

Abstract
Purpose
Thyroid eye disease (TED) is a condition that causes the tissue behind the eye to become inflamed and can result in excessive fatty tissue accumulation in the orbit. Two subpopulations of fibroblasts reside in the orbit: those that highly express Thy1 (Thy1+) and those with little or no Thy1 (Thy1–). Thy1– orbital fibroblasts (OFs) are more prone to lipid accumulation than Thy1+ OFs. The purpose of this study was to investigate the mechanisms whereby Thy1– OFs more readily accumulate lipid.
Methods
We screened Thy1+ and Thy1– OFs for differences in microRNA (miRNA) expression. The effects of increasing miR-130a levels in OFs was investigated by measuring lipid accumulation and visualizing lipid deposits. To determine if adenosine monophosphate-activated protein kinase (AMPK) is important for lipid accumulation, we performed small interfering RNA (siRNA)-mediated knockdown of AMPKβ1. We measured AMPK expression and activity using immunoblotting for AMPK and AMPK target proteins.
Results
We determined that miR-130a was upregulated in Thy1– OFs and that miR-130a targets two subunits of AMPK. Increasing miR-130a levels enhanced lipid accumulation and reduced expression of AMPKα and AMPKβ in OFs. Depletion of AMPK also increased lipid accumulation. Activation of AMPK using AICAR attenuated lipid accumulation and increased phosphorylation of acetyl-CoA carboxylase (ACC) in OFs.
Conclusions
These data suggest that when Thy1– OFs accumulate in TED, miR-130a levels increase, leading to a decrease in AMPK activity. Decreased AMPK activity promotes lipid accumulation in TED OFs, leading to excessive fatty tissue accumulation in the orbit.
Keywords: miR-130a, thyroid eye disease, Graves’ orbitopathy, fibroblast, lipid accumulation, AMP-activated protein kinase
Thyroid eye disease (TED) is a manifestation of autoimmune thyroid disease characterized by extensive tissue remodeling in the orbit. Approximately 40% to 50% of patients with Graves’ disease will also be diagnosed with TED.1,2 Of those that present with symptoms of TED, approximately 3% to 6% will go on to develop sight-threatening disease.1 There is no current therapy to prevent TED from occurring in autoimmune thyroid disease patients. Corticosteroids and orbital decompression surgeries to minimize symptoms of TED have been the standard of care for decades. Recently, a new biological drug has been approved by the US Food and Drug Administration to treat TED; teprotumumab (Tepezza; Horizon Therapeutics, Dublin, Ireland)3 has shown promising results in a majority of patients, but some patients are refractory, it must be administered intravenously, and it has a significant cost burden.4 TED is a complex disorder with varied manifestations and is still incompletely understood. Uncovering the mechanisms involved in the pathophysiology of this disease will provide novel targets for therapies that would benefit a diverse set of patients.
The extensive orbital tissue remodeling observed in TED is caused by the activation and differentiation of fibroblasts in the orbit.2,5,6 Orbital fibroblasts (OFs) are a heterogeneous population of cells that can proliferate and differentiate into both myofibroblasts that contribute to scarring and adipocytes that accumulate into excessive amounts of fat tissue. Fibroblasts with high levels of Thy1 surface expression (Thy1+) more readily form myofibroblasts, and fibroblasts with little or no Thy1 (Thy1–) form adipocytes when activated.7–9 OFs can be activated by infiltrating T cells that produce prostaglandins such as 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), an endogenous ligand of peroxisome proliferator-activated receptor gamma (PPARγ), a transcription factor that promotes adipogenesis and lipid accumulation.5,10
Adenosine monophosphate-activated protein kinase (AMPK) is a central player in energy homeostasis and regulates lipid accumulation. AMPK activation reduces lipid accumulation by promoting oxidation of fatty acids and limiting lipid synthesis.11 AMPK is a heterotrimeric complex consisting of a catalytic AMPKα subunit and regulatory AMPKβ and AMPKγ subunits.12,13 AMPK is activated by AMP or adenosine diphosphate (ADP) binding, which leads to the activating phosphorylation of the AMPKα subunit.11 When AMPK has been activated, it phosphorylates and inactivates lipid anabolic enzymes such as acetyl-CoA carboxylase (ACC) and sterol-regulatory-binding-protein 1c (SREBP1c).13
One of the mechanisms for controlling protein expression is through expression of microRNAs (miRNAs). Over 2000 small non-coding miRNAs have been identified that are thought to regulate up to 60% of protein encoding genes.14 miRNAs can regulate multiple target genes by reducing target protein translation and/or decreasing mRNA stability.14 Different cell types and tissues have varied miRNA expression, and the expression of miRNAs can also be affected by aging and disease processes.15
This study set out to investigate differences in Thy1+ and Thy1– OFs in order to understand the molecular mechanisms driving lipid accumulation in Thy1- OFs. We found that miR-130a is elevated in Thy1– OFs and TED tissue and that miR-130a targets AMPK. This novel mechanism could provide a target for new therapeutics to benefit TED patients.
Methods
Cell Culture
Orbital tissue explants were collected during surgical decompression required for the management of TED as well as non-TED eye surgeries at the Flaum Eye Institute at the University of Rochester Medical Center. Informed written consent was obtained from individuals prior to surgery following the guidelines and approval of the University of Rochester Medical School Research Subjects Review Board. Non-orbital fat tissues were provided as scrap waste tissue from the Surgical Pathology Clinical Laboratory at the University of Rochester Medical Center after patient identifying information was removed. Fat tissues were processed as previously described.16 Each OF strain represents fibroblasts isolated from an individual patient. The Table describes key characteristics of the patient population. OFs were used for experiments between passages 3 and 9. Fibroblast strains were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT, USA) and antibiotics (Invitrogen). To initiate adipogenesis, OFs were transferred to DMEM containing 3% FBS and treated with 5-µM 15d-PGJ2 (Cayman Chemical, Ann Arbor, MI, USA), an endogenous PPARγ ligand known to promote adipogenesis,17,18 every second day for 8 to 12 days.
Table.
Demographic Data
| Age (y) | Sex | Smoking Status | Steroid Use |
|---|---|---|---|
| TED patients | |||
| 69 | F | Yes | No |
| 56 | F | Former | No |
| 58 | F | Yes | Oral |
| 53 | F | No | No |
| 27 | F | No | Oral |
| 47 | F | No | Oral |
| 34 | M | Former | No |
| 49 | M | Former | No |
| Non-TED patients | |||
| 58 | F | No | No |
| 45 | F | No | No |
| 48 | F | No | No |
| 57 | M | No | No |
miRNA Extraction and Quantitative PCR
Total cell RNA, including miRNAs were isolated with an miRNeasy Mini Kit (Qiagen, Hilden, Germany). RNA concentrations were determined with the DeNovix DS-11 spectrophotometer (DeNovix Inc., Wilmington, DE, USA). cDNA was generated using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA), and miRNA expression was quantified via real-time quantitative PCR using a TaqMan Universal PCR Master Mix (Applied Biosystems) and the BioRad CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA). Specific primers for hsa-miR-130a-3p were obtained from Applied Biosystems. Expression of miR-130a was normalized to the expression of either U6 small nuclear RNA (snRNA) or miR-16-5p (Applied Biosystems), and the expression of these RNAs was similar in all treatment conditions. The data were then further normalized by setting the control values to one.
Luciferase Reporter Assays
Human embryonic kidney (HEK293FT) cells were used in reporter assays as previously described.19 The reporter plasmid contained the PRKAB1 3′UTR (AMPKβ) cloned downstream of the Renilla luciferase open reading frame in the psiCheck2.1 vector (Promega Corporation, Madison, WI, USA). The human PRKAB1 3′UTR was amplified from human OF cDNA by PCR using KOD polymerase (Novagen Inc., Madison, WI, USA) using the following primers: PRKAB1 3′UTR fwd: 5′-TACTCGAGAGAGCTGGGGGCGGATGG-3′; PRKAB1 3′UTR rev: 5′-TAGCGGCCGCGAAACTTGTCCATCAAGG-3′, where the underlined nucleotides indicate restriction sites for XhoI and NotI. The 3′UTR sequence was verified by DNA sequencing and then inserted into the psiCheck2.1 vector. After introduction of plasmid, cells were further incubated for 18 hours, and then cells were lysed directly in plates using Promega Dual-Glo Luciferase Reagent. Firefly and Renilla luciferase readings were measured on a Varioskan Flash luminescent plate reader (Thermo Fisher Scientific, Waltham, MA, USA). Renilla activity levels were normalized to firefly activity, and values are reported as relative light units.
Introduction of miR-130a Mimics
OFs were cultured in DMEM containing 3% FBS. OFs were then treated with the hsa-miR-130 mirVana miRNA mimic (Ambion Inc., Austin, TX, USA) mixed with Invitrogen Lipofectamine 2000 transfection reagent in Invitrogen Opti-MEM I at a final concentration of 100 nM. Some cultures were treated with 5-µM 15d-PGJ2 every second day for 8 to 12 days. In these instances, miRNA-130a mimic was added 24 hours before application of the 15d-PGJ2. The mimic was reintroduced to cultures on day 4 with the method described above.
AdipoRed Assay
Fibroblasts were plated into 24-well plates and treated as described. After treatment, cells were analyzed for triglyceride accumulation using the AdipoRed Assay Reagent (Lonza Walkersville, Inc., Walkersville, MD, USA) following the manufacturer's instructions. Culture medium was removed, and the cells were washed in 1× PBS. Then, cells were incubated with the diluted AdipoRed reagent for 10 minutes. The cell–AdipoRed complexes were excited at 485 nm in a Varioskan Flash plate reader, and the fluorescence at 572 nm was quantified.
Western Blot Analysis
OFs were lysed and processed as described previously.20 Antibodies targeting p-AMPKα (rabbit anti-p-AMPKα), AMPKα (rabbit anti-AMPKα), p-AMPKβ (rabbit anti-p-AMPKβ), AMPKβ1/2 (rabbit anti-AMPKβ1/2), p-ACC (rabbit anti-p-ACC), ACC (rabbit anti-ACC), and β-tubulin (rabbit anti-β-tubulin) were all obtained from Cell Signaling Technology (Danvers, MA, USA), and AMPKβ (mouse anti-AMPKβ) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Fluorescent Staining
OFs were fixed with 4% paraformaldehyde for 20 minutes, stained with Invitrogen HCS LipidTOX green neutral lipid stain, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) nucleic acid binding dye (Invitrogen). Fluorescence was visualized on a ZOE Fluorescent Cell Imager (Bio-Rad) utilizing the same settings for each image in the experiment.
Gene Expression Knockdown Using Small Interfering RNA
Knockdown experiments were done as described previously.16 Three different PRKAB1 (the gene for AMPKβ) small interfering RNAs (siRNAs) were used in experiments and yielded similar results. PRKAB1 siRNA (catalog nos. 4392420 and 4390824) and a non-specific, negative control siRNA (Negative Control #1) were purchased from Ambion (Ambion). The third PRKAB1 siRNA was purchased from Santa Cruz Biotechnology (catalog no. sc38925).
Statistical Analysis
Data were analyzed using Prism 7 (GraphPad Software, San Diego, CA, USA). Statistical significance is denoted by P values of P < 0.05 (* or #); P < 0.01 (** or ##); P < 0.001 (*** or ###), or P < 0.0001 (**** or ####).
Results
miR-130a Is Elevated in TED
miRNAs are critical regulators of inflammation and cellular differentiation, two processes that play an important role in TED pathophysiology. We profiled miRNA expression in Thy1– and Thy1+ OFs using a low-density qPCR array of the 88 most common miRNAs. One of the most striking differences was found in miR-130a expression. miR-130a was approximately threefold higher in Thy1– TED OFs compared with the Thy1+ subpopulation (Fig. 1A). Thy1– OFs are more prone to accumulate lipid and form adipocytes; therefore, we next investigated whether miR-130a is associated with TED fat by measuring miR-130a levels in TED orbital fat tissue. miR-130a was increased by approximately 3.5-fold in TED orbital fat tissue compared to non-TED fat, suggesting that this miRNA might contribute to TED (Fig. 1B). Furthermore, these differences were maintained when explanted fibroblasts were grown out of these tissues. OFs expressed more miR-130a than fibroblasts explanted from non-orbital tissues (Fig. 1C). Not only did OFs have higher basal levels of miR-130a, but the levels of miR-130a increased threefold when OFs were exposed to 5-µM 15d-PGJ2 for 8 days (Fig. 1D).
Figure 1.
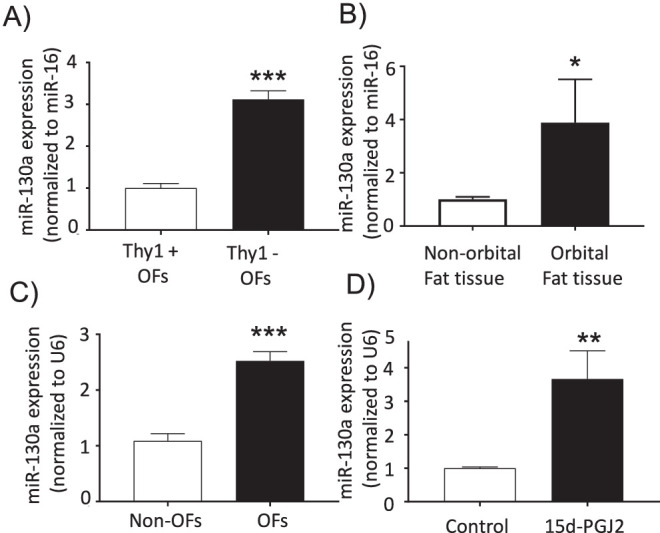
miR-130a levels were increased in the orbit and in TED OFs treated with the PPARγ ligand 15d-PGJ2. (A) Thy1+ and Thy1– OFs were analyzed for miR-130a levels (n = 3; ***P < 0.001 using Student's t-test). (B) Fat tissues from different depots were analyzed for miR-130a. The level of miR-130a in non-orbital fat tissue was normalized to 1.0 (n = 4; *P < 0.05 using the Mann–Whitney non-parametric test for data that included outliers). (C) Fibroblasts were grown out of explanted fat tissues from the orbit and non-orbit fat depots. Expression of miR-130a was analyzed, and the level in non-orbital fibroblasts was normalized to 1.0 (n = 4 different non-OF strains; n = 11 different OF strains; ***P < 0.001 using Student's t-test). (D) OFs were treated with 5-µM 15d-PGJ2 or dimethylsulfoxide (DMSO) as control every second day for 8 to 10 days. Control OF miR-130a levels were normalized to 1.0 (n = 7 different TED OF strains; ****P < 0.0001 using Student's t-test).
miRNA-130a Enhances Lipid Accumulation in OFs
Because miR-130a was upregulated in Thy1– OFs and miR-130a also appears to increase when exposed to 15d-PGJ2, we investigated whether miR-130a might be involved in lipid accumulation. OFs were transfected with miR-130a and lipid levels were measured with the AdipoRed assay (Fig. 2A). OFs were treated with 5-µM 15d-PGJ2 every second day for 10 to 12 days and then lipid levels were measured. We found that 15d-PGJ2 led to an approximately twofold increase in lipids (white bars) in control samples. Interestingly, miR-130a mimic led to a small increase in lipids even without treatment with 15d-PGJ2. In OFs treated with miR-130a mimic and 15d-PGJ2, there was a fivefold increase in lipid levels compared with controls. These results indicate that miR-130a enhances 15d-PGJ2-mediated lipid accumulation.
Figure 2.
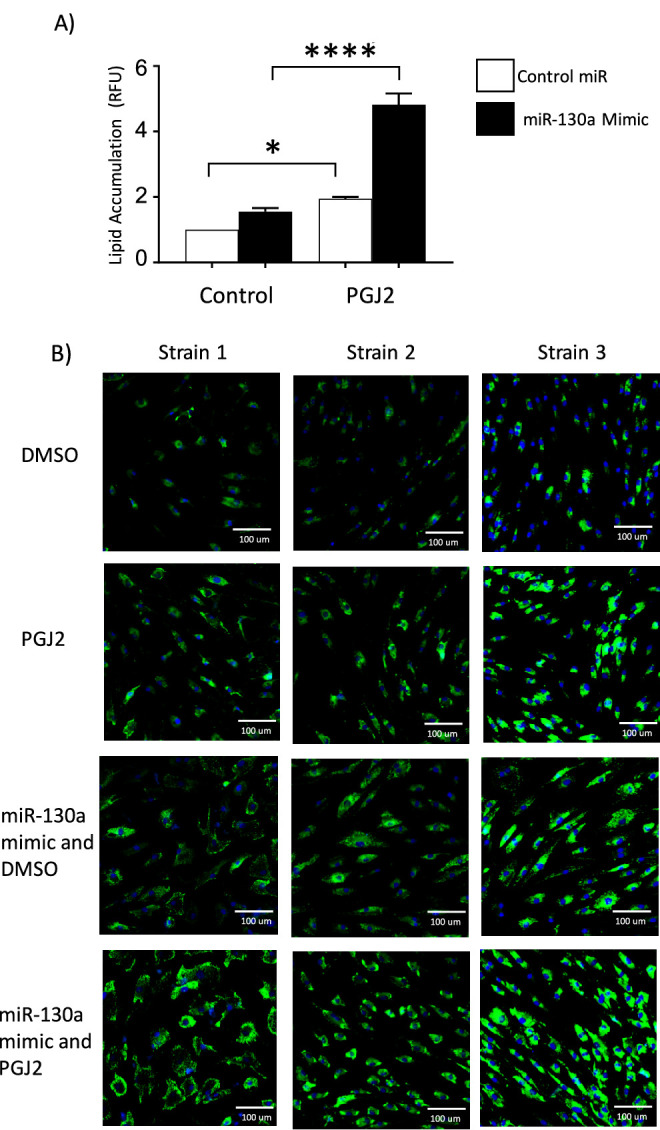
miR-130a enhances lipid accumulation. TED OFs were treated with either control miRNA or a miR-130a mimic. OFs were then treated with either DMSO as control or 5-µM 15d-PGJ2 every second day for 8 to 10 days. (A) Lipid accumulation was measured in four OF strains using the AdipoRed assay (n = 4 different OF strains; *P < 0.05, ****P < 0.0001 when treated cells were compared with DMSO-treated control using one-way ANOVA and Tukey's post hoc analysis). (B) Lipid levels were detected by LipidTOX staining (green), and nuclei were visualized with DAPI (blue).
To further visualize the effects of the miR-130a mimic on the OFs, we performed fluorescent staining for lipids using LipidTOX (green) and DAPI to label the cell nuclei (blue). OF strains were treated with 5-µM 15d-PGJ2 or vehicle, as described above for 10 to 12 days. Cells were then fixed, stained, and imaged (Fig. 2B). Vehicle-treated cells showed a low level of lipid staining that was not present in all cells. The lipid staining was intensified in the fibroblasts treated with 15d-PGJ2 compared with control. Fibroblasts transfected with the miR-130a mimic showed increased lipid staining when compared with vehicle-treated cells. The combined treatment with miR-130a mimic and 15d-PGJ2 led to the largest increase in lipid staining.
miR-130a Targets AMPK, Reducing AMPK Activity
To investigate the mechanism behind miR-130a and increased lipid accumulation in OFs, we searched for genes involved in lipid metabolism with putative binding sights for miR-130a using TargetScan 7.2.21 Two different subunits of AMPK, AMPKα (PRKAA1) and AMPKβ (PRKAB1), contain conserved target seed sequences for miR-130a (Fig. 3A). To test if the PRKAB1 3′UTR (for AMPKβ1) is directly targeted by miR-130a, we cloned the human PRKAB1 3′UTR into a luciferase reporter construct. The construct was introduced into HEK293FT cells with either a non-specific control miRNA or a miR-130a mimic (Fig. 3B). Adding miR-130a to the reporter cells significantly decreased PRKAB1 3′UTR luciferase activity when compared with control miRNA, indicating that miR-130a can target AMPK.
Figure 3.

miR-130a targets AMPKα and AMPKβ. (A) Target seed sequences for miR-130a on PRKAA1 (AMPKα) and PRKAB1 (AMPKβ) are bolded. (B) Reporter assay demonstrated decreased PRKAB1 luciferase activity when miR-130a was added (n = 4 wells per group; ***P < 0.001 using Student's t-test). (C) Two strains of OFs were transfected with miR-130a mimic. After 8 days, cell lysate was collected, and protein expression was analyzed with western blotting. Relative expression graphs are five different OF strains (**P < 0.01, ****P < 0.0001 using Student's t-test).
Next, the ability of miR-130a to target AMPK and AMPK activity in OFs was tested. Here, control or miR-130a miRNAs were introduced into OFs, and AMPK expression and activity were analyzed by measuring AMPK, phosphorylated AMPKα, and ACC. OFs were transfected with miR-130a, and, after 8 days, cells were lysed and proteins were analyzed by western blot. The miR-130a mimic led to a 40% decrease in p-AMPKα, a 25% decrease in total AMPKα, a 50% decrease in p-AMPKβ, a 50% decrease in AMPKβ, and a 50% decrease in p-ACC (Fig. 3C). These data demonstrate that miR-130a expression reduces AMPK levels and activity in human OFs.
15d-PGJ2 Reduces AMPK Activity
Next, we examined the role of 15d-PGJ2 in AMPK activity by treating OFs with 15d-PGJ2 for 24 hours and then analyzing p-AMPK and p-ACC levels. (Fig. 4). AMPKα phosphorylation was reduced by 15d-PGJ2 by 20%, and ACC phosphorylation was reduced 40% (Fig. 4A). These results demonstrate that 15d-PGJ2 reduces AMPK activity. To provide further evidence that 15d-PGJ2 can reduce AMPK levels, HEK293FT cells transfected with the PRKAB1 3′UTR luciferase reporter were treated with 15d-PGJ2. After 24 hours, cells were lysed and luciferase activity measured. We observed that 5-µM 15d-PGJ2 led to a 40% decrease in luciferase reporter activity, and 20-µM 15d-PGJ2 led to a 60% decrease in activity (Fig. 4B).
Figure 4.

15d-PGJ2 decreased AMPK activity. (A) OFs were treated with 5-µM 15d-PGJ2 or left as an untreated control. After 24 hours, cells were lysed and protein analyzed by western blotting. Relative expression graphs are four different OF strains (**P < 0.01, ***P < 0.001, ****P < 0.0001 using Student's t-test). (B) Reporter assay demonstrated a dose-dependent decrease in PRKAB1 luciferase activity when 15d-PGJ2 was added (n = 4 wells per group; **P < 0.01, ****P < 0.0001 using Student's t-test).
AMPK Activity Controls Lipid Accumulation in OFs
To further examine the effect of AMPK on lipid accumulation, OFs were treated with 5-µM 15d-PGJ2 for 10 to 12 days in the presence of PRKAB1 siRNA (the gene for AMPKβ) to reduce AMPK activity or in the presence of an AMPK activator (Fig. 5). PRKAB1 siRNA reduced the protein expression of AMPKβ (an ∼ 80% decrease) in two different OF strains. Additionally, AMPKα levels also decreased by 50% in PRKAB1 siRNA-treated samples (Fig. 5A). AMPKα, AMPKβ, and AMPKγ form a heterotrimeric complex to become active11–13; therefore, the reduction of AMPKβ and AMPKα due to PRKAB1 siRNA suggests there is less activity from AMPK complexes in these OFs. Next, OFs were pretreated for 24 hours with PRKAB1 siRNA and then stimulated with 15d-PGJ2 to induce lipid production. Treating OFs for 10 to 12 days every second day with 15d-PGJ2 led to an approximate threefold increase in lipid accumulation as measured by the AdipoRed assay. In the presence of the PRKAB1 siRNA, lipid accumulation was significantly increased, by ∼30%, compared to control siRNA (Fig. 5B).
Figure 5.

Increasing or decreasing AMPK activity has the opposite effect on lipid accumulation. (A) OFs were transfected with PRKAB1 siRNA (to reduce AMPKβ) or a control siRNA. After 24 hours, OFs were lysed, and the protein expression of AMPKα and AMPKβ was assessed with western blotting. The PRKAB1 siRNA led to a decrease in AMPKβ and AMPKα. Relative expression graphs are three different OF strains (**P < 0.01 using Student's t-test). (B) OFs were transfected with control or PRKAB1 siRNA. After 24 hours, DMSO (vehicle control) or 5-µM 15d-PGJ2 was added every 2 days for 8 to 10 days, and lipid accumulation was measured using the AdipoRed assay (n = 4 different OF strains; ****P < 0.001 when compared with control siRNA; ##P < 0.01 when control siRNA OFs treated with 5-µM 15d-PGJ2 were compared with PRKAB1 siRNA OFs treated with 5-µM 15d-PGJ2). Data were analyzed with one-way ANOVA and Tukey's post hoc analysis. (C) OFs were treated with four different conditions: (1) DMSO, (2) 5-µM 15d-PGJ2, (3) DMSO and 400-µM AICAR, and (4) 5-µM 15d-PGJ2 and 400-µM AICAR. Treatments were added every second day for 8 to 10 days. OFs were analyzed for lipids using the AdipoRed assay (n = 4 different OF strains; ****P < 0.0001 compared with DMSO control; ####P < 0.0001 comparing 5-µM 15d-PGJ2 alone with 5-µM 15d-PGJ2 and 400-µM AICAR using one-way ANOVA and Tukey's post hoc analysis). (D) OFs were treated with 400-µM AICAR or left as untreated control. After 24 hours, cells were lysed, and protein was analyzed by western blotting. Relative expression graphs are five different OF strains (*P < 0.05, ***P < 0.001, ****P < 0.0001 using Student's t-test).
To examine the importance of AMPK activity in controlling OF lipid accumulation, we used the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) to determine if sustained AMPK activation would block lipid accumulation. OFs were treated with 400-µM AICAR concomitantly with 5-µM 15d-PGJ2 to determine if activation of AMPK would reduce lipid accumulation. Lipid accumulation due to 15d-PGJ2 alone increased threefold compared with control. AICAR attenuated lipid accumulation due to 15d-PGJ2 (Fig. 5C). To demonstrate the effect of AICAR on AMPK activity, p-AMPKα and p-ACC expression were detected by western blotting. OFs were treated with 400-µM AICAR or vehicle for 24 hours. AICAR increased the phosphorylation of AMPKα fourfold and ACC sixfold (Fig. 5D).
Discussion
OFs from TED patients display heterogeneity in Thy1 expression, and the level of Thy 1 expression dictates the readiness of the cell to undergo adipogenesis and accumulate lipid.7–9 We discovered that there are differences in the miRNA profiles of OFs with low and high expression levels of Thy1, with miR-130a being expressed to a greater extent in Thy1– fibroblasts. These results suggest that miR-130a may play a role in TED orbital tissue remodeling, especially in terms of excessive lipid accumulation and adipogenesis.
To our knowledge, our findings are the first to demonstrate that miR-130a can promote lipid accumulation through the inhibition of AMPK. miR-130a can target genes for two AMPK subunits, which leads to both diminished AMPK expression and activity. These results provide a mechanism whereby Thy1– OFs are primed for lipid accumulation and the adipogenic process. miR-130a targets and attenuates AMPK activity, releasing AMPK inhibition of lipogenic gene activity and fatty acid synthesis, and miR-130a presents as a novel target for preventing lipid accumulation in TED.
miR-130a is expressed in many different types of cells and regulates cellular functions through a variety of targets.22 For example, miR-130a levels are upregulated by cigarette smoke,23 and smoking increases the chance of developing TED by around sevenfold.24,25 It will be interesting to determine if smoking increases the levels of miR-130a in TED and Graves’ disease patients. Interestingly, miRNA profiling of TED and non-TED orbital tissue determined that miR-130a is elevated in TED orbital tissue.26 There is also evidence suggesting that miR-130a contributes to obesity and inflammation. In Crohn's disease, a disease associated with intense inflammation in the digestive system, miR-130a is upregulated in colon tissue.27 Also, miR-130a is elevated in obese humans,28,29 and circulating miR-130a increases as body mass index increases.30 Circulating miRNAs are currently being evaluated for biomarkers in a variety of diseases.31–33 Patients who are at a greater risk for developing severe TED may present with higher circulating miR-130a levels; if so, this could provide a novel biomarker to screen Graves’ disease patients who may be more susceptible to developing severe TED. miR-130a is also implicated in contributing to the fibrotic process,22,34 increasing proliferation and collagen production.35 It will be interesting to determine if miR-130a also increases proliferation, fibrosis, and collagen production in TED OFs; therefore, targeting and reducing miR-130a could be even more beneficial to reducing TED pathology and progression.
It has been shown that miR-130a blocks adipogenesis in human adipose-derived stromal cells by targeting and decreasing expression of PPARγ.36 However, we observe significant lipid accumulation in human OFs when miR-130a is elevated. In addition to differences in cell types studied, another difference is that we used the natural PPARγ ligand 15d-PGJ2, whereas others have used a traditional adipogenic cocktail that included hydrocortisone and the non-selective phosphodiesterase inhibitor isobutylmethylxanthine. Therefore, the differences could be due to pathways altered by steroid signaling and cAMP pathways. Furthermore, miR-130a likely has additional targets and effects depending on both cellular and physiological conditions.
Using TargetScan,21 we identified two AMPK subunits as potential miR-130a targets; however, miR-130a is predicted to target hundreds of different genes and many of these could also play a role in lipid metabolism and TED. For example, several genes involved in the Wnt signaling pathway are predicted targets of miR-130a, including Wnt1, Wnt2b, FZD6, LRP6 (a Wnt co-receptor), and TCF4. High levels of miR-130a blunted Wnt signaling by targeting Wnt2b, FZD6, and LRP6 in breast cancer cells37 and by targeting Wnt1 in lung epithelial cells.23 Reductions in Wnt signaling are important for adipogenesis38; therefore, miR-130a may disrupt Wnt signaling to promote lipid accumulation in TED OFs. In support of this concept, other studies have revealed that Wnt signaling is disrupted in TED orbital tissue.39,40 Growth factor receptor-bound protein 10 (Grb10) is also decreased by miR-130a.41 A decrease in Grb10 leads to increases in mTOR signaling and lipid accumulation. Therefore, in addition to AMPK, other targets of miR-130a may promote lipid accumulation in TED.
Here, we have shown that AMPKβ1 knockdown significantly increases lipid accumulation in TED OFs, suggesting an important role for AMPK in regulating fatty acid levels. AMPK phosphorylates ACC to inhibit the utilization of acetyl-CoA units for the biosynthesis of fatty acids.42 AMPKα2 knockout mice show increased adiposity and adipocyte hypertrophy, suggesting a key role for AMPK in lipid homeostasis.43 AMPK also regulates lipid levels in other tissues, including skeletal muscle.44 This is interesting because muscle tissue is often disrupted with excessive lipid deposits in TED. We have also shown that AICAR, an AMPK activator, efficiently blocked lipid accumulation in OFs. AICAR led to a dramatic increase in phosphorylation of ACC in OFs; however, AICAR may have AMPK-independent effects on adipogenesis and lipid accumulation. 3T3-L1 pre-adipocytes treated with AICAR show impairments in mitotic clonal expansion, a very early step in adipogenic commitment.45 AICAR may inhibit adipogenesis through increases in the Wnt/β-catenin pathway, which may not be AMPK specific.46 In addition to the effect of AMPK on lipid accumulation, AMPK may also phosphorylate and inhibit PPARγ, leading to a reduction in the transcriptional program of adipogenesis.47,48
The discovery of many AMPK-activating compounds suggests that AMPK is an exciting pharmaceutical target12 and that activating AMPK may prove beneficial in TED. Metformin is a widely used pharmaceutical, the most commonly prescribed drug for combating type 2 diabetes.11 Metformin acts as an indirect activator of AMPK by modestly inhibiting Complex I in the mitochondrial respiratory chain, which increases the ratio of AMP to adenosine triphosphate to activate AMPK.11,12 Metformin was shown to prevent lipid accumulation in TED OFs undergoing adipogenesis and to phosphorylate AMPK; however, this study did not directly test whether inhibiting AMPK was responsible for changes in lipid accumulation.49 Nevertheless, metformin may be a promising drug that could be further tested in TED patients. Additionally, ezetimibe, a cholesterol-lowering drug, activates AMPK and reduces lipid accumulation during adipogenesis,50 and it may be useful in treating TED. Taken together, our study shows for the first time, to our knowledge, that miR-130a is elevated in human OFs prone to adipogenesis and promotes lipid accumulation. miR-130a also targets AMPK to promote lipid accumulation. This highlights potential pathways that could be targeted in future studies to limit the excessive orbital fat tissue accumulation seen in TED.
Acknowledgments
Supported by grants from the National Institutes of Health (EY027308, HL133761) and by an unrestricted grant from Research to Prevent Blindness.
Disclosure: C.L. Hammond, None; E. Roztocil, None; M.O. Gonzalez, None; S.E. Feldon, None; C.F. Woeller, None
References
- 1. Bahn RS. Current insights into the pathogenesis of Graves’ ophthalmopathy. Horm Metab Res. 2015; 47(10): 773–778. [DOI] [PubMed] [Google Scholar]
- 2. Patel A, Yang H, Douglas RS. A new era in the treatment of thyroid eye disease. Am J Ophthalmol. 2019; 208: 281–288. [DOI] [PubMed] [Google Scholar]
- 3. Inserro A. FDA approves biologic teprotumumab, first drug for thyroid eye disease. Available from: https://www.ajmc.com/view/research-highlights-efficacy-of-teprotumumab-for-thyroid-eye-disease. Accessed January 15, 2021.
- 4. Douglas RS, Kahaly GJ, Patel A, et al.. Teprotumumab for the treatment of active thyroid eye disease. N Engl J Med. 2020; 382(4): 341–352. [DOI] [PubMed] [Google Scholar]
- 5. Lehmann GM, Feldon SE, Smith TJ, Phipps RP. Immune mechanisms in thyroid eye disease. Thyroid. 2008; 18(9): 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuriyan AE, Phipps RP, Feldon SE. The eye and thyroid disease. Curr Opin Ophthalmol. 2008; 19(6): 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehmann GM, Woeller CF, Pollock SJ, et al.. Novel anti-adipogenic activity produced by human fibroblasts. Am J Physiol Cell Physiol. 2010; 299(3): C672–C681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koumas L, Smith TJ, Phipps RP. Fibroblast subsets in the human orbit: Thy-1+ and Thy-1- subpopulations exhibit distinct phenotypes. Eur J Immunol. 2002; 32(2): 477–485. [DOI] [PubMed] [Google Scholar]
- 9. Kuriyan AE, Woeller CF, O'Loughlin CW, Phipps RP, Feldon SE. Orbital fibroblasts from thyroid eye disease patients differ in proliferative and adipogenic responses depending on disease subtype. Invest Ophthalmol Vis Sci. 2013; 54(12): 7370–7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feldon SE, O'Loughlin CW, Ray DM, Landskroner-Eiger S, Seweryniak KE, Phipps RP. Activated human T lymphocytes express cyclooxygenase-2 and produce proadipogenic prostaglandins that drive human orbital fibroblast differentiation to adipocytes. Am J Pathol. 2006; 169(4): 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017; 66(6): 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016; 48: e224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q, Liu S, Zhai A, Zhang B, Tian G. AMPK-mediated regulation of lipid metabolism by phosphorylation. Biol Pharm Bull. 2018; 41(7): 985–993. [DOI] [PubMed] [Google Scholar]
- 14. Sundermeier TR, Palczewski K. The impact of microRNA gene regulation on the survival and function of mature cell types in the eye. FASEB J. 2016; 30(1): 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. SanGiovanni JP, SanGiovanni PM, Sapieha P, De Guire V. miRNAs, single nucleotide polymorphisms (SNPs) and age-related macular degeneration (AMD). Clin Chem Lab Med. 2017; 55(5): 763–775. [DOI] [PubMed] [Google Scholar]
- 16. Woeller CF, O'Loughlin CW, Pollock SJ, Thatcher TH, Feldon SE, Phipps RP. Thy1 (CD90) controls adipogenesis by regulating activity of the Src family kinase, Fyn. FASEB J. 2015; 29(3): 920–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995; 83(5): 803–812. [DOI] [PubMed] [Google Scholar]
- 18. Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995; 83(5): 813–819. [DOI] [PubMed] [Google Scholar]
- 19. Woeller CF, O'Loughlin CW, Roztocil E, Feldon SE, Phipps RP. Salinomycin and other polyether ionophores are a new class of antiscarring agent. J Biol Chem. 2015; 290(6): 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hammond CL, Roztocil E, Phipps RP, Feldon SE, Woeller CF. Proton pump inhibitors attenuate myofibroblast formation associated with thyroid eye disease through the aryl hydrocarbon receptor. PLoS One. 2019; 14(9): e0222779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li L, Bounds KR, Chatterjee P, Gupta S. MicroRNA-130a, a potential antifibrotic target in cardiac fibrosis. J Am Heart Assoc. 2017; 6(11): e006763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu Y, Guan S, Ge Y, Yang Y, Cao Y, Zhou J. Cigarette smoke promotes chronic obstructive pulmonary disease (COPD) through the miR-130a/Wnt1 axis. Toxicol In Vitro. 2020; 65: 104770. [DOI] [PubMed] [Google Scholar]
- 24. Thornton J, Kelly SP, Harrison RA, Edwards R. Cigarette smoking and thyroid eye disease: a systematic review. Eye (Lond). 2007; 21(9): 1135–1145. [DOI] [PubMed] [Google Scholar]
- 25. Gillespie EF, Smith TJ, Douglas RS. Thyroid eye disease: towards an evidence base for treatment in the 21st century. Curr Neurol Neurosci Rep. 2012; 12(3): 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jang SY, Chae MK, Lee JH, Lee EJ, Yoon JS. Role of miR-146a in the regulation of inflammation in an in vitro model of Graves’ orbitopathy. Invest Ophthalmol Vis Sci. 2016; 57(10): 4027–4034. [DOI] [PubMed] [Google Scholar]
- 27. Zhao J, Wang H, Zhou J, et al.. MiR-130a-3p, a preclinical therapeutic target for Crohn's disease [published online ahead of print October 6, 2020]. J Crohns Colitis, 10.1093/ecco-jcc/jjaa204. [DOI] [PubMed] [Google Scholar]
- 28. Shi C, Huang F, Gu X, et al.. Adipogenic miRNA and meta-signature miRNAs involved in human adipocyte differentiation and obesity. Oncotarget. 2016; 7(26): 40830–40845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012; 91(1): 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Signorelli SS, Volsi GL, Pitruzzella A, et al.. Circulating miR-130a, miR-27b, and miR-210 in patients with peripheral artery disease and their potential relationship with oxidative stress. Angiology. 2016; 67(10): 945–950. [DOI] [PubMed] [Google Scholar]
- 31. Wang H, Peng R, Wang J, Qin Z, Xue L. Circulating microRNAs as potential cancer biomarkers: the advantage and disadvantage. Clin Epigenetics. 2018; 10: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Chen J, Sen S. MicroRNA as biomarkers and diagnostics. J Cell Physiol. 2016; 231(1): 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huang W. MicroRNAs: biomarkers, diagnostics, and therapeutics. Methods Mol Biol. 2017; 1617: 57–67. [DOI] [PubMed] [Google Scholar]
- 34. Bertero T, Cottrill KA, Annis S, et al.. A YAP/TAZ-miR-130/301 molecular circuit exerts systems-level control of fibrosis in a network of human diseases and physiologic conditions. Sci Rep. 2015; 5: 18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu L, Wang J, Lu H, et al.. MicroRNA-130a and -130b enhance activation of hepatic stellate cells by suppressing PPARγ expression: a rat fibrosis model study. Biochem Biophys Res Commun. 2015; 465(3): 387–393. [DOI] [PubMed] [Google Scholar]
- 36. Lee EK, Lee MJ, Abdelmohsen K, et al.. miR-130 suppresses adipogenesis by inhibiting peroxisome proliferator-activated receptor gamma expression. Mol Cell Biol. 2011; 31(4): 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Poodineh J, Sirati-Sabet M, Rajabibazl M, Mohammadi-Yeganeh S. MiR-130a-3p blocks Wnt signaling cascade in the triple-negative breast cancer by targeting the key players at multiple points. Heliyon. 2020; 6(11): e05434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prestwich TC, Macdougald OA. Wnt/beta-catenin signaling in adipogenesis and metabolism. Curr Opin Cell Biol. 2007; 19(6): 612–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tao W, Ayala-Haedo JA, Field MG, Pelaez D, Wester ST. RNA-sequencing gene expression profiling of orbital adipose-derived stem cell population implicates HOX genes and WNT signaling dysregulation in the pathogenesis of thyroid-associated orbitopathy. Invest Ophthalmol Vis Sci. 2017; 58(14): 6146–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ezra DG, Krell J, Rose GE, Bailly M, Stebbing J, Castellano L. Transcriptome-level microarray expression profiling implicates IGF-1 and Wnt signalling dysregulation in the pathogenesis of thyroid-associated orbitopathy. J Clin Pathol. 2012; 65(7): 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xiao F, Yu J, Liu B, et al.. A novel function of microRNA 130a-3p in hepatic insulin sensitivity and liver steatosis. Diabetes. 2014; 63(8): 2631–2642. [DOI] [PubMed] [Google Scholar]
- 42. Peng IC, Chen Z, Sun W, et al.. Glucagon regulates ACC activity in adipocytes through the CAMKKbeta/AMPK pathway. Am J Physiol Endocrinol Metab. 2012; 302(12): E1560–E1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes. 2004; 53(9): 2242–2249. [DOI] [PubMed] [Google Scholar]
- 44. Wu W, Feng J, Jiang D, et al.. AMPK regulates lipid accumulation in skeletal muscle cells through FTO-dependent demethylation of N6-methyladenosine. Sci Rep. 2017; 7: 41606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Habinowski SA, Witters LA. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun. 2001; 286(5): 852–856. [DOI] [PubMed] [Google Scholar]
- 46. Lee H, Kang R, Bae S, Yoon Y. AICAR, an activator of AMPK, inhibits adipogenesis via the WNT/β-catenin pathway in 3T3-L1 adipocytes. Int J Mol Med. 2011; 28(1): 65–71. [DOI] [PubMed] [Google Scholar]
- 47. Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans. 2003; 31(pt 1): 224–227. [DOI] [PubMed] [Google Scholar]
- 48. Moreno M, Lombardi A, Silvestri E, et al.. PPARs: nuclear receptors controlled by, and controlling, nutrient handling through nuclear and cytosolic signaling. PPAR Res. 2010; 2010: 435689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Han YE, Hwang S, Kim JH, Byun JW, Yoon JS, Lee EJ. Biguanides metformin and phenformin generate therapeutic effects via AMP-activated protein kinase/extracellular-regulated kinase pathways in an in vitro model of Graves’ orbitopathy. Thyroid. 2018; 28(4): 528–536. [DOI] [PubMed] [Google Scholar]
- 50. Lee YS, Park JS, Lee DH, Han J, Bae SH. Ezetimibe ameliorates lipid accumulation during adipogenesis by regulating the AMPK-mTORC1 pathway. FASEB J. 2020; 34(1): 898–911. [DOI] [PubMed] [Google Scholar]