

Keratinocytes control tissue homeostasis and skin inflammation via immunosuppressive glucocorticoids.
Abstract
Glucocorticoids (GC), synthesized by the 11β-hydroxylase (Cyp11b1), control excessive inflammation through immunosuppressive actions. The skin was proposed to regulate homeostasis by autonomous GC production in keratinocytes. However, their immunosuppressive capacity and clinical relevance remain unexplored. Here, we demonstrate the potential of skin-derived GC and their role in the regulation of physiological and prevalent inflammatory skin conditions. In line with 11β-hydroxylase deficiency in human inflammatory skin disorders, genetic in vivo Cyp11b1 ablation and long-term GC deficiency in keratinocytes primed the murine skin immune system resulting in spontaneous skin inflammation. Deficient skin GC in experimental models for inflammatory skin disorders led to exacerbated contact hypersensitivity and psoriasiform skin inflammation accompanied by decreased regulatory T cells and the involvement of unconventional T cells. Our findings provide insights on how skin homeostasis and pathology are critically regulated by keratinocyte-derived GC, emphasizing the immunoregulatory potential of endogenous GC in the regulation of epithelial immune microenvironment.
INTRODUCTION
The skin epithelial immune microenvironment deploys several mechanisms to regulate internal and environment-derived stress signals. While tolerance is induced toward innocuous factors, invasion of pathogens and contact with damaging substances activate keratinocytes and skin-resident immune cells to initiate proinflammatory processes resulting in skin-associated inflammation. Mechanisms for resolution and tissue recovery are necessary to maintain a balanced homeostasis and prevent the development of cutaneous immunopathologies (1).
The immunosuppressive effect of glucocorticoids (GC) is widely accepted and exploited in the therapy of inflammatory diseases (2). Despite their pleiotropic and adverse side effects, GC represent the mainstay in clinical therapy for more than 70 years, as diverse inflammatory disorders are still efficiently treated using synthetic GC (3). However, the potential impact of endogenous GC in the regulation of local inflammation in several immune-related diseases remains unclear. The skin and other so-called extra-adrenal organs have been reported to produce immunoregulatory GC independent from the adrenal cortex (4–9). Several studies have shown that de novo GC synthesis in the skin is regulated by a local network equivalent to the hypothalamus-pituitary-adrenal (HPA) axis and its regulatory elements (10–13). GC synthesis involves cytochrome P450 enzymes, including the 11β-hydroxylase (CYP11B1) catalyzing the final hydroxylation of 11-deoxycortisol to cortisol, respectively 11-deoxycorticosterone to corticosterone (14). Local de novo GC synthesis in the skin allows the integumentary system to react toward tissue insults and inflammation independent from the central stress response by the HPA axis. In this regard, cutaneous GC were shown to be involved in epithelial development, differentiation, and homeostasis, and recent studies also linked deficient skin GC synthesis to inflammatory skin diseases (15–18). However, the capacity and scope of skin-derived GC regarding their immunoregulatory function under physiological and immunopathological conditions and their clinical relevance remain to be investigated.
Here, we addressed this question and generated an inducible in vivo knockout (KO) model with keratinocyte-specific deletion of the critical enzyme Cyp11b1. Cyp11b1 depletion resulted in a substantial reduction of keratinocyte-produced GC and spontaneously primed skin draining lymph node (dLN) cells through skin-derived antigen-presenting cells (APCs). Abrogation of keratinocyte de novo GC synthesis also facilitated sensitization and inflammation in a contact hypersensitivity (CHS) model, associated with an enhanced interleukin-17A (IL-17A) response and increased myeloid cell infiltration. However, atopic dermatitis (AD)–like skin pathology developed nevertheless in both KO mice and littermate controls, although IL-4–mediated pruritus was reduced in the absence of keratinocyte-derived GC. In contrast, psoriasiform inflammation in skin of KO mice was exacerbated and associated with increased cytotoxicity and tissue damage. High-dimensional flow cytometry coupled with algorithm-guided computational analysis revealed decreased regulatory T (Treg) cells and the involvement of interferon-γ (IFN-γ)– and IL-17A–expressing innate-like, unconventional, and invariant T cells in sustained psoriasiform skin inflammation in KO mice. Most notably, long-term Cyp11b1 deficiency in the skin alone resulted in the development of a spontaneous skin inflammation involving type 1 and 17 cytokines, accompanied by mononuclear phagocyte infiltration. Our study thus emphasizes the important immunoregulatory role of skin-derived GC in the regulation of the cutaneous immune system and provides mechanistic insight on the critical role of skin GC in maintaining homeostasis under physiological conditions and in different prevalent inflammatory skin diseases.
RESULTS
Corticosteroidogenic key enzymes are expressed in human and mouse skin
The skin is considered as an extra-adrenal source of GC since its expression of enzymes involved in de novo synthesis, such as CYP11B1, allows autonomous and local production of GC. In addition, the de novo synthesis in the skin is subjected to a local regulatory network, which resembles the regulatory elements of the central HPA axis. It is proposed that skin GC synthesis contributes to skin homeostasis and that deficient steroidogenic enzyme expression and GC synthesis may be involved in the pathogenesis of inflammatory skin diseases. We here show that epidermal expression of not only the critical enzyme CYP11B1 but also CYP11A1, responsible for the generation of the steroid precursor pregnenolone, is abolished in human psoriatic skin lesions compared to healthy donor skin (Fig. 1, A and B). Similar results, obtained from laser capture microdissected epidermis of healthy donor and patient skin sections, demonstrate decreased CYP11B1 and HSD11B1 expression in both AD- and psoriasis-involved lesioned skin, while CYP11A1 transcripts were still detected in AD skin (Fig. 1C). These results confirm previous studies indicating that epidermal steroidogenesis is dysregulated in prevalent inflammatory skin disorders, such as AD and psoriasis (17, 18).
Fig. 1. Corticosteroidogenic key enzymes are expressed in human and mouse skin.
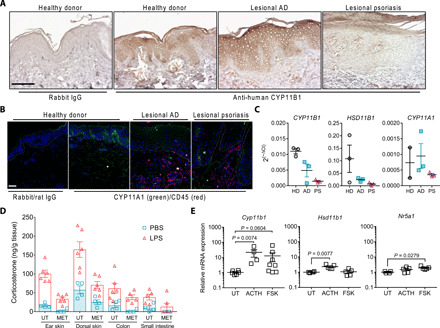
(A) Immunohistochemistry with anti-human CYP11B1 or rabbit immunoglobulin G (IgG) of human skin from healthy donors or patients with lesional AD or psoriasis. Representative images from healthy donors (n = 5), patients with lesional AD (n = 8), or lesional psoriasis (n = 10). Scale bar, 100 μm. (B) Immunofluorescence for CYP11A1 (green) and CD45 (red) or rabbit and rat isotype IgG on frozen human skin sections from healthy donors or patients with lesional AD or psoriasis. Cell nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Representative images from three individual donors and patients are shown. White dashed line indicates dermal-epidermal junction. Scale bar, 50 μm. (C) Reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis of CYP11B1, HSD11B1, and CYP11A1 in laser capture microdissected epidermis from frozen human skin sections of healthy donors (HD), patients with lesional AD (AD) or psoriasis (PS). Expression was normalized to GAPDH, and data are depicted as 2(−∆Ct). Columns represent means ± SEM (n = 2 to 3 individuals per group) of one experiment. (D) Corticosterone radioimmunoassay from untreated (UT) or metyrapone (MET)–treated ex vivo mouse tissue culture in response to phosphate-buffered saline (PBS) or LPS. Symbols represent individual animals. Columns show means ± SEM (n = 4 to 6 per group), pooled from two independent experiments. (E) RNA expression in immortalized C57BL/6 keratinocytes that were untreated or treated with 1 μM ACTH or 20 μM forskolin (FSK) for overnight. Expression was normalized to Actb, and data are depicted as fold change to untreated samples. Data represent means ± SEM (n = 4 to 8 per group), pooled from two to three independent experiments. Statistical significance for (E) was determined using the Kruskal-Wallis test with Dunn’s multiple comparisons test and the ordinary one-way analysis of variance (ANOVA) with Dunn’s multiple comparisons test for Hsd11b1 expression analysis.
We next continued to investigate de novo GC synthesis in mouse skin to experimentally assess its in vivo relevance. To elucidate the capacity of mouse skin to produce GC and to assess its responsiveness toward inflammatory stimuli, we analyzed ex vivo corticosterone production in skin biopsies from untreated and lipopolysaccharide (LPS)–treated mice. Our results show that local ex vivo GC synthesis in mouse skin biopsy and other extra-adrenal organs was reduced using the GC synthesis inhibitor metyrapone, while in vivo LPS challenge further enhanced local GC synthesis indicating the skin’s sensitive steroidogenic responsiveness toward bacterial endotoxin–triggered inflammatory stimuli (Fig. 1D). Furthermore, expression of steroidogenic enzymes was validated in mouse skin (fig. S1A) and primary immortalized mouse keratinocytes, demonstrating that the expression of Cyp11b1, Hsd11b1, and the transcriptional regulator Sf-1 (Nr5a1) was inducible upon treatment with adenosine 3′,5′-monophosphate/protein kinase A signaling activators using adrenocorticotropic hormone (ACTH) and forskolin (Fig. 1E). Together, consistent with previous reports, we demonstrate keratinocyte expression of key steroidogenic enzymes and the steroidogenic responsiveness of the skin under homeostatic and inflammatory conditions, raising the question of the physiological impact, resp. benefit of local corticosteroidogenesis in human and mouse skin.
Genetic ablation of keratinocyte-specific Cyp11b1 abrogates de novo GC synthesis in the skin
To specifically address the role of keratinocyte-derived de novo GC synthesis in the regulation of tissue homeostasis and under inflammatory conditions of the skin, we generated a genetic model system that allows us to specifically target Cyp11b1 expression in keratinocytes to compromise skin GC synthesis. To do so, two LoxP (L2) sites, flanking exons 3, 4, and 5 of the Cyp11b1 locus (Cyp11b1L2/L2), were inserted into the locus, to allow for Cre recombinase–mediated deletion of Cyp11b1 (Fig. 2A). Cyp11b1L2/L2 mice were backcrossed to mice carrying the tamoxifen-inducible Cre recombinase transgene under the Krt14 promoter (K14-CreERTam) to generate K14-CreERTamCyp11b1L2/L2 animals (Fig. 2B and fig. S1, B and C). Cre recombinase–negative littermates of the K14-CreERTamCyp11b1L2/L2 mouse line were used as controls for subsequent experiments (fig. S1D). For all experimental animals, the dorsal skin hair was gently clipped, and all mice received topical application of tamoxifen to the back and ear skin for five consecutive days (Fig. 2C). Specific deletion product resulting from successful tamoxifen-induced Cre-mediated recombination was exclusively detected in the skin of Cre+ mice (referred to as KO) but not in other extra-adrenal tissue or in Cre− littermate control mice (referred to as L2/L2) (Fig. 2D and fig. S1E). Accordingly, Cyp11b1 expression in the skin was significantly decreased in KO mice compared to controls, while Hsd11b1 expression, which mediates the reactivation of inactive, serum-derived GC, was not significantly altered (Fig. 2E). These results indicate that skin of KO mice is deficient for de novo GC synthesis while leaving the capability to provide local GC through reactivation of inactive, serum-derived GC unaffected. Accordingly, serum GC levels were also not altered after the consecutive tamoxifen application (Fig. 2F). In line with keratinocyte-specific Cyp11b1 deletion in the skin, local ex vivo corticosterone production in skin explant cultures from KO mice was significantly reduced compared to controls, while additional ex vivo treatment with the steroid precursor pregnenolone further increased skin de novo corticosterone production in control skin explant cultures in contrast to KO skin explant cultures (Fig. 2G). Furthermore, the bioactivity of ex vivo secreted corticosterone in the supernatant from dorsal and ear skin explant cultures of control and KO mice was analyzed. Therefore, human embryonic kidney (HEK) 293T cells, transfected with the GC response element (GRE) luciferase reporter, were treated with the supernatants to assess their potential in activating the GC receptor (GR) signaling. Bioactive GC secretion was significantly decreased in KO skin explant cultures irrespective of biopsy source compared to controls. Metyrapone-mediated inhibition of GC synthesis further confirmed the ex vivo production of specific, local, and adrenal-independent corticosterone in skin explants (Fig. 2H). Together, this model system represents a unique and valid tool to study the role of skin-derived GC synthesis in the regulation of skin immune homeostasis under physiological and pathological conditions.
Fig. 2. Genetic ablation of keratinocyte-specific Cyp11b1 abrogates de novo GC synthesis in the skin.
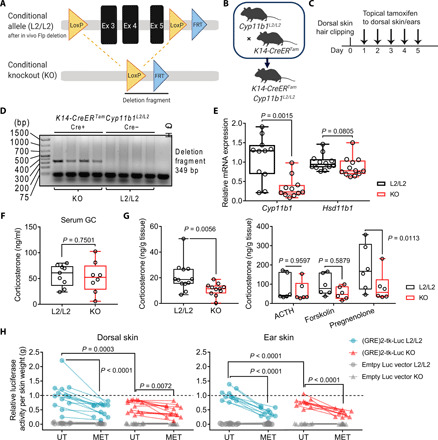
(A) Scheme of Cre/LoxP strategy for Cyp11b1 exons 3 to 5 excision. (B) Generation of mice with inducible Cyp11b1 deletion in keratinocytes (K14-CreERTamCyp11b1L2/L2) by breeding Cyp11b1L2/L2 mice with K14-CreERTam mice. (C) Experimental protocol for in vivo Cyp11b1 deletion in the skin. (D) Agarose gel electrophoresis of the PCR analysis of genomic Cyp11b1 excision from dorsal skin of control (L2/L2) and KO animals. Deletion fragment (349 base pairs) indicates successful Cyp11b1 in vivo deletion. bp, base pairs. (E) Cyp11b1 and Hsd11b1 expression in ear skin. Expression was normalized to Actb. Data are presented as 2(−∆Ct) and shown as fold change over L2/L2 controls. Dots represent individual animals (n = 10 to 13 per group), pooled from three independent experiments. (F and G) Corticosterone radioimmunoassay of blood serum (F) and supernatant of untreated (G, left) or adrenocorticotropin (ACTH), forskolin, or pregnenolone-treated dorsal skin ex vivo cultures (G, right). Dots represent individual animals (n = 6 to 12 per group), pooled from three (F), four (G, left), or two (G, right) independent experiments. (H) GRE luciferase (Luc) reporter assay with HEK 293T cells using supernatant of untreated or metyrapone-treated ex vivo skin culture from L2/L2 or KO animals. Empty luciferase vector–transfected cells served as controls. Normalized relative luciferase activity was depicted as fold change over the mean of untreated L2/L2 controls (dashed line). Paired dots represent skin biopsies from one individual animal (n = 10 to 13 per group). Data are pooled from two to three independent experiments. Box plots (E to G) show the 25th to 75th percentiles with whiskers indicating minimum to maximum values. Statistical significance was determined using unpaired two-tailed t test (F), two-tailed Mann-Whitney test (E and G, left) and ordinary two-way ANOVA with Sidak’s multiple comparisons test (G, right and H). (A) and (B) were created with biorender.com.
Skin GC deficiency facilitates skin APC emigration toward dLNs
We hypothesized that local GC synthesis in the skin may fine-tune the epithelial microenvironment by regulating skin-resident immune cells and shaping barrier immunity and immunopathology. A large proportion of skin-resident immune cells is represented by APCs, which consist of several heterogeneous dendritic cell (DC) and macrophage populations populating the epidermal and dermal areas of the skin (19, 20). Trafficking to skin dLNs to prime peripheral immune cells represents one of their major functions. Since keratinocyte-derived GC may have paracrine modes of action within the skin, we hypothesized that skin APCs are likely to be targeted by locally secreted GC. We thus investigated whether keratinocyte-secreted GC may affect the residency and trafficking of skin APC toward dLN. Tamoxifen-treated K14-CreERTamCyp11b1L2/L2 mice and littermate controls were used to analyze APC populations in skin and dLN. Our results indicate that epidermal major histocompatibility complex II (MHCII)+ APC (fig. S2A) were less abundant in epidermal sheets of KO skin as observed by immunofluorescence. Flow cytometry analysis of specifically CD11c+ MHCII+ APC did not show reduced CD11b+ skin APC in KO ear skin compared to controls, whereas a significant decrease was observed within the CD11b− subset, which include conventional DC type 1 (cDC1) cells (Fig. 3, A and B, and fig. S2, A and B) (20, 21). In line with altered cDC1 frequencies, increased keratinocyte proliferation in tamoxifen-treated K14-CreERTamCyp11b1L2/L2 mice was observed by means of Ki-67 staining indicating epithelial activation (fig. S2C). Furthermore, substantial increase of migratory CD11c+ MHCIIhi APC, but not resident CD11c+ MHCIIint APC, in skin dLN suggest increased skin APC trafficking toward dLN in KO mice compared to controls (Fig. 3C and fig. S2D). In line with the reduced CD11b− APC subset in the skin, our results suggest that the increase of migratory skin APC in the dLN of KO mice is likely due to the increased abundance of the CD11b− CD11c+ MHCIIhi APC subset (Fig. 3D). To specifically confirm the enhanced migration of skin-derived APC toward dLN in KO mice, we performed a previously described in vivo migration assay by painting the skin of mice with fluorescein isothiocyanate (FITC) and the sensitizer dibutyl phthalate (DBP) (22, 23). The dLN of untreated or FITC-treated control and KO mice were then analyzed for skin-emigrated FITC+ CD11c+ MHCIIhi APC. Our results show that skin-derived FITC+ APC in dLN of KO mice were elevated compared to control mice, confirming an increased trafficking of skin-resident APC from skin with deficient de novo GC synthesis (Fig. 3E and fig. S2E). In this regard, the CD11b− subset of skin-derived FITC+ CD11c+ MHCIIhi APC was increased in dLN, as evidenced by higher frequency and total cell numbers, confirming that abrogation of epidermal GC synthesis facilitates emigration of skin-resident APC (Fig. 3F). In line with increased dLN migration of skin-resident APC cells, type 1 and 17 proinflammatory cytokine expression, but also lineage regulators, was elevated in dLN of KO mice (fig. S2E). We also observed increased T cell numbers with central memory phenotype (CD44+ CD62L+) in dLN of KO mice, indicating that peripheral T cells were primed upon in vivo Cyp11b1 depletion in the skin (fig. S2F). Collectively, these findings indicate that cutaneous abrogation of keratinocyte-mediated de novo GC synthesis increased skin immune sensitivity and responsiveness and consequently facilitated spontaneous dLN priming by skin-resident APC.
Fig. 3. Skin GC deficiency facilitates skin APC emigration toward dLNs.

(A and B) Flow cytometry plot (left) and quantification (right) of CD11c+ MHCII+ skin APC (A) and CD11b+/CD11b− subsets of CD11c+MHCII+ cells (B) as frequency of live, single ear cells. Columns (A and B) show means ± SD with n = 7 mice per group. PE, phycoerythrin; FITC, fluorescein isothiocyanate. (C and D) Flow cytometry plot (left) and quantification (right) of migratory CD11c+ MHCIIhi skin APC and resident CD11c+ MHCIIint APC (C) and CD11b+/CD11b− subsets of CD11c+ MHCIIhi cells (D) in skin dLNs, depicted as frequency of live, single dLN cells (left) and total cell numbers (right). Columns (C) and stacked columns (D) show means ± SD with n = 11 to 12 mice per group. (E and F) Flow cytometry plot (left) and quantification (right) of migratory CD11c+ MHCIIhi FITC+ skin-derived APC in skin dLN (E) and CD11b+/CD11b− subsets out of CD11c+ MHCIIhi FITC+ APC (F), depicted as frequency of single, live cells and total cell numbers. Columns (E) and stacked columns (F) show means ± SD with n = 6 to 10 mice per group. Flow cytometry plots are representative and quantification graphs are pooled from two (A, B, E, and F) or three (C and D) independent experiments. Symbols represent individual animals. Statistical significance was determined using unpaired two-tailed t test (A to F) and Mann-Whitney test [(C) for CD11c+ MHCIIint and (D) for CD11b+ subset].
Abrogation of skin de novo GC synthesis aggravates CHS
We next investigated whether abrogation of de novo–synthesized GC in the skin, and consequent skin sensitization and immune priming may also affect inflammatory processes in acute skin disease models. The hapten FITC was used as a contact allergen to induce a classical CHS, an established experimental model for allergic contact dermatitis (23, 24). Skin GC–deficient K14-CreERTamCyp11b1L2/L2 mice (KO) and littermate controls (L2/L2) were sensitized with FITC on their back skin or left untreated as nonsensitized controls. Five days later, all animals received topical application of FITC to their ears (Fig. 4A). Only FITC-sensitized mice responded with a FITC-specific CHS reaction. However, KO mice exhibited an aggravated CHS response with increased ear swelling, edema, and cellular infiltration compared to L2/L2 littermate controls (Fig. 4, B to D). The aggravated skin inflammation in KO mice was associated with an increased infiltration of monocytic and granulocytic phagocytes and an inflammation-induced increase in Il17a expression (fig. S3, A and B, and Fig. 4, E to G). Notably, steroidogenic enzyme transcript levels were not induced upon CHS challenge, suggesting that CHS-induced skin inflammation does not substantially alter their transcriptional expression (fig. S3C). Although no skin pathology was observed for nonsensitized KO mice, Il17a transcript levels in the skin were elevated, indicating a potential for enhanced susceptibility to skin inflammation in the absence of de novo–synthesized skin GC (Fig. 4F). Together, we demonstrate that Cyp11b1 deficiency in keratinocytes and subsequent abrogation of de novo–synthesized GC in the skin not only increased the susceptibility to skin sensitization and dLN priming against contact allergens (Fig. 2) but also resulted in the aggravation of skin inflammation during the CHS effector response, associated with enhanced Il17a expression and increased recruitment of inflammatory myeloid monocytes and granulocytes. Our results therefore suggest that keratinocyte-derived GC pose an immunomodulatory potential in regulating skin sensitization but also restrict inflammatory processes during an acute CHS response.
Fig. 4. Abrogation of skin de novo GC synthesis aggravates CHS.
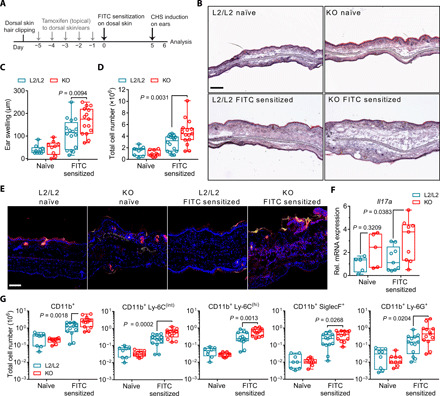
(A) Experimental protocol for FITC skin sensitization and CHS induction following in vivo Cyp11b1 deletion in the skin. (B) Hematoxylin and eosin staining of frozen ear sections from naïve or FITC-sensitized controls (L2/L2) or KO mice 24 hours after CHS induction. Representative images of three independent experiments. Scale bar, 200 μm. (C and D) Ear swelling and ear cell numbers of naïve and sensitized mice. Dots represent individual animals (n = 9 to 16 per group), pooled from three to four independent experiments. (E) Anti–Ly-6G (red) and DAPI (blue) immunofluorescence of frozen ear sections from naïve or sensitized mice. Yellow-stained areas represent FITC treatment–induced fluorescence. Representative images of three independent experiments. Scale bar, 100 μm. (F) RT-qPCR analysis of Il17a expression in ear skin. Expression was normalized to Actb and shown as fold change over naïve L2/L2 mice. Dots represent individual animals (n = 5 to 9 per group), pooled from three independent experiments. (G) Flow cytometry analysis of myeloid granulocytes and monocyte subsets depicted as total cell numbers per ear. Dots represent individual animals (n = 9 to 16 per group), pooled from three independent experiments. Box plots (C, D, F, and G) show the 25th to 75th percentiles with whiskers indicating minimum to maximum values. Statistical differences were determined using ordinary two-way ANOVA with Sidak’s multiple comparisons test.
Skin-derived GC promote pruritus but do not alter AD-like skin inflammation
Our results demonstrate that deficient de novo GC synthesis in keratinocytes predisposes to skin inflammation in regard of CHS reactions. We further continued to investigate the importance of skin GC in prevalent inflammatory skin disorders, such as AD and psoriasis. Given alterations in CYP11B1 expression in the context of AD lesional skin (Fig. 1C), we next tested the immunoregulatory impact of skin-derived GC in the pathogenesis of experimental AD. To do so, tamoxifen-treated K14-CreERTamCyp11b1L2/L2 mice and Cre− littermate controls were skin-sensitized with ovalbumin (OVA) on a developing AD-like skin lesion induced by the vitamin D analog MC903 or on a vehicle-treated skin as previously reported (Fig. 5A) (25–27). Using this treatment regimen, both KO and control mice exhibited similar clinical and histological features of AD-like skin inflammation, characterized by skin edema and myeloid cell infiltration to sites of topical MC903 treatment (Fig. 5, B to G, and fig. S4, A and B). Furthermore, local GC synthesis in skin of MC903-treated littermate controls was substantially increased compared to vehicle-treated controls, whereas skin GC production in KO mice was still reduced indicating the capability of AD-like skin inflammation to potently trigger local de novo GC synthesis (fig. S4C). Serum GC levels in both vehicle- and MC903-treated KO mice were not significantly different (fig. S4C). Likewise, the increase of activated skin dLN T cells and the OVA-specific IL-2 response in ex vivo restimulated skin dLN cultures from MC903-treated KO and control mice were similar (fig. S4, D and E, and Fig. 5H). However, we found KO mice to exhibit dampened OVA-specific IL-4 responses and significantly reduced IL-4 protein in AD-like skin compared to L2/L2 littermate controls (Fig. 5, H and I). In line with IL-4 as an activator of itch-sensory pathways (28), reduced IL-4 production in the absence of skin de novo GC synthesis was associated with reduced pruritus as measured by episodes of scratching, suggesting that keratinocyte-derived GC signaling is involved in the IL-4–mediated itch-sensory pathway (Fig. 5E). In conclusion, our findings provide evidence that local de novo–synthesized GC in the skin do not restrict the pathogenesis of MC903-induced experimental AD and associated AD-like skin inflammation but do promote the IL-4–mediated itch-sensory pathway.
Fig. 5. AD-like skin inflammation is not restricted by de novo–produced skin GC.
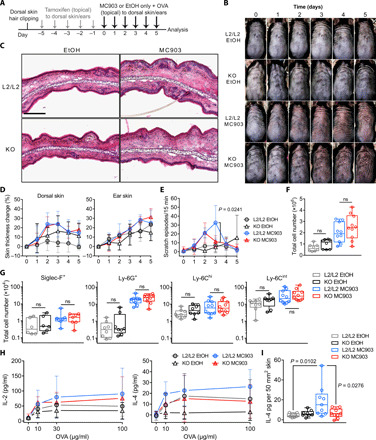
(A) Experimental protocol for OVA skin sensitization on vehicle-treated (EtOH) skin or MC903-induced AD-like skin. (B) Dorsal skin images of four individual control (L2/L2) and KO mice during treatment period. Representative images of three independent experiments. (C) Hematoxylin and eosin staining of frozen ear skin sections. Representative images of three independent experiments; scale bar, 100 μm. (D) Skin thickness change during treatment period as percentage of the base line skin thickness (day 0, untreated). Data show pooled means ± SD of three independent experiments (n = 8 to 10 mice per group). (E) Scratch episodes during treatment period. Data show pooled means ± SD of three independent experiments (n = 6 to 8 mice per group). (F) Total cell numbers per ear after treatment period. Data are pooled from three independent experiments (n = 8 to 10 mice per group). (G) Flow cytometry quantification of monocytes and granulocytes from ear skin single cells. Data are pooled from two to three independent experiments (n = 6 to 10 per group). (H) OVA-specific IL-2 and IL-4 protein levels in cell-free supernatants from dLN restimulated cultures. Data show pooled means ± SD of three independent experiments (n = 6 to 10 mice per group). (I) IL-4 protein levels from individual dorsal skin biopsies. Data are pooled from three independent experiments (n = 8 to 9 per group). Box plots (F, G, and I) show the 25th to 75th percentiles with whiskers indicating minimum to maximum values with dots representing individual animals. Statistical differences were determined using repeated-measures (RM) two-way ANOVA with Tukey’s multiple comparisons test (D and E) and ordinary two-way ANOVA with Sidak’s multiple comparisons test (F to H) and with Tukey’s multiple comparisons test (I). ns, not significant. Photo credit: Truong San Phan, University of Konstanz.
Loss of keratinocyte de novo GC synthesis exacerbates psoriasiform inflammation
Our results have shown that de novo GC synthesis in keratinocytes is able to regulate skin sensitization and consequent inflammation. Given that ablation of skin GC synthesis resulted in a spontaneous skin dLN priming and enhanced Il17a induction in the CHS response, we next investigated the impact of keratinocyte-derived GC on the pathogenesis of psoriasis, a chronic, immune-mediated inflammatory skin disease driven by IL-17 effector cytokine responses. We therefore used an experimental model of psoriasis that is induced by topic application of Aldara (ALD) creme, which contains the potent Toll-like receptor 7/8 ligand imiquimod to activate cutaneous APC in vivo (29). ALD-induced psoriasiform skin inflammation closely resembles the human plaque psoriasis that is mediated through the IL-23/IL-17 axis (30). We topically applied ALD over 8 days to the back or ear skin of control and KO mice after tamoxifen-induced Cyp11b1 deletion (Fig. 6A). Our results revealed that both pathogenesis and progression of psoriasiform dermatitis was significantly exacerbated in KO mice, with increased abscess and scale formation compared to ALD-treated L2/L2 littermate controls (Fig. 6B). In addition, clinical scoring at days 6 to 7 indicated the onset of remission with decreasing signs of inflammation in ALD-treated L2/L2 control mice, whereas aggravation of skin inflammation in KO mice was sustained without signs of amelioration (Fig. 6, B to E). Histological analysis further revealed abnormal abscess formation in KO mice, which was, however, not associated with epidermal hyperproliferation as an psoriatic hallmark but was rather followed by tissue-damaging processes leading to increased leukocyte infiltration and to the loss of an intact epidermis (Fig. 6F). Compared to control mice, the psoriasiform skin inflammation in ear and dorsal skin of KO mice featured increased tissue damage and cell death (Fig. 6, G and H, and fig. S5, A and B). Decreased frequencies of myeloid phagocytes, which are known to initially infiltrate psoriasiform skin lesions, in line with increased cell death, further demonstrate the advanced progression of psoriasiform skin inflammation in KO mice compared to littermate controls, while untreated KO mice exhibited a developing spontaneous skin inflammation as evidenced by clinical scoring and increased myeloid cell infiltration (Fig. 6, C and D and G to I, and fig. S5A). Besides, KO skin biopsies from ALD-treated mice were still deficient in the local de novo production of GC compared to skin biopsies of ALD-treated littermate controls, confirming the long-lasting targeting in the K14-CreERTamCyp11b1L2/L2 mice (fig. S5C). Our results demonstrate that in vivo abrogation of de novo skin GC synthesis exacerbated pathogenesis and progression of ALD-induced psoriasiform skin inflammation and thus highlight the endogenous potential of keratinocyte-derived GC in the regulation of type 17–mediated skin inflammation.
Fig. 6. Loss of keratinocyte de novo GC synthesis exacerbates ALD-induced psoriasiform inflammation.
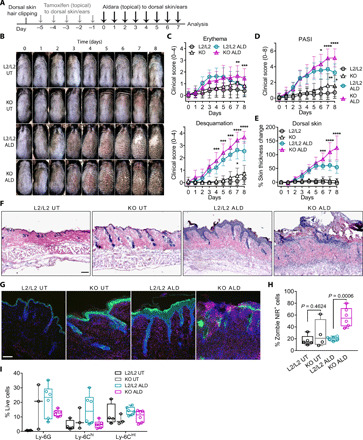
(A) Experimental protocol for ALD-induced psoriasiform skin inflammation. (B) Dorsal skin images of individual untreated and ALD-treated control (L2/L2) and KO mice. Representative images (n = 6 to 8 mice per group) of two independent experiments. (C) Clinical erythema (top) and desquamation score (bottom) and (D) PASI score (sum of erythema and desquamation score) of untreated and ALD-treated dorsal skin during the treatment period. Data represent means ± SD (n = 6 to 8 mice per group), pooled from two independent experiments. (E) Skin thickness change of untreated and ALD-treated mice as percentage of the respective base line skin thickness (day 0). Data show means ± SD (n = 8 to 11 mice per group), pooled from three independent experiments. (F) Hematoxylin and eosin staining of frozen dorsal skin sections from untreated and ALD-treated mice. Representative images of two independent experiments. Scale bar, 300 μm. (G) Anti–Ly-6G (red) and anti–keratin 14 (green) immunofluorescence of frozen dorsal skin sections. Representative images of two independent experiments. Scale bar, 100 μm. (H and I) Flow cytometry frequencies of single dead cells (H) and single live monocyte subsets and granulocytes (I) from ears of indicated mice. Box plots show the 25th to 75th percentiles with whiskers indicating minimum to maximum values. Dots represent individual animals, pooled from two independent experiments with n = 4 to 6 per group (H) and n = 3 to 6 per group (I). Statistical differences were determined using RM two-way ANOVA with Tukey’s multiple comparisons test (C to E) and ordinary two-way ANOVA with Sidak’s multiple comparisons test (H). Photo credit: Truong San Phan, University of Konstanz.
Sustained psoriasiform inflammation in KO skin is associated with reduced Treg cells and involves invariant and unconventional T cells
IL-17 effector cytokines are key drivers in the pathogenesis of psoriasis and are primarily produced by several innate-like and γδ T cells in the model of ALD-induced experimental psoriasis (31, 32). Since GC activate various immunoregulatory mechanisms and exert pleiotropic effects in different immune cell types, we aimed to uncover those T cell populations, which were associated with the aggravated psoriasiform skin inflammation in mice with deficient de novo GC synthesis in the skin. Therefore, we used computational flow cytometry analysis using t-distributed stochastic neighbor embedding (tSNE) algorithm–based visualization and FlowSOM algorithm–based meta clustering to identify T cell populations in ears of untreated and ALD-treated control and KO mice (Fig. 7A). Our comprehensive analysis showed that CD4−CD8− αβ T cells (CD4−CD8− T cells), CD8+ γδ T cell receptor–positive (TCR+) cells (CD8 γδ T cells), and several NK1.1-expressing T cell (NKT) subsets were dominantly recruited in ALD-treated KO ears, whereas CD4+ T helper (TH) and CD4+ Foxp3+ Treg cells were most notably decreased (Fig. 7B). These results indicate that the exacerbated psoriasiform inflammation in KO skin was maintained because of the unique recruitment of innate-like, unconventional, and invariant T cells and the reduction of Treg cells. Analysis of the inflammatory profile of each population additionally revealed the presence of IL-17A+ and IFN-γ+ T cells (Fig. 7C and fig. S7, A and B). Accordingly, increased dermal γδ TCRint T cells (γδ T cells), mostly representing Vγ4+ γδ T cells, are described to represent the major source of IL-17A in psoriasiform skin inflammation, which was consistently observed in ALD-treated control mice (Fig. 7C) and reported previously (31, 32). However, ALD-treated KO mice showed increased IFN-γ+ γδ T cells and CD8+ γδ T cells with IL-17A expression in ear skin immune cell infiltrates (Fig. 7C and fig. S7, A to E). Our results therefore suggest that psoriasis-like inflammation in skin lacking keratinocyte-derived de novo–synthesized GC results in reduced Treg cell numbers and specifically promotes IFN-γ– and IL-17A–expressing innate-like, unconventional γδ T cells and invariant NK1.1+ T cells to exacerbate psoriasiform skin inflammation, which is associated with increased cytotoxicity and epithelial damage.
Fig. 7. Psoriasiform inflammation in KO skin is associated with reduced Treg cells and increased IL-17A+CD8+ γδ T cells.
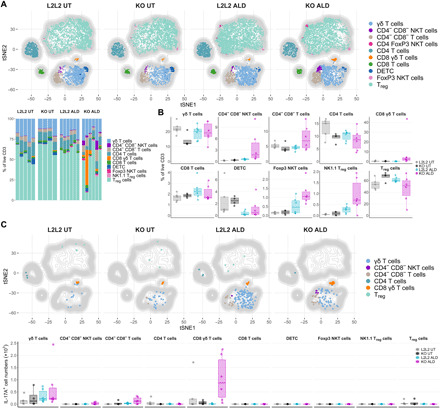
(A and B) Computational flow cytometry analysis of single CD45+CD11b−CD3+ T cells from untreated and ALD-treated ears of control (L2/L2) and KO mice are visualized using tSNE algorithm with overlaid distribution of cell populations defined by FlowSOM algorithm–based clustering (top, A). tSNE map depicts aggregated samples (each downsampled to 1000 cells) with n = 4 to 6 mice per group from two independent experiments. Frequencies of FlowSOM-defined clusters as stacked bar graph (bottom) and as box plots (B) showing the 25th to 75th percentiles with whiskers indicating the range to the smallest and largest data point until the 1.5 × interquartile range (IQR). (C) tSNE maps as in (A) visualizing T cell clusters with overlaid distribution of IL-17A+ cells and total IL-17A+ cells per ear presented as box plots (bottom) showing the 25th to 75th percentiles with whiskers indicating the range to the smallest and largest data point until the 1.5 × IQR. Stacked bar graph (A) and dots in box plots (B and C) represent individual animals (n = 4 to 6 per group), pooled from two independent experiments. DETC, dendritic epidermal T cell; NKT, NK1.1+ T cells.
Long-term deficiency of skin de novo GC synthesis results in spontaneous type 1 and 17 skin inflammation
Our ALD experiments demonstrated that even untreated KO mice exhibited clinical signs of skin inflammation starting around days 7 to 8 after tamoxifen application (Fig. 6, B to D). Skin sensitization and dLN priming, observed directly after genetic in vivo ablation of Cyp11b1 in keratinocytes, appear to contribute to the development of a spontaneous skin inflammation in KO mice (Fig. 3, A to D, and fig. S2, C, E, and F). Further analysis revealed that 10 to 14 days after tamoxifen application, affected KO mice exhibited ear skin thickening, epidermal hyperproliferation, and myeloid cell infiltration involving Ly-6C+ monocyte subsets and Ly-6G+ neutrophils (Fig. 8, A and B and fig. S8A). In addition, RNA expression analysis from ears of KO mice demonstrated elevated levels of several type 1 and 17 signature cytokines and the Cxcl9 chemokine, consistent with the myeloid phagocyte infiltration in contrast to ear skin of littermate controls (Fig. 8C). These results indicate that long-term deficiency of GC de novo synthesis in keratinocytes alone is capable to promote the activation of type 1 and 17 immune response, which subsequently can develop into a spontaneous skin inflammation consequently involving the recruitment of inflammatory myeloid cells. Since the given immunophenotype of the spontaneous inflammation in KO skin resembles, to a certain degree, the psoriasiform skin inflammation, we suggest that endogenous and locally derived keratinocyte GC specifically regulate skin immune pathologies and potently counteract IL-17–driven skin inflammation to maintain tissue homeostasis.
Fig. 8. Long-term deficiency of skin de novo GC synthesis results in spontaneous type 1 and 17 skin inflammation.
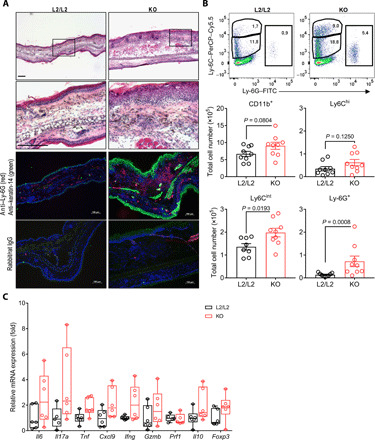
(A) Hematoxylin and eosin staining (top) and anti–Ly-6G (red) with anti–keratin-14 (green) (middle) or rat/rabbit IgG isotype immunofluorescence (bottom) on frozen ear skin sections of control (L2/L2) and KO mice. Representative images of three independent experiments. Scale bar, 100 μm. (B) Flow cytometry plots (top) and quantification (bottom) of myeloid granulocytes and monocyte subsets, isolated from ears 14 days after tamoxifen treatment, depicted as total cell numbers per ear. Flow cytometry plots are representative, and columns show means ± SEM with dots representing individual animals (n = 8 to 9 per group), pooled from three independent experiments. (C) RT-qPCR analysis of indicated target gene expressions in ears of L2/L2 and KO mice 10 days after tamoxifen treatment. Target genes were normalized to Actb and shown as fold change over L2/L2 controls. Box plots show the 25th to 75th percentiles with whiskers indicating minimum to maximum values. Dots represent individual animals (n = 5 to 6 per group), pooled from two independent experiments. Statistical differences were determined using two-tailed unpaired t test for CD11b+, Ly-6Cint, and Ly-6Chi cells (B) and two-tailed Mann-Whitney test for Ly-6G+ cells (B). PerCP, peridinin-chlorophyll-protein.
DISCUSSION
The concept of local GC synthesis in the regulation of the skin immune microenvironment has gained increasing scientific and clinical attention since the first demonstration of extra-adrenal GC synthesis at this epithelial barrier (4, 5, 17). Keratinocytes were shown to produce GC in the skin, tightly regulated by a local HPA-like axis via steering hormones and neuropeptides. Rodent and human skin were shown to be capable in producing GC, while patients with inflammatory skin diseases were suspected to have a defective steroidogenesis. Yet, a clear demonstration that local GC synthesis in the skin regulates local immune responses has been missing.
We here further provide evidence for CYP11B1 expression in healthy human skin and its down-regulation in patients with inflammatory skin disorders (Fig. 1, A and B). Whether this down-regulation is involved in the pathogenesis of skin disorders, or represents a secondary effect, is still unclear and requires further investigation using extensive genome analyses, such as analyzing genetic polymorphisms (single-nucleotide polymorphism) at relevant steroidogenic genes, including the comparison of healthy skin and noninvolved skin of patients with inflammatory skin diseases. Besides de novo synthesis, also reactivation of inactive GC by 11β-hydroxysteroid dehydrogenase type 1 (HSD11B1) occurs in various tissues. A study using Hsd11b1-deficient keratinocytes in the skin suggested an immunosuppressive role for locally reactivated GC (33). However, the observed effects were only minor and suggest that peripheral reactivation of adrenal-derived inactive GC does not substantially contribute to the local regulation of extensive skin inflammations.
The skin translates and communicates environmental signals through a variety of endocrine systems by the sustained synthesis of different steroids. Since autonomous de novo GC synthesis may play a pivotal role in skin homeostasis and in the regulation of the local skin immune system, we introduced an inducible model for Cyp11b1 deficiency in the skin, demonstrating that keratinocyte-specific abrogation of de novo GC synthesis results in decreased GC secretion. This enabled us to investigate how endogenous GC may regulate the epithelial immune microenvironment in vivo under physiological and inflammatory conditions.
Deficient skin GC synthesis affected the residency/emigration behavior of skin APC and resulted in skin dLN priming. Although Langerhans cells (LCs) seem to be reduced in the epidermis of KO skin (fig. S2A), general CD11b+ skin APC abundancy was not altered (Fig. 3B). Since we did not proceed with the detailed analysis of the heterogeneous CD11b+ skin APC populations, it is very likely that this difference is perished within the residual skin APCs as LCs only represent a small population among skin-resident CD11b+ DCs and macrophages. In contrast to the CD11b+ skin APC, APC trafficking was most pronounced for CD11b− skin APC, which mainly consists of XC-chemokine receptor 1–expressing cDC1 cells. They have been shown to cross-present keratinocyte-derived self-antigens and to contribute to type 1 and 17 immune and CD8+ T cell priming (19–21, 34). Although acute inflammation was initially not observed upon Cyp11b1 deletion, abrogation of cutaneous GC synthesis resulted in skin dLN priming through skin-resident APC. Further studies are required to uncover the specific GC-mediated regulation of the heterogeneous, skin-resident DC, and macrophage populations. In-depth phenotyping and understanding the underlying mechanisms would reveal their individual and subset-specific in vivo GC sensitivity and provide previously unknown ways to control specific arms of the adaptive immunity through keratinocyte-derived GC.
We further investigated the role of skin GC synthesis under pathological conditions using in vivo skin inflammation models. The CHS, as an experimental model of human allergic contact dermatitis, includes a series of immunological reactions. Initiated during the skin sensitization, APCs contribute to T cell priming in skin dLN. Hapten rechallenge subsequently results in CHS with myeloid cell–driven skin inflammation during the elicitation phase. Here, FITC reapplication on sensitized mice continuously induced a robust CHS reaction. The preceding increase in skin sensitization and dLN priming in KO mice resulted in an aggravated skin inflammation associated with increased myeloid cell infiltration. Despite the use of DBP as a vehicle, which contributes to TH2 polarization, type 2 effector responses seemed to be neglectable, since CHS responses were overwhelmed by type 1 and 17 responses, as described in previous studies for most CHS reactions (23, 24). Although type IV hypersensitivity reactions are T cell–mediated, CHS skin inflammation is mostly driven by innate immune cells (23, 24). In particular, inflammatory monocytes, macrophages, and neutrophils were shown to represent responsive targets for GC treatment in allergic contact dermatitis (35). Our results accordingly demonstrated pronounced myeloid phagocyte infiltration in CHS-challenged KO skin. Enhanced skin inflammation was associated with elevated Il17a transcripts. We here revealed keratinocyte-derived GC as a potent regulator of myeloid cell–driven CHS inflammation by restraining IL-17–dependent responses in vivo.
We further investigated the impact of keratinocyte-derived GC in an experimental model of AD. The MC903-induced AD-like skin condition relies on the elicitation of epithelial-derived thymic stromal lymphopoietin and the induction of IL-4 through innate immune cells to induce TH2 polarization (25–27). Although our results indicate that keratinocyte-derived GC do not restrict the developing AD-like skin inflammation, reduced IL-4 levels associated with reduced pruritus in KO mice suggest that skin-derived GC promote itch in this model system. GC signaling was associated with type 2 responses at various sites (36–39). Recent studies also revealed that basophil-derived IL-4 and DC are required for optimal TH2 polarization to food antigens in the skin that predisposed to intestinal food allergy (26, 27). Whether impaired IL-4 production in KO mice represents a consequence of defective TH2 polarization due to insufficient cDC2 cells or because of competitively primed dLN environment remains unclear. Despite the successful treatments of asthma and allergies using synthetic GC, they seem to preferentially suppress type 1 and 17–dependent reactions, while type 2 responses are spared or even enhanced (2, 38). While our data suggest that skin-derived GC promote itch in this model system, topical MC903 treatment on both dorsal and ear skin promote systemic immune responses affecting adrenal steroidogenesis that, given its magnitude, may mask immunoregulatory effects of skin-derived GC. In this regard, our results show that serum GC levels of tamoxifen-treated naïve mice were not altered (Fig. 2F), but at a later time point, serum GC levels in ethanol-treated KO mice were reduced, while in MC903-treated KO mice they were increased (fig. S4C). Although not significantly different, our data may indicate that adrenal GC synthesis in KO mice is likely affected by the MC903 treatment and may contribute to compensate the GC level imbalance within the skin. The role of adrenal steroidogenesis in this regard is still unclear, but it is very likely that the “cross-talk” between the local skin HPA axis and the elements of the systemic HPA axis are of relevance and should be considered in further investigations (40, 41). Aside from that, there is still incomprehension on the role and impact of the MC903-induced local skin GC response in the regulation or promotion of type 2 responses. Further studies are therefore warranted to identify the role of skin-derived GC in the pathogenesis of AD and to reveal the underlying mechanisms.
To investigate the relevance of IL-17–driven skin pathology in KO mice, ALD was used to induce experimental psoriasis in vivo. We demonstrated that deficient skin GC synthesis exacerbated psoriasiform skin inflammation, associated with abnormal abscess formation and tissue damage, while keratinocyte-mediated GC synthesis in control mice contributed to disease regression. The unique involvement of IFN-γ– and IL-17A–expressing unconventional γδ T cells, invariant NK1.1+ T cells, and CD4−CD8− αβ T cells in the sustained skin inflammation of ALD-treated KO mice presents a novel composition of skin T cells and proposes that they likely represent the most sensitive GC targets. Rare and distinct, CD8+ γδ T cells were described to represent a subset of γδ T cells (42, 43). Here, they seem to majorly contribute to the dysregulated inflammatory process in ALD-treated KO skin by the production of IL-17A. Similar to other epithelial-resident CD8+ γδ T cells, they exhibit a pathogenic potential and have cytotoxic properties (44, 45). In contrast, the increase of Treg cells in psoriasiform skin inflammation was described to be pivotal for disease regression and remission (46). While we accordingly observed the disease-dependent increase of Treg cells in ALD-treated controls, decreased Treg cells in ALD-treated KO mice indicate insufficient immune regulation in skin with deficient GC synthesis. Our findings thus propose that the lack of immunosuppressive skin GC sustains psoriasiform skin inflammation through the accumulation of inflammatory T cells and reduced Treg cell numbers and indicates the potency of endogenous skin GC.
In addition, untreated KO mice exhibited a developing skin pathology with granulocytic skin infiltrates and elevated proinflammatory gene expressions over time (Fig. 8, B and C), indicating that long-term deficiency of cutaneous GC synthesis results in a spontaneous type 1 and 17 skin inflammation. We thus conclude that early skin dLN priming upon Cyp11b1 ablation relevantly affects the skin immune system in a pathogenic way. However, additional comprehensive analyses are required to reveal the involved skin-infiltrating immune cell populations in detail.
Still, it remains speculative whether the trigger of this spontaneous skin inflammation is of microbial nature or an endogenous mediator. Overreactions toward skin commensals may involve pathogen-associated molecular patterns, while autoreactive processes resulting in sterile (auto)inflammation may derive from damage-associated molecular patterns (47). In this regard, systemic LPS administration strongly promoted skin GC synthesis (Fig. 1D). GC-mediated actions through the GR and the GC-induced leucin zipper (GILZ) were described to efficiently inhibit these signaling cascades at several sites (48). Deficient skin GC supply may facilitate proinflammatory processes, which are usually suppressed under steady-state conditions. A break in self-tolerance represents another mechanism, since tolerogenic DC and peripheral Treg cell induction depends on the GC-mediated GILZ induction (49, 50). Local GC synthesis may represent an essential mechanism in the repertoire of keratinocytes to induce peripheral tolerance and to regulate local immune cells and inflammation to maintain epithelial homeostasis. Further studies are required to reveal the role of cutaneous DCs and macrophages with regard of tolerance induction and microbiota-dependent responses in a skin microenvironment lacking keratinocyte-derived GC.
In this study, we demonstrated the broad role of keratinocyte-derived GC and their relevance in the regulation of tissue homeostasis and prevalent inflammatory skin conditions. Detailed mechanisms of skin GC–mediated suppression in individual target cells remain yet unclear as GR-exerted actions comprise a variety of target genes and proteins. Full elucidation of these processes and the ability to further modify local de novo GC synthesis in the skin will complement our understanding of endogenous, skin-derived GC and their potential in controlling key players in skin physiology and inflammation with significant impact on the current clinical corticosteroid therapies for inflammatory and autoimmune diseases.
MATERIALS AND METHODS
Study design
The objective of this study aims to unravel the in vivo relevance of endogenous, skin-derived GC in the regulation of the local immune homeostasis in physiological conditions and prevalent inflammatory skin disorders. Differential expression of steroidogenic enzymes, involved in the de novo GC synthesis, was characterized in human skin sections of healthy donors or patients with inflammatory skin disorders using immunohistochemistry and RNA expression analysis of laser capture microdissected epidermal sections. To experimentally assess the in vivo potency and relevance of endogenous, skin-derived GC, we generated an inducible mouse model with conditional deletion of Cyp11b1 in keratinocytes and investigated its impact on skin immune homeostasis and in prevalent inflammatory skin disorders (allergic contact dermatitis, AD, and psoriasis) by applying different skin disease models (FITC-induced CHS, MC903-induced AD, and ALD-induced psoriasiform skin inflammation). Detailed description of the methodology and measurement techniques of the skin disease models are described in separate sections below. Pathological and clinical parameters of the dorsal skin and the ears were assessed, and immunophenotyping was performed using histological analysis, RNA expression analysis, enzyme-linked immunosorbent assay (ELISA), and flow cytometry including computational flow cytometry analysis. Skin biopsy punches from dorsal skin and ears were used to analyze corticosterone synthesis or were used for immune cell infiltrate analysis. Immunophenotype analysis also included skin dLNs for RNA analysis or restimulation experiments. Skin biopsies from thorax or abdomen of healthy donors were obtained from plastic surgeries. Lesional skin biopsies were taken from patients with AD or psoriasis vulgaris. Diagnosis of AD and psoriasis vulgaris was confirmed by D. Simon and S. Abraham, specialized clinician at the Department of Dermatology at the University Hospital Bern (Switzerland) and at the Department of Dermatology at the University Hospital of the Technical University Dresden (Germany). Human sample sizes were chosen on the basis of the availability of voluntaries. The conducted experiments were approved by the Ethics Committee of the University Hospital of the Technical University Dresden (EK147042017) and by the Ethics Committee of the University Hospital Bern (KEK Bern 61/05), and written informed consent was obtained from all patients. All experiments were performed in accordance with the Declaration of Helsinki. C57BL/6 wild-type mice were bred in the animal facility of the University of Konstanz under specific pathogen–free conditions and housed in individually ventilated cages. All animal experiments were conducted according to the animal experimentation regulations of Germany and were approved by the Ethics Review Committee of the Regional Council Freiburg. Mice with the expression of the fusion protein between the Cre recombinase and a tamoxifen-specific estrogen receptor under the control of the human K14 promoter (K14-CreETam) were provided by M. Leverkus (Mannheim, Germany) and described before (51, 52). Mice with LoxP sites flanking exons 3, 4, and 5 of Cyp11b1 were generated at the Institute Clinique de la Souris (Cyp11b1L2/L2) on a C57BL/6 background. Floxed Cyp11b1 mice were bred with K14-CreETam mice for generating inducible KO mice with epidermal-specific Cyp11b1 deletion (referred as K14-CreERTamCyp11b1L2/L2). All in vivo experiments were conducted using mice with an age between 7 and 14 weeks. Back hair was gently clipped, and the dorsal skin was treated daily with 100 μl of tamoxifen and the ears with 20 μl of tamoxifen (Sigma-Aldrich) in ethanol solution (25 mg/ml) for five consecutive days to induce CreERTam activity. Littermates with Cyp11b1 LoxP alleles but lacking Cre recombinase expression were also treated with tamoxifen and included as controls in all experiments. Except for the experimental model of AD and psoriasis, both male and female animals were included in all experiments. Mice with similar age were sex-matched and equally distributed into experimental groups. Sample sizes for animal studies were estimated according to preliminary and published studies or were determined using G*Power (3.1.9.2) with α set to 0.05 and an adequate power level of 0.95. Data were excluded if analysis was not possible and samples fail in quality controls because of unspecific product amplification in reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis, insufficient cell numbers, or technical errors in flow cytometry data collection. Most of the experiments with statistical analysis were performed at least two times, if not stated otherwise. Experiments were independently reproduced and pooled as stated in the figure legends. All replication attempts of the experiments were successful and showed similar findings. During experiments, no randomization was done as all animals were sex- and age-matched and underwent specific in vivo treatments. Randomization was applied on the photo images for clinical psoriasis area and severity index (PASI) scoring. Investigators were blinded to the experimental groups per genotype when skin thickness was assessed for CHS and AD induction experiments, during AD-associated scratch counting and for the clinical PASI scoring.
LPS challenge
C57BL/6 wild-type mice were intraperitoneally injected with either 200 ng of LPS (Sigma-Aldrich) or with phosphate-buffered saline (PBS) and were euthanized 6 hours after treatment. Indicated organs were extracted for ex vivo culture and subsequent corticosterone radioimmunoassay.
FITC painting and in vivo APC migration assay
FITC (Sigma-Aldrich) was dissolved in a solution of acetone and DBP (1:1) to a concentration of 1% (w/v). After tamoxifen treatment for 5 days, mice were topically treated with 30 μl of 1% FITC solution to the dorsal skin, while control mice were left untreated. Mice were euthanized 24 hours after sensitization, and skin dLNs were isolated for flow cytometry or mRNA expression analysis.
FITC-induced CHS
K14-CreERTamCyp11b1L2/L2 mice were treated with tamoxifen as described above, and Cre+ mice and littermate controls were sensitized with 400 μl of 0.5% (w/v) FITC solution (dissolved in acetone and DBP, 1:1) on the dorsal skin, while untreated groups with Cre+ mice and littermate controls served as nonsensitized controls. After 5 days, the ear thickness of both ears from all mice was measured using a dial thickness gauge (7301, Mitutoyo). Thereafter, 20 μl of 0.5% FITC solution was applied to both ears of all mice. All mice were euthanized 24 hours after challenge, and the ear thickness was remeasured to assess the CHS-induced ear swelling [skin thickness (after elicitation) − skin thickness (baseline) × 10 = thickness change in micrometers]. Investigators were blind to the genotype/treatment groups when ear skin thickness was assessed. Ears were further used for flow cytometry analysis, immunohistology, or RNA expression analysis.
MC903-induced experimental model of developing AD-like skin condition
K14-CreERTamCyp11b1L2/L2 male mice were used for the induction of AD-like skin inflammation using the vitamin D analog MC903 (calcipotriol, Tocris Bioscience, Bristol, UK). After gentle hair clipping, all mice were treated with tamoxifen as described above. For additional 5 days, Cre+ mice and littermate controls were either treated with 2 nmol of MC903 in 20 μl of 100% ethanol to the ears and 10 nmol of MC903 in 100 μl of 100% ethanol to the dorsal skin or received ethanol only as vehicle treatment. Ears and back skin of all mice were topically treated with 100 and 200 μg of OVA in 20 and 100 μl of PBS, respectively. Before the topical treatment, skin thickness of ears and back skin was measured using a dial thickness gauge (7301, Mitutoyo). Skin thickness change for each animal was calculated as percentage of basal skin thickness from day 0. Digital photographic dorsal skin images were taken from all mice for all days.
AD-associated itching (scratching behavior)
Clinical manifestation of itching was assessed by analyzing the scratching behavior. All experimentally involved investigators were blind to the genotype/treatment groups during the scoring. All mice were individually observed for a period of 15 min, and scratch episodes were counted. Scratching was counted when a hindlimb was used for scratching toward the ears and the back skin. One scratch episode was considered whenever a hindlimb was lifted for scratching and relocated back to the ground, regardless of how many scratching strokes.
ALD-induced experimental model of psoriasiform skin inflammation
K14-CreERTamCyp11b1L2/L2 male mice were used for the induction of experimental psoriasis skin inflammation. After back hair clipping and tamoxifen treatment as described above, mice topically received 55 mg of ALD creme containing 5% imiquimod (Meda) on the dorsal skin or 7 mg on each ear for eight consecutive days. Mice without ALD treatment served as untreated controls. During the treatment period, dorsal skin thickness was daily assessed for all mice using a dial thickness gauge (7301, Mitutoyo). Skin thickness change was calculated as percent of basal skin thickness from individual mice at day 0. Digital photographic dorsal skin images were taken from all mice for all days. All images were used to score erythema and scaling to assess the severity of psoriasiform skin lesions according to a modified PASI with scores ranging from 0 to 4. Calculations for the affected area were not considered since the treated area was equal for all mice. The overall PASI score was calculated as the sum of the erythema and the scaling score. Dorsal skin images of all mice were randomized before the scoring and were scored by three independent investigators who were blinded to the experimental groups.
Mouse dendritic cell isolation
Brachial, axillary, and auricular dLNs were perforated using two 26-gauge syringe needles and digested for 30 min at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich) containing 10% fetal bovine serum (FBS), collagenase IV (0.75 mg/ml) (Sigma-Aldrich), deoxyribonuclease I (40 μg/ml) (Roche), and 3 mM CaCl2. Digestion was stopped using 30 μl of 0.5 M EDTA, and cells were filtered through a 40-μm cell strainer to obtain single-cell suspension.
Mouse ear skin cell isolation
Dorsal and ventral sides of ear skins were manually separated using forceps and digested in serum-free DMEM (Sigma-Aldrich) containing Liberase TL (Thermolysin Low) Research Grade (0.25 mg/ml; Roche) for 2 hours at 37°C. Digested tissue was smashed and filtered through a 70-μm cell strainer to obtain single-cell suspension. Cells were washed and resuspended in complete DMEM containing 10% FBS, 1% l-glutamine, and gentamicin.
Tissue ex vivo culture
Indicated organs were isolated after mice were euthanized. The colon and the small intestine were washed with PBS and longitudinally halved. The halves were further cut into small slices and incubated in phenol red–free DMEM with 10% charcoal-stripped FBS, l-glutamine, and gentamicin overnight. Ear skin was halved along the ventral/dorsal side and incubated by floating on medium overnight. Dorsal skin was cleaned by carefully removing subcutaneous fat and excised using 8-mm-diameter biopsy punches (BP-80F, Kai Medical). Skin biopsies were incubated by floating on medium overnight. Tissue ex vivo cultures were treated with metyrapone (300 μg/ml) (M2696, Sigma-Aldrich), 1 μM ACTH (A0298, Sigma-Aldrich), 20 μM forskolin (F3917, Sigma-Aldrich), and 3 μM pregnenolone (700142P, Sigma-Aldrich) or were left untreated overnight. All incubation steps were performed at 37°C and 5% CO2. Supernatants were collected for corticosterone radioimmunoassay.
Cell culture
Spontaneously immortalized primary C57BL/6 wild-type keratinocytes were cultured in complete supplemented Stemline Keratinocyte Medium II (Sigma-Aldrich) containing the Stemline Keratinocyte Growth Supplement (Sigma-Aldrich), l-glutamine, and gentamicin and for experiments in nonsupplemented Stemline Keratinocyte Medium II. HEK 293T cells (American Type Culture Collection) were cultured in phenol red–free DMEM containing 10% charcoal-stripped FBS, l-glutamine, and gentamicin. For treatments, keratinocytes were seeded at 3 × 105 per well and incubated overnight. Cells were washed and treated with either 20 μM forskolin (F3917, Sigma-Aldrich) or 1 μM ACTH (A0298, Sigma-Aldrich) overnight. Supernatants were collected for the GC bioactivity assay, and cells were harvested for mRNA expression analysis.
Flow cytometry
Single-cell suspensions from skin and dLN were prepared from the tissue, and live cells were counted using trypan blue staining. Before the staining, cells were blocked using 3% bovine serum albumin (BSA) and anti-CD16/32 TruStain FcX PLUS antibody (156603, BioLegend) and stained using a combination of surface marker–detecting antibodies. Dead cells were excluded using DNA-intercalating 4′,6-diamidino-2-phenylindole (DAPI) (422801, BioLegend). For fixation and intracellular staining, the Foxp3/Transcription Factor Staining Buffer Set (00-5521-00, eBioscience) was used according to the manufacturer’s protocol. If applicable, dead cell staining was performed before surface marker staining using either the Zombie NIR or the Violet Fixable Viability Kit (423105 and 423113, BioLegend) whenever samples were fixed. Fluorochrome-conjugated antibodies were used for flow cytometry analysis and were purchased from BioLegend, eBioscience, and BD Biosciences (table S1). Photomultiplier tube voltages were manually set, and compensation was calculated using single-staining controls with mouse lymphocytes or anti-rat, anti-hamster, anti-mouse immunoglobulin G (IgG), κ and negative-control CompBeads (BD Biosciences). Flow cytometry was performed with LSRFortessa (BD Biosciences) and the FACSDiva Software (BD Biosciences). Data were analyzed using FlowJo software (TreeStar). Total cell numbers were calculated from the acquired flow cytometry data and the counted viable cells using the hemocytometer with trypan blue staining.
Unsupervised analysis and automated FlowSOM cluster identification
High-dimensional flow cytometry analysis of T cell subsets in the skin was performed following the workflow of Nowicka et al. and Brummelman et al. (53, 54) using an adapted R script from the following: https://github.com/BecherLab/High-dimensional-single-cell-Analysis-for-Cytometry-/blob/master/pipeline_FlowSOM.R.
Compensation-corrected flow cytometry standard (FCS) files were cleaned and preprocessed, and CD3+CD11b− population was manually gated from single live CD45+ cells and exported using FlowJo software (TreeStar) (fig. S7A). Data transformation was performed by applying the inverse hyperbolic arcsinh function with individually set cofactors using Cyt3 (MATLAB). Transformed FCS files were uploaded into an RStudio environment, and the scales were further transformed using the matrixStats package to normalize the expression of all markers between values of 0 and 1 with low (1%) and high (99%) percentiles as a limit. Afterward, each FCS sample was normalized through downsampling to 1000 cells and combined. Combined data were visualized using the tSNE algorithm with the Rtsne package (fig. S7B) (55). FlowSOM algorithm–based clustering with 100 self-organizing map codes (by default) was applied on the combined dataset to enable the identification of small populations (fig. S7C) (56). Metaclustering was performed using the ConsensusClusterPlus package with k = 30, distribution of clusters were visualized using tSNE maps, and frequencies were depicted as stacked bar graphs (fig. S7, D and E). Clusters were manually merged and annotated to 10 identified cell populations based on the expression heatmap (fig. S7F).
RNA isolation and quantitative real-time PCR analysis
Total RNA from mouse skin or dLN was extracted using the SV Total RNA Isolation System (Promega) or peqGOLD TriFast (Peqlab, VWR) according to the manufacturer’s instruction. Laser capture microdissected RNA from human frozen sections was isolated with the ReliaPrep RNA Cell Miniprep System (Promega). Isolated RNA was reverse-transcribed into complementary DNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time qPCR was performed using the StepOnePlus Real-time PCR System (Applied Biosystems) with the FAST SYBR Green Master Mix (Applied Biosystems). Transcript levels were normalized to Actb for mouse or GAPDH for human transcripts and depicted as 2−∆Ct (ΔCt = Ct target gene − Ct reference gene) or presented as fold change to untreated controls. A full list of primer sequences used in this manuscript can be found in table S2.
Mouse epidermal sheet preparation
Euthanized mice were treated with depilation cream (Balea) for 5 min to reduce skin hair. Dorsal skin biopsies (8 mm) or ear skin halves were snap-frozen with liquid nitrogen or immediately used for epidermal sheet preparation. Skin tissue was washed with PBS and attached to microscope slides using double-sided adhesive tape with the epidermis sticking to the tape. Slides were incubated in PBS with 10 mM EDTA at 37°C for 2 to 3 hours, and the dermis was removed from the epidermis. The remaining epidermis was fixed using cold acetone for 10 min for further immunofluorescence staining.
Mouse skin frozen section
Mouse ears and dorsal skin biopsies were embedded in Tissue-Tek O.C.T Compound (4583 Sakura) and frozen using liquid nitrogen. Cryosections with 12- or 8-μm thickness were cut using the Hyrax C50 Cryostat (Carl Zeiss Microscopy) at −20°C chamber temperature. Sections were mounted onto SuperFrost microscope slides (AAAA000080, Thermo Fisher Scientific) for hematoxylin and eosin (H&E) staining or on HistoBond+ adhesive microscope slides (0810431, Paul Marienfeld) for immunostainings. Slides were shortly submerged into deionized water to remove the cryo-embedding medium and fixed with cold acetone for 10 min for further staining procedures.
GC bioactivity assay
HEK 293T cells were transiently transfected with the GRE luciferase reporter (GRE2-tk-luc) or an empty luciferase vector pGL3-luc and the GR-expressing vector SVGR1 through calcium phosphate precipitation for 6 hours. Cells were washed afterward and recovered overnight. Thereafter, cells from different transfection plates were trypsinized, washed, pooled, and seeded into 96-well plates. After 6 hours, cells were treated with supernatants from either ex vivo tissue or keratinocyte culture. After overnight incubation, cells were lysed and luciferase activity was assessed by adding luciferin and adenosine 5′-triphosphate using a luminescent detecting plate reader (Infinite 200 PRO, Tecan). The β-galactosidase assay using o-nitrophenyl-β-d-galactopyranoside substrate was performed to correct for transfection efficiencies.
Skin lysis for tissue lysate ELISA
Frozen dorsal skin punch biopsies were transferred to tubes containing radioimmunoprecipitation assay buffer and the cOmplete Protease Inhibitor Cocktail (11697498001, Roche). Tissue was lysed with a metal bead using the homogenizer TissueLyser II (Qiagen) for 4 min at 30 Hz. The supernatant was transferred to a new tube and centrifuged. Debris-free supernatant was taken for further IL-4 ELISA analysis.
Sandwich ELISA
Sandwich ELISA was performed according to BioLegend’s instructions with minor adaptions. Plates were coated with anti–IL-2 (503702, BioLegend) or anti–IL-4 (504101, BioLegend) capture antibodies in coating buffer [0.1 M NaHCO3 and 0.03 M Na2CO3 (pH 9.5)] at 4°C overnight and washed off. After blocking, samples and recombinant protein standards were plated and incubated at 4°C overnight. After washing, secondary detection antibodies, biotin anti–IL-2 (503804, BioLegend) or biotin anti–IL-4 (504201, BioLegend), were added to the plate and incubated at room temperature for 1 hour. After washing, streptavidin peroxidase conjugate (189733, Merck) was added to the plates for 30 min. Plates were washed, and trimethylboron substrate (421101, BioLegend) was used for development. Reactions were stopped using sulfuric acid, and optical density was measured at 450 nm with a 570-nm reference wavelength using a plate reader (Infinite 200 PRO, Tecan).
Corticosterone radioimmunoassay
Mouse blood sera (1:100 diluted) and supernatants from tissue ex vivo cultures were harvested and boiled at 90°C for 15 min and centrifuged at full speed for 10 min at 4°C. Duplicates of 100 μl from each sample were used, and 100 μl of anti-corticosterone antibody (AB1297, Merck) diluted in radioimmunoassay buffer (50 mM tris-HCl, 0.1 M NaCl, and 0.1% NaN3) was added to each sample, and the mixture was incubated for 15 min at 4°C while shaking. Afterward, 100 μl of radioactive hot corticosterone-[1,2,6,7-3H(N)] (NET399250UC, PerkinElmer) in radioimmunoassay buffer was added, and samples were incubated at 4°C overnight. After the addition of 100 μl of dextran/charcoal mixture, samples were mixed followed by another incubation of 10 min at 4°C. After centrifugation, the supernatant was added to scintillation vials containing MicroScint-40 (6013647, PerkinElmer). Counts per minute were measured using a beta-counter (LS-6500, Beckman Coulter). Corticosterone concentration was calculated from a corticosterone standard using the sigmoidal nonlinear regression fit with four-parameter logistic and X as the log of concentration. Total corticosterone concentration was normalized to the dry tissue weight and depicted as nanograms per gram tissue.
Laser capture microdissection
Human skin sections (10-μm thickness) were placed on PEN (polyethylene naphthalate) membrane–covered slides (415190-9041-000, Carl Zeiss Microscopy), which were heated at 180°C for 4 hours and irradiated with ultraviolet (UV) light for 30 min before their use. Sections were dried at −20°C, fixed with 70% ethanol for 2 min, and stained with cresyl violet acetate solution (C5042, Sigma-Aldrich). Slides were shortly air-dried after exposure to an increasing gradient of ethanol solutions. Sections were microscopically analyzed using the Axio Observer Z1 (Carl Zeiss Microscopy). Laser capture microdissection was conducted using a UV laser of the PALM MicroBeam (Carl Zeiss Microscopy) and the PALM RoboSoftware (Carl Zeiss Microscopy). Epidermal areas of the skin sections were therefore labeled, and the RoboLPC function was used to microdissect and catapult the tissue into the cap of a 0.2-ml AdhesiveCap tube (Carl Zeiss Microscopy). Laser capture microdissected epidermal areas from three serial sections of one individual donor or patient were collected for further RNA extraction.
Histological analysis
Frozen mouse skin sections were used for H&E staining. Serial sections from the same biopsy were used for consequent immunohistochemistry staining and immunofluorescence. Formalin-fixed, paraffin-embedded human skin sections underwent deparaffinization and hydration steps. Heat-induced epitope retrieval using tris-EDTA (pH 9) buffer was performed for 15 min. Sections were blocked with 5% BSA, 10% goat serum, 0.05% Tween 20, and 0.1% Triton X-100 in PBS for 1 hour. Afterward, endogenous biotin was blocked using an avidin/biotin blocking system (SIG-31126, BioLegend) according to the manufacturer’s protocol, followed by the primary antibody staining with rabbit anti-human CYP11B1 (HPA056348, Sigma-Aldrich) or rabbit IgG (Jackson ImmunoResearch Laboratories, Inc.) as an isotype control at 4°C overnight. After washing off the primary antibody, sections underwent peroxidase block using 3% H2O2 and 1% sodium azide in PBS for 1 hour. After several washing steps, secondary antibody staining was performed with biotin-SP-goat anti-rabbit antibody (Jackson ImmunoResearch Laboratories, Inc.) for 3 hours, followed by an amplification step using a VECTASTAIN ABC kit (PK-4000, Vectastain Laboratories Inc.) according to the manufacturer’s protocol. Peroxidase substrate staining was done with the ImmPACT DAB Substrate (SK-4105, Vectastain Laboratories Inc.). Sections were washed, counterstained with hematoxylin, dehydrated, and nonaqueously mounted. Ki-67 immunohistochemistry staining was performed on frozen mouse ear sections with 8-μm thickness. Following the blocking steps as described above, sections were stained with rabbit anti-mouse Ki-67 (D3B5, Cell Signaling Technology) overnight. Secondary antibody staining and development were performed as described above. Human frozen skin sections were blocked with 5% BSA, 1% anti–Fcγ-receptor antibody, 5% mouse serum, and 10% goat serum in PBS and stained with rabbit anti-human CYP11A1 (Sigma-Aldrich) and rat anti-human CD45 (eBioscience) overnight. For mouse epidermal sheet immunofluorescence, tissue was blocked with 5% BSA, 1% anti–Fcγ-receptor antibody, 5% mouse serum, 10% goat serum, and 0.1% saponin in PBS. Sheets were stained with rat anti-mouse MHCII (107601, BioLegend) or with rabbit anti-mouse Ki-67 (D3B5, Cell Signaling Technology) at 4°C overnight. For immunofluorescence of mouse ear sections, tissue sections were blocked with 5% BSA, 1% anti–Fcγ-receptor antibody, 5% mouse serum, and 10% goat serum in PBS and stained with rat anti-mouse Ly-6G (14-5931-82, eBioscience) and/or with rabbit anti-keratin 14 (SAB4501657, Sigma-Aldrich). For isotype staining controls, rat IgG2b, κ (400601, BioLegend), rat IgG2a, κ (14-4321-82, eBioscience), or rabbit IgG (011-000-003, Jackson ImmunoResearch Laboratories, Inc.) were used during primary antibody staining. In general, secondary antibody staining for immunofluorescence was performed with goat anti-rabbit IgG (H + L) conjugated to Alexa Fluor 488 (A-11008, Invitrogen) or with goat anti-rat IgG (H + L) conjugated to Alexa Fluor 568 (A-11077, Invitrogen). Immunofluorescence samples were covered with DAPI-containing mounting medium (F6057, Sigma-Aldrich). Immunofluorescence slides were analyzed with the epifluorescence microscope Axio Observer Z1 (Carl Zeiss Microscopy), and images were taken using AxioVision Software (Carl Zeiss Microscopy). H&E and immunohistochemistry stainings were analyzed using an Axio Vert.A1 microscope (Carl Zeiss Microscopy) with an Axiocam 105 color camera (Carl Zeiss Microscopy), and images were taken using ZEN software (Carl Zeiss Microscopy). ImageJ (National Institutes of Health) was used to process histological images, and brightness/contrast adjustments were equally applied to all samples.
Tissue lysis and conventional PCR for genotyping
Mouse skin or tail biopsies were lysed [50 mM KCl, 1.5 mM MgCl2, 10 mM tris-HCl, 0.45% NP-40, 0.45% Tween 20, and proteinase K (0.1 mg/ml)] at 55°C overnight. Lysis was stopped at 95°C for 10 min, and DNA was precipitated with 100% ethanol and sodium acetate (3 M, pH 5.2) on ice for 1 hour. Lysates were centrifuged, and DNA pellets were washed twice with 70% ethanol by resuspending the pellet. The pellet was air-dried and dissolved in nuclease-free water. Purified genomic DNA was used for PCR amplification to detect CreERTam fusion gene and myogenin, LoxP site, or the excision product. PCRs to detect CreERTam fusion gene and myogenin contained 10× reaction buffer, deoxynucleoside triphosphate (dNTP) (400 μM, New England Biolabs), primers (400 nM, New England Biolabs), and Taq polymerase (2.5 U, New England Biolabs) and were run with 35 cycles (95°, 50°, and 72°C, 30 s each step). PCRs to detect LoxP site or the excision product contained 10× reaction buffer (New England Biolabs), dNTP (200 μM, New England Biolabs), primers (500 nM, New England Biolabs), and Taq polymerase (0.625 U, New England Biolabs) and were run with 30 to 33 cycles (94°, 60°, and 72°C, 30 s each step). PCR products and size references (Thermo Fisher Scientific) were loaded on a 2 to 3% agarose gel in tris-acetate-EDTA buffer, separated by gel electrophoresis, and visualized using an imager (ImageQuantLAS400, GE Healthcare Life Sciences).
Statistical analysis
Statistical differences were determined using GraphPad Prism software (GraphPad Inc., v7). If appropriate, normal distribution of the data was tested using the D’Agostino-Pearson normality test, the Shapiro-Wilk normality test, and the Kolmogorov-Smirnov test. Only if all tests were passed (P > 0.05), the data were considered as normally distributed. Significance level α was set to 0.05 for all statistical analyses with *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.001 if not stated otherwise. Details of each statistical test applied on the data are indicated in the corresponding figure legend. Details regarding the preprocessing steps and the computational workflow for the FlowSOM analysis of the flow cytometry data are stated in the section “Unsupervised analysis and automated FlowSOM cluster identification.”
Acknowledgments
We grieve for our collaborator and friend M. Leverkus who passed away too early. We are deeply indebted to M. Leverkus and D. Panayotova-Dimitrova for provision of the K14-CreERTam mice and technical advice. We thank D. Dietrich and S. Beneke for the use of the scintillation counter, the flow cytometry core facility FlowKon of the University of Konstanz for technical support, and S. Okret for the GRE reporter constructs. We also thank M. Hipp, F. Schmitt-Hoffner, D. Goly, D. Eichbichler, and A. Wöhr for technical assistance. Funding: This study was supported by the German Science Foundation (DFG, BR 3369/9-1) to T.B. Author contributions: T.S.P. conceptualized the project, designed and conducted the experiments, analyzed the data, performed bioinformatic analyses, and wrote the manuscript. L.S. and J.M. performed the experiments and analyzed the data. V.M.M. helped in performing the experiments and data analysis. P.Z. and S.M. reviewed the computational FlowSOM analysis and provided intellectual input and technical advices for data analysis. D.S., D.K., and S.A. recruited human research volunteers and provided human skin sections and intellectual input. D.F.L. and M.N. provided key protocols and intellectual input and helped in designing the experiments. T.B. conceptualized the project, acquired funding, supervised the overall study, and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Data were not deposited to any database repositories. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabe0337/DC1
REFERENCES AND NOTES
- 1.Dainichi T., Kitoh A., Otsuka A., Nakajima S., Nomura T., Kaplan D. H., Kabashima K., The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol. 19, 1286–1298 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Cain D. W., Cidlowski J. A., Immune regulation by glucocorticoids. Nat. Rev. Immunol. 17, 233–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhen T., Cidlowski J. A., Antiinflammatory action of glucocorticoids––New mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Phan T. S., Merk V. M., Brunner T., Extra-adrenal glucocorticoid synthesis at epithelial barriers. Genes Immun. 20, 627–640 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Slominski A., Zbytek B., Nikolakis G., Manna P. R., Skobowiat C., Zmijewski M., Li W., Janjetovic Z., Postlethwaite A., Zouboulis C. C., Tuckey R. C., Steroidogenesis in the skin: Implications for local immune functions. J. Steroid Biochem. Mol. Biol. 137, 107–123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hostettler N., Bianchi P., Gennari-Moser C., Kassahn D., Schoonjans K., Corazza N., Brunner T., Local glucocorticoid production in the mouse lung is induced by immune cell stimulation. Allergy 67, 227–234 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Taves M. D., Gomez-Sanchez C. E., Soma K. K., Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 301, E11–E24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller M., Cima I., Noti M., Fuhrer A., Jakob S., Dubuquoy L., Schoonjans K., Brunner T., The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J. Exp. Med. 203, 2057–2062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed A., Schmidt C., Brunner T., Extra-adrenal glucocorticoid synthesis in the intestinal mucosa: Between immune homeostasis and immune escape. Front. Immunol. 10, 1438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slominski M., Ermak A., Mihm G., ACTH Receptor, CYP11A1, CYP17 and CYP21A2 genes are expressed in skin. J. Clin. Endocrinol. Metab. 12, 2746–2749 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Slominski A. T., Manna P. R., Tuckey R. C., Cutaneous glucocorticosteroidogenesis: Securing local homeostasis and the skin integrity. Exp. Dermatol. 23, 369–374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skobowiat C., Dowdy J. C., Sayre R. M., Tuckey R. C., Slominski A., Cutaneous hypothalamic-pituitary-adrenal axis homolog: Regulation by ultraviolet radiation. Am. J. Physiol. Endocrinol. Metab. 301, E484–E493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ermak G., Slominski A., Production of POMC, CRH-R1, MC1, and MC2 receptor mRNA and expression of tyrosinase gene in relation to hair cycle and dexamethasone treatment in the C57BL/6 mouse skin. J. Invest. Dermatol. 108, 160–165 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Miller W. L., Auchus R. J., The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sevilla L. M., Pérez P., Roles of the glucocorticoid and mineralocorticoid receptors in skin pathophysiology. Int. J. Mol. Sci. 19, 1906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bigas J., Sevilla L. M., Carceller E., Boix J., Pérez P., Epidermal glucocorticoid and mineralocorticoid receptors act cooperatively to regulate epidermal development and counteract skin inflammation article. Cell Death Dis. 9, 588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hannen R. F., Michael A. E., Jaulim A., Bhogal R., Burrin J. M., Philpott M. P., Steroid synthesis by primary human keratinocytes; implications for skin disease. Biochem. Biophys. Res. Commun. 404, 62–67 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Hannen R., Udeh-Momoh C., Upton J., Wright M., Michael A., Gulati A., Rajpopat S., Clayton N., Halsall D., Burrin J., Flower R., Sevilla L., Latorre V., Frame J., Lightman S., Perez P., Philpott M., Dysfunctional skin-derived glucocorticoid synthesis is a pathogenic mechanism of psoriasis. J. Invest. Dermatol. 137, 1630–1637 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Guilliams M., Dutertre C.-A., Scott C. L., Govern N. M., Sichien D., Chakarov S., Van Gassen S., Chen J., Poidinger M., De Prijck S., Tavernier S. J., Low I., Irac S. E., Mattar C. N., Sumatoh H. R., Low G. H. L., Chung T. J. K., Chan D. K. H., Tan K. K., Hon T. L. K., Fossum E., Bogen B., Choolani M., Chan J. K. Y., Larbi A., Luche H., Henri S., Saeys Y., Newell E. W., Lambrecht B. N., Malissen B., Ginhoux F., Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45, 669–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kashem S. W., Haniffa M., Kaplan D. H., Antigen-presenting cells in the skin. Annu. Rev. Immunol. 35, 469–499 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Malissen B., Tamoutounour S., Henri S., The origins and functions of dendritic cells and macrophages in the skin. Nat. Rev. Immunol. 14, 417–428 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Randolph G. J., Angeli V., Swartz M. A., Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat. Rev. Immunol. 5, 617–628 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Larson R. P., Zimmerli S. C., Comeau M. R., Itano A., Omori M., Iseki M., Hauser C., Ziegler S. F., Dibutyl phthalate (DBP) induced thymic stromal lymphopoietin (TSLP) is required for Th2 contact hypersensitivity responses. J. Immunol. 184, 2974–2984 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan D. H., Igyártó B. Z., Gaspari A. A., Early immune events in the induction of allergic contact dermatitis. Nat. Rev. Immunol. 12, 114–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li M., Hener P., Zhang Z., Kato S., Metzger D., Chambon P., Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. U.S.A. 103, 11736–11741 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noti M., Kim B. S., Siracusa M. C., Rak G. D., Kubo M., Moghaddam A. E., Sattentau Q. A., Comeau M. R., Spergel J. M., Exposure to food allergens through inflamed skin promotes intestinal food allergy through the thymic stromal lymphopoietin-basophil axis. J. Allergy Clin. Immunol. 133, 1390–1399.e1-6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hussain M., Borcard L., Walsh K. P., Rodriguez M. P., Mueller C., Kim B. S., Kubo M., Artis D., Noti M., Basophil-derived IL-4 promotes epicutaneous antigen sensitization concomitant with the development of food allergy. J. Allergy Clin. Immunol. 141, 223–234.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Oetjen L. K., Mack M. R., Feng J., Whelan T. M., Niu H., Guo C. J., Chen S., Trier A. M., Xu A. Z., Tripathi S. V., Luo J., Gao X., Yang L., Hamilton S. L., Wang P. L., Brestoff J. R.; M. Laurin Council, Brasington R., Schaffer A., Brombacher F., Hsieh C.-S., Gereau R. W. IV, Miller M. J., Chen Z.-F., Hu H., Davidson S., Liu Q., Kim B. S., Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 171, 217–228.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilliet M., Conrad C., Geiges M., Cozzio A., Thürlimann W., Burg G., Nestle F. O., Dummer R., Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch. Dermatol. 140, 1490–1495 (2004). [DOI] [PubMed] [Google Scholar]
- 30.van der Fits L., Mourits S., Voerman J. S. A., Kant M., Boon L., Laman J. D., Cornelissen F., Mus A.-M., Florencia E., Prens E. P., Lubberts E., Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol. 182, 5836–5845 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Cai Y., Shen X., Ding C., Qi C., Li K., Li X., Jala V. R., Zhang H.-g., Wang T., Zheng J., Yan J., Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity 35, 596–610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pantelyushin S., Haak S., Ingold B., Kulig P., Heppner F. L., Navarini A. A., Becher B., Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Invest. 122, 2252–2256 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terao M., Itoi S., Matsumura S., Yang L., Murota H., Katayama I., Local glucocorticoid activation by 11β-hydroxysteroid dehydrogenase 1 in keratinocytes: The role in hapten-induced dermatitis. Am. J. Pathol. 186, 1499–1510 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Henri S., Poulin L. F., Tamoutounour S., Ardouin L., Guilliams M., de Bovis B., Devilard E., Viret C., Azukizawa H., Kissenpfennig A., Malissen B., CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells. J. Exp. Med. 207, 189–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tuckermann J. P., Kleiman A., Moriggl R., Spanbroek R., Neumann A., Illing A., Clausen B. E., Stride B., Förster I., Habenicht A. J. R., Reichardt H. M., Tronche F., Schmid W., Schütz G., Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J. Clin. Invest. 117, 1381–1390 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jabara H. H., Ahern D. J., Vercelli D., Geha R. S., Hydrocortisone and IL-4 induce IgE isotype switching in human B cells. J. Immunol. 147, 1557–1560 (1991). [PubMed] [Google Scholar]
- 37.Jabara H. H., Brodeur S. R., Geha R. S., Glucocorticoids upregulate CD40 ligand expression and induce CD40L-dependent immunoglobulin isotype switching. J. Clin. Invest. 107, 371–378 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elenkov I. J., Glucocorticoids and the Th1/Th2 balance. Ann. N. Y. Acad. Sci. 1024, 138–146 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Wu C. Y., Sarfati M., Heusser C., Fournier S., Rubio-Trujillo M., Peleman R., Delespesse G., Glucocorticoids increase the synthesis of immunoglobulin E by interleukin 4-stimulated human lymphocytes. J. Clin. Invest. 87, 870–877 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jozic I., Stojadinovic O., Kirsner R. S., Tomic-Canic M., Stressing the steroids in skin: Paradox or fine-tuning? J. Invest. Dermatol. 134, 2869–2872 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Slominski A. T., Manna P. R., Tuckey R. C., On the role of skin in the regulation of local and systemic steroidogenic activities. Steroids 103, 72–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kadivar M., Petersson J., Svensson L., Marsal J., CD8αβ + γδ T cells: A novel T cell subset with a potential role in inflammatory bowel disease. J. Immunol. 197, 4584–4592 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Garcillán B., Marin A. V. M., Jiménez-Reinoso A., Briones A. C., Muñoz-Ruiz M., García-León M. J., Gil J., Allende L. M., Martínez-Naves E., Toribio M. L., Regueiro J. R., γδ T lymphocytes in the diagnosis of human T cell receptor immunodeficiencies. Front. Immunol. 6, 20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodman T., Lefrançois L., Expression of the γ-δ T-cell receptor on intestinal CD8+ intraepithelial lymphocytes. Nature 333, 855–858 (1988). [DOI] [PubMed] [Google Scholar]
- 45.Gaballa A., Stikvoort A., Önfelt B., Mattsson J., Sundin M., Uhlin M., T-cell frequencies of CD8+ γδ and CD27+ γδ cells in the stem cell graft predict the outcome after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 54, 1562–1574 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Hartwig T., Zwicky P., Schreiner B., Yawalkar N., Cheng P., Navarini A., Dummer R., Flatz L., Conrad C., Schlapbach C., Becher B., Regulatory T cells restrain pathogenic T helper cells during skin inflammation. Cell Rep. 25, 3564–3572.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Mills K. H. G., TLR-dependent T cell activation in autoimmunity. Nat. Rev. Immunol. 11, 807–822 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Beaulieu E., Morand E. F., Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat. Rev. Rheumatol. 7, 340–348 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Bereshchenko O., Coppo M., Bruscoli S., Biagioli M., Cimino M., Frammartino T., Sorcini D., Venanzi A., Sante M. D., Riccardi C., GILZ promotes production of peripherally induced treg cells and mediates the crosstalk between glucocorticoids and TGF-β signaling. Cell Rep. 7, 464–475 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Calmette J., Ellouze M., Tran T., Karaki S., Ronin E., Capel F., Pallardy M., Bachelerie F., Krzysiek R., Emilie D., Schlecht-Louf G., Godot V., Glucocorticoid-induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo. J. Immunol. 193, 5863–5872 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Panayotova-Dimitrova D., Feoktistova M., Ploesser M., Kellert B., Hupe M., Horn S., Makarov R., Jensen F., Porubsky S., Schmieder A., Zenclussen A. C., Marx A., Kerstan A., Geserick P., He Y.-W., Leverkus M., cFLIP regulates skin homeostasis and protects against TNF-induced keratinocyte apoptosis. Cell Rep. 5, 397–408 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Vasioukhin V., Degenstein L., Wise B., Fuchs E., The magical touch: Genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc. Natl. Acad. Sci. U.S.A. 96, 8551–8556 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nowicka M., Krieg C., Weber L. M., Hartmann F. J., Guglietta S., Becher B., Levesque M. P., Robinson M. D., CyTOF workflow: Differential discovery in high-throughput high-dimensional cytometry datasets [version 4; peer review: 2 approved]. F1000Res. 6, 748 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brummelman J., Haftmann C., Núñez N. G., Alvisi G., Mazza E. M. C., Becher B., Lugli E., Development, application and computational analysis of high-dimensional fluorescent antibody panels for single-cell flow cytometry. Nat. Protoc. 14, 1946–1969 (2019). [DOI] [PubMed] [Google Scholar]
- 55.van der Maaten L., Hinton G., Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2625 (2008). [Google Scholar]
- 56.Van Gassen S., Callebaut B., Van Helden M. J., Lambrecht B. N., Demeester P., Dhaene T., Saeys Y., FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87, 636–645 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabe0337/DC1