

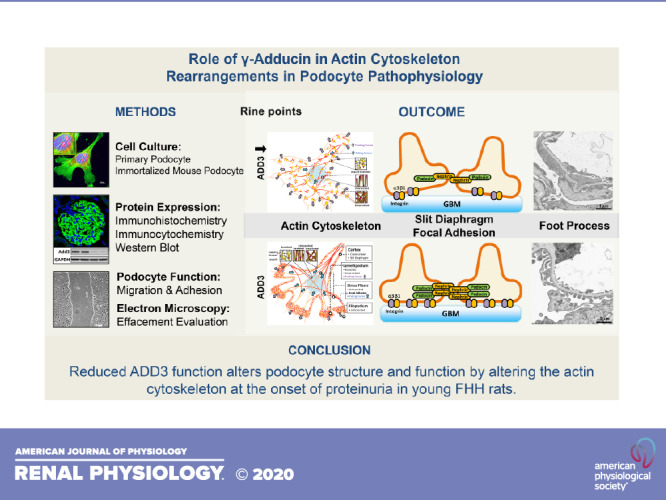
Keywords: γ-adducin, actin cytoskeleton, glomerulus, podocyte, renal injury
Abstract
We recently reported that the enhanced susceptibility to chronic kidney disease (CKD) in the fawn-hooded hypertensive (FHH) rat is caused, at least in part, by a mutation in γ-adducin (ADD3) that attenuates renal vascular function. The present study explored whether Add3 contributes to the modulation of podocyte structure and function using FHH and FHH.Add3 transgenic rats. The expression of ADD3 on the membrane of primary podocytes isolated from FHH was reduced compared with FHH.Add3 transgenic rats. We found that F-actin nets, which are typically localized in the lamellipodia, replaced unbranched stress fibers in conditionally immortalized mouse podocytes transfected with Add3 Dicer-substrate short interfering RNA (DsiRNA) and primary podocytes isolated from FHH rats. There were increased F/G-actin ratios and expression of the Arp2/3 complexes throughout FHH podocytes in association with reduced synaptopodin and RhoA but enhanced Rac1 and CDC42 expression in the renal cortex, glomeruli, and podocytes of FHH rats. The expression of nephrin at the slit diaphragm and the levels of focal adhesion proteins integrin-α3 and integrin-β1 were decreased in the glomeruli of FHH rats. Cell migration was enhanced and adhesion was reduced in podocytes of FHH rats as well as in immortalized mouse podocytes transfected with Add3 DsiRNA. Mean arterial pressures were similar in FHH and FHH.Add3 transgenic rats at 16 wk of age; however, FHH rats exhibited enhanced proteinuria associated with podocyte foot process effacement. These results demonstrate that reduced ADD3 function in FHH rats alters baseline podocyte pathophysiology by rearrangement of the actin cytoskeleton at the onset of proteinuria in young animals.
INTRODUCTION
Chronic kidney disease (CKD) is a worldwide health burden, and hypertension is one of the major risk factors for the development of CKD. The prevalence of CKD has steadily increased over the past decades (1). The Medicare costs for the treatment of 500,000 patients with CKD and end-stage renal disease exceed $120 billion per year (2, 3). Despite the intense investigation, the genetic susceptibility and underlying molecular mechanisms of hypertension-related CKD are still not well defined.
The fawn-hooded hypertensive (FHH) rat is a genetic model of progressive proteinuria, focal segmental glomerulosclerosis, systemic hypertension, and CKD. This strain is susceptible to the adverse renal effects of hypertension and uninephrectomy (4–7). We recently identified an inactivating recessive mutation in γ-adducin (ADD3) in FHH rats. This mutation is associated with reduced ADD3 membrane expression, disrupted actin cytoskeleton in vascular smooth muscle cells (VSMCs), and impaired renal hemodynamics (5). Transfer of a portion of the renal failure-1 (Rf-1) region, where the wild-type Add3 is localized, and knockin of wild-type Add3 restored renal hemodynamics and attenuated proteinuria in FHH.1BN congenic and FHH.Add3 transgenic strains with aging and following induction of DOCA-salt hypertension (4, 5). These results are in line with our previous reports that knockdown of ADD3 expression in the renal and cerebral arterioles, using an Add3 Dicer-substrate short interfering RNA (DsiRNA), impaired the myogenic response of these arterioles ex vivo and enhanced peak potassium currents in VSMCs isolated from positively transfected vessels (8). To this end, our previous findings demonstrated that the enhanced susceptibility to CKD following the development of hypertension in the FHH rat is associated with a mutation in ADD3 that attenuates renal vascular function, possibly by destabilizing the actin cytoskeleton in VSMCs.
Interestingly, we also found that young FHH rats exhibited enhanced glomerular permeability to albumin (Palb), which was rescued in FHH.Add3 transgenic rats (5, 9). Indeed, impaired renal hemodynamics contributes to glomerular hyperfiltration and vascular pole-associated glomerulosclerosis, which could promote podocyte injury and enhance Palb in FHH rats after they develop hypertension (10). This hypothesis was also true in other animal models (9, 11–16). However, the actin cytoskeleton also plays an essential role in protecting podocytes from hypertension-induced glomerular injury that initiates the progression to CKD. Human genetic studies have demonstrated that mutations in genes regulating the actin structure and function in podocytes are associated with proteinuria and glomerular disease (17–25). Considering that mutant or wild-type ADD3 is globally expressed in FHH or FHH.Add3 transgenic rats, it remains to be determined whether the glomerular injury in FHH rats is solely a consequence of impaired renal hemodynamics or acts synergistically with a direct effect of actin disruption on podocyte function. Thus the present study explored the role of Add3 in podocyte structure and function by comparing actin dynamics and distribution of actin-related proteins to the integrity of the slit diaphragm and focal adhesions in the renal cortex, glomeruli, and primary podocytes isolated from FHH and FHH.Add3 transgenic rats. We also compared mean arterial pressures (MAP), proteinuria, and the development of foot process effacement (FPE) in young rats at 16 wk of age.
MATERIALS AND METHODS
Animals
Experiments were performed using 8- to 16-wk-old male FHH and FHH.Add3 transgenic rats that we generated previously (5). The rats were obtained from our in-house colonies maintained at the University of Mississippi Medical Center (UMMC). All animals were housed under standard laboratory animal conditions, including a 12-h light/dark cycle with free access to standard diets and water. The UMMC laboratory animal care facility is approved by the American Association for the Accreditation of Laboratory Animal Care. All experimental procedures were conducted in accordance with and approved by the Institutional Animal Care and Use Committees of the UMMC and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Measurement of Mean Arterial Pressure and Proteinuria
Rats were implanted with telemetry probes (HD-S10, Data Sciences International, St. Paul, MN) for measurements of MAP, as we have previously described (5, 15). Briefly, the animals were anesthetized with isoflurane, and a telemetry probe was implanted into the femoral artery. Baytril (10 mg/kg sc) and Rimadyl (5 mg/kg sc) were given to the animals before and the next morning after surgery to prevent infection and for pain relief. After a 1-wk recovery period, MAP was recorded in separate groups of 12- and 16-wk-old male FHH and FHH.Add3 rats for 3 consecutive days using a Ponemah v5.20 software (Data Sciences International). Urine was collected overnight separate groups of 8-, 12-, and 16-wk-old male FHH and FHH.Add3 rats in metabolic cages. Proteinuria was measured using the Bradford method with BSA as the standard (Bio-Rad Laboratories, Hercules, CA).
Immortalized Mouse Podocyte Cell Culture
A conditionally immortalized mouse podocyte cell line was kindly provided by Dr. Pin-Lan Li (Department of Pharmacology and Toxicology, Virginia Commonwealth University School of Medicine, Richmond, VA). Undifferentiated podocytes were maintained under permissive conditions in dishes precoated with collagen type I (Corning, Corning, NY) in RPMI-1640 medium (ThermoFisher Scientific, Waltham, MA) supplemented with 10% FBS (ThermoFisher Scientific), 100 U/mL penicillin, 100 µg/mL streptomycin, and 10 U/mL recombinant mouse interferon (IFN)-γ (ThermoFisher Scientific) at 33°C. The morphology of the actin cytoskeleton was compared in differentiated transfected podocytes grown under nonpermissive conditions (26–30) at 37°C without IFN-γ for 6–10 days. The cells were validated by >95% positively staining with antibodies against nephrin, podocin, and synaptopodin.
Transfection of Add3 DsiRNA
Differentiated conditionally immortalized mouse podocytes were transfected with Add3 DsiRNA as previously reported (8). Briefly, podocytes were seeded into collagen type I-coated six-well plates to reach 80–85% confluency. The cells were then replaced with culture medium containing 10% FBS but without antibiotics or IFN-γ and transfected with 25 nM of an Add3 DsiRNA that we have previously described (sense: 5'-rGrGrArGrCrArGrGrArCrCrArCrArUrCrArUrArArUrCrATC-3' and antisense: 5'-rGrArUrGrArUrUrArUrGrArUrGrUrGrGrUrCrCrUrGrCrUrCrCrUrU-3') using DharmaFECT siRNA transfection reagent 2 (ThermoFisher Scientific). The cells were then incubated with 5% CO2 at 37°C for 48 h before Western blot or immunocytochemistry. A DS NC1 duplex (Integrated DNA Technologies, Coralville, IA) was used as a negative control (8).
Isolation of Primary Podocytes
Glomeruli isolation.
After MAP and proteinuria were measured, glomeruli were isolated from 12-wk-old male FHH and FHH.Add3 transgenic rats using the sieving method we reported previously (9). Briefly, rats were euthanized using 4% isoflurane. The kidneys were removed, and the renal cortex was separated from the medulla and passed through stainless steel sieves with pore sizes from 250 to 106 μm (USA standard sieve no. 60 and no. 140, respectively, ThermoFisher Scientific) into a sterile petri dish. The glomeruli were collected on a 63-μm stainless steel sieves and washed off from the screen with ice-cold PBS (ThermoFisher Scientific). The mixture was centrifuged at 600 g for 5 min. The pellet containing glomeruli was frozen in liquid nitrogen for Western blot analysis.
Isolation of primary podocytes.
Glomeruli were isolated from 8-wk-old male FHH and FHH.Add3 transgenic rats, as described above. The pellet was resuspended with RPMI-1640 medium supplemented with 10% FBS and antibiotics plated into a collagen type I-coated plate. After 10 days of incubation at 37°C with 5% CO2 and changing medium every 2 days, the outgrowing cells were trypsinized and filtered through a 40-μm cell strainer and replated for following experiments. This nonenzymatic sieving protocol yielded a high purity of isolated primary podocytes (31), and the cells were validated by >95% positively staining with antibodies against nephrin, podocin, and synaptopodin.
Immunocytochemistry
Transfected conditionally immortalized mouse podocytes or primary podocytes isolated from FHH and FHH.Add3 rats were seeded on autoclaved glass coverslips that were precoated with Cell-Tak Cell and Tissue Adhesive (3.5 µg/cm2, Corning). Podocytes were fixed with prewarmed 3.7% paraformaldehyde and permeabilized 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO). After being blocked with 1% BSA, the cells were stained with Alexa Fluor 488 phalloidin (1:40, A12379, ThermoFisher Scientific) to observe the shape and structure of F-actin and costained with Alexa Fluor 594 deoxyribonuclease I (1:200, D12372, ThermoFisher Scientific) for G-actin or incubated with rabbit anti-Add3 (1:200, sc-25733, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-actin-related protein 2/3 complex subunit 1B (ARPC1B; 1:200, ab227082, Abcam, Cambridge, MA), rabbit anti-podocin (1:100, P0372, Sigma-Aldrich), mouse anti-synaptopodin (SYNPO; 1:50, sc-515842, Santa Cruz Biotechnology), and rabbit anti-nephrin (1:1,000, PA5-20330, ThermoFisher Scientific) followed by Alexa Fluor 488 goat anti-rabbit (A-11070, ThermoFisher Scientific), Alexa Fluor 555 goat anti-rabbit (A-21428, ThermoFisher Scientific), or Alexa Fluor 555 goat anti-mouse (A-21424, ThermoFisher Scientific) secondary antibodies. Nephrin synthetic peptide (1:60, PEP-0450, ThermoFisher Scientific) was used to validate the specificity of the primary antibodies. A drop of antifade mounting medium with DAPI (H-1200, Vector Laboratories, Burlingame, CA) was applied to the slides and coverslipped. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope (Nikon, Melville, NY). ARPC1B, an Arp2/3 complex component (32), and nephrin average intensity and coverage area as well as F/G-actin ratios (of positively stained areas) were measured using NIS-Elements imaging software 4.6 (Nikon). Experiments using mouse podocytes were repeated three times, and triplicate wells were used at each experiment. Primary podocytes were isolated from six to nine rats per strain.
Western Blot Analysis
Conditionally immortalized mouse podocytes and primary rat podocytes were harvested with ice-cold RIPA buffer (Sigma-Aldrich) containing protease and phosphatase inhibitors (PPI; ThermoFisher Scientific), agitated, and incubated for 30 min at 4°C to allow the cells to be completely lysed. Renal cortical tissue and isolated glomeruli were homogenized in lysis buffer (RIPA + PPI) using a ground glass homogenizer followed with a FastPrep-24 homogenizer utilizing bead-beating technology (MP Biomedicals, Santa Ana, CA) as we have described previously (33). Homogenized podocytes, glomeruli, and the renal cortex were then sonicated and centrifuged at 9,000 g for 15 min at 4°C. The supernatants were collected, and protein concentrations in the homogenates were measured using the Bradford method (Bio-Rad Laboratories) as we previously described (15, 34, 35). Renal cortical membrane protein was obtained from resuspension of the pellets with RIPA + PPI after ultracentrifugation of supernatants from 11,000-g centrifugation at 100,000 g for 1 h at 4°C (Sorvall WX 80+ Ultracentrifuge, ThermoFisher Scientific). Membrane protein concentrations were determined. Aliquots of podocytes, glomeruli, renal cortical homogenates, and renal cortical membrane protein were separated by electrophoresis on 4–20% SDS-polyacrylamide gels, transferred to 0.2-μm nitrocellulose membranes using a Trans-Blot Turbo Transfer apparatus (Bio-Rad), and blocked with 5% nonfat milk in Tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h at room temperature. After being washed three times with TBST, membranes were incubated overnight at 4°C with the following primary antibodies: mouse anti-Add3 (1:200, sc-365177, Santa Cruz Biotechnology), rabbit anti-nephrin (1: 2,000, PA5-20330, ThermoFisher Scientific), rabbit anti-podocin (1: 1,000, P0372, Sigma-Aldrich), rabbit anti-ARPC1B (1:1,000, ab227082, Abcam), rabbit anti-integrin-α3 (ITGA3; 1:1,000, PA5-82356, ThermoFisher Scientific), rabbit anti-integrin-β1 (ITGB1; 1:2,000, ab179471, Abcam), rabbit anti-RhoA (1:5,000, ab187027, Abcam), rabbit anti-SYNPO (1:1,000, ab101883, Abcam), mouse anti-Rac1 (1:750, ab33186, Abcam), and rabbit anti-CDC42 (1:10,000, ab187643, Abcam), followed by incubation at room temperature for 1 h with the following horseradish peroxidase-conjugated secondary antibodies: goat anti-rabbit (ab6271, Abcam) or rabbit anti-mouse (ab97046, Abcam). Membranes were then exposed to a SuperSignal West Dura substrate (Thermo Scientific), images of immunoreactive bands were captured, and relative intensities were analyzed using a ChemiDoc Imaging System (Bio-Rad). GAPDH (1: 2,500, no. 2118S, Cell Signaling Technology, Danvers, MA) was used as a loading control. Add3 blocking peptide (1:20, sc-365177P, Santa Cruz Biotechnology) and nephrin synthetic peptide (1:60, PEP-0450, ThermoFisher Scientific) were used to validate the specificity of the primary antibodies.
Immunohistochemistry
Formalin-fixed kidneys were embedded in paraffin, and 3-μm sections were prepared. Paraffin sections were deparaffinized with xylene, followed by rehydration with 100%, 95%, and 70% ethanol, respectively. After permeabilization with 0.1% trypsin at 37°C, the sections were placed in citrate buffer and heated at 98°C for 20 min for antigen retrieval, followed by a rinse with methanol at −20°C for 5 min. Sections were blocked in 10% goat serum (G9023, Sigma-Aldrich) that matched the host species for raising secondary antibodies and then incubated at 4°C overnight with the following primary antibodies: rabbit anti-Add3 (1:200, sc-25733, Santa Cruz Biotechnology), rabbit anti-nephrin (1:500, PA5-20330, ThermoFisher Scientific), rabbit anti-ITGA3 (1:100, PA5-82356, ThermoFisher Scientific), rabbit anti-ITGB1 (1:500, ab179471, Abcam), and mouse anti-SYNPO (1:50, sc-515842, Santa Cruz Biotechnology), followed by incubation at room temperature for 1 h with the following secondary antibodies: Alexa Fluor 488 goat anti-rabbit (A-11070, ThermoFisher Scientific) or Alexa Fluor 555 goat anti-mouse (A-21424, ThermoFisher Scientific). The slides were applied with an antifade mounting medium with DAPI (Vector Laboratories) and coverslipped. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope or a Nikon Eclipse 55i fluorescence microscope equipped with a DS-Fil1 color camera (Nikon). The percentage of ADD3-, nephrin-, ITGA3-, ITGB1-, and SYNPO-positive areas in individual glomeruli was quantified and compared using NIS-Elements imaging software 4.6 (Nikon) as we have previously described (15, 36, 37).
Adhesion Assay
Podocyte adhesion was compared using a modified protocol provided by a Calcein AM Cell Viability Assay Kit (no. 786‐1387, G-Bioscience, St. Louis, MO) and described in a previous report (38). Briefly, differentiated podocytes were trypsinized, resuspended in serum-free RPMI-1640 with 5 µM calcein AM, and incubated at 37°C for 30 min. Cells were resuspended one more time, and 5 × 104 cells/well were seeded in a 96-well plate precoated with collagen type I (0.05 mg/mL, no. 354236, Corning) at 37°C for 1 h to allow cells to adhere. Following incubation, cells were gently washed three times with prewarmed serum-free RPMI-1640 medium to remove nonadherent cells. The attached live cells that hydrolyzed nonfluorescent calcein AM to fluorescent calcein were assessed using a SYNERGY H1 microplate reader (BioTek Instruments, Winooski, VT) at wavelengths of excitation and emission at 485 and 530 nm, respectively. Cell images were also captured using a Lionheart FX Automated Microscope (BioTek).
Migration Assay
Podocyte migration was compared using a modified wound-healing assay (39, 40). Briefly, differentiated podocytes (5 × 105 cells/well) were seeded in collagen type I precoated six-well plate in RPMI-1640 medium supplemented with 10% FBS. A wound was created with a sterilized 200-μL tip after the cells were incubated at 37°C with 5% CO2 for 24 h when reaching a confluent monolayer. Cell debris was removed by washing with serum-free RPMI-1640 medium, and cells were photographed at 0 and 24 h after wounding using a Lionheart FX Automated Microscope (BioTek). Migratory rates were calculated as (A − B)/A × 100% (41) using ImageJ software, where A and B are the open wound areas at 0 and 24 h, respectively.
Transmission Electron Microscopy
Twelve-week-old FHH and FHH.Add3 transgenic rats were euthanized with isoflurane, and the kidneys were removed. Before fixation of the kidney for immunohistochemistry or homogenizing for Western blots, a 1-mm3 block of tissue was cut immediately and immersed in 4% formaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (PB; pH 7.4) overnight at 4°C followed by postfixation with 1% osmium tetroxide in 0.1 M PB. The samples were sent to the UMMC Electron Microscopy division of the Pathology Department to be prepared and imaged for transmission electron microscopy. Twelve images were examined per kidney, and four rats per group were studied.
Statistical Analysis
All data in the text are presented as means ± SE. An unpaired t test was used to compare the significance of differences in corresponding values between two groups. Two-way ANOVA for repeated measurements and the Holm-Sidak test were used to compare the significance of the differences in MAP and proteinuria between and within strains at different ages. P < 0.05 was accepted as statistically significant.
RESULTS
Comparison of MAP and Protein Excretion in Young FHH and FHH.Add3 Transgenic Rats
Baseline MAP was similar in FHH and FHH.Add3 transgenic rats at 12 and 16 wk of age (Fig. 1A). Protein excretion was not different in two strains when they were at 8 wk of age. However, proteinuria (Fig. 1B) was significantly higher in FHH rats at 12 wk (35.30 ± 5.61 mg/day) and 16 wk (126.56 ± 10.68 mg/day) of age compared with FHH.Add3 transgenic rats at the same ages (19.10 ± 1.55 and 39.67 ± 8.51 mg/day, respectively). These results are consistent with our recent reports in different groups of FHH and FHH.Add3 transgenic rats that were continually measured MAP and protein excretion from 12 to 22 wk of age (5).
Figure 1.

Comparison of mean arterial pressure (MAP) and protein excretion in young fawn-hooded hypertensive (FHH) and FHH.Add3 transgenic rats. A: MAP was compared in male FHH and FHH.Add3 transgenic rats at 12 and 16 wk of age. B: proteinuria was compared in FHH and FHH.Add3 transgenic rats at 8, 12, and 16 wk of age. Mean values ± SE are presented; n = 4–9 rats per group. *P < 0.05 from the corresponding values in age-matched FHH rats. †P < 0.05 from the corresponding values in male rats at 12 and 16 wk of age within a strain versus values measured at 8 wk of age. ADD3, γ-adducin.
ADD3 Expression Is Reduced in Podocytes and Diminished in Renal Cortical Membrane Fractions in FHH Rats
We confirmed that ADD3 was expressed in podocytes and tubular cells (Fig. 2A), consistent with previous reports (5, 42, 43). Podocytes could be identified by the morphology and positively express SYNPO. We found that the expression of ADD3 was barely detectable in the glomeruli of FHH rats with a mean fluorescence intensity (Fig. 2B) of 23.61 ± 1.18 artificial units (AU); however, it was significantly enhanced in the glomeruli of FHH.Add3 rats (60.52 ± 2.16 AU). Similarly, the percentage of the ADD3-positive area (Fig. 2C) was diminished in FHH (37.61 ± 0.66%) compared with FHH.Add3 rats (61.37 ± 0.85%). The expression of ADD3 in renal cortical membrane fractions was only found in FHH.Add3 rats but not in FHH rats (Fig. 2D).
Figure 2.

γ-Adducin (ADD3) expression is reduced in podocytes and diminished in renal cortical membrane fractions in fawn-hooded hypertensive (FHH) rats. Shown is a comparison of ADD3 expression and distribution in male FHH and FHH.Add3 transgenic rats. A: representative images of ADD3 expression in the renal cortex. Arrows denote ADD3-positive-stained podocytes that express synaptopodin (SYNPO). B: quantitation of ADD3 mean fluorescence intensity per glomerulus. C: quantitation of ADD3-positive area per glomerulus. D: representative image of ADD3 expression in renal cortical membrane fractions by Western blot. M, molecular mass. Six sections per rat and 30 glomeruli per section were examined. Images were captured using a fluorescence microscope with ×20 objective (total magnification of ×850). Mean values ± SE are presented; n = 6 male rats per group. *P < 0.05 from the corresponding values in age-matched FHH rats.
Validation of Primary Podocytes Isolated From FHH and FHH.Add3 Transgenic Rats
Primary podocytes were isolated from 8-wk-old FHH and FHH.Add3 transgenic rats according to protocols described by Mundel et al. (28) and Krtil et al. (31). Freshly isolated glomeruli on day 0 (D0) were placed onto a collagen type I-coated plate. Outgrowth of cobblestone-like undifferentiated podocytes occurred from the decapsulated glomeruli, and they became fully differentiated arborized cells after 10 days in culture (Fig. 3A). Primary podocytes were further confirmed by immunocytochemistry demonstrating that they positively express nephrin, SYNPO, and podocin (Fig. 3B). The expression of ADD3 was markedly reduced in primary podocytes of FHH relative to FHH.Add3 transgenic rats, as detected by immunostaining and Western blots (Fig. 3C), consistent with its expression pattern in primary VSMCs isolated from these two strains of rats that we have previously reported (5).
Figure 3.

Validation of primary podocytes isolated from fawn-hooded hypertensive (FHH) and FHH.Add3 transgenic rats. A: representative images of freshly isolated glomeruli on day 0 (D0), cobblestone-like undifferentiated podocytes outgrowth from the decapsulated glomerulus on D5, and fully differentiated arborized podocytes on D10. Images were captured using a Lionheart FX Automated Microscope with ×10 objective (total magnification of ×350). B: representative images of nephrin-, synaptopodin (SYNPO)-, and podocin-positive primary podocytes by immunocytochemistry. C, top: representative images of γ-adducin (ADD3) expression and distribution in primary podocytes. Podocytes were isolated from 6–9 male rats in each strain. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×60 oil immersion objective and a ×3 digital zoom (total magnification of ×3,960). C, bottom: representative images and quantitation of ADD3 expression in podocytes by Western blot. BP, blocking peptide; M, molecular mass. Mean values ± SE are presented. *P < 0.05 from the corresponding values in age-matched FHH rats.
Validation of Conditionally Immortalized Mouse Podocytes and Efficiency of Knockdown of Expression of ADD3 With Add3 DsiRNA
Conditionally immortalized mouse podocytes were validated that they express nephrin, SYNPO, and podocin by immunocytochemistry (Fig. 4A). Figure 4B shows the efficiency of the knockdown of expression of ADD3 in conditionally immortalized mouse podocytes 36 h posttransfection with Add3 DsiRNA. ADD3-positive cells were 70.78 ± 1.41% less in Add3 DsiRNA-transfected cells compared with NC1 negative control cells. High efficiency of knockdown of ADD3 was also confirmed by Western blot analysis (Fig. 4C).
Figure 4.

Validation of conditionally immortalized mouse podocytes and the efficiency of knockdown of the expression of γ-adducin (ADD3) with Add3 Dicer-substrate short interfering RNA (DsiRNA). A: representative images of nephrin-, synaptopodin (SYNPO)-, and podocin-positive conditionally immortalized mouse podocytes by immunocytochemistry. B and C: representative images and quantitation of ADD3 immunostaining (B) and Western blots (C) of the efficiency of knockdown of the expression of ADD3 in conditionally immortalized mouse podocytes 36 h posttransfection with Add3 DsiRNA. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×20 objective (total magnification of ×440). Experiments were repeated 4–5 times in triplicate in each experiment. Mean values ± SE are presented. *P < 0.05 from the corresponding values in NC1 negative controls (Ctrl).
Reduced ADD3 Expression Enhances F-Actin Nets Formation in Podocytes
As shown in Fig. 5A, the actin cytoskeleton displayed branched irregular polygonal shapes with punctate and branched F-actin networks (F-actin nets) in the cell bodies of mouse podocytes treated with Add3 DsiRNA, which replaced unbranched stress fibers. The F-actin nets that are typically localized in the lamellipodia in podocytes under physiological conditions (44) were observed in the NC1 negative control cells that expressed ADD3, which also displayed parallel F-actin stress fibers in the same localization of the cells. On the other hand, the distribution of G-actin was reduced, and the F/G actin ratio was enhanced in mouse podocytes treated with Add3 DsiRNA. Similarly, we also found abundant branched F-actin net formation within the cell bodies in FHH podocytes, but the morphology of the stress fibers remained unbranched and parallel in podocytes of FHH.Add3 rats (Fig. 5B). The F/G-actin ratio was also enhanced in FHH podocytes (8.49 ± 0.42) compared with those from FHH.Add3 rats (3.05 ± 0.12). We found that expression of ARPC1B was increased throughout FHH podocytes and the Arp2/3 complex was mainly localized in the lamellipodia of podocytes of FHH.Add3 rats (Fig. 5C). High expression of ARPCIB was also confirmed by Western blot (Fig. 5C).
Figure 5.

Downregulation of γ-adducin (ADD3) enhances F-actin net formation in podocytes. A: representative images (left) and quantitation (right) of immunostaining for comparison of the expression and distribution of F/G-actin and the F/G-actin ratio in Add3 Dicer-substrate short interfering RNA (DsiRNA) and NC1 scrambled siRNA transfected immortalized mouse podocytes B: representative images (left) and quantitation (right) of immunostaining for comparison of the expression and distribution of F/G-actin and F/G-actin ratio in primary podocytes isolated from FHH and FHH.Add3 transgenic rats. C: representative images of immunostaining for comparison of the expression and distribution of ARPC1B (left) and quantitation with Western blots (right) in primary podocytes isolated from male FHH and FHH.Add3 transgenic rats. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×60 oil immersion objective and a ×3 digital zoom (total magnification of ×3,960). Experiments using conditionally immortalized mouse podocytes were repeated 3 times in triplicate in each experiment. Podocytes were isolated from 6–9 male rats in each strain. ARPC1B, actin-related protein 2/3 complex subunit 1B. Mean values ± SE are presented. *P < 0.05 from the corresponding values in age-matched FHH rats or NC1 negative controls (Ctrl).
Reduced ADD3 Expression Alters the Rho Family of Small GTPases in Podocytes in FHH Rats
As shown in Fig. 6A, the percentage of the fluorescent SYNPO-positive area was significantly lower in the glomeruli obtained from FHH rats (33.63 ± 0.64%) compared with FHH.Add3 rats (66.92 ± 0.61%). SYNPO and RhoA protein expression was higher in the renal cortex (by 1.34- and 1.74-fold, respectively; Fig. 6B) as well as in primary podocytes (by 1.61- and 1.84-fold, respectively; Fig. 6C) in FHH.Add3 than FHH rats. In contrast, Rac1 and CDC42 protein expression was lower in the renal cortex by 0.63- and 0.62-fold, respectively (Fig. 6D) in FHH.Add3 than FHH rats. Rac1 and CDC42 protein expression was also lower in primary podocytes of FHH.Add3 than FHH rats (Fig. 6E).
Figure 6.

Reduced γ-adducin (ADD3) expression alters the Rho family of small GTPases levels in podocytes in fawn-hooded hypertensive (FHH) rats. A: representative images (left) and quantitation (right) of synaptopodin (SYNPO) immunostaining in the glomeruli of male FHH and FHH.Add3 rats. Six sections per rat and 30 glomeruli per section were examined; n = 6 rats per group. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×20 objective and ×1 digital zoom (left, total magnification of ×440) and ×4.5 digital zoom (right, total magnification of ×1,980). B: representative images (left) and quantitation (right) of SYNPO and RhoA expression in the renal cortex of male FHH and FHH.Add3 rats. C: representative images (left) and quantitation (right) of SYNPO and RhoA expression in the primary podocytes isolated from male FHH and FHH.Add3 rats. D: representative images (left) and quantitation (right) of Rac1 and CDC42 expression in the renal cortex of male FHH and FHH.Add3 rats. E: representative images (left) and quantitation (right) of Rac1 and CDC42 expression in primary podocytes isolated from male FHH and FHH.Add3 rats. Mean values ± SE are presented; n = 6–9 rats in each strain. *P < 0.05 from the corresponding values in age-matched FHH rats.
Reduced ADD3 Expression Attenuates Slit Diaphragm Protein Expression in FHH Rats
The nephrin antibody was validated by Western blot analysis in the renal cortex of FHH and FHH.Add3 rats (Fig. 7A) and by immunostaining using conditionally immortalized mouse podocytes (Fig. 7B) with and without a blocking peptide (BP). We found that the percentage of the nephrin-positive area in the glomeruli (Fig. 7C) and the mean intensity of nephrin in primary podocytes (Fig. 7D) were significantly lower in FHH rats compared with FHH.Add3 rats (1.93- and 1.78-fold, respectively). Additionally, nephrin and podocin protein expression were higher by 1.53- and 1.46-fold, respectively, in the renal cortex (Fig. 7E) of FHH.Add3 than FHH rats.
Figure 7.

Reduced γ-adducin (ADD3) expression attenuates slit diaphragm protein expression in fawn-hooded hypertensive (FHH) rats. Validation of the nephrin antibody by Western blot in the renal cortex of male FHH and FHH.Add3 rats is shown. A and B: immunostaining using conditionally immortalized mouse podocytes (A) with and without the blocking peptide (BP; B). M, molecular mass. C: representative images (left) and quantitation (right) of nephrin immunostaining in the glomeruli of male FHH compared with FHH.Add3 rats. Six sections per rat and 30 glomeruli per section were examined; n = 6 rats per group. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×20 objective and ×1 digital zoom (left, total magnification of ×440) and ×4.5 digital zoom (right, total magnification of ×1,980). D: representative images (left) and quantitation (right) of nephrin immunostaining in primary podocytes of FHH compared with FHH.Add3 rats. Images were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×60 oil immersion objective and a ×3 digital zoom (total magnification of ×3,960). E: representative images (left) and quantitation (right) of nephrin and podocin expression by Western blot in the renal cortex of male FHH and FHH.Add3 rats. Mean values ± SE are presented. *P < 0.05 from the corresponding values in age-matched FHH rats.
Reduced ADD3 Expression Affects Focal Adhesion Integrins in FHH Rats
As shown in Fig. 8, A and B, the percentage of ITGA3- and ITGB1-positive areas was significantly lower in the glomeruli of FHH rats (26.50 ± 0.62% and 29.46 ± 0.67%, respectively) compared with FHH.Add3 rats (64.86 ± 0.70% and 57.32 ± 0.66%, respectively). Protein expression of ITGA3 and ITGB1 was 1.34- and 1.28-fold higher in the glomeruli obtained from FHH.Add3 than FHH rats (Fig. 8C).
Figure 8.

Reduced γ-adducin (ADD3) expression affects focal adhesion integrins in fawn-hooded hypertensive (FHH) rats. A: representative images (left) and quantitation (right) of integrin-α3 (ITGA30) immunostaining in the glomeruli obtained from male FHH compared with FHH.Add3 rats. Six sections per rat and 30 glomeruli per section were examined; n = 6 rats per group. B: representative images (left) and quantitation (right) of integrin-α3 (ITGB1) expression in the renal cortex of male FHH and FHH.Add3 rats. Images in A and B were captured using a Nikon C2 laser scanning confocal on an Eclipse Ti2 inverted microscope with ×20 objective and ×1 digital zoom (left, total magnification of ×440) and ×4.5 digital zoom (right, total magnification of ×1,980). C: representative images (left) and quantitation (right) of ITGA3 and ITGB1 expression in the glomeruli of male FHH and FHH.Add3 rats. Mean values ± SE are presented. *P < 0.05 from the corresponding values in age-matched FHH rats.
Reduced ADD3 Expression Diminishes Podocyte Adhesion and Enhances Podocyte Migration
Cell adhesion was decreased in the podocyte of FHH compared with FHH.Add3 rats as detected by mean fluorescence intensities of 919.67 ± 29.44 (100%) and 1,369.08 ± 38.98 (148.87 ± 4.27%), respectively (Fig. 9A, left). Cell adhesion was also decreased in mouse podocyte transfected with Add3 DsiRNA versus NC1 control, with mean fluorescence intensities of 2,157.75 ± 89.27 (100%) and 2,978.83 ± 101.16 (138.05 ± 4.69%), respectively (Fig. 9A, middle and right). In contrast, the migration rate of primary podocytes was significantly higher in FHH (65.98 ± 1.19%) than FHH.Add3 rats (42.54 ± 1.38%; Fig. 9B).
Figure 9.

Reduced γ-adducin (ADD3) expression is associated with diminished podocyte adhesion, enhanced podocyte migration, and podocyte foot process effacement (FPE) in fawn-hooded hypertensive (FHH) rats. A: podocyte adhesion was decreased in male FHH compared to FHH.Add3 rats (left) and in mouse podocyte transfected with Add3 Dicer-substrate short interfering RNA (DsiRNA) vs. NC1 control (Ctrl; middle). Representative images of fluorescent adhesive cells are presented on the right. B: representative images (left) and quantitation (right) demonstrate that migration rate of primary podocytes was significantly higher in male FHH than FHH.Add3 rats. C: representative images show that knockin of wild-type Add3 improved podocyte ultrastructure and rescued FPE observed in age-matched FHH rats. Black arrows denote the area of FPE in FHH rats. Blue arrows denote intact slit diaphragms in FHH.Add3 rats. Magnification: ×32,100. Experiments using conditionally immortalized mouse podocytes and primary podocytes were repeated 3–4 times in triplicate in each experiment. Podocytes were isolated from 4–6 male rats in each strain. Mean values ± SE are presented. *P < 0.05 from the corresponding values in age-matched FHH rats or NC1 negative controls.
Reduced ADD3 Expression Is Associated With FPE in FHH Rats at 12 wk of Age
Twelve-week-old FHH rats exhibited enhanced FPE observed with transmission electron microscopy, whereas knockin of wild-type Add3 improved podocyte ultrastructure and rescued FPE (Fig. 9C).
DISCUSSION
Diabetes and hypertension, especially when superposed with aging, raise the risk for the development of CKD involving several potential underlying mechanisms (37, 45–50). However, only half of these patients develop CKD, indicating there is a strong genetic component. The genes and pathways involved are unknown, and there is an urgent need to identify these factors to develop new therapies to prevent CKD.
Podocytes are specialized and terminally differentiated visceral epithelial cells that form a crucial component of the glomerular filtration barrier. Podocytes have a complex cellular architecture consisting of the cell body, primary processes (microtubule-driven membrane extensions), and foot processes (FPs; the actin-driven membrane extensions) (51). The formation of interdigitated FPs enwraps the glomerular capillaries to oppose hydrostatic pressure in the glomerular capillary, which is the natural driving force behind macromolecular filtration (52). Actin proteins form the main cytoskeleton of the FPs, which is determined by the integrity of a slit diaphragm and focal adhesions anchoring the podocyte to its environment (53). Actin is the most abundant protein expressed in eukaryotic cells. It exists as a globular monomer (G-actin) and a filamentous polymer (F-actin). F-actin is a polar polymer and an essential element of the actin filament that undergoes rapid polymerization and depolymerization, which controls the stability of the actin cytoskeleton and regulates cell contractility, motility, endocytosis, intracellular trafficking, and maintenance of cell and subcellular organelle morphology (54). The Arp2/3 complex initials the branches of actin organization found at the leading edge of motile cells and at the site of clathrin-mediated endocytosis (54, 55). Actin branch elongation is limited by interaction with capping proteins that block growth of the barbed ends of F-actin filament (56). In podocytes (Fig. 10A), unbranched actin stress fibers in the cell body provide pulling forces and connect the cell cytoskeleton to the extracellular matrix via focal adhesion sites. Branched and cross-linked F-actin nets make up the lamellipodia and protrude actin structure to engine cell movement by pushing the cell membrane. Actin at the cell cortex that coats the plasma membrane at the back and sides of the cell is essential for slit diaphragm to maintain paracellular integrity (57). Physiological FPs are well balanced by the pulling and pushing forces of unbranched actin stress fibers in the cell body and branched F-actin nets in the lamellipodium of podocyte (57). Dysregulation of the actin cytoskeleton leads to the retraction of FPs and FPE, damage of the slit diaphragm, and attenuation of focal adhesions (cell-matrix) resulting in disruption of podocyte barrier function (44, 58–60). Reduced ADD3 expression, such as podocytes of FHH rats, enhances formation of F-actin nets in the cell body that replaces unbranched stress fibers that leads to an imbalance of pushing/pulling forces and promotes podocyte FPE (Fig. 10B).
Figure 10.

Schematic diagram illustrating alteration of actin-based structure in podocytes in fawn-hooded hypertensive (FHH) rats. A: physiological foot processes are well balanced by the pulling and pushing forces of unbranched actin stress fibers in the cell body and branched F-actin nets in the lamellipodium of podocytes. B: reduced γ-adducin (ADD3) expression in FHH rats enhances the formation of branched F-actin nets in the cell body, which replaces unbranched stress fibers and leads to an imbalance of pushing/pulling forces, and promotes podocyte foot process effacement. Arp2/3, actin-related protein 2/3 complex.
Multiple factors have been suggested directly or indirectly contribute to diminished podocyte and glomerular function and the development of CKD, including impaired renal hemodynamics, renal susceptibility genes, and environmental stresses. Impaired renal hemodynamics results in hyperfiltration and/or increased transmission of pressure to the glomerulus and plays a vital role in the development of hypertensive and diabetic nephropathy (61, 62). Glomerular hyperfiltration promotes vascular pole-associated glomerular injury in a sequence of enlarged afferent arterioles, dilated primary branches of an afferent arteriole, formation of tuft adhesions (dilated primary branches of an afferent arteriole apposes to Bowman’s capsule), and pseudocyst formation of associated podocytes/retraction of FPs. These events promote the effacement of podocyte FPs, loss of the slit diaphragm, and reduction in focal adhesions. These detrimental effects result in podocyte detachment from the glomerular basement membrane (10), which enhances albumin permeability of the glomerulus to develop proteinuria and CKD (62). Unlike the indirect detrimental effects on podocytes induced by glomerular hyperfiltration, mutations in genes associated with CKD and other insults (toxic, infectious, etc.) could directly contribute to podocyte damage (57). Additionally, human genetic studies have also demonstrated that mutations in several genes regulating the actin structure and function, such as α-actinin-4, actin-binding (ACTN4) (63, 64), heterodimer with ADD3, actin-binding (ADD1) (18), heterodimer with ADD1, actin-binding (ADD2) (18), inverted formin-2, actin assembly regulator (INF2) (17, 65), CD2-associated protein, cortical actin assembly activator (CD2AP) (21), and myosin 1e, actin-dependent molecular motor (Myo1E) (23, 24), are associated with proteinuria and glomerular disease.
We recently reported that an inactivating recessive mutation in Add3 in FHH rats, a genetic hypertension and CKD model, contributes to reduced ADD3 membrane expression, disrupted actin cytoskeleton in VSMCs, diminished vascular reactivity in both cerebral and renal arterioles, and impaired renal hemodynamics (5, 66). Indeed, impaired renal hemodynamics leads to glomerular and podocyte injury in FHH rats at 25 wk of age (10); however, this strain develops some evidence of focal and segmental glomerulosclerosis as early as 8 wk of age (67–69) long before the development of hypertension. In a recent report, we found that mean blood pressure was similar at ∼115 mmHg in 12-wk old FHH and FHH.Add3 rats, whereas protein excretion and Palb were significantly enhanced in FHH than FHH.Add3 transgenic rats at this age (5). Considering that globally knocking in the wild-type ADD3 reversed not only impairments of vascular function but also glomerular function, it is possible that wild-type ADD3 rescued glomerular function in FHH.Add3 rats may represent synergistic events of correction of downstream pathological consequences from elevated glomerular capillary pressure in combination with a direct effect of rearrangement of the actin cytoskeleton in podocytes.
Add3 belongs to the adducin gene family, which plays an essential role in actin cytoskeleton arrangements (5, 43). The tetrameric ADD3 proteins interact with actin, spectrin, and bind the plasma membrane (Fig. 11A) (43, 70, 71). ADD3 caps the fast-growing end of F-actin and prevents the addition of G-actin for polymerization (72). ADD3-actin complexes form contractile units with myosin and are regulated by myosin light chain (MLC) regulated protein (Fig. 11B). A deficiency in ADD3 promotes actin polymerization and dissociation of actin-spectrin-adducin-myosin complexes from the membrane (Fig. 11C). These events increase activation of the actin-nucleating Arp2/3 protein and new branch formation off existing actin filaments. Reduced actin cytoskeleton stabilization by increasing or decreasing F/G-actin ratio diminishes MLC phosphorylation causing impaired vasoconstriction in VSMCs (73–75). Formation of more branched F-actin nets occurs under stress with actin cross-linking proteins involving the loss of actin cytoskeleton stability and MLC phosphorylation (73–75).
Figure 11.

schematic diagram of the relationship of γ-adducin (ADD3) and the actin cytoskeleton. A: adducin interacts with actin and spectrin binding to the membrane and caps F-actin elongation. B: adducin-actin complex forms constrictive units with myosin and is regulated by myosin light chain regulatory protein. C: Loss of adducin function leads to dissociation of the protein complex of ADD3/F-actin/myosin from the membrane, activation of the actin-nucleating actin-related protein 2/3 (Arp2/3) complex, F-actin polymerization, and F-actin net formation.
The current study was designed to extend our previous work demonstrating that male FHH rats exhibited more impaired renal hemodynamics than FHH.Add3 rats; we used male rats and first confirmed that ADD3 is expressed in podocytes and tubular cells to a lower extent in FHH than FHH.Add3 transgenic rats. These results are in line with previous reports showing that ADD3 is expressed in blood vessels, tubules and podocytes in the kidney (8, 42, 43). Similar to our previous findings in VSMCs, there was reduced ADD3 expression in the membrane in primary podocytes isolated from FHH in comparison with FHH.Add3 transgenic rats (5). We found that F-actin nets, which are typically localized in the lamellipodia, replaced unbranched stress fibers in conditionally immortalized mouse podocytes transfected with Add3 DsiRNA and primary podocytes isolated from FHH rats; both have reduced ADD3 membrane expression. As an actin capping protein, loss of ADD3 releases its anchor at the ends of F-actin, which leads to dissociation of actin-spectrin-adducin-myosin complexes from the cell membrane and actin nucleation and paradoxical polymerization, which was detected as an increased F/G-actin ratio and increased numbers of the Arp2/3 complex in our study. The Arp2/3 proteins initiate the growth of branched actin filaments, which plays a crucial role in cell movement at the plasma membrane (54). Under physiological condition, the Arp2/3 complex mainly localizes in the lamellipodia of podocytes (54, 59). In the present study, we found that ARPC1B, a component of Arp2/3 complex, was expressed throughout FHH podocytes and replaced most of the stress fibers in the cell bodies (Fig. 5, B and C, and Fig. 10B).
Physiological FPs in podocytes are well balanced by the pulling and pushing forces of unbranched actin stress fibers in the cell body and branched F-actin nets in the lamellipodium of podocyte (57). RhoA, Rac1, and CDC42 are small GTPases that belong to the Rho family and are abundantly expressed in podocytes. Adducin is regulated by protein kinases A and C and tyrosine and Rho kinases (43). Rho kinase, a downstream effector protein of the small GTPase family, phosphorylates ADD1 and plays a vital role in the regulation of cell motility (76, 77). Activation of Rho kinases by RhoA promotes actin: myosin contraction and stress fiber formation (60, 78). Conversely, the subclasses Rac1 and CDC42 promote the formation of lamellipodia (60). The podocyte adapter protein SYNPO induces stress fibers by stabilizing RhoA and suppresses filopodia by disrupting CDC42-IRSp53-Mena signaling complexes (78). Importantly, previous studies showed that either too much or too little podocyte motility results in FPE (79–82). In our case, we found podocyte migration was enhanced in FHH than FHH.Add3 rats. Prominent stress fibers in cultured podocytes reflect intact FPs in vivo, whereas the loss of actin stress fibers and reorganization into cortical actin in vitro equals FP effacement in vivo (83). We found that FHH rats exhibited reduced SYNPO and RhoA but enhanced Rac1 and CDC42 expression in the renal cortex, glomeruli and podocytes. These results are in line with the actin morphology and localization in podocytes of FHH rats in the present study.
The actin cytoskeletal dynamics are essential for an efficient FP formation to maintain podocyte function and the glomerular filtration barrier. Podocyte FPs are actin-driven membrane extensions that comprise two distinct hubs: an intercellular interaction structure slit diaphragm and a cell and glomerular basement membrane interaction structure focal adhesion. Slit diaphragms, localized at the podocyte cell cortex, are composed of cross-linked unbranched actin filaments (57). Nephrin spans the slit diaphragm across adjacent FPs with the help of podocin anchoring nephrin to the plasma membrane. Nephrin interacts with the COOH terminus of podocin. Nephrin is further anchored to the actin cytoskeleton by the scaffolding protein CD2-associated protein. As a transmembrane protein, nephrin not only physically links to the actin cytoskeleton but also functionally modulates actin dynamics, involving cofilin-related actin polymerization process and the regulation of lamellipodia formation (84, 85). Integrins play an essential role in the formation and integrity of focal adhesions in podocytes. They interact with the actin-associated proteins, such as paxillin, talin, and α-actinin, as well as other cell adhesion molecules, such as cadherins, to provide physical reinforcement for the podocytes to oppose mechanical stress, transduce cell signals, and maintain cell motility (57). We previously found that nephrin was lost in DOCA/salt-hypertensive FHH versus FHH.Add3 rats (5). The present study further found that expression of nephrin at the cell cortex (slit diaphragm) was reduced in primary podocytes isolated from FHH rats compared with FHH.Add3 rats. Moreover, the expression of nephrin and podocin was reduced in FHH rats starting from 12 wk of age when they first appeared to have significant enhanced proteinuria compared with age-matched FHH.Add3 rats (5). Additionally, at 12 wk of age, focal adhesion proteins ITGA3 and ITGB1 were reduced and podocyte adhesion was diminished in FHH rats, and this genetic hypertension and CKD strain exhibited FPE, consistent with previous findings in other model systems (82, 86).
In summary, the current study demonstrated that reduced membrane expression of ADD3 in podocytes rearranges the localization and morphology of the actin cytoskeleton that replaces unbranched stress fibers with branched F-actin nets. This alternation is associated with attenuated slit diaphragm and focal adhesion protein expression that enhances cell migration, reduces cell adhesion, and promotes podocyte FPE. However, in the face of our previous work demonstrating that downregulation of ADD3 impaired renal hemodynamics in male FHH and global Add3 knockout rats (5, 6), we cannot determine whether the primary or initial factor that causes renal injury in FHH rats is vascular (VSMCs) or glomerular (podocytes) injury. Moreover, whether there are sex differences in this regard needs to be further studied. There is no change in blood pressure and protein excretion in FHH versus global Add3 transgenic and global Add3 knockout versus wild-type rats at 8 wk of age, but the FHH and Add3 knockout rats already exhibit impaired myogenic responses of isolated afferent arterioles (5) and glomerulosclerosis (67–69). These results imply that the glomerular injury in FHH rats is related to a synergistic interaction of vascular dysfunction with a direct effect of actin disruption on podocyte function. Additional work using tissue-specific Add3 genetically modified animal models is definitely needed to sort out this point. Nevertheless, our results provide new insight into why individuals who have genetic defects in actin cytoskeleton-related genes, such as Add3, are more susceptible to the development of CKD, especially with aging and hypertension.
GRANTS
This work was supported by National Institutes of Health Grants AG 050049, AG 057842, P20GM 104357, and HL 138685 and American Heart Association Grants 16GRNT31200036 and 20PRE35210043.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.F. conceived and designed research; W.G., Y.L., L.F., B.Z., J.R.J., S.W., H.Z., X.F., and B.V.N. performed experiments; W.G., Y.L., R.J.R., and F.F. analyzed data; W.G., Y.L., R.J.R., and F.F. interpreted results of experiments; W.G., Y.L., L.F, and F.F. prepared figures; W.G., Y.L., and F.F. drafted manuscript; W.G., Y.L., L.F., S.W., H.Z., X.F., B.V.N., T.Z., R.J.R., and F.F. edited and revised manuscript; W.G., Y.L., L.F., B.Z., J.R.J., S.W., H.Z., X.F., B.V.N., T.Z., R.J.R., and F.F. approved final version of manuscript.
REFERENCES
- 1.Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, Hobbs FD. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS One 11: e0158765, 2016. doi: 10.1371/journal.pone.0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins AJ, Foley RN, Herzog C, Chavers B, Gilbertson D, Herzog C, , et al. US Renal Data System 2012 Annual Data Report. Am J Kidney Dis 61: A7e1-476, 2013. doi: 10.1053/j.ajkd.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 3.Saran R, Robinson B, Abbott KC, Bragg-Gresham J, Chen X, Gipson D, Gu H, Hirth RA, Hutton D, Jin Y, Kapke A, Kurtz V, Li Y, McCullough K, Modi Z, Morgenstern H, Mukhopadhyay P, Pearson J, Pisoni R, Repeck K, Schaubel DE, Shamraj R, Steffick D, Turf M, Woodside KJ, Xiang J, Yin M, Zhang X, Shahinian V. US Renal Data System 2019 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis 75: A6–A7, 2020. doi: 10.1053/j.ajkd.2019.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Burke M, Pabbidi M, Fan F, Ge Y, Liu R, Williams JM, Sarkis A, Lazar J, Jacob HJ, Roman RJ. Genetic basis of the impaired renal myogenic response in FHH rats. Am J Physiol Renal Physiol 304: F565–F577, 2013. doi: 10.1152/ajprenal.00404.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan F, Geurts AM, Pabbidi MR, Ge Y, Zhang C, Wang S, Liu Y, Gao W, Guo Y, Li L, He X, Lv W, Muroya Y, Hirata T, Prokop J, Booz GW, Jacob HJ, Roman RJ. A mutation in gamma-adducin impairs autoregulation of renal blood flow and promotes the development of kidney disease. J Am Soc Nephrol 31: 687–700, 2020. doi: 10.1681/ASN.2019080784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan L, Gao W, Nguyen BV, Jefferson JR, Liu Y, Fan F, Roman RJ. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am J Physiol Renal Physiol 319: F624–F635, 2020. doi: 10.1152/ajprenal.00239.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simons JL, Provoost AP, De Keijzer MH, Anderson S, Rennke HG, Brenner BM. Pathogenesis of glomerular injury in the fawn-hooded rat: effect of unilateral nephrectomy. J Am Soc Nephrol 4: 1362–1370, 1993. [DOI] [PubMed] [Google Scholar]
- 8.Fan F, Pabbidi MR, Ge Y, Li L, Wang S, Mims PN, Roman RJ. Knockdown of Add3 impairs the myogenic response of renal afferent arterioles and middle cerebral arteries. Am J Physiol Renal Physiol 312: F971–F981, 2017. doi: 10.1152/ajprenal.00529.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan F, Chen CC, Zhang J, Schreck CM, Roman EA, Williams JM, Hirata T, Sharma M, Beard DA, Savin VJ, Roman RJ. Fluorescence dilution technique for measurement of albumin reflection coefficient in isolated glomeruli. Am J Physiol Renal Physiol 309: F1049–1059, 2015. doi: 10.1152/ajprenal.00311.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kriz W, Hosser H, Hahnel B, Simons JL, Provoost AP. Development of vascular pole-associated glomerulosclerosis in the Fawn-hooded rat. J Am Soc Nephrol 9: 381–396, 1998. [DOI] [PubMed] [Google Scholar]
- 11.Fan F, Ge Y, Lv W, Elliott MR, Muroya Y, Hirata T, Booz GW, Roman RJ. Molecular mechanisms and cell signaling of 20-hydroxyeicosatetraenoic acid in vascular pathophysiology. Front Biosci 21: 1427–1463, 2016. doi: 10.2741/4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan F, Muroya Y, Roman RJ. Cytochrome P450 eicosanoids in hypertension and renal disease. Curr Opin Nephrol Hypertens 24: 37–46, 2015. doi: 10.1097/MNH.0000000000000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan F, Roman RJ. Effect of cytochrome P450 metabolites of arachidonic acid in nephrology. J Am Soc Nephrol 28: 2845–2855, 2017. doi: 10.1681/ASN.2017030252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge Y, Fan F, Didion SP, Roman RJ. Impaired myogenic response of the afferent arteriole contributes to the increased susceptibility to renal disease in Milan normotensive rats. Physiol Rep 5: e13089, 2017. doi: 10.14814/phy2.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang C, He X, Murphy SR, Zhang H, Wang S, Ge Y, Gao W, Williams JM, Geurts AM, Roman RJ, Fan F. Knockout of dual-specificity protein phosphatase 5 protects against hypertension-induced renal injury. J Pharmacol Exp Ther 370: 206–217, 2019. doi: 10.1124/jpet.119.258954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Zhang C, Liu Y, Gao W, Wang S, Fang X, Guo Y, Li M, Liu R, Roman RJ, Sun P, Fan F. Influence of dual-specificity protein phosphatase 5 on mechanical properties of rat cerebral and renal arterioles. Physiol Rep 8: e14345, 2020. doi: 10.14814/phy2.14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42: 72–76, 2010. [Erratum in Nat Genet 42: 361, 2010]. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrandi M, Cusi D, Molinari I, Del Vecchio L, Barlassina C, Rastaldi MP, Schena FP, Macciardi F, Marcantoni C, Roccatello D, Peters LL, Armelloni S, Min L, Giardino L, Mattinzoli D, Camisasca C, Palazzo F, Manunta P, Ferrari P, Bianchi G. alpha- and beta-Adducin polymorphisms affect podocyte proteins and proteinuria in rodents and decline of renal function in human IgA nephropathy. J Mol Med 88: 203–217, 2010. doi: 10.1007/s00109-009-0549-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrandi M, Molinari I, Rastaldi MP, Ferrari P, Bianchi G, Manunta P. Rostafuroxin protects from podocyte injury and proteinuria induced by adducin genetic variants and ouabain. J Pharmacol Exp Ther 351: 278–287, 2014. doi: 10.1124/jpet.114.217133. [DOI] [PubMed] [Google Scholar]
- 20.Ferrandi M, Molinari I, Torielli L, Padoani G, Salardi S, Rastaldi MP, Ferrari P, Bianchi G. Adducin- and ouabain-related gene variants predict the antihypertensive activity of rostafuroxin, part 1: experimental studies. Sci Transl Med 2: 59ra86, 2010. doi: 10.1126/scitranslmed.3001815. [DOI] [PubMed] [Google Scholar]
- 21.Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER, Shaw AS. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science 300: 1298–1300, 2003. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 22.Lanzani C, Citterio L, Glorioso N, Manunta P, Tripodi G, Salvi E, Carpini SD, Ferrandi M, Messaggio E, Staessen JA, Cusi D, Macciardi F, Argiolas G, Valentini G, Ferrari P, Bianchi G. Adducin- and ouabain-related gene variants predict the antihypertensive activity of rostafuroxin, part 2: clinical studies. Sci Transl Med 2: 59ra87, 2010. doi: 10.1126/scitranslmed.3001814. [DOI] [PubMed] [Google Scholar]
- 23.Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M, Bettoni S, Morigi M, Delledonne M, Pecoraro C, Abbate I, Capobianchi MR, Hildebrandt F, Otto E, Schaefer F, Macciardi F, Ozaltin F, Emre S, Ibsirlioglu T, Benigni A, Remuzzi G, Noris M, PodoNet C, PodoNet Consortium. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med 365: 295–306, 2011. doi: 10.1056/NEJMoa1101273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanna-Cherchi S, Burgess KE, Nees SN, Caridi G, Weng PL, Dagnino M, Bodria M, Carrea A, Allegretta MA, Kim HR, Perry BJ, Gigante M, Clark LN, Kisselev S, Cusi D, Gesualdo L, Allegri L, Scolari F, D'Agati V, Shapiro LS, Pecoraro C, Palomero T, Ghiggeri GM, Gharavi AG. Exome sequencing identified MYO1E and NEIL1 as candidate genes for human autosomal recessive steroid-resistant nephrotic syndrome. Kidney Int 80: 389–396, 2011. doi: 10.1038/ki.2011.148. [DOI] [PubMed] [Google Scholar]
- 25.Santin S, Garcia-Maset R, Ruiz P, Gimenez I, Zamora I, Pena A, Madrid A, Camacho JA, Fraga G, Sanchez-Moreno A, Cobo MA, Bernis C, Ortiz A, de Pablos AL, Pintos G, Justa ML, Hidalgo-Barquero E, Fernandez-Llama P, Ballarin J, Ars E, Torra R, FSGS Spanish Study Group. Nephrin mutations cause childhood- and adult-onset focal segmental glomerulosclerosis. Kidney Int 76: 1268–1276, 2009. doi: 10.1038/ki.2009.381. [DOI] [PubMed] [Google Scholar]
- 26.Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxid Redox Signal 18: 1537–1548, 2013. doi: 10.1089/ars.2012.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong J, Li G, Zhang Q, Ritter J, Li W, Li PL. D-ribose induces podocyte NLRP3 inflammasome activation and glomerular injury via AGEs/RAGE Pathway. Front Cell Dev Biol 7: 259, 2019. doi: 10.3389/fcell.2019.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mundel P, Reiser J, Borja AZ, Pavenstädt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res 236: 248–258, 1997. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 29.Zhang C, Xia M, Boini KM, Li CX, Abais JM, Li XX, Laperle LA, Li PL. Epithelial-to-mesenchymal transition in podocytes mediated by activation of NADPH oxidase in hyperhomocysteinemia. Pflugers Arch 462: 455–467, 2011. doi: 10.1007/s00424-011-0981-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Conley SM, Li G, Yuan X, Li PL. Rac1 GTPase inhibition blocked podocyte injury and glomerular sclerosis during hyperhomocysteinemia via suppression of nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 inflammasome activation. Kidney Blood Press Res 44: 513–532, 2019. doi: 10.1159/000500457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krtil J, Pláteník J, Kazderová M, Tesař V, Zima T. Culture methods of glomerular podocytes. Kidney Blood Press Res 30: 162–174, 2007. doi: 10.1159/000102520. [DOI] [PubMed] [Google Scholar]
- 32.Kahr WH, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, Lo RW, Li L, Li R, Li Q, Thoeni C, Pan J, Leung G, Lara-Corrales I, Murchie R, Cutz E, Laxer RM, Upton J, Roifman CM, Yeung RS, Brumell JH, Muise AM. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun 8: 14816, 2017. doi: 10.1038/ncomms14816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Zhang H, Liu Y, Li L, Guo Y, Jiao F, Fang X, Jefferson JR, Li M, Gao W, Gonzalez-Fernandez E, Maranon RO, Pabbidi MR, Liu R, Alexander BT, Roman RJ, Fan F. Sex differences in the structure and function of rat middle cerebral arteries. Am J Physiol Heart Circ Physiol 318: H1219–H1232, 2020. doi: 10.1152/ajpheart.00722.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan F, Geurts AM, Pabbidi MR, Smith SV, Harder DR, Jacob H, Roman RJ. Zinc-finger nuclease knockout of dual-specificity protein phosphatase-5 enhances the myogenic response and autoregulation of cerebral blood flow in FHH.1BN rats. PLoS One 9: e112878, 2014. doi: 10.1371/journal.pone.0112878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Y, Wang S, Liu Y, Fan L, Booz GW, Roman RJ, Chen Z, Fan F. Accelerated cerebral vascular injury in diabetes is associated with vascular smooth muscle cell dysfunction. Geroscience 42: 547–561, 2020. doi: 10.1007/s11357-020-00179-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muroya Y, Fan F, Regner KR, Falck JR, Garrett MR, Juncos LA, Roman RJ. Deficiency in the formation of 20-hydroxyeicosatetraenoic acid enhances renal ischemia-reperfusion injury. J Am Soc Nephrol 26: 2460–2469, 2015. doi: 10.1681/ASN.2014090868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muroya Y, He X, Fan L, Wang S, Xu R, Fan F, Roman RJ. Enhanced renal ischemia-reperfusion injury in aging and diabetes. Am J Physiol Renal Physiol 315: F1843–F1854, 2018. doi: 10.1152/ajprenal.00184.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen J, Gui D, Chen Y, Mou L, Liu Y, Huang J. Astragaloside IV improves high glucose-induced podocyte adhesion dysfunction via alpha3beta1 integrin upregulation and integrin-linked kinase inhibition. Biochem Pharmacol 76: 796–804, 2008. doi: 10.1016/j.bcp.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 39.Bi D, Toyama K, Lemaitre V, Takai J, Fan F, Jenkins DP, Wulff H, Gutterman DD, Park F, Miura H. The intermediate conductance calcium-activated potassium channel KCa3.1 regulates vascular smooth muscle cell proliferation via controlling calcium-dependent signaling. J Biol Chem 288: 15843–15853, 2013. doi: 10.1074/jbc.M112.427187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grada A, Otero-Vinas M, Prieto-Castrillo F, Obagi Z, Falanga V. Research techniques made simple: analysis of collective cell migration using the wound healing assay. J Invest Dermatol 137: e11–e16, 2017. doi: 10.1016/j.jid.2016.11.020. [DOI] [PubMed] [Google Scholar]
- 41.Yanagida-Asanuma E, Asanuma K, Kim K, Donnelly M, Young Choi H, Hyung Chang J, Suetsugu S, Tomino Y, Takenawa T, Faul C, Mundel P. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am J Pathol 171: 415–427, 2007. doi: 10.2353/ajpath.2007.070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He XC, Wang SX, Guo Y, Gao WJ, Roman RJ, Fan F. Down regulation of gamma-adducin diminishes glomerular function and promotes hypertension related chronic kidney disease. Hypertension 74: A130, 2019. doi: 10.1161/hyp.74.suppl_1.130. [DOI] [Google Scholar]
- 43.Matsuoka Y, Li X, Bennett V. Adducin: structure, function and regulation. Cell Mol Life Sci 57: 884–895, 2000. doi: 10.1007/PL00000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17: 428–437, 2007. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Centers for Disease Control and Prevention. Chronic Kidney Disease in the United States. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention, 2019. [Google Scholar]
- 46.Csipo T, Fulop GA, Lipecz A, Tarantini S, Kiss T, Balasubramanian P, Csiszar A, Ungvari Z, Yabluchanskiy A. Short-term weight loss reverses obesity-induced microvascular endothelial dysfunction. Geroscience 40: 337–346, 2018. doi: 10.1007/s11357-018-0028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HJ, Feliers D, Barnes JL, Oh S, Choudhury GG, Diaz V, Galvan V, Strong R, Nelson J, Salmon A, Kevil CG, Kasinath BS. Hydrogen sulfide ameliorates aging-associated changes in the kidney. Geroscience 40: 163–176, 2018. doi: 10.1007/s11357-018-0018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lv W, Booz GW, Fan F, Wang Y, Roman RJ. Oxidative stress and renal fibrosis: recent insights for the development of novel therapeutic strategies. Front Physiol 9: 105, 2018. doi: 10.3389/fpls.2018.00105, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol 820: 65–76, 2018. doi: 10.1016/j.ejphar.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lv W, Fan F, Wang Y, Gonzalez-Fernandez E, Wang C, Yang L, Booz GW, Roman RJ. Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol Genomics 50: 20–34, 2018. doi: 10.1152/physiolgenomics.00039.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reiser J, Altintas MM. Podocytes. F1000Res 5: 114, 2016. doi: 10.12688/f1000research.7255.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol 74: 299–323, 2012. doi: 10.1146/annurev-physiol-020911-153238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sever S, Schiffer M. Actin dynamics at focal adhesions: a common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int 93: 1298–1307, 2018. doi: 10.1016/j.kint.2017.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94: 235–263, 2014. doi: 10.1152/physrev.00018.2013. [DOI] [PubMed] [Google Scholar]
- 55.Weinberg J, Drubin DG. Clathrin-mediated endocytosis in budding yeast. Trends Cell Biol 22: 1–13, 2012. doi: 10.1016/j.tcb.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Achard V, Martiel JL, Michelot A, Guerin C, Reymann AC, Blanchoin L, Boujemaa-Paterski RA. primer"-based mechanism underlies branched actin filament network formation and motility. Curr Biol 20: 423–428, 2010. doi: 10.1016/j.cub.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 57.Garg P. A Review of podocyte biology. Am J Nephrol 47, Suppl 1: 3–13, 2018. doi: 10.1159/000481633. [DOI] [PubMed] [Google Scholar]
- 58.Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte's response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol 304: F333–347, 2013. doi: 10.1152/ajprenal.00478.2012. [DOI] [PubMed] [Google Scholar]
- 59.Schell C, Huber TB. The evolving complexity of the podocyte cytoskeleton. J Am Soc Nephrol 28: 3166–3174, 2017. doi: 10.1681/ASN.2017020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian X, Ishibe S. Targeting the podocyte cytoskeleton: from pathogenesis to therapy in proteinuric kidney disease. Nephrol Dial Transplant 31: 1577–1583, 2016. doi: 10.1093/ndt/gfw021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bidani AK, Griffin KA, Williamson G, Wang X, Loutzenhiser R. Protective importance of the myogenic response in the renal circulation. Hypertension 54: 393–398, 2009. doi: 10.1161/HYPERTENSIONAHA.109.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burke M, Pabbidi MR, Farley J, Roman RJ. Molecular mechanisms of renal blood flow autoregulation. Curr Vasc Pharmacol 12: 845–858, 2014. doi: 10.2174/15701611113116660149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng D, DuMontier C, Pollak MR. The role of alpha-actinin-4 in human kidney disease. Cell Biosci 5: 44, 2015. doi: 10.1186/s13578-015-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, Appel GB, Pollak MR. Mutational and biological analysis of alpha-actinin-4 in focal segmental glomerulosclerosis. J Am Soc Nephrol 16: 3694–3701, 2005. doi: 10.1681/ASN.2005070706. [DOI] [PubMed] [Google Scholar]
- 65.Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, Gilbert-Dussardier B, Touchard G, Karras A, Presne C, Grunfeld JP, Legendre C, Joly D, Rieu P, Mohsin N, Hannedouche T, Moal V, Gubler MC, Broutin I, Mollet G, Antignac C. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 22: 239–245, 2011. doi: 10.1681/ASN.2010050518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang S, Travis O, He X, Fan F, Roman R. Down-regulation of gamma-adducin disrupts the actin cytoskeleton in fhh rats and may contribute to the development of hypertension-induced renal injury. FASEB J 32: 721.710, 2018. doi: 10.1096/fasebj.2018.32.1_supplement.721.10. [DOI] [Google Scholar]
- 67.Kreisberg JI, Karnovsky MJ. Focal glomerular sclerosis in the fawn-hooded rat. Am J Pathol 92: 637–652, 1978. [PMC free article] [PubMed] [Google Scholar]
- 68.Kuijpers MH, de Jong W. Spontaneous hypertension in the fawn-hooded rat: a cardiovascular disease model. J Hypertens Suppl 4: S41–44, 1986. [PubMed] [Google Scholar]
- 69.Kuijpers MH, Gruys E. Spontaneous hypertension and hypertensive renal disease in the fawn-hooded rat. Br J Exp Pathol 65: 181–190, 1984. [PMC free article] [PubMed] [Google Scholar]
- 70.Joshi R, Bennett V. Mapping the domain structure of human erythrocyte adducin. J Biol Chem 265: 13130–13136, 1990. [PubMed] [Google Scholar]
- 71.Joshi R, Gilligan DM, Otto E, McLaughlin T, Bennett V. Primary structure and domain organization of human alpha and beta adducin. J Cell Biol 115: 665–675, 1991. doi: 10.1083/jcb.115.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Winder SJ, Ayscough KR. Actin-binding proteins. J Cell Sci 118: 651–654, 2005. doi: 10.1242/jcs.01670. [DOI] [PubMed] [Google Scholar]
- 73.Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol 295: C576–C587, 2008. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noguchi TQ, Morimatsu M, Iwane AH, Yanagida T, Uyeda TQ. The role of structural dynamics of actin in class-specific myosin motility. PLoS One 10: e0126262, 2015. doi: 10.1371/journal.pone.0126262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stricker J, Falzone T, Gardel ML. Mechanics of the F-actin cytoskeleton. J Biomech 43: 9–14, 2010. doi: 10.1016/j.jbiomech.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fukata Y, Oshiro N, Kaibuchi K. Activation of moesin and adducin by Rho-kinase downstream of Rho. Biophys Chem 82: 139–147, 1999. doi: 10.1016/S0301-4622(99)00113-1. [DOI] [PubMed] [Google Scholar]
- 77.Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsuura Y, Kaibuchi K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol 145: 347–361, 1999. doi: 10.1083/jcb.145.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol 173: 383–394, 2006. doi: 10.1083/jcb.200511093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen CA, Chang JM, Chang EE, Chen HC, Yang YL. TGF-beta1 modulates podocyte migration by regulating the expression of integrin-beta1 and -beta3 through different signaling pathways. Biomed Pharmacother 105: 974–980, 2018. doi: 10.1016/j.biopha.2018.06.054. [DOI] [PubMed] [Google Scholar]
- 80.Gao X, Xu H, Liu H, Rao J, Li Y, Zha X. Angiopoietin-like protein 3 regulates the motility and permeability of podocytes by altering nephrin expression in vitro. Biochem Biophys Res Commun 399: 31–36, 2010. doi: 10.1016/j.bbrc.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 81.Kruger C, Burke SJ, Collier JJ, Nguyen TT, Salbaum JM, Stadler K. Lipid peroxidation regulates podocyte migration and cytoskeletal structure through redox sensitive RhoA signaling. Redox Biol 16: 248–254, 2018. doi: 10.1016/j.redox.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reiser J, Oh J, Shirato I, Asanuma K, Hug A, Mundel TM, Honey K, Ishidoh K, Kominami E, Kreidberg JA, Tomino Y, Mundel P. Podocyte migration during nephrotic syndrome requires a coordinated interplay between cathepsin L and alpha3 integrin. J Biol Chem 279: 34827–34832, 2004. doi: 10.1074/jbc.M401973200. [DOI] [PubMed] [Google Scholar]
- 83.Wang L, Ellis MJ, Gomez JA, Eisner W, Fennell W, Howell DN, Ruiz P, Fields TA, Spurney RF. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int 81: 1075–1085, 2012. doi: 10.1038/ki.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garg P, Verma R, Cook L, Soofi A, Venkatareddy M, George B, Mizuno K, Gurniak C, Witke W, Holzman LB. Actin-depolymerizing factor cofilin-1 is necessary in maintaining mature podocyte architecture. J Biol Chem 285: 22676–22688, 2010. doi: 10.1074/jbc.M110.122929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Venkatareddy M, Cook L, Abuarquob K, Verma R, Garg P. Nephrin regulates lamellipodia formation by assembling a protein complex that includes Ship2, filamin and lamellipodin. PLoS One 6: e28710, 2011. doi: 10.1371/journal.pone.0028710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pozzi A, Zent R. Integrins in kidney disease. J Am Soc Nephrol 24: 1034–1039, 2013. doi: 10.1681/ASN.2013010012. [DOI] [PMC free article] [PubMed] [Google Scholar]