous cell carcinoma. Cancer Res. 79, 6084–6100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain M., Arvanitis C., Chu K., Dewey W., Leonhardt E., Trinh M., Sundberg C. D., Bishop J. M., Felsher D. W., Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297, 102–104 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Dammert M. A., Brägelmann J., Olsen R. R., Böhm S., Monhasery N., Whitney C. P., Chalishazar M. D., Tumbrink H. L., Guthrie M. R., Klein S., Ireland A. S., Ryan J., Schmitt A., Marx A., Ozretić L., Castiglione R., Lorenz C., Jachimowicz R. D., Wolf E., Thomas R. K., Poirier J. T., Büttner R., Sen T., Byers L. A., Reinhardt H. C., Letai A., Oliver T. G., Sos M. L., MYC paralog-dependent apoptotic priming orchestrates a spectrum of vulnerabilities in small cell lung cancer. Nat. Commun. 10, 3485 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang C. V., MYC on the path to cancer. Cell 149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rekhtman N., Neuroendocrine tumors of the lung: An update. Arch. Pathol. Lab. Med. 134, 1628–1638 (2010). [DOI] [PubMed] [Google Scholar]
- 40.George J., Walter V., Peifer M., Alexandrov L. B., Seidel D., Leenders F., Maas L., Müller C., Dahmen I., Delhomme T. M., Ardin M., Leblay N., Byrnes G., Sun R., De Reynies A., McLeer-Florin A., Bosco G., Malchers F., Menon R., Altmüller J., Becker C., Nürnberg P., Achter V., Lang U., Schneider P. M., Bogus M., Soloway M. G., Wilkerson M. D., Cun Y., McKay J. D., Moro-Sibilot D., Brambilla C. G., Lantuejoul S., Lemaitre N., Soltermann A., Weder W., Tischler V., Brustugun O. T., Lund-Iversen M., Helland Å., Solberg S., Ansén S., Wright G., Solomon B., Roz L., Pastorino U., Petersen I., Clement J. H., Sänger J., Wolf J., Vingron M., Zander T., Perner S., Travis W. D., Haas S. A., Olivier M., Foll M., Büttner R., Hayes D. N., Brambilla E., Fernandez-Cuesta L., Thomas R. K., Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 9, –1048 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito T., Kudoh S., Ichimura T., Fujino K., Hassan W. A. M. A., Udaka N., Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: Significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum. Cell 30, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Augert A., Eastwood E., Ibrahim A. H., Wu N., Grunblatt E., Basom R., Liggitt D., Eaton K. D., Martins R., Poirier J. T., Rudin C. M., Milletti F., Cheng W.-Y., Mack F., Pherson D. M., Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal. 12, eaau2922 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oser M. G., Sabet A. H., Gao W., Chakraborty A. A., Schinzel A. C., Jennings R. B., Fonseca R., Bonal D. M., Booker M. A., Flaifel A., Novak J. S., Christensen C. L., Zhang H., Herbert Z. T., Tolstorukov M. Y., Buss E. J., Wong K.-K., Bronson R. T., Nguyen Q.-D., Signoretti S., Kaelin W. G. Jr., The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev. 33, 1718–1738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldelli P., Meldolesi J., The transcription repressor REST in adult neurons: Physiology, pathology, and diseases. eNeuro 2 10.1523/ENEURO.0010-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Drouin-Ouellet J., Lau S., Brattås P. L., Ottosson D. R., Pircs K., Grassi D. A., Collins L. M., Vuono R., Sjöland A. A., Westergren-Thorsson G., Graff C., Minthon L., Toresson H., Barker R. A., Jakobsson J., Parmar M., REST suppression mediates neural conversion of adult human fibroblasts via microRNA-dependent and -independent pathways. EMBO Mol. Med. 9, 1117–1131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charbord J., Poydenot P., Bonnefond C., Feyeux M., Casagrande F., Brinon B., Francelle L., Aurégan G., Guillermier M., Cailleret M., Viegas P., Nicoleau C., Martinat C., Brouillet E., Cattaneo E., Peschanski M., Lechuga M., Perrier A. L., High throughput screening for inhibitors of REST in neural derivatives of human embryonic stem cells reveals a chemical compound that promotes expression of neuronal genes. Stem Cells 31, 1816–1828 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Ran Y., Hossain F., Pannuti A., Lessard C. B., Ladd G. Z., Jung J. I., Minter L. M., Osborne B. A., Miele L., Golde T. E., γ-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol. Med. 9, 950–966 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim Y. H., Girard L., Giacomini C. P., Wang P., Hernandez-Boussard T., Tibshirani R., Minna J. D., Pollack J. R., Combined microarray analysis of small cell lung cancer reveals altered apoptotic balance and distinct expression signatures of MYC family gene amplification. Oncogene 25, 130–138 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Cardnell R. J., Li L., Sen T., Bara R., Tong P., Fujimoto J., Ireland A. S., Guthrie M. R., Bheddah S., Banerjee U., Kalu N. N., Fan Y.-H., Dylla S. J., Johnson F. M., Wistuba I. I., Oliver T. G., Heymach J. V., Glisson B. S., Wang J., Byers L. A., Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 8, 73419–73432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Owonikoko T. K., Dahlberg S. E., Sica G. L., Wagner L. I., Wade J. L. III, Srkalovic G., Lash B. W., Leach J. W., Leal T. B., Aggarwal C., Ramalingam S. S., Randomized phase II trial of cisplatin and etoposide in combination with veliparib or placebo for extensive-stage small-cell lung cancer: ECOG-ACRIN 2511 study. J. Clin. Oncol. 37, 222–229 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y., Yu H., Yoo S., Lee E., Laganà A., Parekh S., Schadt E. E., Wang L., Zhu J., A network analysis of multiple myeloma related gene signatures. Cancer 11, 1452 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramírez F., Dündar F., Diehl S., Grüning B. A., Manke T., deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li S., Wan C., Zheng R., Fan J., Dong X., Meyer C. A., Liu X. S., Cistrome-GO: A web server for functional enrichment analysis of transcription factor ChIP-seq peaks. Nucleic Acids Res. 47, W206–W211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Doench J. G., Fusi N., Sullender M., Hegde M., Vaimberg E. W., Donovan K. F., Smith I., Tothova Z., Wilen C., Orchard R., Virgin H. W., Listgarten J., Root D. E., Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bray N. L., Pimentel H., Melsted P., Pachter L., Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc2578/DC1