

Abstract
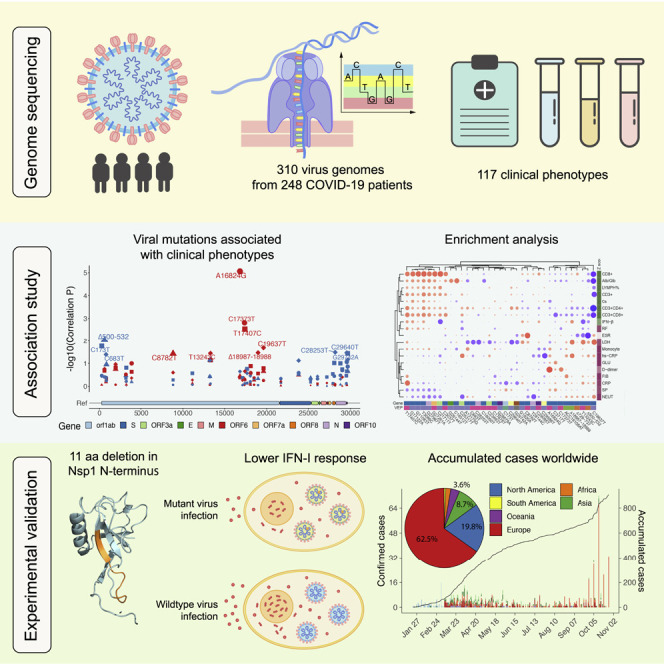
The SARS-CoV-2 virus, the causative agent of COVID-19, is undergoing constant mutation. Here, we utilized an integrative approach combining epidemiology, virus genome sequencing, clinical phenotyping, and experimental validation to locate mutations of clinical importance. We identified 35 recurrent variants, some of which are associated with clinical phenotypes related to severity. One variant, containing a deletion in the Nsp1-coding region (Δ500-532), was found in more than 20% of our sequenced samples and associates with higher RT-PCR cycle thresholds and lower serum IFN-β levels of infected patients. Deletion variants in this locus were found in 37 countries worldwide, and viruses isolated from clinical samples or engineered by reverse genetics with related deletions in Nsp1 also induce lower IFN-β responses in infected Calu-3 cells. Taken together, our virologic surveillance characterizes recurrent genetic diversity and identified mutations in Nsp1 of biological and clinical importance, which collectively may aid molecular diagnostics and drug design.
Keywords: genomic epidemiology, genetic variants, SARS-CoV-2, association, COVID-19, Nsp1, reverse genetics, deletion, clinical phenotype, interferon
Graphical abstract
Bringing together epidemiological, genetic, and clinical data, Lin et al. identify viral mutations that associate with clinical phenotypes. The 500-532 locus in the Nsp1 coding region is a deletion hotspot in the SARS-CoV-2 genome, and deletion variants lead to lower IFN-I response. Prevalence of 500-532 deletion variants is accumulating worldwide.
Introduction
The disease COVID-19, caused by a novel coronavirus now named SARS-CoV-2, was first reported on 30 December 2019 (Wu et al., 2020), with whole-genome sequencing of some of the first cases following rapidly after (Lu et al., 2020b; Zhu et al., 2020). The World Health Organization (WHO) declared COVID-19 a new pandemic on 11 March 2020. As of 9 November 2020, there have been 50,317,256 confirmed cases and 1,255,857 deaths reported worldwide. As an RNA virus, SARS-CoV-2 evolves rapidly and accumulates genetic diversity in relatively short periods. Estimates of the mutation rates suggest a substitution rate between 1.23 × 10−4 and 6.58 × 10−3 initially (van Dorp et al., 2020). There is a possibility that mutations accumulated during a pandemic could produce measurable effects on the infected population or complicate epidemic control efforts, highlighting a need to monitor the genetic diversity and epidemiology of SARS-CoV-2 throughout the pandemic. Several molecular epidemiologic studies, such as the ones carried out in Guangdong (Lu et al., 2020a) and Shanghai (Zhang et al., 2020b) in China and in the United States (Fauver et al., 2020), have provided valuable insights. Notably, the Spike D614G mutation was associated with increased viral load in COVID-19 patients (Korber et al., 2020). However, our knowledge of the relationship between SARS-CoV-2 genetic diversity and the clinical implications of genomic variants remains largely unexplored.
Collaborating with 20 hospitals and institutes, we analyzed epidemiological, genetic, and clinical data collected in Sichuan province of China across the outbreak period from 22 January to 20 February 2020. Sequencing of SARS-CoV-2 genome using a Nanopore MinION device provided flexible and real-time surveillance of genetic diversity in eight cities in Sichuan and Wuhan city of Hubei province. Furthermore, we investigated the phylogenetic evolution of viral mutations and their association with 117 clinical phenotypes. From this analysis, we identified Δ500-532, an in-frame deletion in Nsp1 (non-structural protein 1) N terminus coding region present in more than 20% of our sequenced samples, and found its association with higher RT-PCR cycle thresholds in the SARS-CoV-2 test and lower IFN-β levels in the serum of infected patients. The 500-532 locus is the fifth most frequent deletion region across the SARS-CoV-2 genome, and deletion variants in this locus were found in 37 countries. Biological importance of seven major types of deletion was characterized experimentally, and Calu-3 cell infection model supported our observed association of reduced type I interferon response. Our study links the genetic variants of SARS-CoV-2 to a potentially important clinical phenotype of COVID-19, which may aid molecular diagnostics and drug design for SARS-CoV-2.
Results
Epidemiological and genomic surveillance of COVID-19 outbreak
Located in southwest China, Sichuan is the fourth most populous Chinese province (83.41 million people) and has 24 cities including Chengdu, the province capital with a population size of 16.6 million (Figure 1 A). During the period between January and the end of February 2020, there were 538 COVID-19 cases confirmed by qPCR tests in Sichuan province, and 28.8% of these cases were from Chengdu (Figure 1A; Table S1). The mortality rate was estimated at 0.55% in Sichuan, lower than the national level (5.50% reported on 22 April) in China. The first case of “atypical” pneumonia (later confirmed as COVID-19) in Sichuan was reported on the 21 January 2020. Intense surveillance was initiated on 24 January. Confirmed cases in Sichuan increased exponentially, peaking on 30 January 2020 with 38 newly confirmed cases per day (Figure 1B). Since 5 March 2020, the daily reported number of cases in Sichuan was no more than one, with the origin of cases switching to imports from abroad (235 imported cases reported until 9 November 2020). More than two-thirds of the cases were imported from provinces outside Sichuan, mostly from Hubei (81.4%). Despite possible underestimation due to the asymptomatic cases, the number of confirmed cases is in proportion to the population size of 24 cities in Sichuan (Pearson R = 0.79, p = 2.18 × 10−5), with an average of 7.8 cases per million. In one city, Garzê (65.2 cases per million), the frequency of cases is much higher, where local transmission was linked to religious gatherings (Figure 1C), suggesting that social distancing may be a key factor in preventing virus spread.
Figure 1.

Spatial and temporal epidemiology of SARS-CoV-2 in Sichuan province
(A) Geographic distribution of confirmed and sequenced COVID-19 cases in Sichuan province, China. Confirmed case numbers were colored on the map and the size of the circles is proportional to the number of sequenced cases.
(B) Time series of the PCR-confirmed COVID-19 cases in Sichuan by the date of symptom onset. Cases are classified according to whether they were sequenced in this study. The dashed line indicates the cumulative number of the cases. The right panel shows the age and gender distribution of reported COVID-19 cases.
(C) The relationship between the number of confirmed cases and the population size of 21 cities in Sichuan province. Pearson correlation was applied and the gray area around the regression line indicates the 95% confidence interval. Color scale indicates cases per million population.
(D) The frequency of each SNP (blue) and indel (red) in the SARS-CoV-2 genome that was identified in the Sichuan cohort. The y axis indicates positions in the SARS-CoV-2 genome.
To monitor the genetic diversity of SARS-CoV-2, we undertook a collaborative investigation of the molecular epidemiology in two cohorts, a discovery cohort in Sichuan and a validation cohort in Wuhan, Hubei province, where most imported cases were from. We collected epidemiological and clinical data of 538 COVID-19 patients from 21 cities in Sichuan, from which we sequenced 141 clinical samples from 88 patients (Figure 1A; Table S1), representing 16.4% of all confirmed cases in Sichuan (Figure S1A; Table S1). Out of the 88 patients, 44% were female and the mean age was 45.7 (SD = 18.4; Table S2). The sample collection time spanned the majority of the outbreak period in Sichuan province, which was between 21 January and 8 March 2020 (Figure 1B). To investigate whether recurrent genetic variants in Sichuan were imported or occurred locally due to the founder effect, we further screened 169 clinical samples from 160 COVID-19 patients between 3 February and 29 March 2020 in Wuhan, the mean age of whom was 50.6 (SD = 18.9) and 44% of whom were female (Figure S1B; Table S3).
The genomes were generated using a combination of multiplex PCR amplification followed by nanopore sequencing on a MinION device or untargeted metagenomic sequencing on Illumina NextSeq (Figure S1C). For nanopore sequencing, the version 1 ARTIC primer set (Quick, 2020) was used and low coverage in non-random regions was spotted (Figure S1C), potentially due to varied efficiency of amplification (Lu et al., 2020a). We thus included an additional primer set that generates 1 kb amplicons to improve the coverage (Table S4). Using this protocol, we were able to generate SARS-CoV-2 genomes rapidly at four sequencing sites and achieved an average of 80.1% coverage with more than ten reads (mean depth = 0.39 million reads per sample, Figures S1D–S1F). A total of 310 near- or partial-complete genomes from 248 COVID-19 patients were obtained from Sichuan or Wuhan cohort (Figure S1; Tables S1, S2, and S3).
Genetic variants and phylogenetic analyses of SARS-CoV-2 genome
To identify the genetic variations of the SARS-CoV-2 virus, we used the ARTIC bioinformatic pipeline to identify single nucleotide polymorphisms (SNPs) and insertions and deletions (indels). In total, 104 SNPs and 18 indels were found across the 88 SARS-CoV-2 genomes in Sichuan cohort (Figure 1D; Table S5). The majority of variants were singletons; i.e., were observed in only one genome. Of the variation identified, 31 SNPs and 4 indels were present in more than two patients (denoted as “recurrent variants” hereafter; Figure 2 A).
Figure 2.

Recurrent genetic variants and phylogenetic analysis in the SARS-CoV-2 genomes
(A) Percentage of samples with the 35 recurrent genetic variants we studied in different continents or regions. The effect of the variants was predicted by VEP (Ensembl Variant Effect Predictor).
(B) Visualization of the corresponding time-scaled maximum clade credibility tree based on estimated maximum likelihood phylogeny of SARS-CoV-2 genome sequences from Sichuan (colored dots) and genomes from other countries or Chinese provinces (gray dots). The clusters (A–D) are highlighted in colors corresponding to that in Figure S2B, and samples with Δ500-532 are colored in red. All nodes with posterior probabilities lower than 0.5 have been collapsed into polytomies and their range of divergence dates are shaded in gray.
(C) The daily number of locally transmitted (red) and imported (gray) COVID-19 cases in Sichuan province. The accumulated locally transmitted case number was shown in the red line.
(D) Posterior distributions of the tMRCAs (time to most recent common ancestry) of the five phylogenetic clusters (A–D) from the molecular clock analysis. Distributions (orange) are truncated at the upper and lower limits of the 95% HPD (highest posterior density) intervals; the vertical red lines indicate the median estimates. Blue shading and horizontal red lines indicate the sampling period over which genomes in each cluster were collected. Dots indicate the collection dates of the genomes, colored by sampling location from Sichuan (red) and other regions (gray).
To investigate whether these 35 recurrent genetic variants identified in Sichuan cohort were imported or occurred locally, we compared them with the validation cohort consisting of 169 samples from Wuhan and with the public GISAID (https://www.gisaid.org/) data. We first used 81,391 high-quality public genome sequences deposited in the GISAID by 9 November 2020. Of the 35 SNPs, 29 (82.8%) were also present in other continents; some variants (C3037T, G26144T, C8782T, T28144C, G11083T, and C18060T) are prevalent in Europe, Africa, Oceania, and America (Figures 2A and S2A), suggesting that the majority of SNPs have been circulating worldwide. Next, a comparison with the Wuhan cohort revealed an additional four variants present in both Sichuan and Wuhan, but not in the GISAID data from other regions. These recurrent variants include three indels: Δ500-532, ACC18108AT, and Δ729-737; and one SNP: T13243C (Figures 2A and S2A); indicating that these variants may have been imported from Wuhan, consistent with the travel records of patients in Sichuan (Table S2). The remaining two recurrent variants, Δ18987-18988 and A16824G, were only observed in Sichuan but not in Wuhan, which may be due to local evolution or importation from other under-sampled regions. Six variants that were only identified in our study, including Δ500-532, Δ729-737, C3717T, A16824G, ACC18108AT, and Δ18987-18988, were validated using RT-PCR followed by Sanger sequencing (Figures S3A–S3G). The other 26 were cross validated using Illumina sequencing (Figure S3H), confirming the accuracy of our sequencing and variant calling approaches.
To understand the phylogenetic history and transmission of SARS-CoV-2, we used a rigorous evolutionary analysis method (Lu et al., 2020a) consisting of maximum likelihood (ML) and Bayesian molecular clock approaches. We added our 88 new virus genomes from Sichuan to 250 curated genome sequences that were sampled from 31 regions or countries (Table S6) used previously (Lu et al., 2020a). In the phylogenetic tree (Figure 2B), SARS-CoV-2 genomes obtained from Sichuan cohort distributed among viral lineages sampled from other Chinese provinces or other countries, supporting the epidemiological records that the majority of reported cases were linked to travel (Tables S1 and S2). Notably, two deletions in Nsp1 coding region, Δ500-532 and Δ729-737, were observed in 23 (26%) and 2 (2.2%) cases in Sichuan and 39 (24%) and 4 (3%) cases in Wuhan, respectively. Only one sample in Sichuan harbored both Δ500-532 and Δ729-737. We therefore focused on the genomes with Δ500-532 and found them distributed sparsely in the different clusters in the phylogenetic tree (Figure 2B). We further performed haplotype analysis (Leigh et al., 2015; Rozas et al., 2017; Tang et al., 2020) on Nsp1 variants and identified 14 different haplotypes in Wuhan and Sichuan, 5 out of which were from multiple cities and contained this deletion (Figure S2D). Taken together, our results suggest that the Δ500-532 occurred in multiple cities and might be imported multiple times from Wuhan, consistent with the fact that 83.3% of patients in Sichuan with this deletion had a travel history to Wuhan (Tables S1 and S2).
To identify local groups of cases, we constructed an ML phylogenetic tree and observed that four clusters (denoted A–D) include 13 cases from the Sichuan cohort (Figure S2B), which had posterior probability higher than 80% (Figure 2B; Table S7). These clusters represent 14.7% of the Sichuan cohort and provide a signature that is consistent with local transmission: clusters C and D only contain sequences sampled from one city in Sichuan, in line with their epidemiological records of local transmission (Table S7). Cluster B includes one sample from Japan, and others were all from Guang’an city, and their travel records supported that this cluster was introduced to Sichuan from different cities (Table S7). Next, we estimated the dates of the most recent common ancestor (tMRCA) of clusters A–D (Figures 2C and 2D). Clusters A–C have earlier tMRCAs around the second half of January 2020, consistent with the start of the epidemic in Sichuan, whereas cluster D has a later tMRCA, in line with the time of the epidemic peak in Sichuan province (Figure 2C). We further estimated the median tMRCA of the COVID-19 pandemic, which was 29 November 2019 (with a 95% highest posterior density [HPD] from 14 November to 12 December 2019; Figure 2D). Taken together, our phylogenetic analyses support the epidemiological time series that the majority of reported cases were linked to travel rather than local transmission, and SARS-CoV-2 lineages including Δ500-532 were imported multiple times from Wuhan to Sichuan.
Clinical implications of genetic variants of SARS-CoV-2
To investigate the potential clinical implications of the identified 35 recurrent genetic variants (31 SNPs and 4 indels) of SARS-CoV-2, we included 117 clinical phenotypes in our study (Table S8) and performed association analysis. First, we compared the clinical phenotypes between patients who developed severe or non-severe symptoms and identified traits that were associated with COVID-19 severity (Figure 3 A). Generally, patients who developed severe symptoms manifested lymphocytopenia (reduced CD3+, CD8+ cell number, or LYMPH%) and increased level of C-reactive protein (CRP, hs-CRP) and D-dimer, in agreement with clinical guidelines and previous studies (Li et al., 2020; Liu et al., 2020a; Zhang et al., 2020b; Zhou et al., 2020). Higher serum glucose (GLU), monocyte, neutrophil counts (NEUT), respiratory frequency (RF), fibrinogen (FIB), and systolic pressure (SP) were also seen in the severe cases, while higher serum calcium, albumin/globulin ratio (Alb/Glb), and serum IFN-β level were observed in the non-severe cases (Figure 3A). LDH (lactate dehydrogenase) and ESR (erythrocyte sedimentation rate), which are among traits recommended for COVID-19 diagnosis (Li et al., 2020; Liu et al., 2020a; Tan et al., 2020; Zhou et al., 2020), were also taken into account. We then classified 19 severity-related phenotypes into (11) severe and (8) non-severe traits (Figure 3A; STAR methods).
Figure 3.

Associations of 35 recurrent genetic variants with clinical phenotypes
(A) Volcano plot of the significant traits that are associated with COVID-19 severity. The x axis shows the difference of Z score between patients that developed severe or non-severe symptoms. The y axis shows –log10(p value) of t test between these two groups. Red indicates indicating severe related traits, and blue, non-severe related traits. Severe related traits include CRP (C-reactive protein), hs-CRP (high-sensitivity CRP), GLU (serum glucose), SP (systolic pressure), D-dimer, RF (respiratory frequency), FIB (fibrinogen), NEUT (neutrophils counts), Monocyte (monocyte counts), and LDH (lactate dehydrogenase); and non-severe related include CD3+, CD3+CD4+, CD3+CD8+, CD8+ cell counts, LYMPH% (percentage of lymphocytes), Ca (serum calcium), Alb/Glb (albumin and globulin ratio), and IFN-b (serum IFN-β). The traits with p value less than 0.05 are shown.
(B) Manhattan plot of 35 genetic variants that were associated with CRP, D-dimer, ESR (erythrocyte sedimentation rate), and LDH. Genome position and –log10 (correlation p value) were shown. Different symbols indicate that correlated phenotypes and size of the symbols is in proportion to the absolute (abs) value of correlation efficient (r). Red or blue indicates a positive or negative correlation, respectively.
(C) Heatmap of correlation coefficients between clinical phenotypes and genetic variants. The clustering was based on Pearson correlation coefficients. Intensity of red and blue indicates correlation efficient, red indicates positive correlation, and blue, negative correlation. Intensity of purple and green indicates higher or lower mean values in the severe group.
(D) Δ500-532 is significantly enriched with non-severe traits but not with severe traits. p values were adjusted using permutation.
(E) Dot plot shows summed normalized enrichment score (NES) and p values of t test between the group with or without a certain variant for Ct values in qPCR tests. The dots in the top right corner have more significant p values for Ct values of N (red) or ORF (blue) genes and a higher enrichment score.
A unified pre-processing strategy (see STAR methods) was used to normalize each clinical parameter and regress out confounding factors such as age and gender. The genetic variants were processed into continuous values between 0 and 1, reflecting the dosage/percentage of each SNP or indel in each case. Pearson correlation coefficient was then calculated for each processed clinical phenotype and the recurrent variants (Figure S4; Data S1). Forty-one pairs of virus variant-clinical phenotypes exhibited association with p value less than 0.01 (Table S9). Notably, the phenotypes identified from these pairs were significantly enriched with the 19 severity-related traits (p = 0.012, hypergeometric test), indicating that these variants might be related to COVID-19 outcome. Four traits (LDH, CRP, D-dimer, and ESR), which are the typically recommended traits for COVID-19 diagnosis (Li et al., 2020; Liu et al., 2020a; Tan et al., 2020; Zhou et al., 2020), were shown in Figure 3B. Several loci, notably Nsp13 A16824G and Nsp1 Δ500-332, showed significant correlations, positively with D-dimer and negatively with ESR, respectively (Figure 3B), suggesting the potential impact of these virus variants on clinical phenotypes. Similarly, we assessed the general relationship between the 19 severity-related phenotypes and the 35 recurrent genetic variants. Correlation coefficients between the phenotypes and variants were used in the bi-clustering analysis (Figure 3C). Interestingly, the clinical phenotypes were clustered into two groups, with lower or higher mean values in the severe cases, suggesting that some variants may have consistent correlations with severity-related traits.
To validate this, we adapted a ranked enrichment analysis method similar to GSEA (Subramanian et al., 2005) to distinguish whether a variant is enriched with non-severe or severe traits (see STAR methods; Table S10). For example, Δ500-532, an in-frame deletion of Nsp1 coding region, is negatively correlated with ESR, serum IFN-β level, and CD3+CD8+ T cell counts in the blood (Figure 3C), also showed significant enrichment with non-severe (p adjust = 0.0071) but not severe traits (Figure 3D).
To further assess the potential clinical relevance of these variants, we classified the cases into two groups, with or without a certain virus variant, and compared gender, age, Ct values, and the 117 clinical phenotypes between the two groups of cases. The Ct value (cycle threshold value in qPCR test for SARS-CoV-2 ORF or N genes) was included in the analysis due to its indication of the viral load. Moreover, it is found negatively associated with COVID-19 severity (Liu et al., 2020b) and was used to assess the effect of SARS-CoV-2 genetic variants such as D614G in Spike protein (Korber et al., 2020). Notably, patients who were infected with virus harboring Δ500-532 had the highest Ct values among all tested 35 variants (Figure 3E, p < 4.4 × 10−5; Data S2).
Prevalence of various deletions in Nsp1 worldwide
Given the prevalence of Nsp1 Δ500-532 in both Sichuan and Wuhan and its association with higher Ct values, lower serum IFN-β, and non-severe traits, we next assessed its prevalence worldwide. We calculated the frequency of deletion for each base pair of the genome. The 500-532 locus is the fifth most frequent deleted region across the SARS-CoV-2 genome (Figure 4 A), whereas the 729-737 locus is not a deletion hotspot. Indeed, the 500-532 locus is significantly higher than the average deletion per base in the genome (Wilcox test, p = 2.88 × 10−41). Our sequencing results support the deletion with high sequence depth and the deletion was validated by Sanger sequencing (Figures 4B, S6A, and S6B). Notably, we retrieved 24 types of deletion variants in 500-532 locus from 922 cases in 37 countries from GISAID (Figure 4C; Table S11). 62.5% and 19.8% of these cases were from Europe and North America, respectively. The number of accumulated cases increased sharply from September to November 2020 (Figure 4D), and the major variants are the Δ518-520 (Δ1aa), Δ518-523 (Δ2aa), and Δ509-523 (Δ5aa), circulating mainly in Europe and North America (Figures 4D and 4E). Therefore, our results revealed a hotspot of deletion in the SARS-CoV-2 genome, and the associated case number is steadily increasing worldwide.
Figure 4.

Prevalence of deletion variants in the 500-532 locus in SARS-CoV-2 genome
(A) Percentage of deletion in the SARS-CoV-2 genome. The red arrow indicates the 500-532 locus.
(B) Sashimi plot indicates the sequencing reads that were mapped around the identified Δ500-532 region (see Figures S6B and S6C).
(C) The alignment of amino acid residues of mutation neighboring 500-532 locus in Nsp1 coding region. Identified cases number (N) and the number of countries were shown. WT, wild-type SARS-CoV-2 Nsp1 sequence. Stars indicate the deletion mutants that were further tested experimentally (related to Figures 5, 6, and 7).
(D) The confirmed (left y axis) or accumulated (right y axis) cases with Nsp1 deletion mutants. The pie chart shows the percentages of cases identified in each continent.
(E) Geographic distribution of COVID-19 cases with the deletion variants worldwide. Color intensity on the map indicates confirmed cases in that region. The pie charts indicate the proportions of different deletion variants in each country, and the size of pie charts indicate the number of cases with the deletion mutants.
Structural and functional implications of the deletions in Nsp1 of SARS-CoV-2
Comparing the amino acid sequences of Nsp1 of SARS-CoV and SARS-CoV-2, we found 84.4% identity in the same length of 180 residues, and most amino acid substitutions are of similar types (Figure S6), indicating that Nsp1 of SARS-CoV-2 may have a similar function as that of SARS-CoV. Δ500-532 results in an 11-amino-acid residue deletion of A79PHGHVMVELV89 in the N terminus of SARS-CoV-2 Nsp1. Next, we assessed the potential effect of the deletions on the structure of Nsp1 protein. Three recent cryo-EM studies (Schubert et al., 2020; Thoms et al., 2020; Yuan et al., 2020) showed that C-terminal domain of SARS-CoV-2 Nsp1 binds to 40S ribosomal subunit and interferes with mRNA entry. However, none of the studies provided structural details for N-terminal domain of this protein. Previous studies reported a novel beta-barrel structure for the N-terminal domain of SARS-CoV Nsp1 by nuclear magnetic resonance (Almeida et al., 2007; Wathelet et al., 2007), which was roughly matched in the cyro-EM studies (Thoms et al., 2020; Yuan et al., 2020). In this predicted globular N-terminal domain structure, residues in the region corresponding to the Δ500-532 form a central beta-strand, removal of which would likely cause complete destruction of the barrel fold (Figure 5 A).
Figure 5.

Nsp1 mutants retained ribosomal binding ability while affecting mRNA metabolic process
(A) Mapping of 500-532 locus (A79PHGHVMVELV89, orange) onto the predicted 3D structure of N terminus of SARS-CoV-2 Nsp1 (Almeida et al., 2007; Wathelet et al., 2007).
(B) SARS-CoV-2 Nsp1 mutant proteins bind to 40S ribosomal subunit. HEK293T cells were transfected with Nsp1-expressing (N-terminally tagged with FLAG) or control (FLAG-GFP) plasmid and immunoprecipitated with anti-FLAG antibody. Immunoblots were stained with anti-S6 or anti-FLAG antibodies.
(C) PCA (principal-component analysis) on transcriptome of Nsp1-expressing compared to GFP-expressing HEK239T cells.
(D) GO enrichment analysis of genes that were upregulated in the mutant compared to WT Nsp1-expressing cells.
(E) Heatmap of differentially expressed genes enriched in mRNA metabolic process. Genes coding for ribosomal proteins (RPs) were shown.
To understand whether the deletion affects Nsp1 binding to 40S, we transfected HEK293T cells with plasmids expressing the full-length, wild-type (WT) Nsp1 protein and its mutant forms of Δ500-523 (Δ11aa), and Δ509-523 (Δ5aa) and Δ518-520 (Δ1aa, the most prevalent deletion type identified in 16 countries; Figure 4C), and found that mutant SARS-CoV-2 Nsp1 retain the binding ability to 40S ribosomal subunit (Figure 5B). The transcriptome analysis of HEK293T cells transfected with Δ11aa and Δ5aa showed that Nsp1-expressing cells are distinct from the GFP control, with WT Nsp1 highly different from the other samples and two deletion mutants clustered more closely (Figure 5C). Genes that were significantly upregulated in the mutant Nsp1- compared to WT Nsp1-expressing cells were enriched in the biological processes of “peptide biosynthetic/metabolic process,” “ribonucleoprotein complex biogenesis,” “protein targeting to membrane/ER,” “mRNA metabolic process,” and “nonsense-mediated decay” (Figure 5D), in line with the reported functions of SARS-CoV-2 Nsp1 (Banerjee et al., 2020; Schubert et al., 2020; Thoms et al., 2020). Notably, many genes in mRNA metabolic process pathway encoding for ribosomal proteins (RPs) were downregulated in WT compared to the GFP control. In contrast, two deletions showed a distinct expression pattern from WT and were more akin to that of GFP control (Figure 5E). These results suggest that these N-terminal deletions in Nsp1 may affect its function in mRNA and protein metabolic processes.
Since Δ500-532 is negatively correlated with serum IFN-β level in the infected patients, we next assessed how the deletions affect type I interferon (IFN-I) response. In addition to the tested Δ500-532 (Δ11aa), and Δ509-523 (Δ5aa) and Δ518-520 (Δ1aa), we further included Δ3 aa (Δ509-517), Δ2 aa (Δ518-523), and Δ1 aa (Δ515-523 and Δ521-523), a total of seven major deletion types in the overexpression studies. Consistently, all deletion variants showed reduced IFN-I response in the transfected HEK293T and A549 (a human lung cell line) cells at both transcription and protein levels (Figures 6A, 6B, and S7). To further understand the potential underlying mechanism, we next performed IFNB1-promoter-controlled luciferase reporter assays and found that all deletion mutants suppressed IFNB1 response and its downstream signaling via reduced IFN-stimulated response element (ISRE)-mediated promoter activities (Figures 6C and 6D). We further performed transcriptome analysis on transfected A549 cells. PCA (principal-component analysis) revealed that the GFP control deviated from the Nsp1-expressing cells in principal component 1 (PC1), whereas the WT Nsp1-expressing cells were distinct from the other four types of deletion, which are clustered closely in PC2 (Figure 6E), indicating that the deletions may have a similar impact on the transcriptome that are different from the WT Nsp1. Interestingly, long deletions separate from short deletions in PC2 and have more differentially expressed genes (Figures 6E and 6F). Indeed, when comparing these deletions to WT Nsp1, the significantly downregulated genes were enriched in “defense response to virus,” “viral genome replication,” “regulation of transcription by RNA polymerase II,” and “response to type I interferon” (Figure 6F), supporting that IFN-I signaling was downregulated in the deletion mutants (Figures 6F and 6G).
Figure 6.

Nsp1 mutants downregulate IFN-1 response
(A) Relative mRNA expression level of IFNB1, IFNA1, and IFNA2 to GAPDH in SARS-Cov-2 wild-type (WT) or mutant Nsp1-expressing A549 cells.
(B) Concentration of IFN-β in the supernatant of Nsp1-expressing A549 cells. GFP and SARS-CoV Nsp1 were used as negative and positive controls. Each dot represents a single biological replicate. Median and range of 75th percentile are also shown in the boxplots. ∗ indicates statistical significance between mutant and WT Nsp1, and + indicates significance between GFP controls. Mann-Whitney U test was performed.
(C and D) IFNB1-promoter (C) and interferon-stimulated response element (ISRE)-mediated promoter (D) controlled luciferase reporter assays. HEK293T cells were transiently transfected with luciferase plasmid only (Ctrl) or along with GFP- or Nsp1-expressing plasmids, with (+) or without (–) IFN-β treatment in (D). Promoter activities were determined as the ratio between a firefly luciferase and internal control of Renilla luciferase. Mean and SEM are shown. ∗ indicates significance between mutant and WT Nsp1. Mann-Whitney U test was performed.
(E) PCA on transcriptome of A549 cells transfected with Nsp1 variants or GFP control.
(F) GO enrichment analysis of the genes that were significantly downregulated in mutant compared to WT Nsp1-expressing A549 cells.
(G) Interferon signaling pathways were significantly downregulated in Δ500-532 mutant compared to WT Nsp1-expressing A549 cells. The pathway was generated in IPA (Ingenuity Pathway Analysis). Green indicates downregulation, red indicates upregulation, and gray indicates genes that were in the dataset but not differentially expressed. The intensity of red or green indicates the log2(fold-change) of each gene.
Nsp1 deletions lead to lower type I interferon response in SARS-CoV-2-infected cells
To investigate the impact of deletions in Nsp1 on SARS-CoV-2 virus, we used viruses isolated from clinical samples, including isolates having WT, Δ518-520 (Δ1aa) and Δ509-517 (Δ3aa) Nsp1 in the experimental infections of Calu-3 cells. All three viruses showed an over 100-fold increase in genome copy numbers measured from cell supernatants from 1 to 72 h post-infection. With both MOI 2 and 0.2, Δ518-520 and Δ509-517 showed similar growth kinetics as WT (Figure 7 A). And with both MOI 2 and 0.2, virus isolate with WT Nsp1 induced a higher level of IFNB1 response at its peak at 48 h post-infection, with more than 2-fold higher level than Δ518-520 and Δ509-517 mutant viruses (Figure 7B).
Figure 7.

Growth kinetics and regulation of IFN-I response in wild-type and Nsp1 deletion virion
(A) Extracellular viral RNA copy number produced by Calu-3 cells infected with virus isolates with wild-type (WT) Nsp1, Δ518-520 (Δ1aa), or Δ509-517 (Δ3aa), at MOI 2 (left) or MOI 0.2 (right). SARS-CoV-2 RdRp gene was quantified.
(B) Relative expression level of IFNB1 (to ACTB) in Calu-3 cells infected with virus isolates at MOI 2 (left) or MOI 0.2 (right).
(C) Extracellular viral RNA copy number produced by Calu-3 cells infected with WT or 2 independent clones of Δ500-532 (Δ11aa) recombinant SARS-CoV-2 mutants at MOI 2. SARS-CoV-2 RdRp (left) and E (right) genes were quantified.
(D) Viral progeny determined by plaque assay in Vero E6 cells.
(E) Comparison of plaque phenotypes of cell supernatant harvested at 48 h post infection (from the growth curve D in Vero E6 cells).
(F) IFNB1 transcript level (normalized to GAPDH) in Calu-3 cells infected with WT, mutant rSARS-CoV-2 (Δ500-532 D1 or Δ500-532 D2), or mock infected.
(G) Concentration of IFN-β in the supernatant of infected Calu-3 cells. Mean and SEM are shown. Significance between WT and mutant viruses was indicated; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Multiple t tests were performed.
To assess whether these effects are solely attributed to the deletion in Nsp1, we further utilized a reverse genetics system (Thi Nhu Thao et al., 2020) to engineer two infectious clones of recombinant SARS-CoV-2 with Δ500-532 (Δ11aa) deletion. With MOI 2, both WT and deletion mutants produced viable viral progeny and showed similar growth kinetics in Calu-3 cells (Figures 7C and 7D), similar to what was observed with the clinical isolates having Δ518-520 and Δ509-517. However, the plaque phenotype of the mutant was reduced compared to the WT virus at 48 h post-infection (Figure 7E). Importantly, cells infected with WT rSARS-CoV-2 produced more than three times higher IFNB1 transcript than Δ500-532 mutants at the peak of its expression at 48 h post-infection (Figure 7F), similar to what we observed with clinical isolates (Figure 6F). Additionally, the concentration of IFN-β was also reduced in the supernatant of cells infected with mutant rSARS-CoV-2 (Figure 7G). Taken together, Nsp1 deletion mutants are replication competent, presenting similar growth kinetics as WT virus in Calu-3 cells, and induced lower IFN-I response. These results support our observed association between Δ500-532 and reduced serum IFN-β from our association studies.
Discussion
Despite the exceptional magnitude and rapid expansion of the global COVID-19 pandemic, it remains a challenge to reveal the biological and clinical implications of genomic variations of SARS-CoV-2. Through tracking the molecular evolution and clinical features of SARS-CoV-2-infected patients in two cohorts in Sichuan and Hubei provinces of China, we identified recurrent variants that are associated with important clinical phenotypes. Since the sample size in this study is still limited, their associations must be interpreted in the context of currently available epidemiological and clinical data. We believe a larger cohort with international collaborations will further validate and discover more associations with important biological insights.
For the deletion of Δ500-532 in Nsp1 N-terminal coding region, it was found correlated with lower viral load (evident by higher Ct value in qPCR test), enrichment with non-severe traits, and lower serum IFN-β, indicating its clinical importance. Moreover, we found that there are many types of Nsp1 deletion mutants worldwide, all within this 500-532 locus, involving 922 cases from 37 countries, suggesting that it is a hotspot of deletion due to potential convergent evolution. We further revealed that the deletion in Nsp1 causes reduced IFN-I response in overexpression studies and the infected human cell model. All tested seven major types of deletion rendered a lower level of IFN-I response in overexpressing cells, and this reduction is related to inhibition of IFNB1 promoter activity, which further suppresses IFN-I downstream signaling. Importantly, IFN-I response was also significantly reduced in Calu-3 cells infected with mutant viruses isolated from clinical samples or engineered using reverse genetics. Similar to its closely related coronaviruses, SARS-CoV-2 Nsp1 binds to 40S ribosomal subunit through anchoring of its C-terminal helices to the mRNA channel (Thoms et al., 2020), while its N-terminal is flexibly disposed and thought dispensable for this interaction. Interestingly, N-terminal domain of SARS-CoV was reported to be involved in suppressing innate immune factors or in the regulation of cellular mRNA (Narayanan et al., 2008). The deletion identified in our study that is likely important to Nsp1 N-terminal structure does not diminish Nsp1-ribosome binding ability, but indeed significantly suppressed IFN-I response. Although we found that genes coding for ribosomal proteins (RPS/RPL) and pathways related to mRNA/protein metabolism were differentially regulated in mutant Nsp1-expressing cells compared to the WT Nsp1, it remains to be examined how the deletion, particularly the key residues in the N-terminal domain affects host cell translation or mRNA decay. Interestingly, several other Nsp1 mutants were also identified in different geographical areas (Benedetti et al., 2020; Islam et al., 2020). These results indicate that SARS-CoV-2 is undergoing profound genomic changes and evolution and that Nsp1 is likely important for viral phenotypes. Indeed, in our plaque assays, we found that our Δ500-532 mutant rSARS-CoV-2 presented a smaller plaque phenotype than the WT rSARS-CoV-2. Animal infection models will be needed to further characterize the differences in infectivity and pathogenesis of the mutant.
As the pandemic continues to unfold with a rapid increase in cases worldwide, SARS-CoV-2 surveillance necessitates a global, coordinated strategy. Despite limited sample size that was enrolled, our study provides an integrated view of clinical and biological implications of SARS-CoV-2 genetic variants, which may provide valuable clues to the immediate application of these genomic markers for molecular epidemiological investigation. Future longitudinal studies will allow exploration of causal relationships between these variants and SARS-CoV-2 pathogenesis, and such studies could offer important information for effective drug design for SARS-CoV-2.
Limitations of study
Due to the rapid and effective implementation of control measures in China, the number of cases enrolled in this study with both Δ500-532 Nsp1 deletion and detailed clinical records was limited. Although increase in Ct values of qPCR tests was observed with these cases, whether viruses harboring this mutilation generally cause less pathology remains to be further examined in a larger number of clinical cases or in animal models. Since the N-terminal domain is not found binding to 40S, how the deletion affects the functions of Nsp1 leading to lower IFN-I response in the infected cells also requires further characterization.
STAR★methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| RPMI Medium | HyClone | Cat# SH30809.01 |
| DMEM | HyClone | Cat# SH30243.01 |
| Penicillin-Streptomycin (SCU) | Thermo Fisher Scientific | Cat# 15140122-100 mL |
| Penicillin-Streptomycin (UH) | Sigma-Aldrich | Cat# P0781 |
| Penicillin-Streptomycin (LUMC) | Sigma-Aldrich | Cat# P4458-100 ML |
| Fetal Bovine Serum (SCU) | Cell Box | Cat# SAG-01U-02 |
| Fetal Bovine Serum (UH) | GIBCO | Cat# 10270-106 |
| Fetal Bovine Serum (LUMC) | Thermo Fisher Scientific | Cat# A3382001 |
| G418 | TransGen Biotech | Cat# FG401-01 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | Cat# 25300054-100 mL |
| MEM-Eagle-EBSS w/HEPES | Lonza | Cat# BE12-136F |
| MEM NEAA (SCU) | Lonza | Cat# BE13-114E |
| MEM NEAA (UH) | Sigma-Aldrich | Cat# M7145 |
| Eagle’s MEM (UH) | Sigma-Aldrich | Cat# M2279 |
| L-Glutamine | Sigma-Aldrich | Cat# G7513-100 ML |
| Sodium Pyruvate (100 mM) | Thermo Fisher Scientific | Cat# 11360039-100 mL |
| TRIzol™ Reagent | Invitrogen | Cat# 15596026; Cat# 15596018 |
| 1-Bromo-3-chloropropane | Sigma-Aldrich | Cat# B9673 |
| Q5® Hot Start High-Fidelity 2 × Master Mix | New England Biolabs | Cat# M0494L |
| Random hexamers | Invitrogen | Cat# N8080127 |
| dNTPs mix (10 mM each) | Invitrogen | Cat# 18427013 |
| RNaseOUT RNase Inhibitor | Invitrogen | Cat# 10777019 |
| SSIV Reverse Transcriptase | Invitrogen | Cat# 18090200 |
| Nuclease-free water | Invitrogen | Cat# 4387936 |
| Agencourt AMPure XP Beads | Invitrogen | Cat# A63880 |
| Ethanol | Sangon | Cat# A500737 |
| Isopropanol | Sangon | Cat# A507048 |
| NEB Blunt/TA Ligase Master Mix | New England Biolabs | Cat# M0367L |
| NEBNext End repair / dA-tailing Module | New England Biolabs | Cat# E7546 |
| 2 × SYBR Green Master Mix | Bio-Rad Laboratories | Cat# 1708884 |
| R1 RNA Cartridge | BiOptic | Cat# C105210 |
| S1 DNA Cartridge | BiOptic | Cat# C105202 |
| ANTI-FLAG® M2 Affinity Gel | Millipore | Cat# A2220 |
| Cell lysis buffer | Cell Signaling Technology | Cat# 9803 |
| Protein Loading Dye | Sangon | Cat# C508320 |
| Protease Inhibitor Mini Tablets, EDTA-free | Thermo Fisher Scientific | Cat# A32955 |
| Clarity Max Western ECL Substrate, 100 ml | Bio-Rad Laboratories | Cat# 1705062 |
| Recombinant Human IFN-β (SCU) | Peprotech | Cat# 300-02BC |
| Human IFN-beta 1a (LUMC) | Bio-techne | Cat# 11410-2 |
| Lipofectamine™ 2000 Transfection Reagent | Invitrogen | Cat# 11668019 |
| MyTaq Red DNA polymerase | Bioline | Cat# BIO-21110 |
| TaqMan™ Universal Master Mix II, no UNG | Thermo Fisher Scientific | Cat# 4440040 |
| TriPure Isolation Reagent | Roche | Cat# 11667165001 |
| TaqMan™ Fast Virus 1-Step Master Mix | Thermo Fisher Scientific | Cat# 4444436 |
| Antibodies | ||
| Anti-S6 monoclonal antibodies | Cell Signaling Technology | Cat# 2217; RRID: AB_331355 |
| Anti-FLAG antibodies | Cell Signaling Technology | Cat# 14793; RRID: AB_2572291 |
| Oligonucleotides | ||
| Primers used in this study | See Table S4 | N/A |
| Commercial kits and assays | ||
| Qubit dsDNA HS Assay Kit | Invitrogen | Cat# Q32854 |
| Native Barcoding Expansion 1-12 | Oxford Nanopore Technologies | Cat# EXP-NBD104 |
| Native Barcoding Expansion 13-24 | Oxford Nanopore Technologies | Cat# EXP-NBD114 |
| Ligation Sequencing Kit | Oxford Nanopore Technologies | Cat# SQK-LSK109 |
| Direct cDNA Sequencing kit | Oxford Nanopore Technologies | Cat# SQK-DCS109 |
| RevertAid RT Reverse Transcription Kit | Thermo Fisher Scientific | Cat# K1691 |
| Human ELISA Kit for Interferon Beta (IFN-β) (SCU) | Cloud-Clone Corp. | Cat# SEA222Hu |
| Human IFN-beta Quantikine ELISA Kit (LUMC) | Bio-techne | Cat# DIFNB0 |
| miRNeasy Micro Kit | QIAGEN | Cat# 217084 |
| Dual Luciferase Reporter Assay Kit | Vazyme | Cat# DL101-01 |
| iTaqTM Universal SYBR® Green One-Step Kit | Bio-Rad Laboratories | Cat# 172-5151 |
| mMESSAGE mMACHINE T7 Kit 25 Rxn | Ambion | Cat# AM1344 |
| Amaxa Solution T kit | Lonza | Cat# VVCA-1002 |
| EXPRESS One-Step Superscript™ qRT-PCR Kit, universal | Thermo Fisher Scientific | Cat# 11781200 |
| 18 s TaqMan™ Gene Expression Assay ID: Hs99999901_s1 | Thermo Fisher Scientific | Cat# 4331182 |
| IFNB TaqMan™ Gene Expression Assay ID: Hs01077958_s1 | Thermo Fisher Scientific | Cat# 4448489 |
| ACTB TaqMan™ Gene Expression Assay ID: Hs01060665_g1 | Thermo Fisher Scientific | Cat# 4331182 |
| GAPDH TaqMan™ Gene Expression Assay ID: Hs02786624_g1 | Thermo Fisher Scientific | Cat# 4331182 |
| Software and algorithms | ||
| BWA (version v0.7.17-r1188) | (Li, 2013) | https://arxiv.org/abs/1303.3997 |
| STAR (version 2.7.1a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| SeqTK (version 1.3-r114-dirty) | N/A | https://github.com/lh3/seqtk |
| Guppy (version v3.4.5) | Nanoporetech | https://denbi-nanopore-training-course.readthedocs.io/en/latest/basecalling/basecalling.html |
| qcat (version v1.1.0) | Nanoporetech | https://github.com/nanoporetech/qcat |
| MAFFT (version v7.4) | (Katoh et al., 2002) | https://mafft.cbrc.jp/alignment/software/ |
| PhyML (version v3.3) | (Guindon et al., 2010) | http://www.atgc-montpellier.fr/phyml/ |
| BEAST (version v1.10.4) | (Drummond and Rambaut, 2007) | http://beast.community |
| DnaSP | (Rozas et al., 2017) | https://bioinformaticshome.com/tools/descriptions/DnaSP.html |
| PopART | (Leigh et al., 2015) | http://popart.otago.ac.nz/index.shtml |
| Tracer (version v1.7.1) | (Rambaut et al., 2018) | http://tree.bio.ed.ac.uk/software/tracer/ |
| htseq (version 0.11.2) | (Anders et al., 2015) | https://htseq.readthedocs.io |
| ARTIC (version SOP-v1.1.0) | N/A | https://artic.network/ |
| R (version 3.6.0) | N/A | https://www.r-project.org |
| msa (version 1.16.0) | (Bodenhofer et al., 2015) | http://www.bioconductor.org/packages/release/bioc/html/msa.html |
| ComplexHeatmap (version 2.0.0) | (Gu et al., 2016) | https://jokergoo.github.io/ComplexHeatmap-reference/book/ |
| caret (version 6.0-84) | N/A | https://cran.r-project.org/web/packages/caret/index.html |
| fgsea (version 1.10.0) | (Korotkevich et al., 2019) | http://www.bioconductor.org/packages/release/bioc/html/fgsea.html |
| clusterProfiler (version 3.10.1) | (Yu et al., 2012) | http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| bestNormalize (version 1.5.0) | (Peterson and Cavanaugh, 2020) | https://cran.r-project.org/web/packages/bestNormalize/index.html |
| DESeq2 (version 1.22.2) | (Love et al., 2014) | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| vcfR (version 1.10.0) | (Knaus and Grünwald, 2017) | https://cran.r-project.org/web/packages/vcfR/index.html |
| gggenes (version 0.4.0) | N/A | https://cran.r-project.org/web/packages/gggenes/index.html |
| Deposited data | ||
| GISAID | N/A | EPI_ISL_451312 – EPI_ISL_451399; EPI_ISL_454904 – EPI_ISL_455014; EPI_ISL_455363 – EPI_ISL_455411 |
| phylogeny | N/A | http://chenlulab.com/ncov |
| RNA-seq NCBI BioProject | N/A | PRJNA634718 |
Resource availability
Lead contact
Further information and requests for data, resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lu Chen (luchen@scu.edu.cn).
Materials availability
All constructs and plasmids generated in this study will be made available on request sent to the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The genome sequences were deposited to GISAID (EPI_ISL_451312 – EPI_ISL_451399; EPI_ISL_454904 – EPI_ISL_455014; EPI_ISL_455363 – EPI_ISL_455411) and the phylogeny results are accessible via web address http://chenlulab.com/ncov. The RNA-seq data using nanopore were deposited to NCBI BioProject (PRJNA634718). The codes for variant fraction calculation, clinical trait normalization, association study and ranked enrichment analysis are available at https://github.com/LuChenLab/COVID-19.git. Additional Supplemental Items are available from Mendeley Data at https://doi.org/10.17632/w75cdpknmc.1.
Experimental model and subject details
Ethics approval and collaborating institutes
This study was approved by The Biomedical Research Ethics Committee of West China Hospital and the document IDs are NO.2020 (100), NO.2020 (193) and NO.2020 (267). These ethical statements were approved by all collaborating institutions listed below. Written consents were obtained from patients or their guardian(s). Samples were obtained for routine diagnostic purposes, and the residual clinical diagnostic samples were provided for research and surveillance purposes.
Collaborating institutes include 19 hospitals from Sichuan and Wuhan Chain Medical Labs, an Independent Diagnostic Testing Facility (IDTF) from Wuhan, Hubei. The involved hospitals in Sichuan are as follows: West China Hospital, West China Second University Hospital, Public Health Clinical Center of Chengdu and Jianyang people’s Hospital in Chengdu, Dazhou Central Hospital in Dazhou, Deyang people’s Hospital in Deyang, Yuechi people’s Hospital, Wusheng County People’s Hospital and Guang’an people’s Hospital in Guang’an, Daofu County People’s Hospital and People’s Hospital of Ganzi Tibetan Autonomous Prefecture in Ganzi Tibetan Autonomous Prefecture, Mianyang 404 hospital in Mianyang, Luzhou infectious diseases hospital in Luzhou, Nanchong Central Hospital in Nanchong, Neijiang Second People’s Hospital in Neijiang, Yibin Second People’s Hospital in Yibin, Suining Central Hospital in Suining, Ziyang first people’s Hospital in Ziyang, Ya’an people’s Hospital in Ya’an.
Due to the restrictions during the COVID-19 outbreak period, the samples from COVID-19 patients from 18 hospitals were transported to West China Hospital for further processing. The transportation was approved by the Sichuan Health Committee and complying with National regulations in China and Guidelines for the Safe Transport of Infectious Substances and Diagnostic Specimens by the WHO.
Cell lines used in this study
We used the following cell lines in this study: (a) HEK293T, a female human embryonic kidney cell line; (b) A549, a male human lung epithelial cell line; (c) Calu-3, a male human lung epithelial cell line; (d) Vero E6, a female African green monkey kidney cell line; and (e) BHK-21, a baby hamster kidney cell line. All cell lines were obtained from ATCC.
Cell culture conditions
HEK293T and A549 cells were maintained in DMEM (HyClone) and RPMI medium (HyClone) supplemented with 10% Fetal Bovine Serum (FBS; Cell-Box) and 1% penicillin-streptomycin (Thermo Fisher Scientific).
Calu-3 cells were maintained in Eagle’s minimal essential medium (EMEM; Lonza), further supplemented with 8% FCS (Bodinco), 2 mM L-glutamine, 100IU/mL of penicillin and 100 μg/mL of streptomycin (Sigma-Aldrich), 1 × non-essential amino acids (NEAA; Lonza), 1mM of Sodium Pyruvate (Thermo Fisher). Vero E6 and baby hamster kidney (BHK-21) cells were cultured as described previously (Salgado-Benvindo et al., 2020; Nedialkova et al., 2010). Cell cultures were maintained at 37°C in an atmosphere of 5% CO2 and 95% to 99% humidity.
SARS-CoV-2 viral infection conditions
All experiments involving infectious SARS-CoV-2 virus were performed in designated Biosafety Level 3 (BSL-3) workspaces in accordance with institutional guidelines in University of Helsinki (UH) or Leiden University Medical Center (LUMC).
Method details
Sample collection and related clinical records
Biological sampling
Throat swabs, sputum, anal swabs and blood specimens were routinely collected from suspected and confirmed COVID-19 patients for the presence of SARS-CoV-2 virus RNA at the hospitals and IDTF. Specimens from the same patient were analyzed for real-time surveillance purposes. Two cohorts of patients were enrolled in this study: a discovery cohort in Sichuan including 141 samples from 88 patients in Sichuan which were processed for Nanopore sequencing or Illumina sequencing, and a validation cohort of 169 samples from 160 patients in Wuhan, which were used for Nanopore sequencing.
Viral RNA isolation, sample processing and quantitative real-time PCR test for COIVD-19
After collection, the samples were placed into a tube containing 2 mL viral RNA preservation solution. Total RNA was extracted using QIAamp viral RNA mini kit (QIAGEN, Germany) following the manufacturer’s protocol.
Real-time RT-PCR was performed by amplifying two target genes, including open reading frame 1ab (ORF1ab) and nucleocapsid protein (N) using RT-PCR kit (Sansure Biotech Inc, China) on a Real-time PCR thermal cycler (ABI 7500 system, Applied Biosystems instruments, USA).
Clinical classification of COVID-19 cases
The severity status of confirmed COVID-19 cases was categorized as mild, moderate, severe, and critical according to the Diagnosis and Treatment Scheme for COVID-19 released by the National Health Commission of China (Version 7). The detailed characteristics of the four categories have been described in a previous study(Lu et al., 2020a). In this study, we classified mild and moderate cases to the non-severe group, and severe and critical cases to the severe group. Ten out of the 88 sequenced samples belonged to the severe group.
Clinical data collection
The following clinical traits were collected in this study (see also Table S8).
Complete blood count (CBC). blood was collected in tubes with EDTA anticoagulant (BD). Classification of blood composition was performed using SYSMEXXN-10, Sysmex, Japan instrument. The chromogenic substrate method is used to detect antithrombin III.
Biochemical examination. blood was collected in tubes without anticoagulants (BD). Biochemical examination was performed using Cobas c702, Roche, Germany instrument by UV spectrophotometry.
Coagulation test. blood was collected in tubes with Sodium citrate (BD). Automatic coagulation analysis was carried out by coagulation method, hair color substrate method or immunity method using SYSMEXCS-5100, Sysmex, Japan including TT (Thrombin time, s), APTT (thromboplastin time, s), PT (prothrombin time, s), FIB (fibrinogen, g/L), FDP (fibrin degradation products, μg/L), DD (D-dimer, mg/L).
Circulating levels of inflammatory cytokines and chemokines were determined, including IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, IL-17A, IL-17E, IL-17F, IL-21, IL-22, IL-23, IL-33, TNF-α, IP-10, IFN-γ, IFN-α2, IFN-β in the serum were measured using a multiplexed flow cytometric assay on a Luminex system (MAGPIX with xPONENT) according to the instructions of the manufacturer (MILLIPLEX Analyst 5.1). Samples were measured in duplicates. Based on the standard curves, the coefficient of variation (CV) was calculated and did not exceed 20%.
Chest imaging. We applied the artificial intelligent system, named as AI-assisted Chest CT Diagnostic System for COVID-19 Pneumonia, and used the quantitative measurements and prognosis of COVID-19(Zhang et al., 2020a).
SARS-CoV-2 genome sequencing
Design and validation of the sequencing primer scheme for Nanopore sequencing
Multiplex-PCR combined with the nanopore sequencing was used, following the general version 1 protocol as described in (https://artic.network/ncov-2019). For 400 bp amplicons pool, primer set version V1 was published previously (with ∼90 bp overlapping between context amplicons. We noticed non-randomly low coverage in some regions of the genome using this published protocol, potentially due to various amplification efficiency, primer dimer formation or genetic divergence from the reference genome (Lu et al., 2020a). We therefore optimized our diagnostic assays by adding primer sets that creating amplicons of ∼1 kb. This primer set was designed using Primal Scheme (with ∼50 bp overlapping between context amplicons (Table S4). For 400bp amplicons, there were 196 primers divided evenly into 2 pools; whereas for 1kb amplicons, there were 36 primers in Pool 1 and 34 primers in Pool 2. The primer pool stocks were generated by mixing equivalent volume (e.g., 5 μL) of stock primers (100 μM). Odd number primers were mixed as Pool 1 and even number primers as Pool 2. Primers were used at a final concentration of 0.015 μM each.
Sequencing of SARS-CoV-2 genome
Virus genomes were generated by two different approaches, (i) untargeted metagenomic sequence: 11 samples were sequenced using Illumina NextSeq 500. (ii) multiplex PCR approach followed by Nanopore sequencing: MinION library preparation was performed according to “PCR tiling of nCoV virus” on Nanopore Protocol (Quick, 2020). When the same patient was sampled multiple times or was sequencing in two platforms, we only retained the single highest-quality genome per patient to avoid confounding the downstream analyses.
Briefly, for the nanopore sequencing, the multiplex PCR was performed with a mixture of two pooled primer sets generating amplicons of 400 bp and 1 kb using the cDNA reverse transcribed with random primers was used as a template. After 35 rounds of amplification, the PCR products were collected and purified using Ampure XP beads, followed with end-repairing, barcoding and adaptor ligations. Around 50 fmol of final library DNA was loaded onto the MinION (SQK-LSK109). The nanopore sequencing platform takes less than 24 h to run, achieving on average 80.1% coverage of the virus genome with more than 10 reads and mean depth of 0.39 million reads per sample.
Nsp1 overexpression studies
Nsp1 overexpression and qRT-PCR
The open reading frames for SARS-CoV or SARS-CoV-2 Nsp1 were ordered codon optimized and cloned into a mammalian expression plasmid, pcDNA3.1, in frame with a N-terminal FLAG tag. The shorter deletion mutants of Nsp1 were constructed through PCR site-directed mutagenesis. HEK293T and A549 cells were transfected with plasmids expressing Nsp1 variants or GFP control using Lipofectamine 2000 (Invitrogen). Twenty-four h after transfection, cells were harvested or further selected with G418 (Transgen Biotech, China) for another 24 h and then harvested for RNA extraction. Total RNA was isolated using the standard TRIzol (Invitrogen) extraction method. cDNA was obtained using the RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific). qRT-PCR was performed using a Bio-Rad CFX96 real-time PCR apparatus and SYBR green Master mix (Bio-Rad Laboratories). Primer sequences were listed in Table S4. The relative expression levels of each gene (IFNB1, IFNA1, IFNA2) were normalized to GAPDH.
Co-immunoprecipitation
HEK293T cells were harvested at 48 h post-transfection as described above and lysed with Cell Lysis Buffer (Cell Signaling Technology). Cell lysates were used for immunoprecipitation using anti-FLAG M2 affinity gel (Millipore) according to the manufacturer’s instructions. Eluted proteins and Cell lysates (input) were analyzed by 10% SDS-PAGE and immunoblotting using following antibodies: anti-S6 monoclonal antibodies (Cell Signaling Technology, Cat No. 2217) and anti-FLAG antibodies (Cell Signaling Technology, Cat No. 14793). The chemiluminescence signal was detected using a ChemiDOCTMMP Imaging System (Bio-Rad Laboratories).
ELISA
Culture supernatants and whole cell lysate of transfected HEK293T or A549 cells were harvest at 24 h post-transfection. The concentration of IFN-β was measured using human enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions (Cloud-Clone Corp., China). CYTATION 3 Imaging reader (Biotek) was used to read the signals.
Luciferase assay
For reported gene expression, we constructed the dual luciferase plasmids based on pGL4.7 vector (Promega, Cat No. E6881). IFNB1 or Interferon Stimulated Response Element (ISRE) promoter sequence were cloned into the luciferase reporter plasmids. For IFNB1 promoter activity, HEK293T cells were co-transfected with reporter plasmids and GFP or Nsp1 variants plasmids. After 48 h, luciferase assays were performed according to the manufacturer’s instructions (Vazyme). For ISRE promoter activity, after 24 h post-transfection, human IFN-β (Peprotech) were added into the cell medium, luciferase assays were performed at 24 h post-treatment. Promoter activities were determined as the ratio between a Renilla luciferase that is under the control of IFNB1 promoter or IRSE-promoter and an internal control of Firefly luciferase. All experiments were repeated at least three times.
RNA-sequencing
HEK293T and A549 cells were harvested at 24 h post-transfection as described. Total RNA extraction was using miRNeasy Micro Kit (QIAGEN, German) according to the manufacture’s instruction. The concentration of RNA was measured by Qubit 3 (Invitrogen, USA), and RNA integrity was determined using Qsep1 (BiOptic, China) capillary gel electrophoresis (CGE) with an R1 RNA cartridge or Bioanalyzer 2100 system (Agilent Technologies, CA, USA) with RNA Nano 6000 Assay Kit.
Library preparation and sequencing were performed in Novogene Corporation. Ribosomal RNA was removed using Epicenter Ribo-zero rRNA Removal Kit (Epicenter, USA), and rRNA-free residue was cleaned up by ethanol precipitation. Sequencing libraries were then generated using the rRNA-depleted RNA by NEBNext® Ultra Directional RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations. The library was sequenced on an HiSeq2500 (Illumina, USA) with 150 bp paired-end reads.
Viral infection studies
Reverse genetics
Deletion of 11aa (Δ500-532) in SARS-CoV-2 Nsp1 was engineered in a bacterial artificial chromosome (BAC) vector containing the full-length cDNA copy of BetaCoV/Wuhan/IVDC-HB-01/2019 strain (by two-step en passant recombineering in Escherichia coli (Tischer et al., 2010). Two BACs were isolated independently and used for in vitro transcription and virus launching as described previously(Ogando et al., 2020). The presence of deletion was confirmed by Sanger sequencing of Nsp1-coding region and by PCR using primers amplifying over this region.
Ten micrograms of synthetic mRNA of SARS-CoV-2 N protein were generated as defined before (and co-electroporated with 5 μg of full-length RNA into 5 × 106 BHK-21 cells in Nucleofection T solution (Lonza) using the Amaxa nucleofector 2b (program A-031). Then, transfected cells were mixed with Vero E6 cells at a 1:1 ratio and plated for supernatant harvesting and checking viral replication by immunofluorescence microscopy. Cells were incubated at 37°C and supernatants were collected when full cytopathic effect was observed at 3 days post transfection. Virus was passaged once at low MOI (multiplicity of infection). in Vero E6 in order to obtain working viral stock solutions. Virus progeny titers were determined by plaque assay in Vero E6 cells (and presence of engineered deletion in viral progeny was confirmed by sequencing.
rSARS-CoV-2 infection and qRT-PCR
Calu-3 cells were infected with recombinant SARS-CoV-2 (rSARS-CoV-2) wild type or deletion mutant at an MOI of 2 during 1 h at 37°C with shaking. After removal of the inoculum, cells were washed twice with PBS and maintained in EMEM with 2% FCS, L-glutamine, NEAA and antibiotics. Supernatants and cells were collected after 1, 6, 24, 48 and 72 h p.i. for qRT-PCR. Extracellular RNA was isolated from 200 μL of supernatant and intracellular RNA was isolated from cell pellets, both using TriPure (Roche) isolation reagent according to the manufacturer’s instruction. Equine a rteritis virus (EAV) in AVL lysis buffer (QIAGEN) was spiked into the Tripure reagent as an internal control for RNA isolation and amplification.
Viral genome copies were quantified by TaqMan multiplex real-time PCR using TaqMan Fast Virus 1-step master mix (Thermo Fisher Scientific) and a CFX384 TouchTM 802 Real Time PCR detection system (BioRad). Primers and probes targeting SARS-CoV-2 Nsp12 (RdRp) and E gene were used as described previously(Salgado-Benvindo et al., 2020; Corman et al., 2020). A standard curve of 10-fold serial dilutions of a T7 RNA polymerase-generated in vitro transcript containing the RT-qPCR target sequences was used for absolute quantification. Each sample was analyzed in triplicate. And Two independent experiments were performed for each assay.
Isolation of live viruses
The virus isolates #34 and C1P1 were isolated from nasopharyngeal samples of COVID-19 patients in Calu-1 cells at University of Helsinki (UH). The isolate #34 contains three nucleotide deletion Δ518-520 (GTG) corresponding to the amino acid V85. The isolate FIN-1-CACO was initially isolated in Vero-E6 cells (Haveri et al., 2020) followed by four passages in CACO-2 cells. During the passaging a deletion of nine nucleotides (Δ509-517) corresponding to three amino acids (G82HV84) occurred. The isolate C1P1 has intact (non-deleted, wildtype) genome and was used in the experiments as a control. All virus isolates were passaged once in Vero E6 cells and ultra-centrifugated through 30% sucrose cushion at 141 000 g for 90 min prior to the experiments. The infectivity of the purified virus isolates was measured using plaque titration assay.
Infection of clinical viral isolates and qRT-PCR
Calu-3 cells were seeded at the density of 150 000 cells per well in 24-well plates and maintained in Eagle’s MEM supplemented with non-essential amino acids, 20% fetal bovine serum, L-glutamine, penicillin and streptomycin, until the cell layer was semi-confluent. The cells were washed once with cell culture media and inoculated with a given virus at MOI of 0.2 or 2, followed by 1 h incubation in 36°C in a 5% CO2 atmosphere. Thereafter, the cells were washed once with cell culture medium and growth medium (Eagle’s MEM supplemented with non-essential amino acids, 2% fetal bovine serum, L-glutamine, penicillin and streptomycin) was added to the cultures. The cell culture supernatant and the cells were harvested immediately after infection 1, 24, 48 and 72 h post infection.
Viral loads in cell culture supernatants were quantified using previously described RT-PCR protocol (Corman et al., 2020) using SARS-CoV RdRp specific primers and probe, and SARS-CoV-2 RdRp control transcript was used as a standard.
IFNB1 qRT-PCR
Calu-3 cells infected with rSARS-CoV-2 or clinical viral isolates were collected at multiple time points post infection. RNA was extracted from the samples using Trizol reagent according to the manufacturer’s protocol. The real time PCR was performed using iTaq™ Universal SYBR® Green One-Step Kit on a BioRad CFX384 TouchTM 802 Real Time PCR detection system (LUMC) or TaqMan Gene Expression Assay on an Agilent Stratagene Mx3005P instrument (UH).
IFN-β ELISA
Levels of IFN-β produced during infection were determined using 50 μL of cell supernatant by Immunoassay according to manufacturer-s instruction (Bio-techne). At the end of the assay formaldehyde was added to each well to a final concentration of 3% for inactivation. In order to calculate IFN-β concentrations a standard curve method was used. Dilutions of commercial Human IFN-beta 1a were used on each assay as positive controls. Samples were normalized to background (well with EMEM-2%FCS medium) and absorbance at 450, 540 and 570nm was measured using a monochromatic filter in a multimode plate reader (Envision; Perkin Elmer).
Quantification and statistical analysis
Variant calling, validation and phylogenetic analysis
The calling and fraction calculation of genetic variants
For Illumina sequencing data, we mapped to the short reads to the SARS-CoV-2 genome using bwa v0.7.17-r1188 (Li and Durbin, 2009). The variants were called when a position has a depth > = 10 and different from the reference genome. For Nanopore sequencing data, the ARTIC bioinformatics pipeline for COVID (https://artic.network/ncov-2019) was used to call single nucleotide changes, deletions and insertions relative to the reference sequence. The final consensus genomes were generated for each sample based on the variants called in each position.
The ARTIC only output the variants with high support fraction (minimum SupportFraction of 0.40 in our data), to obtain the exact fraction of each variant from 0 to 1, we processed the raw reads using the Guppy software v3.4.5 (https://nanoporetech.com/nanopore-sequencing-data-analysis), and output basecalled fastq files were concatenated and demultiplexed using qcat v1.1.0 (https://github.com/nanoporetech/qcat). SeqTK (https://github.com/lh3/seqtk) was then used to trim 30 bp from both ends to eliminate primer sequences. The resulting fastq files from Nanopore sequencing were mapped to the SARS-CoV-2 genome using bwa v0.7.17-r1188 (Li and Durbin, 2009). For each base pair of the genome, we further calculated the fraction of evidence supported for seven possible outcomes, that is, A, T, C, G, insertion, deletion and N which means no reads covering this site from aligned bam file. The fraction was then calculated only when a position has more than 10 reads.
Validation of genetic variants
For SNPs and indels identified only in our study but not in public datasets, we designed primers (Table S4; Figure S3A) for PCR validation flanking of six variants (Δ500-532, Δ729-737, C3717T, A16824G, ACC18108AT and Δ18987-18988) using Primer3Plus (Untergasser et al., 2007). PCR reaction was carried out using the cDNA template from patient samples with corresponding variants. The PCR products were visualized using Qsep1 (BiOptic, China) capillary gel electrophoresis (CGE) with an S2 DNA cartridge. Sanger sequencing was carried out to confirm the deletions and SNPs (Figures S3B–S3G). For the 35 recurrent variants, we further identified whether they were also presented in the sequences generated using the Illumina platform (Figure S3H).
Curation of whole-genome sequence datasets
We downloaded the 81391 high quality genome sequences available on GISAID (Global Initiative on Sharing All Influenza Data; https://www.gisaid.org) by 9 November 2020 with acknowledgment to all the contributors. Sequences that are too short or with too many gaps, replicates of the same patient or without sampling date were removed. Reference genome and annotation of SARS-CoV-2 (MN908947) were downloaded from GenBank (www.ncbi.nlm.nih.gov/nuccore/MN908947) and other related viruses were downloaded from GenBank or GISAID.
Phylogenetic analysis
We downloaded 250 sequences representing the global diversity of the virus while minimizing the impact of sampling bias from a phylogenetic of Guangdong province (Lu et al., 2020a). We constructed a maximum likelihood (ML) phylogenetic tree using PhyML (Guindon et al., 2010). Then we checked the consistency between the evolutionary distances of the samples in the ML tree and the actual sampling dates using TempEst (Rambaut et al., 2016), which showed a high correlation (Figure S2C, r = 0.528), indicating the high accuracy of the tMRCA analysis. We performed the phylogenetic analysis using their method with identical parameters in https://github.com/laduplessis/SARS-CoV-2_Guangdong_genomic_epidemiology/ which outputs the maximum clade credibility (MCC) tree and tMRCA (time to the most recent common ancestor) statistics for each taxon set of observed phylogenetic clusters A-D (Table S7) based on Bayesian coalescent tree analysis using BEAST v1.10.4 (Ayres et al., 2012).
Association analysis
Pre-processing and association analysis of 117 clinical traits
All traits were normalized using the following steps: 1) transformation: first all data were transformed with inverse normal transformation using bestNormalize R package; 2) regress out confounding covariates: potential covariates were tested (age, gender) for association with phenotypes using stepwise linear regression using caret R package (https://topepo.github.io/caret/), adjustment for covariates was thus only undertaken given evidence of the association between covariate and phenotype; 3) standardization: traits passed the previous steps and then were standardized with a mean of zero and SD of 1; 4) variant fraction: to account for the genotype uncertainty that might arise from the sequencing, we used genotype frequency for each genetic variant (SNP or deletion), which was expressed on a quantitative scale between 0 and 1, representing the dosage of the virus variants in each case; 5) association: pearson correlation coefficient is calculated between the pre-processed traits and recurrent variants.
Ranked clinical traits enrichment analysis
To identify the potential genetic variants that are related to the severity of COVID-19, we used an enrichment analysis, akin to Gene Set Enrichment Analysis (GSEA) for interpreting gene expression data(Subramanian et al., 2005). The goal of this enrichment analysis is to determine whether members of a trait set (S) that associated with a variant tend to randomly distribute or occur toward the top (or bottom) of the list (L) that are non-severe or severe related. When a trait set related to the phenotypic distinction, severe or non-severe in this case, tends to show the latter distribution. First, we defined two lists (L) of traits: non-severe related traits (n = 8) and severe related traits (n = 11) using the following criteria: a) Nonsevere related traits were selected when a trait has a significantly higher value in non-severe group from the 117 phenotypes (p < 0.05, t test). This includes CD3+, CD3+CD4+, CD3+CD8+, CD8+, LYMPH%, IFN-β, Alb/Glb and Ca; b) Severe related traits were similarly selected when a trait has a significantly higher value in the severe group. This includes CRP, D-dimer, FIB, GLU, NEUT, hs-CRP, Monocyte, Respiratory frequency and Systolic pressure. Elevated LDH and ESR levels were typical phenotypes in severe COVID-19, they were added into severe-related traits, albeit not significant in our test. Second, all clinical traits of each variant are ranked based on the difference between samples with (fraction > 0.2) and without the variant. Finally, we followed GSEA(Subramanian et al., 2005) to calculated an enrichment score (ES) which is the maximum deviation from zero encountered in the random walk, a normalized ES (NES), a p value for the significance level of ES, and adjusted p value was calculated using permutation.
Differential gene expression analysis
Raw reads from HEK293T or A549 RNA-sequencing were processed and mapped to the human genome (hg38) with the gene annotation of Ensembl v93 (ftp.ensembl.org/pub/release-93/) using STAR (version 2.7.1a) (Dobin et al., 2013). Secondary alignments and reads aligned on less than 80% of their length were filtered out. Expression was quantified by the number of reads which mapped on a given gene using htseq version 0.11.2 (Anders et al., 2015). Differentially expressed genes (DEG, adjust.p value < 0.05) were determined using DESeq2 version 1.22.2 (Love et al., 2014). GO biological process and pathway enrichment analyses on DEGs were performed on DEGs using enrichGO from clusterProfiler version 3.10.1 (Yu et al., 2012). IPA (Ingenuity Pathway Analysis) was used to visualize changes in the interferon signaling pathway.
Acknowledgments
Lu Chen was supported by the National Key Research and Development Program of China (2017YFA0106800) and the National Science Fund for Excellent Young Scholars (81722004). This work was supported by the National Key Research and Development Program of China (2016YFA0201400 and 2016YFD0500301), the Sichuan Provincial Program for Diagnostic and Treatment of COVID-19 (2020YFS0004, 2020YFS0002, and 2020YFS0010), the Science and Technology Bureau of Chengdu (2020-YF05-00310-SN), the Special Funds for Prevention and Control of COVID-19 of Sichuan University (2020scunCoV-10007), the special research fund on COVID-19 of West China Hospital Sichuan University (HX-2019-nCoV-004), the Sichuan Science and Technology Program (2020FYS0015 and 2020FYS0017), National Natural Science Foundation of China (82041033), Academy of Finland (329323 and 335858), Finland, Jane and Aatos Erkko Foundation, Finland, Research Funds of University of Helsinki and Helsinki University Hospital, Finland (TYH 2018322), and the European Union Horizon 2020 programme VEO (Versatile emerging infectious disease observatory no. 874735). L. Cheng was supported by the Marie Curie individual fellowship (663830).
We thank Department of Clinical Research Management, West China Biobanks for sample and clinical data collection, and Biomarker Technologies Corporation, Beijing, China for their assistance in sample sequencing. And we kindly acknowledge Volker Thiel and colleagues (Institute of Virology and Immunology, Bern, Switzerland) for sharing their SARS-CoV-2 reverse genetics system, and Sanna Mäki for her assistance in experiments involving clinical viral isolates.
Author contributions
Lu Chen, B.Y., J.G, W.L., and J.-w.L. designed and directed the study. M.W., Y. Zhou, W.L., and B.Y. undertook clinical sample collection and processing and analyses of clinical records. B.D., C.C., Yaoyao Zhang, H.-c.W., and Y. Zhao undertook library construction and sequencing. C.T., R.Z., Yiming Zhang, X.X., D.Z., Y. Li, M.X., J. Song, and L. Cheng performed genetic and bioinformatics analyses. C.T. and Z.L. performed epidemiological analyses. H.-c.W. and C.-l.L. performed the overexpression experiments, and Z.Z., R.Y., D.W., L.Z., S.-s.C., and X.Z. assisted in the sequencing and overexpression experiments. S.K., L.L., O.V., and T.S. analyzed the clinical viral isolates. H.L., L.W., and Y. Luo constructed plasmids encoding SARS-CoV-2 genome. E.J.S. and N.S.O. designed and performed reverse genetics analyses. Lu Chen and J.-w.L. analyzed the bioinformatic and experimental data and wrote the manuscript. Lu Chen, J.-w.L., G.L., J.G., B.D., M.W., C.T., Y. Zhou, L. Cheng, T.C., and N.S.O. edited the manuscript. Other authors were involved in one of the processes of coordination, collection, processing and sequencing of the clinical samples, bioinformatics analysis, and experimental validation. All authors read and approved the contents of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 29, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2021.01.015.
Supplemental information
References
- Almeida M.S., Johnson M.A., Herrmann T., Geralt M., Wüthrich K. Novel beta-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:3151–3161. doi: 10.1128/JVI.01939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayres D.L., Darling A., Zwickl D.J., Beerli P., Holder M.T., Lewis P.O., Huelsenbeck J.P., Ronquist F., Swofford D.L., Cummings M.P. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 2012;61:170–173. doi: 10.1093/sysbio/syr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A.K., Blanco M.R., Bruce E.A., Honson D.D., Chen L.M., Chow A., Bhat P., Ollikainen N., Quinodoz S.A., Loney C. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell. 2020;183:1325–1339.e21. doi: 10.1016/j.cell.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F., Snyder G.A., Giovanetti M., Angeletti S., Gallo R.C., Ciccozzi M., Zella D. Emerging of a SARS-CoV-2 viral strain with a deletion in nsp1. J. Transl. Med. 2020;18:329. doi: 10.1186/s12967-020-02507-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhofer U., Bonatesta E., Horejš-Kainrath C., Hochreiter S. msa: an R package for multiple sequence alignment. Bioinformatics. 2015;31:3997–3999. doi: 10.1093/bioinformatics/btv494. [DOI] [PubMed] [Google Scholar]
- Corman V.M., Landt O., Kaiser M., Molenkamp R., Meijer A., Chu D.K., Bleicker T., Brünink S., Schneider J., Schmidt M.L. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauver J.R., Petrone M.E., Hodcroft E.B., Shioda K., Ehrlich H.Y., Watts A.G., Vogels C.B.F., Brito A.F., Alpert T., Muyombwe A. Coast-to-Coast Spread of SARS-CoV-2 during the Early Epidemic in the United States. Cell. 2020;181:990–996.e5. doi: 10.1016/j.cell.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Haveri A., Smura T., Kuivanen S., Österlund P., Hepojoki J., Ikonen N., Pitkäpaasi M., Blomqvist S., Rönkkö E., Kantele A. Serological and molecular findings during SARS-CoV-2 infection: the first case study in Finland, January to February 2020. Euro Surveill. 2020;25 doi: 10.2807/1560-7917.ES.2020.25.11.2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M.R., Hoque M.N., Rahman M.S., Alam A.S.M.R.U., Akther M., Puspo J.A., Akter S., Sultana M., Crandall K.A., Hossain M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020;10:14004. doi: 10.1038/s41598-020-70812-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus B.J., Grünwald N.J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017;17:44–53. doi: 10.1111/1755-0998.12549. [DOI] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G., Sukhov V., Sergushichev A. Fast gene set enrichment analysis. BioRxiv. 2019 doi: 10.1101/060012. [DOI] [Google Scholar]
- Leigh J.W., Bryant D., Nakagawa S. popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 2015;6:1110–1116. [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013 https://arxiv.org/abs/1303.3997 [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Xu S., Yu M., Wang K., Tao Y., Zhou Y., Shi J., Zhou M., Wu B., Yang Z. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020;146:110–118. doi: 10.1016/j.jaci.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Zhang J., Yang Y., Ma H., Li Z., Zhang J., Cheng J., Zhang X., Zhao Y., Xia Z. The role of interleukin-6 in monitoring severe case of coronavirus disease 2019. EMBO Mol. Med. 2020;12:e12421. doi: 10.15252/emmm.202012421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Yan L.-M., Wan L., Xiang T.-X., Le A., Liu J.-M., Peiris M., Poon L.L.M., Zhang W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020;20:656–657. doi: 10.1016/S1473-3099(20)30232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J., du Plessis L., Liu Z., Hill V., Kang M., Lin H., Sun J., Francois S., Kraemer M.U.G., Faria N.R. Genomic Epidemiology of SARS-CoV-2 in Guangdong Province, China. Cell. 2020;181:997–1003.e9. doi: 10.1016/j.cell.2020.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K., Huang C., Lokugamage K., Kamitani W., Ikegami T., Tseng C.T., Makino S. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 2008;82:4471–4479. doi: 10.1128/JVI.02472-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkova D.D., Gorbalenya A.E., Snijder E.J. Arterivirus Nsp1 modulates the accumulation of minus-strand templates to control the relative abundance of viral mRNAs. PLoS Pathog. 2010;6:e1000772. doi: 10.1371/journal.ppat.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogando N.S., Zevenhoven-Dobbe J.C., van der Meer Y., Bredenbeek P.J., Posthuma C.C., Snijder E.J. The Enzymatic Activity of the nsp14 Exoribonuclease Is Critical for Replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020;94:20. doi: 10.1128/JVI.01246-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R.A., Cavanaugh J.E. Ordered quantile normalization: a semiparametric transformation built for the cross-validation era. J. Appl. Stat. 2020;47:2312–2327. doi: 10.1080/02664763.2019.1630372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J. Protocols.io; 2020. nCoV-2019 sequencing protocol. [Google Scholar]
- Rambaut A., Lam T.T., Max Carvalho L., Pybus O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evol. 2016;2:vew007. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J., Ferrer-Mata A., Sánchez-DelBarrio J.C., Guirao-Rico S., Librado P., Ramos-Onsins S.E., Sánchez-Gracia A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- Salgado-Benvindo C., Thaler M., Tas A., Ogando N.S., Bredenbeek P.J., Ninaber D.K., Wang Y., Hiemstra P.S., Snijder E.J., van Hemert M.J. Suramin Inhibits SARS-CoV-2 Infection in Cell Culture by Interfering with Early Steps of the Replication Cycle. Antimicrob. Agents Chemother. 2020;64:20. doi: 10.1128/AAC.00900-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert K., Karousis E.D., Jomaa A., Scaiola A., Echeverria B., Gurzeler L.A., Leibundgut M., Thiel V., Mühlemann O., Ban N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020;27:959–966. doi: 10.1038/s41594-020-0511-8. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C., Huang Y., Shi F., Tan K., Ma Q., Chen Y., Jiang X., Li X. C-reactive protein correlates with computed tomographic findings and predicts severe COVID-19 early. J. Med. Virol. 2020;92:856–862. doi: 10.1002/jmv.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Wu C., Li X., Song Y., Yao X., Wu X., Duan Y., Zhang H., Wang Y., Qian Z. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020;7:1012–1023. doi: 10.1093/nsr/nwaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi Nhu Thao T., Labroussaa F., Ebert N., V’kovski P., Stalder H., Portmann J., Kelly J., Steiner S., Holwerda M., Kratzel A. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature. 2020;582:561–565. doi: 10.1038/s41586-020-2294-9. [DOI] [PubMed] [Google Scholar]
- Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer B.K., Smith G.A., Osterrieder N. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 2010;634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- Untergasser A., Nijveen H., Rao X., Bisseling T., Geurts R., Leunissen J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71–W74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp L., Acman M., Richard D., Shaw L.P., Ford C.E., Ormond L., Owen C.J., Pang J., Tan C.C.S., Boshier F.A.T. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020;83:104351. doi: 10.1016/j.meegid.2020.104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Peng L., Park J.J., Hu Y., Devarkar S.C., Dong M.B., Shen Q., Wu S., Chen S., Lomakin I.B., Xiong Y. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell. 2020;80:1055–1066.e6. doi: 10.1016/j.molcel.2020.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K., Liu X., Shen J., Li Z., Sang Y., Wu X., Zha Y., Liang W., Wang C., Wang K. Clinically Applicable AI System for Accurate Diagnosis, Quantitative Measurements, and Prognosis of COVID-19 Pneumonia Using Computed Tomography. Cell. 2020;182:1360. doi: 10.1016/j.cell.2020.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Tan Y., Ling Y., Lu G., Liu F., Yi Z., Jia X., Wu M., Shi B., Xu S. Viral and host factors related to the clinical outcome of COVID-19. Nature. 2020;583:437–440. doi: 10.1038/s41586-020-2355-0. [DOI] [PubMed] [Google Scholar]
- Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., Xiang J., Wang Y., Song B., Gu X. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequences were deposited to GISAID (EPI_ISL_451312 – EPI_ISL_451399; EPI_ISL_454904 – EPI_ISL_455014; EPI_ISL_455363 – EPI_ISL_455411) and the phylogeny results are accessible via web address http://chenlulab.com/ncov. The RNA-seq data using nanopore were deposited to NCBI BioProject (PRJNA634718). The codes for variant fraction calculation, clinical trait normalization, association study and ranked enrichment analysis are available at https://github.com/LuChenLab/COVID-19.git. Additional Supplemental Items are available from Mendeley Data at https://doi.org/10.17632/w75cdpknmc.1.