

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a deadly malignancy with dire prognosis due to aggressive biology, lack of effective tools for diagnosis at early stage, and limited treatment options. Detection of PDAC using conventional radiographic imaging is limited by the dense, hypovascular stromal component and relatively scarce neoplastic cells within the tumor microenvironment (TME). The CC motif chemokine 2 (CCL2) and its cognate receptor CCR2 (CCL2/CCR2) axis is critical in fostering and maintaining this kind of TME by recruiting immunosuppressive myeloid cells such as the tumor-associated macrophages, thereby presenting an opportunity to exploit this axis for both diagnostic and therapeutic purposes. We engineered CCR2-targeting ultrasmall copper nanoparticles (Cu@CuOx) as nano-vehicles not only for targeted PET imaging by intrinsic radiolabeling with 64Cu but also for loading and delivery of chemotherapy drug gemcitabine to PDAC. This 64Cu radiolabeled nano-vehicle allowed sensitive and accurate detection of PDAC malignancy in autochthonous genetically engineered mouse models. The ultrasmall Cu@CuOx showed efficient renal clearance, favorable pharmacokinetics and minimal in vivo toxicity. Systemic administration of gemcitabine loaded Cu@CuOx effectively suppressed the progression of PDAC tumors in a syngeneic xenograft mouse model and prolonged survival. These CCR2-targeted ultrasmall nanoparticles offer a promising image-guided therapeutic agent and show great potential for translation.
Keywords: ultrasmall copper nanoparticle, CCR2, pancreatic ductal adenocarcinoma, PET, cancer treatment
Graphical Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a devastating human cancer and a growing challenge to the healthcare system in the United States. Most PDAC patients at advanced-stage live less than one year, and only ~9% patients can live up to 5 years.1 Clinically, the combination of surgery and adjuvant systemic chemotherapy is commonly used to treat PDAC patients. However, less than 15% of patients diagnosed at early stage will respond. Most patients will eventually suffer disease relapse.2–4 The majority of patients are diagnosed at locally advanced or metastatic stages that preclude the potential for successful tumor removal.5 Combination chemotherapies can prolong survival but are not curative.6, 7 Due to the absence of useful biomarker for early detection and treatment, the rapid local and distant metastasis, and dearth of effective therapy, the mortality of PDAC patient is high. Conventional cytotoxic treatments, such as single or multi-agent chemotherapy regimens using gemcitabine (Gem),8–11 or multidrug regimens,6, 12, 13 have been evaluated in clinical trials, however, long term outcome remains poor.
Resistance to chemotherapy remains a daunting medical challenge in the treatment of PDAC. The tumor microenvironment (TME) of PDAC has a desmoplastic stroma featuring an abundance of fibroblasts, leukocytes, endothelial cells, and extracellular matrix proteins.14 This unique stroma poses physical and biological barrier to drug delivery. Notably, infiltrative leukocytes within the TME include bone-marrow-derived myeloid cells such as tumor-associated macrophages, which not only stifle the effect of chemotherapy and anti-tumor immunity but also promote tumor growth and metastasis.14–16 The chemokine ligand type 2 (CCL2, also named as monocyte chemoattractant protein-1, MCP-1) and its receptor, chemokine receptor type 2 (CCR2), form a signaling axis that plays a key role in recruiting CCR2+ inflammatory monocytes that egressed from bone marrow to tumors and changed their phenotype to immunosuppressive tumor-associated macrophages.17–19 The CCL2/CCR2 axis has been involved in the pathogenesis of many diseases such as inflammation, atherosclerosis, and tumor growth and metastasis.17, 18, 20–27 Importantly, pre-clinical and phase I/II clinical studies have revealed that when coupled with CCR2 inhibition, therapeutic response to chemotherapy could be improved.18, 28 However, these studies utilize small molecule inhibitors against CCR2, which are typically cytostatic and may become ineffective upon emergence of CCR2-independent adaptive response,17 underscoring the need to improve on the efficacy of CCR2-targeting strategies.
In the past decade, nanotechnology has led to advances in cancer therapy by delivering chemotherapeutic drugs directly to targeted tumor sites, prolonging drug release, and enhancing drug metabolism.11, 29–32 To date, lots of nanoparticles have been used for PDAC imaging or delivery of therapeutic drugs.33–37 Many nanoparticles showed encouraging results via the enhanced permeability and retention (EPR) effect despite the stromal barriers within TME of mice.38 Some nanoparticles had been used in PDAC patients although the treatment efficiency need significant improvement.39, 40 To further improve the diagnosis accuracy and treatment efficiency, the targeting ultrasmall nanoparticles have drawn significant interest as an emerging platform due to accurate tumor detection, favorable in vivo pharmacokinetics, and rapid renal clearance, and potential for translational cancer theranostics.41–46 Previously, we reported the in vivo biodegradable copper nanoparticles intrinsically radiolabeled with 64Cu (64Cu-CuNCs) for sensitive and accurate PET imaging of triple negative breast cancer.47 In contrast to other drug delivery systems,48, 49 the pH-sensitive degradation makes it an useful platform not only for imaging but also to rapidly unload therapeutics for improved treatment outcome. The fast and robust synthesis may be able to address the need for repeated treatment and empower the potential for future translation.50
Moreover, we have developed a CCR2 imaging agent using ECL1i peptide and demonstrated its targeting specificity to monocytes and macrophages in multiple animal models.22, 51–53 In this study, we reconstructed the ultrasmall 64Cu radiolabeled copper nanoparticles (64Cu-Cu@CuOx) with ECL1i to target tumor stroma and loaded gemcitabine (64Cu-Cu@CuOx-ECL1i-Gem) for PET guided drug delivery into PDAC tumors. We assessed its specificity and sensitivity for targeted PET imaging in two genetically engineered pancreatic cancer mouse models. We then assessed treatment efficacy in a syngeneic xenograft mouse model and related in vivo toxicity.
Results and discussion
Synthesis of the ultrasmall copper nanoparticles
The synthesis of 64Cu radiolabeled CCR2-targeting Cu@CuOx (64Cu-Cu@CuOx-ECL1i) was modified from previous procedures (Figure 1a) with high yield and radiolabeling specific activity (Figures 1 and S1–6).44, 45, 47, 54 Scanning transmission electron microscopy (STEM) image of Cu@CuOx-ECL1i showed uniformity of these nanoparticles with an average diameter of 2.7 ± 0.2 nm (Figure 1b). Dynamic light scattering (DLS) measurement demonstrated a renal-clearable hydrodynamic diameter (5.3 ± 0.1 nm, Figure 1c) and neutral surface charge (ξ-potential: −3.7 ± 0.7 mV). Previously, we demonstrated approximately 180 TA-PEGs on the surface of Cu@CuOx.47 Based on our optimized targeting strategy, we started the synthesis with a 1:2 molar ratio (TA-PEG-ECL1i: TA-PEG-OMe) for Cu@CuOx surface conjugation. High performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry (ICP-MS) measurement determined there were approximately 60 ± 1.3 copies of TA-PEG-ECL1i on each Cu@CuOx, consistent with our previous work (Table S1 and Figure S4). Though surface oxidation was observed on Cu@CuOx, there was little effect on the conjugation of TA-PEG-ECL1i.47 Fast protein liquid chromatography (FPLC) analysis demonstrated the integrity and chemical purity of Cu@CuOx-ECL1i after synthesis (Figure S5).
Figure 1.
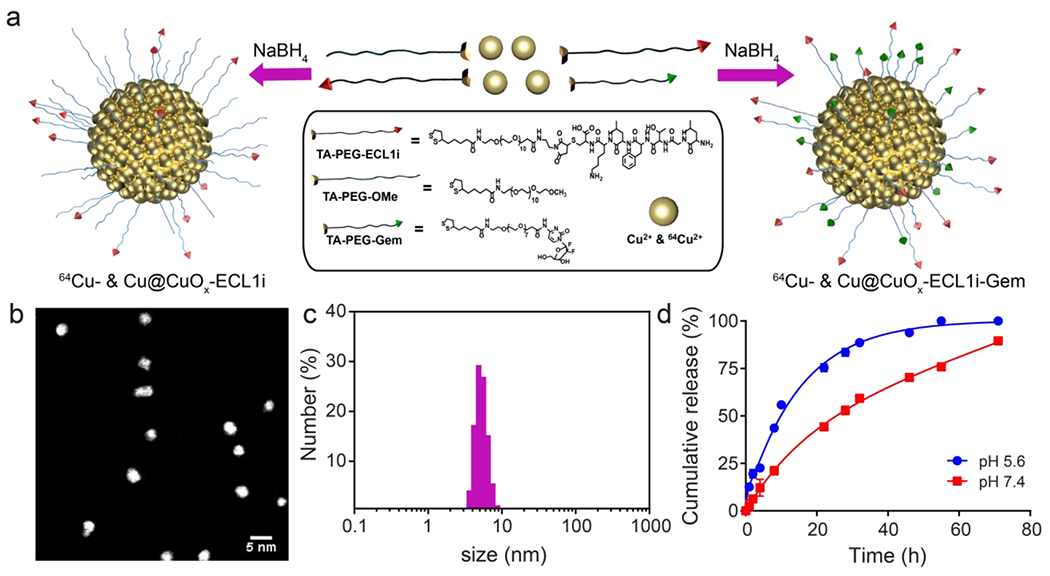
Synthesis and characterization of ultrasmall copper nanoparticles (Cu@CuOx). (a) Schematic diagram of the synthesis of Cu@CuOx-ECL1i, Cu@CuOx-ECL1i-Gem, and 64Cu radiolabeled nanoparticles, (b) STEM of Cu@CuOx-ECL1i, (c) Number average hydrodynamic diameter of Cu@CuOx-ECL1i, (d) In vitro TA-PEG-Gem release profiles of Cu@CuOx-ECL1i-Gem under physiological and acidic conditions.
Gemcitabine loaded nanoparticles (Cu@CuOx-ECL1i-Gem) were synthesized following the same protocol using TA-PEG-Gem: TA-PEG-ECL1i at 2:1 molar ratio. The ξ-potential and hydrodynamic diameter of Cu@CuOx-ECL1i-Gem were −4.8 ± 2.4 mV and 4.9 ± 0.3 nm, respectively, demonstrating a minimal effect of Gem loading on Cu@CuOx surface charge and size. Quantitative measurement showed there were 134.9 ± 6.7 copies of Gem and 53.0 ± 7.0 copies of TA-PEG-ECL1i per nanoparticle, with a loading efficiency of 81.0 ± 4.1% and 63.3 ± 8.4 %, respectively. Gem is known to undergo rapid deamination in blood to become the inactive metabolite 2’,2’-difluorodeoxyuridine. In vitro cytotoxicity assay showed that TA-PEG modified gemcitabine (TA-PEG-Gem) had comparable IC50 value (48.8 μM) to that of gemcitabine (19.7 μM) (Figure S7). Thus, this modification strategy and covalent conjugation of the prodrug onto Cu@CuOx not only improved drug stability but also prolonged circulation half-life for enhanced drug delivery and treatment outcome.55 Moreover, the in vitro release profiles showed gradual release of TA-PEG-Gem in both physiological and acidic conditions, which was similar with the dissolution kinetics of the ultrasmall Cu@CuOx,47 revealing the drug release was attributed to the dissolution of the nanoparticles (Figure 1d). Therefore, upon reaching the tumor in vivo, acidic conditions within tumor microenvironment would lead to the gradual dissolution of Cu@CuOx to Cu(II) and subsequent release of gemcitabine within the tumors for improved treatment efficacy. With this strategy, the Cu@CuOx-ECL1i-Gem were expected to accumulate in the tumoral regions on account of CCR2 targeting and release drug on-site for enhanced treatment effect.
Pharmacokinetics of 64Cu- Cu@CuOx-ECL1i and 64Cu- Cu@CuOx-ECL1i-Gem
In vivo pharmacokinetics of 64Cu-Cu@CuOx-ECL1i and 64Cu-Cu@CuOx-ECL1i-Gem were assessed through biodistribution studies using C57BL/6 mice. As shown in Figure 2, both 64Cu-Cu@CuOx-ECL1i and 64Cu-Cu@CuOx-ECL1i-Ge m showed rapid blood clearance with comparable half-lives (t1/2 = 1.27 h for 64Cu-Cu@CuOx-ECL1i and t1/2 = 0.81 h for 64Cu-Cu@CuOx-ECL1i-Gem), consistent with our previous reports.44, 47 At 4 h post injection, 66.8% of 64Cu-Cu@CuOx-ECL1i and 77.4% of 64Cu-Cu@CuOx-ECL1i-Gem were cleared from blood in contrast to the data obtained at 1 h. At 24 h, the blood retentions of both nanoparticles were less than 2.0 percent injected dose per gram (%ID/g), providing a low background for enhanced tumor uptake contrast.
Figure 2.
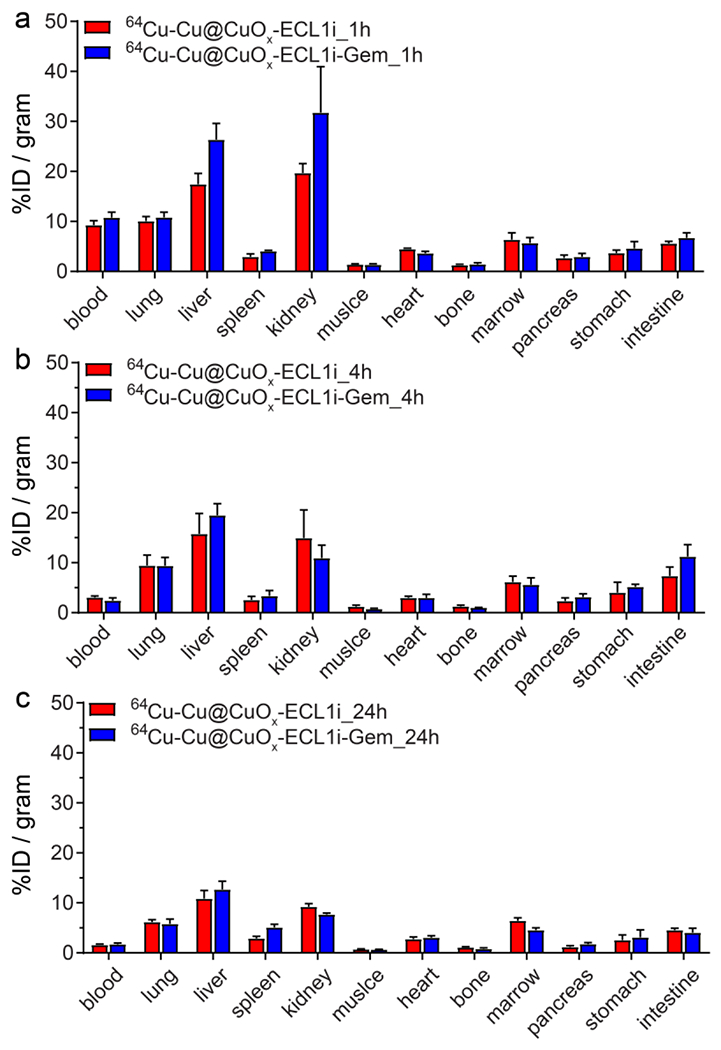
Biodistribution of 64Cu-Cu@CuOx-ECL1i and 64Cu-Cu@CuOx-ECL1i-Gem in wildtype C57BL/6 mice (n = 4/group) at (a) 1h, (b) 4 h and (c) 24 h post injection.
In contrast to high mononuclear phagocyte system (MPS) retention of metal nanoparticles with sizes more than 10 nm, the retentions of 64Cu-Cu@CuOx-ECL1i and 64Cu-Cu@CuOx-ECL1i-Gem in both the liver and spleen were significantly lower due to their ultrasmall sizes.42, 56–58 Compared to 64Cu-Cu@CuOx-ECL1i, the accumulations of 64Cu-Cu@CuOx-ECL1i-Gem were slightly higher at all three time points during the 24 h study. This is mainly due to the conjugation of relatively more hydrophobic gemcitabine comparing to hydrophilic ECL1i peptide on the surface of Cu@CuOx, which led to a more hydrophobic surface and related hepatic and splenic accumulations as reported by others.59, 60
Both nanoparticles revealed efficient renal clearance during the 24 h study. Due to their smaller size, the kidney clearance of 64Cu-Cu@CuOx-ECL1i-Gem (31.8 ± 9.2 %ID/g) at 1 h post injection was approximately 60% more than that of 64Cu-Cu@CuOx-ECL1i (19.7 ± 1.9 %ID/g). This rapid urinary clearance of the drug loaded nanoparticles is particularly helpful to minimize potential toxicity caused by the retention of nanostructures in non-targeted organs.61 In other major organs, the two nanoparticles demonstrated comparable localization during the 24 h study, in agreement with our previous reports.44, 45, 47
Assessment of tumor targeting specificity of 64Cu-Cu@CuOx-ECL1i in vitro and in autochthonous PDAC mouse models
To estimate the CCR2 targeting specificity of 64Cu-Cu@CuOx-ECL1i, we first assessed the in vitro cell uptake using both a PDAC tumor cells (KI, PDAC cells derived from KRASG12D/INK4A deficient mice)62 and a human monocytic THP-1 cell line.63 As shown in Figure S8, after incubation at 0 °C for 30 mins, the cell uptake of 64Cu-Cu@CuOx-ECL1i was nearly 5-fold as much as that of the non-targeted 64Cu-Cu@CuOx (64Cu-Cu@CuOx-NT) in both KI and THP-1 cells (p<0.001, n=4-6). Moreover, with 100 times excess of non-radioactive Cu@CuOx-ECL1i as blockade, 86.9 % and 80.4% uptake of 64Cu-Cu@CuOx-ECL1i were blocked in THP-1 (p<0.0001, n=4-6) and KI (p<0.001, n=4-6) cells, respectively, suggesting the CCR2 targeting specificity of 64Cu-Cu@CuOx-ECL1i. Moreover, the uptake of 64Cu-Cu@CuOx-ECL1i in THP-1 and KI cells were also blocked by 32.8% (p<0.01, n=4-6) and 66.3 % (p<0.01, n=4-6), respectively, with 1000-fold excess of ECL1i peptide, further confirming the CCR2 targeting specificity of 64Cu-Cu@CuOx-ECL1i.
We next assessed the CCR2 targeting specificity of 64Cu-Cu@CuOx-ECL1i with PET/CT in two genetically engineered mouse models including the p48-CRE; LSL-KRasG12D/wt; p53flox/flox (KPPC) and p48-CRE; LSL-KrasG12D/wt; p53flox/wt (KPC) mice. These models are characterized by a stromal-rich TME and aggressive biology that highly mimics human PDAC, and are the most commonly used models in PDAC research.64, 65 In these models, neoplastic progression occurs spontaneously, along with desmoplastic changes at different paces, with invasive PDAC foci typically detected in 6-week-old KPPC mice and ~15-week-old KPC mice. The 24 h PET image revealed increased retention of 64Cu-Cu@CuOx-ECL1i in the tumors of 7-9-week-old KPPC mice in contrast to the minimal non-specific retention in those imaged with 64Cu-Cu@CuOx-NT (Figure 3). Quantitative uptake analysis showed that 64Cu-Cu@CuOx-ECL1i tumor uptake (11.16 ± 1.22 %ID/g, n=5) was almost 3-fold as much as that acquired with 64Cu-Cu@CuOx-NT (4.43 ± 1.74 %ID/g, p<0.0001, n=4). In wild type (WT) control littermate mice, the accumulation of 64Cu-Cu@CuOx-ECL1i (2.90 ± 0.14 %ID/g, p<0.0001, n=4) in pancreas was 4 times less of that obtained in KPPC mice. Competitive receptor blocking using a 50-fold excess of non-radioactive Cu@CuOx-ECL1i significantly decreased tumor accumulation of 64Cu-Cu@CuOx-ECL1i (5.14 ± 0.70 %ID/g, p<0.0001, n=5) by more than 50%. Moreover, blocking study using 50-fold excess of Cu@CuOx-NT showed little effect on 64Cu-Cu@CuOx-ECL1i tumor uptake (9.84 ± 2.67 %ID/g, n=4, p>0.05, Figure S9). All these data validated the specificity of 64Cu-Cu@CuOx-ECL1i targeting CCR2 in KPPC tumors.
Figure 3.
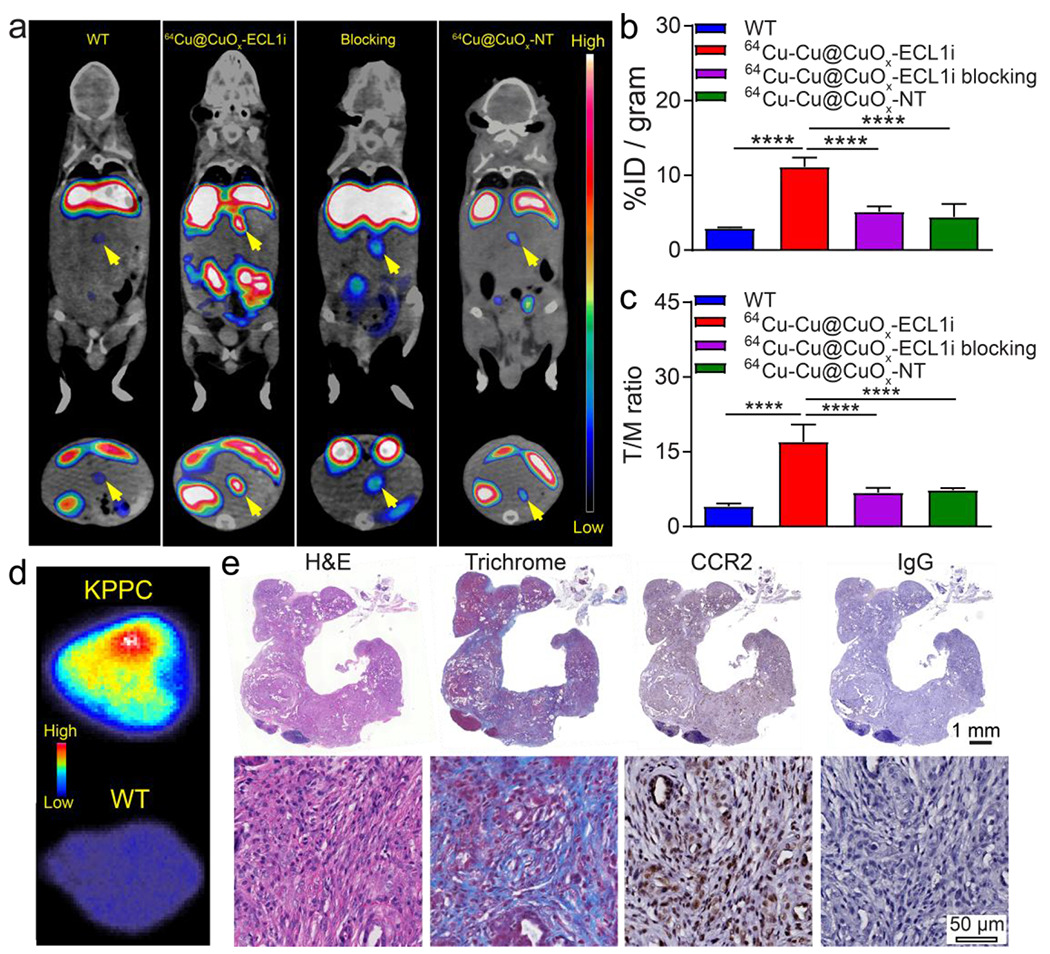
(a) Representative In vivo PET/CT images of 64Cu-Cu@CuOx-ECL1i in wild type littermate, 7-9-week-old KPPC mice, KPPC mice with 50-fold blocking dose, and 64Cu-Cu@CuOx-NT in KPPC mice at 24 h post injection (yellow arrow: pancreas/pancreatic tumor). Quantitative analysis of (b) tumor uptakes and (c) tumor/muscle (T/M) ratio that showed significant difference between KPPC mice, wild type littermates, blocking mice, and 64Cu-Cu@CuOx-NT (**** p<0.0001, n=4-5/group). (d) Autoradiography of 64Cu-Cu@CuOx-ECL1i uptake in KPPC tumor showing intense and heterogeneous distribution compared to the low accumulation in a wild type mouse. (e) H&E, trichrome, CCR2 (brown), and control IgG immunostaining showing the histology, fibrosis, and CCR2 expression of the KPPC tumor.
Due to the significant tumor accumulation and low non-specific localization at the surrounding tissue, the tumor/muscle (T/M) contrast ratio of 64Cu-Cu@CuOx-ECL1i in KPPC tumors was 17.03 ± 3.43 (n=5) at 24 h, highlighting its sensitivity for tumor localization. The significantly decreased T/M ratio in the blocked KPPC tumors (6.83 ± 0.95, p<0.001, n=5) further confirmed the tumor targeting specificity of 64Cu-Cu@CuOx-ECL1i.
Following PET imaging, tumors were immediately collected and fixed for autoradiography and histopathological characterization. As shown in Figure 3d, ex vivo autoradiography of 64Cu-Cu@CuOx-ECL1i in KPPC tumor showed intense and heterogeneous distribution across the tumor while minimal retention was determined in the pancreas of WT littermates, which further confirmed the PET data. Hematoxylin and eosin (H&E) and trichrome staining showed extensive fibrotic regions in the KPPC tumor (Figure 3e), representing the desmoplastic stromal formation during malignancy development and progression. 3,3’-diaminobenzidine (DAB) immunostaining showed elevated expression of CCR2 across the tumor while the control IgG staining had little signal, which further supported the PET imaging (Figure 3e).
The PDAC targeting efficiency of 64Cu-Cu@CuOx-ECL1i was further assessed in a more chronic tumor model using approximately 22-23-week-old KPC mice, when PDAC tumors are fully developed and recapitulate major pathological characteristics of human PDAC, such as significant development of fibrosis and massive accumulation of macrophages.64, 65 Consistent with the data acquired in the KPPC model, targeting 64Cu-Cu@CuOx-ECL1i showed a strong PET signal in the KPC tumor versus little retention in WT littermates, and non-targeting 64Cu-Cu@CuOx-NT showed low non-specific tumor accumulation suggesting targeting specificity (Figure 4a). Quantitative analysis showed the uptake of 64Cu-Cu@CuOx-ECL1i at 5.47 ± 0.75 %ID/g (n=5) in the KPC tumors, which was statistically higher than the non-targeting 64Cu-Cu@CuOx-NT in KPC mice (2.01 ± 0.29 %ID/g, p<0.0001, n=4) and 64Cu-Cu@CuOx-ECL1i in WT mice (1.43 ± 0.20 %ID/g, p<0.0001, n=4) (Figure 4b). The T/M ratio of 64Cu-Cu@CuOx-ECL1i (8.71 ± 1.94, n=5) was more than twice as that acquired with 64Cu-Cu@CuOx-NT (3.51 ± 0.21, p<0.001, n=5) in the KPC tumors, reconfirming the advantage of targeted imaging. Whole tissue H&E and trichrome staining revealed significant tumor progression and fibrosis. Ex vivo autoradiography acquired right after PET imaging revealed accumulation of 64Cu-Cu@CuOx-ECL1i in the fibrotic region, indicating binding to the tumor stroma (Figure S10). Interestingly, tumor uptake and the T/M ratio of 64Cu-Cu@CuOx-ECL1i in the KPC mice were much lower than those in the KPPC mice, suggesting the advanced stroma in KPC tumors which limits the access and binding to CCR2+ cells. In contrast to a 111In-labeled anti-claudin-4 monoclonal antibody, though the tumor accumulations were comparable, 64Cu-Cu@CuOx-ECL1i revealed higher contrast,66 reasonably due to the faster systemic clearance of the ultrasmall nanoparticles generating a low background comparing to the extended blood retention of antibodies.
Figure 4.
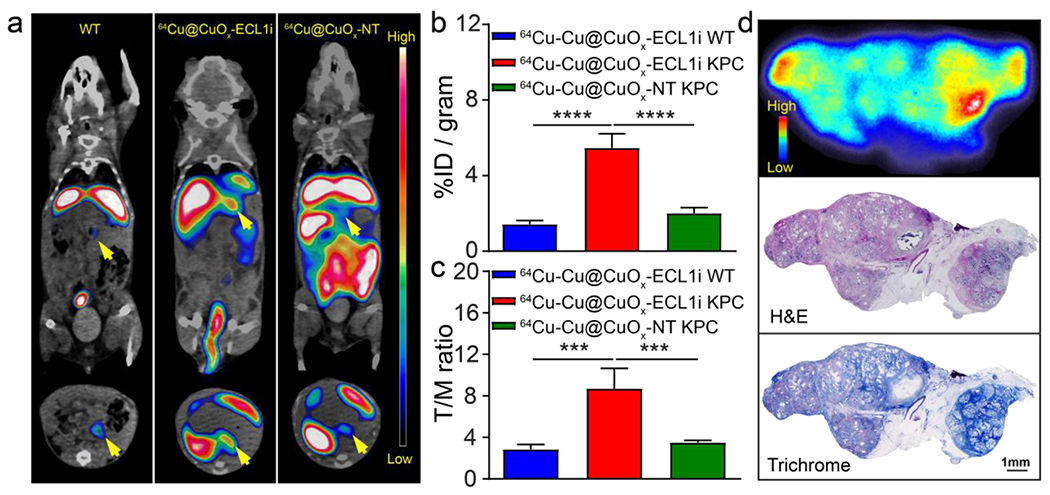
(a) Representative In vivo PET/CT images of 64Cu-Cu@CuOx-ECL1i in 22-23-week-old KPC mice and wild type littermates, and 64Cu-Cu@CuOx-NT in KPC mice at 24 h post injection (yellow arrow: pancreas/pancreatic tumor). Quantitative analysis of (b) tumor uptakes and (c) tumor/muscle ratio of 64Cu-Cu@CuOx-ECL1i in WT and KPC mice, and 64Cu-Cu@CuOx-NT in KPC mice showing significant difference among KPC mice, wild type littermate, and 64Cu-Cu@CuOx-NT (*** p<0.001, **** p<0.0001, n=4-5/group). (d) Autoradiography showing intensive but heterogeneous 64Cu-Cu@CuOx-ECL1i distribution across the KPC tumor. H&E, and trichrome staining of KPC tumor showing the correlation between fibrotic regions (blue) and 64Cu-Cu@CuOx-ECL1i tumor uptake.
We also compared 64Cu-Cu@CuOx-ECL1i imaging to 18F-FDG PET/CT in both KPPC and KPC mouse models. Though 18F-FDG signals were detected in both models (Figure S11), quantitative analysis showed 18F-FDG tumor uptake of 5.94 ± 0.17 %ID/g in KPPC mice which was approximately half the value of 64Cu-Cu@CuOx-ECL1i (p<0.0001, n=4-5/group). In the KPC model, 18F-FDG uptake (4.91 ± 0.10 %ID/g, n=4-5/group) was consistent with that of 64Cu-Cu@CuOx-ECL1i. However, the T/M ratios of 18F-FDG in KPPC (1.75 ± 0.26, p<0.0001) and KPC (1.84 ± 0.17, p<0.0001) mice were 9 and 3 times lower than those acquired with 64Cu-Cu@CuOx-ECL1i, respectively. This was likely due to the non-specific nature of 18F-FDG uptake, leading to high background and low contrast ratio, which further highlighted the significance of CCR2 targeting 64Cu-Cu@CuOx-ECL1i for PDAC diagnosis.
CCR2 targeted Gem delivery using Cu@CuOx-ECL1i-Gem as a therapeutic agent in PDAC
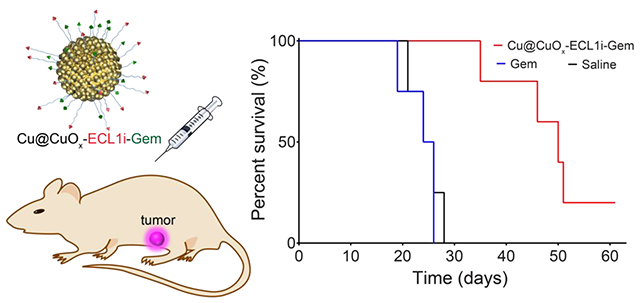
Gemcitabine has been widely used for advanced and metastatic PDAC treatment alone and combined with other agents.8 However, these therapeutic regimens have not dramatically improved long-term outcomes.11, 13, 17, 28, 67 The suppressive immune cells, including CCR2+ myeloid cells, in the TME may contribute to chemoresistance. Recent clinical trial from our institution showed that pharmacologic CCR2 inhibition augments the clinical response to FOLFIRINOX chemotherapy to enable successful surgical resection for patients with borderline-resectable or locally advanced PDAC,28 at least in part by alleviating the immunosuppressive TME. On this premise, we hypothesize that elimination, rather than inhibiting CCR2 receptor, of CCR2+ myeloid cells using Cu@CuOx-ECL1i-Gem may be a more effective approach, which we tested in a syngeneic immunocompetent xenograft (KI) model.62 We had to resort to this model because our KPC cells have the propensity to ulcerate when grown subcutaneously, precluding prolonged treatment. As shown in Figure 5a, the implanted tumors of KI mice treated with Cu@CuOx-ECL1i, low dose Gem (7 mg/kg) via intravenous (IV) administration, and high dose Gem (100 mg/kg) via intraperitoneal (IP) administration at day 10 post tumor implantation did not show any treatment benefit in contrast to mice administrated with saline. The negligible treatment effect of Cu@CuOx-ECL1i suggested the Gem delivery vehicle itself had no therapeutic effect given the concerns of potential copper cytotoxicity.68 Thus, no survival benefit was determined in these groups compared to saline (median survival: Cu@CuOx-ECL1i = 25 days, Gem low dose = 25 days; Gem high dose = 26 days; saline = 25 days) (Figure 5b).
Figure 5.
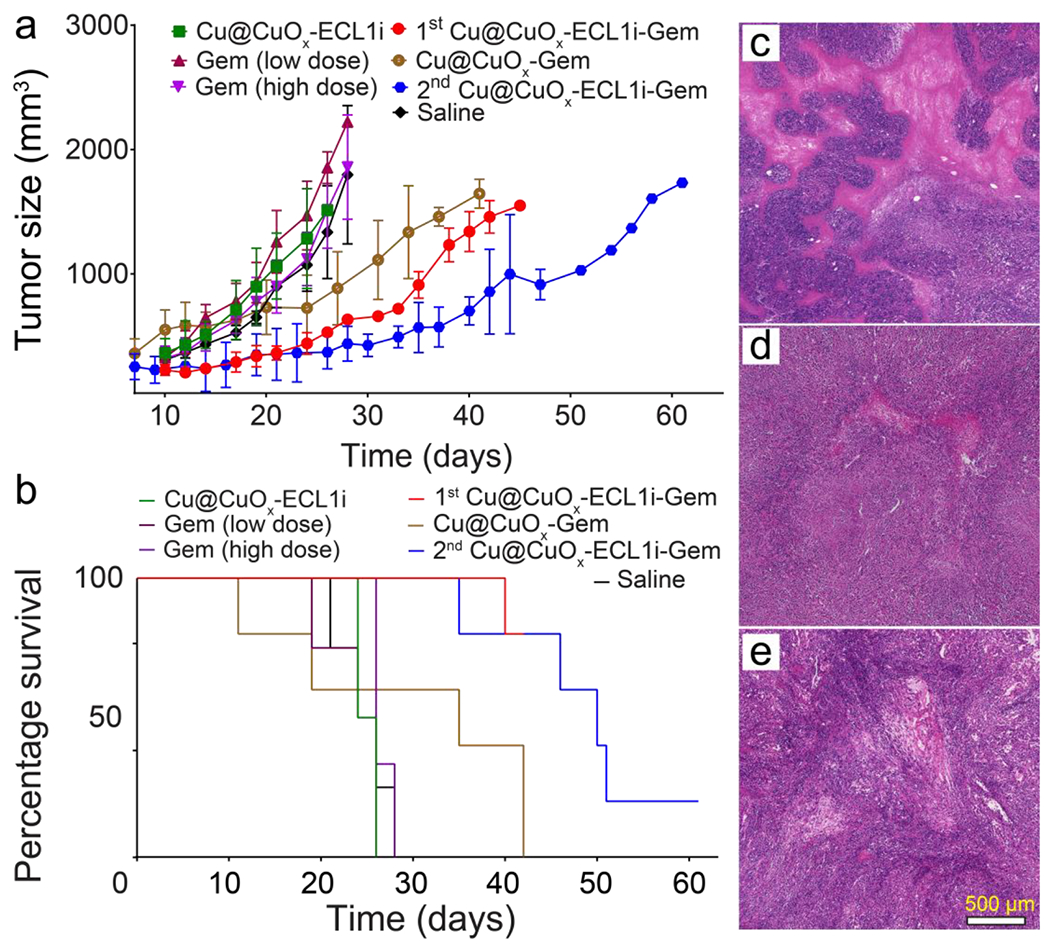
The tumor growth (a) and mouse survival (b) curves of KI implanted mice after treating with Cu@CuOx-ECL1i-Gem, Cu@CuOx-ECL1i, Cu@CuOx-Gem, gemcitabine (7 mg/kg body weight, IV), gemcitabine (100 mg/kg body weight, IP), and saline. 1st treatment with Cu@CuOx-ECL1i-Gem started at 10 days post tumor implantation. The Cu@CuOx-Gem and 2nd treatment began at 7 days post tumor implantation. H&E staining of the tumor slices from mice treated with Cu@CuOx-ECL1i-Gem (c), gemcitabine (100 mg/kg body weight) (d), and saline (e).
Based on our PET imaging results, we first assessed treatment efficiency using the CCR2 targeted Cu@CuOx-ECL1i-Gem (Cu: 10 mg/kg, Gem loading: 3.4±0.8 mg/kg). As shown in figure 5a, the KI tumor growth was effectively prevented in contrast to the other control groups. At day 21, the tumor sizes of Cu@CuOx-ECL1i-Gem group (363.2 ± 56.3 mm3, n=5) were approximately two times smaller than those measured in saline group (897.4 ± 170.5 mm3, p<0.05, n=4). This was reasonably due to the targeted delivery of Gem via the CCR2 mediated tumor retention of Cu@CuOx-ECL1i-Gem abovementioned, which also significantly extended the median survival of KI mice to more than 42 days (Figure 5b).
In a repeated effort to further assess the treatment efficiency of Cu@CuOx-ECL1i-Gem, KI mice were treated with non-targeting Cu@CuOx-Gem and Cu@CuOx-ECL1i-Gem at day 7 post tumor implantation. Though Cu@CuOx-Gem showed tumor inhibition due to the low, non-specific delivery of Gem, the CCR2 targeting Cu@CuOx-ECL1i-Gem revealed substantial tumor inhibition with a statistical difference (p < 0.01, n=5/group) in tumor size observed at day 33. Importantly, the effective tumor inhibition prolonged the median survival of KI mice treated with Cu@CuOx-ECL1i-Gem to 51 days compared to 35 days with Cu@CuOx-Gem, highlighting the significance of targeted Gem delivery for PDAC treatment.34 Moreover, in contrast to the results obtained with Cu@CuOx-ECL1i-Gem treatment starting at day 10, the median survival was extended more than 20% (9 days), emphasizing the importance of early intervention. Histopathological characterization of the tumors treated with Cu@CuOx-ECL1i-Gem revealed a large necrotic region, in comparison, the tumors in mice receiving only Gem and saline had less necrotic levels, further confirming the effective tumor treatment by Cu@CuOx-ECL1i-Gem. The effective tumor growth inhibition of Cu@CuOx-ECL1i-Gem may not only be attributed to the effective delivery of gemcitabine to tumor, but also the syngeneic effect of CCR2 targeting directed inhibition of tumor-associated macrophages recruitment.69
Cu@CuOx-ECL1i-Gem in vivo toxicity
Cu is an essential element for bodies, but a surplus of Cu intake may cause adverse health problems. Therefore, we assessed the in vivo toxicity of the Cu nanoparticle scaffolds in WT CD1 mice at dosages of 1, 10, 15, and 20 mg Cu/kg body weight via IV injection. The mice receiving 20 mg/kg Cu@CuOx-NT died 2-3 days following administration, while the other groups of mice survived with no behavior changes, no hepatic or renal deficiencies as measured by serum, and no difference in liver and kidney weight as a percentage of body weight (Figure S12). Histopathologic examination showed no obvious lesions in major organs including lung, heart, liver, kidney, and bone marrow. Thus, 15 mg Cu/kg body weight was determined to be a safety threshold for one-time administration. We next determined the cumulative toxicity of Cu@CuOx-ECL1i and Cu@CuOx-ECL1i-Gem (Cu mass=10 mg/kg) in WT C57BL/6 mice following administration twice per week for 2 weeks. Complete blood count and white blood cell differential measurements, as well as serum biochemistries, validated that the dosage regimen had no adverse influence on hematopoietic, hepatic, and renal functions (Figure 6a and 6b). Gross necropsy and histopathologic examination showed no appreciable differences in bone marrow, liver, and kidney for Cu@CuOx-ECL1i-Gem compared to the mice administrated with saline. Bone marrow samples from control and treated animals revealed no differences in cellularity or composition of the marrow. Microscopic examination revealed liver parenchyma with preserved architecture and no significant portal inflammation, steatosis or fibrosis in both control and treated groups. Likewise, kidney samples from control and treated animals revealed renal parenchyma with no significant glomerular sclerosis, tubular damage, inflammation or interstitial fibrosis (Figure 6c). Comparison of heart samples from control and treated animals revealed no inflammation or myocyte necrosis in either group while lung specimens revealed no significant acute or chronic inflammation or fibrosis (Figure S13).
Figure 6.
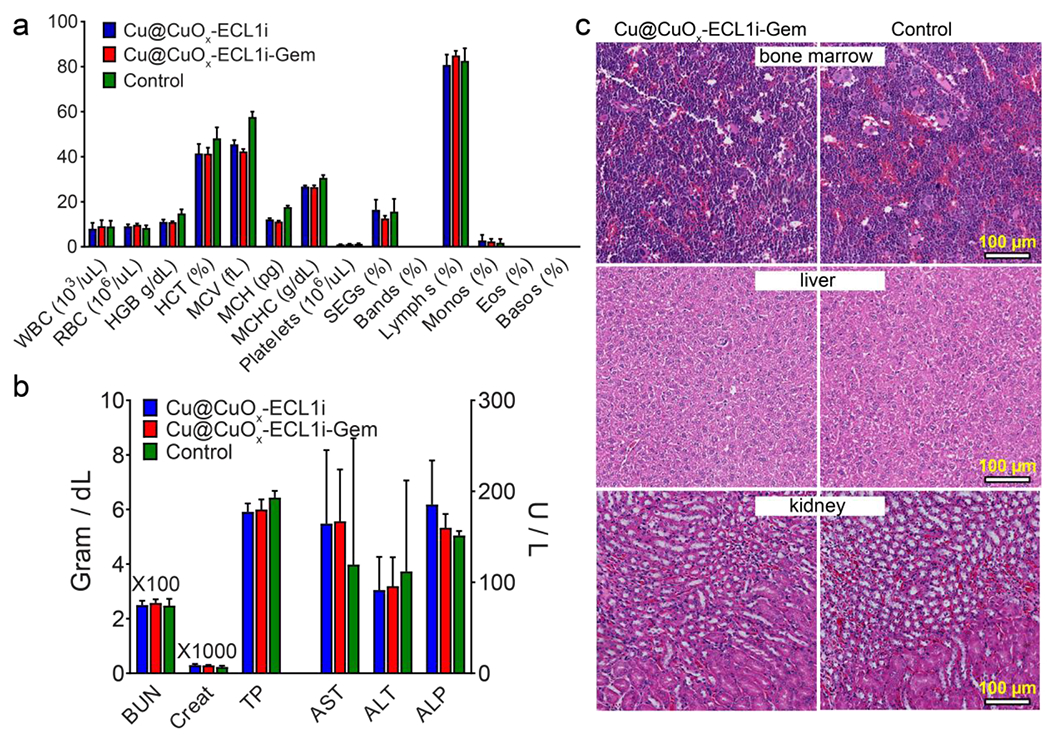
Hematological (a) and serum biochemical (b) tests of C57BL/6 mice after 2 weeks receiving successive treatment (twice per week) of 10 mg/kg (Cu dose) Cu@CuOx-ECL1i and Cu@CuOx-ECL1i-Gem, and 100 μL saline for control group. (c) The H&E staining of bone marrow, liver, and kidney showed no significant histological differences between Cu@CuOx-ECL1i-Gem and the control group.
The overall histological findings supported the safety of Cu@CuOx-ECL1i-Gem for PDAC treatment in mice. This also highlighted the advantages of effective renal clearance and the degradable nature of Cu@CuOx as we previously reported.47 This rapid renal clearance led to low retention of unbound Cu@CuOx-ECL1i-Gem in major organs, reducing the long-term toxicity. The gradual dissolution of Cu@CuOx could effectively decrease the toxicological effect of Cu for improved biocompatibility.
Conclusion
In summary, we developed a CCR2-targeting ultrasmall nanoparticle for PDAC imaging and therapy. The straightforward synthesis of 64Cu-Cu@CuOx-ECL1i enabled high radiolabeling specific activity for sensitive PET imaging, scale-up synthesis, and drug loading capability for effective drug delivery. The ultrasmall nanoparticles exhibited desirable biodistribution and rapid systemic clearance to decrease toxicity. In both KPPC and KPC mouse models, 64Cu-Cu@CuOx-ECL1i demonstrated specific tumor detection and low nonspecific retention. The high T/M ratio of 64Cu-Cu@CuOx-ECL1i in comparison with 18F-FDG highlighted its potential for early and sensitive diagnosis of PDAC malignancy. The CCR2 targeted therapy of Cu@CuOx-ECL1i-Gem led to substantial tumor necrosis, effective inhibition of tumor growth, and prolonged survival. Preliminary toxicity evaluations demonstrated the biocompatibility of both Cu@CuOx-ECL1i and Cu@CuOx-ECL1i-Gem for PDAC imaging and therapy.
Our studies also had limitations. The PDAC imaging was done with 64Cu-Cu@CuOx-ECL1i while the treatments were with Cu@CuOx-ECL1i-Gem, based on the study design. Although these two platforms did not show significant differences in physicochemical properties and in vivo pharmacokinetics, it may be necessary to investigate the theranostic potential of 64Cu-Cu@CuOx-ECL1i-Gem for PDAC in future studies. Additionally, the current treatment regimen needs further optimization such as dosage and frequency to enhance the therapeutic outcomes. It would also be necessary to study the treatment efficacy combining Cu@CuOx-ECL1i-Gem with other clinically used chemotherapeutic agents including paclitaxel or nab-paclitaxel for improved outcome.
Taken together, the CCR2-targeting nanoparticles demonstrated the potential for PDAC imaging guided therapy and warranted further investigation for future translation.
EXPERIMENTAL SECTION
Materials and methods.
Chemicals. Lipoamido-dPEG®8-TFP ester (TA-PEG-TFP ester), MAL-dPEG®11-lipoamide (TA-PEG-maleimide) and m-dPEG®12-Lipoamide (TA-PEG-OMe) were purchased from Quanta BioDesign, Ltd. and used without further purification. The CCR2 peptide (LGTFLKC) was customized by CPC Scientific (Sunnyvale, CA). The 64Cu (half-life = 12.7 h, β+ = 17%, β− = 40%) was generated at the Cyclotron Facility of Washington University. All other reagents were purchased from Millipore-Sigma, TCI America, or Fisher Scientific and were used without further purification. Water was purified with an E-Pure filtration system (Barnstead International, Dubuque, IA) with a resistivity of 18.2 MΩ cm. 1HNMR spectra were acquired using a 400 MHz NMR spectrometer (Varian/Agilent, Santa Clara, CA). Matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) was operated in a reflector mode with positive ion detection to collect data with a Bruker AutoFlex Speed. JEOL JEM-2100F Field Emission Electron Microscope was used for higher-resolution TEM imaging and scanning transmission electron microscopy (STEM) imaging was operated at an accelerating voltage of 200 kV. Nanoparticle suspension was dropped onto an ultrathin carbon film supported by a lacey carbon film on a copper grid and dried in air for TEM and STEM. The hydrodynamic size and ζ-potential were measured using Zetasizer Nano ZS (Malvern instruments). Radiochemical purity was measured by instant radio-thin layer chromatography (radio-TLC) (Bioscan). RP-HPLC analysis and preparation were conducted on an Agilent 1200 system with Phenomenex® Luna 5 μm 100 Å C18 LC column and Vydac 218TP™ 5 μm 300 Å C18 semipreparative column, respectively. ICP-MS was performed on a PerkinElmer Elan DRCII instrument. FPLC was evaluated on AKTA FPLC system (GE Healthcare) with Superdex® 200 Increase 10/300 GL gel filtration column eluting by phosphate buffered saline (PBS). UV absorbance was obtained in a Victor 3 1420 multilabel counter (Perkin Elmer).
Synthesis of TA-PEG-ECL1i.
ECL1i peptide (50.0 mg, 64.0 μmol) and TA-PEG-maleimide (51.4 mg, 58.1 μmol) were mixed in 25 mL 0.1 M pH = 7.0 phosphate buffer solution. After overnight stirring at R.T., TA-PEG-ECL1i was purified by RP-HPLC using H2O and acetonitrile as mobile phase and lyophilized into a white powder. The overall yield was approximately 55%. MALDI-TOF m/z: [M + H]+ Cal. for C75H129N11O24S3H 1664.8; Found 1664.9, [M + Na]+ Cal. for C75H129N11O24S3Na 1686.8; Found 1686.9, [M + K]+ Cal. for C75H129N11O24S3K 1702.8; Found 1702.9.
Synthesis of TA-PEG-Gem.
Gemcitabine hydrochloride (50 mg, 0.17 mmol) and N,N-diisopropylethylamine (DIPEA, 29 μL, 0.17 mmol) were mixed in 200 μL anhydrous DMSO. After gemcitabine was dissolved, TA-PEG-TFP ester (130 mg, 0.17 mmol) in 200 μL anhydrous DMSO was added, followed by 29 μL DIPEA. The mixture was stirred overnight at 80 ⁰C. Next, the mixture was precipitated into diethyl ether twice, and washed with saturated NaHCO3 and brine. The product was obtained as a pale yellow oil (yield = 80%). 1H NMR (D2O, 400 MHz) δ (ppm): 8.26 (d, J = 7.7 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 6.29 (t, J = 7.4 Hz, 1H), 4.42 – 4.33 (m, 1H), 4.15 (s, 1H), 4.06 (d, J = 12.8 Hz, 1H), 3.89 (dd, J = 12.8, 5.9 Hz, 3H), 3.70 – 3.60 (m, 30H), 3.39 (t, J = 5.1 Hz, 2H), 3.25 – 3.16 (m, 3H), 2.82 (t, J = 5.7 Hz, 2H), 2.49 (dd, J = 12.9, 6.4 Hz, 1H), 2.28 (d, J = 7.5 Hz, 2H), 1.98 (dd, J = 12.9, 6.4 Hz, 1H), 1.73 – 1.63 (m, 4H), 1.42 (d, J = 7.6 Hz, 2H). MALDI-TOF m/z: [M + Na]+ Cal. for C36H60N4O14S2F2Na 875.4; Found 875.4, [M + K]+ Cal. for C36H60N4O14S2F2K 913.3; Found 913.3.
Synthesis of 64Cu-Cu@CuOx-ECL1i and 64Cu-Cu@CuOx-ECL1i-Gem.
The synthesis of copper nanoparticles was modified from our previous method. Typically, CuCl2 (376 μL, 10 mM), TA-PEG ligands (400 μL, 2.5 mM (64Cu-Cu@CuOx-ECL1i used 1:2 molar ratio of TA-PEG-ECL1i and TA-PEG-OMe, 64Cu-Cu@CuOx-ECL1i-Gem used 1:2 molar ratio of TA-PEG-ECL1i and TA-PEG-Gem), and 64CuCl2 (0.1 M NH4OAc, pH 5.5, ca. 18.5 MBq μL−1) were mixed in 2 mL water and stirred for 15 mins at RT. Then, sodium borohydride (450 μL, 20 mM) was added to the reaction vial with rapid stirring for another 25 mins. The prepared nanoparticles were then immediately centrifuged with a centrifugal filter unit (Amicon Ultra, 10 kDa NMWL, 7500 g, 15 mins) and washed 3 times with water. The final product was diluted with sterile water and immediately delivered for PET imaging. The scale up nanoparticles for treatment were filtered through 0.22 μm filter (Corning® 50 mL Tube Top Vacuum Filter System) prior to centrifugation, which was injected through IV immediately after purification. Radiochemical purity was measured by instant radio-thin layer chromatography (iTLC or radio-TLC) using glass microfiber chromatography paper impregnated with a silica gel (Agilent Technology) and 10% ammonium acetate and methanol (1:1 volume ratio) mixture as developing solution (Radio-TLC, BioScan).
Cell culture.
KI cells derived from KRASG12D/INK4A deficient mice, and THP1 cells were cultured in Dulbecco modified Eagle medium with L-glutamine (Mediatech, Manassas, VA) and 10% fetal bovine serum (Sigma, St. Louis, MO), gentamicin (50 μg/ml), and amphothericin B (0.25 μg/ml). All cells were cultured at 37°C, 5% CO2, and 95% humidity.
Mouse tumor model.
Conditional p48-CRE; LSL-KRasG12D/wt; and p53flox/flox 1-Cre strains were interbred to obtain KPPC and KPC animals on a mixed 129/SvJae/C57Bl/6 background. For the KI model, KI cell line (obtained from a PDX1-Cre/KRASG12D/INK4Aflox/floxmouse62) was expanded and inoculated subcutaneously into syngeneic 8-week-old female FVBN/J mice. All studies were conducted in compliance with the institutional animal care and use committee (IACUC) guidelines of the Washington University.
Cytotoxicity study.
The KI Cells were seeded into 96-well plates with 5000 cells per well. 24 hrs later, the medium was replaced by fresh medium containing TA-PEG-Gem and Gem at the indicated concentration (range, 0-500 μM), respectively. All experimental groups were performed at least three times. After 3 days of continuous drug treatment, cell viability was determined using the WST-1 cell cytotoxicity assay (Roche, Basel, Switzerland) following the manufacturer’s protocol. Briefly, 10 μl WST-1 reagent was added in each well and then incubated for additional 4 h. The absorbance of the solution was measured using a microplate reader at 440 nm. IC50 was computed using GraphPad Prism 7.03 software (GraphPad Software, San Diego, CA).
Cell uptake studies.
10,000 THP1 or KI cells were incubated in the medium with 0.074 MBq 64Cu-Cu@CuOx-NT, 64Cu-Cu@CuOx-ECL1i, and 64Cu-Cu@CuOx-ECL1i in the presence of 100-fold of non-radioactive Cu@CuOx-ECL1i or 1000-fold of ECL1i peptide at 0 °C for 30 mins, respectively. Each group was performed at least in triplicate. The unbonded nanoparticles were removed by washing the cells with PBS for 4 times. The radioactively was measured in a PerkinElmer 1480 automatic gamma counter and recorded as counts per minute (cpm).
Immunofluorescence and Microscopy.
KI cells were incubated on coverslips (Chemglass Inc, CLS-1760-012) in 24 well plates for 24 h at 37 °C. After being washed with phosphate-buffered solution (PBS) and fixed with 10% neutral-buffered formalin (Sigma, St. Louis, MO) for 15 mins at room temperature, the cells were blocked in 10% BSA for 30 mins (ThermoFisher, Waltham, MA). Then the cells were stained with mouse anti-CCR2 antibody (Abcom, Cambridge, MA, 1:100 dilution) or 10% BSA for 1 h at room temperature, washed by PBS three times, and incubated with donkey anti-mouse Cy5 antibody (Jackson ImmunoResearch, West Grove, PA, 1:300 dilution) and DAPI (ThermoFisher, 1:1000 dilution) for 1 h. The samples were visualized on macroscope camera (Leica, DC7000T) after putting the coverslip on the slide with mounting medium.
Biodistribution.
All animal experiments were carried out in compliance with IACUC guidelines of the Washington University. Biodistribution studies were performed using female wild type C57BL/6 mice (Charles River Laboratories, Wilmington, MA). All the mice were anesthetized with inhaled isoflurane and adminstrated with about 370 kBq of 64Cu-Cu@CuOx-ECL1i or 64Cu-Cu@CuOx-ECL1i-Gem in 100 μL saline (APP pharmaceuticals, Schaumburg, IL) via the tail vein. At 1, 4, and 24 h post injection, the mice were re-anesthetized and euthanatized by cervical dislocation (n = 4/time point). Major Organs of interest from mice were harvested and weighed to acquire the weight of the organs. Then the organs were counted in a Beckman 8000 gamma counter (Beckman, Fullterton, CA) to obtain the radioactivity in these organs. The 64Cu-Cu@CuOx-ECL1i or 64Cu-Cu@CuOx-ECL1i-Gem standards were prepared and counted along with the samples to calculate percentage of the injected dose per gram of tissue (%ID/g). A nonlinear regression analysis was used to calculate the mean blood half-lives of the two nanoparticles (Prism, version 7.03, Graphpad).
Micro-PET/CT imaging.
KPPC mice at 7-9-week-old, KPC mice at 22-23-week old, and their wild type littermates at the same age were anesthetized and injected with 3.7 MBq 64Cu-Cu@CuOx-ECL1i/64Cu-Cu@CuOx-NT in 100 μL saline via the tail vein. Small animal PET scans were carried out on Inveon PET/CT system (Siemens, Malvern, PA) at 24 h post injection (60 min frame). The attenuation, scatter, normalization and camera dead time were all corrected for micro-PET images for co-registration with micro-CT images. The Inveon PET/CT scanner is periodically calibrated using a normalization phantom of known activity concentration to ensure its quantitative accuracy. Micro-PET images were reconstructed with the maximum a posteriori (MAP) algorithm. Data quantification was carried out using Inveon Research Workplace. Tumor accumulation was computed as percent injected dose per gram (%ID/g) of tissue in three-dimensional region-of-interests without the correction for partial volume effect. PET blocking study was done by co-injection of 50-fold non-radioactive Cu@CuOx-ECL1i/Cu@CuOx-NT for competition.
Autoradiography.
The mice were perfused with saline transcardially and the tumors were collected and sliced immediately following PET/CT scan. The slices were covered by a phosphor-imaging film plate and exposed overnight. The film was then imaged with a GE Typhoon FLA 9500 Biomolecular Imager.
Cu toxicity study.
8-9 weeks old CD1 mice were IV administrated with 1, 10, 15, and 20 mg/kg body weight of Cu@CuOx-NT (n = 5/group). Saline was injected as the control group. The mice receiving 20 mg/kg of body weight of Cu@CuOx-NT died 2-3 days post treatment. One-week post administration, the mice in other groups were submitted to Division of Comparative Medicine (DCM) research animal diagnostic laboratory in Washington University in St. Louis for clinical pathologic and histopathologic evaluation. The mouse blood was collected by cardiocentesis immediately after euthanasia by CO2 inhalation. Hematology was performed on blood samples anticoagulated with EDTA using commercially supplied tubes (Microvette 100, Sarstedt AG, Numbrecht, Germany). The complete blood count was performed using the Hemavet 1700 Veterinary Multispecies Hematology System (Drew Scientific, Miami Lakes, FL). Blood smears of each sample were prepared, dried, fixed in methanol, and stained using Wright-Giemsa stain for microscopic evaluation. Serum measurements of blood urea nitrogen (BUN), creatinine, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and total protein (TP) were determined using the Liasys 330 liquid reagent chemistry analyzer (AMS Diagnostics, Weston, FL). After weighing the mice, kidneys and livers were harvested and fixed in 10% neutral buffered formalin. Following fixation, the tissues were trimmed, paraffin-embedded, and prepared as 5-micron sections and then stained with hematoxylin and eosin for standard histopathologic evaluation. 8-week-old male and female C57BL/6 mice were randomly treated with (1) Cu@CuOx-ECL1i-Gem; (2) Cu@CuOx-ECL1i; and (3) saline (n = 5/group) for 2 successive weeks. The Cu mass was equivalent to 10 mg/kg of body weight. 3 days post treatment, all the mice were submitted to DCM research animal diagnostic laboratory for evaluation.
Treatment.
A KI xenograft model was used to determine tumor growth trends and treatment efficacy of Cu@CuOx-ECL1i-Gem. 10 days after tumor implantation, mice were randomized into 5 groups (n = 5/group): (1) Cu@CuOx-ECL1i-Gem (Cu mass: 10 mg/kg body weight, Gem: 3.4 ± 0.8 mg/kg body weight, IV injection, twice per week); (2) Cu@CuOx-ECL1i (same Cu@CuOx-NT dose as (1), IV injection, twice per week); (3) gemcitabine 7 mg/kg body weight (IV injection, twice per week); (4) gemcitabine 100 mg/kg body weight (IP injection, once per week); (5) saline (100 μL, IV injection, twice per week). For the 2nd treatment study, the mice started dose administration at 7 days post implantation. The dose regimens of Cu@CuOx-Gem and Cu@CuOx-ECL1i-Gem were the same as the 1st treatment study. The Cu mass was limited to a dosage of 10 mg/kg for all nanoparticle groups, and the relative loaded gemcitabine dose was 3.4 ± 0.8 mg/kg body weight. The low dose gemcitabine group was comparable, in terms of gemcitabine dose, to the dosing regimen of Cu@CuOx-ECL1i-Gem. The high dose gemcitabine group was comparable with the standard human dosing regimen.70 Mouse weight and tumor volume were monitored 2-3 times each week. Tumor mass was calculated as (length × width2)/2. The treatment was continued for 6 successive weeks or until the mice died or were sacrificed and deemed as dead when their tumor volume reached over 2000 mm3. After treatment, the tumors were collected for H&E staining.
Immunohistochemistry of tumor tissues.
Tumor serial sections with 5 μm thickness were prepared from Histochoice®-fixed (24 h), paraffin-embedded specimens. A series of xylenes and graded alcohols were used to deparaffinize and rehydrate the sections for staining with hematoxylin and eosin to assess morphology of the tissues. Consecutive sections of rehydrated tumor underwent antigen retrieval pre-treatment (Diva Decloaker, 1x) as well as hydrogen peroxide quenching before being treated with serum for 1 hour to block nonspecific binding (Vectastain; Vector Laboratories, Burlingame, CA). The slides were stained with primary antibody overnight at 4C (anti-CCR2, 1:1000 in blocking serum, Novus Biologicals). After secondary antibody was used (Vector Laboratories, Burlingame, CA), a diaminobenzidine-based immunostaining was carried out. Digital images of the stained sections were obtained using both scanning light and light microscopes (Nanozoomer, Hamamatsu, and Leica).
Statistical analysis.
Statistical analysis was done with GraphPad Prism (version 6.07). Group variation was reported as mean ± standard deviation. Differences between two or multiple groups were calculated using the unpaired two-tailed Student’s t-test or one-way ANOVA. A significant difference was reported when a P value was less than 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by grant P50CA196510 from the National Cancer Institute; Washington University in St. Louis School of Medicine, the Department of Surgery, and Siteman Cancer Center; University of Rochester, University of North Carolina at Chapel Hill (Lineberger Comprehensive Cancer Center), and Johns Hopkins Medicine (The Sidney Kimmel Comprehensive Cancer Center). This work was partially supported by the National Cancer Institute (R01CA235672). This work was partially performed at the Nano Research Facility (NRF), a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation under Grant No. ECS-0335765. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. NRF is part of the School of Engineering and Applied Science at Washington University in St. Louis. This work was performed with the support from the Siteman Cancer Center Small Animal Imaging Core at Washington University in St. Louis. We appreciate the staff members from Washington University cyclotron facility for 64Cu production.
Experiments were performed in part through the use of Washington University Center for Cellular Imaging (WUCCI) supported by Washington University School of Medicine, The Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505) and the National Institute for Neurological Disorders and Stroke (NS086741). The authors acknowledge financial support from Washington University in St. Louis and the Institute of Materials Science and Engineering for the use of instruments and staff assistance.
Footnotes
Synthesis of TA-PEG-ECL1i and TA-PEG-Gem (S1); MALDI-TOF mass spectrum of TA-PEG-ECL1i (S2); 1H NMR and MALDI-TOF mass spectrum of TA-PEG-Gem (S3); Characterization of Cu nanoparticles (Table S1); HPLC spectra of TA-PEG-ECL1i and TA-PEG-Gem, and calculation of Cu@CuOx-ECL1i-Gem loading capacity (S4); FPLC analysis of Cu@CuOx-ECL1i (S5); Radio-TLC of 64Cu-Cu@CuOx-ECL1i (S6); Cell viability and IC50 of Gem and TA-PEG-Gem in KI cells (S7); In vitro cell uptake in THP1 and KI cells (S8); 64Cu-Cu@CuOx-ECL1i PET blocked with 50 times of Cu@CuOx-NT (S9); Amplified H&E and trichrome staining of KPPC tumor (S10); 18F-FDG PET characteristics in PDAC models (S11); In vivo toxicity evaluation of Cu nanoparticles (S12 and S13).
The authors declare no conflict of interest.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2020. CA Cancer J. Clin 2020, 70, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Vivaldi C; Fornaro L; Vasile E FOLFIRINOX Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med 2019, 380, 1187–1188. [DOI] [PubMed] [Google Scholar]
- (3).Neoptolemos JP; Palmer DH; Ghaneh P; Psarelli EE; Valle JW; Halloran CM; Faluyi O; O’Reilly DA; Cunningham D; Wadsley J; Darby S; Meyer T; Gillmore R; Anthoney A; Lind P; Glimelius B; Falk S; Izbicki JR; Middleton GW; Cummins S; et al. Comparison of Adjuvant Gemcitabine and Capecitabine with Gemcitabine Monotherapy in Patients with Resected Pancreatic Cancer (ESPAC-4): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet 2017, 389, 1011–1024. [DOI] [PubMed] [Google Scholar]
- (4).Oettle H; Neuhaus P; Hochhaus A; Hartmann JT; Gellert K; Ridwelski K; Niedergethmann M; Zulke C; Fahlke J; Arning MB; Sinn M; Hinke A; Riess H Adjuvant Chemotherapy with Gemcitabine and Long-Term Outcomes among Patients with Resected Pancreatic Cancer: The CONKO-001 Randomized Trial. JAMA 2013, 310, 1473–1481. [DOI] [PubMed] [Google Scholar]
- (5).Rahib L; Smith BD; Aizenberg R; Rosenzweig AB; Fleshman JM; Matrisian LM Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- (6).Conroy T; Desseigne F; Ychou M; Bouche O; Guimbaud R; Becouarn Y; Adenis A; Raoul JL; Gourgou-Bourgade S; de la Fouchardiere C; Bennouna J; Bachet JB; Khemissa-Akouz F; Pere-Verge D; Delbaldo C; Assenat E; Chauffert B; Michel P; Montoto-Grillot C; Ducreux M; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med 2011, 364, 1817–1825. [DOI] [PubMed] [Google Scholar]
- (7).Von Hoff DD; Ervin T; Arena FP; Chiorean EG; Infante J; Moore M; Seay T; Tjulandin SA; Ma WW; Saleh MN; Harris M; Reni M; Dowden S; Laheru D; Bahary N; Ramanathan RK; Tabernero J; Hidalgo M; Goldstein D; Van Cutsem E; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel Plus Gemcitabine. N. Engl. J. Med 2013, 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Burris HA; Moore MJ; Andersen J; Green MR; Rothenberg ML; Madiano MR; Cripps MC; Portenoy RK; Storniolo AM; Tarassoff P; Nelson R; Dorr FA; Stephens CD; VanHoff DD Improvements in Survival and Clinical Benefit with Gemcitabine as First-Line Therapy for Patients with Advanced Pancreas Cancer: A Randomized Trial. J. Clin. Oncol 1997, 15, 2403–2413. [DOI] [PubMed] [Google Scholar]
- (9).Moore MJ; Goldstein D; Hamm J; Figer A; Hecht JR; Gallinger S; Au HJ; Murawa P; Walde D; Wolff RA; Campos D; Lim R; Ding K; Clark G; Voskoglou-Nomikos T; Ptasynski M; Parulekar W; National Cancer Institute of Canada Clinical Trials, G. Erlotinib Plus Gemcitabine Compared with Gemcitabine Alone in Patients with Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol 2007, 25, 1960–1966. [DOI] [PubMed] [Google Scholar]
- (10).Sudo K; Nakamura K; Yamaguchi T S-1 in the Treatment of Pancreatic Cancer. World J. Gastroenterol 2014, 20, 15110–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Von Hoff DD; Ramanathan RK; Borad MJ; Laheru DA; Smith LS; Wood TE; Korn RL; Desai N; Trieu V; Iglesias JL; Zhang H; Soon-Shiong P; Shi T; Rajeshkumar NV; Maitra A; Hidalgo M Gemcitabine Plus Nab-Paclitaxel Is an Active Regimen in Patients with Advanced Pancreatic Cancer: A Phase I/II Trial. J. Clin. Oncol 2011, 29, 4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mahaseth H; Brutcher E; Kauh J; Hawk N; Kim S; Chen Z; Kooby DA; Maithel SK; Landry J; El-Rayes BF Modified FOLFIRINOX Regimen with Improved Safety and Maintained Efficacy in Pancreatic Adenocarcinoma. Pancreas 2013, 42, 1311–1315. [DOI] [PubMed] [Google Scholar]
- (13).Adamska A; Domenichini A; Falasca M Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci 2017, 18, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Neesse A; Bauer CA; Ohlund D; Lauth M; Buchholz M; Michl P; Tuveson DA; Gress TM Stromal Biology and Therapy in Pancreatic Cancer: Ready for Clinical Translation? Gut 2019, 68, 159–171. [DOI] [PubMed] [Google Scholar]
- (15).Mei L; Du W; Ma WW Targeting Stromal Microenvironment in Pancreatic Ductal Adenocarcinoma: Controversies and Promises. J. Gastrointest. Oncol 2016, 7, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).von Ahrens D; Bhagat TD; Nagrath D; Maitra A; Verma A The Role of Stromal Cancer-Associated Fibroblasts in Pancreatic Cancer. J. Hematol. Oncol 2017, 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nywening TM; Belt BA; Cullinan DR; Panni RZ; Han BJ; Sanford DE; Jacobs RC; Ye J; Patel AA; Gillanders WE; Fields RC; DeNardo DG; Hawkins WG; Goedegebuure P; Linehan DC Targeting Both Tumour-Associated CXCR2+ Neutrophils and CCR2+ Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut 2018, 67, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sanford DE; Belt BA; Panni RZ; Mayer A; Deshpande AD; Carpenter D; Mitchem JB; Plambeck-Suess SM; Worley LA; Goetz BD; Wang-Gillam A; Eberlein TJ; Denardo DG; Goedegebuure SP; Linehan DC Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res 2013, 19, 3404–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhu Y; Herndon JM; Sojka DK; Kim KW; Knolhoff BL; Zuo C; Cullinan DR; Luo J; Bearden AR; Lavine KJ; Yokoyama WM; Hawkins WG; Fields RC; Randolph GJ; DeNardo DG Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Shen ZY; Chen TX; Ma XH; Ren WZ; Zhou ZJ; Zhu GZ; Zhang A; Liu YJ; Song JB; Li ZH; Ruan HM; Fan WP; Lin LS; Munasinghe J; Chen XY; Wu AG Multifunctional Theranostic Nanoparticles Based on Exceedingly Small Magnetic Iron Oxide Nanoparticles for T1-Weighted Magnetic Resonance Imaging and Chemotherapy. ACS Nano 2017, 11, 10992–11004. [DOI] [PubMed] [Google Scholar]
- (21).Liu Y; Pierce R; Luehmann HP; Sharp TL; Welch MJ PET Imaging of Chemokine Receptors in Vascular Injury-Accelerated Atherosclerosis. J. Nucl. Med 2013, 54, 1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Williams JW; Elvington A; Ivanov S; Kessler S; Luehmann H; Baba O; Saunders BT; Kim KW; Johnson MW; Craft CS; Choi JH; Sorci-Thomas MG; Zinselmeyer BH; Brestoff JR; Liu Y; Randolph GJ Thermoneutrality but Not UCP1 Deficiency Suppresses Monocyte Mobilization into Blood. Circ. Res 2017, 121, 662–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Qian BZ; Li J; Zhang H; Kitamura T; Zhang J; Campion LR; Kaiser EA; Snyder LA; Pollard JW CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature 2011, 475, 222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhao L; Lim SY; Gordon-Weeks AN; Tapmeier TT; Im JH; Cao Y; Beech J; Allen D; Smart S; Muschel RJ Recruitment of a Myeloid Cell Subset (Cd11b/Gr1 Mid) via CCL2/CCR2 Promotes the Development of Colorectal Cancer Liver Metastasis. Hepatology 2013, 57, 829–839. [DOI] [PubMed] [Google Scholar]
- (25).Badiyan SN; Olsen JR; Lee AY; Yano M; Menias CO; Khwaja S; Wang-Gillam A; Strasberg SM; Hawkins WG; Linehan DC; Myerson RJ; Parikh PJ Induction Chemotherapy Followed by Concurrent Full-Dose Gemcitabine and Intensity-Modulated Radiation Therapy for Borderline Resectable and Locally Advanced Pancreatic Adenocarcinoma. Am. J. Clin. Oncol 2016, 39, 1–7. [DOI] [PubMed] [Google Scholar]
- (26).Grossman JG; Nywening TM; Belt BA; Panni RZ; Krasnick BA; DeNardo DG; Hawkins WG; Goedegebuure SP; Linehan DC; Fields RC Recruitment of CCR2+ Tumor Associated Macrophage to Sites of Liver Metastasis Confers a Poor Prognosis in Human Colorectal Cancer. Oncoimmunology 2018, 7, e1470729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).English SJ; Sastriques SE; Detering L; Sultan D; Luehmann H; Arif B; Heo GS; Zhang X; Laforest R; Zheng J; Lin CY; Gropler RJ; Liu Y CCR2 Positron Emission Tomography for the Assessment of Abdominal Aortic Aneurysm Inflammation and Rupture Prediction. Circ. Cardiovasc. Imaging 2020, 13, e009889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nywening TM; Wang-Gillam A; Sanford DE; Belt BA; Panni RZ; Cusworth BM; Toriola AT; Nieman RK; Worley LA; Yano M; Fowler KJ; Lockhart AC; Suresh R; Tan BR; Lim KH; Fields RC; Strasberg SM; Hawkins WG; DeNardo DG; Goedegebuure SP; et al. Targeting Tumour-Associated Macrophages with CCR2 Inhibition in Combination with FOLFIRINOX in Patients with Borderline Resectable and Locally Advanced Pancreatic Cancer: A Single-Centre, Open-Label, Dose-Finding, Non-Randomised, Phase 1b Trial. Lancet Oncol. 2016, 17, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Min Y; Caster JM; Eblan MJ; Wang AZ Clinical Translation of Nanomedicine. Chem. Rev 2015, 115, 11147–11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Pelaz B; Alexiou C; Alvarez-Puebla RA; Alves F; Andrews AM; Ashraf S; Balogh LP; Ballerini L; Bestetti A; Brendel C; Bosi S; Carril M; Chan WC; Chen C; Chen X; Chen X; Cheng Z; Cui D; Du J; Dullin C; et al. Diverse Applications of Nanomedicine. ACS Nano 2017, 11, 2313–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Elsabahy M; Heo GS; Lim SM; Sun G; Wooley KL Polymeric Nanostructures for Imaging and Therapy. Chem. Rev 2015, 115, 10967–11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Huang J; Qian W; Wang L; Wu H; Zhou H; Wang AY; Chen H; Yang L; Mao H Functionalized Milk-Protein-Coated Magnetic Nanoparticles for MRI-Monitored Targeted Therapy of Pancreatic Cancer. Int. J. Nanomedicine 2016, 11, 3087–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lee GY; Qian WP; Wang L; Wang YA; Staley CA; Satpathy M; Nie S; Mao H; Yang L Theranostic Nanoparticles with Controlled Release of Gemcitabine for Targeted Therapy and MRI of Pancreatic Cancer. ACS Nano 2013, 7, 2078–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Manzur A; Oluwasanmi A; Moss D; Curtis A; Hoskins C Nanotechnologies in Pancreatic Cancer Therapy. Pharmaceutics 2017, 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Teng WZ; Jia F; Han HJ; Qin ZH; Jin Q; Ji J Polyamino Acid-Based Gemcitabine Nanocarriers for Targeted Intracellular Drug Delivery. Polym. Chem 2017, 8, 2490–2498. [Google Scholar]
- (37).Anselmo AC; Mitragotri S Nanoparticles in the Clinic: An Update. Bioeng. Transl. Med 2019, 4, e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tanaka HY; Kano MR Stromal Barriers to Nanomedicine Penetration in the Pancreatic Tumor Microenvironment. Cancer Sci. 2018, 109, 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Brachi G; Bussolino F; Ciardelli G; Mattu C Nanomedicine for Imaging and Therapy of Pancreatic Adenocarcinoma. Front. Bioeng. Biotechnol 2019, 7, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Meng H; Nel AE Use of Nano Engineered Approaches to Overcome the Stromal Barrier in Pancreatic Cancer. Adv. Drug Deliver. Rev 2018, 130, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gao F; Cai P; Yang W; Xue J; Gao L; Liu R; Wang Y; Zhao Y; He X; Zhao L; Huang G; Wu F; Zhao Y; Chai Z; Gao X Ultrasmall [64Cu]Cu Nanoclusters for Targeting Orthotopic Lung Tumors Using Accurate Positron Emission Tomography Imaging. ACS Nano 2015, 9, 4976–4986. [DOI] [PubMed] [Google Scholar]
- (42).Jiang XY; Du BJ; Huang YY; Zheng J Ultrasmall Noble Metal Nanoparticles: Breakthroughs and Biomedical Implications. Nano Today 2018, 21, 106–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shen S; Jiang D; Cheng L; Chao Y; Nie K; Dong Z; Kutyreff CJ; Engle JW; Huang P; Cai W; Liu Z Renal-Clearable Ultrasmall Coordination Polymer Nanodots for Chelator-Free 64Cu-Labeling and Imaging-Guided Enhanced Radiotherapy of Cancer. ACS Nano 2017, 11, 9103–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhao Y; Detering L; Sultan D; Cooper ML; You M; Cho S; Meier SL; Luehmann H; Sun G; Rettig M; Dehdashti F; Wooley KL; DiPersio JF; Liu Y Gold Nanoclusters Doped with 64Cu for CXCR4 Positron Emission Tomography Imaging of Breast Cancer and Metastasis. ACS Nano 2016, 10, 5959–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhao YF; Sultan D; Detering L; Luehmann H; Liu YJ Facile Synthesis, Pharmacokinetic and Systemic Clearance Evaluation, and Positron Emission Tomography Cancer Imaging of 64Cu-Au Alloy Nanoclusters. Nanoscale 2014, 6, 13501–13509. [DOI] [PubMed] [Google Scholar]
- (46).Miller-Kleinhenz J; Guo XX; Qian WP; Zhou HY; Bozeman EN; Zhu L; Ji X; Wang YA; Styblo T; O’Regan R; Mao H; Yang L Dual-Targeting Wnt and uPA Receptors Using Peptide Conjugated Ultra-Small Nanoparticle Drug Carriers Inhibited Cancer Stem-Cell Phenotype in Chemo-Resistant Breast Cancer. Biomaterials 2018, 152, 47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Heo GS; Zhao Y; Sultan D; Zhang X; Detering L; Luehmann HP; Zhang X; Li R; Choksi A; Sharp S; Levingston S; Primeau T; Reichert DE; Sun G; Razani B; Li S; Weilbaecher KN; Dehdashti F; Wooley KL; Liu Y Assessment of Copper Nanoclusters for Accurate in Vivo Tumor Imaging and Potential for Translation. ACS Appl. Mater. Interfaces 2019, 11, 19669–19678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kamaly N; Yameen B; Wu J; Farokhzad OC Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev 2016, 116, 2602–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lu Y; Zhang E; Yang J; Cao Z Strategies to Improve Micelle Stability for Drug Delivery. Nano Res. 2018, 11, 4985–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lai WF; Wong WT; Rogach AL Development of Copper Nanoclusters for in Vitro and in Vivo Theranostic Applications. Adv. Mater 2020, 32, e1906872. [DOI] [PubMed] [Google Scholar]
- (51).Li W; Luehmann HP; Hsiao HM; Tanaka S; Higashikubo R; Gauthier JM; Sultan D; Lavine KJ; Brody SL; Gelman AE; Gropler RJ; Liu Y; Kreisel D Visualization of Monocytic Cells in Regressing Atherosclerotic Plaques by Intravital 2-Photon and Positron Emission Tomography-Based Imaging-Brief Report. Arterioscler. Thromb. Vasc. Biol 2018, 38, 1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Auvynet C; de Chanville CB; Hermand P; Dorgham K; Piesse C; Pouchy C; Carlier L; Poupel L; Barthelemy S; Felouzis V; Lacombe C; Sagan S; Salomon B; Deterre P; Sennlaub F; Combadiere C ECL1i, d(LGTFLKC), a Novel, Small Peptide That Specifically Inhibits CCL2-Dependent Migration. Faseb J. 2016, 30, 2370–2381. [DOI] [PubMed] [Google Scholar]
- (53).Heo GS; Kopecky B; Sultan D; Ou M; Feng G; Bajpai G; Zhang X; Luehmann H; Detering L; Su Y; Leuschner F; Combadiere C; Kreisel D; Gropler RJ; Brody SL; Liu Y; Lavine KJ Molecular Imaging Visualizes Recruitment of Inflammatory Monocytes and Macrophages to the Injured Heart. Circ. Res 2019, 124, 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zhao YF; Sultan D; Detering L; Cho SH; Sun GR; Pierce R; Wooley KL; Liu YJ Copper-64-Alloyed Gold Nanoparticles for Cancer Imaging: Improved Radiolabel Stability and Diagnostic Accuracy. Angew. Chem. Int. Ed 2014, 53, 156–159. [DOI] [PubMed] [Google Scholar]
- (55).Moysan E; Bastiat G; Benoit JP Gemcitabine versus Modified Gemcitabine: A Review of Several Promising Chemical Modifications. Mol. Pharm 2013, 10, 430–444. [DOI] [PubMed] [Google Scholar]
- (56).Liu YJ; Welch MJ Nanoparticles Labeled with Positron Emitting Nuclides: Advantages, Methods, and Applications. Bioconjug. Chem 2012, 23, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sun X; Cai W; Chen X Positron Emission Tomography Imaging Using Radiolabeled Inorganic Nanomaterials. Acc. Chem. Res 2015, 48, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wang QY; Wang SY; Hu X; Li FY; Ling DS Controlled Synthesis and Assembly of Ultra-Small Nanoclusters for Biomedical Applications. Biomater. Sci 2019, 7, 480–489. [DOI] [PubMed] [Google Scholar]
- (59).Nie S Understanding and Overcoming Major Barriers in Cancer Nanomedicine. Nanomedicine (Lond) 2010, 5, 523–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Brannon-Peppas L; Blanchette JO Nanoparticle and Targeted Systems for Cancer Therapy. Adv. Drug Deliv. Rev 2004, 56, 1649–1659. [DOI] [PubMed] [Google Scholar]
- (61).Du BJ; Jiang XY; Das A; Zhou QH; Yu MX; Jin RC; Zheng J Glomerular Barrier Behaves as an Atomically Precise Bandpass Filter in a Sub-Nanometre Regime. Nature Nanotechnol. 2017, 12, 1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Aguirre AJ; Bardeesy N; Sinha M; Lopez L; Tuveson DA; Horner J; Redston MS; DePinho RA Activated Kras and Ink4a/Arf Deficiency Cooperate to Produce Metastatic Pancreatic Ductal Adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Parker LC; Whyte MK; Vogel SN; Dower SK; Sabroe I Toll-Like Receptor (TLR)2 and TLR4 Agonists Regulate CCR Expression in Human Monocytic Cells. J. Immunol 2004, 172, 4977–4986. [DOI] [PubMed] [Google Scholar]
- (64).Jiang H; Hegde S; Knolhoff BL; Zhu Y; Herndon JM; Meyer MA; Nywening TM; Hawkins WG; Shapiro IM; Weaver DT; Pachter JA; Wang-Gillam A; DeNardo DG Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med 2016, 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hingorani SR; Wang L; Multani AS; Combs C; Deramaudt TB; Hruban RH; Rustgi AK; Chang S; Tuveson DA Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [DOI] [PubMed] [Google Scholar]
- (66).Torres JB; Knight JC; Mosley MJ; Kersemans V; Koustoulidou S; Allen D; Kinchesh P; Smart S; Cornelissen B Imaging of Claudin-4 in Pancreatic Ductal Adenocarcinoma Using a Radiolabelled Anti-Claudin-4 Monoclonal Antibody. Mol. Imaging Biol 2018, 20, 292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kim G Nab-Paclitaxel for the Treatment of Pancreatic Cancer. Cancer Manag. Res 2017, 9, 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Gaetke LM; Chow-Johnson HS; Chow CK Copper: Toxicological Relevance and Mechanisms. Arch. Toxicol 2014, 88, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Tian L; Yi X; Dong Z; Xu J; Liang C; Chao Y; Wang Y; Yang K; Liu Z Calcium Bisphosphonate Nanoparticles with Chelator-Free Radiolabeling to Deplete Tumor-Associated Macrophages for Enhanced Cancer Radioisotope Therapy. ACS Nano 2018, 12, 11541–11551. [DOI] [PubMed] [Google Scholar]
- (70).Yip-Schneider MT; Wu HB; Stantz K; Agaram N; Crooks PA; Schmidt CM Dimethylaminoparthenolide and Gemcitabine: A Survival Study Using a Genetically Engineered Mouse Model of Pancreatic Cancer. BMC Cancer 2013, 13, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.