

TO THE EDITOR:
In addition to preserving homeostasis and contributing to immunity, platelets may exhibit a dysregulated state1,2 in acute respiratory distress syndrome (ARDS) and potentially exacerbate lung injury.3-5 Although thrombocytopenia has been associated with increased mortality in ARDS,6 and particularly in patients infected with H1N1 influenza,7 analysis of quantitative changes in platelet activation levels in ARDS and of how these may differ between the various etiologies of ARDS is limited.3
Coronavirus disease 2019 (COVID-19) is clinically associated with a high incidence of thrombosis8-10 and with a high burden of pulmonary and systemic platelet-rich microthrombi.11,12 Interestingly, in contrast to a cohort of patients with H1N1-influenza infection with ARDS of equal severity, patients with COVID-19 ARDS displayed a 9 times higher burden of alveolar capillary platelet-rich microthrombi.11 Patients with COVID-19 who were autopsied demonstrated an abundance of platelet-rich thrombi in the pulmonary and systemic microvasculature despite having received anticoagulation therapy.12 Clinically, thromboelastography (TEG) studies have also demonstrated a hypercoagulable profile in patients with severe COVID-199,13 despite prophylactic anticoagulation. TEG profiles remained hypercoagulable, with parameters suggesting a significant hypercoagulable effect from platelet activity and fibrin.13
Multiple study groups14-16 including ours17 have reported dramatically increased platelet activation, platelet reactivity, and platelet-leukocyte aggregates in COVID-19 compared with those in healthy blood donors (controls). In one study,15 patients with severe COVID-19 harbored a higher degree of platelet activation and platelet-monocyte aggregation compared with patients with COVID-19 that was not severe. In addition, plasma from patients with severe COVID-19 increased the ex vivo activation of platelets and monocytes from healthy controls, a phenomenon inhibited by abciximab, a platelet aggregation inhibitor. Platelet activation correlated with COVID-19 severity in that study.15 A second study demonstrated an 89-fold increase in platelet hyperreactivity with low-dose stimulation by thromboxane A2 (TXA2) receptor agonist compared with that in controls.16 A third study14 demonstrated increases in platelet activation, platelet reactivity, and platelet-leukocyte aggregates in patients with COVID-19 compared with healthy controls. These studies demonstrate that platelet reactivity is enhanced in patients with COVID-19 and posit that platelet hyperreactivity may be a primary driver of thrombosis in patients with severe COVID-19, contributing to organ failure and death. Whether this degree of platelet hyperreactivity is unique to COVID-19 or is also encountered during ARDS not related to COVID-19 was addressed in our study.
COVID-19 patients without (n = 11) and with ARDS (n = 9) and patients with ARDS unrelated to COVID-19 etiology (n = 8) who were admitted to the Cheikh Zaid Hospital of Abulcasis University (Rabat, Morocco) were included in this study. All patients with COVID-19 were studied (on average) at 7.2 ± 0.6 hours after receiving a nasopharyngeal swab that showed positivity for SARS-CoV-2 (COVID-19). Ten healthy blood donors were used as additional controls. Recruitment was approved by the Ethics Committee of Cheikh Zaid Hospital (CEFCZ/PR/2020/PR04) and complies with the Declaration of Helsinki. All participants gave their written informed consent. Patients who were receiving medications that interfere with platelet function within 2 weeks before the experiment started were excluded from the study. COVID-19 diagnosis was confirmed at the time of admission by reverse transcription polymerase chain reaction analysis of samples from the nasopharyngeal swabs, and patients were subsequently divided into nonsevere and severe COVID-19 groups on the basis of clinical criteria using the American Thoracic Society guidelines for community-acquired pneumonia.18 Patients with nonsevere COVID-19 were not included in this study. Etiologies of ARDS unrelated to COVID-19 were diverse and included pneumococcal pneumonia (n = 1), carbon monoxide poisoning (n = 1), post-cardiac surgery (n = 2), rhinovirus infection (n = 1), and cerebrovascular accident (n = 3). ARDS was defined according to Berlin criteria.19 Sex- and age-matched healthy blood donors were used as controls. When platelet samples were acquired, all patients with ARDS had an arterial oxygen partial pressure:fractional inspired oxygen (PaO2:FiO2) ratio ≤150 mmHg and were given pressure-controlled ventilation with tidal volumes of ∼6 to 8 mL/kg ideal body weight. Patients with COVID-19 ARDS were maintained on a positive end-expiratory pressure of 8 to 14 cm H2O. Only 1 patient had renal failure (ARDS with carbon monoxide poisoning), and none required renal replacement therapy at the time of sample acquisition. Table 1 provides basic demographic and clinical data for patients. Supplemental Tables 1 and 2 provide comparative analysis of the clinical, hematologic, and biochemical parameters of patients with ARDS with or without COVID-19, and comorbidities are listed in supplemental Table 3.
Table 1.
Comparison of healthy donors, patients with COVID-19 (without or with ARDS), and patients with ARDS unrelated to COVID-19
| Index | ARDS | P* | non-ARDS | P* | ||
|---|---|---|---|---|---|---|
| No COVID-19 | COVID-19 | COVID-19 | Healthy donors | |||
| No. of patients | 8 | 9 | 11 | 10 | ||
| Sex | ||||||
| Female | 2 | 4 | 6 | 5 | ||
| Male | 6 | 5 | 5 | 5 | ||
| Survivors | 7 | 9 | 11 | 10 | ||
| Age, y | 57 ± 23.98 | 57 ± 24.04 | >.99 | 47.54 ± 13.99 | 45.40 ± 18.58 | .7674 |
| Weight, kg | 85.43 ± 22.40 | 71.17 ± 16.70 | .2219 | 71.36 ± 10.75 | 74.00 ± 15.64 | .6546 |
| Hospitalization time, d | 9.14 ± 5.96 | 12 ± 4.53 | .2930 | 9.00 ± 3.07 | 0 + 0 | NA |
| Mechanical ventilation | Yes | Yes | NA | No | No | NA |
| FiO2, % | 50 ± 20 | 62.86 ± 7.56 | .1463 | NA | NA | NA |
| PaO2:FiO2, mm Hg | <150 | <150 | NA | NA | NA | NA |
| Values at admission | ||||||
| Platelet count, × 109/L | 243.0 ± 74.4 | 159.9 ± 76.51 | .0467 | 194.55 ± 82.07 | 273.20 ± 64.27 | .0253 |
| Lymphocyte count, × 109/L | 2.13 ± 1.032 | 1.28 ± 0.6438 | .0621 | 1.27 ± 0.76 | 2.15 ± 1.20 | .0568 |
| Platelet:lymphocyte ratio | 144 ± 92.06 | 141.2 ± 72.24 | .9465 | 165.24 ± 53.39 | 177.82 ± 124.41 | .7627 |
| ALT, U/L | 34.94 ± 15.61 | 33.27 ± 13.61 | .1016 | 34.11 ± 15.56 | 16.74 ± 5.88 | .0036 |
| AST, U/L | 35.43 ± 12.05 | 42.98 ± 10.74 | .2069 | 35.11 ± 17.82 | 15.95 ± 3.93 | .0036 |
| LDH, U/L | 417.9 ± 202.4 | 649.1 ± 245.6 | .0639 | 583.27 ± 258.68 | 314.80 ± 110.45 | .0068 |
| C-reactive protein, mg/L | 16.16 ± 8.79 | 18.52 ± 13.67 | .6989 | 13.78 ± 7.99 | 8.16 ± 3.82 | .0576 |
| D-dimers, mg/L | 0.39 ± 0.29 | 1.31 ± 0.57 | .0017 | 0.97 ± 0.54 | 0.40 ± 0.28 | .0076 |
| Fibrinogen, mg/dL | 260.62 ± 92.49 | 439.50 ± 76.29 | .0005 | 274.64 ± 102.02 | 272.70 ±76.31 | .9615 |
Data are presented as mean ± standard deviation.
ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; NA, not applicable.
Statistical analysis: unpaired Student t test was used to calculate P values.
Blood samples were collected from all patients, and platelets were isolated according to an established protocol.17 In brief, platelet-rich plasma was obtained by centrifuging acid citrate dextrose solution and anticoagulated blood (1:5 ratio) at 200g for 15 minutes. Platelets were then pelleted from platelet-rich plasma (1000g for 10 minutes) and washed with Hanks balanced salt solution-Hanks sodium citrate buffer to a final concentration of 250 × 109/L. When platelets of patients (n = 5) were tested, no spontaneous aggregation of platelets in the presence of 137 mM NaCl was observed. Platelet suspensions were stimulated or not with α-thrombin (0.05 and 2 U/mL) under continuous stirring (1000 rpm) at 37°C. Platelet aggregation was monitored and recorded using an 8-channel optical aggregometer (SD Medical Innovation, Frouard, France).
Our findings demonstrate that platelets from patients with COVID-19 are activated much more efficiently than platelets from healthy controls and from patients with ARDS without COVID-19 (Figure 1A) in response to a suboptimal α-thrombin concentration (0.05 U/mL). These differences were abolished at a higher concentration of α-thrombin (2 U/mL). Furthermore, the time required to achieve 50% aggregation (T50) was significantly reduced for platelets obtained from patients with COVID-19 ARDS (T50 = 1.8 ± 0.2 minutes) compared with platelets obtained from patients with ARDS unrelated to COVID-19 (T50 = 3.1 ± 0.4 minutes) (P < .05) (Figure 1B). Plasma samples at the time of admission were also analyzed for thromboxane B2 (TXB2, a stable metabolite of TXA2), platelet factor 4 (PF4), soluble CD40 ligand (sCD40L), and serotonin as surrogate markers of in vivo platelet activation. Results (Figure 1C-F) indicate that plasma from patients with ARDS unrelated to COVID-19 have increased levels of TXB2 and PF4 and have equivalent levels of sCD40L and serotonin relative to levels in healthy controls. In contrast, patients with COVID-19 (who did or did not have ARDS) demonstrated significantly increased levels of all 4 mediators relative to those in healthy controls as well as to those in patients with ARDS without COVID-19. These results suggest that during COVID-19, platelets release more soluble mediators and are hyperresponsive to α-thrombin relative to platelets from patients with ARDS without COVID-19 and from healthy controls.
Figure 1.
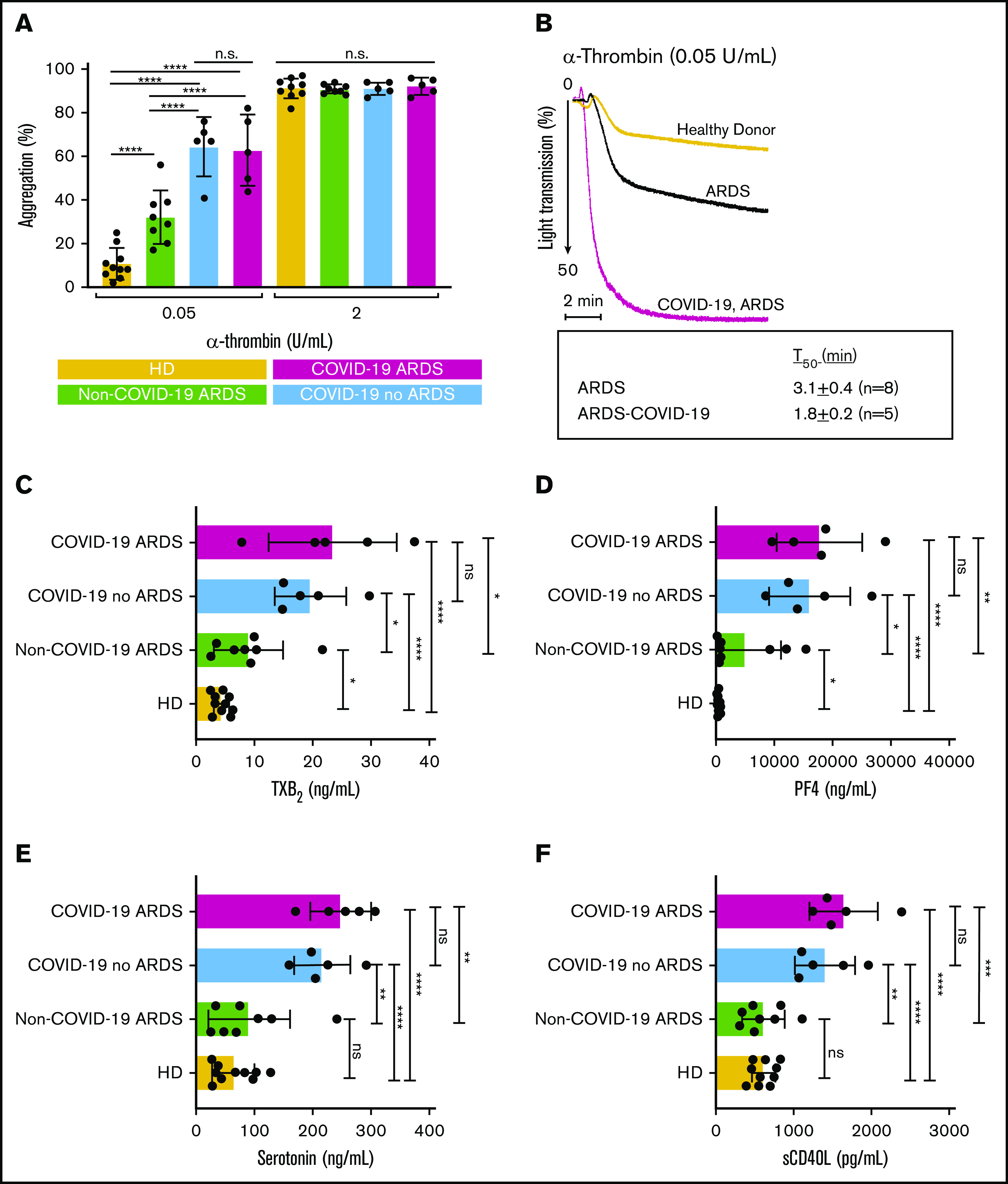
Enhanced platelet hyperreactivity is associated with COVID-19. Platelets were isolated from healthy donors (HDs), patients with ARDS unrelated to COVID-19, and patients with COVID-19 (with or without ARDS). (A) Quantification of platelet maximal aggregation for healthy donors (n = 10), for patients with ARDS unrelated to COVID-19 (n = 8), and for patients infected with SARS-CoV-2 (without ARDS [n = 5] and with ARDS [n = 5]) in response to α-thrombin activation. Statistical analysis was performed using ordinary one-way analysis of variance with Sidak’s multiple comparisons test. (B) Representative aggregation traces of washed platelets from healthy donors, patients with ARDS without COVID-19, and patients with ARDS and COVID-19 who were treated with α-thrombin (0.05 U/mL) under continuous stirring at 37°C. Times to reach 50% (T50) aggregation are indicated below graph. Levels of (C) TBX2, (D) PF4, (E) serotonin, and (F) sCD40L measured in plasma at time of platelet isolation were determined using commercial enzyme-linked immunosorbent assay kits. Statistical analysis was performed using paired Student t test. ns, not significant. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Overall, our findings demonstrate that platelet hyperreactivity is distinctly increased in patients with COVID-19 compared with patients who have ARDS unrelated to COVID-19. To the best of our knowledge, this is the first report that suggests a heightened platelet reactivity in COVID-19 patients compared with patients with ARDS unrelated to COVID-19. This difference in platelet hyperreactivity may be of clinical and therapeutic significance, given the high incidence of pulmonary and systemic vascular thrombosis in patients with severe COVID-198,9 who are frequently resistant to anticoagulation with heparin.9 This unusually resistant hypercoagulable state associated with a high incidence of thrombosis is likely mediated by platelet hyperreactivity, a phenomenon observed in other etiologies of ARDS but, as demonstrated in this study, occurring with a uniquely high intensity during COVID-19. Mirroring our findings in COVID-19, the existing evidence that links H1N1 influenza ARDS to increased platelet hyperreactivity,20 increased levels of platelet-derived chemokines,21 and a higher incidence of clinical thrombosis22 suggests that quantifying platelet hyperreactivity in COVID-19 in clinical practice may be of prognostic and therapeutic23 value.
We recognize that our study is limited by the number of participants, the heterogeneity of patients with ARDS unrelated to COVID-19, and the use of the single platelet agonist α-thrombin. A larger group of patients and additional platelet agonists are required to complement our observations. The strength of this study included using strict exclusion criteria and ruling out any influence of antiplatelet medications.
Our findings generate 2 testable hypotheses: (1) assess whether a platelet hyperreactivity threshold exists during the course of COVID-19 disease beyond which inhibitors of platelet activation and aggregation mediators may be of clinical benefit, and (2) determine the pathophysiologic role of mediators harboring extensive vasoactive, bronchoactive, and systemic effects (eg, serotonin,17,24 histamine,25 thromboxane14) released by platelets and other inflammatory cells (eg, mast cells) during COVID-19.
Acknowledgments: This study was supported by the Cheikh Zaid Foundation and by grants from the New Frontier Research Fund (NFRN-2019-00004) (L.F. and E.B.). F.P. is the recipient of a Fonds de Recherche du Québec en Santé (FRQS) fellowship award, and E.B. is recipient of a senior award from FRQS.
Supplementary Material
The full-text version of this article contains a data supplement.
Footnotes
For original data, please contact Younes Zaid at younes_zaid@yahoo.ca.
Contribution: Y.Z., F.G., E.B., F.J., and L.F. conceived and designed the study; Y.Z., F.P., W.E., L.C., and A.C. helped design the study, performed experiments, and helped with data extraction; Y.Z., F.G., F.P., A.C.M., F.J., E.B., and L.F. contributed critical reagents, biospecimens, and instruments; Y.Z., F.G., F.P., F.J., E.B., and L.F. wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Louis Flamand, Centre de Recherche du Centre Hospitalier Universitaire de Québec–Université Laval, 2705 Laurier Blvd, Room T1-49, Québec, QC G1V 4G2, Canada; e-mail: louis.flamand@crchudequebec.ulaval.ca.
References
- 1.Sreeramkumar V, Adrover JM, Ballesteros I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. 2014;346(6214):1234-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. 2016;7:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Middleton EA, Rondina MT, Schwertz H, Zimmerman GA. Amicus or adversary revisited: platelets in acute lung injury and acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2018;59(1):18-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grommes J, Alard JE, Drechsler M, et al. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med. 2012;185(6):628-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest. 2009;119(11):3450-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Liu Z, Wang Z, et al. Thrombocytopenia is associated with acute respiratory distress syndrome mortality: an international study. PLoS One. 2014;9(4):e94124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Delgado JC, Rovira A, Esteve F, et al. Thrombocytopenia as a mortality risk factor in acute respiratory failure in H1N1 influenza. Swiss Med Wkly. 2013;143:w13788. [DOI] [PubMed] [Google Scholar]
- 8.Moll M, Zon RL, Sylvester KW, et al. VTE in ICU patients with COVID-19. Chest. 2020;158(5):2130-2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maatman TK, Jalali F, Feizpour C, et al. Routine venous thromboembolism prophylaxis may be inadequate in the hypercoagulable state of severe coronavirus disease 2019. Crit Care Med. 2020;48(9):e783-e790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klok FA, Kruip MJHA, van der Meer NJM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapkiewicz AV, Mai X, Carsons SE, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020;24:100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuriditsky E, Horowitz JM, Merchan C, et al. Thromboelastography profiles of critically ill patients with coronavirus disease 2019. Crit Care Med. 2020;48(9):1319-1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hottz ED, Azevedo-Quintanilha IG, Palhinha L, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. 2020;136(11):1330-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comer SP, Cullivan S, Szklanna PB, et al. COVID-19 induces a hyperactive phenotype in circulating platelets [published online ahead of print 5 August 2020]. medRxiv. doi:10.1101/2020.07.24.20156240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaid Y, Puhm F, Allaeys I, et al. Platelets can associate with SARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020;127(11):1404-1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Metlay JP, Waterer GW, Long AC, et al. Diagnosis and treatment of adults with community-acquired pneumonia. An official clinical practice guideline of the American Thoracic Society and Infectious Diseases Society of America. Am J Respir Crit Care Med. 2019;200(7):e45-e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ranieri VM, Rubenfeld GD, Thompson BT, et al. ; ARDS Definition Task Force . Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526-2533. [DOI] [PubMed] [Google Scholar]
- 20.Rondina MT, Brewster B, Grissom CK, et al. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest. 2012;141(6):1490-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lê VB, Schneider JG, Boergeling Y, et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med. 2015;191(7):804-819. [DOI] [PubMed] [Google Scholar]
- 22.Harms PW, Schmidt LA, Smith LB, et al. Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol. 2010;134(1):27-35. [DOI] [PubMed] [Google Scholar]
- 23.Sugiyama MG, Gamage A, Zyla R, et al. Influenza virus infection induces platelet-endothelial adhesion which contributes to lung injury. J Virol. 2015;90(4):1812-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cloutier N, Allaeys I, Marcoux G, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A. 2018;115(7):E1550-E1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selig WM, Patterson CE, Henry DP, Rhoades RA. Role of histamine in acute oleic acid-induced lung injury. J Appl Physiol. 1986;61(1):233-239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.