

SUMMARY
The emergence of cancer from diverse normal tissues is thought to be governed by a common set of fundamental processes. However, these processes are not fully defined. Here, we show that forced expression of glucose-6-phosphate dehydrogenase (G6PD) affords immortalized mouse and human cells anchorage-independent growth in vitro and tumorigenicity in animals. Mechanistically, G6PD augments NADPH levels by simulating NAD+ kinase-mediated biosynthesis of NADP+ besides converting NADP+ to NADPH, bolstering antioxidant defense. G6PD also increases intracellular nucleotide precursor levels through the production of ribose and NADPH, promoting cell proliferation. Supplementation of antioxidants or nucleosides suffices to convert mouse and human cells into a tumorigenic state in vitro and in animals, and supplementation of both is required when their overlapping metabolic consequences are minimized. These results suggest that normal cells have a limited capacity for redox balance and nucleotide synthesis, and overcoming this limit might represent a key aspect of oncogenic transformation.
Keywords: Oncogenic transformation, cancer metabolism, pentose phosphate pathway, G6PD, NAD kinase, NADPH, redox regulation, nucleotide synthesis
Graphical Abstract
In Brief
The basic elements required for oncogenic transformation remain unclear. By analyzing glucose-6-phosphate dehydrogenase (G6PD)-mediated oncogenic transformation, Zhang et al. show that the upregulation of antioxidant defense and nucleotide production suffice to transform murine and human cells. Therefore, oncogenic transformation may involve overcoming a limited redox balance capacity and nucleotide precursor availability.
INTRODUCTION
Cancer encompasses over 100 diseases that arise in various cell types and tissues of the human body via a microevolution process, during which normal cells acquire a number of heritable genetic and epigenetic alterations through largely random events. Despite this extraordinary complexity and diversity, the pathogenesis of cancer has been rationalized to represent a common set of biological and biochemical principles. However, these principles are incompletely understood (Hanahan and Weinberg, 2011; Vogelstein and Kinzler, 2004). Experimental conversion of normal rodent and human cells to a malignant state has been instrumental in our understanding of tumorigenesis (Hahn and Weinberg, 2002). This approach has revealed two fundamental steps: immortalization and transformation (Hahn and Weinberg, 2002). The former involves inactivation of cell cycle inhibitors (e.g., Rb and p53) and stabilization of the telomere, affording normal cells unlimited replicative potential. The latter can be effectuated by a mutated proto-oncogene or tumor suppressor, which is thought to provide a constitutive mitogenic signal. Since the discovery of an oncogenic HRAS allele in the early 1980s (Reddy et al., 1982; Tabin et al., 1982), a plethora of cellular proto-oncogenes and tumor suppressors have been identified (Vogelstein et al., 2013). However, as most of these cancer-critical genes encode cell surface receptors, intracellular signaling components, or transcription factors, their mutations often act pleiotropically to elicit a multiplicity of changes in cell physiology. As such, the basic elements of oncogenic transformation remains unclear.
A widespread trait of tumor cells, which is brought about directly or indirectly by oncogenic mutations, is the reprogramming of cellular metabolism (Cairns et al., 2011; DeBerardinis and Chandel, 2016; Pavlova and Thompson, 2016; Vander Heiden et al., 2009; Vander Heiden and DeBerardinis, 2017; Zhu and Thompson, 2019). A prominent feature of this reprogramming, as observed nearly a century ago, is a marked increase in the consumption of glucose and the preferential conversion of glucose to lactate, even when tumor cells are exposed to ambient oxygen (aerobic glycolysis or the Warburg effect). Tumor cells also display alterations in the uptake and utilization of many other nutrients including glutamine, serine, glycine, branched-chain amino acids, acetate, fatty acids, proteins, etc. Some alterations are inconsequential; others support, although not directly contribute to, tumorigenesis; and a few may be regarded as transforming (Vander Heiden and DeBerardinis, 2017). The latter include mutations in genes that encode tricarboxylic acid cycle (TCA)-associated enzymes isocitrate dehydrogenase, succinate dehydrogenase, and fumarate hydratase. Nevertheless, these mutations result in the production of oncometabolites including (D)-2-hydroxyglutarate, succinate, or fumarate, all of which impact activities of α-ketoglutarate-dependent dioxygenases leading to changes in epigenome and cellular signaling (Kaelin and McKnight, 2013). Moreover, these mutations alone do not appear to suffice for oncogenic transformation. Therefore, it remains an open question which metabolic activity per se, if any, can be a transforming event.
Of note, upregulation of a few wild-type metabolic enzymes, which likely elicits a relatively narrow range of biochemical outcomes, has been reported to transform murine cells (Auvinen et al., 1992; Kuo et al., 2000; Zhang et al., 2012). Among them is glucose-6-phosphate dehydrogenase (G6PD) (Kuo et al., 2000), the committed enzyme of the oxidative branch of the pentose phosphate pathway (PPP) (Figure S1A) (Stincone et al., 2015). G6PD is regulated by various oncogenes and tumor suppressors including ATM, NRF2, mTORC1, TAp73, and p53 (Cosentino et al., 2011; Du et al., 2013; Duvel et al., 2010; Jiang et al., 2011; Jiang et al., 2013b; Mitsuishi et al., 2012) and is implicated in the pathogenesis of human cancers (Patra and Hay, 2014; Stincone et al., 2015). Here, we set out to characterize the role of G6PD in oncogenic transformation and investigate the underlying mechanism. We find that G6PD can transform immortalized human fibroblastic and epithelial cells as well as murine cells, affording these cells anchorage-independent growth and rendering them tumorigenic in animals. In addition to converting NADP+ to NADPH, G6PD stimulates de novo NADP+ biosynthesis, thereby upregulating redox balance capacity. G6PD also promotes nucleotide synthesis. Moreover, supplementing immortalized mouse and human cells with antioxidants and nucleosides suffices to convert them to a tumorigenic state. These findings define metabolic activities that can drive malignant transformation.
RESULTS
G6PD mediates oncogenic transformation of immortalized murine and human cells
To characterize G6PD-mediated transformation, we expressed human G6PD in the spontaneously immortalized murine fibroblast NIH3T3 cells via retroviral transduction and generated polyclonal, mass-infected cell populations (3T3/G6PD cells) (Figure 1A). In parallel, we infected NIH3T3 cells with retroviruses encoding the oncogene H-RasV12, an enzymatically inactive G6PD with mutations in G6P- and NADP+-binding sites (G6PDm1), or only the drug selection gene (Figure 1A). Relative to control cells expressing only the drug selection gene, 3T3/G6PD cells, which displayed ~5-fold higher G6PD activity, proliferated noticeably faster on adherent plates, akin to 3T3/H-RasV12 cells (Figure 1B). By contrast, 3T3/G6PDm1 cells, which showed no increase in G6PD activity (Figure 1A), grew similarly to control cells (Figure 1B).
Figure 1. G6PD mediates oncogenic transformation of immortalized murine and human cells in vitro.
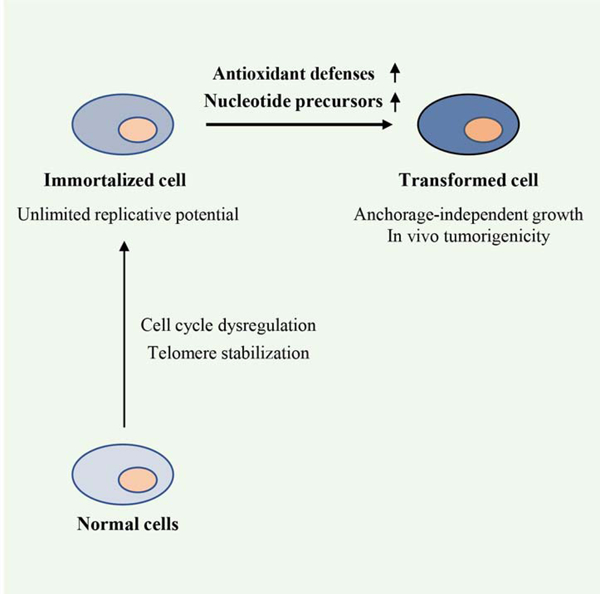
(A–D) NIH3T3 cells expressing only the drug selection gene (Ctrl), H-RasV12, G6PD, or G6PDm1 were assayed for G6PD enzymatic activity (A, top), protein expression (A, bottom), adherent proliferation (B), and soft agar colony formation with colony numbers (C) and representative colony images (D) shown.
(E–H) BJ (E and F) and PHMLEB (G and H) cells harboring control vector, H-RasV12, G6PD, or G6PDm1 were tested for protein expression and G6PD enzymatic activity (E and G) and soft agar colony formation (F and H).
(I and J) PHMLEB cells, and PHMLEB/G6PD cells previously un-grown (initial or I) or grown (PG) in soft agar were assayed for G6PD protein levels and enzymatic activity (I), and (re)tested for soft agar colony formation (J).
Data are means ± SD of representative result (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
The ability to survive and grow without anchoring to extracellular matrix is a hallmark of the tumorigenic state (Freedman and Shin, 1974). As expected, 3T3/H-RasV12 cells readily generated colonies in soft-agar medium, while control NIH3T3 cells yielded virtually no colonies (Figures 1C, 1D, and S1B). Similar to H-RasV12 and consistent with a previous report (Kuo et al., 2000), G6PD permitted NIH3T3 cells to grow in soft agar medium, generating colonies that were numerous and relatively large in size (Figures 1C, 1D, and S1B). By contrast, 3T3/G6PDm1 cells yielded colonies that were substantially less and smaller (Figures 1C, 1D, and S1B). These results indicate that G6PD enzymatic activity confers on immortalized murine cells anchorage-independent growth.
Compared to murine cells, human cells are more refractory to oncogenic transformation (Hahn and Weinberg, 2002; Schinzel and Hahn, 2008). To evaluate the effect of G6PD on human cells, we generated immortalized human foreskin fibroblast BJ cells by serially introducing SV40 T antigens and the human telomerase catalytic subunit hTERT (Hahn et al., 1999) (Figure S1C). Ectopic expression of G6PD in immortalized BJ cells (hereafter referred to as BJ cells), which increased total G6PD activity by ~2-folds (Figure 1E), afforded these cells anchorage-independent growth (Figures 1F and S1D). By contrast, G6PDm1 showed no transforming activity.
To extend this analysis to other cell types, we used human mammary epithelial cells (HMECs) that were immortalized with SV40 T antigens and hTERT (PHMLEB cells) (Elenbaas et al., 2001). Forced expression of G6PD, but not G6PDm1, increased overall G6PD activity and adherent proliferation of PHMLEB cells (Figures 1G and S1E) and conferred on them anchorage-independent growth (Figures 1H and S1F). During soft agar growth, there was a selection for higher G6PD activity. Specifically, compared to initial PHMLEB/G6PD cells, PHMLEB/G6PD cells isolated from soft agar clones contained ~120% and ~80% higher G6PD protein level and activity, respectively (Figure 1I), and, when re-plated in soft agar medium, gave rise to ~200% more colonies (Figures 1J and S1G). These results indicate that elevated G6PD activity permits anchorage-independent growth of human fibroblastic and epithelial cells.
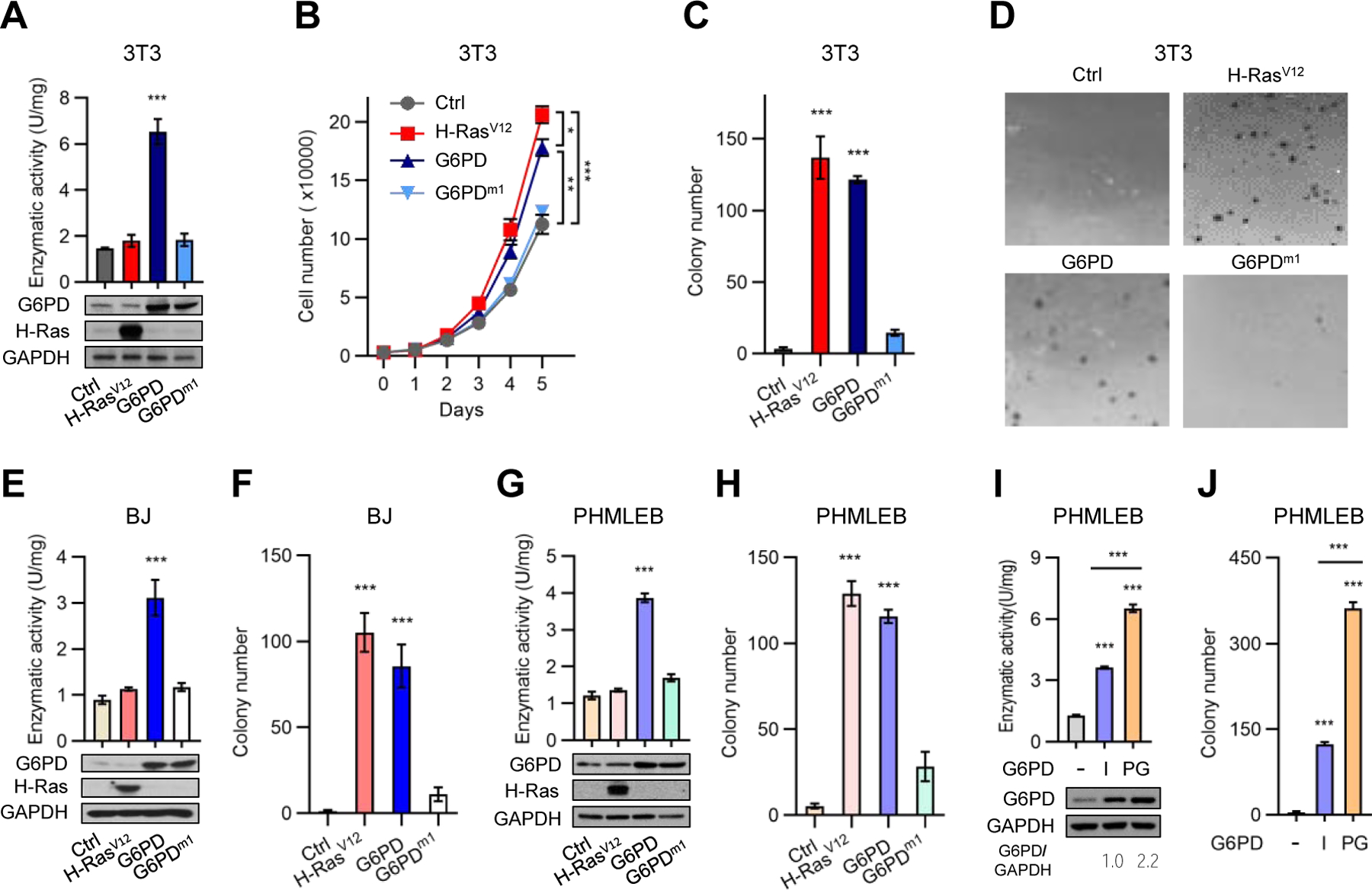
To ascertain G6PD-mediated transformation, we transplanted various NIH3T3 and BJ cells subcutaneously in immunodeficient nude mice. As expected, tumors were detected in all animals transplanted with H-RasV12-expressing NIH3T3 and BJ cells, whereas no tumors were detected in animals transplanted with control cells (Figures 2A–2F). Of note, tumors were observed in all six animals transplanted with 3T3/G6PD in ~3 weeks (Figures 2A–2C) and in all six animals translated with BJ/G6PD cells in ~7 weeks (Figures 2D–2F). These tumors progressed at a slower pace compared to those derived from the corresponding H-RasV12-expressing cells (Figures 2A and 2D) and contained a lower percentage of proliferating cells (Figure 2G). H-RasV12 can activate the angiogenesis program in addition to mitogenic signaling pathways (Serban et al., 2008). Consistently, tumors produced by 3T3/H-RasV12 and BJ/H-RasV12 cells contained more blood vessels (Figure 2H), which might in part account for their faster growth.
Figure 2. G6PD renders immortalized murine and human cells tumorigenic in animals.
(A–F) NIH3T3 (A–C) and BJ (D–F) cells harboring H-RasV12, G6PD, or control vector were subcutaneously injected into athymic nude mice. Shown are growth of tumors over time (A and D) and images (B and E) and weights (C and F) of tumors at the end point of experiment. (G and H) Tumors generated by H-RasV12- and G6PD-expressing cells were analyzed with immunohistochemistry for proliferative cells by Ki-67 staining (G) and for blood density by endothelial marker CD31 staining (H). Shown are representative images of Ki-67 staining (Left) and percentages of proliferating cells (right) (G), and representative images of CD31 staining (Left) and blood vessels density normalized by that of H-Rasv12 (right) (H). Scale bars, 40 μm in (G), 200 μm in (H).
Individual data and means ± SD are shown. ***P < 0.001.
For comparison, we evaluated other PPP enzymes. Upon overexpression, the second NADPH-generating enzyme in the oxidative PPP, 6-phosphogluconate dehydrogenase (6PGD), and non-oxidative PPP enzymes transketolase (TKT) and transaldolase (TAL), did not significantly increase adherent proliferation of PHMELB cells (Figures S1H and S1I). These enzymes also showed minimal or no transforming activity (Figures S1J and S1K). Nevertheless, forced expression of each enzyme moderately enhanced, while knocking down TKT or TAL by small hairpin RNA (shRNA) markedly inhibited, G6PD-mediated transformation (Figures S1L and S1M). Therefore, G6PD is unique among PPP enzymes in its potent transforming capability, which nevertheless depends on an intact PPP.
A survey of public databases showed that G6PD expression was significantly increased in human cancers including liver, colorectal, renal, breast, and skin cancers, correlating with poor prognosis (Figure S2), For example, among patients with liver cancer, the median survival time was ~7 years for those with low G6PD expression, but only ~1 year for those with high G6PD expression (Figure S2B and Table S1). Thus, upregulation of G6PD might contribute to the progression of human tumors.
G6PD enhances redox balance capacity and nucleotide precursor availability
To investigate the metabolic changes that underlie G6PD-mediated transformation, we performed liquid chromatography-mass spectrometry (LC-MS)-based metabolomics on control NIH3T3 and 3T3/G6PD cells. Relative to control cells, 3T3/G6PD cells contained higher abundance of metabolites in the PPP and the nucleotide biosynthesis pathway, as well as phosphoribosylpyrophosphate (PRPP)—which links these two processes (Figures 3A and S3A–S3D). 3T3/G6PD cells also showed a modest but statistically significant increase in glucose (but not glutamine) consumption (Figures 3B and S3E), higher ratios of reduced to oxidized NADP (NADPH/NADP+; Figure S3F) and reduced to oxidized glutathione (GSH/GSSG; Figure S3G), and more robust DNA synthesis (Figure S3H). These results suggest that G6PD overexpression enhances glucose flux through the PPP to promote redox homeostasis and nucleotide biosynthesis.
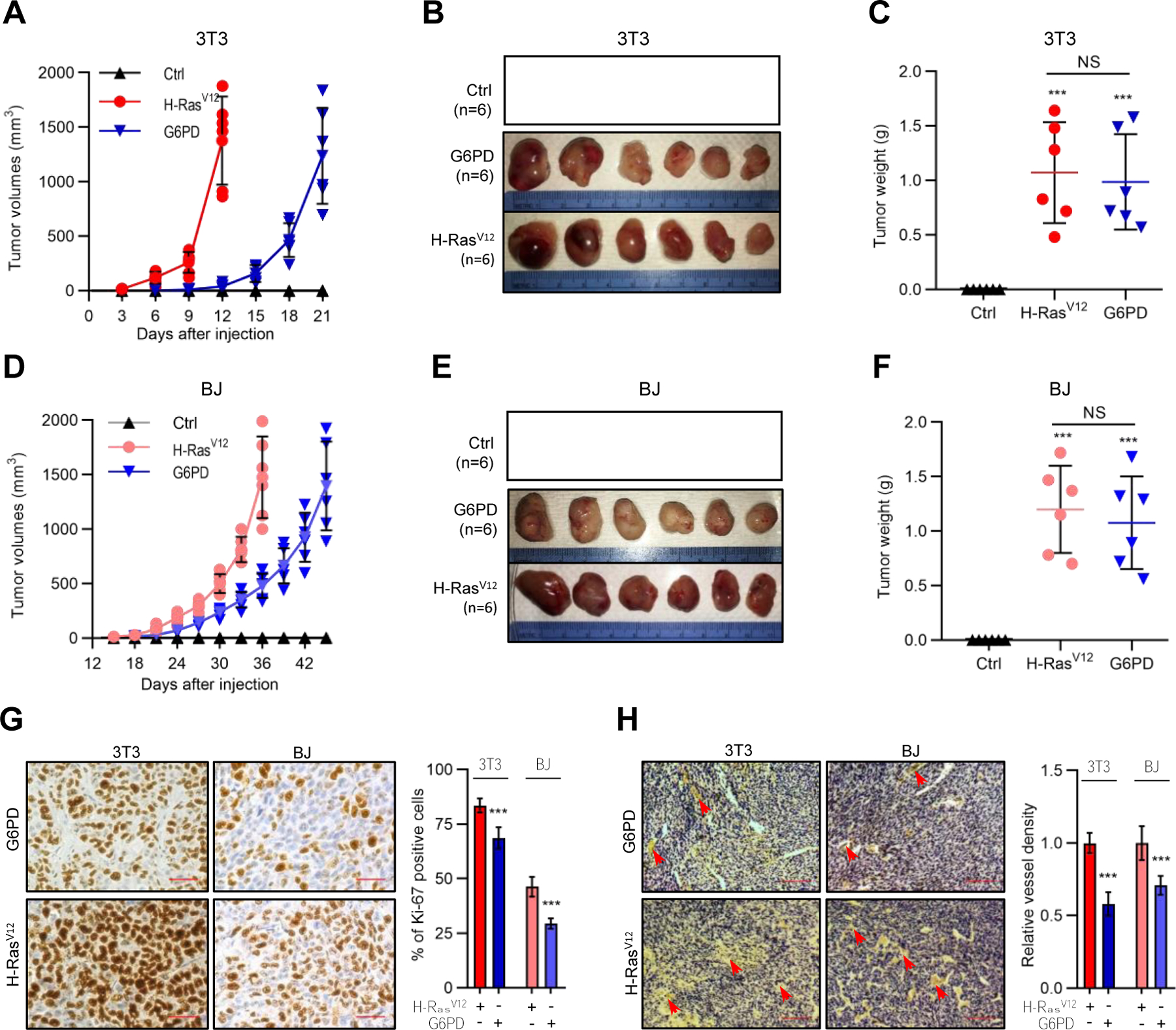
Figure 3. G6PD-induced anchorage-independent growth is associated with enhanced antioxidant capacity and nucleotide precursor availability.
(A) Volcano plot of metabolites detected by metabolomics analysis of 3T3/G6PD cells versus control NIH3T3 cells (see also Figure S3A), with red dots representing metabolites with change ≥ 2 fold and q-value < 0.05 and blue dots representing metabolites with change < 2-fold. m5U, ribothymidine.
(B and C) NIH3T3 and 3T3/G6PD cells grown under matrix-attached or -detached conditions for 4 hr were analyzed for glucose consumption (B, normalized by the number of viable cells) and the NADPH/NADP+ ratio (C).
(D–K) PHMLEB (D–G) and NIH3T3 (H–K) cells harboring H-RasV12, G6PD, G6PDm1, or control vector were cultured under matrix-attached condition (0 hr) or matrix-detached conditions for 6 or 12 hr. Cells were assayed for cell death (D and H), the NADPH/NADP+ ratio (E and I), ROS content (F and J), and ATP levels (G and K, normalized by protein concentration).
(L–N) PHMLEB and PHMLEB/G6PD cells expressing control (Ctrl), TKT, or TALDO1 (TAL) shRNA were cultured under matrix-attached condition (0 hr) or matrix-detached condition for 6 or 12 hr. Cells were assayed for protein expression (L), the NADPH/NADP+ ratio (M), and ROS content (N).
Data are means ± SD of representative result (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Considering that G6PD permits anchorage-independent growth (Figures 1 and S1B–S1G), we also examined how control NIH3T3 and 3T3/G6PD cells might respond differently to matrix detachment in their metabolism. Matrix-detached control cells showed a mild increase in PPP metabolites (~30–60%; Figure S3C) and a modest reduction in the NADPH/NADP+ ratio (~20%; Figure 3C). In contrast, they accumulated PRPP and various nucleotides to high levels (~200–500%; Figure S3D). This was likely resulted from a decline in consumption, as DNA synthesis slowed down dramatically in these cells (Figure S3H). Matrix-detached 3T3/G6PD cells also displayed a reduction in the NADPH/NADP+ ratio, but they still kept it at a level substantially higher than that in NIH3T3 cells (Figure 3C). In addition, matrix-detached 3T3/G6PD cells maintained PPP intermediates, PRPP, and nucleotides at higher abundance (Figures S3C and S3D), while showing ~50% increase in DNA synthesis (Figure S3H). Therefore, G6PD ameliorates oxidative stresses and promotes DNA synthesis under matrix-detachment conditions.
Untransformed fibroblasts and epithelial cells fail to grow in suspension due to detachment-induced apoptosis (anoikis), which is associated with an increase in oxidative stress and a consequential decline in ATP production (Schafer et al., 2009). Suppression of anoikis is crucial for oncogene-induced transformation. G6PD, but not G6PDm1, afforded PHMELB cells significant protection against cell death during prolonged culture in suspension (6–12 hr; Figure 3D). This was accompanied by a higher NADPH/NADP+ ratio, less reactive oxygen species (ROS), and more ATP content (Figures 3E–3G). Similarly, G6PD, but not G6PDm1, promoted survival of, as well as redox balance and ATP production in, NIH3T3 cells (Figures 3H–3K). Of note, the effects of G6PD were nearly as strong as those elicited by H-RasV12 in both PHMELB and NIH3T3 cells (Figures 3D–3K). These effects of G6PD were dependent on nonoxidative PPP enzymes TKT and TAL (Figures 3L–3N), indicating the importance of the flux from ribose-5 phosphate (R5P) back to glycolysis in sustaining oxidative PPP activity. Collectively, these results show that G6PD-mediated tumorigenesis is associated with enhanced antioxidant capacity and nucleotide precursor availability.
G6PD increases total NADP+ and NADPH pools
Intracellular NADPH concentration is substantially higher than that of NADP+ (Ying, 2008). By increasing the conversion of NADP+ to NADPH, forced G6PD expression is expected to decrease NADP+ levels and increase the NADPH/NADP+ ratio, but such changes should only slightly enlarge the NADPH pool. Surprisingly, 3T3/G6PD cells contained more NADP+ than control cells, as shown by the LC-MS-based metabolomics (Figure S3A). Moreover, combined with an extraction method that minimizes the interconversion of NADP+ and NADPH (Lu et al., 2018), an LC-MS analysis revealed that NADPH levels were markedly elevated in 3T3/G6PD cells relative to NIH3T3 cells (~3-fold; Figure 4A). An enzyme-based assay confirmed that G6PD expanded both NADP+ and NADPH pools, while maintaining a higher NADPH/NADP+ ratio in PHMLEB (Figures 4B to 4D) and NIH3T3 (Figures 4E to 4G) cells grown under matrix-attached or -detached conditions. For comparison, we examined the effect of 6PGD. Overexpression of 6PGD increased the NADPH/NADP+ ratio in matrix-attached cells as strongly as G6PD did (Figures 4H and 4I), but it enlarged the NADPH pool only slightly and reduced, rather than increase, the NADP+ pools (Figure 4J). Therefore, expanding the total NADP+ and NADPH pool appears to be a unique property of G6PD. Upon matrix detachment, PHMELB/6GPD cells were unable to maintain NADPH/NADP+ ratio as effectively as PHMELB/G6PD cells did (compared Figures 4I with 4D), suggesting that augmented NADP+ biogenesis contributes to redox balance under stress conditions.
Figure 4. G6PD associates with and stimulates the activity of NADK1 increasing total NADP+ and NADPH pools.
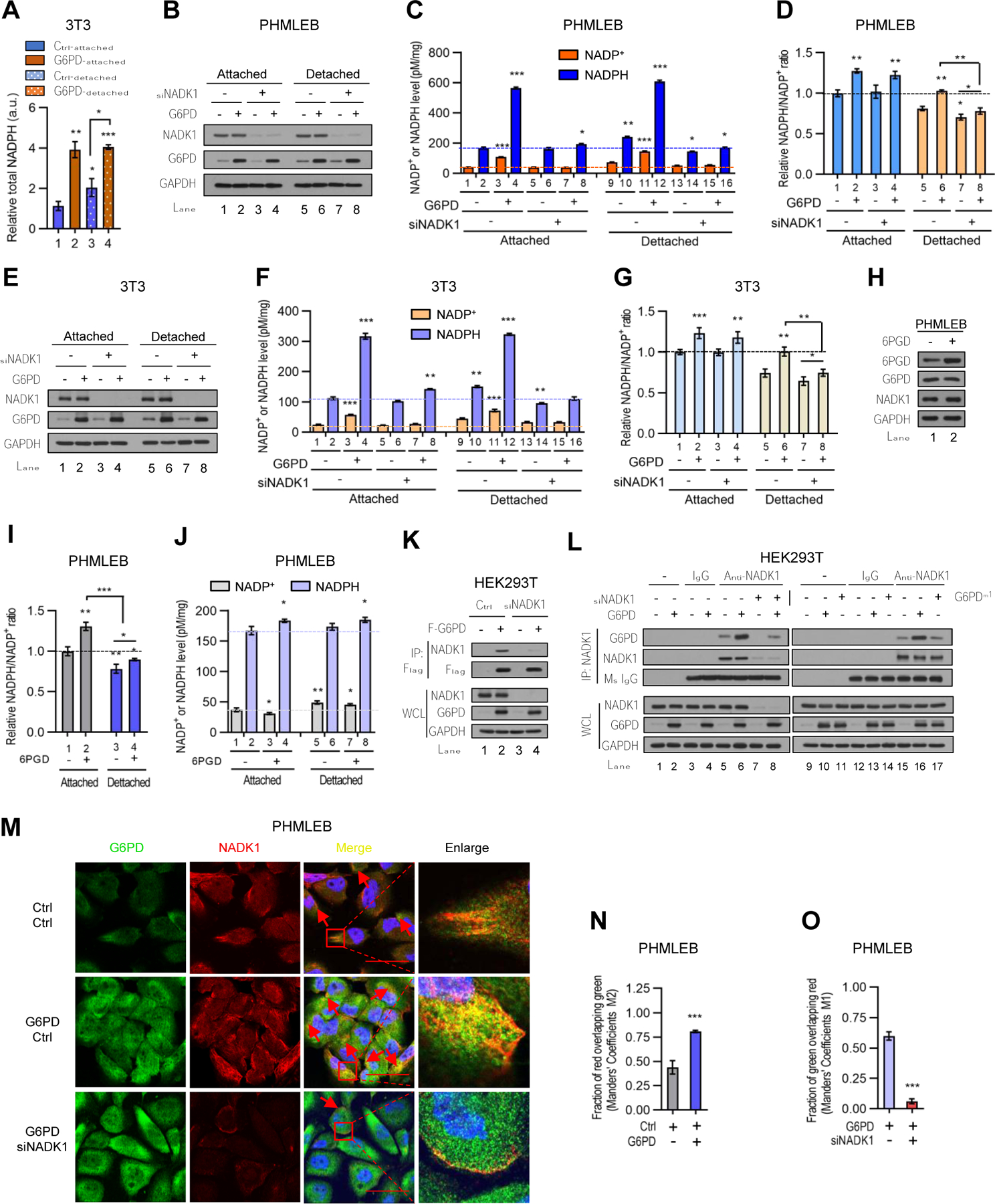
(A) Control and G6PD-overexpressing NIH3T3 cells grown under matrix-attached or -detached conditions for 4 hr were analyzed for total NADPH pool size measured by LC-MS. The data were normalized by protein concentration.
(B–G) Control (−) or G6PD-overexpressing PHMLEB (B–D) and NIH3T3 (E–G) cells were transfected with control (−) or NADK1 siRNA and cultured under matrix-attached or -detached conditions for 6 hr. Cells were assayed for protein expression (B and E), cellular NADPH and NADP+ levels (C and F, normalized by protein concentration), and the NADP+/NADPH ratio (D and G).
(H–J) PHMLEB cells stably transduced with control (−) or 6PGD lentiviral vector were grown under matrix-attached or -detached condition for 6 hr and assayed for protein expression (H), the NADPH/NADP+ ratio (I), and total NADPH and NADP+ levels (J, normalized by protein concentration).
(K and L) Control (−) and NADK1-knockdown HEK293T cells were transfected with control, Flag-G6PD, or Flag-G6PDm1 plasmid as indicated. Cell lysates were incubated without antibody (−), or with control mouse IgG or an anti-Flag (K) or anti-NADK1 (L) antibody.
Immunoprecipitates and whole cell lysates (WCL) were analyzed by Western blot. (M–O) Immunofluorescence analysis of G6PD and NADK1 in PHMLEB cells transfected with control or G6PD plasmid (M and N), or with G6PD plasmid plus control or NADK1 siRNA (M and O), for the localization of G6PD (green) and NADK1 (red). Scale bar, 42 μm. The colocalization of G6PD and NADK1 was quantified by Manders’ M1/M2 coefficient analysis.
Data are means ± SD of representative result (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
G6PD enhances NADP+ synthesis by directly activating NADK1
The most straightforward way to enhance the total NADP+ and NADPH pool is via NAD kinase (NADK), which converts NAD+ to NADP+ (Magni et al., 2006). To evaluate whether G6PD-mediated increase in NADP+ and NADPH abundance is dependent on NADK, we knocked down the cytosolic isoform of NADK (NADK1) using small interfering RNA (siRNA). In NADK1-knockdown PHMLEB cells (Figure 4B), G6PD was no longer able to expand NADP+ and NADPH pools under matrix-attached or -detached conditions (Figure 4C), even though it was still able to augment the NADPH/NADP+ ratio, at least under attached conditions (Figure 4D), indicating retained G6PD catalytic activity. Similarly, in NADK1-knockdown NIH3T3 cells, G6PD was unable to expand NADP+ and NADPH pool sizes, although it still increased the NADPH/NADP+ ratio (Figures 4E–4G).
Their functional connection prompted us to evaluate whether G6PD can bind to and activate NADK1. We observed that both Flag-tagged G6PD and endogenous G6PD interacted with endogenous NADK1 in human embryonic kidney 293T (HEK293T) cells (Figure 4K, and Figure 4L, lanes 5 and 15). Moreover, endogenous G6PD colocalized with NADK1 in the cytoplasm (Figure 4M). Forced G6PD expression increased the interaction of G6PD and NADK1 (Figure 4L, lanes 6 and 16) and augmented their co-localization (Figures 4M and 4N). By contrast, knockdown of NADK1 reduced the G6PD-NADK1 interaction (Figure 4L, lanes 7 and 8) and co-localization (Figures 4M and 4O).
To evaluate the effect of G6PD on the activity of NADK1, we used an in vitro assay in which NADK1-mediated conversion of NAD+ to NADP+ was coupled with G6PD-mediated conversion of NADP+ to NADPH (Hoxhaj et al., 2019). By testing different concentrations of substrates and enzymes, we identified conditions where the enzymatic activity of NADK1, but not G6PD, was rate-limiting for the final production of NADPH (Figures 5A, S4A, and S4B). Under these conditions, even though NADK1 activity was rate limiting, the production of NADPH was accelerated in a dose-dependent manner by recombinant human G6PD protein that was purified from E. coli (Figures 5B and S4C) or HEK293T cells (Figures 5C and S4D–S4F). A pull-down assay confirmed that G6PD bound to NADK1 in the reaction mixture (Figures 5D and 5E).
Figure 5. G6PD counteracts the autoinhibitory N-terminal region of NADK1 and drives the NAD+-NADP+ flux in cells.
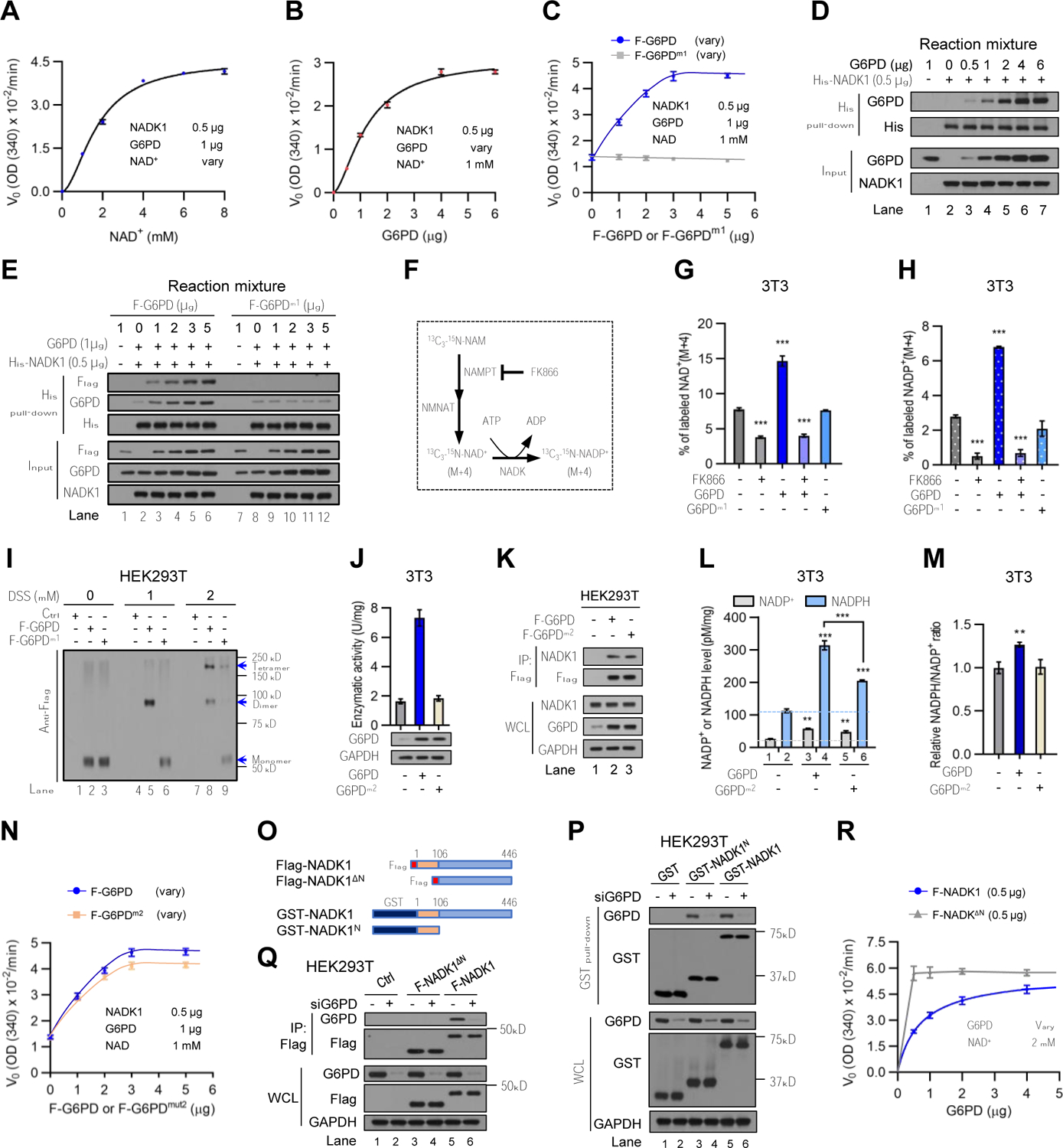
(A and D) Rate (V0) of NAD+ to NADPH conversion in the presence of G6PD purified from E coli (1 μg) and 6xHis-NADK1 (0.5 μg) were measured by OD340 as a function of NAD+ concentration (data from Figure S4A) (A). Interaction of 6xHis-NADK1 with G6PD in the reaction mixtures was analyzed by a pull-down assay with nickel affinity gel (D).
(B) Rate (V0) of NAD+ to NADPH conversion in the presence of NAD+ (1 mM) and 6xHis-NADK1 (0.5 μg) as a function of G6PD concentration (Figure S4C).
(C and E) Rate (V0) of NAD+ to NADPH conversion in the presence of 1 μg of G6PD purified from E. coli, different amounts of Flag-G6PD or Flag-G6PDm1 purified from HEK293T cells, and 6xHis-NADK1 (0.5 μg) (Figures S4E and S4F) (C), and interaction of 6xHis-NADK1 with Flag-G6PD or Flag-G6PDm1 in the reaction mixture (E).
(F) Schematic diagram of isotopic tracing for in vivo NADK function. NAM, nicotinamide; NAMPT, nicotinamide phosphoribosyltransferase; NMNAT, nicotinamide mononucleotide adenylyl transferase.
(G and H) Control, 3T3/G6PD, and 3T3/G6PDm1 cells were treated with vehicle or the NAMPT inhibitor FK866 (0.1 μM) for 16 hr. Cells were incubated for 1 hr with 13C3-15N-nicotinamide, and percentages of M+4 isotopomers of NAD+(G) and NADP+ (H) were analyzed by LC-MS.
(I) HEK293T cells transfected with control, Flag-G6PD, or Flag-G6PDm1 plasmid were treated with the indicated concentrations of DSS. Cell lysates were analyzed by Western blot. G6PD/G6PDm1 monomer, dimer, and tetramer are indicated. Endogenous p53 was used as a control for cross-linking (Figure S4G)
(J, L, and M) NIH3T3 cells harboring G6PD, G6PDm2, or control vector were assayed for G6PD enzymatic activity and protein expression (J), total cellular NADP+ and NADPH levels (L, normalized by protein concentration), and the NADPH/NADP+ ratio (M).
(K) HEK293T cells were transfected with control, Flag-G6PD, or Flag-G6PDm2 plasmid. Cell lysates were immunoprecipitated with an antibody against the Flag epitope. Immunoprecipitates and WCL were analyzed by Western blot.
(N) Rate (V0) of conversion of NAD+ to NADPH in the presence of 1 μg of G6PD purified from E. coli, different amounts of Flag-G6PD or Flag-G6PDm2 purified from HEK293T cells, and 6xHis-NADK1 (0.5 μg) (Figures S4J and S4K).
(O) Schematic diagram of Flag-tagged or GST fusion of NADK1, NADK1∆N, and NADK1N. Amino acid positions are labeled.
(P and Q) HEK293T cells were treated with control (−) or G6PD siRNA and transfected with control, GST-NADK1N, GST-NADK1, Flag-NADK1∆N, or Flag-NADK1 plasmid. Cell lysates were incubated with glutathione agarose (P) or anti-FLAG mAb affinity gel (Q). The precipitated proteins and WCL were analyzed by Western blot.
(R) Rate (V0) of conversion of NAD+ to NADPH in the presence of different amount of G6PD purified from E. coli, 0.5 μg of Flag-NADK1∆N (F-NADK1∆N) or Flag-NADK1 (F-NADK1) purified from HEK293T cells, and 2 mM of NAD+ (Figures S4N and S4O).
Data are means ± SD of representative result (n = 3). **P < 0.01, ***P < 0.001.
To assess the effect of G6PD on NADK function in cells, we performed an isotope tracing experiment to determine the metabolic flux from NAD+ to NADP+. In many cell lines, the NADK substrate NAD+ is made mainly from nicotinamide (NAM) via the salvage pathway (Liu et al., 2018). We cultured cells in medium supplemented with 2,6,7-13C3-(pyridyl-15N)-NAM and measured M+4 mass isotopomers of NAD+ and NADP+ by LC-MS (Figure 5F) (Hoxhaj et al., 2019; Liu et al., 2018). Forced G6PD expression increased the abundance of M+4 NAD+ and NADP+ by ~90% and ~150%, respectively (Figures 5G and 5H). This effect was blocked by FK866, a small molecule inhibitor of the salvage enzyme nicotinamide phosphoribosyltransferase (NAMPT) (Figures 5G and 5H), underscoring the specificity of the assay. Therefore, G6PD activates NADK1, promoting NADP+ biosynthesis.
In contrast to wild-type G6PD, forced expression of G6PDm1 failed to increase the association of G6PD with NADK1 (Figure 4L, lane 17) or stimulate the NAD+-NADP+ flux (Figures 5G and 5H). In vitro, recombinant G6PDm1 protein was unable to interact with and activate NADK1 (Figures 5C, 5E, and S4F). Wild-type G6PD forms a dimer or tetramer (Figures 5I and S4G), which is required for its enzymatic activity (Au et al., 2000). The NADP+-binding site mutated in G6PDm1 (Lys366) is a “structural” site whose binding to NADP+ stabilizes the dimer/tetramer conformation (Au et al., 2000). As expected, G6PDm1 was present largely as monomers in cells (Figures 5I and S4G). These results suggest that the active G6PD dimer/tetramer, but not the inactive monomer, interacts with and activates NADK1.
To ascertain that G6PD activates NADK1 through protein-protein interaction, we used another catalytically-inactive mutant of G6PD in which a G6P-binding reside (Lys171) was replaced with Gln (K171Q or m2) (Chen et al., 2019; Kotaka et al., 2005). As expected, G6PDm2 showed no enzymatic activity (Figure 5J) but retained the dimeric/tetrameric structure (Figure S4H). G6PDm2 interacted with NADK1 as strongly as G6PD did (Figure 5K). Forced expression of G6PDm2 enlarged the total NADP+ and NADPH pool sizes (Figure 5L). This effect was somewhat weaker than that of G6PD, especially for the NADPH pool (Figure 5L), and G6PDm2 did not elevate the NADPH/NADP+ ratio (Figure 5M), presumably because G6PDm2 could not convert NADP+ to NADPH. Nevertheless, purified G6PDm2 protein interacted with, and stimulated the activity of, NADK1 almost as effectively as G6PD did in vitro (Figures 5N, and S4I–S4L). Therefore, G6PDm2 is fully functional as an NADK1 activator.
NADK1 normally exists in an auto-inhibitory conformation, with its N-terminal region restraining the C-terminal kinase domain (Hoxhaj et al., 2019). We generated NADK1 mutations that lacked (∆N) or contained only (N) the N-terminal region (Figure 5O). Reciprocal immunoprecipitation assays showed that G6PD interacted with NADK1N (Figure 5P), but not NADK1∆N (Figure 5Q), indicating that the N-terminal region of NADK1 is both necessary and sufficient for G6PD association. Consistently, the enzymatic activity of NADK1∆N, which was substantially higher than the activity of wild-type NADK1 (Hoxhaj et al., 2019), was not further simulated by G6PD (Figures 5R and S4M–S4O). Therefore, G6PD likely activates NADK1 by counteracting the autoinhibition of the N-terminal region. Collectively, these results indicate that besides its well-established role in reducing NADP+ to NADPH, G6PD activates NADK1 through a direct protein-protein interaction, augmenting NADP+ biosynthesis (Figure S4P).
Exogenous antioxidants and nucleosides suffice for oncogenic transformation
Given that G6PD promotes both antioxidant defense and nucleotide synthesis, we investigated whether one or both outcomes are responsible for its transforming activity. We used ROS scavengers to facilitate redox balance. Of note, when supplemented with N-acetyl cysteine (NAC), PHMLEB cells readily formed colonies in soft agar medium (Figures 6A and S5A). Similarly, supplementation with a cell-permeable derivative of Vitamin E (Trolox) conferred on PHMLEB cells anchorage-independent growth (Figure S5B). To increase intracellular nucleotide levels, we used a mix of four ribonucleosides (A, G, U, and C) and four deoxyribonucleoside (dA, dG, dT, and dC), which can be taken up by cells. The exogenous nucleosides also enabled PHMLEB cells to produce soft agar colonies (Figures 6A and S5A). The combined treatment of NAC and nucleosides, with each at the optimal dose, led to the formation of more colonies (Figures 6A and S5A), an effect comparable to that of G6PD overexpression (Figure 1H). Moreover, NAC, Trolox, and nucleosides individually permitted NIH3T3 cells to generate soft agar colonies (Figures 6B, and S5C–S5F), and NAC and nucleosides in combination further increased the number of colonies (Figures 6B, S5C, and S5G) to levels achieved by G6PD overexpression (compared Figures 6B with 1C).
Figure 6. Supplementation with NAC and nucleosides is sufficient for oncogenic transformation of immortalized cells.
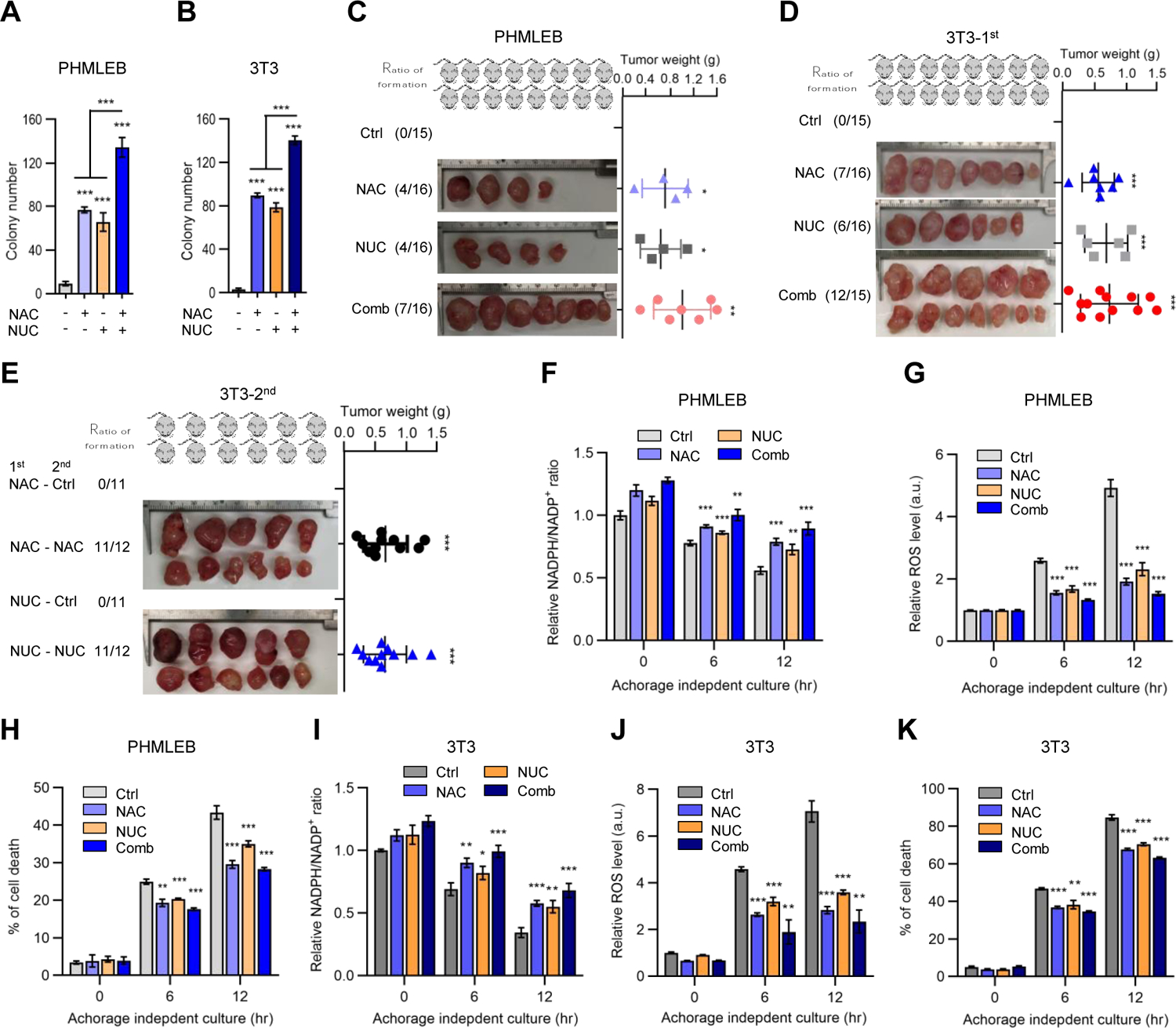
(A and B) Soft agar colonies formed by PHMLEB (A) and NIH3T3 (B) cells treated with vehicle (Ctrl), NAC (0.25 mM), nucleosides (NUC, 20 μg/mL each, 160 μg/mL total), or both NAC and NUC. (B) is replotted from Figures S5D, S5F, and S5G of same conditions.
(C and D) Athymic nude mice were injected with four million of PHMLEB (C) or NIH3T3 (D) cells and supplemented with vehicle, NAC, and/or nucleosides. Shown are frequency of tumor formation (left) and tumor images (middle) and weights (right) at the end of the experiment (see also Figures S6A–S6G).
(E) Frequency of tumor formation (left) and tumor images (middle) and weights (right) at the end of the experiment for 2nd inoculation of four million cells from tumors that were generated with the supplementation of NAC or nucleosides in (D), in presence or absence of the initial treatment (Figures S6L and S6M).
(F–K) PHMLEB (F–H) and NIH3T3 (I–K) cells were cultured under matrix-attached (0 hr) or - detached (6 and 12 hr) conditions in presence of vehicle, NAC (0.25 mM), NUC (160 μg/mL), or both NAC and NUC. Cells were assayed for the NADPH/NADP+ ratio (F and I), ROS levels (G and J), and cell death (H and K).
Data are means ± SD of representative result (n = 3, or as indicated). *P < 0.05, **P < 0.01, ***P < 0.001.
To determine whether NAC and nucleosides enable tumorigenicity in animals, we transplanted athymic nude mice subcutaneously with PHMLEB cells. Mice were fed with drinking water containing NAC, injected intravenously with nucleosides, or both. Strikingly, supplementation of NAC or nucleosides alone led to the generation of tumors at the site of transplantation in 4 out of 16 injected mice, while supplementation of both led to the formation of tumors in 7 out of 16 mice (Figures 6C and S6A–S6C). An RT-PCR analysis for the human GAPDH gene confirmed that these tumors were originated from PHMLEB cells (Figure S6D). By contrast, control mice that were fed with normal drinking water and injected with saline did not produce any detectable tumors (Figure 6C).
In parallel, we transplanted NIH3T3 cells into athymic nude mice and treated these mice with NAC, nucleosides, or both. Tumors were detected at the injection sites in 7 and 6 out of 16 mice supplemented with NAC and nucleosides, respectively, and in 12 out of 15 mice supplemented with both (Figures 6D and S6E–S6G). We repeated this experiment by transplanting less NIH3T3 cells and again observed tumor formation in mice treated with NAC (3 out of 7 mice), nucleosides (4 out of 8 mice), or both (6 out of 8 mice), but not in control mice (Figures S6H–S6K).
In these experiments, the appearance of tumor was relatively fast (as short as one month), arguing against the possibility that supplementation of antioxidants or nucleosides might enable PHMLEB and NIH3T3 cells to acquire oncogenic mutations, which in turn drove tumorigenesis. To further rule out this possibility, we re-inoculated cells derived from initial tumors in new hosts. Cells derived from initial tumors with NAC or nucleoside supplementation could produce new tumors in the presence, but not in the absence, of the same supplement (Figures 6E, S6L, and S6M). New tumors arose faster relative to initial tumors (compare Figures S6L and S6M with S6E and S6F). This was expected as cells with proliferative advantages would be selected for during the initial tumorigenesis process, but such changes were insufficient for oncogenic transformation. Cells derived from initial tumors with both NAC and nucleosides supplementation could form tumors in the new host in the absence of these supplements, but these tumors grew much slower and failed to reach a large size (Figures S6N and S6O). By contrast, the same cells formed large tumors at a fast pace when NAC and nucleosides were again provided (Figures S6N and S6O). Therefore, the generation of initial tumors in the presence of NAC or nucleosides was primarily or exclusively due to the upregulation in antioxidant capacity and nucleotide precursor availability engendered by these supplements.
Both antioxidants and nucleosides are required for oncogenic transformation when their overlapping metabolic consequences are minimized
As expected, NAC increased the NADPH/NADP+ ratio and reduced ROS levels in, and promoted the survival of, matrix-detached PMLEB (Figures 6F–6H) and NIH3T3 (Figures 6I–6K) cells. Also as expected, exogenous nucleosides increased intracellular nucleotide levels (Figure S7A) and enhanced DNA synthesis (Figure 7A). Less obviously, we found that NAC augmented DNA synthesis in matrix-detached cells (~35%; Figure 7A), and that exogenous nucleosides increased intracellular NADPH/NADP+ ratio (Figures 6F and 6I), reduced ROS levels (Figures 6G and 6J), and promoted survival (Figures 6H and 6K) of matrix-detached PMLEB and NIH3T3 cells. Thus, there is a crosstalk between redox balance and nucleotide synthesis.
Figure 7. Both antioxidants and nucleosides are required for oncogenic transformation when their overlapping metabolic consequences are minimized.
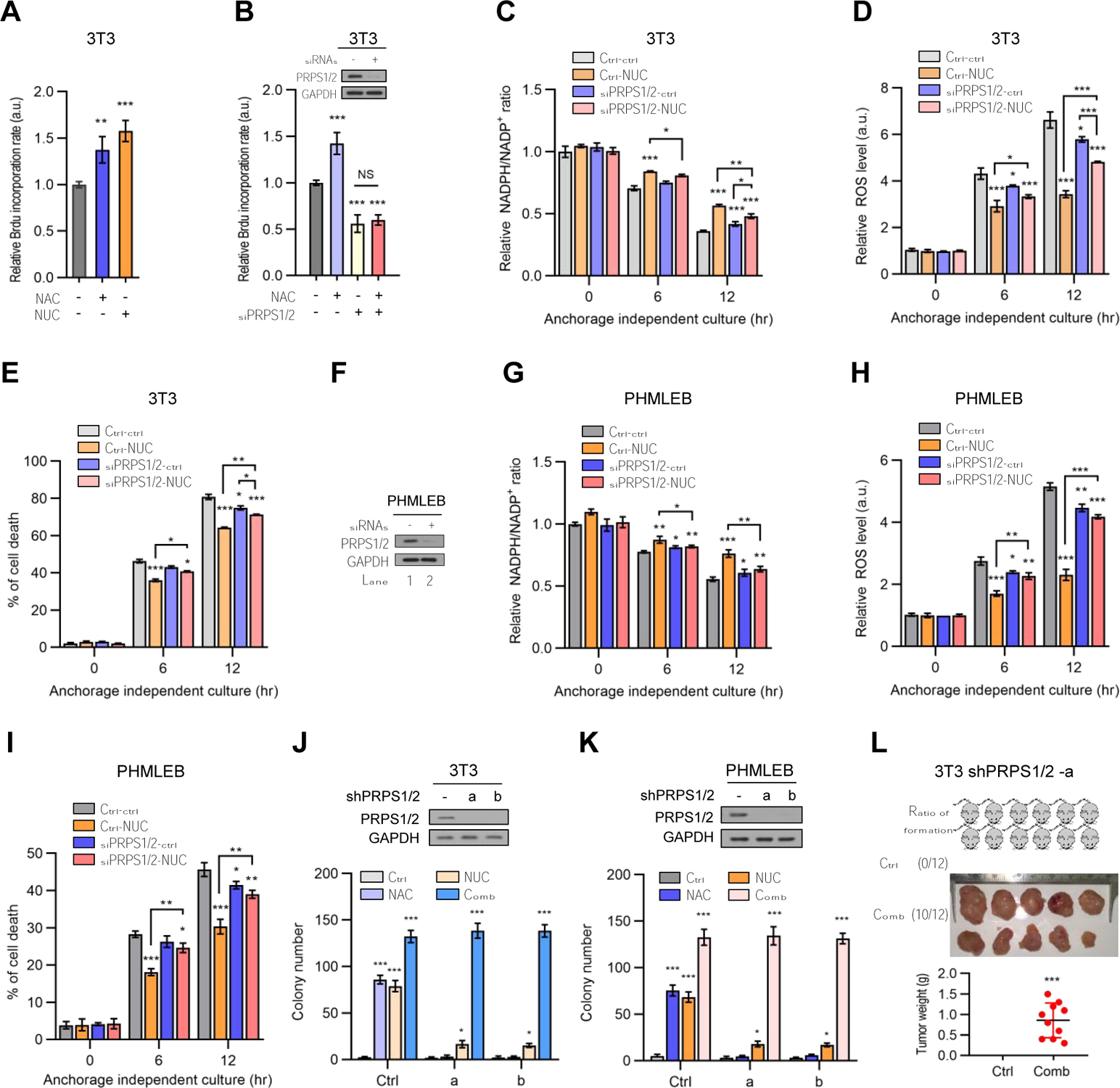
(A) NIH3T3 cells cultured under matrix-detached conditions for 4 hr in presence of vehicle, NAC (0.25 mM), or NUC (160 μg/mL) were assayed for DNA synthesis by BrdU incorporation. The data were normalized by cell number.
(B) NIH3T3 cells were treated with control siRNA or a combination of PRPS1 and PRPS2 siRNAs. Cells were cultured under matrix-detached conditions in the presence or absence of NAC (0.25 mM) for 4 hr and assayed for protein expression by Western blot (top) and DNA synthesis by BrdU incorporation (bottom).
(C–I) Control and PRPS1/2-knockdown NIH3T3 (C–E) and PHMLEB (F–I) cells were cultured under matrix-attached (0 hr) or -detached (6 and 12 hr) conditions in presence or absence of NUC (160 μg/mL). Cells were assayed for PRPS1/2 protein expression (F), the NADPH/NADP + ratio (C and G), ROS levels (D and H), and cell death (E and I).
(J and K) NIH3T3 (J) or PHMLEB (K) cells stably expressing a control shRNA or a combination of PRPS1/2 shRNAs were treated with vehicle (Ctrl), NAC (0.25 mM), NUC (160 μg/mL), or both NAC and NUC. Cells were assayed for protein expression (top) and soft agar colony formation (bottom) (see also Figures S7C–S7E).
(L) Four million of NIH3T3 PRPS1/2 knockdown cells were subcutaneously injected into athymic nude mice. Mice were treated with vehicle control or both NAC and NUC. Shown are the frequency of tumor formation (top, left) and tumor images (top, right) and weights (bottom) at the end of the experiments.
Data are means ± SD of representative result (n = 3, or as indicated). *P < 0.05, **P < 0.01, ***P < 0.001.
While such a crosstalk might be mediated by diverse signaling machinery, it is possible the direct metabolic connections between these processes also play a role. One point of intersection is phosphoribosylpyrophosphate synthetase (PRPS), the enzyme that generates PRPP from R5P and hence links the PPP with nucleotide synthesis (Figure S1A). Of note, knockdown of both isoforms of PRPS (PRPS1 and PRPS2), which reduced DNA synthesis as expected, also rendered NAC incapable of promoting DNA synthesis in matrix-detached cells (Figure 7B) or enhancing cell proliferation of adherent cells (Figure S7B). At the same time, knockdown of PRPS1/2 rendered nucleosides ineffective in maintaining the NADPH/NADP+ ratio and redox balance in, and survival of, matrix-detached NIH3T3 (Figures 7C–7E) and PHMLEB (Figures 7F–7I) cells. Thus, the overlapping effects of NAC and nucleosides on redox balance and DNA synthesis were largely abolished upon PRPS knockdown.
We subsequently assessed the transforming potential of NAC and/or nucleosides for PRPS-deficient cells. PHMELB and NIH3T3 cells devoid of PRPS1/2 were unable to grow in soft-agar medium with the supplementation of NAC or nucleosides alone (Figures 7J, 7K, and S7C–S7E). These cells showed impairment in proliferation, but this defect could be rescued by nucleosides or nucleosides plus NAC (Figure S7B), Therefore, the lack of transforming activity of nucleosides on PRPS1/2-knockdown cells was not due to a defect in proliferation. Of note, in the presence of both NAC and nucleosides, PRPS1/2-deficient PHMELB and NIH3T3 cells formed soft-agar colonies as effectively as the corresponding PRPS1/2-proficient cells (Figures 7J, 7K, and S7E). To confirm the transforming activity of NAC and nucleosides in combination on PRPS1/2-deficient cells, we transplanted PRPS1/2-knockdown NIH3T3 cells in athymic nude mice. When supplemented with the same doses of NAC plus nucleoside, PRPS1/2-knockdown NIH3T3 produced tumors with a frequency (10 out of 12 injected mice) and growth rate similar to those of control NIH3T3 cells (compared Figures 7L and S7F with 6D and S6G). Therefore, when the overlapping effects of NAC and nucleosides are minimized, both of them are required for oncogenic transformation.
To extend this analysis, we knocked down G6PD in PHMLEB and NIH3T3 cells. The knockdown cells displayed no obvious defects and was not strongly influenced by NAC and/or nucleosides when grown on adherent plates (Figure S7G). Interestingly, however, NAC and nucleosides in combination, but not individually, conferred on G6PD-knockdown PHMLEB and NIH3T3 cells the ability to grow in soft-agar medium (Figures S7G–S7J). Collectively, these results suggest that a combination of robust antioxidative capacity and augmented nucleoside synthesis permits tumorigenesis.
DISCUSSION
Various immortalized murine and human cells can be converted to the tumorigenic state through the introduction of oncogenes, yet the key elements that underlie oncogenic transformation are incompletely understood. Extending a previous finding (Kuo et al., 2000), here we show that G6PD permits neoplastic transformation of human fibroblasts, as well as human epithelial cells where most tumors arise. Its ability to effectuate anchorage-independent growth in vitro and tumor formation in animals indicates that G6PD can largely meet, or substitute for, the mitogenic changes required for oncogenic transformation. Metabolic analyses showed that the outcomes of G6PD overexpression include augmented antioxidant defense capacity and nucleotide precursor availability. Mimicking these outcomes, supplementation of exogenous antioxidants and nucleosides confers on immortalized mouse and human cells tumorigenicity. These observations suggests that enabling survival and proliferation under adverse conditions may be an essential part of oncogenic transformation.
ROS have long been thought to aid in tumor initiation and progression by accelerating genetic mutations, inducing inflammation, and/or activating proliferative pathways (Finkel, 2011). However, high levels of ROS also damage DNA, proteins, lipids, and other essential cellular components. To survive, cells including incipient and frank cancer cells must maintain ROS below a certain threshold. Due to the oxidizing extracellular environment, cells largely depend on intrinsic systems to maintain the reducing intracellular environment. This task can become especially challenging under stress conditions when nutrient uptake and utilization may be impaired, as demonstrated by matrix detachment-induced cell death (Schafer et al., 2009). As such, the capability to maintain redox balance in untransformed cells may be limited, representing compromises between supporting cell survival under physiological stress conditions and preventing cell survival under severe stress conditions experienced by incipient tumors. Overcoming this limit may represent a fundamental element of oncogenic transformation. The effect of a robust antioxidant defense is unlikely restricted to the formation of primary tumors, as NAPDH-producing enzymes are further upregulated during metastasis (Piskounova et al., 2015). Thus, tumorigenic transformation and progression may be closely related to an increasingly higher antioxidant capacity.
Most cellular antioxidant systems use NADPH as the ultimate electron donor (Ying, 2008). Given that the ratio of NADPH to NADP+ is generally high in resting cells, a further increase in cellular antioxidant capacity is dependent on de novo biosynthesis of NADP+. However, an increase in NADP+, even acutely, may inhibit crucial metabolic processes such as folate metabolism (Chen et al., 2019). Although the oxidative PPP is a major source of NADPH in many cells (Zhang et al., 2017), this is usually interpreted in terms of G6PD- and 6GPD-mediated reduction of NADP+. Here we show that G6PD can directly activate NADK1. This bolsters de novo NADP+ biosynthesis and, at the same time, ensures swift conversion of the newly synthesized NADP+ to NADPH, thereby minimizing the deleterious consequence of high NADP+ abundance. The G6PD-NADK connection likely allows spatial and temporal coupling of NADP+ biosynthesis with its reduction to NADPH, stimulating the production of NADP+ where and when it is needed. A recent study showed that NADK1 is activated by AKT-mediated phosphorylation (Hoxhaj et al., 2019). Therefore, NADK1 activation may be a focal point of regulation for cellular redox state.
While a robust antioxidant capacity enables tumor cells to survival under stress conditions, the relentless and autonomous cell proliferation characteristic of the malignant state is ultimately dependent on the supply of biomaterials. Although proliferating mammalian cells can import a wild range of nutrients including sugar, lipids, amino acids, and even proteins, they mostly rely on de novo synthesis for nucleotides. Nucleotides are needed not only for the generation of DNA and RNA, but also for net production of various activate metabolites involved in bioenergetics (e.g., ATP and GTP), redox regulation (e.g., NAD+, NADP+, FAD, and FMN), anabolic process (e.g., UDP-glucose), and regulatory functions (e.g., ADP ribosylation) (Lane and Fan, 2015). Nucleotide synthesis entails multiple metabolites including amino acids, ribose, one-carbon donors in a series of energetically demanding pathways across different cellular compartments (Lane and Fan, 2015; Zhu and Thompson, 2019). Therefore, the cellular capacity for nucleotide synthesis may also be limited in untransformed cells especially under stress conditions, and prevailing over this limit is likely another prerequisite for transformation. As for antioxidant defense, the role of augmented nucleotide synthesis likely extends beyond transformation, as it is also implicated in avoidance of senescence induced by oncogenes (Aird et al., 2013) and evasion of proliferation arrest under metabolic stress conditions (Lunt et al., 2015).
NAC and nucleosides elicit overlapping effects on redox balance and nucleotide synthesis. By enhancing antioxidant capability, NAC might make more R5P and NADPH available for nucleotide production. Moreover, exogenous nucleosides might reduce the usage of R5P and one-carbon units for de novo nucleotide biosynthesis, thereby directing them for NADPH regeneration. These scenarios would provide an explanation for the role of PRPS1/2 in mediating the overlapping effects of NAC and nucleosides. When their metabolic outcomes are largely separated as in PRPAS1/2-knockdown and likely also G6PD-knockdown cells, both NAC and nucleotides are required for malignant transformation, indicating redox balance and nucleotide synthesis are important for transformation.
Accumulating evidence suggests that a robust antioxidant defense and nucleotide synthesis is an integral part of oncogenic signaling. For example, KRAS directs glycolytic intermediates to the nonoxidative branch of the PPP to support nucleotide biosynthesis (Ying et al., 2012), while promoting glutamine flux through the cytoplasmic malic enzyme ME1 (Son et al., 2013). c-Myc stimulates nucleotide synthesis via PRPS2 (Cunningham et al., 2014), while markedly increases glutaminolysis that traverses through the mitochondrial malic enzyme ME2 (Wise et al., 2008). Both ME1 and ME2 are important for NADPH production (Jiang et al., 2013a). Nevertheless, the potent transforming activity of H-RasV12, especially in vivo, indicates that Ras and likely some other oncogenes can impact key steps of tumorigenesis beyond the initial transformation.
Although metabolic reprogramming is prevalent in tumor cells, its phenotypical demarcation with the core hallmark capacities of cancer is not always clear (Hanahan and Weinberg, 2011). Metabolic alterations, such as mutations in the TCA cycle enzymes, can have a profound impact on histone/DNA methylation and hypoxia-induced factors (HIFs)-mediated transcriptional program (Kaelin and McKnight, 2013), which in turn can alter genome stability, angiogenesis, and invasion. By revealing that upregulated redox balance capacity and nucleotide precursor availability suffice for transformation, the current study supports the notion that metabolic reprogramming itself may be a core hallmark capacity of cancer. The importance of redox regulation is consistent with observations that dietary antioxidants increase cancer incidence in animal models and clinical trials (Klein et al., 2011; Sayin et al., 2014). The importance of nucleotide synthesis is in accordance with the success of many cancer drugs that interfere with de novo nucleotide biosynthesis (Chabner and Roberts, 2005; Christopherson et al., 2002). Therefore, while immortalization endows normal cells with an unlimited lifespan, we propose that malignant transformation may confer on immortalized cells the ability to survive and proliferate under adverse conditions.
Limitations of Study
Although knockdown of PRPS1/2 minimizes the overlapping metabolic consequences of exogenous antioxidant and nucleoside, the underlying mechanism remains to be determined. Moreover, we cannot exclude the possibility that exogenous antioxidants and nucleotides may elicit a common, more basic biological or biochemical effect that ultimately drives tumorigenesis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiaolu Yang (xyang@pennmedicine.upenn.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERMENTAL MODEL AND SUBJECT DETAILS
Mice and In Vivo Studies
Two million of NIH3T3 cells or immortalized BJ cells expressing H-RasV12, G6PD, or control vector were injected subcutaneously into the flank of male athymic nude mice (4–5 weeks old). For treatment with or without NAC and/or nucleosides, the indicated number of NIH3T3 cells, four million of PHMLEB cells, or four million of NIH3T3 cells in which PRPS1/2 were knockdown by shRNAs were injected subcutaneously into the flank of male athymic nude mice. For re-transplanting cells from tumors that were initially formed by NIH3T3 cells in the presence of NAC and/or nucleosides, three representative tumors from each group were selected and dissected immediately after mice were euthanized. The tumors were washed with 1x HBSS (Fisher Scientific), and same amount of materials from each tumor within the same group was collected. The tumor masses from the same group were mixed together, cut to small pieces, and digested by 1 x collagenase (Fisher Scientific) for 20 min at 37 °C. The mixture of tumor cell aggerates were filtrated with 100 μm cell strainers (VWR, Radnor, PA). Collected filtrates were incubated at 37 °C for 10 min for further digestion and filtered with 40 μm cell strainer (VWR). Collected filtrates of single cells were added with equal volume of 2% FBS/HBSS solution to stop digestion. After centrifuge and further washed by 10% FBS/DMEM medium, tumor cells were counted in the presence of 0.4% Trypan blue solution (Fisher Scientific) to exclude dead cells, and four million of viable cells were immediately injected into the flank of male athymic nude mice.
For treatment with or without NAC and/or nucleosides, mice were fed with normal drinking water or drinking water containing 1 g/L NAC, and I.P. injected three times a week with saline or saline containing nucleosides (mixed at an equal molar ratio) at a dose of 2.4 mg/25 g mice. The treatment started a week prior to the injection of cells and continued until the mice were euthanized. After tumor nodules were visible, tumor volumes (V) were measured every three days based on the formula V = W2 × L × 0.5, where W represents the largest tumor diameter in centimeters and L the next largest tumor diameter (Zhang et al., 2015). After mice were euthanized, tumors were dissected out, imaged, weighted or used for further characterization.
The mice were housed at 22 – 24 °C on a 12 hr light/dark cycle and fed with a standard diet (LabDiet 5010). The health status of the mice was monitored twice a week by their behaves and body weights. All experiments involving animals were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Cell culture, transfection and infection
Immortalized mouse embryonic fibroblast NIH3T3 cells, human primary foreskin fibroblast BJ cells, and human embryonic kidney HEK293T cells were purchased from ATCC. Immortalized human mammary epithelial (PHMLEB) cells were a gift from Dr. R. Weinberg (Elenbaas et al., 2001). NIH3T3 and HEK293T cells were cultured in DMEM medium (Fisher Scientific) supplemented with 10% Fetal bovine serum (FBS) (Sigma). PHMLEB cells were cultured in MEGM™ BulletKit™ Growth Media (Lonza). Primary and immortalized BJ cells were cultured in EMEM medium supplemented with 10% FBS. Cells were maintained at 37 °C in a humidified incubator with 5% CO2.
For anchorage-independent or matrix-detached culture, plates were pre-coated with 1.2% ploy-HEMA (W/V) (dissolved in 95% ethanol in water solution). The cells were seeded at the same density as the corresponding cells cultured on anchorage-dependent or matrix-attached conditions. When indicated, cells were treated with NAC (0.25 mM), nucleosides (a total concentration of 160 μg/mL, 20 μg/mL for each nucleoside), or both.
DNA plasmids were transfected into cells using Lipofectamine 2000 (Fisher Scientific) following the manufacturer’s protocol. Retroviral vectors were packed with pCL-Ampho and pCMV-VSV-G plasmids, and lentiviral vectors were packed with pCMV-dR8.91 and pCMV-VSV-G plasmids. Medium of retroviruses and lentiviruses packed by HEK293T cells were then collected, filtered, and concentrated for infection of target cell lines. For transfecting siRNAs, lipofectamine RNAimax (Fisher Scientific) was used following the manufacturer’s instruction.
METHOD DETAILS
IHC for Ki-67 and CD31
Formalin-fixed paraffin-embedded (FFPE) tissue blocks of xenograft tumors were stained for Ki67 and CD31. Briefly, tissue slides were subjected to antigen retrieval using Novocastra Epitope Retrieval Solutions, pH6 (Leica, Wetzlar, Germany), followed by incubation with primary antibodies at 4 °C overnight. To neutralize endogenous peroxidase, Dako Dual Endogenous Enzyme Block (DAKO, Denmark) was used. A biotin goat anti-mouse secondary antibody (BD Pharmingen) was used for Ki67, followed by incubation with Streptavidin-Horseradish-Peroxidase (SAV-HRP, BD Pharmingen). For CD31, the secondary antibody was anti-mouse/anti-Rabbit Envision Flex HRP labeled Polymer (DAKO). Three representative xenograft tumors from each group were analyzed. Ten histologically similar fields were randomly selected from each slide for analysis. Proliferation of tumor cells were quantified by calculating the percentage of cells stained positively for Ki67 at 20x magnification. The vessel density of tumor was quantified by calculating the area of cells positively stained for CD31 at 10x magnification with Image Pro Plus7, and normalized to that in tumors derived from H-RasV12-expressing cells.
Analysis of G6PD expression in patient samples
G6PD mRNA levels in median-centered Log2 intensity for cancer patients and corresponding controls were extracted from Oncomine dataset (https://www.oncomine.org/). For colon cancer, Gaedcke Colorectal, Hong Colorectal, and Sabates-Bellver Colon datasets were included. For liver cancer, Roessler Liver, Roessler Liver 2, and Wurmbach Liver datasets were included. For melanoma, Talantov Melanoma dataset was used. For renal cancer, Cutcliffe Renal, Jones Renal, and Yusenko Renal datasets were included. The data was then analyzed as shown. The survival curves of G6PD high/low patients with liver, colorectal, renal, and breast cancers were extracted from G6PD pathology dataset (https://www.proteinatlas.org/ENSG00000160211-G6PD/pathology) of the Human Protein Atlas. The G6PD expression cut off values (in Fragments Per Kilobase Million (FPKM)), percentage of 5 years survival rates for each type of cancer patients with high/low G6PD expression, and p value are summarized in Table S1.
Soft agar colony formation
Cells were seeded at a proper density (5,000 cells/well for NIH3T3 cells, 8,000 cells/well for PHMLEB cells, and 7,500 cells/well for immortalized BJ cells) in the top layer of 0.36% soft agar premixed with culture medium supplemented with 10% FBS in 6-well plates, and incubated at 37 °C for 2 weeks for NIH3T3 cells, 2.5 weeks for PHMLEB cells, and 3 weeks for BJ cells. These conditions were chosen for easy detection of colony formation by G6PD-expressing cells. For treatment, culture medium with 10% FBS was supplemented with NAC (0.25 mM unless otherwise indicated), Trolox (0.5 mM), nucleosides (a total concentration of 160 μg/mL, 20 μg/mL for each nucleoside), or the indicated combinations for 3 (for NIH3T3) or 3.5 (for PHMLEB) weeks. The premixed 0.36% soft agar supplemented with 10% FBS with or without the indicated treatments was replenished on the top once a week. By the end of the experiment, the colonies were either isolated for additional assays or stained with 0.05% crystal violet in 4% PFA solution for imaging and quantification.
Western blot analysis and RT-PCR
Cell lysates were prepared in RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 10 mM Tris-HCl pH 7.5, and 5 mM EDTA) plus a protease inhibitor cocktail). Equivalent amounts of protein were separated by SDS-PAGE and transferred to a PVDF membrane (Millipore, Billerica, MA). Proteins were detected using an enhanced chemiluminescence system (Amersham Biosciences, Piscataway, NJ).
Tumors dissected from mice for RNA isolation were homolyzed and lysed by TRIzol (Fisher Scientific) following the manufacturer’s protocol. RNAs were reverse transcribed by a High-Capacity cDNA Reverse Transcription Kit (Fisher Scientific). 2X GoTaq® Master Mixes (Promega) was used to amplify human GAPDH gene by PCR.
Immunoprecipitation and GST pull-down
HEK293T cells were lysed in the IP lysis buffer (50 mM HEPES, pH7.4, 150 mM NaCl, 1.5 mM MgCl2, 4 μM MG132, 10% glycerol, 0.5% NP-40, and 0.5% Triton X-100) plus protease inhibitors by gentle sonication. Cell lysates were immunoprecipitated with Anti-FLAG® M2 Affinity Gel or anti-NADK1 plus Protein A/G beads, or incubated Pierce™ Glutathione Agarose for GST pull-down. Immunoprecipitates, GST pull-down proteins, and whole cell lysates were separated by SDS-PAGE followed by Western blot.
Immunofluorescence imaging and quantification
Control and G6PD-overexpressing PHMLEB cells grown on coverslips were treated with siRNA for 48 hr. Cells were washed with PBS and fixed with 4% paraformaldehyde (PFA). After being treated with 0.1 % Triton X-100, cells were incubated overnight with primary antibodies, and then with Alexa Fluor-conjugated anti-mouse and anti-rabbit secondary antibodies (Fisher Scientific). The coverslips were mounted with mounting buffer containing 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA), and images were acquired with a confocal microscope (Carl Zeiss, White Plains, NY). The colocalization/overlapping M1 & M2 coefficients (Manders’) were calculated with JACoP plugin of Fiji ImageJ at 60x following the algorithm’s general guidelines (Manders et al., 1993).
Protein purification
For protein purification from HEK293T cells, Flag-tagged G6PD, G6PDm1, G6PDm2, NADK1, and Flag-NADK1∆N in the pRK5 plasmid were transfected via Lipofectamin 2000. 48 hr later, cells were harvested using IP lysis buffer (20 mM Tris-HCl at pH 7.4, 150 mM NaCl, 0.5% Triton X-100, 0.5% NP-40, and 10% glycerol). Lysates were sonicated for 10 seconds and centrifuged at 17,000 x g at 4 °C for 15 min. Supernatants were incubated with anti-Flag M2 Affinity Gel at 4 °C overnight. Gel was sequentially washed with lysis buffers containing 50, 200, 400, 200, and 50 mM KCl, respectively (once each), and then with Tris buffer (100 mM Tris-HCl with 10% glycerol, pH 8.0). Recombinant proteins were eluted using 0.2 mg/ml 3xFlag peptide at 4 °C for 1 hr and concentrated by Amicon Ultra-0.5 Centrifugal Filter (Sigma-Aldrich). Protein concentration was determined by SDS-PAGE and Coomassie Blue staining, along with a protein standard (BSA).
Protein crosslinking
For protein cross-linking, control, Flag-tagged G6PD, G6PDm1, or G6PDm2 in the pRK5 plasmid were transfected in HEK293T cells via Lipofectamin 2000 as indicated. 48 hr later, the cells were collected and treated with control (0 mM) or different concentrations (1 and 2 mM) of cross-linker DSS for 45 minutes on ice and then lysed with RIPA buffer and analyzed by Western blot. Endogenous p53 was used as a control for cross-linking.
G6PD and NADK1 enzyme activity
G6PD enzymatic activity was determined by a Glucose 6 Phosphate Dehydrogenase Activity Fluorometric Assay Kit (Abcam) following the manufacturer’s protocols. Enzymatic activities were normalized on the basis of total protein, which was determined by a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA). The data are expressed in arbitrary enzyme units per mg of total protein.
NADK1 activity was assayed as described (Hoxhaj et al., 2019) with modifications. Approximately 0.5 μg of purified 6xHis-NADK1, Flag-NADK1∆N, or Flag-NADK1 was subjected to the assay that coupled NADK-mediated production of NADP+ from NAD+ with G6PD-mediated reduction of NADP+ to NADPH, which was measured as a change in A340nm over time. Measurements of 0.5 μg NADK1 enzymatic velocity at different concentrations of NAD+ (0, 1, 2, 4, 6, and 8 mM) were performed in a 100 μl reaction mixture containing 10 mM ATP, 10 mM glucose-6-phosphate, 1 μg human G6PD purified from E coli. (Sigma), 10 mM MgCl2, and 100 mM Tris-HCl (pH 8.0). Measurement of 1 μg G6PD (Sigma) enzymatic velocity at different concentrations of its substrate NADP+ (0, 0.5, 1, 2, and 4 mM) was performed in a 100 μl reaction mixture containing 10 mM glucose-6-phosphate, 10 mM MgCl2, and 100 mM Tris-HCl (pH 8.0). NAD+ at 1 mM was used to determine the impact of G6PD on NADK1 enzymatic activities. Under this condition, the production of NADPH is limited by the activity of NADK1, but not G6PD. Measurement of the NADK1 enzymatic velocity impacted by different amount of G6PD (Sigma) (0, 0.5, 1, 2, 4, and 6 μg) was performed in a 100 μl reaction mixture containing 10 mM ATP, 10 mM glucose-6-phosphate, 0.5 μg of purified His-tag NADK1, 10 mM MgCl2, 1 mM NAD+, and 100 mM Tris-HCl (pH 8.0). Measurement of the NADK1 enzymatic velocity impacted by different amount of Flag-G6PD, Flag-G6PDm1, or Flag-G6PDm2 (0, 1, 2, 3, and 5 μg) was performed in a 100 μl reaction mixture containing 1 μg G6PD (Sigma), 10 mM ATP, 10 mM glucose-6-phosphate, 0.5 μg of purified His-tag NADK1, 10 mM MgCl2, 1 mM NAD+, and 100 mM Tris-HCl (pH 8.0). Measurement of the enzymatic velocity of Flag-NADK1∆N or Flag-NADK1 impacted by different amount of G6PD (0, 0.5, 1, 2, and 4 μg) was performed in a 100 μl reaction mixture containing 10 mM ATP, 10 mM glucose-6-phosphate, 0.5 μg of purified Flag-NADK1∆N or Flag-NADK1, 10 mM MgCl2, 2 mM NAD+, and 100 mM Tris-HCl (pH 8.0). For all assays, OD340nm was measured at 37 °C every 2 minutes for 20 min. Initial velocity was determined by the slope of initial linear portion of enzyme kinetic curve.
In vitro pull-down assay
The reaction mixtures after NADK1 enzymatic assay were subjected to a pulldown assay. 90% of the reaction mixture (270 μl) was incubated with HIS-Select® Nickel Affinity Gel for pulldown of 6xHis-tagged NADK1. After washing, the beads-bound proteins were analyzed by Western blot, along with the remaining 30 μl reaction mixture to determine the amount of NADK1 and G6PD.
Glucose and glutamine consumption and global metabolomic analysis
The rates of glucose and glutamine consumption were determined using an YSI7100 Multiparameter Bioanalytical System (YSI Life Sciences, Suwanee, GA) and normalized by cell number. Relative rates of glucose and glutamine usage were normalized by that of the control. Metabolite analysis was performed as previously described (Hui et al., 2017). Cells were washed twice with ice-cold PBS, immediately covered with −20 °C Solvent A (40:40:20 methanol: acetonitrile: water with 0.1 M formic acid solution, precooled on ice) for the extraction of metabolites, and scraped down. The mixtures were collected in a microcentrifuge tube and incubated on dry ice for 5 min. The volume of the extraction solution (in μl) was 50 times of the cell volume in packed cell volume (PCV). Solvent B (15% (W/V) NH4HCO3 in water solution) was added to solvent A in a ratio of 8.8 μl solvent B per 100 μl solvent A and mixed, followed by centrifugation at 16,000 x g and 4 °C for 15 min. The supernatant was transferred to liquid chromatography mass spectrometry (LC-MS) autosampler vials for analysis. In brief, a quadrupoleorbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific) operating in negative ion mode was coupled to hydrophilic interaction chromatography via electrospray ionization and used to scan from m/z 73 to 1,000 at 1 Hz and 140,000 resolution. LC separation was achieved on a XBridge BEH Amide column (2.1 mm × 150 mm, 2.5 μm particle size, 130Å pore size; Waters) using a gradient of solvent A (20 mM ammonium acetate and 20 mM ammonium hydroxide in 95:5 water: acetonitrile, pH 9.45) and solvent B (acetonitrile). Flow rate was 150 μl min−1 . The gradient was: 0 min, 85% B; 2 min, 85% B; 3 min, 80% B; 5 min, 80% B; 6 min, 75% B; 7 min, 75% B; 8 min, 70% B; 9 min, 70% B; 10 min, 50% B; 12 min, 50% B; 13 min, 25% B; 16 min, 25% B; 18 min, 0% B; 23 min, 0% B; 24 min, 85% B; 30 min, 85% B. Data were analyzed using the MAVEN software35.
Focused metabolite analysis
Cells were washed twice with PBS, and metabolites were extracted from cells by scraping into 500 μl of ice cold methanol/water (4:1, v/v) containing 500 ng of internal standard ([13C4]succinate, [13C6]citrate, [13C3]lactate, [13C4]fumarate). Samples were collected and sonicated for 30 s and centrifuged at 13,000 x g and 4 °C for 15 minutes. The supernatant was then transferred to another tube and incubated at room temperature for 1h. Samples were evaporated to dryness under nitrogen after incubation and suspended in 100 μl of mobile phase A (400 mM 1,1,1,3,3,3-hexafluoro-2-propanol and 100 mM DIPEA in water). The LC-MS was performed as described previously (Guo et al., 2016). Abundance of metabolites was normalized based on total protein content.
NADPH quantification by LC-MS
Measurements of NADPH by LC-MS were made using a previously described method (Lu et al., 2018). For matrix-attached conditions, cells were seeded in standard 6-well plates and cultured in DMEM medium with 10% dialyzed FBS for the indicated time. Medium was aspirated, and metabolites were extracted with 600 μl Solvent A (40:40:20 methanol: acetonitrile: water with 0.1 M formic acid solution, precooled on ice). After 30 seconds, extraction was quenched with 75 μl Solvent B (15% (W/V) NH4HCO3 in water solution). For matrix-detached conditions, cells were seeded in ploy-HEMA pre-coated 6-well plates and cultured in the same medium as above for the indicated time. Upon harvesting, cells were gently collected into 1.5mL Eppendorf tubes by pipetting and centrifuged at 1000 x g for ~30 seconds. The media was aspirated, and cell pellets were extracted with 200 μl Solvent A. After 30 seconds, extracts were quenched with 25 μl Solvent B. For both conditions, total time from cell collection to extract quenching was ~ 1 min to minimize disturbance of cell metabolism. Samples were then incubated at −80 °C for 30 min, followed by centrifugation at ~16,000 x g and 4 °C to remove insoluble cell components. The resulting supernatants were analyzed directly by LC-MS. Metabolites were analyzed using a quadrupole-orbitrap mass spectrometer (Q Exactive Plus, Fisher Scientific) operating in negative ion mode, coupled via electronspray-ionization to hydrophilic interaction chromatography (HILIC) with LC separation on a XBridge BEH Amide column. Metabolite abundances were normalized to extraction dilution and packed cell volumes for each condition. Data were analyzed using the MAVEN software suite (Clasquin et al., 2012).
NADP+, NAPDH, and ATP quantification by colorimetric assays
NADP+ and NADPH were also quantified using the NADP/NAPDH quantitation colorimetric assays with a NADPH standard (BioVision, Inc, San Francisco, CA) and normalized by protein concentration. The NADP/NADPH ratio was measured by a NADP/NADPH-Glo™ assays kit (Promega, Madison, WI), following the manufacturers’ protocol, and the relative NADPH/NADP+ ratio is normalized by that of control cells (or treatment control) under matrix-attached condition. ATP levels were detected by an ATP colorimetric assays kit (Abcam) and normalized by protein concentration, and the relative ATP levels is normalized by that of control cells (or treatment control) under matrix-attached condition.
Analysis of NAD+-NADP+ flux
[13C3-15N]-nicotinamide (13C3-15N-NAM) was used to determine the NAD+-NADP+ flux as described previously (Hoxhaj et al., 2019; Liu et al., 2018). Briefly, cells were treated with vehicle or the NAMPT inhibitor FK866 for 16 hr in DMEM media with 10% dialyzed FBS. Cells were washed once with nicotinamide-free DMEM and incubated in the same medium containing 4 mg/L of 13C3-15N-nicotinamide and 10% dialyzed FBS for 1 hr. The nicotinamide-free medium was prepared using powdered DMEM supplemented with standard DMEM concentrations of folic acid, niacinamide, pyridoxal, riboflavin, thiamine, glucose, sodium pyruvate, and, in the place of nicotinamide, 13C3-15N-nicotinamide, with pH adjusted to 7.2. The metabolites were collected and immediately analyzed by LC-MS to determine the ratio of labeling.
ROS quantification
ROS quantification was done as previously described (Du et al., 2013; Jiang et al., 2013a). Briefly, cells were treated with 10 mM DCFDA (2’,7’-dichlorodihydrofluorescein) for 30 min before harvested. Cells were washed twice with PBS, stained with PI for 2 minutes, and immediately analyzed by flow cytometry. The DCFDA signals were measured by a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA) after excluded dead cells by gating out PI-positive cells. ROS levels are normalized by that of control cells (or treatment control) under matrix-attached conditions.
Cell death assay
NIH3T3 cells or PHMLEB cells were cultured under matrix-attached or -detached conditions for indicated time. Cells were harvested, stained by an Annex V/PI kit (BD Biosciences) following the manufacture’s protocol, and analyzed by a BD Accuri C6 flow cytometer. Cells with Annexin V-positive staining were characterized as dead cells, including early apoptotic (Annexin V-positive, PI-negative), later apoptotic, and necrotic cells (both Annexin V-positive, PI-positive).
Cell proliferation and BrdU incorporation assays
Number of viable cells was determined by a Cell Counting Kit-8 (CCK-8) (Sigma) following the manufacturers’ protocol. Relative cell number was normalized by the number of viable cells on day 0. BrdU incorporation was measured by a BrdU Cell Proliferation Assay Kit (BioVision, Milpitas, California) following the manufacturer’s protocol. The incorporation rate was normalized by the total number of viable cells in each sample determined by a CCK-8 kit. Relative BrdU incorporation rate was normalized by that of control cells under matrix-attached conditions.
Quantification and Statistical Analysis
The heat-map, volcano analysis and pathway enrichment analysis were preformed using the online algorithm following manual’s instructions (https://software.broadinstitute.org/morpheus/ and http://www.metaboanalyst.ca/). Statistical analyses were performed using two-way ANOVA or Student’s t-test (GraphPad Prism 8). Detailed statistics can be found in the figures and figure legends. No other methods were used to determine whether the data met assumptions of the statistical approach or not.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or SOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 6xHis tag | Cell Signaling | 2365 |
| 6PGD | Cell Signaling | 13389S |
| Biotin goat MS IgG (IHC) | BD Pharmingen | 553999 |
| CD31 | Novartis | NBP2–44342 |
| Anti-mouse/Anti-Rabbit Envision Flex HRP labeled Polymer (IHC) | DAKO | K8000 |
| Flag tag | Cell Signaling | 14793S |
| G6PD (WB) | Cell Signaling | 12263S |
| G6PD (IF, IHC, IP) | Proteintech | 66373–1-Ig |
| GAPDH | Santa Cruz | sc-47724 |
| Goat anti-mouse IgG (IF 488) | Fisher Scientific | A32723 |
| Goat anti-rabbit (IF 594) | Fisher Scientific | A32731 |
| GST tag | Santa Cruz | Sc-138 |
| HRP-anti-mouse IgG (WB) | Cell Signaling | 7076S |
| HRP-anti-rabbit IgG (WB) | Cell Signaling | 7074S |
| Ki67 | BD Pharmingen | 550609 |
| NADK1(WB) | Cell Signaling | 55948S |
| NADK1(IP) | Santa Cruz | sc-100347 |
| NADK1(IF) | Proteintech | 15548–1-AP |
| Mouse IgG (IP) | Santa Cruz | Sc-2025 |
| p53 | Santa Cruz | Sc-126 |
| PRPS1/2 | Santa Cruz | sc-100822 |
| H-RAS | Santa Cruz | sc-35 |
| TALDO1 | Abcam | ab137629 |
| TERT | Santa Cruz | sc-377511 |
| TKT | Cell Signaling | 8616 |
| Chemicals, Recombined Proteins, and Inhibitors | ||
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) | Sigma-Aldrich | 238813 |
| (−)-Riboflavin (cell culture) | Sigma-Aldrich | R9504 |
| 2′,7′-Dichlorofluorescin diacetate (DCFDA) | Sigma-Aldrich | D6883 |
| 3X FLAG® Peptide | Sigma-Aldrich | F4799 |
| 4,6-diamidino-2-phenylindole (DAPI) | Vector Laboratories | H-1200 |
| 6xHis-tagged human NADK1 protein | Sigma-Aldrich | SRP8020 |
| β-NAD | Sigma-Aldrich | N7004 |
| β-NADH phosphate disodium salt (NADP+) | Sigma-Aldrich | N5755 |
| β-NADPH | Sigma-Aldrich | N7505 |
| Adenosine triphosphate salt (ATP) | Sigma-Aldrich | A1852 |
| REAGENT or SOURCES | SOURCE | IDENTIFIER |
| ANTI-FLAG® M2 Affinity Gel | Sigma-Aldrich | A2220 |
| BamH1 (restriction enzyme) | New England Biolabs | R3136 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A2153 |
| Crystal violet | Sigma-Aldrich | C0775 |
| Collagenase/ Hyaluronidase (10 x) | Fisher Scientific | NC9694308 |
| D-G6P | Sigma-Aldrich | G7250 |
| Dako Dual Endogenous Enzyme Block | DAKO | S2003 |
| Dialyzed FBS | Sigma-Aldrich | F0392 |
| Disuccinimidyl suberate (DSS) | Sigma-Aldrich | S1885 |
| Dulbecco’s MEM (DMEM) Base (w/o L-Glutamine, L-Cystine, Glucose, Phenol Red, Sodium Pyruvate) | United States Biological | D9815 |
| Dpn1 (restriction enzyme) | New England Biolabs | R0176 |
| FBS | Sigma-Aldrich | TMS-013 |
| FK 866 hydrochloride | Tocris | 4808 |
| Folic acid (cell culture) | MP Biomedicals | 0219466505 |
| Glucose Solution (cell culture) | Fisher Scientific | A2494001 |
| HBSS (1x) | Fisher Scientific | 14025092 |
| HIS-Select® Nickel Affinity Gel | Sigma-Aldrich | P6611 |
| Human G6PD protein (From E. coli) | Sigma-Aldrich | SRP6505 |
| lipofectamine 2000 | Fisher Scientific | 11668019 |
| lipofectamine RNAimax | Fisher Scientific | 13778075 |
| Low temperature melting agarose | Lonza | 50101 |
| MEGM™ BulletKit™ Growth Media | Lonza | CC-3162 |
| MgCl2 | Sigma-Aldrich | 1374248 |
| Mouse diet | LabDiet | 5010 |
| Nicotinamide-2,6,7-13C3-(pyridyl-15N) | Sigma-Aldrich | 809799 |
| Nucleosides | ||
| Deoxy Adenosine monohydrate | DN-1001 | |
| Deoxy Cytidine | DN-1002 | |
| Deoxy Guanosine | DN-1003 | |
| Thymidine | ChemGenes Corp | DN-1004 |
| Ribo Adenosine | RP-1183 | |
| Ribo Cytidine | RP-1184 | |
| Ribo Guanosine | RP-1185 | |
| Ribo Uridine | RP-1186 | |
| Novocastra Epitope Retrieval Solutions | Leica | AR9640 |
| N-Acetyl Cysteine (NAC) | Sigma-Aldrich | A9165 |
| Paraformaldehyde (PFA) | Sigma-Aldrich | P6148 |
| Pierce™ Glutathione Agarose | Fisher Scientific | 16100 |
| Poly (2-hydroxyethyl methacrylate) (ploy-HEMA) | Sigma-Aldrich | P3932 |
| Propidium Iodide (PI) | Sigma-Aldrich | P4864 |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| REAGENT or SOURCES | SOURCE | IDENTIFIER |
| Protein A/G agarose beads | Santa Cruz | sc-2003 |
| SalI (restriction Enzyme) | New England Biolabs | R0138 |
| Sodium pyruvate solution (cell culture) | Sigma-Aldrich | S8636 |
| Streptavidin-Horseradish-Peroxidase | BD Pharmingen | 554066 |
| T4 ligase | Fisher Scientific | EL0014 |
| Thiamine pyrophosphate (cell culture) | Sigma-Aldrich | C8754 |
| Tris-HCl | Sigma-Aldrich | 93363 |
| Trypan blue solution | Fisher Scientific | 15–250-061 |
| Critical Commercial Assays | ||
| Glucose 6 Phosphate Dehydrogenase Activity Assay Kit (Fluorometric) | Abcam | ab176722 |
| Bio-Rad protein assay kit I | Bio-Rad | 5000001 |
| NADP/NADPH Quantitation Colorimetric Kit | BioVision | K347 |
| NADP/NADPH-GloTM assays kit | Promega | G9081 |
| ATP Assay Kit (Colorimetric/Fluorometric) |
Abcam | ab83355 |
| FITC Annexin V Apoptosis Detection Kit II | BD Pharmingen | 556570 |
| Cell Counting Kit-8 | Sigma | 96992 |
| TRIzol | Fisher Scientific | 15596 |
| High-Capacity cDNA Reverse Transcription Kit | Fisher Scientific | 43688 |
| 2X GoTaq® Master Mixes | Promega | M7122 |
| BrdU Cell Proliferation Assay Kit | BioVision | K306 |
| Enhanced chemiluminescence system | Amersham Biosciences | RPN2232 |
| Oligonucleotides | ||
| siRNA for G6PD (h) | Santa Cruz | sc-60667 |
| siRNA for PRPS1/2 (m) | Santa Cruz | sc-62895 |
| siRNA for PRPS1/2 (h) | Santa Cruz | sc-62894 |
| shRNA for G6PD (m) | UPENN HTS core | TRCN0000041446 |
| shRNA for G6PD (h) | Open Biosystem | RHS3979–9595543 |
| shRNA-1 for PRPS1 (m) | UPENN HTS core | TRCN0000024886 |
| shRNA-2 for PRPS1 (m) | UPENN HTS core | TRCN0000024888 |
| shRNA-2 for PRPS1 (h) | UPENN HTS core | TRCN0000367679 |
| shRNA-3 for PRPS1 (h) | UPENN HTS core | TRCN0000355958 |
| shRNA-2 for PRPS2 (m) | UPENN HTS core | TRCN0000025539 |
| shRNA-3 for PRPS2 (m) | UPENN HTS core | TRCN0000025542 |
| shRNA-1 for PRPS2 (h) | UPENN HTS core | TRCN0000010131 |
| shRNA-5 for PRPS2 (h) | UPENN HTS core | TRCN0000010133 |
| shRNA for TALDO1 (h) | Sigma-Aldrich | SHCLNG-NM_006755 |
| shRNA for TKT (h) | Sigma-Aldrich | SHCLNG-NM_001064 |
| Forward primer for human GAPDH | 5’ GTCTCCTCTGACTTCAACAGCG 3’ | |
| Reverse primer for human GAPDH | 5’ ACCACCCTGTTGCTGTAGCCAA 3’ | |
| REAGENT or SOURCES | SOURCE | IDENTIFIER |
| Experimental Models: Organism/Strains | ||
| Athymic nude mice | Homozygous for Foxn1nu | Jackson Laboratory |
| Cell lines | ||
| NIH3T3 (unknown) | ATCC | ATCC® CRL-1658™ |
| BJ (male) | ATCC | ATCC® CRL-2522™ |
| PHMLEB (female) | R. A. Weinberg | Ref (Elenbaas et al., 2001) |
| HEK293T (female) | ATCC | ATCC® CRL-11268™ |
| Plasmids | ||
| 6PGD | pReceiver-Lv151 | EX-A6274-Lv151 |
| Ampho | pCL (Novus Biologicals) | NBP2–29541 |
| dR8.91 | pCMV (Nova lifetech) | PVT2323 |
| Flag-control | pRK5 | Ref (Du et al., 2013) |
| Flag-G6PD | pRK5 | Ref (Du et al., 2013) |
| Flag-G6PDm1 (R365A and K366A) | pRK5 (Site-directed mutagenesis) | N/A |
| Flag-G6PDm2 (K171Q) | PRK5 (Site-directed mutagenesis) | N/A |
| Flag-NADK1 | pRK5 | N/A |
| Flag-NADK1∆N | pRK5 | N/A |
| G6PD | pReceiver-Lv105 | EX-Z2649-Lv105 |
| G6PD | pBABE-puro | Ref (Du et al., 2013) |
| G6PDm1 (R365A and K366A) | pBABE-puro (Site-directed mutagenesis) | N/A |
| G6PDm2 (K171Q) | pBABE-puro (Site-directed mutagenesis) | N/A |
| GST-control | pRK5 | Ref (Guo et al., 2014) |
| GST-NADK1 | pRK5 | N/A |
| GST-NADK1N | pRK5 | N/A |
| hRASV12 | pBABE-puro (Addgene) | #9051 |
| hTERT | pBABE-hygro (Addgene) | #1773 |
| NADK1 | pReceiver-Lv152 | EX-Z7851-Lv152 |
| T antigens | pBABE-neo (Addgene) | #10891 |
| TALDO1 | pReceiver-Lv152 | EX-C0584-Lv152 |
| TKT | pReceiver-Lv105 | EX-M0713-Lv105 |
| VSV-G | pCMV (Addgene) | #8454 |
| Software and Algorithms | ||
| MORPHEUS | Broad Institute | https://software.broadinstitute.org/morpheus/ |
| Image Pro plus7 | Media Cybernetics | N/A |
| ZEN lite | Carl Zeiss Microscopy | N/A |
| BD Accuri C6 | BD Pharmingen | N/A |
| MetaboAnalyst | Open source project | https://www.metaboanalyst.ca/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Fiji ImageJ | Open source project | https://imagej.net/Using_Fiji |
| JACoP plugin | JACoP_.java | https://imagej.nih.gov/ij/plugins/track/jacop2.html |
| MAVEN software35 | Maven Software | N/A |
| GraphPad Prism 8 | Graph Pad Software | N/A |
Highlights.
G6PD can effectively transform immortalized fibroblasts and epithelial cells
Forced G6PD expression bolsters antioxidant defense and nucleotide synthesis
G6PD activates NAD kinase to stimulate NADP+ biosynthesis and converts NADP+ to NADPH
Exogenous antioxidants and nucleosides suffice to transform murine and human cells
ACKNOWLEDGEMENTS
We thank R. Weinberg for PHMLEB cells, and D. Harischandra, S. Ghaisas, S. Yu, L. Huang, A. Mancuso, and W. Prall for technical assistance. This study was supported by NIH grants R01CA182675, R01CA184867, R01CA235760, R01CA243520, and R50CA211437, and U.S. Department of Defense grant W81XWH-15-1-0678.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERETS
J.D.R. is a paid consultant of Pfizer; a founder, director, and stockholder of Farber Partners; a founder and stockholder in Toran Therapeutics; inventor of patents in the area of G6PD and NADPH metabolism held by Princeton University; and a director of the Princeton University-PKU Shenzhen collaboration. I.A.B. is a founder of Proteoform Bio and a paid consultant for Calico, Chimerix, PTC Therapeutics, Takeda Pharmaceuticals, Vivo Capital and Scitemex LLC.
REFERENCES
- Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, et al. (2013). Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep 3, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au SW, Gover S, Lam VM, and Adams MJ (2000). Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency. Structure 8, 293–303. [DOI] [PubMed] [Google Scholar]
- Auvinen M, Paasinen A, Andersson LC, and Holtta E (1992). Ornithine decarboxylase activity is critical for cell transformation. Nature 360, 355–358. [DOI] [PubMed] [Google Scholar]
- Cairns RA, Harris IS, and Mak TW (2011). Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95. [DOI] [PubMed] [Google Scholar]
- Chabner BA, and Roberts TG Jr. (2005). Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer 5, 65–72. [DOI] [PubMed] [Google Scholar]
- Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, and Rabinowitz JD (2019). NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 1, 404–415. [PMC free article] [PubMed] [Google Scholar]
- Christopherson RI, Lyons SD, and Wilson PK (2002). Inhibitors of de novo nucleotide biosynthesis as drugs. Acc Chem Res 35, 961–971. [DOI] [PubMed] [Google Scholar]
- Clasquin MF, Melamud E, and Rabinowitz JD (2012). LC-MS data processing with MAVEN: a metabolomic analysis and visualization engine. Curr Protoc Bioinformatics Chapter 14, Unit14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino C, Grieco D, and Costanzo V (2011). ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J 30, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Moreno MV, Lodi A, Ronen SM, and Ruggero D (2014). Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 157, 1088–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, and Yang X (2013). TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol 15, 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, and Weinberg RA (2001). Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15, 50–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T (2011). Signal transduction by reactive oxygen species. J Cell Biol 194, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman VH, and Shin SI (1974). Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell 3, 355–359. [DOI] [PubMed] [Google Scholar]
- Guo L, Giasson BI, Glavis-Bloom A, Brewer MD, Shorter J, Gitler AD, and Yang X (2014). A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol Cell 55, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Worth AJ, Mesaros C, Snyder NW, Glickson JD, and Blair IA (2016). Diisopropylethylamine/hexafluoroisopropanol-mediated ion-pairing ultra-high-performance liquid chromatography/mass spectrometry for phosphate and carboxylate metabolite analysis: utility for studying cellular metabolism. Rapid Commun Mass Spectrom 30, 1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, and Weinberg RA (1999). Creation of human tumour cells with defined genetic elements. Nature 400, 464. [DOI] [PubMed] [Google Scholar]
- Hahn WC, and Weinberg RA (2002). Modelling the molecular circuitry of cancer. Nat Rev Cancer 2, 331–341. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, Gao P, Selfors LM, Asara JM, and Manning BD (2019). Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science 363, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Mancuso A, Wellen KE, and Yang X (2013a). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, and Yang X (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, and Yang X (2013b). A critical role of glucose-6-phosphate dehydrogenase in TAp73-mediated cell proliferation. Cell Cycle 12, 3720–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr., and McKnight SL (2013). Influence of metabolism on epigenetics and disease. Cell 153, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Thompson IM Jr., Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, et al. (2011). Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306, 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotaka M, Gover S, Vandeputte-Rutten L, Au SW, Lam VM, and Adams MJ (2005). Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr D Biol Crystallogr 61, 495–504. [DOI] [PubMed] [Google Scholar]
- Kuo W, Lin J, and Tang TK (2000). Human glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3 cells and induces tumors in nude mice. Int J Cancer 85, 857–864. [DOI] [PubMed] [Google Scholar]
- Lane AN, and Fan TW (2015). Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43, 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Su X, Quinn WJ 3rd, Hui S, Krukenberg K, Frederick DW, Redpath P, Zhan L, Chellappa K, White E, et al. (2018). Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab 27, 1067–1080 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Wang L, Chen L, Hui S, and Rabinowitz JD (2018). Extraction and Quantitation of Nicotinamide Adenine Dinucleotide Redox Cofactors. Antioxid Redox Signal 28, 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, Muralidhar V, Hosios AM, Israelsen WJ, Gui DY, Newhouse L, Ogrodzinski M, Hecht V, Xu K, Acevedo PN, et al. (2015). Pyruvate kinase isoform expression alters nucleotide synthesis to impact cell proliferation. Mol Cell 57, 95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magni G, Orsomando G, and Raffaelli N (2006). Structural and functional properties of NAD kinase, a key enzyme in NADP biosynthesis. Mini Rev Med Chem 6, 739–746. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, and Aten JA (1993). Measurement of co‐localization of objects in dual‐colour confocal images. J. Microsc. 169, 375–382. [DOI] [PubMed] [Google Scholar]
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H (2012). Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22, 66–79. [DOI] [PubMed] [Google Scholar]
- Patra KC, and Hay N (2014). The pentose phosphate pathway and cancer. Trends Biochem Sci 39, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy EP, Reynolds RK, Santos E, and Barbacid M (1982). A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149–152. [DOI] [PubMed] [Google Scholar]
- Sayin VI, Ibrahim MX, Larsson E, Nilsson JA, Lindahl P, and Bergo MO (2014). Antioxidants accelerate lung cancer progression in mice. Sci Transl Med 6, 221ra215. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, and Brugge JS (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel AC, and Hahn WC (2008). Oncogenic transformation and experimental models of human cancer. Front Biosci 13, 71–84. [DOI] [PubMed] [Google Scholar]
- Serban D, Leng J, and Cheresh D (2008). H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res 102, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Olin-Sandoval V, Gruning NM, Kruger A, Tauqeer Alam M, et al. (2015). The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90, 927–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, and Chang EH (1982). Mechanism of activation of a human oncogene. Nature 300, 143–149. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, and Kinzler KW (2004). Cancer genes and the pathways they control. Nat Med 10, 789–799. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr., and Kinzler KW (2013). Cancer genome landscapes. Science 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105, 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10, 179–206. [DOI] [PubMed] [Google Scholar]
- Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, et al. (2012). Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Geng L, Talmon G, and Wang J (2015). MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. J Biol Chem 290, 6215–6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen L, Liu L, Su X, and Rabinowitz JD (2017). Chemical Basis for Deuterium Labeling of Fat and NADPH. J Am Chem Soc 139, 14368–14371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, and Thompson CB (2019). Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 20, 436–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.