

Abstract
Although the first-line rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone regimen (R-CHOP) substantially improved outcomes for patients with diffuse large B-cell lymphoma (DLBCL), 40% of the patients suffered from relapsed/refractory disease and had poor survival outcomes. The detailed mechanism underlying R-CHOP resistance has not been well defined. For this review, we conducted a thorough search for literature and clinical trials involving DLBCL resistance. We discussed DLBCL biology, epigenetics, and aberrant signaling of the B-cell receptor (BCR), phosphatidylinositol 3-kinase (PI3K)/Akt, nuclear factor kappa light chain enhancer of activated B-cells (NF-κB), and the Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) pathways as defining mechanisms of DLBCL heterogeneity and R-CHOP resistance. The cell of origin, double- or triple-hit lymphoma and double-protein-expression, clonal evolution, tumor microenvironment, and multi-drug resistance help to contextualize DLBCL resistance in an (epi)genetically and biologically comparative manner. With better understanding of the biological and molecular landscape of DLBCL, a more detailed classification system and tailored treatments will ideally become available to further improve the prognosis of DLBCL patients.
Keywords: Diffuse large B-cell lymphoma, Tumor microenvironment, Multi-drug resistance, Genetic heterogeneity
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL), comprising approximately 25% of the total mature NHL cases in the United States and 45.8% in China.[1,2] Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) has been considered as a standard first-line therapy following improvements in the prognosis of patients with DLBCL, resulting in a 10-year overall survival (OS) of 43.5%.[3] However, 20% of the patients treated with R-CHOP had refractory disease, and approximately 20% of the patients who initially benefited from R-CHOP reported relapsed cases.[4] The 30% to 50% of patients with primary or secondary resistance to R-CHOP had significantly poorer survival outcomes and a median OS for about 6.3 months.[5] The mechanisms of R-CHOP resistance have not been adequately elucidated. For this review, we conducted a thorough search for literature and clinical trials involving DLBCL resistance and recapitulated the mechanisms of R-CHOP resistance regarding tumor biology and molecular pathways. Moreover, we addressed practical agents targeting each pathway, thus paving the road for future hierarchical classification and tailored treatments.
Tumor biology of R-CHOP resistance
Cell of origin
Gene expression profiling (GEP) analysis has defined activated B-cell-like (ABC) and germinal center B-cell-like (GCB) as the two major subtypes of DLBCL (approximately 50% and 30%, respectively), according to cell of origin (COO). GEP studies have shown that genes defining the ABC subgroup, such as interferon regulatory factor 4 (IRF4), FLICE-like inhibitory protein (FLIP), and B-cell lymphoma 2 (BCL-2), are normally induced during B cell proliferation and activation as characteristics of plasma cells.[6] In contrast, the GCB subgroup exhibits genes that are preferentially expressed in germinal center B-cells, such as cluster of differentiation 10 (CD10), LIM domain only 2 (LMO2), and BCL-6.[6] Intriguingly, recurrent enhancer of zeste 2 (EZH2) mutation, phosphatase and tensin homolog (PTEN) deletion, BCL-2 translocation/mutation, and cREL amplification, which are common in the GCB subtype, are seldom described in the ABC subtype.[7] The GCB subgroup had a significantly better 3-year OS than the non-GCB subgroup when treated with R-CHOP (85% vs. 69%), thereby indicating distinct mechanisms of R-CHOP resistance in the ABC subgroup.[8,9] At the same time, the GCB and ABC subtypes of DLBCL are also associated with an umbrella entity, encompassing biologically and genetically distinct aberrations. Thus, a more precise intra-subgroup classification is recommended.
According to a novel classification system proposed by Schmitz et al[9] based on the co-occurrence of gene aberrations, 23.1% of the patients with ABC-DLBCL exhibited an MCD subtype (MYD88 plus CD79B mutations), 13.6% exhibited BN2 (BCL-6 fusions plus NOTCH2 mutations), and 6.1% exhibited N1 (NOTCH1 mutations). Meanwhile, 37.2% of the patients with GCB-DLBCL exhibited the EZB subtype (EZH2 mutations and BCL-2 translocations) and 11.6% exhibited BN2 while the remaining 51.1% were unclassified. After R-CHOP treatment, patients with the N1 or MCD subtype had significantly inferior outcomes compared to those with the EZB or BN2 subtype. The predicted 5-year OS rates for the MCD, N1, BN2, and EZB subtypes were 26%, 36%, 65%, and 68%, respectively.[9] Drugs that target B-cell receptor-dependent nuclear factor kappa light chain enhancer of activated B-cells (NF-κB) activation (such as Bruton tyrosine kinase [BTK] inhibitors) may be investigated in the BN2 and MCD subtypes while immune-checkpoint inhibitors appear promising in the N1 subtype due to their prominent T-cell gene-expression signature.[9]
Additionally, Chapuy et al[10] identified the following five robust DLBCL clusters with discrete outcomes and coordinated genetic signatures: C1: a low-risk ABC-DLBCL subset of extrafollicular origin with frequent mutations in the NF-κB pathway members (eg, BCL-10 and tumor necrosis factor alpha-induced protein 3 [TNFAIP3]), NOTCH2, and BCL-6 activation; C2: an ABC/GCB-independent subset with 17p loss and TP53 inactivation; C3: a GCB-DLBCL subset with frequent mutations in epigenetic enzymes (eg, lysine methyltransferase 2D [KMT2D], CREB-binding protein [CREBBP], and EZH2), BCL-2, and phosphatase and tensin homolog (PTEN); C4: a GCB-DLBCL subset with mutations in NF-κB modifiers (eg, caspase recruitment domain-containing protein 11 [CARD11], NFKB inhibitor epsilon [NFKBIE], and NFKB inhibitor alpha [NFKBIA]), CD83, and STAT3; C5: an ABC-DLBCL subset with concordant CD79B (48%)/MYD88L265P (50%) mutations and frequent gain of BCL-2, which is associated with extranodal tropism. Accordingly, patients with the C3 or C5 subtype had significantly inferior survival outcomes compared to that of C1, C2, or C4 when treated with R-CHOP, and BTK inhibitors might improve the prognosis of the C5 subtype while epigenetics-modifying agents might be active in the C3 subtype.[10]
Clonal evolution
A gain in R-CHOP resistance in DLBCL is increasingly suspected to correlate with the process of clonal evolution. Melchardt et al[11] described the following three major patterns of clonal evolution using high-throughput sequencing (HTS) of 104 genes in 28 DLBCL patients: large global change, subclonal selection, and minimal or no change. Fluctuations of subclones harboring the gene mutations related to R-CHOP resistance (eg, BCL-2 and proto-oncogene serine/threonine-protein kinase 1) were observed as a result of chemoimmunotherapy. More non-synonymous gene mutations in DLBCL patients were associated with a shorter median OS. It was suggested that R-CHOP selected the subclones that expressed gene mutations (eg, tumor protein 53 [TP53]) that favored tumor progression.[11] Likewise, Rizzo et al[12] described the following two patterns of clonal evolution in relapsed DLBCL employing the HTS of variability, diversity, and joining (VDJ) rearrangements on the immunoglobulin heavy chain: an early divergent mode and a late-divergent mode with divergence at the phylogenic degree. No significant antigen selection in the complementary determining region of the VDJ locus has been observed under immunochemotherapy pressure, suggesting that oncogenic selection rather than antigen selection is a major impetus in R-CHOP resistance.[13]
Under the stress of rituximab treatment, resistant subclones may evolve to lose the surface CD20 antigen (at the RNA or protein level) or to develop genetic mutations of the membrane-spanning four-domains, subfamily A, member I gene.[14] Moreover, regarding the action mechanisms of rituximab, resistant lymphoma subclones could be selected by FcrR single nucleotide polymorphisms to diminish the rituximab-induced antibody-dependent cellular cytotoxicity.[14]
Tumor microenvironment
The tumor microenvironment is considered to be a defining feature in B-cell differentiation and DLBCL tumorigenesis, including immune cells, stromal cells, and extracellular components [Figure 1]. The response to R-CHOP was affected by the pretreatment of tumor microenvironment concerning fibrosis, angiogenesis, and immune cells.[15] The tumor microenvironment was differentially constructed at the gene and protein levels when comparing R-CHOP-sensitive DLBCL with resistant DLBCL.[16]CD37 deficiency, programmed cell death-ligand 1 (PD-L1), and CD47 upregulation on malignant B-cells revealing immune evasion were observed in resistant cases.[17–19] However, it was found that CD47 upregulation correlated with poorer prognosis in patients with non-GCB DLBCL compared to the GCB subtype, thereby indicating that a distinct microenvironment existed in different COOs.[20] A CD47 blockade by Hu5F9-G4 can synergize with rituximab to overcome R-CHOP resistance in DLBCL.[19] Janus kinase-signal transducer and activator of transcription 3 (JAK-STAT3) signaling propagates several levels of intimate interaction between tumor cells and the immunological microenvironment to regulate angiogenesis, inflammation, immunosuppression, and oncogenesis.[21]
Figure 1.
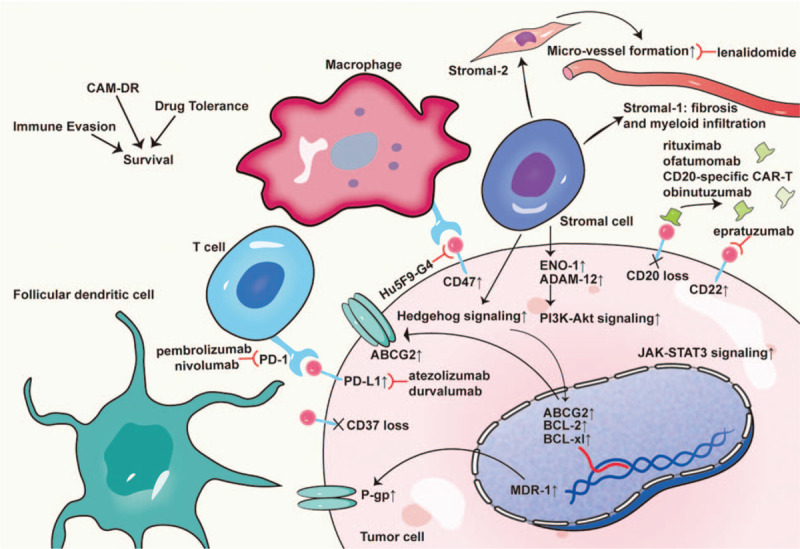
R-CHOP-resistant DLBCL microenvironment. This is the schematic representation of the defining features in the tumor microenvironment of DLBCL. The tumor microenvironment contains immune cells, stromal cells, and extracellular components (eg, matrix, chemokines, cytokines, exosomes, and blood vessels). Stromal cells of the tumor microenvironment include mesenchymal stromal cells, lymphoma-associated macrophages, myeloid-derived suppressor cells, and dendritic cells. Immune cells of the DLBCL microenvironment include cytotoxic T cells, follicular dendritic cells, regulatory T cells, natural killer cells, macrophages, and reticulum cells, which are essential for the multiple cycles of B-cell proliferation, mutation, selection, and immune evasion. The response to R-CHOP is affected by the tumor microenvironment in terms of cell adhesion to the stromal cells, angiogenesis, multi-drug resistance upregulation, and immune evasion. ABCG-2: ATP binding cassette subfamily G member 2; ENO-1: Enolase 1; CAM-DR: Cell adhesion-mediated drug resistance; CAR-T: Chimeric antigen receptor-T cell; DLBCL: Diffuse large B cell lymphoma; P-gp: P-glycoprotein; PD-1: Programmed cell death protein 1; PD-L1: Programmed death-ligand 1; R-CHOP: Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone.
The apoptosis-protective adhesion to stromal cells, termed cell adhesion-mediated drug resistance (CAM-DR), is another underlying mechanism of R-CHOP resistance in DLBCL.[22] CAM-DR was revealed to correlate with rituximab resistance through ADAM metalloproteinase domain 12 (ADAM-12) upregulation and phosphatidylinositol 3-kinase (PI3K)-Akt signaling modulation.[23] However, as per findings reported by Lenz et al,[15] genetically identified non-malignant signatures of stromal-1 (fibrosis and myeloid infiltration) and stromal-2 (vessel formation) in pretreatment biopsies were found to be prognostically favorable and unfavorable for patients with DLBCL who received R-CHOP, respectively. This finding was in concordance with a report by Cardesa-Salzmann et al[24] which stated that increased microvessel densities independently signified an inferior OS for patients with de novo DLBCL treated with R-CHOP.
Multi-drug resistance
Multi-drug resistance (MDR) describes acquired cross-resistance to a wide variety of structurally and functionally unrelated agents. The integrated membrane protein P-glycoprotein (Pgp) ATP binding cassette-1 serves as an ATP-dependent efflux pump for the active removal of its substrates through the lipid bilayer and for the reduction in intracellular cytotoxic concentration, thereby resulting in MDR.[25] Pgp is encoded by MDR-1, one of the most investigated MDR genes located on chromosome 7q21.12, where significant polymorphisms exist.[26] In addition to Pgp, other transporters in the ATP-binding cassette transporter family have been identified to confer drug resistance, such as multidrug-resistance-related protein-1 (MRP-1) and ATP-binding cassette subfamily G member 2 (ABCG2).[25] Doxorubicin, vincristine, and prednisone are substrates for Pgp and are known to induce MDR-1 expression.[27] DLBCL patients with high expression of Pgp, MRP-1, or ABCG2 demonstrated a significantly poor outcomes.[28,29]
Targeting molecular mechanisms in R-CHOP resistance
The findings reported by Schmitz et al[9] and Chapuy et al[10] suggested a different targeted therapies for genetically distinct DLBCL subsets. For example, the perturbation of proximal B-cell receptor (BCR) signaling is suggested for the C5 and MCD subtypes, BCL-2 inhibitors for C5, NF-κB signaling blockade for C1 and BN2, and inhibition of JAK-STAT3 signaling for C4.[9,10] Herein, we summarized the most investigated gene and protein aberrations in DLBCL as well as the cross-linked signaling pathway intricacies at the intersection of DLBCL biology and the clinic. These abnormalities are further elucidated by the perturbation of epigenetics in this study. Existing resistance to R-CHOP necessitates the use of alternative strategies and individualized therapies. In this context, rational drugs under investigation targeting R-CHOP resistance are further enumerated and discussed on a molecular basis [Figure 2].
Figure 2.

Targeting molecular pathways in R-CHOP-resistant DLBCL. This is the schematic representation of the BCR, PI3K-Akt, NF-κB, and JAK-STAT3 molecular signaling pathways in DLBCL. Aberrant expression of these pathways have emerged as top candidate targets implicated in R-CHOP resistance. These abnormalities are partially elucidated by epigenetic abnormalities. BCR: B cell receptor; BTK: Bruton's tyrosine kinase; CARD11: Caspase-associated recruitment domain 11; DLBCL: Diffuse large B cell lymphoma; EZH2: Enhancer of zeste 2; FOXO: Forkhead box O; HDAC: Histone deacetylase; JAK-STAT3: Janus kinase/signal transducer and activator of transcription 3; MALT-1: Mucosa-associated lymphoid tissue lymphoma translocation-1; MAPK: Mitogen-activated protein kinase; NEMO: NF-kappa B essential modulator; NF-κB: Nuclear factor kappa light chain enhancer of activated B cells; PI3K-Akt: Phosphatidylinositol 3-kinase/Akt; PKCβ: Protein kinase C β; PTEN: Phosphatase and tensin homolog; PDK-1: Phosphatidylinositol-dependent kinase 1; PRDM1: Positive regulatory domain containing 1; R-CHOP: Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone; SYK: Spleen tyrosine kinase; SFK: Src family of protein tyrosine kinases; IRAK: Interleukin 1 receptor associated kinase.
Double- or triple-hit lymphoma and double-expressor lymphoma
Double-hit lymphoma (DHL) and triple-hit lymphoma (THL) were embraced as a new category of high-grade B-cell lymphoma by the 2016 World Health Organization classification of tumors with MYC/8q24 translocation accompanied by rearrangement of BCL-2/18q21, BCL-6/3q27, or both.[30] Approximately 80% to 90% of the DHLs or 19% to 34% of the DLBCLs are also double-expressor lymphomas (DELs) with dual overexpression of c-MYC and BCL-2 detected by immunohistochemistry.[31] Patients with DHL, THL, or DEL were generally considered to have a poor outcomes in the era of R-CHOP; however, the prognostic value of BCL-2, BCL-6, or MYC remains controversial.[32,33]MYC plays a dominant role in proliferation, cell growth, apoptosis, and oncogenic transcription[34] while BCL-2 maintains cellular viability via apoptosis inhibition.[35] The dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin plus rituximab regimen was shown to be a better choice for DLBCL patients with MYC translocation, although no randomized controlled trials have confirmed this so far.[36] Landsburg et al[37] reported that ibrutinib monotherapy was highly effective in relapsed/refractory (rr) DEL. Regarding the overexpression of BCL-2 in DHL or DEL, BCL-2 inhibitor ABT-199 was demonstrated to result in an overall response rate (ORR) of 38% in rrDLBCL in a phase I trial.[38] In a HOVON phase II trial,[39] R-CHOP plus lenalidomide appeared to provide benefits to patients with MYC translocation with a 2-year OS and disease-free survival (DFS) of 73% and 75%, respectively. A previous study showed that the XPO1 inhibitor could decrease c-MYC expression, and combined use of the BCL-2 and XPO1 inhibitors was highly effective in eradicating DHL cells in vitro and prolonging host survival in vivo in a mouse DHL model.[40]
BCR signaling pathway
The BCR signaling pathway exhibits a functionally determinant role in B-cell activation, proliferation, and differentiation. The constitutive activation of PI3K and phosphatidylinositol-dependent kinase 1 (PDK-1) is essential for the survival of ABC-DLBCL cells with chronic active BCR signaling.[41,42] Signaling adapters caspase-associated recruitment domain 11 (CARD11), BIMP3, CARD-containing MAGUK protein 1 (CARMA1), BCL-10, and mucosa-associated lymphoid tissue lymphoma translocation-1 (MALT-1) comprise the CARMA1–BCL-10–MALT-1 (CBM) complex, but it is an integral whole, contributes to the upregulation of constitutive NF-κB activation in ABC-DLBCL.[43] Additionally, the MALT-1 subunit of the CBM complex features indirect NF-κB-promoting activity via proteolysis of the NF-κB inhibitors CYLD, RELB, A20, and BCL-10.[44] Indeed, chronic-active BCR signaling plays a pivotal in the survival of almost all ABC-DLBCLs and driven by frequent activating mutations of the immunoreceptor tyrosine-based activation motifs in CD79B and CD79A or of the coiled-coil domain in CARD11.[44] In contrast, GCB-DLBCLs are prone to survive with a BCR-negative immunophenotype.[45]
Previous studies have provided a framework for targeted inhibition of the BCR signaling pathway in BCR-dependent ABC-DLBCL.[44,46–52] Apart from COO algorithms, genetic assessments to confirm the exact lesion of the molecular pathways are recommended. For example, BTK inhibition is lethal for the upstream CD79 mutant cells but dispensable for the downstream CARD11-mutant DLBCL, which would be killed by downstream blockage of the NF-κB pathway.[53]
PI3K-Akt signaling pathway
PI3K-Akt signaling activation is enabled by sequential phosphorylation of PI3K, phosphatidylinositol-4,5-bisphosphate (PIP2), and Akt/protein kinase B.[54]Akt directly activates NF-κB signaling via phosphorylated IκBs at Thr23[55] and indirectly activates NF-κB signaling by stimulating the mitogen-activated protein kinase (MAPK) family.[56]PTEN serves as a major negative regulator of PI3K-Akt signaling by dephosphorylating PIP3, and PI3K/Akt dysregulation was indicated in 55% of the GCB-DLBCLs and 14% of the non-GCB DLBCLs with PTEN deficiency.[57]
The PI3K-Akt signaling pathway is involved in rituximab action, chronic activation of BCR signaling, and CAM-DR. High levels of phosphorylated Akt had an adverse prognostic impact on patients with DLBCL treated with R-CHOP.[58] Conceivably, components of the PI3K-Akt signaling pathway exhibit validated targets of DLBCL. Everolimus and temsirolimus are rapamycin analogs that target the raptor mammalian target of rapamycin. In a phase II trial of everolimus for rrDLBCL, an ORR of 30% was reported.[59] Furthermore, single-agent ibrutinib or copanlisib for inhibition of BTK or PI3K triggered simultaneous activation of the other pathway, thereby highlighting the significance of combined therapy.[42]
NF-κB signaling pathway
NF-κB signaling is activated by canonical (activates p50/rel and p50/relA) and non-canonical (activates p52/relB) pathways with both anti-apoptotic and apoptotic functions.[60] As downstream effectors of chronic active BCR signaling, the sustained activity of NF-κB signaling exerts a prominent tumor survival feature in ABC-DLBCL but not in GCB-DLBCL.[61] Both p50 and p65 activation proved to be unique mechanisms of R-CHOP resistance in ACB-DLBCL.[62,63] ABC-DLBCLs facilitate the canonical NF-κB signaling pathway by aberration of MYD88, BCL-10, CARD11, CD79A, CD79B, cyclin D2, CCR7, IRF4, FLIP, NFKBIA, TRAF2, and TNFAIP3.[44,64] Cells with MYD88 mutations exhibit constitutive activation of canonical NF-κB signaling.[60] Therefore, the target components of NF-κB signaling are of therapeutic importance. Lenalidomide and its analog thalidomide, which have antiangiogenic and pleiotropic immunomodulatory functions, exert direct tumor toxicity by binding to cereblon to inhibit downstream NF-κB signaling.[65] The regimen of lenalidomide plus R-CHOP appeared to mitigate the inferior survival of non-GCB DLBCLs in two phase II trials of 112 patients with newly diagnosed DLBCL.[66]
JAK-STAT3 signaling pathway
JAKs belong to the cytosolic tyrosine kinase family. JAKs are phosphorylated and activated upon cytokine binding. Subsequently, STAT proteins are activated and transition to the nucleus to facilitate transcription of downstream genes (eg, IL-6, IL-10, cyclin D1, cyclin D2, MYC, BCL-xl, and BCL-2), thereby influencing cell viability, immunosuppression, angiogenesis, and oncogenesis.[67] The STAT3-BCL-2-IL-10 loop is involved in R-CHOP resistance.[68]STAT3 expression was detected in 37% of the DLBCLs and in 54% of the ABC-DLBCLs treated with R-CHOP, thereby indicating poor survival, especially for the ABC subtype.[69] Conceivably, activation of the JAK-STAT3 signaling pathway of ABC-DLBCL suggests promising therapeutic targets, including JAK, STAT3, and IL-10 receptors.[70] According to results from a phase Ib trial, STAT3 antisense oligonucleotide AZD9150 showed efficacy in patients with rrNHL.[71]
Epigenetics
Epigenetic modifications confer crucial information to the transcription phenotypes and biological behaviors of tumorigenesis and drug resistance. Mutations of epigenetic modifiers (EP300, KMT2D, and SETDB1) were observed at both diagnosis and relapse, suggesting their roles as driver mutations of tumorigenesis in DLBLC.[13] Methylation and acetylation are the most investigated epigenetic patterns underlying R-CHOP resistance in DLBCL. Distinct epigenetic profiles contributing to the ABC and GCB phenotypes were detected and helped to redefine the molecular subtypes of DLBCL.[72] The prominent feature of ABC-DLBCLs and JAK activation was found to correlate with phosphorylation of histone H3 tyrosine 41 rather than STAT regulation.[73] MicroRNAs are also involved in drug resistance in DLBCL,[74] and the abnormal epigenetic profiling of microRNAs represents a putative mechanism of the positive regulatory domain-containing 1 (PRDM1) repression in DLBCL with wild-type PRDM1.[75]
Notably, unlike the aberrant genes and pathways mentioned above, epigenetic abnormalities are reversible. CD20 repression and rituximab resistance detected in the RRBL1 cell line were successfully reversed by the histone-deacetylase inhibitor trichostatin A.[76] The class-1 histone deacetylase inhibitor panobinostat increased STAT3 acetylation at lys685 and selectively killed STAT3-positive DLBCL cells in vitro.[77] A distinct epigenetic modifier of BCL-6 with ZF-KRAB fusions repressed BCL-6 expression and led to cell death in DLBCL.[78] Subsequent investigation revealed that DNA methyltransferase azacitidine followed by R-CHOP proved feasible in a phase I trial of 12 high-risk patients with newly diagnosed DLBCL.[79] The efficacy of tazemetostat, a single-agent inhibitor of histone methyltransferase EZH2 for the treatment of rrDLBCL, was also evaluated in trials.[80]
Conclusions
At present, we are well equipped to predict outcomes for DLBCL patients treated with R-CHOP using genetic, immunohistochemical, serum, and imaging markers. However, proper classification of DLBCL patients and identification of R-CHOP non-responders at diagnosis are critical. The BCR, PI3K-Akt, NF-κB, and JAK-STAT3 molecular signaling pathways are established mechanisms underlying R-CHOP resistance. The COO, clonal evolution, tumor microenvironment, MDR, and epigenetics are important for biological considerations. Novel target agents based on these mechanisms are currently being used in preclinical and clinical trials of DLBCL. Although there is only modest evidence to support their roles as first-line alternatives for R-CHOP, they must not be underestimated in the management of resistant DLBCL. Ideally, with better understanding of the biological and molecular landscape of DLBCL, a more detailed classification system and tailored treatments will be available in the near future.
Conflicts of interest
None.
Footnotes
How to cite this article: Wang L, Li LR. R-CHOP resistance in diffuse large B-cell lymphoma: biological and molecular mechanisms. Chin Med J 2021;134:253–260. doi: 10.1097/CM9.0000000000001294
References
- 1.Teras LR, DeSantis CE, Cerhan JR, Morton LM, Jemal A, Flowers CR. 2016 US lymphoid malignancy statistics by World Health Organization subtypes: 2016 US Lymphoid Malignancy Statistics by World Health Organization Subtypes. CA Cancer J Clin 2016; 66:443–459. doi: 10.3322/caac.21357. [DOI] [PubMed] [Google Scholar]
- 2.Chinese Society of Hematology CMA Chinese guidelines for diagnosis and treatment of diffuse large B cell lymphoma (in Chinese). Chin J Hematol 2013; 34:816.doi: 10.3760/cma.j.issn.0253-2727.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 3.Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: A study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 2010; 116:2040–2045. doi: 10.1182/blood-2010-03-276246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu JQ, Song YP, Su LP, Zhang MZ, Li W, Hu Y, et al. Three-year follow-up on the safety and effectiveness of rituximab plus chemotherapy as first-line treatment of diffuse large B-cell lymphoma and follicular lymphoma in real-world clinical settings in China: A prospective, multicenter, noninterventional study. Chin Med J 2018; 131:1767–1775. doi: 10.4103/0366-6999.237401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017; 130:1800–1808. doi: 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 7.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: Optimizing outcome in the context of clinical and biologic heterogeneity. Blood 2015; 125:22–32. doi: 10.1182/blood-2014-05-577189. [DOI] [PubMed] [Google Scholar]
- 8.Fu K, Weisenburger DD, Choi WWL, Perry KD, Smith LM, Shi X, et al. Addition of rituximab to standard chemotherapy improves the survival of both the germinal center B-cell–like and non–germinal center B-cell–like subtypes of diffuse large B-cell lymphoma. J Clin Oncol 2008; 26:4587–4594. doi: 10.1200/JCO.2007.15.9277. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med 2018; 378:1396–1407. doi: 10.1056/NEJMoa1801445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 2018; 24:679–690. doi: 10.1038/s41591-018-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melchardt T, Hufnagl C, Weinstock DM, Kopp N, Neureiter D, Trankenschuh W, et al. Clonal evolution in relapsed and refractory diffuse large B-cell lymphoma is characterized by high dynamics of subclones. Oncotarget 2016; 7:51494–51502. doi: 10.18632/oncotarget.9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rizzo D, Viailly P-J, Mareschal S, Bohers E, Picquenot J-M, Penther D, et al. Oncogenic events rather than antigen selection pressure may be the main driving forces for relapse in diffuse large B-cell lymphomas. Am J Hematol 2017; 92:68–76. doi: 10.1002/ajh.24584. [DOI] [PubMed] [Google Scholar]
- 13.Morin RD, Assouline S, Alcaide M, Mohajeri A, Johnston RL, Chong L, et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin Cancer Res 2016; 22:2290–2300. doi: 10.1158/1078-0432.CCR-15-2123. [DOI] [PubMed] [Google Scholar]
- 14.Zou L, Song G, Gu S, Kong L, Sun S, Yang L, et al. Mechanism and treatment of rituximab resistance in diffuse large B-cell lymphoma. Curr Cancer Drug Targets 2019; 19:681–687. doi: 10.2174/1568009619666190126125251. [DOI] [PubMed] [Google Scholar]
- 15.Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 2008; 359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linderoth J, Eden P, Ehinger M, Valcich J, Jerkeman M, Bendahl P-O, et al. Genes associated with the tumour microenvironment are differentially expressed in cured versus primary chemotherapy-refractory diffuse large B-cell lymphoma. Br J Haematol 2008; 141:423–432. doi: 10.1111/j.1365-2141.2008.07037.x. [DOI] [PubMed] [Google Scholar]
- 17.Xu-Monette ZY, Li L, Byrd JC, Jabbar KJ, Manyam GC, Maria de Winde C, et al. Assessment of CD37 B-cell antigen and cell of origin significantly improves risk prediction in diffuse large B-cell lymphoma. Blood 2016; 128:3083–3100. doi: 10.1182/blood-2016-05-715094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiyasu J, Miyoshi H, Hirata A, Arakawa F, Ichikawa A, Niino D, et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015; 126:2193–2201. doi: 10.1182/blood-2015-02-629600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 Blockade by Hu5F9-G4 and rituximab in non-Hodgkin's lymphoma. N Engl J Med 2018; 379:1711–1721. doi: 10.1056/NEJMoa1807315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouwstra R, He Y, de Boer J, Kooistra H, Cendrowicz E, Fehrmann RSN, et al. CD47 expression defines efficacy of rituximab with CHOP in non-germinal center B-cell (non-GCB) diffuse large B-cell lymphoma patients (DLBCL), but not in GCB DLBCL. Cancer Immunol Res 2019; 7 (10):1663–1671. doi: 10.1158/2326-6066.CIR-18-0781. [DOI] [PubMed] [Google Scholar]
- 21.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment: Tumour immunology. Nat Rev Immunol 2007; 7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 22.Mraz M, Zent CS, Church AK, Jelinek DF, Wu X, Pospisilova S, et al. Bone marrow stromal cells protect lymphoma B-cells from rituximab-induced apoptosis and targeting integrin (-4-(-1 (VLA-4) with natalizumab can overcome this resistance. Br J Haematol 2011; 155:53–64. doi: 10. 1111/j. 1365-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin H, Zhong F, Ouyang Y, Wang Q, Ding L, He S. Upregulation of ADAM12 contributes to accelerated cell proliferation and cell adhesion-mediated drug resistance (CAM-DR) in non-Hodgkin's lymphoma. Hematology (Amsterdam, Netherlands) 2017; 22:527–535. doi: 10.1080/10245332.2017.1312205. [DOI] [PubMed] [Google Scholar]
- 24.Cardesa-Salzmann TM, Colomo L, Gutierrez G, Chan WC, Weisenburger D, Climent F, et al. High microvessel density determines a poor outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus chemotherapy. Haematologica 2011; 96:996–1001. doi: 10.3324/haematol.2010.037408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: From genomics to mechanism. Oncogene 2003; 22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 26.Schwab M, Eichelbaum M, Fromm MF. Genetic polymorphisms of the human MDR1 drug transporter. Annu Rev Pharmacol Toxicol 2003; 43:285–307. doi: 10.1146/annurev.pharmtox.43.100901.140233. [DOI] [PubMed] [Google Scholar]
- 27.Kerb R, Hoffmeyer S, Brinkmann U. ABC drug transporters: Hereditary polymorphisms and pharmacological impact in MDR1, MRP1 and MRP2. Pharmacogenomics 2001; 2:51–64. doi: 10.1517/14622416.2.1.51. [DOI] [PubMed] [Google Scholar]
- 28.Kim JE, Singh RR, Cho-Vega JH, Drakos E, Davuluri Y, Khokhar FA, et al. Sonic hedgehog signaling proteins and ATP-binding cassette G2 are aberrantly expressed in diffuse large B-Cell lymphoma. Mod Pathol 2009; 22:1312–1320. doi: 10.1038/modpathol.2009.98. [DOI] [PubMed] [Google Scholar]
- 29.Ohsawa M, Ikura Y, Fukushima H, Shirai N, Sugama Y, Suekane T, et al. Immunohistochemical expression of multidrug resistance proteins as a predictor of poor response to chemotherapy and prognosis in patients with nodal diffuse large B-cell lymphoma. Oncology 2005; 68:422–431. doi: 10.1159/000086984. [DOI] [PubMed] [Google Scholar]
- 30.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127:2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkozy C, Traverse-Glehen A, Coiffier B. Double-hit and double-protein-expression lymphomas: aggressive and refractory lymphomas. Lancet Oncol 2015; 16:555–567. doi: 10.1016/S1470-2045(15)00005-4. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Zhang X, Zhang T, Song Z, Hu G, Li W, et al. Prognostic significance of BCL-2 and BCL-6 expression in MYC-positive DLBCL. Clin Lymphoma Myeloma Leuk 2018; 18:e381–e389. doi: 10.1016/j.clml.2018.06.010. [DOI] [PubMed] [Google Scholar]
- 33.Islam S, Paek AL, Hammer M, Rangarajan S, Ruijtenbeek R, Cooke L, et al. Drug-induced aneuploidy and polyploidy is a mechanism of disease relapse in MYC/BCL2-addicted diffuse large B-cell lymphoma. Oncotarget 2018; 9:36875–36890. doi: 10.18632/oncotarget.26251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008; 8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 35.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988; 335:440.doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 36.Casan JM, Barraclough A, Shortt J, Hawkes EA. Dose-adjusted EPOCH-R therapy in MYC-rearranged diffuse large B-cell lymphoma: Not yet the standard of care. Lancet Haematol 2019; 6:e119.doi: 10.1016/S2352-3026(19)30013-4. [DOI] [PubMed] [Google Scholar]
- 37.Landsburg DJ, Hughes ME, Koike A, Bond D, Maddocks KJ, Guo L, et al. Outcomes of patients with relapsed/refractory double-expressor B-cell lymphoma treated with ibrutinib monotherapy. Blood Adv 2019; 3:132–135. doi: 10.1182/bloodadvances.2018026401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davids MS, Seymour JF, Gerecitano JF, Kahl BS, Pagel JM, Wierda WG, et al. Phase I study of ABT-199 (GDC-0199) in patients with relapsed/refractory (r/r) non-Hodgkin lymphoma (NHL): Responses observed in diffuse large B-cell (DLBCL) and follicular lymphoma (FL) at higher cohort doses. Clin Adv Hematol Oncol 2014; 12: 8 Suppl 16: 18–19. [PubMed] [Google Scholar]
- 39.Chamuleau MED, Burggraaff CN, Nijland M, Bakunina K, Mous R, Lugtenburg PJ, et al. Treatment of patients with MYC rearrangement positive large B-cell lymphoma with R-CHOP plus lenalidomide: Results of a multicenter HOVON phase II trial. Haematologica 2019; doi: 10.3324/haematol.2019.238162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Azizian NG, Dou Y, Pham LV, Li Y. Simultaneous targeting of XPO1 and BCL2 as an effective treatment strategy for double-hit lymphoma. J Hematol Oncol 2019; 12:119.doi: 10.1186/s13045-019-0803-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kloo B, Nagel D, Pfeifer M, Grau M, Düwel M, Vincendeau M, et al. Critical role of PI3K signaling for NF-(B–dependent survival in a subset of activated B-cell–like diffuse large B-cell lymphoma cells. Proc Natl Acad Sci U S A 2011; 108:272–277. doi: 10.1073/pnas.1008969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K, et al. Simultaneous inhibition of PI3Kdelta and PI3Kalpha induces ABC-DLBCL regression by blocking BCR-dependent and -independent activation of NF-kappaB and AKT. Cancer Cell 2017; 31:64–78. doi: 10.1016/j.ccell.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010; 463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fontan L, Kabaleeswaran V, Volpon L, Osborne Michael J, Beltran E, Garcia M, et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 2012; 22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W-G, Cui W-L, Wang L, Zhu F, Wan X-C, Ping B, et al. Loss of B-cell receptor expression defines a subset of diffuse large B-cell lymphoma characterized by silent BCR/PI3K/AKT signaling and a germinal center phenotype displaying low-risk clinicopathologic features. Am J Surg Pathol 2015; 39:902–911. doi: 10.1097/PAS.0000000000000396. [DOI] [PubMed] [Google Scholar]
- 46.Merli M, Ferrario A, Maffioli M, Olivares C, Stasia A, Arcaini L, et al. New uses for brentuximab vedotin and novel antibody drug conjugates in lymphoma. Expert Rev Hematol 2016; 9:767–780. doi: 10.1080/17474086.2016.1205949. [DOI] [PubMed] [Google Scholar]
- 47.Nagel D, Spranger S, Vincendeau M, Grau M, Raffegerst S, Kloo B, et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 2012; 22:825–837. doi: 10.1016/j.ccr.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Flinn IW, Bartlett NL, Blum KA, Ardeshna KM, LaCasce AS, Flowers CR, et al. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Eur J Cancer 2016; 54:11–17. doi: 10.1016/j.ejca.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 49.Wilson WH, Gerecitano JF, Goy A, De Vos S, Kenkre VP, Barr PM, et al. The bruton's tyrosine kinase (BTK) inhibitor, ibrutinib (PCI-32765), has preferential activity in the ABC subtype of relapsed/refractory de novo diffuse large B-cell lymphoma (DLBCL): Interim results of a multicenter, open-label, phase 2 study. Blood 2012; 120:686. [Google Scholar]
- 50.Szydlowski M, Kiliszek P, Sewastianik T, Jablonska E, Bialopiotrowicz E, Gorniak P, et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood 2016; 127:739–748. doi: 10.1182/blood-2015-06-654111. [DOI] [PubMed] [Google Scholar]
- 51.Palanca-Wessels MC, Salles GA, Czuczman MS, Assouline SE, Flinn IW, Sehn LH, et al. Final results of a phase I study of the anti-CD79b antibody-drug conjugate DCDS4501A in relapsed or refractory (R/R) B-cell non-Hodgkin lymphoma (NHL). Blood 2013; 122:4400.doi: 10.1182/blood.V122.21.4400.4400. [Google Scholar]
- 52.Crump M, Leppä S, Fayad L, Lee JJ, Rocco AD, Ogura M, et al. Randomized, double-blind, phase III trial of enzastaurin versus placebo in patients achieving remission after first-line therapy for high-risk diffuse large B-cell lymphoma. J Clin Oncol 2016; 34:2484–2492. doi: 10.1200/JCO.2015.65.7171. [DOI] [PubMed] [Google Scholar]
- 53.Nagel D, Bognar M, Eitelhuber AC, Kutzner K, Vincendeau M, Krappmann D. Combinatorial BTK and MALT1 inhibition augments killing of CD79 mutant diffuse large B cell lymphoma. Oncotarget 2015; 6:42232–42242. doi: 10.18632/oncotarget.6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 55.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-(B by the Akt/PKB kinase. Curr Biol 1999; 9: Suppl 1: 601–604. doi: 10.1016/S0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 56.García MG, Alaniz LD, Cordo Russo RI, Alvarez E, Hajos SE. PI3K/Akt inhibition modulates multidrug resistance and activates NF-(B in murine lymphoma cell lines. Leuk Res 2009; 33:288–296. doi: 10.1016/j.leukres.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 57.Pfeifer M, Grau M, Lenze D, Wenzel S-S, Wolf A, Wollert-Wulf B, et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 2013; 110:12420–12425. doi: 10.1073/pnas.1305656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hasselblom S, Hansson U, Olsson M, Torén L, Bergström A, Nilsson-Ehle H, et al. High immunohistochemical expression of p-AKT predicts inferior survival in patients with diffuse large B-cell lymphoma treated with immunochemotherapy: p-AKT Expression in DLBCL. Br J Haematol 2010; 149:560–568. doi: 10.1111/j.1365-2141.2010.08123.x. [DOI] [PubMed] [Google Scholar]
- 59.Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia 2011; 25:341–347. doi: 10.1038/leu.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu L, Li L, Medeiros LJ, Young KH. NF-(B signaling pathway and its potential as a target for therapy in lymphoid neoplasms. Blood Rev 2017; 31:77–92. doi: 10.1016/j.blre.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor (B activity is required for survival of activated B cell–like diffuse large B cell lymphoma cells. J Exp Med 2001; 194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cai Q, Tu M, Xu-Monette ZY, Sun R, Manyam GC, Xu X, et al. NF-(B p50 activation associated with immune dysregulation confers poorer survival for diffuse large B-cell lymphoma patients with wild-type p53. Mod Pathol 2017; 30:854–876. doi: 10.1038/modpathol.2017.5. [DOI] [PubMed] [Google Scholar]
- 63.Zhang M, Xu-Monette ZY, Li L, Manyam GC, Visco C, Tzankov A, et al. RelA NF-κB subunit activation as a therapeutic target in diffuse large B-cell lymphoma. Aging 2016; 8:3321–3340. doi: 10.18632/aging.101121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagel D, Vincendeau M, Eitelhuber AC, Krappmann D. Mechanisms and consequences of constitutive NF-(B activation in B-cell lymphoid malignancies. Oncogene 2014; 33:5655–5665. doi: 10.1038/onc.2013.565. [DOI] [PubMed] [Google Scholar]
- 65.Ma LY, Su L. Application of lenalidomide on diffused large B-cell lymphoma: Salvage, maintenance, and induction treatment. Chin Med J 2018; 131:2510–2513. doi: 10.4103/0366-6999.243567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Castellino A, Chiappella A, LaPlant BR, Pederson LD, Gaidano G, Macon WR, et al. Lenalidomide plus R-CHOP21 in newly diagnosed diffuse large B-cell lymphoma (DLBCL): Long-term follow-up results from a combined analysis from two phase 2 trials. Blood Cancer J 2018; 8:108.doi: 10.1038/s41408-018-0145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Calò V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, et al. STAT proteins: From normal control of cellular events to tumorigenesis. J Cell Physiol 2003; 197:157–168. doi: 10.1002/jcp.10364. [DOI] [PubMed] [Google Scholar]
- 68.Park YH, Sohn SK, Kim JG, Lee M-H, Song HS, Kim MK, et al. Interaction between BCL2 and interleukin-10 gene polymorphisms alter outcomes of diffuse large B-cell lymphoma following rituximab plus CHOP chemotherapy. Clin Cancer Res 2009; 15:2107–2115. doi: 10.1158/1078-0432.CCR-08-1588. [DOI] [PubMed] [Google Scholar]
- 69.Huang X, Meng B, Iqbal J, Ding BB, Perry AM, Cao W, et al. Activation of the STAT3 signaling pathway is associated with poor survival in diffuse large B-cell lymphoma treated with R-CHOP. J Clin Oncol 2013; 31:4520–4528. doi: 10.1200/JCO.2012.45.6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Béguelin W, Sawh S, Chambwe N, Chan FC, Jiang Y, Choo JW, et al. IL10 receptor is a novel therapeutic target in DLBCLs. Leukemia 2015; 29:1684–1694. doi: 10.1038/leu.2015.57. [DOI] [PubMed] [Google Scholar]
- 71.Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J Immunother Cancer 2018; 6:119.doi: 10.1186/s40425-018-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shaknovich R, Geng H, Johnson NA, Tsikitas L, Cerchietti L, Greally JM, et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 2010; 116:e81–e89. doi: 10.1182/blood-2010-05-285320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rui L, Drennan AC, Ceribelli M, Zhu F, Wright GW, Huang DW, et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A 2016; 113:E7260–E7267. doi: 10.1073/pnas.1610970113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu X, Li Z. New insights into MicroRNAs involves in drug resistance in diffuse large B cell lymphoma. Am J Transl Res 2015; 7:2536–2542. PMID: 26885255. [PMC free article] [PubMed] [Google Scholar]
- 75.Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A, et al. Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in Diffuse large B cell lymphomas. Am J Pathol 2010; 177:1470–1479. doi: 10.2353/ajpath.2010.091291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tomita A, Hiraga J, Kiyoi H, Ninomiya M, Sugimoto T, Ito M, et al. Epigenetic regulation of CD20 protein expression in a novel B-cell lymphoma cell line, RRBL1, established from a patient treated repeatedly with rituximab-containing chemotherapy. Int J Hematol 2007; 86:49–57. doi: 10.1532/IJH97.07028. [DOI] [PubMed] [Google Scholar]
- 77.Gupta M, Han JJ, Stenson M, Wellik L, Witzig TE. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012; 26:1356–1364. doi: 10.1038/leu.2011.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo H, Schmidt JA, Lee Y-S, Oltz EM, Payton JE. Targeted epigenetic repression of a lymphoma oncogene by sequence-specific histone modifiers induces apoptosis in DLBCL. Leuk Lymphoma 2017; 58:445–456. doi: 10.1080/10428194.2016.119097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov 2013; 3:1002–1019. doi: 10.1158/2159-8290.CD-13-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morschhauser F, Salles G, McKay P, Tilly H, Schmitt A, Gerecitano J, et al. Interim report from a phase 2 multicenter study of Tazemetostat, an EZH2 inhibitor, in patients with relapsed or refractory B-cell non-Hodgkin lymphomas. Hematol Oncol 2017; 35:24–25. doi: 10.1002/hon.2437_3. [Google Scholar]