

Abstract
There have been recent extensive studies and rapid advancement on the pathogenesis underlying idiopathic pulmonary fibrosis (IPF), and intricate pathogenesis of IPF has been suggested. The purpose of this study was to clarify the logical relationship between these mechanisms. An extensive search was undertaken of the PubMed using the following keywords: “etiology,” “pathogenesis,” “alveolar epithelial cell (AEC),” “fibroblast,” “lymphocyte,” “macrophage,” “epigenomics,” “histone,” acetylation,” “methylation,” “endoplasmic reticulum stress,” “mitochondrial dysfunction,” “telomerase,” “proteases,” “plasminogen,” “epithelial-mesenchymal transition,” “oxidative stress,” “inflammation,” “apoptosis,” and “idiopathic pulmonary fibrosis.” This search covered relevant research articles published up to April 30, 2020. Original articles, reviews, and other articles were searched and reviewed for content; 240 highly relevant studies were obtained after screening. IPF is likely the result of complex interactions between environmental, genetic, and epigenetic factors: environmental exposures affect epigenetic marks; epigenetic processes translate environmental exposures into the regulation of chromatin; epigenetic processes shape gene expression profiles; in turn, an individual's genetic background determines epigenetic marks; finally, these genetic and epigenetic factors act in concert to dysregulate gene expression in IPF lung tissue. The pathogenesis of IPF involves various imbalances including endoplasmic reticulum, telomere length homeostasis, mitochondrial dysfunction, oxidant/antioxidant imbalance, Th1/Th2 imbalance, M1–M2 polarization of macrophages, protease/antiprotease imbalance, and plasminogen activation/inhibition imbalance. These affect each other, promote each other, and ultimately promote AEC/fibroblast apoptosis imbalance directly or indirectly. Excessive AEC apoptosis and impaired apoptosis of fibroblasts contribute to fibrosis. IPF is likely the result of complex interactions between environmental, genetic, and epigenetic factors. The pathogenesis of IPF involves various imbalances centered on AEC/fibroblast apoptosis imbalance.
Keywords: Alveolar epithelial cell, Apoptosis, Idiopathic pulmonary fibrosis, Fibroblast, Pathogenesis
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, age-related, progressive lung disease characterized by progressive lung fibrogenesis and the histological picture of usual interstitial pneumonia (UIP). It is associated with increased cough and dyspnea and impaired quality of life. The median survival of patients with IPF is 3 to 5 years from diagnosis.[1]
The paradigm about disease pathogenesis has shifted from belief that chronic inflammation is the direct cause of IPF[2,3] to the idea that oxidative stress plays an important role in the onset and progression of IPF[4] to more recent suggestion that repetitive micro-injuries and dysfunction of the alveolar epithelial cells (AECs) injury lead to uncontrolled activation and proliferation of fibroblasts, and excessive accumulation of extracellular matrix (ECM), by generating crucial profibrotic signaling and mediators.[5,6] Correspondingly, the therapy for IPF has also undergone a transition from treatment with anti-inflammatory and immunosuppressant drugs (eg, prednisone and azathioprine) to treatment with antioxidant drugs (eg, N-acetylcysteine [NAC]) and to treatment with more recent antifibrotic drugs (nintedanib and pirfenidone). Unfortunately, it gradually became evident that anti-inflammatory and antioxidative therapy was ineffective in improving survival in patients with IPF.[7,8] Although pirfenidone and nintedanib are effective at reducing lung function decline, neither is curative for the disease.[9,10] Does the ineffectiveness of anti-inflammatory and antioxidative therapy mean that inflammation and oxidant/antioxidant imbalance are not particularly important in the pathogenesis of IPF?
Various mechanisms underlying IPF have been identified. A body of evidence indicates that IPF is related to possible triggers or environmental risk factors, genetic predisposition, and epigenetics,[11–13] and these various mechanisms — such as endoplasmic reticulum (ER) stress,[14–16] telomere length homeostasis,[17] mitochondrial dysfunction,[18] oxidant/antioxidant imbalance,[19] T helper type 1 cell (Th1)/Th2 imbalance,[20–22] M1–M2 polarization of macrophages,[23–25] protease/antiprotease imbalance,[26–28] plasminogen activation/inhibition imbalance,[29] AEC/fibroblast apoptosis imbalance,[30–33] epithelial-mesenchymal transition (EMT), and transforming growth factor (TGF)-β — are involved in the pathogenesis of IPF. What is the relationship during environmental risk factors, genetic predisposition, and epigenetics, and what are their underlying mechanisms for initiating IPF? What relationships and interactions exist among various mechanisms mentioned above?
In the present review, we begin with a discussion on the role of possible triggers, genetic instability, and epigenetic changes in the pathogenesis of IPF and their interplay, followed by the contribution of various imbalances centered around an AEC/fibroblast apoptosis imbalance in the pathogenesis of IPF and their interaction and end with opinions on potential therapeutic targets based on the above-mentioned complex pathogenesis.
Etiology: Environmental, Genetic, and Epigenetic Factors
Possible triggers/environmental risk factors
Although the exact cause of IPF remains unclear, toxic exposures, including cigarette smoking, environmental and occupational exposures, infection, and gastroesophageal reflux, appear to be important contributing factors.[11–13]
Genetic predisposition
Several genetic mutations have been implicated in familial IPF, which broadly are subdivided into two categories: genes related to surfactant protein processing and trafficking (SFTPA1,[34,35]SFTPA2,[36]SFTPB,[37]SFTPC,[38] ATP-binding cassette-type 3[39]) and genes that maintain telomere length (TERT and TERC).[40]
The mutation results in an aberrant surfactant protein that cannot be correctly processed, resulting in protein misfolding, accumulation, induction of ER stress, and apoptosis of AECs.[41–43] Mutations in TERT or TERC lead to loss of telomerase activity. Consequently, telomeres shorten successively with each cell division, and when they achieve a critical length they activate p53-dependent apoptosis or replicative senescence.[44] Additionally, a genome-wide association study found that a single-nucleotide polymorphism in the promoter region of the mucin 5B gene[45–47] and a loss-of-function polymorphism in TOLLIP[46] which is a gene encoding the inhibitor of toll-like receptor 4 (TLR4) are strongly associated with IPF. Some of these mutations have been reported not only in several familial IPF cases but also in sporadic cases of IPF,[38,40,48] which suggests that sporadic and familial cases of IPF probably reflect a continuum of genetic risk and that sporadic IPF is also a disease with a genetic predisposition.
Epigenetics
Epigenetic factors contribute to the dysregulation of gene expression in IPF lung, which leads to changes in gene expression without a change in the gene-coding sequences. The most common epigenetic mechanisms include DNA methylation and histone modifications and non-coding RNA (ncRNA) regulation.
Methylation of DNA
Methylation of DNA is usually associated with decreased gene expression. For example, Thy1, a receptor that inhibits the differentiation of fibroblasts to myofibroblasts, is not expressed in fibroblastic foci in vivo.[49] Loss of Thy1 occurs through epigenetic silencing caused by hypermethylation of cytosine-guanine islands in the gene promoter. As a result, such epigenetic change leads to the aggressive behavior of IPF fibroblasts.
Histone modifications
In quiescent cells, genomic DNA is wrapped around histones to form nucleosomes, restricting transcriptional access to the DNA. Methylation, acetylation, phosphorylation, and ubiquitylation of histone tails occur at specific residues and control gene expression by regulating DNA accessibility to RNA polymerase II and transcription factors. For example, acetylation of histones results in the relaxation of chromatin, facilitating gene transcription. Histone acetylation and deacetylation on lysine residues by histone acetyltransferases and histone deacetylation by histone deacetylases (HDACs) are closely associated with active and repressive chromatin states and increased and decreased transcription factor binding to specific gene promoters, respectively.[50] Prostaglandin (PG)E2, a cyclooxygenase (COX)-dependent arachidonic acid metabolite in the lung, exerts an important antifibrotic effect by promoting the survival of AECs while increasing the sensitivity of fibroblasts/myofibroblasts to apoptosis, whereas fibroblasts from IPF patients are unable to up-regulate the COX-2 enzyme and are thereby deficient in PGE2 production. Defective histone acetylation is responsible for the diminished expression of COX-2.[51] One study found aberrant expression and activity of histone deacetylases in sporadic IPF and observed that compared with control lungs, protein levels of HDACs are significantly elevated in IPF lungs, and apoptosis resistance in IPF fibroblasts is mediated by enhanced activity of HDAC enzymes.[52] Pan-HDAC inhibition by LBH589 may present a novel therapeutic option for patients with IPF. Huang et al[53] found that increased histone deacetylase expression was partially responsible for the down-regulation of the factor-associated suicide (Fas) death receptor in fibrotic fibroblasts, another reason for apoptosis resistance in fibroblasts.
ncRNA regulation
ncRNAs are often considered a part of the epigenome. ncRNAs are functional RNA molecules that are not translated into proteins, including transfer RNAs, ribosomal RNAs, small nucleolar RNAs, microRNAs (miRNAs), and long non-coding RNAs.[54] miRNAs are the most extensively studied family of small ncRNAs. miRNAs are short (∼22 nt) single-stranded ribonucleic acids functioning as post-transcriptional regulators of gene expression that play important roles by binding to specific sequences, blocking translation, or causing degradation of the target mRNA, which results in gene silencing. Through various mechanisms, some miRNAs (eg, miR-21,[55,56] miR-31,[57] miR-145,[58] miR-154,[59] and miR-199a[60]) play a role in promoting fibrosis during the pathogenesis of IPF, and their expression levels are often elevated, while others (such as let-7d,[56,61] miR-9,[62] miR-18a,[63] miR-26a,[56] miR-27b,[64] miR-29,[65] miR-30a,[56,66] miR-155,[67] miR-200,[68] miR-221,[69] miR-323a,[70] miR-326,[71] miR-338,[72] miR-375,[73] and miR-486[74]) prevent fibrosis, and their expression levels are reduced. One recent study demonstrated that the long-intervening non-coding RNAs (lincRNAs) LINC00960 and LINC01140 can regulate fibroblast proliferation and inflammation, while changes in LINC01140 expression may mediate a reduced inflammatory response in IPF fibroblasts.[75] While the mechanisms by which these lincRNAs regulate these responses are unknown, it is speculated that the biological actions of lincRNAs may be mediated through domains containing conserved sequences, which interact with proteins and/or base pairs with RNA/DNA.[76]
Interactions between environmental, genetic, and epigenetic factors
IPF is likely the result of complex interactions between environmental, genetic, and epigenetic factors.
First, environmental exposures strongly affect epigenetic marks.[77] Epigenetic processes translate environmental exposures into the regulation of chromatin. Cigarette smoke, the main environmental risk factor for IPF, influences the methylation of specific promoters in genes involved in the pathogenesis of IPF, such as WNT7A.[78] One study revealed how cigarette smoke influences histone modifications and chromatin accessibility.[79] Additionally, epigenetic processes shape the gene-expression profiles involved in disease pathophysiology. In turn, an individual's genetic background determines epigenetic marks in two ways: by direct inheritance[80] and by genetic variants. Genome-wide studies demonstrate a strong genetic component to inter-individual variation in methylation and histone modification profiles.[81] Finally, genetic and epigenetic factors act in concert to dysregulate gene expression in IPF lung [Figure 1].
Figure 1.
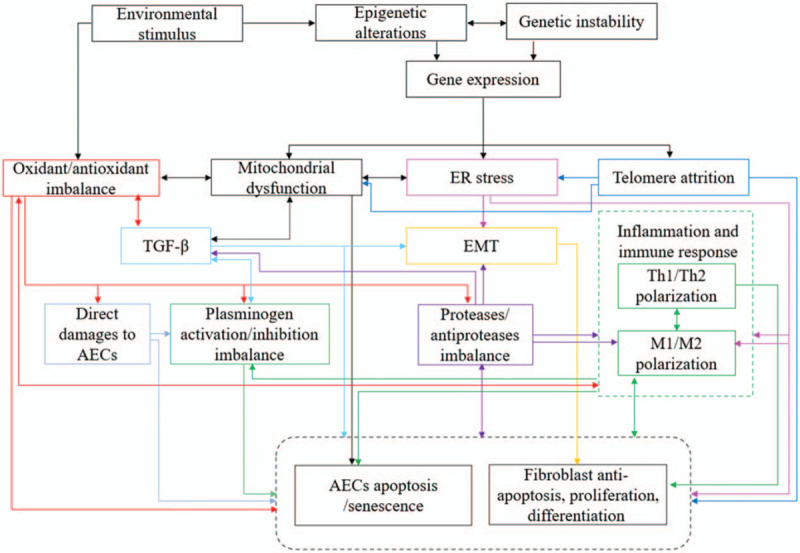
The etiology and pathogenesis of IPF: various imbalances centered on AECs/fibroblasts apoptosis imbalance. AECs: Alveolar epithelial cells; EMT: Epithelial-mesenchymal transition; ER: Endoplasmic reticulum; IPF: Idiopathic pulmonary fibrosis; TGF-β: Transforming growth factor-β; Th: T helper.
Various Imbalances Centered on an Apoptosis Imbalance in AECS and Fibroblasts
ER stress
Accumulation of mutant and misfolded proteins in the ER has been shown to induce severe ER stress. Reduced clearance of misfolded proteins (eg, proteasomal dysfunction, impaired autophagy), disturbances in redox homeostatic, nutrient deprivation, and environmental insults (eg, viral infection) can bring about the aggregation of unfolded or misfolded proteins, exacerbating ER stress, which triggers the activation of the unfolded protein response (UPR) to alleviate ER stress and sustain the cellular homeostasis.[82] However, when stress is overloaded or prolonged, the UPR becomes maladaptive and triggers apoptotic cell death through activating cell death pathways such as caspase-4, c-Jun NH2 terminal kinase, and C/EBP homologous protein.[83–85] ER stress may cause fibrosis through AEC apoptosis, EMT, myofibroblast differentiation, and M2 macrophage polarization.[14–16]
Telomere length homeostasis
A telomere is a region of tandem repeats of short DNA sequences at the ends of chromosomes, which are important for their stability and allow the complete replication of the ends. Telomere length homeostasis is essential for proper cellular function.[86] Telomeres shorten with every cell division owing to incomplete replication of telomere caps on the ends of chromosomes. Telomerase, a specialized RNA-directed DNA polymerase, elongates telomere sequences at the termini of chromosome DNA, preventing progressive telomere loss and maintaining chromosome stabilization. Mutations in the essential telomerase genes, found in both sporadic and familial IPF cases, result in telomerase loss of function and reduced telomerase activity, thereby accelerating the telomere shortening.[40] In addition, other factors such as smoking,[87] chronic inflammation, and cumulative oxidative stress can cause telomere shortening.[88]
When telomeres shorten to a critical length, they can be sensed as double-stranded DNA breaks, activating the DNA damage sensor and checkpoint inhibitor p53, which leads to apoptosis or replicative senescence,[17] impairing normal lung epithelial homeostasis.
Mitochondria-regulated homeostasis
Contents of mitochondrial dysfunction
Mitochondrial dysfunction, which means mitochondria-regulated homeostasis is broken, in IPF includes reduced efficiency of electron transport chain (ETC) function along with excessive production of reactive oxygen species (ROS), decreased mitochondrial biogenesis, and impaired mitophagy — a key pathway for mitochondria turnover and the removal of dysfunctional mitochondria.
Mechanisms of mitochondrial dysfunction
First, telomere shortening stimulates the DNA damage sensor and checkpoint inhibitor p53; activated p53 then further impairs mitochondrial biogenesis by repressing expression of a key mediator of mitochondrial biogenesis, Peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1α, resulting in inefficient oxidative phosphorylation and increased ROS production.[89] Second, phosphatase and tensin homolog-induced putative kinase 1 (PINK1) labels the dysfunctional mitochondrion for trafficking to the autophagosome. ER stress then causes downregulation of PINK1 expression in AECs, resulting in an accumulation of dysmorphic mitochondria, deficient mitophagy, reduced ETC activity, and increased ROS production.[90] As a consequence, ROS produced by dysfunctional mitochondria can cause further ER stress, thus forming a vicious circle.[91] Third, oxidative stress can cause mitochondrial dysfunction. Excessive ROS promote mitochondrial DNA (mtDNA) damage,[92] but additionally, that mtDNA damage can augment further increases in ROS production,[18,93] leading to a vicious feed-forward cycle that worsens mitochondrial dysfunction. In addition, a profibrotic environment promotes mitochondrial dysfunction in pulmonary epithelial cells. One study found that TGF-β down-regulates mitochondrial ETC function, leading to increased ROS production.[94] Increased ROS can further activate latent TGF-β, thus creating a vicious feed-forward cycle with the potential to recruit fibroblasts and promote fibrogenesis.[95] Mitochondrial dysfunction can also activate TGF-β through metabolic reprogramming: the metabolic shift from the highly efficient method of ATP production to the less efficient method of glycolysis due to mitochondrial dysfunction — and correspondingly, fatty acid oxidation increases in response to the shift to glycolysis in fibroblasts or alveolar macrophages in fibrotic lung tissue — resembles the cancer-associated Warburg effect.[96–100] Glycolytic flux increases lactate production and lowers the local tissue pH, causing increased activation of TGF-β, which induces differentiation of fibroblasts to myofibroblasts.[96]
The consequences of mitochondrial dysfunction
To summarize, telomere shortening, ER stress, oxidative stress, and a profibrotic environment (TGF-β) promote mitochondrial dysfunction in pulmonary epithelial cells; as a consequence, mitochondrial dysfunction can further stimulate ER stress, oxidative stress, and TGF-β, creating a vicious feed-forward cycle. In addition, mitochondrial dysfunction promotes AEC apoptosis and senescence.[18]
Oxidant/antioxidant imbalance
Both endogenous and exogenous sources included in the oxidative stress and generation of ROS
One theory regarding the pathogenesis of IPF is that an oxidant/antioxidant imbalance, known as oxidative stress, exists in the alveolar regions of affected lungs. In lungs from IPF patients, ROS are produced by inflammatory cells (lymphocytes, macrophages, and neutrophils) and parenchymal cells (eg, myofibroblasts and fibroblasts) in response to cytokines, growth factors, and exogenous oxidizing agents such as air pollutants and cigarette smoke.[101,102] Some factors, such as TGF-β, inflammation, and mitochondrial dysfunction, can induce the sustained production of ROS.[18,93]
Effects of oxidant/antioxidant imbalance
ROS further amplify the profibrotic TGF-β downstream signal and promote inflammation.[19] Therefore, reciprocal promotion of TGF-β and ROS, of inflammation and ROS, and of mitochondrial dysfunction and ROS results in a perverse and vicious cycle for fibrosis. ROS also cause direct damages to AECs, predisposing individuals to lung fibrosis.[4] Importantly, ROS may contribute to a protease/antiprotease imbalance because they can directly induce matrix metalloproteinase (MMP) transcription,[103] activate MMPs,[104] and inactivate protease inhibitors.[105] In addition, mitochondrial ROS promote AEC cell apoptosis and senescence, while fibroblasts are resistant to apoptosis.[1,92,106,107] One reason for this is that ROS activate the persistent platelet-derived growth factor (PDGF) receptor production that is implicated in fibroblast differentiation and proliferation in fibrosis.[1,108] Considered together, ROS result in other pathophysiological consequences, including TGF-β activation, inflammation, protease/antiprotease imbalance, AEC damage, apoptosis and senescence, and fibroblast differentiation; all of these are implicated in driving lung fibrosis [Figure 1].
Inflammation and immunity
Th1/Th2 imbalance
The toll-like receptor (TLR) signaling pathway and T-cell differentiation mediated by fate-specifying cytokines
Although the role of inflammation in the pathogenesis of IPF remains controversial, evidence supports the fact that immune responses play an important role in the initiation or progression of IPF. Pathogen-associated molecular patterns and damage-associated molecular patterns are recognized by TLRs expressed in monocytes, macrophages, dendritic cells (DCs), mast cells, neutrophils, eosinophils, basophils, and epithelial cells, thereby initiating an innate immune response, which activates nuclear factor-κB (NF-κB) to produce proinflammation cytokines or activates interferon (IFN) regulatory factors to produce type I IFNs. Cytokines produced by these cells prime the differentiation of naïve T cells into Th1, Th2, Th17, or regulatory T cell (Treg) phenotypes according to the antigenic stimulation involved[109] [Figure 2].
Figure 2.
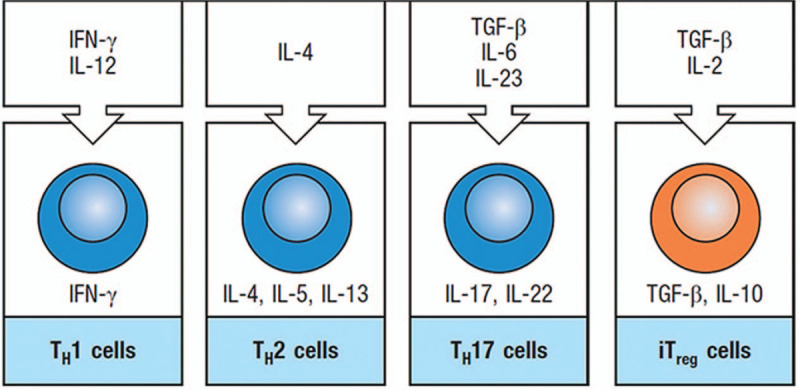
The differentiation of naïve T cells into specific phenotypes induced by fate-specifying cytokines. IFN-γ: Interferon-γ; IL: Interleukin; iTreg: Inducible regulatory T population; TGF-β: Transforming growth factor-β; TH: T helper.
The existence and causes of Th1/Th2 imbalance in IPF
T-cell activation in IPF patients is likely skewed toward a Th2 response.[20–22] Studies have shown that Th1 cells and secretory cytokine (IFNγ) levels decrease in the bronchoalveolar lavage or circulation of patients with IPF,[110] whereas Th2 cells and associated cytokines (interleukin [IL]-4, IL-5, and IL-13) increase in the lungs or circulation of patients with IPF.[22,111] Th1/Th2 imbalance toward a Th2 phenotype is induced by damaged tissues or mediators that promote Th2 differentiation. One recent study showed an elevated level of galectin-1 in bronchoalveolar lavage of patients with IPF, which contributed to a Th1/Th2 imbalance toward a Th2 phenotype by selectively inducing apoptosis on Th1 and Th17 cells, but not on naive, Th2, or regulatory FoxP3+ T cells.[112,113] PGE2 can direct Th2 differentiation by inhibiting IL-12 production by monocytes cultured in the presence of IL-4 and granulocyte-macrophage colony-stimulating factor.[114] In addition, a decrease in a suppressor of cytokine signaling (SOCS)-1 expression is involved in IPF, because SOCS-1 may inhibit Th2 differentiation as an inhibitor of profibrotic cytokines, such as IL-4.[115]
Th1 and Th2 cells take on opposite roles in fibrogenesis
Th1 cells and their secretory cytokines (IFNγ) are thought of as being antifibrotic. For instance, one study showed that IL-12 exerted an antifibrotic effect by promoting the production of IFNγ in a bleomycin-induced animal model of IPF.[116] Inhibiting Th1 differentiation by targeting of the transcription factor T-bet increased bleomycin-induced lung injury.[117] By contrast, Th2 and associated cytokines (IL-4, IL-5, and IL-13) exhibit a fibrogenic property by stimulating fibroblast proliferation, fibroblast differentiation into myofibroblasts, and collagen production.[118,119] Therefore, the imbalance between Th1 and Th2 is actually an imbalance between profibrosis and antifibrosis.
M1–M2 polarization of macrophages
Macrophages can be broadly classified as M1 and M2 according to their phenotype and role.[120] The M1–M2 polarization of macrophages and the Th1–Th2 polarization of T cells are well-correlated processes with positive feedback between each other. A high amount of IFN-γ produced by Th1 cells is the major inducing factor in the activation of M1 macrophages, whereas high amounts of IL-4 and IL-13 produced by Th2 cells can activate M2 macrophages, which suppress inflammation and confer a tissue repair function.[121–124] An overzealous or prolonged M2 polarization results in excessive amounts of profibrotic mediators (TGF-β1, PDGF, and tissue inhibitors of metal proteinase 1 [TIMP1], and CCL18), which promote fibroblast accumulation and the differentiation of fibroblasts into myofibroblasts, and collagen production and deposition. Thus, M2 macrophages act as the key effector arm of Th2-driven type 2 responses in fibrosis.[23–25] ER stress has been reported to regulate skew toward M2 polarization.[125,126]
Protease/antiprotease imbalance
MMPs are a family of endopeptidases with 23 members in humans. These enzymes are responsible for ECM degradation and for cleaving membrane receptors and various bioactive mediators (such as cytokines, growth factors, and chemokines).[127] It is well known that an imbalance of MMPs and their inhibitors, the TIMPs, mainly due to overexpression of MMPs, is implicated in the pathogenesis of IPF.[128] Most MMPs, such as MMP1,[129] MMP3,[130] MMP7,[129–131] MMP-8,[130,131] MMP-9,[130,131] MMP12,[131] MMP14,[132] and MMP28,[133] are overexpressed in IPF lungs compared with controls and play a profibrotic role, contributing to the progression of IPF. Protease and antiprotease imbalance may cause fibrosis by promoting EMT,[26] by inflammation,[27] by promoting M1–M2 polarization,[28] and by activating TGF-β signaling. It is worth noting that not all MMPs have harmful effects, and sporadic MMPs may have beneficial effects; for example, MMP19 upregulated in AECs plays a protective role in IPF.[134]
Plasminogen activation/inhibition imbalance
Plasmin plays an antifibrotic role in the pathogenesis of IPF by promoting fibrinolysis as well as degradation of ECM. Plasmin has been shown previously to induce COX-2, PGE2, and hepatocyte growth-promoting factor (HGF) synthesis in AECs and fibroblasts, which are important effectors of antifibrotic actions. By contrast, plasminogen activator inhibitor 1 (PAI-1) plays a profibrotic role in the pathogenesis of IPF as a primary inhibitor of urokinase-type and tissue-type plasminogen activators, respectively, which convert plasminogen into plasmin.[135] One mechanism whereby increased PAI-1 promotes lung fibrosis involves increasing the sensitivity of fibroblasts to TGF-β.[136] PAI-1 expression is increased relative to the plasminogen activators, contributing to plasminogen activation/inhibition imbalance. AEC damage causes PAI-1 overexpression in AECs and lung macrophages.[29] Evidence shows that there is a positive feedback loop between PAI-1 and TGF-β.[136] Moreover, ROS, IL-1β, and tumor necrosis factor (TNF) promote expression of PAI-1.[137]
AEC/fibroblast apoptosis imbalance
Homeostasis of AECs and fibroblasts
In the normal repair of AECs, activated fibroblasts produce ECM and provide a provisional scaffold for AEC migration, proliferation, and re-epithelization. After the AEC damage has been repaired, fibroblasts undergo apoptosis in order to restore normal cellular homeostasis and maintain tissue architecture and organ function. Fibroblast apoptosis is essential in normal wound healing but is dysregulated in IPF.
Imbalance of AEC and fibroblast apoptosis in IPF
In IPF, the phenotypes of fibroblasts and AECs change. Considering AECs as an example, aberrantly activated AECs, which are characterized by cells undergoing apoptosis, senescence, or damage, secrete cell-type-specific profibrotic and proinflammatory cytokines. As a result, the dysregulated crosstalk and abnormal mediators between them lead to AEC apoptosis, fibroblast anti-apoptosis, and abnormal ECM.
Down-regulated factors promoting the survival of AECs and inducing fibroblast apoptosis
For instance, in IPF, PGE2[138] produced by AECs and fibroblasts, and HGF and keratinocyte growth factor produced by fibroblasts,[139] reduce. Studies showed that both miR-30a and miR-29 in AECs can suppress AEC apoptosis[140] and promote apoptosis of lung fibroblasts in an IPF animal model.[141] Their expression decreases in AECs during IPF development. Interestingly, the conclusions of the research on fibroblast growth factor (FGF) expressed by both fibroblasts and AECs in the lung are controversial. Some studies report that FGF expression levels decrease (eg, FGF-10[142]) and FGF plays a pathological role (fibroblast invasion) in IPF, while other studies report that FGF expression levels increase (eg, FGF-10,[143] FGF-1,[144] basic FGF,[145] FGF-9 and FGF-18,[146] and FGF-23[147]) and FGF plays a protective role (AEC survival and fibroblast apoptosis). A possible explanation for these findings is that, on the one hand, FGF may have a potential dual nature in the lung, while, on the other hand, this heterogeneity may originate from different specimen types and different microenvironments (in vivo or ex vivo).
Overexpressed factors inducing the apoptosis of AECs and promoting proliferation or anti-apoptosis of fibroblasts are overexpressed
TGF-β and ROS overexpressed by fibroblasts promote AEC apoptosis while increasing resistance to apoptosis in fibroblasts.[1,92,106,107] AECs can also excessively secrete profibrotic mediators, including TGF-β, connective tissue growth factor (CTGF), PDGF, and some Th2 cytokines (eg, IL-17E) [Figure 3]. These mediators not only promote AEC apoptosis via an autocrine mechanism but also incite overactivation and proliferation of fibroblasts.[30,148,149]
Figure 3.
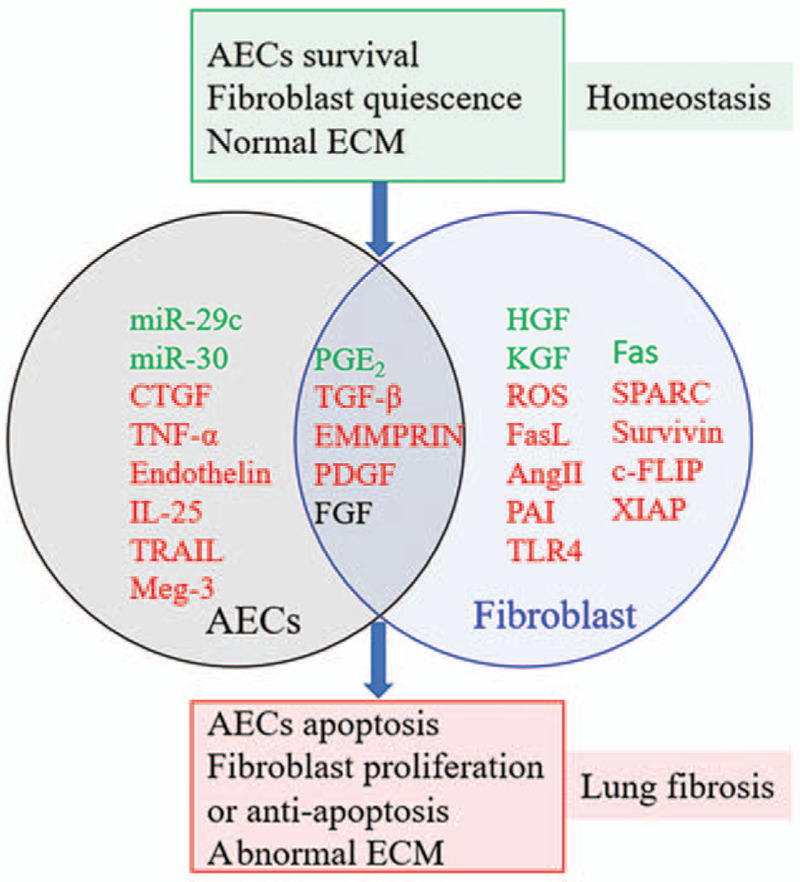
Profibrotic phenotype of AECs and fibroblasts and abnormal mediators produced by AECs or fibroblasts contribute to fibrosis. The mediators in the black circle named AECs on the left indicate the mediators secreted by AECs. The mediators in the blue circle named fibroblasts on the right indicate the mediators secreted by fibroblasts. Mediators marked in the green font indicate whose expression is downregulated. Mediators identified in the red font indicate whose expression is upregulated. The mediators marked in the black font indicate whether their expression level is downregulated or upregulated is still controversial. AECs: Alveolar epithelial cells; Ang II: Angiotensin II; c-FLIP: Cellular FLICE-like inhibitory protein; CTGF: Connective tissue growth factor; ECM: Extracellular matrix; EMMPRIN: ECM metalloproteinase inducer; Fas: Factor-associated suicide; FasL: Factor-associated suicide ligand; FGF: Fibroblast growth factor; HGF: Hepatocyte growth-promoting factors; IL-25: Interleukin-25; KGF: Keratinocyte growth factor; Meg-3: Maternally expressed gene 3; miR: MicroRNA; PAI: Plasminogen activator inhibitor; PDGF: Platelet-derived growth factor; PGE2: Prostaglandin E2; ROS: Reactive oxygen species; SPARC: Secreted protein acidic and rich in cysteine; TGF-β: Transforming growth factor-β; TLR4: Toll-like receptors 4; TNF-α: Tumor necrosis factor-α; TRAIL, TNF-related apoptosis-inducing ligand; XIAP: X-linked inhibitor of apoptosis.
Other overexpressed factors inducing apoptosis of AECs
In addition to the factors mentioned above (TGF-β, ROS, CTGF, PDGF, and IL-17E), the following further elaborate on other factors that can promote AEC apoptosis. Angiotensin-II[150] and factor-associated suicide ligand (FasL)[151] overexpressed by fibroblasts may directly or indirectly induce apoptosis of adjacent AECs. The lincRNA maternally expressed gene 3 increased in AECs of IPF[31] is known to induce apoptosis by activating pro-apoptotic protein P53.[152] TNF-related apoptosis-inducing ligand, a member of the TNF superfamily, which is upregulated in AECs of IPF lung tissue samples, has been shown to induce apoptosis via DR4 and DR5 receptor binding.[153] Numerous studies prove that telomere attrition,[154] unchecked ER stress,[107] and mitophagy deficiency[18] can induce AEC apoptosis in lungs with IPF [Figure 3].
Other factors promoting proliferation or anti-apoptosis of fibroblasts
In addition to the factors mentioned above (TGF-β, ROS, CTGF, PDGF, and IL-17E), the following further elaborate on other factors that can promote the proliferation or anti-apoptosis of fibroblasts. Increased expression of inhibitors of apoptosis in fibroblasts, such as surviving,[155] the Fas death receptor, cellular FLICE-like inhibitory protein,[156] PAI-1,[157] secreted protein acidic and rich in cysteine (SPARC),[158] and X-linked inhibitor of apoptosis (XIAP),[159] protects lung fibroblasts from apoptosis. Studies demonstrate decreased Fas expression in fibroblasts during histone modifications,[53] and ECM metalloproteinase inducer overexpression[160] in fibroblasts from IPF can induce an apoptosis-resistant phenotype of fibroblasts in IPF. Apoptosis resistance in fibroblasts is partially mediated by TLR4 activation, which results in the transcription of pro-survival signaling factors via NF-κB and PI3K-Akt activation.[161,162] Of note, injured or activated AECs produce virtually mediators contributing to apoptosis-resistant phenotypes of fibroblasts, such as TNF-α, endothelin 1, CXCL12,[163] IL-25, and IL-17BR (IL-25's receptor).[32] The underlying mechanism is as follows: increased levels of SPARC, TGF-β, and endothelin-1 in IPF lung promote fibroblast resistance to apoptosis by activating the Akt signal pathway.[158,164] Both TGF-β and endothelin-1 induce XIAP expression to inhibit Fas-mediated apoptosis in fibroblasts.[165] CXCL12, a potent chemoattractant for local fibroblasts, is essential for EMT and fibroblast-to-myofibroblast differentiation.[33] One study demonstrated that the mRNA and protein levels of IL-25 and IL-17BR (IL-25's receptor) are significantly higher in IPF patients and that IL-25 potentiates the expression of CTGF in AECs and the recruitment and proliferation of lung fibroblasts.[32]
AEC/fibroblasts apoptosis imbalance is the core of IPF
In summary, the pathogenesis of IPF involves various imbalances including ER, telomere length homeostasis, mitochondrial dysfunction, oxidant/antioxidant imbalance, Th1/Th2 imbalance, M1–M2 polarization of macrophages, protease/antiprotease imbalance, plasminogen activation/inhibition imbalance, and AEC/fibroblast apoptosis imbalance. As can be seen from the above description and Figure 1, among them, AEC/fibroblast apoptosis imbalance is the core of IPF, because although other imbalances affect each other and promote each other, they ultimately promote AEC/fibroblast apoptosis imbalance directly or indirectly.
Excess AEC apoptosis induces alveolar basement membrane destruction, leading to inefficient re-epithelialization and the recruitment of fibroblasts to the site of damage to promote excess ECM deposition and pulmonary architecture destruction.[166] An alternative mechanism is that the apoptotic AECs can directly trigger progressive fibrosis by inducing a response in neighboring cells. Studies have revealed that uptake of apoptotic AECs by macrophages contributes to fibrosis through the increased expression of TGFβ, a growth factor with both anti-inflammatory and profibrotic activities.[167,168]
Fibroblast apoptosis is the primary mechanism by which fibroblasts are removed, whereas fibroblasts differentiate into myofibroblasts in profibrotic microenvironments owing to impaired apoptosis of fibroblasts, resulting in the aggregation of activated myofibroblasts. Myofibroblasts are the major actors in the fibrogenetic process and are characterized by expression of α-smooth muscle actin and synthesis of varying amounts of ECM, including collagens, glycoproteins, and proteoglycans, which will ultimately lead to lung architecture destruction and fibrosis.[169] Indeed, myofibroblast accumulation and activation are hallmarks of the pathobiology of UIP, which is a typical pathological manifestation of IPF.[6,170]
Views on Therapeutic Approaches Based on Complex Pathogenesis
Therapy targeting inflammation and immune response
The following evidence could help explain why anti-inflammatory drugs are mostly ineffective in IPF: First, B cells are the antibody-producing cells of the adaptive immune system and B cell abnormalities are involved in the pathogenesis of IPF,[171] but lung diseases involving humoral immunity mediated by B cells are usually insufficiently responsive or refractory to glucocorticosteroid therapy.[172] Second, studies show that glucocorticosteroid treatment is not sufficient to inhibit the activity of alveolar macrophages, instead, it activates a class of M2 macrophages and causes M1–M2 polarization of macrophages.[173,174] In addition, a recent study demonstrates that activated and non-proliferating lymphocytes and mature DCs are involved in the pathogenesis of IPF,[175] glucocorticosteroid-based therapy has a poor activity on differentiated lymphocytes and especially mature DCs.[176] As the research progresses, we will find that inflammation and immune response is still important in the pathogenesis of IPF, but it is complicated and needs to be further studied.
Therapy targeting oxidant/antioxidant imbalance
As shown in Figure 4, oxidant mainly refers to the following ROS: 1O2, O2−, HO2., HO., and H2O2. Endogenous ROS are mainly produced through the ETC in mitochondria and by chemical reactions catalyzed by NADPH oxidase (NOX). The antioxidant defense system includes small-molecular-weight antioxidants (eg, vitamin E/C, glutathione [GSH]), superoxide dismutase, and enzymes that catalyze H2O2 metabolism (GSH peroxidase, GSH reductase, and catalase).[177]
Figure 4.
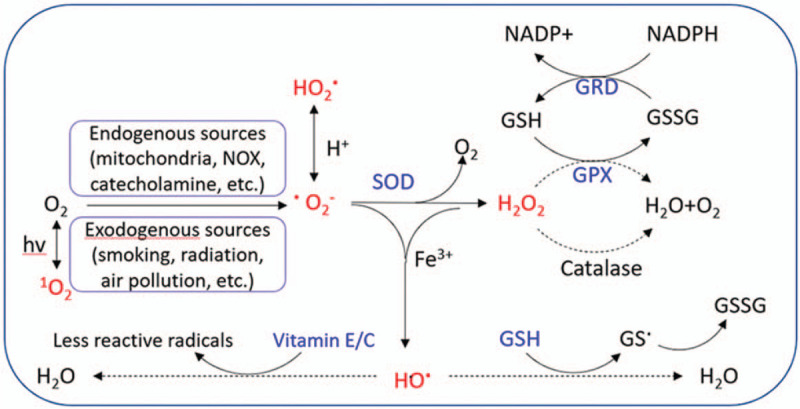
Source and metabolic pathway of reactive oxygen species (ROS). The chemical molecules marked in the red font are ROS, and the substances marked in the blue font belong to the antioxidant system. GPX: GSH peroxidase; GRD: GSH reductase; GSH: Glutathione; GSSG: Oxidized glutathione; NOX: NADPH oxidase; SOD: Superoxide dismutase; hv: Irradiation with light.
NAC is an antioxidant that not only can be metabolized into GSH precursor, cysteine, to supplement intracellular GSH. Clinical trials or meta-analysis indicate NAC offers no benefit for the preservation of forced vital capacity compared with placebo in IPF.[8,178,179] This by no means implies that oxidant/antioxidant imbalance is no longer important in the pathogenesis of IPF. As can be seen from Figure 4, GSH is just one branch of the body's antioxidant defense system.
One study found that mitochondrial catalase–overexpressed transgenic mice are protected against lung fibrosis in part via the prevention of AEC mtDNA damage.[92] Evidence has also demonstrated that NOX-4 is a significant enzyme that catalyzes the generation of ROS in the pathogenesis of IPF.[180,181] Directly reducing ROS production by inhibition of NOX or rapidly scavenging ROS produced in mitochondria by targeting mitochondrial ROS seems to be more specific and efficient than general antioxidant treatment for IPF. They are promising therapeutic targets, but their safety and effectiveness in humans are yet to be confirmed.
Treatment plan targeting multiple points centered on one point
These evolving concepts of disease pathogenesis of IPF are the bases for emerging pharmacotherapies for this complex disease. As shown in Figure 1, AEC/fibroblast apoptosis imbalance is at the core of the pathogenesis of IPF, so the treatment plan for IPF should be mainly based on the adjustment of AEC/fibroblast apoptosis/proliferation imbalance, taking into account various other imbalances. Downregulation of factors leading to apoptosis of AECs and anti-apoptosis of fibroblasts may be a potential treatment. For instance, AT-406, a blockade of XIAP and the cellular IAP (cIAP), has been shown to restore apoptotic sensitivity of fibroblasts from IPF,[165] and animal studies have shown AT-406 to be of benefit in treating IPF.[182] The resistance to apoptosis of fibroblasts could be caused by downregulating the expression of miR-29c as well as Fas receptor by TGF-β. Therefore, the upregulation of miR-29 expression in IPF lungs could restore the sensitivity to apoptosis of lung fibroblasts and inhibit ECM production.[141] Additionally, FasL is up-regulated in lung tissues of IPF; therefore, specific inhibition of the Fas-FasL pathway in AECs may prevent the development of pulmonary fibrosis. Up-regulation of factors reducing apoptosis of AECs and restoring the sensitivity to apoptosis of fibroblasts is another potential treatment. For example, miR-30a expression decreases in AECs, and the up-regulation of miR-30a can suppress AEC apoptosis.
As mentioned above, fibrosis may be heterogeneous and multifactorial in etiology and pathogenesis.
Therefore, attempts to prevent or counteract a single upstream pathway may not be enough to inhibit the progression of fibrosis. Future research should focus on treatment methods that target multiple mechanisms, with adjustment of AEC/fibroblast apoptosis imbalance as the central focus and adjustment of various other imbalances as the secondary points of focus.
Conclusions
IPF is likely the result of complex interactions between environmental, genetic, and epigenetic factors.
First, environmental exposures strongly affect epigenetic marks, and epigenetic processes translate environmental exposures into the regulation of chromatin. Second, epigenetic processes shape the gene expression profiles involved in disease pathophysiology. In turn, an individual's genetic background determines epigenetic marks in two ways: by direct inheritance and by genetic variants. Finally, genetic and epigenetic factors act in concert to dysregulate gene expression in IPF lung. The pathogenesis of IPF involves various imbalances centered on AEC/fibroblast apoptosis imbalance, including ER, telomere length homeostasis, mitochondrial dysfunction, oxidant/antioxidant imbalance, Th1/Th2 imbalance, M1–M2 polarization of macrophages, protease/antiprotease imbalance, and plasminogen activation/inhibition imbalance. Among them, AEC/fibroblast apoptosis imbalance is the core of IPF, because although other imbalances affect each other and promote each other, they eventually lead to dysregulated crosstalk between AECs and fibroblasts. As a result, the dysregulated crosstalk and abnormal mediators between them result in AEC apoptosis, fibroblast anti-apoptosis, and abnormal ECM. It is worth noting that the ineffectiveness of anti-inflammatory and antioxidative therapies for IPF patients in no way means that inflammation and oxidant/antioxidant imbalances are no longer important in the pathogenesis of IPF. Fibrosis may be heterogeneous and multifactorial in etiology and pathogenesis. Therefore, attempts to prevent or counteract a single upstream pathway may not be enough to inhibit the progression of fibrosis. Future research should focus on treatment methods that target multiple mechanisms, with adjustment of AEC/fibroblast apoptosis imbalance as the central focus and adjustment of various other imbalances as the secondary points of focus.
Funding
This study was supported by a grant from the National Natural Science Foundation of China (No. 81970083).
Conflicts of interest
None.
Footnotes
How to cite this article: Wang Q, Xie ZL, Wu Q, Jin ZX, Yang C, Feng J. Role of various imbalances centered on alveolar epithelial cell/fibroblast apoptosis imbalance in the pathogenesis of idiopathic pulmonary fibrosis. Chin Med J 2021;134:261–274. doi: 10.1097/CM9.0000000000001288
Qing Wang and Zhao-Liang Xie were contributed equally to this work.
References
- 1.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017; 3:17074.doi: 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 2.Keogh BA, Crystal RG. Alveolitis: the key to the interstitial lung disorders. Thorax 1982; 37:1–10. doi: 10.1136/thx.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med 2001; 345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 4.Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med 2005; 172:417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012; 380:680–688. doi: 10.1016/S0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 6.Selman M, King TE, Pardo A. American Thoracic Society, European Respiratory Society, American College of Chest Physicians Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001; 134:136–151. doi: 10.7326/0003-4819-134-2-200101160-00015. [DOI] [PubMed] [Google Scholar]
- 7.Raghu G, Anstrom KJ, King TE, Jr, Lasky JA, Martinez FJ. Idiopathic Pulmonary Fibrosis Clinical Research Network Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012; 366:1968–1977. doi: 10.1056/NEJMoa1113354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Jr, Raghu G. Idiopathic Pulmonary Fibrosis Clinical Research Network Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370:2093–2101. doi: 10.1056/NEJMoa1401739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skandamis A, Kani C, Markantonis SL, Souliotis K. Systematic review and network meta-analysis of approved medicines for the treatment of idiopathic pulmonary fibrosis. J Drug Assess 2019; 8:55–61. doi: 10.1080/21556660.2019.1597726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleetwood K, McCool R, Glanville J, Edwards SC, Gsteiger S, Daigl M, et al. Systematic review and network meta-analysis of idiopathic pulmonary fibrosis treatments. J Manag Care Spec Pharm 2017; 23: 3-b Suppl: S5–S16. doi: 10.18553/jmcp.2017.23.3-b.s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spagnolo P, Sverzellati N, Rossi G, Cavazza A, Tzouvelekis A, Crestani B, et al. Idiopathic pulmonary fibrosis: an update. Ann Med 2015; 47:15–27. doi: 10.3109/07853890.2014.982165. [DOI] [PubMed] [Google Scholar]
- 12.Macneal K, Schwartz DA. The genetic and environmental causes of pulmonary fibrosis. Proc Am Thorac Soc 2012; 9:120–125. doi: 10.1513/pats.201112-055AW. [DOI] [PubMed] [Google Scholar]
- 13.Lee JS, Song JW, Wolters PJ, Elicker BM, King TE, Jr, Kim DS, et al. Bronchoalveolar lavage pepsin in acute exacerbation of idiopathic pulmonary fibrosis. Eur Respir J 2012; 39:352–358. doi: 10.1183/09031936.00050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2012; 302:L721–L729.doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta 2013; 1832:940–947. doi: 10.1016/j.bbadis.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Wang Y, Pandupuspitasari NS, Wu G, Xiang X, Gong Q, et al. Endoplasmic reticulum stress, a new wrestler, in the pathogenesis of idiopathic pulmonary fibrosis. Am J Transl Res 2017; 9:722–735. [PMC free article] [PubMed] [Google Scholar]
- 17.Vancheri C, du Bois RM. A progression-free end-point for idiopathic pulmonary fibrosis trials: lessons from cancer. Eur Respir J 2013; 41:262–269. doi: 10.1183/09031936.00115112. [DOI] [PubMed] [Google Scholar]
- 18.Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J Clin Invest 2017; 127:405–414. doi: 10.1172/JCI87440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schamberger AC, Schiller HB, Fernandez IE, Sterclova M, Heinzelmann K, Hennen E, et al. Glutathione peroxidase 3 localizes to the epithelial lining fluid and the extracellular matrix in interstitial lung disease. Sci Rep 2016; 6:29952.doi: 10.1038/srep29952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kazufumi M, Sonoko N, Masanori K, Takateru I, Akira O. Expression of bcl-2 protein and APO-1 (Fas antigen) in the lung tissue from patients with idiopathic pulmonary fibrosis. Microsc Res Tech 1997; 38:480–487. doi: 10.1002/(SICI)1097-0029(19970901)38:5<480::AID-JEMT4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 21.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A 2014; 111:367–372. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune mechanisms in pulmonary fibrosis. Am J Respir Cell Mol Biol 2016; 55:309–322. doi: 10.1165/rcmb.2016-0121TR. [DOI] [PubMed] [Google Scholar]
- 24.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016; 44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol 2017; 79:593–617. doi: 10.1146/annurev-physiol-022516-034356. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, et al. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 2011; 179:1733–1745. doi: 10.1016/j.ajpath.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Prieto E, González-López A, Cabrera S, Astudillo A, Gutiérrez-Fernández A, Fanjul-Fernandez M, et al. Resistance to bleomycin-induced lung fibrosis in MMP-8 deficient mice is mediated by interleukin-10. PLoS One 2010; 5:e13242.doi: 10.1371/journal.pone.0013242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gharib SA, Johnston LK, Huizar I, Birkland TP, Hanson J, Wang Y, et al. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J Leukoc Biol 2014; 95:9–18. doi: 10.1189/jlb.1112587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Osterholzer JJ, Christensen PJ, Lama V, Horowitz JC, Hattori N, Subbotina N, et al. PAI-1 promotes the accumulation of exudate macrophages and worsens pulmonary fibrosis following type II alveolar epithelial cell injury. J Pathol 2012; 228:170–180. doi: 10.1002/path.3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol 2014; 306:L786–L796. doi: 10.1152/ajplung.00243.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gokey JJ, Snowball J, Sridharan A, Speth JP, Black KE, Hariri LP, et al. MEG3 is increased in idiopathic pulmonary fibrosis and regulates epithelial cell differentiation. JCI Insight 2018; 3:e122490.doi: 10.1172/jci.insight.122490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Luo S, Li B, Dai H, Zhang J. Feature article: IL-25 contributes to lung fibrosis by directly acting on alveolar epithelial cells and fibroblasts. Exp Biol Med (Maywood) 2019; 244:770–780. doi: 10.1177/1535370219843827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011; 378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- 34.Takezaki A, Tsukumo SI, Setoguchi Y, Ledford JG, Goto H, Hosomichi K, et al. A homozygous SFTPA1 mutation drives necroptosis of type II alveolar epithelial cells in patients with idiopathic pulmonary fibrosis. J Exp Med 2019; 216:2724–2735. doi: 10.1084/jem.20182351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nathan N, Giraud V, Picard C, Nunes H, Dastot-Le Moal F, Copin B, et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum Mol Genet 2016; 25:1457–1467. doi: 10.1093/hmg/ddw014. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 2009; 84:52–59. doi: 10.1016/j.ajhg.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Jiang J, Chen WJ, Su LX, Xie LX. Genetic characterization of a Chinese family with familial idiopathic pulmonary fibrosis. Chin Med J (Engl) 2012; 125:1945–1951. [PubMed] [Google Scholar]
- 38.Varmus H. Ten years on--the human genome and medicine. N Engl J Med 2010; 362:2028–2029. doi: 10.1056/NEJMe0911933. [DOI] [PubMed] [Google Scholar]
- 39.Coghlan MA, Shifren A, Huang HJ, Russell TD, Mitra RD, Zhang Q, et al. Sequencing of idiopathic pulmonary fibrosis-related genes reveals independent single gene associations. BMJ Open Respir Res 2014; 1:e000057.doi: 10.1136/bmjresp-2014-000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Courtwright AM, El-Chemaly S. Telomeres in interstitial lung disease: the short and the long of it. Ann Am Thorac Soc 2019; 16:175–181. doi: 10.1513/AnnalsATS.201808-508CME. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, Speer M, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 42.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci 2003; 116:683–692. doi: 10.1242/jcs.00267. [DOI] [PubMed] [Google Scholar]
- 43.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol 2005; 32:521–530. doi: 10.1165/rcmb.2005-0009OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armanios M. Syndromes of telomere shortening. Annu Rev Genomics Hum Genet 2009; 10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur A, Mathai SK, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med (Lausanne) 2017; 4:154.doi: 10.3389/fmed.2017.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013; 1:309–317. doi: 10.1016/S2213-2600(13)70045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2020; 201:564–574. doi: 10.1164/rccm.201905-1017OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364:1503–1512. doi: 10.1056/NEJMoa1013660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol 2008; 39:610–618. doi: 10.1165/rcmb.2007-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 2000; 20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol 2009; 29:4325–4339. doi: 10.1128/MCB.01776-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Korfei M, Skwarna S, Henneke I, MacKenzie B, Klymenko O, Saito S, et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax 2015; 70:1022–1032. doi: 10.1136/thoraxjnl-2014-206411. [DOI] [PubMed] [Google Scholar]
- 53.Huang SK, Scruggs AM, Donaghy J, Horowitz JC, Zaslona Z, Przybranowski S, et al. Histone modifications are responsible for decreased Fas expression and apoptosis resistance in fibrotic lung fibroblasts. Cell Death Dis 2013; 4:e621.doi: 10.1038/cddis.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol 2007; 14:103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- 55.Yamada M, Kubo H, Ota C, Takahashi T, Tando Y, Suzuki T, et al. The increase of microRNA-21 during lung fibrosis and its contribution to epithelial-mesenchymal transition in pulmonary epithelial cells. Respir Res 2013; 14:95.doi: 10.1186/1465-9921-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu B, Jiang T, Hu X, et al. Downregulation of microRNA-30a in bronchoalveolar lavage fluid from idiopathic pulmonary fibrosis patients. Mol Med Rep 2018; 18:5799–5806. doi: 10.3892/mmr.2018.9565. [DOI] [PubMed] [Google Scholar]
- 57.Wang CJ, Li BB, Tan YJ, Zhang GM, Cheng GL, Ren YS. MicroRNA-31/184 is involved in transforming growth factor-β-induced apoptosis in A549 human alveolar adenocarcinoma cells. Life Sci 2020; 242:117205.doi: 10.1016/j.lfs.2019.117205. [DOI] [PubMed] [Google Scholar]
- 58.Yang S, Cui H, Xie N, Icyuz M, Banerjee S, Antony VB, et al. miR-145 regulates myofibroblast differentiation and lung fibrosis. FASEB J 2013; 27:2382–2391. doi: 10.1096/fj.12-219493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang H, Gu Y, Li T, Zhang Y, Huangfu L, Hu M, et al. Integrated analyses identify the involvement of microRNA-26a in epithelial-mesenchymal transition during idiopathic pulmonary fibrosis. Cell Death Dis 2014; 5:e1238.doi: 10.1038/cddis.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi Y, Gochuico BR, Yu G, Tang X, Osorio JC, Fernandez IE, et al. Syndecan-2 exerts antifibrotic effects by promoting caveolin-1-mediated transforming growth factor-β receptor I internalization and inhibiting transforming growth factor-β1 signaling. Am J Respir Crit Care Med 2013; 188:831–841. doi: 10.1164/rccm.201303-0434OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pandit KV, Corcoran D, Yousef H, Yarlagadda M, Tzouvelekis A, Gibson KF, et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010; 182:220–229. doi: 10.1164/rccm.200911-1698OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dai WJ, Qiu J, Sun J, Ma CL, Huang N, Jiang Y, et al. Downregulation of microRNA-9 reduces inflammatory response and fibroblast proliferation in mice with idiopathic pulmonary fibrosis through the ANO1-mediated TGF-β-Smad3 pathway. J Cell Physiol 2019; 234:2552–2565. doi: 10.1002/jcp.26961. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Ye H, Xiang F, Song LJ, Zhou LL, Cai PC, et al. miR-18a-5p Inhibits sub-pleural pulmonary fibrosis by targeting TGF-β receptor II. Mol Ther 2017; 25:728–738. doi: 10.1016/j.ymthe.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zeng X, Huang C, Senavirathna L, Wang P. Liu L. miR-27b inhibits fibroblast activation via targeting TGFβ signaling pathway. BMC Cell Biol 2017; 18:9.doi: 10.1186/s12860-016-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herrera J, Beisang DJ, Peterson M, Forster C, Gilbertsen A, Benyumov A, et al. Dicer1 deficiency in the idiopathic pulmonary fibrosis fibroblastic focus promotes fibrosis by suppressing microRNA biogenesis. Am J Respir Crit Care Med 2018; 198:486–496. doi: 10.1164/rccm.201709-1823OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mao C, Zhang J, Lin S, Jing L, Xiang J, Wang M, et al. MiRNA-30a inhibits AECs-II apoptosis by blocking mitochondrial fission dependent on Drp-1. J Cell Mol Med 2014; 18:2404–2416. doi: 10.1111/jcmm.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chi L, Xiao Y, Zhu L, Zhang M, Xu B, Xia H, et al. microRNA-155 attenuates profibrotic effects of transforming growth factor-beta on human lung fibroblasts. J Biol Regul Homeost Agents 2019; 33:1415–1424. doi: 10.23812/19-41A. [DOI] [PubMed] [Google Scholar]
- 68.Moimas S, Salton F, Kosmider B, Ring N, Volpe MC, Bahmed K, et al. miR-200 family members reduce senescence and restore idiopathic pulmonary fibrosis type II alveolar epithelial cell transdifferentiation. ERJ Open Res 2019; 5:00138–02019. doi: 10.1183/23120541.00138-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang YC, Liu JS, Tang HK, Nie J, Zhu JX, Wen LL, et al. miR–221 targets HMGA2 to inhibit bleomycin–induced pulmonary fibrosis by regulating TGF–β1/Smad3-induced EMT. Int J Mol Med 2016; 38:1208–1216. doi: 10.3892/ijmm.2016.2705. [DOI] [PubMed] [Google Scholar]
- 70.Ge L, Habiel DM, Hansbro PM, Kim RY, Gharib SA, Edelman JD, et al. miR-323a-3p regulates lung fibrosis by targeting multiple profibrotic pathways. JCI Insight 2016; 1:e90301.doi: 10.1172/jci.insight.90301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Das S, Kumar M, Negi V, Pattnaik B, Prakash YS, Agrawal A, et al. MicroRNA-326 regulates profibrotic functions of transforming growth factor-β in pulmonary fibrosis. Am J Respir Cell Mol Biol 2014; 50:882–892. doi: 10.1165/rcmb.2013-0195OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhuang Y, Dai J, Wang Y, Zhang H, Li X, Wang C, et al. MiR-338∗ suppresses fibrotic pathogenesis in pulmonary fibrosis through targeting LPA1. Am J Transl Res 2016; 8:3197–3205. [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Y, Huang C, Reddy Chintagari N, Bhaskaran M, Weng T, Guo Y, et al. miR-375 regulates rat alveolar epithelial cell trans-differentiation by inhibiting Wnt/β-catenin pathway. Nucleic Acids Res 2013; 41:3833–3844. doi: 10.1093/nar/gks1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ji X, Wu B, Fan J, Han R, Luo C, Wang T, et al. The anti-fibrotic effects and mechanisms of microRNA-486-5p in pulmonary fibrosis. Sci Rep 2015; 5:14131.doi: 10.1038/srep14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hadjicharalambous MR, Roux BT, Csomor E, Feghali-Bostwick CA, Murray LA, Clarke DL, et al. Long intergenic non-coding RNAs regulate human lung fibroblast function: Implications for idiopathic pulmonary fibrosis. Sci Rep 2019; 9:6020.doi: 10.1038/s41598-019-42292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014; 505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 77.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8:253–262. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tennis MA, Vanscoyk MM, Wilson LA, Kelley N, Winn RA. Methylation of Wnt7a is modulated by DNMT1 and cigarette smoke condensate in non-small cell lung cancer. PLoS One 2012; 7:e32921.doi: 10.1371/journal.pone.0032921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen D, Fang L, Li H, Tang MS, Jin C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J Biol Chem 2013; 288:21678–21687. doi: 10.1074/jbc.M113.476630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abramowitz LK, Bartolomei MS. Genomic imprinting: recognition and marking of imprinted loci. Curr Opin Genet Dev 2012; 22:72–78. doi: 10.1016/j.gde.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kasowski M, Kyriazopoulou-Panagiotopoulou S, Grubert F, Zaugg JB, Kundaje A, Liu Y, et al. Extensive variation in chromatin states across humans. Science 2013; 342:750–752. doi: 10.1126/science.1242510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frakes AE, Dillin A. The UPRER: sensor and coordinator of organismal homeostasis. Mol Cell 2017; 66:761–771. doi: 10.1016/j.molcel.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 84.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 2017; 13:477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]
- 85.Bhansali S, Khatri S, Dhawan V. Terminalia Arjuna bark extract impedes foam cell formation and promotes apoptosis in ox-LDL-stimulated macrophages by enhancing UPR-CHOP pathway. Lipids Health Dis 2019; 18:195.doi: 10.1186/s12944-019-1119-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li JS, Miralles Fusté J, Simavorian T, Bartocci C, Tsai J, Karlseder J, et al. TZAP: a telomere-associated protein involved in telomere length control. Science 2017; 355:638–641. doi: 10.1126/science. aah6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sulastri D, Lestari Y, Afriwardi, Desmawati Relationship between body composition and smoking habit with telomere length of Minangkabau ethnicity men, in West Sumatera, Indonesia. Pak J Biol Sci 2017; 20:516–522. doi: 10.3923/pjbs.2017.516.522. [DOI] [PubMed] [Google Scholar]
- 88.Faner R, Rojas M, Macnee W, Agustí A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 186:306–313. doi: 10.1164/rccm.201202-0282PP. [DOI] [PubMed] [Google Scholar]
- 89.Sui B, Hu C, Jin Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology 2016; 17:267–279. doi: 10.1007/s10522-015-9609-5. [DOI] [PubMed] [Google Scholar]
- 90.Bueno M, Brands J, Voltz L, Fiedler K, Mays B, St Croix C, et al. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018; 17:e12720.doi: 10.1111/acel.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, et al. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol 2012; 46:731–739. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 92.Kim SJ, Cheresh P, Jablonski RP, Morales-Nebreda L, Cheng Y, Hogan E, et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic Biol Med 2016; 101:482–490. doi: 10.1016/j.freeradbiomed.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rangarajan S, Bernard K, Thannickal VJ. Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc 2017; 14: Suppl 5: S383–S388. doi: 10.1513/AnnalsATS.201705-370AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carrillo-Gálvez AB, Gálvez-Peisl S, González-Correa JE, de Haro-Carrillo M, Ayllón V, Carmona-Sáez P, et al. GARP is a key molecule for mesenchymal stromal cell responses to TGF-β and fundamental to control mitochondrial ROS levels. Stem Cells Transl Med 2020; 9:636–650. doi: 10.1002/sctm.19-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One 2015; 10:e0121246.doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med 2012; 186:740–751. doi: 10.1164/rccm.201201-0084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Andrianifahanana M, Hernandez DM, Yin X, Kang JH, Jung MY, Wang Y, et al. Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J 2016; 30:3733–3744. doi: 10.1096/fj.201600428R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu J, Li J, Yu Z, Rao H, Wang S, Lan H. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-α-regulated aerobic glycolysis. Pulm Pharmacol Ther 2017; 45:136–141. doi: 10.1016/j.pupt.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 99.Xie N, Cui H, Ge J, Banerjee S, Guo S, Dubey S, et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2017; 313:L834–L844. doi: 10.1152/ajplung.00235.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cho SJ, Moon JS, Lee CM, Choi AM, Stout-Delgado HW. Glucose transporter 1-dependent glycolysis is increased during aging-related lung fibrosis, and phloretin inhibits lung fibrosis. Am J Respir Cell Mol Biol 2017; 56:521–531. doi: 10.1165/rcmb.2016-0225OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bergeron A, Soler P, Kambouchner M, Loiseau P, Milleron B, Valeyre D, et al. Cytokine profiles in idiopathic pulmonary fibrosis suggest an important role for TGF-beta and IL-10. Eur Respir J 2003; 22:69–76. doi: 10.1183/09031936.03.00014703. [DOI] [PubMed] [Google Scholar]
- 102.Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, et al. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J 2005; 19:854–856. doi: 10.1096/fj.04-2882fje. [DOI] [PubMed] [Google Scholar]
- 103.Nelson KK, Melendez JA. Mitochondrial redox control of matrix metalloproteinases. Free Radic Biol Med 2004; 37:768–784. doi: 10.1016/j.freeradbiomed.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 104.Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004; 4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 105.Wu SM, Pizzo SV. Mechanism of hypochlorite-mediated inactivation of proteinase inhibition by alpha 2-macroglobulin. Biochemistry 1999; 38:13983–13990. doi: 10.1021/bi991438i. [DOI] [PubMed] [Google Scholar]
- 106.Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci 2015; 16:21486–21519. doi: 10.3390/ijms160921486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest 2018; 128:64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lei H, Kazlauskas A. A reactive oxygen species-mediated, self-perpetuating loop persistently activates platelet-derived growth factor receptor α. Mol Cell Biol 2014; 34:110–122. doi: 10.1128/MCB.00839-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amati L, Pepe M, Passeri ME, Mastronardi ML, Jirillo E, Covelli V. Toll-like receptor signaling mechanisms involved in dendritic cell activation: potential therapeutic control of T cell polarization. Curr Pharm Des 2006; 12:4247–4254. doi: 10.2174/138161206778743583. [DOI] [PubMed] [Google Scholar]
- 110.Peng SC, Hu X, Wei LQ, Li ZH. The correlation of helper T lymphocyte 1/helper T lymphocyte 2 with clinical and image features in patients with idiopathic pulmonary fibrosis. Zhonghua Nei Ke Za Zhi 2013; 52:489–493. [PubMed] [Google Scholar]
- 111.Passalacqua G, Mincarini M, Colombo D, Troisi G, Ferrari M, Bagnasco D, et al. IL-13 and idiopathic pulmonary fibrosis: Possible links and new therapeutic strategies. Pulm Pharmacol Ther 2017; 45:95–100. doi: 10.1016/j.pupt.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 112.Cedeno-Laurent F, Dimitroff CJ. Galectin-1 research in T cell immunity: past, present and future. Clin Immunol 2012; 142:107–116. doi: 10.1016/j.clim.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Corapi E, Carrizo G, Compagno D, Laderach D. Endogenous galectin-1 in T lymphocytes regulates anti-prostate cancer immunity. Front Immunol 2018; 9:2190.doi: 10.3389/fimmu.2018.02190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaliński P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol 1997; 159:28–35. [PubMed] [Google Scholar]
- 115.Bao Z, Zhang Q, Wan H, He P, Zhou X, Zhou M. Expression of suppressor of cytokine signaling 1 in the peripheral blood of patients with idiopathic pulmonary fibrosis. Chin Med J (Engl) 2014; 127 (11):2117–2120. [PubMed] [Google Scholar]
- 116.Luzina IG, Todd NW, Iacono AT, Atamas SP. Roles of T lymphocytes in pulmonary fibrosis. J Leukoc Biol 2008; 83:237–244. doi: 10.1189/jlb.0707504. [DOI] [PubMed] [Google Scholar]
- 117.Xu J, Mora AL, LaVoy J, Brigham KL, Rojas M. Increased bleomycin-induced lung injury in mice deficient in the transcription factor T-bet. Am J Physiol Lung Cell Mol Physiol 2006; 291:L658–667. doi: 10.1152/ajplung.00006.2006. [DOI] [PubMed] [Google Scholar]
- 118.Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol 2003; 132:168–176. doi: 10.1159/000073718. [DOI] [PubMed] [Google Scholar]
- 119.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol 2015; 15:271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 120.Hume DA. The many alternative faces of macrophage activation. Front Immunol 2015; 6:370.doi: 10.3389/fimmu.2015.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature 2013; 496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 2011; 11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 124.Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 2014; 5:614.doi: 10.3389/fimmu.2014.00614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther 2016; 24:915–925. doi: 10.1038/mt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Y, Zhu J, Zhang L, Zhang Z, He L, Mou Y, et al. Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/transcription factor EC/IL-4 receptor α positive feedback loop in M2 macrophages. J Allergy Clin Immunol 2017; 140:1550–1561.e8. doi: 10.1016/j.jaci.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 127.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 2014; 15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Menou A, Duitman J, Crestani B. The impaired proteases and anti-proteases balance in idiopathic pulmonary fibrosis. Matrix Biol 2018; 68-69:382–403. doi: 10.1016/j.matbio.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 129.Guiot J, Moermans C, Henket M, Corhay JL, Louis R. Blood biomarkers in idiopathic pulmonary fibrosis. Lung 2017; 195:273–280. doi: 10.1007/s00408-017-9993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Craig VJ, Zhang L, Hagood JS, Owen CA. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015; 53:585–600. doi: 10.1165/rcmb.2015-0020TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Todd JL, Vinisko R, Liu Y, Neely ML, Overton R, Flaherty KR, et al. IPF-PRO registry investigators. Circulating matrix metalloproteinases and tissue metalloproteinase inhibitors in patients with idiopathic pulmonary fibrosis in the multicenter IPF-PRO Registry cohort. BMC Pulm Med 2020; 20: doi: 10.1186/s12890-020-1103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiong Y, Zhang J, Shi L, Ning Y, Zhu Y, Chen S, et al. NOGO-B promotes EMT in lung fibrosis via MMP14 mediates free TGF-beta1 formation. Oncotarget 2017; 8:71024–71037. doi: 10.18632/oncotarget.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Maldonado M, Salgado-Aguayo A, Herrera I, Cabrera S, Ortíz-Quintero B, Staab-Weijnitz CA, et al. Upregulation and nuclear location of MMP28 in alveolar epithelium of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2018; 59:77–86. doi: 10.1165/rcmb.2017-0223OC. [DOI] [PubMed] [Google Scholar]
- 134.Yu G, Kovkarova-Naumovski E, Jara P, Parwani A, Kass D, Ruiz V, et al. Matrix metalloproteinase-19 is a key regulator of lung fibrosis in mice and humans. Am J Respir Crit Care Med 2012; 186:752–762. doi: 10.1164/rccm.201202-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ban C, Wang T, Zhang S, Xin P, Liang L, Wang C, et al. Fibrinolytic system related to pulmonary arterial pressure and lung function of patients with idiopathic pulmonary fibrosis. Clin Respir J 2017; 11:640–647. doi: 10.1111/crj.12397. [DOI] [PubMed] [Google Scholar]
- 136.Seo JY, Park J, Yu MR, Kim YS, Ha H, Lee HB. Positive feedback loop between plasminogen activator inhibitor-1 and transforming growth factor-beta1 during renal fibrosis in diabetes. Am J Nephrol 2009; 30:481–490. doi: 10.1159/000242477. [DOI] [PubMed] [Google Scholar]
- 137.Liu RM. Oxidative stress, plasminogen activator inhibitor 1, and lung fibrosis. Antioxid Redox Signal 2008; 10:303–319. doi: 10.1089/ars.2007.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Keerthisingam CB, Jenkins RG, Harrison NK, Hernandez-Rodriguez NA, Booth H, Laurent GJ, et al. Cyclooxygenase-2 deficiency results in a loss of the anti-proliferative response to transforming growth factor-beta in human fibrotic lung fibroblasts and promotes bleomycin-induced pulmonary fibrosis in mice. Am J Pathol 2001; 158:1411–1422. doi: 10.1016/s0002-9440(10)64092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Marchand-Adam S, Marchal J, Cohen M, Soler P, Gerard B, Castier Y, et al. Defect of hepatocyte growth factor secretion by fibroblasts in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2003; 168:1156–1161. doi: 10.1164/rccm.200212-1514OC. [DOI] [PubMed] [Google Scholar]
- 140.Xie T, Liang J, Geng Y, Liu N, Kurkciyan A, Kulur V, et al. MicroRNA-29c prevents pulmonary fibrosis by regulating epithelial cell renewal and apoptosis. Am J Respir Cell Mol Biol 2017; 57:721–732. doi: 10.1165/rcmb.2017-0133OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Matsushima S, Ishiyama J. MicroRNA-29c regulates apoptosis sensitivity via modulation of the cell-surface death receptor, Fas, in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 2016; 311:L1050–L1061. doi: 10.1152/ajplung.00252.2016. [DOI] [PubMed] [Google Scholar]
- 142.Chanda D, Kurundkar A, Rangarajan S, Locy M, Bernard K, Sharma NS, et al. Developmental reprogramming in mesenchymal stromal cells of human subjects with idiopathic pulmonary fibrosis. Sci Rep 2016; 6:37445.doi: 10.1038/srep37445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gupte VV, Ramasamy SK, Reddy R, Lee J, Weinreb PH, Violette SM, et al. Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Respir Crit Care Med 2009; 180:424–436. doi: 10.1164/rccm.200811-1794OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.MacKenzie B, Korfei M, Henneke I, Sibinska Z, Tian X, Hezel S, et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir Res 2015; 16:83.doi: 10.1186/s12931-015-0242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Epstein Shochet G, Brook E, Eyal O, Edelstein E, Shitrit D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am J Physiol Lung Cell Mol Physiol 2019; 316:L1025–L1034. doi: 10.1152/ajplung.00526.2018. [DOI] [PubMed] [Google Scholar]
- 146.Joannes A, Brayer S, Besnard V, Marchal-Sommé J, Jaillet M, Mordant P, et al. FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am J Physiol Lung Cell Mol Physiol 2016; 310:L615–L629.doi: 10.1152/ajplung.00185.2015. [DOI] [PubMed] [Google Scholar]
- 147.Barnes JW, Duncan D, Helton S, Hutcheson S, Kurundkar D, Logsdon NJ, et al. Role of fibroblast growth factor 23 and klotho cross talk in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2019; 317:L141–L154.doi: 10.1152/ajplung.00246.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Camelo A, Dunmore R, Sleeman MA, Clarke DL. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol 2014; 4:173.doi: 10.3389/fphar.2013.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pan LH, Yamauchi K, Uzuki M, Nakanishi T, Takigawa M, Inoue H, et al. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur Respir J 2001; 17:1220–1227. doi: 10.1183/09031936.01.00074101. [DOI] [PubMed] [Google Scholar]
- 150.Abdul-Hafez A, Mohamed T, Omar H, Shemis M, Uhal BD. The renin angiotensin system in liver and lung: impact and therapeutic potential in organ fibrosis. J Lung Pulm Respir Res 2018; 5:00160. [PMC free article] [PubMed] [Google Scholar]
- 151.Nareznoi D, Konikov-Rozenman J, Petukhov D, Breuer R, Wallach-Dayan SB. Matrix metalloproteinases retain soluble FasL-mediated resistance to cell death in fibrotic-lung myofibroblasts. Cells 2020; 9:411.doi: 10.3390/cells9020411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res 2017; 121:575–583. doi: 10.1161/CIRCRESAHA.117.310624. [DOI] [PubMed] [Google Scholar]
- 153.Akram KM, Lomas NJ, Forsyth NR, Spiteri MA. Alveolar epithelial cells in idiopathic pulmonary fibrosis display upregulation of TRAIL, DR4 and DR5 expression with simultaneous preferential over-expression of pro-apoptotic marker p53. Int J Clin Exp Pathol 2014; 7:552–564. [PMC free article] [PubMed] [Google Scholar]
- 154.Arish N, Petukhov D, Wallach-Dayan SB. The role of telomerase and telomeres in interstitial lung diseases: from molecules to clinical implications. Int J Mol Sci 2019; 20:2996.doi: 10.3390/ijms20122996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sisson TH, Maher TM, Ajayi IO, King JE, Higgins PD, Booth AJ, et al. Increased survivin expression contributes to apoptosis-resistance in IPF fibroblasts. Adv Biosci Biotechnol 2012; 3:657–664. doi: 10.4236/abb.2012.326085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Golan-Gerstl R, Wallach-Dayan SB, Zisman P, Cardoso WV, Goldstein RH, Breuer R. Cellular FLICE-like inhibitory protein deviates myofibroblast fas-induced apoptosis toward proliferation during lung fibrosis. Am J Respir Cell Mol Biol 2012; 47:271–279. doi: 10.1165/rcmb.2010-0284RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huang WT, Akhter H, Jiang C, MacEwen M, Ding Q, Antony V, et al. Plasminogen activator inhibitor 1, fibroblast apoptosis resistance, and aging-related susceptibility to lung fibrosis. Exp Gerontol 2015; 61:62–75. doi: 10.1016/j.exger.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chang W, Wei K, Jacobs SS, Upadhyay D, Weill D, Rosen GD. SPARC suppresses apoptosis of idiopathic pulmonary fibrosis fibroblasts through constitutive activation of beta-catenin. J Biol Chem 2010; 285:8196–8206. doi: 10.1074/jbc.M109.025684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Predescu SA, Zhang J, Bardita C, Patel M, Godbole V, Predescu DN. Mouse lung fibroblast resistance to Fas-mediated apoptosis is dependent on the baculoviral inhibitor of apoptosis protein 4 and the cellular FLICE-inhibitory protein. Front Physiol 2017; 8:128.doi: 10.3389/fphys.2017.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hasaneen NA, Cao J, Pulkoski-Gross A, Zucker S, Foda HD. Extracellular matrix metalloproteinase inducer (EMMPRIN) promotes lung fibroblast proliferation, survival and differentiation to myofibroblasts. Respir Res 2016; 17: doi: 10.1186/s12931-016-0334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.He Z, Gao Y, Deng Y, Li W, Chen Y, Xing S, et al. Lipopolysaccharide induces lung fibroblast proliferation through Toll-like receptor 4 signaling and the phosphoinositide3-kinase-Akt pathway. PLoS One 2012; 7:e35926.doi: 10.1371/journal.pone.0035926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hanson KM, Hernady EB, Reed CK, Johnston CJ, Groves AM, Finkelstein JN. Apoptosis resistance in fibroblasts precedes progressive scarring in pulmonary fibrosis and is partially mediated by toll-like receptor 4 activation. Toxicol Sci 2019; 170:489–498. doi: 10.1093/toxsci/kfz103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc 2006; 3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 164.Du Z, Lin Z, Wang Z, Liu D, Tian D, Xia L. SPOCK1 overexpression induced by platelet-derived growth factor-BB promotes hepatic stellate cell activation and liver fibrosis through the integrin α5β1/PI3K/Akt signaling pathway. Lab Invest 2020; 100:1042–1056. doi: 10.1038/s41374-020-0425-4. [DOI] [PubMed] [Google Scholar]
- 165.Ajayi IO, Sisson TH, Higgins PD, Booth AJ, Sagana RL, Huang SK, et al. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to Fas-mediated apoptosis. Am J Respir Cell Mol Biol 2013; 49:86–95. doi: 10.1165/rcmb.2012-0224OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fattman CL. Apoptosis in pulmonary fibrosis: too much or not enough? Antioxid Redox Signal 2008; 10:379–385. doi: 10.1089/ars.2007.1907. [DOI] [PubMed] [Google Scholar]
- 167.Kim KK, Dotson MR, Agarwal M, Yang J, Bradley PB, Subbotina N, et al. Efferocytosis of apoptotic alveolar epithelial cells is sufficient to initiate lung fibrosis. Cell Death Dis 2018; 9:1056.doi: 10.1038/s41419-018-1074-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998; 101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Moore MW, Herzog EL. Regulation and relevance of myofibroblast responses in idiopathic pulmonary fibrosis. Curr Pathobiol Rep 2013; 1:199–208. doi: 10.1007/s40139-013-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med 2004; 55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 171.Xue J, Kass DJ, Bon J, Vuga L, Tan J, Csizmadia E, et al. Plasma B lymphocyte stimulator and B cell differentiation in idiopathic pulmonary fibrosis patients. J Immunol 2013; 191:2089–2095. doi: 10.4049/jimmunol.1203476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Donahoe M, Valentine VG, Chien N, Gibson KF, Raval JS, Saul M, et al. Autoantibody-targeted treatments for acute exacerbations of idiopathic pulmonary fibrosis. PLoS One 2015; 10:e0127771.doi: 10.1371/journal.pone.0127771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Homolka J, Ziegenhagen MW, Gaede KI, Entzian P, Zissel G, Müller-Quernheim J. Systemic immune cell activation in a subgroup of patients with idiopathic pulmonary fibrosis. Respiration 2003; 70:262–269. doi: 10.1159/000072007. [DOI] [PubMed] [Google Scholar]
- 174.Florez-Sampedro L, Song S, Melgert BN. The diversity of myeloid immune cells shaping wound repair and fibrosis in the lung. Regeneration (Oxf) 2018; 5:3–25. doi: 10.1002/reg2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Marchal-Sommé J, Uzunhan Y, Marchand-Adam S, Valeyre D, Soumelis V, Crestani B, et al. Cutting edge: nonproliferating mature immune cells form a novel type of organized lymphoid structure in idiopathic pulmonary fibrosis. J Immunol 2006; 176:5735–5739. doi: 10.4049/jimmunol.176.10.5735. [DOI] [PubMed] [Google Scholar]
- 176.Matyszak MK, Citterio S, Rescigno M, Ricciardi-Castagnoli P. Differential effects of corticosteroids during different stages of dendritic cell maturation. Eur J Immunol 2000; 30:1233–1242. doi: 10.1002/(SICI)1521-4141(200004)30:4<1233::AID-IMMU1233>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 177.Walters DM, Cho HY, Kleeberger SR. Oxidative stress and antioxidants in the pathogenesis of pulmonary fibrosis: a potential role for Nrf2. Antioxid Redox Signal 2008; 10:321–332. doi: 10.1089/ars.2007.1901. [DOI] [PubMed] [Google Scholar]
- 178.Rogliani P, Calzetta L, Cavalli F, Matera MG, Cazzola M. Pirfenidone, nintedanib and N-acetylcysteine for the treatment of idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Pulm Pharmacol Ther 2016; 40:95–103. doi: 10.1016/j.pupt.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 179.Behr J, Bendstrup E, Crestani B, Günther A, Olschewski H, Sköld CM, et al. Safety and tolerability of acetylcysteine and pirfenidone combination therapy in idiopathic pulmonary fibrosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med 2016; 4:445–453. doi: 10.1016/S2213-2600(16)30044-3. [DOI] [PubMed] [Google Scholar]
- 180.Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 2009; 15:1077–1081. doi: 10.1038/nm.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax 2010; 65:733–738. doi: 10.1136/thx.2009.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ashley SL, Sisson TH, Wheaton AK, Kim KK, Wilke CA, Ajayi IO, et al. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am J Respir Cell Mol Biol 2016; 54:482–492. doi: 10.1165/rcmb.2015-0148OC. [DOI] [PMC free article] [PubMed] [Google Scholar]