

Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by the destruction of alveolar tissue (in emphysema) and airway remodeling (leading to chronic bronchitis), which cause difficulties in breathing. It is a growing public health concern with few therapeutic options that can reverse disease progression or mortality. This is in part because current treatments mainly focus on ameliorating symptoms induced by inflammatory pathways as opposed to curing disease. Hence, emerging research focused on upstream pathways are likely to be beneficial in the development of efficient therapeutics to address the root causes of disease. Some of these pathways include mitochondrial function, cytoskeletal structure and maintenance, and airway hydration, which are all affected by toxins that contribute to COPD. Because of the complexity of COPD and unknown targets for disease onset, simpler model organisms have proved to be useful tools in identifying disease-relevant pathways and targets. This review summarizes COPD pathology, current treatments, and therapeutic discovery research, with a focus on the aforementioned pathways that can advance the therapeutic landscape of COPD.
Keywords: airway and alveolar epithelium, cigarette smoke, cytoskeleton, mitochondria, model organisms
INTRODUCTION
Chronic obstructive pulmonary disease (COPD), an illness that diminishes lung function to make breathing difficult, is one of the leading causes of death in the United States and worldwide (38, 62). As of 2010, an estimated 328 million people have COPD worldwide and COPD is predicted to be the third leading cause of death in the world by 2030 (38). Considering its impact on public health, research on what underlies COPD for the development of therapeutics has been underway for decades. Nonetheless, COPD continues to have a lack of therapeutic options that can reverse lung damage and prevent disease progression. Although research on COPD is ongoing and new therapeutics are currently in the pipeline, there continue to be few advances on the root causes of the disease and potential treatments to stop them. In this review, we include a general overview of COPD pathology and some of the therapeutic strategies currently in use. In addition, we discuss emerging COPD research that has the potential to provide new breakthroughs for therapeutic development.
GENERAL PATHOLOGY AND TREATMENT OF STABLE COPD
A major cause of COPD is exposure to tobacco smoke. Continual exposure to the compounds in tobacco smoke induces tissue damage and inflammatory responses that lead to the primary phenotypes of the disease: emphysema and chronic bronchitis. Emphysema is characterized as the destruction of alveolar tissue. As a result, lung elasticity is lost by the degradation of the extracellular matrix (ECM) and alveolar cells are lost through apoptosis and autophagy (60). Apoptosis, inflammation, and ECM breakdown continue to propagate in self-amplifying loops that further the progression of emphysema (60). In the airways, inflammation can occur in both the large and small airways after exposure to tobacco smoke. Chronic smoking also increases the chances of remodeling of the airways (60). Airway remodeling, the overproduction of mucus by airway goblet cells, and the increased difficulty in airway clearance contribute to the airflow obstruction found in chronic bronchitis (31). One of the detriments of COPD is that the disease progresses even after smoking cessation (60).
The 2020 Global Initiative for Chronic Obstructive Lung Disease offers recommendations for pharmacological COPD treatment based on patient symptoms and history of hospitalizations. Patients with mild COPD are generally given short-acting or long-acting β2-agonists and/or antimuscarinic drugs (22). β2-Agonists stimulate β2-adrenergic receptors, leading to increased cAMP levels and protein kinase A activation (22). Antimuscarinic drugs block acetylcholine from binding to muscarinic receptors in the smooth muscle of the airways (22). Through these mechanisms, both types of drugs reduce bronchoconstriction in airway smooth muscle. If symptoms worsen, inhaled corticosteroids can be used in combination with long-acting β2-agonists or antimuscarinic drugs that treat dyspnea and exacerbations (22). Other options available for more severe cases of COPD include the phosphodiesterase-4 (PDE4) inhibitor roflumilast in combination with the drugs stated previously (13). Current treatments for COPD mainly focus on addressing the symptoms of the disease, and evidence also suggests that some patients are resistant to inhaled corticosteroid treatment (13). With the limitations of current treatments, more effective treatment strategies are needed. One potential strategy includes implementing a precision medicine approach that identifies more upstream cellular and molecular pathways undermining the disease (53).
RECENT COPD PIPELINE STRATEGIES
In lieu of developing more disease-specific treatments, we describe some current efforts by other groups in the COPD pipeline to address disease onset (Fig. 1). These efforts are heavily focused on targeting inflammatory pathways, which is relevant considering that inflammation is one of the main contributors to disease pathology. Nonetheless, inflammation in COPD is complex and involves a host of pathways (7). Therefore, targeting only one pathway may not necessarily be effective in treating COPD. In fact, several drug development strategies have failed to show efficacy, possibly due to this issue. Delivery strategies may also contribute to this lack of efficacy. Oral drugs can cause off-target effects as they do not directly encounter the airways as efficiently as inhaled medications. Nevertheless, the following sections describe some significant players in COPD and promising targets that can address inflammatory complications.
Figure 1.

The chronic obstructive pulmonary disease (COPD) pipeline focuses on targeting inducers of inflammation. Cigarette smoke activates numerous downstream pathways and targets that lead to inflammation, including inflammatory mediators, oxidative stress, kinases, and phosphodiesterases. Current therapeutic strategies aim to reduce inflammatory responses by inhibiting specific targets under each major group.
Oxidative Stress
Oxidative stress is thought to be a significant driver of COPD as several disease-related pathways are affected, including inflammation, aging, and DNA damage (9). While oxidative stress impacts many pathways, one that is well studied involves nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor, and its interaction with the negative regulator, Kelch-like ECH-associated protein-1 (KEAP1). Oxidative stress inactivates KEAP1, allowing Nrf2 to translocate into the nucleus, where it activates pathways that increase antioxidant metabolism and attenuate inflammatory responses (16). In the context of COPD, Nrf2 activity increases with cigarette smoke (CS) exposure due to the increase in oxidative stress, but its activity declines with the progression of COPD (52). In a study with a small group of COPD patients, gene expression levels downstream of Nrf2 were higher in bronchial epithelial cells of current smokers versus former smokers, consistent with Nrf2-related anti-inflammatory activity occurring with CS exposure (52). CS extract (CS bubbled into cell media) also affects Nrf2 activity in differentiated primary normal human bronchial epithelial cells (NHBE) grown at air-liquid interface. Several oxidative stress-responsive genes related to the Nrf2 pathways are upregulated with CS extract exposure (49). In addition, Nrf2 protein expression has a biphasic relationship with CS extract exposure, increasing then decreasing with increased concentrations of CS extract (49). This result supports the idea that Nrf2 is important in regulating inflammatory responses from CS initially, but it loses this ability with continual exposure to CS.
Many lines of evidence implicate Nrf2 in COPD pathology; thus, Nrf2 and its associated signaling pathways are being pursued as therapeutic targets. One study identified the receptor for advanced glycation end products (RAGE), a member of the immunoglobulin superfamily, as a deactivator of Nrf2 activity in response to CS exposure (36). An antagonist for RAGE, FPS-ZM1, promoted Nrf2 translocation into the nucleus, allowing for decreased expression of damage-associated-molecular-pattern signaling (36). Other agents that change Nrf2 expression and activity in airway cells include aspirin-triggered resolvin D1, crocin, sulforaphane, and schisandrin B (19, 27, 29, 46). Sulforaphane, a natural compound found in cruciferous vegetables, that activates Nrf2 by binding to KEAP1, proceeded into clinical trials for a variety of diseases (16). Nonetheless, in a trial for COPD, sulfurophane failed to show efficacy because Nrf2 gene expression and inflammatory markers levels did not consistently change after treatment in either alveolar macrophages and bronchial epithelial cells (16). This result was possibly due to poor drug penetration to the lung (9). Something else to note is that this trial measured these parameters after a few weeks of treatment instead of a longer period of time (61). Other COPD drugs in the pipeline are nonelectrophilic, noncovalent drugs that target the Nrf2-KEAP1 protein-protein interaction (PPI) rather than the proteins themselves (16). Nonelectrophilic PPI inhibitors have different pharmacodynamics and off-target effects compared with electrophilic drugs like sulforaphane, thus offering another avenue for drug development on Nrf2 (16).
Kinase-Mediated Pathways
Several types of kinases induce chronic inflammation when activated, making them another therapeutic target group for COPD (8). Indeed, drugs have been or currently are in development that target a variety of kinase groups, including MAPK, receptor tyrosine kinases (RTKs), phosphoinositide 3-kinases (PI3Ks), JAK, and NF-κB (8). However, none have been approved for clinical use, possibly due to issues such as target specificity and loss of efficacy over time (8). For instance, one group that drew attention for clinical development was the p38 MAPK family. Inhibiting p38 reduces inflammation in cellular and animal models, but many failed to show promise in clinical trials (8). An example is the p38α compound AZD7624, which decreases cytokine release in human alveolar macrophages but did not decrease exacerbations in COPD patients (44). However, work on developing and studying more specific inhibitors are underway. One such p38 inhibitor with evidence of efficacy is RV-568. Classified as a narrow-spectrum kinase inhibitor with high potency for the α- and γ-isoforms of p38, RV-568 demonstrated anti-inflammatory activity in monocytes, macrophages, and epithelial cells by inhibiting cytokine (CXCL8 and IL-6) release (15). Animal models also showed promising results, and in a small 14-day clinical trial, lung function improved with drug treatment (15).
Phosphodiesterase Inhibitors
Interest in PDE4 inhibitors arose with the discovery that increasing intracellular cAMP levels can have anti-inflammatory effects. PDE4 subtypes are found in various immune cells and cells affected by immune response (48). When they are inhibited, cAMP signaling allows for the upregulation of cAMP-response element (CRE)-containing genes and the inhibition of NF-κB, which decreases inflammatory responses (48). The consequences of decreased cAMP via PDE upregulation can occur in airway cells and lung tissue as well. In a study with precision lung slices from CS-exposed mice, intracellular cAMP levels were decreased based on fluorescence resonance energy transfer (FRET) sensor measurements (69). Protein and mRNA levels are lower for two paralogs of PDE4 (PDE4B, PDE4D; two of the four PDE4 genes) in these animals as well (69). In another study by authors in the same group, they discovered that, among patients, smokers have increased protein expression of PDE4D in the airway epithelium (68). These works highlight PDE4 as a candidate for drug development against CS-induced inflammation.
As mentioned above, the PDE4 inhibitor roflumilast is used for more severe cases of COPD (22). Nevertheless, its efficacy is limited due to it being an oral drug and causing adverse side effects. Hence, more potent analogs of roflumilast are being developed (41). Several inhaled formulations of PDE4 inhibitors are in the pipeline too, which may improve their efficacy compared with roflumilast (45). One of these drugs, CHF6001 showed promise in clinical trials by decreasing the inflammatory biomarkers leukotriene B4 (LTB4), C-X-C motif ligand 8 (CXCL8), matrix metalloproteinase-9 (MMP-9), tumor necrosis factor-α (TNFα), and macrophage inflammatory protein-1β (MIP-1β)in sputum of patients already on triple therapy with inhaled β-adrenergic agonists, antimuscarinics, and inhaled corticosteroids (55). Similar results appeared in alveolar macrophages and lung tissue from COPD patients with CHF6001 reducing TNFα production (more potently than roflumilast) and activating cAMP response element-binding protein (CREB) (35).
Another drug in clinical trials is ensifentrine/RPL554, a dual PDE3/PDE4 inhibitor. In addition to having anti-inflammatory effects, ensifentrine evokes bronchodilator effects (54). Ensifentrine also improves the effects of other bronchodilators when they are used in combination (54). The forced expiratory volume (volume of air forcefully expelled) in one second, or FEV1, increases with ensifentrine together with salbutamol (β-adrenergic agonist), or ipratropium (antimuscarinic), both short-acting bronchodilators, than each short-acting bronchodilator alone (54). Ensifentrine with tiotropium, a long-acting bronchodilator with antimuscarinic effects, demonstrated similar benefits in FEV1 (54). More clinical trials are planned for ensifentrine, and other PDE4 inhibitors continue to be in the pipeline as well (54).
Inflammatory Mediators
Interleukins serve as inflammatory mediators, and variants such as IL-4, IL-13, IL-9, and IL-5, which promote type 2 immunity, have been implicated in COPD disease progression (10). Interleukins are released from a variety of cell types from exposure to tobacco smoke or other inhaled irritants. Aside from immune cells, tobacco smoke can cause epithelial cells, endothelial cells, and fibroblasts to release these mediators (7). The mediators then increase the number of macrophages, neutrophils, eosinophils, lymphocytes, dendritic cells, and other immune cells that induce inflammatory responses (7).
Targeting these interleukins is of interest for developing therapeutics because of their role in promoting inflammatory responses. In a recent study, the authors found that IL-4, in an elastase-induced emphysema mouse model, is released from basophils and promotes alveolar destruction by increasing metalloproteinase-12 expression (50). Another study demonstrated that IL-9 inhibition in a COPD mouse model also abrogates structural damage from CS exposure (66). In terms of drug development, IL-5 and its receptor IL-5Rα, key players in eosinophil-related inflammation, are targets of blocking antibodies mepolizumab, reslizumab, and benralizumab (10). These drugs are approved for asthma but failed to show improved efficacy in COPD clinical trials (10). Nonetheless, these drugs have not yet been tested for efficacious effects in COPD patient subgroups with eosinophilia. Other approved drugs for asthma target IL-13 and IL-4, but these drugs have not been extensively tested for COPD treatment. Clinical trials are still open for such studies with dupilumab, an IL-4 and IL-13 receptor monoclonal antibody, being a candidate (10).
Despite the interest in developing compounds for eosinophilic inflammation, research is ongoing to elucidate the effects of other inflammatory mediators on disease progression. Higher concentrations of cytokines, such as TNFα, exist in sputum and exacerbations of COPD patients (10). IL-17 appears to have a role in small-airway fibrosis in COPD model mice, and metalloproteinases, particularly MMP-9, are also targets of interest due to their involvement in alveolar wall destruction (7, 63). Chemokines are also generating interest, with CXCL8 and its receptor CXCR2 being the major targets. Interestingly, microRNAs, which likewise regulate inflammation, are involved in COPD, with miR-155 expression being increased in COPD patients compared with nonsmokers (18). MiR-155 knockout mice have lower levels of inflammatory markers and cells, and elastase-induced emphysema is attenuated in these mice. No miRNA-based therapeutics for COPD exist yet, although they have utility in diseases like cancer (24).
OPPORTUNITIES FOR NEW BREAKTHROUGHS
Many of the current drug development strategies for COPD focus on targeting proinflammatory pathways. Although these strategies are helpful in treating inflammation, lung damage remains and continues to be irreversible. Hence, there is a need for research into areas that have the potential to address these issues such as how lung damage occurs farther upstream of inflammation. Delving into these upstream pathways may offer more potential therapeutic targets that may address the root causes of disease (Fig. 2). In addition, conducting research on lung repair and regeneration may present other useful strategies to counter damage already done to the lung.
Figure 2.

Emerging chronic obstructive pulmonary disease (COPD) biology offers potentials for new breakthroughs in therapeutic development. The airway epithelium is composed of ciliated cells (blue), secretory cells (orange), and basal cells (green) on top of a basement membrane (BM) and extracellular matrix (ECM). Its role is to act as a barrier to toxins that enter the airway, including cigarette smoke. Continual exposure to cigarette smoke in the epithelium, however, can contribute to COPD phenotypes. Pathways that are affected by cigarette smoke upstream of inflammatory responses are currently being researched and may be helpful in identifying new targets for drug development. These pathways include airway hydration, the structural integrity of the airway epithelium and its barrier function, mitochondrial dysfunction and mitochondrial reactive oxygen species (ROS) production, extracellular matrix remodeling, and epithelial-to-mesenchymal transition (EMT).
Uncovering new methodologies to assist in conducting these new areas of research could also be of interest. For example, model organisms can help to identify new disease-relevant targets. These targets or other pathways implicated in COPD can be further studied in human cellular disease models, which are also being currently researched and developed. More comments on these points and information on ongoing research are briefly described below.
Mitochondria
Mitochondria are a major source of endogenous reactive oxygen species (ROS) when under stress, making them viable targets for therapeutic research. Studies have focused on how mitochondrial dysfunction arises in the context of COPD and CS exposure (2). Many of these findings suggest that mitochondrial morphology, metabolism, and other functions become abnormal in human and mouse airway epithelial cells from CS. More recent findings highlight the effects of CS on mitochondrial morphology and the impact of these morphological changes. In human airway smooth muscle cells, CS causes mitochondrial fragmentation by decreasing expression of mitochondrial fusion protein Mfn2 and increasing expression of fission protein Drp1 (4). This fragmentation leads to loss of mitochondrial membrane potential and defects in oxidative phosphorylation (4). Interestingly, CS induces proliferation and airway remodeling in these cells, suggesting that the promotion of mitochondrial fission drives cells toward glycolysis for energy (4). Similarly, in primary human alveolar type II cells from smokers and patients with emphysema, mitochondrial membrane potential and mtDNA repair are disrupted, leading to higher mtDNA damage, along with decreased mitochondrial fission (33).
Other proteins of interest affected by CS that contribute to mitochondrial dysfunction in the airway epithelium include Miro1, Nix, and pp66Shc. The expression levels of Miro1, a GTPase that is involved in mitochondrial shape and trafficking, are reduced after exposure to CS extract. Decreases in Miro1 cause defects in mitophagy, an important process in removing damaged mitochondria (58). In the same vein, increases in Nix levels by CS extract induce mitophagy. Nix is involved in mitochondrial clearance and is related to the proapoptotic BH3-only proteins. When it is silenced by siRNA, CS extract-induced defects in ATP levels and mitochondrial membrane potential are ameliorated (64). Finally, pp66Shc is an adaptor protein that is translocated into the mitochondria and generates ROS when phosphorylated by protein kinase C (PKC) isozymes. In airway epithelial cells exposed to CS extract, pp66Shc expression increases, resulting in increased mitochondrial ROS production (65). Interestingly, pp66Shc silencing attenuates this effect on ROS and improves other mitochondrial processes (65). Considering how involved these proteins are in mitochondrial injury, they can be potential therapeutic targets against mitochondrial-induced ROS. The pathways and processes related to these proteins (mitophagy, oxidative phosphorylation, fusion/fission) may also be valuable to examine further to identify additional targets to reduce the impact of ROS.
Structural Integrity of Airway Epithelium
The airway epithelium is the first line of defense against any particulates and bacteria that enter the airway, yet this epithelium must maintain a barrier that is selectively permeable. Toxins such as CS can interrupt this barrier function, leading to the release of inflammatory signals that contribute to the onset of disease (1). On a mechanistic level, one reason for the loss of monolayer integrity is the disruption of cellular junctions. Proteins that comprise tight junctions (such as zonula occludens-1 and occludins) or adherens junctions (like E-cadherin) are downregulated in airway cells exposed to CS, resulting in a loss in barrier function reflected by a decrease in transepithelial resistance (1). Investigations into therapeutic compounds that target junctional proteins are underway, but they are still limited (1).
To further understand barrier function in a related context, studies on cell mechanics and the cytoskeletal elements that interact with these junctions are transpiring. The actin cytoskeleton, which is linked to apical tight junctions and which regulates cell structure, is affected by continual CS exposure in NHBEs (1). Actin assembly increases with more CS exposure, and cortical tension, a measure of cell stiffness, also rises (43). Increased actin polymer levels and cell stiffness may contribute to the tissue remodeling and barrier dysfunction seen in COPD (43). Indeed, other studies are emerging on actin and actin-associated proteins as potential regulators of disease. One study identified other actin-related proteins affected by CS via mass spectrometry: coactosin-like protein (COTL1), microtubule-associated protein RP/EB family member 1 (MARE1), and heat shock protein B1 (HSPB1) (17). Another identified potential protein affected by CS is the family with sequence similarity member 13A (FAM13A), a modulator of RhoA activity and actin dynamics (14). Rac1, a regulator of actin assembly, is also implicated in epithelial-to-mesenchymal transition (EMT) (28). EMT, the process of the airway epithelium becoming more mesenchymal, is also thought to occur in COPD (1). Many proteins feed into this process, and future studies may reveal several cytoskeletal proteins as key players of EMT during COPD.
The extracellular matrix (ECM) is an additional structural component of the airway epithelium that likely contributes to COPD phenotypes. In epithelial and alveolar cells, the breakdown of the ECM by proteases such as MMP-9 is presumed to help cause emphysema by inducing inflammatory responses and airway remodeling (1). Indeed, the concentrations of biomarkers indicating ECM degradation for components like elastin, collagen I, and collagen IV, are higher in COPD patients compared with healthy patients (12). Interestingly, when NHBEs are grown on a scaffold containing ECM from COPD patients, expression for COPD-related genes is altered in these cells compared with those grown on scaffolds from healthy patients (25). The various changes in gene expression point toward a COPD phenotype, suggesting that changes in the ECM can disrupt the integrity of the airway epithelium (25). Although previous studies have attempted to develop therapeutics against the proteases (MMPs, etc.) that degrade the ECM, they have been unsuccessful in procuring an effective drug. Other proteins, such as cytoskeletal proteins that regulate the ECM, may be valuable targets. For example, defects in nonmuscle myosin II (NM II), a force-generating motor protein important in cell structure and shape change, altered ECM remodeling in a mouse model and induced an emphysema-like phenotype (30). With modulators against some isoforms of NM II already discovered like 4-hydroxyacetophenone (4-HAP), they may be interesting candidates to test in therapeutic studies (59).
Airway Hydration
In addition to being a barrier that prevents bacteria and particulates from entering the airway, the epithelium functions to trap these particulates and remove them from the airway altogether. Mucus produced by goblet cells is released on the surface of the airway, forming a layer of apical liquid called the airway surface liquid (ASL), which includes a mucus layer and an aqueous periciliary layer. Mucociliary clearance is propagated by motile cilia along the surface of the epithelium (21). CS appears to affect mucociliary clearance by shortening cilia length, slowing down ciliary beating, increasing mucus secretion, and lowering ASL height (21). This reduction in mucus clearance causes mucus build-up, which can be one of the causes of airway obstruction and infection found in chronic bronchitis (21). Cystic fibrosis (CF) comparably occurs from reduced airway hydration and lack of mucus clearance, making therapeutic targets from CF potentially useful for COPD treatment. Interestingly, cystic fibrosis transmembrane conductance regulator (CFTR), an anion channel that secretes Cl− to maintain airway hydration, is affected by CS exposure in human bronchial epithelial cells, with a recent study by Marklew et al. (39) suggesting that CS leads to its inactivation by internalization trafficking. Overall, CFTR and other ion channels, like the epithelial sodium channel (ENaC), that are responsible for airway hydration may be helpful targets to pursue and studies on their therapeutic potential are ongoing (21, 40).
Pro-Regenerative Strategies
The progression of COPD disrupts repair mechanisms, resulting in irreversible lung damage. This observation has generated an interest in regenerative strategies such as stem cell and tissue engineering treatments to repair areas of the lung that are already damaged. One extensively studied method is the infusion of mesenchymal stromal cells (MSCs) at sights of injury. Interestingly, MSCs demonstrated anti-inflammatory effects in both in vitro and in vivo mouse models, but clinical trials with MSCs have yet to show efficacy (57). MSCs did not affect pulmonary function or quality of life in these trials (57). Nonetheless, making changes to MSC clinical trial design and conducting more studies to understand how MSCs work mechanistically may address the disparity between the preclinical and clinical trial data.
Another therapeutic approach is to target and activate endogenous regenerative mechanisms in the lung that are affected by COPD. Most studies on lung regeneration have been conducted in mouse models, and they have procured relevant pathways, including Wnt/β-catenin, Notch, fibroblast growth factor (FGF), retinoic acid (RA), and Hedgehog signaling (42). Interestingly, Wnt and Notch signaling are both disrupted in airway epithelial cells of COPD patients (42). Gene expression for proteins related to RA transport and activity are altered in COPD lung tissue and in fibroblasts as well (42). Pharmacological activation of these pathways has also led to promising results in in vivo models. For instance, activation of the Wnt/β-catenin signaling pathway with lithium chloride attenuated emphysemic phenotypes by improving lung function and decreasing airspace enlargement in a mouse model (42). In some rat and mouse emphysema models, activating RA receptors (RARs) with all-trans RA (ATRA) induced alveolar regeneration and improved lung function (42). ATRA actually proceeded into clinical trials but failed to improve lung function after treatment (42, 57). Another trial with palovarotene, a selective agonist of RARγ, also failed to show significant efficacy by not improving lung function (42, 57).
Although these trials along with the MSC trials have not yet procured promising treatments, there is still potential for regenerative strategies to be used therapeutically. The regenerative signaling pathways already mentioned (and some not mentioned here) can be furthered studied. More research needs to be done to thoroughly characterize these pathways to identify more promising targets and better understand how they function in lung regeneration.
Using Model Organisms as Discovery Tools
Animal models are heavily utilized to study COPD induced by CS exposure in a variety of models such as mice, guinea pigs, rats, and dogs (20). Although these models are useful in understanding the progression of disease and the effects of specific therapeutic targets, these animal models are rarely used to identify disease-relevant (and potentially novel) pathways in lung disease. In the few cases where they are, the processes to do so appear complex. Hence, the use of simpler model organisms is one way to bypass this complexity and perform genetic screens more easily and/or at a larger scale (Fig. 3). Studies with Drosophila melanogaster and Caenorhabditis elegans have procured pathways or recapitulated known pathways that are relevant to COPD (23, 47). In Drosophila, RNA sequencing of trachea tissue from untreated and CS-exposed larvae revealed that gene expression was altered for 143 genes (47). Interestingly, some of these genes function in Nrf2, TGFβ, and JAK/STAT signaling pathways. These pathways have already been identified as relevant to COPD disease progression, making Drosophila another option to study these pathways in terms of CS exposure (47). As for C. elegans, the impact of CS exposure on genes that regulate innate immunity was investigated through microarray analysis. One of the genes identified, lbp-7, encodes for a lipid-binding protein (23). Interestingly, mRNA levels for the human ortholog of lbp-7, fatty acid binding protein-5 (FABP-5), were lower in COPD patient cells compared with cells from nondiseased smokers (23). More recently, Dictyostelium discoideum, a social amoeba, was used to identify disease-relevant pathways (32). In a genetic selection for suppressors against CS-induced insults, a variety of genes came out as protective against CS (32). This list of suppressors included cytoskeletal, metabolic, protein folding, and translational proteins (32). One of the proteins, adenine nucleotide translocase (ANT), was further studied in human airway epithelial cells. It demonstrated protective effects against CS by improving metabolic function and increasing airway hydration when overexpressed, making it another potential therapeutic target for COPD (32).
Figure 3.
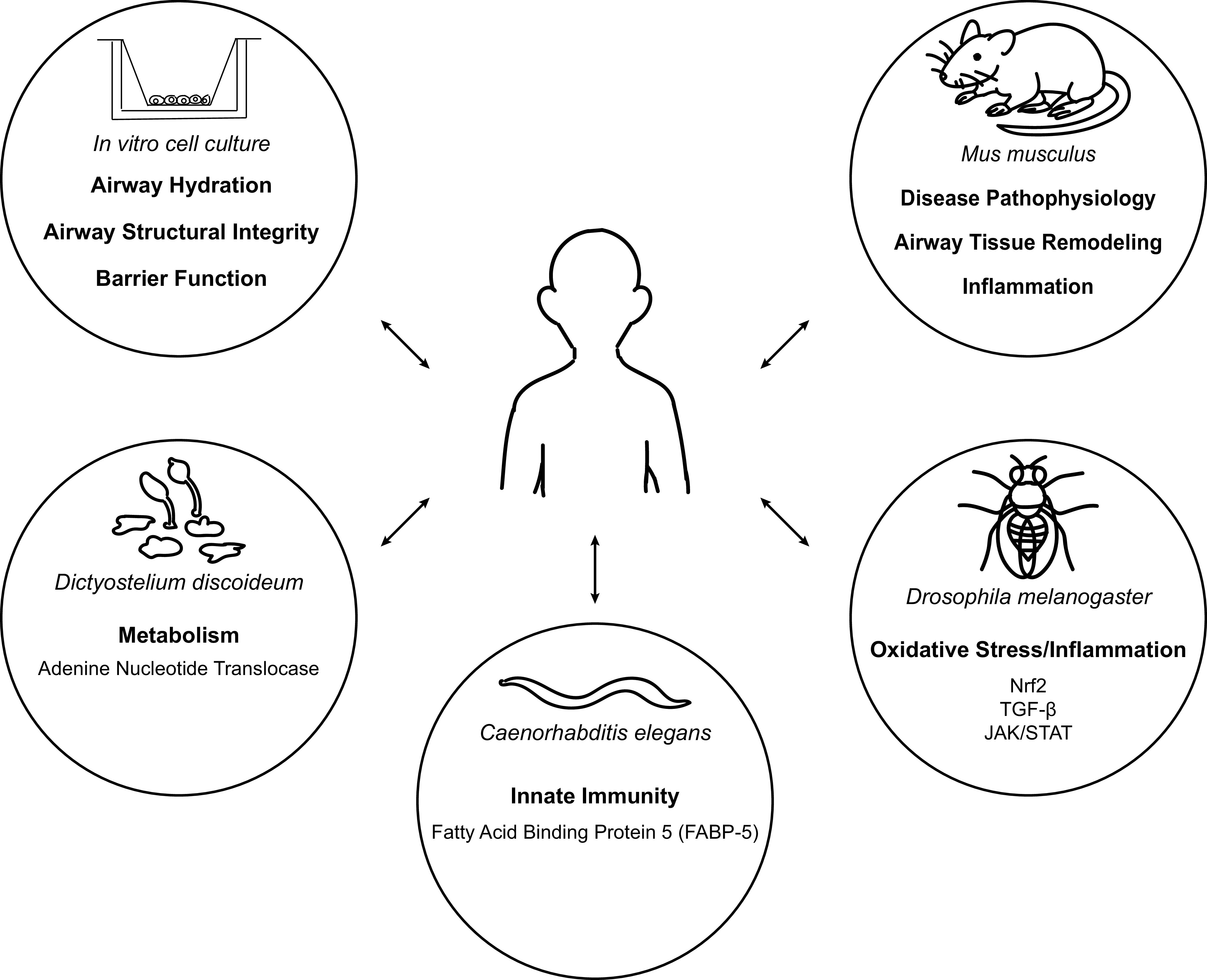
Various model organisms can be utilized to identify new and relevant biology in chronic obstructive pulmonary disease (COPD). Mice have been heavily used to study COPD and understand disease progression, but the processes that come with identifying new essential disease biology are more complex in both human and mouse. Interestingly, there is emerging research on finding additional disease-relevant biology with simpler model organisms. New pathways affected by cigarette smoke have been identified in in vitro cell culture models, Caenorhabditis elegans (23), Drosophila (47), and social amoeba Dictyostelium discoideum (32). By utilizing these simpler models, there is the potential to recapitulate major findings or discover novel research on COPD, which can then be further studied in complex systems such as human and mouse.
Through these model organisms, a variety of relevant pathways have been identified. Whereas some pathways have already been discovered or are known inflammatory pathways, other novel targets such as ANT affect other processes outside of inflammation and may have more causative roles. Indeed, some of these targets are related to previously mentioned research areas (i.e., mitochondria, cellular structure, and airway hydration) that have the potential to address what underlies COPD. These targets can be furthered studied in human disease models, like ANT, to confirm how relevant they are to disease onset or progression.
Using 3-D Human-Based Disease Models for Pharmacological Studies
As mentioned previously, animal models have been useful in understanding disease progression and effects of different treatments. These models, along with immortalized human lung cell lines, have contributed greatly to COPD research, but there are caveats to using them. For example, the lung anatomy and some COPD disease phenotypes vary in mouse and rat models compared with human (67). Treatments that appear to be effective in rodents may not necessarily be effective in humans as a result. Hence, the use of human-based disease models can be used to more effectively understand the cellular mechanisms underlying COPD (including the mechanisms of relevant targets found through model organisms) and evaluate the efficacy of treatments. A variety of these models exist to study the airway epithelium. Most models are composed of primary cells from donor lungs that are grown under submerged conditions or at air-liquid interface (ALI) on Transwell inserts, where they can differentiate into secretory and ciliated cells (26, 67).
Recently, more complex 3-D cell culture models have been developed. Unlike previous models, they allow for more representative human disease modeling by mimicking the structure of the lung and exposing cells to physiological mechanical cues (26, 67). These models include organoids, which are self-assembling structures that are generally derived from stem/progenitor cells and embedded into a 3-D matrix. Organoids can be generated from mouse or human cells, and they can be formed by a variety of cell types, including alveolar cells, airway secretory cells, and airway basal cells (5). Considering that they are derived from stem cells, organoids are useful in studying lung formation and repair and epithelial function. These mechanisms are dysfunctional in COPD; so in a disease context, organoids can aid in identifying and understanding targets that underlie these mechanisms. In addition, organoids can be useful in identifying potential therapeutics. For example, screens have been developed to find compounds that regulate basal cell differentiation and affect the ratio of ciliated and secretory cells, which is disrupted in COPD (5). Although these screens can be done with ALI cultures, organoid screens are faster and many more samples can be evaluated (5). Overall, organoids have the potential to identify new treatments and identify new disease mechanisms.
In a similar vein, precision-cut lung slices (PCLSs), is another 3-D model that mimics the complexity of the lung. They are lung tissue slices that encompass more of the lung’s complexity by containing all the cell types and the ECM composition in a particular area (3, 37). In the context of COPD, PCLSs have been used to study the effects of tobacco smoke exposure on different targets, including the previously mentioned study by Zuo et al. (69). Studying PCLSs from diseased patients also provides useful information on disease and therapeutic mechanisms. For example, frizzled-4 (FZD4), a WNT receptor, was downregulated in COPD PCLSs (56). Pharmacological activation of FZD4 with valproic acid induced Wnt/β-catenin signaling and elastogenic components like insulin-like growth factor 1 (IGF-1) in COPD PCLSs, making it a potential therapeutic target (56). In addition, PCLSs have also been helpful in elucidating mechanisms for current treatments. In a study by Koziol-White et al. (34), the researchers demonstrated that the combined use of a glucocorticoid (budesonide) and a β2-agonist (formoterol) additively promoted bronchodilation as opposed to either drug alone in PCLSs. Other experiments in both human PCLSs and airway smooth muscle cells revealed that budesonide increases cAMP production, which further promotes bronchodilation by adding to the cAMP already released via β2-agonist treatment (34). Overall, these studies are just a few examples of how PCLSs can be used to study disease mechanisms and evaluate therapeutic responses, which will be beneficial for future therapeutic development.
Another model with similar characteristics to organoids and PCLSs is lung-on-a-chip. Lung-on-a-chip is a microfluidic device composed of microchannels lined with human cells that are exposed to a continuous flow of nutrients and growth factors. Many different types of models have been developed by different groups based on diseases and research areas (51). Some lung-on-a-chip models for asthma and COPD were developed in a study by Benam et al. (11). They interestingly demonstrated that their model could be used as a tool to evaluate therapeutic responses (11). An experimental anti-inflammatory bromodomain-containing protein-4 (BRD4) inhibitor suppressed neutrophil adhesion to inflamed cells on the chip. Interestingly, when the same drug was tested on 2-D Transwell cultures, the efficacy of the BRD4 inhibitor was reduced (11). This result suggests that the inhibitor depended on the existence of flow in the model, demonstrating that the microfluidic aspect of the model was also useful in evaluating the mechanism of action for this drug (11). The results from this study and others exhibit the potential for lung-on-a-chip as a tool for drug studies in the future (51). Like organoids and PCLSs, lung-on-a-chip simulates lung tissue more closely, making it valuable in evaluating therapeutic responses and predicting whether treatments will be clinically relevant.
CONCLUSIONS
COPD is a major public health concern, and as it continues to be a global burden, the importance of developing new treatments is apparent. Current treatments are not curative, and while new strategies and drugs are in the pipeline, they still address mostly secondary inflammatory pathways of the disease. An additional major complication in COPD drug development likely comes from the essential dependence on surrogate end points like FEV1 to assess the impact of a therapeutic strategy. Thus, any new therapeutic strategy will ultimately require long-term studies to confirm that the surrogate end points accurately reflect efficacy on disease outcome.
Overall, a tremendous need exists for discovering new core biology and ways to incorporate this into therapeutic development and in a more precision-based approach toward treating patients. The opportunities for new breakthroughs previously mentioned and summarized in Table 1 are several avenues that can be further studied. The idea of using model organisms to discover new lung biology and studying new biology in human disease models is especially promising, considering that the current models are concepts that have not changed for about three decades.
Table 1.
New avenues of research for COPD and potential therapeutic targets
| Research Area | Molecular Targets | References |
|---|---|---|
| Mitochondria and metabolism | Mfn2, Drp1, Miro1, Nix, pp66Shc, ANT | (38, 40–42, 60) |
| Airway structural integrity | Junctional proteins including ZO-1, occludins, and E-cadherin | (43) |
| Airway barrier function | COTL1, MARE1, HSPB1, FAM13A, Rac1 | (44–47) |
| ECM remodeling | MMP-9, NM II | (43, 48–51) |
| Airway hydration | CFTR, ENaC, other ion channels | (52–54) |
ANT, adenine nucleotide translocase; CFTR, cystic fibrosis transmembrane conductance regulator; COPD, chronic obstructive pulmonary disease (COPD); COTL1, coactosin-like protein; ECM, extracellular matrix; ENaC, epithelial sodium channel; FAM13A, family with sequence similarity member 13A; HSPB1, heat shock protein B1; MARE1, microtubule-associated protein RP/EB family member 1; mMMP-9, matrix metalloproteinase-9; NM II, nonmuscle myosin II; ZO-1, zonula occludens-1.
GRANTS
Our work is supported by the National Institutes of Health (HL124099, HL151107, GM66817, and F31 HL145910).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.K.N. prepared figures; J.M.K.N. drafted manuscript; J.M.K.N., D.N.R., and V.K.S. edited and revised manuscript; J.M.K.N., D.N.R., and V.K.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the members of the Robinson and Sidhaye laboratories for helpful discussions. We thank Alexandra Surcel and Amanda Balaban for helpful comments on the review.
REFERENCES
- 1.Aghapour M, Raee P, Moghaddam SJ, Hiemstra PS, Heijink IH. Airway epithelial barrier dysfunction in chronic obstructive pulmonary disease: role of cigarette smoke exposure. Am J Respir Cell Mol Biol 58: 157–169, 2017. doi: 10.1165/rcmb.2017-0200TR. [DOI] [PubMed] [Google Scholar]
- 2.Aghapour M, Remels AHV, Pouwels SD, Bruder D, Hiemstra PS, Cloonan SM, Heijink IH. Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 318: L149–L164, 2020. doi: 10.1152/ajplung.00329.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsafadi HN, Uhl FE, Pineda RH, Bailey KE, Rojas M, Wagner DE, Königshoff M. Applications and approaches for three-dimensional precision-cut lung slices. Am J Respir Cell Mol Biol 62: 681–691, 2020. doi: 10.1165/rcmb.2019-0276TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J Cell Physiol 232: 1053–1068, 2017. doi: 10.1002/jcp.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barkauskas CE, Chung M, Fioret B, Gao X, Katsura H, Hogan BLM. Lung organoids: current uses and future promise. Development 144: 986–997, 2017. doi: 10.1242/dev.140103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes PJ Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 138: 16–27, 2016. doi: 10.1016/j.jaci.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Barnes PJ Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev 68: 788–815, 2016. doi: 10.1124/pr.116.012518. [DOI] [PubMed] [Google Scholar]
- 9.Barnes PJ Oxidative stress-based therapeutics in COPD. Redox Biol 33: 101544, 2020. doi: 10.1016/j.redox.2020.101544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes PJ Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 18: 454–466, 2018. doi: 10.1038/s41577-018-0006-6. [DOI] [PubMed] [Google Scholar]
- 11.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee H, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA, Ingber DE. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 13: 151–157, 2016. doi: 10.1038/nmeth.3697. [DOI] [PubMed] [Google Scholar]
- 12.Bihlet AR, Karsdal MA, Sand JMB, Leeming DJ, Roberts M, White W, Bowler R. Biomarkers of extracellular matrix turnover are associated with emphysema and eosinophilic-bronchitis in COPD. Respir Res 18: 22, 2017. doi: 10.1186/s12931-017-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calverley PMA, Rabe KF, Goehring U-M, Kristiansen S, Fabbri LM, Martinez FJ. M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 374: 685–694, 2009. doi: 10.1016/S0140-6736(09)61255-1. [DOI] [PubMed] [Google Scholar]
- 14.Castaldi PJ, Guo F, Qiao D, Du F, Naing ZZC, Li Y, Pham B, Mikkelsen TS, Cho MH, Silverman EK, Zhou X. Identification of functional variants in the FAM13A chronic obstructive pulmonary disease genome-wide association study locus by massively parallel reporter assays. Am J Respir Crit Care Med 199: 52–61, 2019. doi: 10.1164/rccm.201802-0337OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charron CE, Russell P, Ito K, Lea S, Kizawa Y, Brindley C, Singh D. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-α and -γ selectivity, suppresses COPD inflammation. Eur Respir J 50: 1700188, 2017. doi: 10.1183/13993003.00188-2017. [DOI] [PubMed] [Google Scholar]
- 16.Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen A-L, Kensler TW, Dinkova-Kostova AT. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18: 295–317, 2019. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 17.D'Anna C, Cigna D, Di Sano C, Di Vincenzo S, Dino P, Ferraro M, Bini L, Bianchi L, Di Gaudio F, Gjomarkaj M, Pace E. Exposure to cigarette smoke extract and lipopolysaccharide modifies cytoskeleton organization in bronchial epithelial cells. Exp Lung Res 43: 347–358, 2017. doi: 10.1080/01902148.2017.1377784. [DOI] [PubMed] [Google Scholar]
- 18.De Smet EG, Van Eeckhoutte HP, Avila Cobos F, Blomme E, Verhamme FM, Provoost S, Verleden SE, Venken K, Maes T, Joos GF, Mestdagh P, Brusselle GG, Bracke KR. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol 13: 423–436, 2020. doi: 10.1038/s41385-019-0241-6. [DOI] [PubMed] [Google Scholar]
- 19.Dianat M, Radan M, Badavi M, Mard SA, Bayati V, Ahmadizadeh M. Crocin attenuates cigarette smoke-induced lung injury and cardiac dysfunction by anti-oxidative effects: the role of Nrf2 antioxidant system in preventing oxidative stress. Respir Res 19: 58, 2018. doi: 10.1186/s12931-018-0766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghorani V, Boskabady MH, Khazdair MR, Kianmeher M. Experimental animal models for COPD: a methodological review. Tob Induc Dis 15, 2017. doi: 10.1186/s12971-017-0130-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh A, Boucher RC, Tarran R. Airway hydration and COPD. Cell Mol Life Sci 72: 3637–3652, 2015. doi: 10.1007/s00018-015-1946-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease 2020 report [Online]. Global Initiative for Chronic Obstructive Lung Disease. https://goldcopd.org/wp-content/uploads/2019/11/GOLD-2020-REPORT-ver1.1wms.pdf [20 July 2020]. [Google Scholar]
- 23.Green RM, Gally F, Keeney JG, Alper S, Gao B, Han M, Martin RJ, Weinberger AR, Case SR, Minor MN, Chu HW. Impact of cigarette smoke exposure on innate immunity: a Caenorhabditis elegans model. PLoS One 4: e6860, 2009. doi: 10.1371/journal.pone.0006860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front Genet 10, 2019. doi: 10.3389/fgene.2019.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedström U, Hallgren O, Öberg L, DeMicco A, Vaarala O, Westergren-Thorsson G, Zhou X. Bronchial extracellular matrix from COPD patients induces altered gene expression in repopulated primary human bronchial epithelial cells. Sci Rep 8: 3502, 2018. doi: 10.1038/s41598-018-21727-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiemstra PS, Tetley TD, Janes SM. Airway and alveolar epithelial cells in culture. Eur Respir J 54: 1900742, 2019. doi: 10.1183/13993003.00742-2019. [DOI] [PubMed] [Google Scholar]
- 27.Jia R, Zhang H, Yang Z, Zhao H, Liu F, Wang H, Miao M, Wang Q, Liu Y. Protective effects of Schisandrin B on cigarette smoke-induced airway injury in mice through Nrf2 pathway. Int Immunopharmacol 53: 11–16, 2017. doi: 10.1016/j.intimp.2017.09.030. [DOI] [PubMed] [Google Scholar]
- 28.Jiang J, Zhang S, Shen H, Guan Y, Liu Q, Zhao W, Jia Y, Shen J, Yan X, Xie Q. Rac1 signaling regulates cigarette smoke-induced inflammation in the lung via the Erk1/2 MAPK and STAT3 pathways. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1863: 1778–1788, 2017. doi: 10.1016/j.bbadis.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 29.Jiao Z, Chang J, Li J, Nie D, Cui H, Guo D. Sulforaphane increases Nrf2 expression and protects alveolar epithelial cells against injury caused by cigarette smoke extract. Mol Med Rep 16: 1241–1247, 2017. doi: 10.3892/mmr.2017.6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim H-T, Yin W, Jin Y-J, Panza P, Gunawan F, Grohmann B, Buettner C, Sokol AM, Preussner J, Guenther S, Kostin S, Ruppert C, Bhagwat AM, Ma X, Graumann J, Looso M, Guenther A, Adelstein RS, Offermanns S, Stainier DYR. Myh10 deficiency leads to defective extracellular matrix remodeling and pulmonary disease. Nat Commun 9: 4600, 2018. doi: 10.1038/s41467-018-06833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187: 228–237, 2013. doi: 10.1164/rccm.201210-1843CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kliment CR, Nguyen JMK, Kaltreider MJ, Lu Y, Claypool SM, Radder JE, Sciurba FC, Zhang Y, Gregory AD, Iglesias PA, Sidhaye VK, Robinson DN. Adenine nucleotide translocase regulates the airway epithelium, mitochondrial metabolism and ciliary function. bioRxiv, 2020. doi: 10.1101/2020.05.18.101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosmider B, Lin C-R, Karim L, Tomar D, Vlasenko L, Marchetti N, Bolla S, Madesh M, Criner GJ, Bahmed K. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. EBioMedicine 46: 305–316, 2019. doi: 10.1016/j.ebiom.2019.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koziol-White C, Johnstone TB, Corpuz ML, Cao G, Orfanos S, Parikh V, Deeney B, Tliba O, Ostrom RS, Dainty I, Panettieri RA. Budesonide enhances agonist-induced bronchodilation in human small airways by increasing cAMP production in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 318: L345–L355, 2020. doi: 10.1152/ajplung.00393.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lea S, Metryka A, Li J, Higham A, Bridgewood C, Villetti G, Civelli M, Facchinetti F, Singh D. The modulatory effects of the PDE4 inhibitors CHF6001 and roflumilast in alveolar macrophages and lung tissue from COPD patients. Cytokine 123: 154739, 2019. doi: 10.1016/j.cyto.2019.154739. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Lee J, Hong S-H, Rahman I, Yang S-R. Inhibition of RAGE attenuates cigarette smoke-induced lung epithelial cell damage via RAGE-mediated Nrf2/DAMP signaling. Front Pharmacol 9: 684, 2018. doi: 10.3389/fphar.2018.00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu G, Betts C, Cunoosamy DM, Åberg PM, Hornberg JJ, Sivars KB, Cohen TS. Use of precision cut lung slices as a translational model for the study of lung biology. Respi Res 20: 162, 2019. doi: 10.1186/s12931-019-1131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.López‐Campos JL, Tan W, Soriano JB. Global burden of COPD. Respirology 21: 14–23, 2016. doi: 10.1111/resp.12660. [DOI] [PubMed] [Google Scholar]
- 39.Marklew AJ, Patel W, Moore PJ, Tan CD, Smith AJ, Sassano MF, Gray MA, Tarran R. Cigarette smoke exposure induces retrograde trafficking of CFTR to the endoplasmic reticulum. Sci Rep 9: 13655, 2019. doi: 10.1038/s41598-019-49544-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore PJ, Reidel B, Ghosh A, Sesma J, Kesimer M, Tarran R. Cigarette smoke modifies and inactivates SPLUNC1, leading to airway dehydration. FASEB J 32: 6559–6574, 2018. doi: 10.1096/fj.201800345R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moussa BA, El-Zaher AA, El-Ashrey MK, Fouad MA. Synthesis and molecular docking of new roflumilast analogues as preferential-selective potent PDE-4B inhibitors with improved pharmacokinetic profile. Eur J Med Chem 148: 477–486, 2018. doi: 10.1016/j.ejmech.2018.02.038. [DOI] [PubMed] [Google Scholar]
- 42.Ng-Blichfeldt JP, Gosens R, Dean C, Griffiths M, Hind M. Regenerative pharmacology for COPD: breathing new life into old lungs. Thorax 74: 890–897, 2019. doi: 10.1136/thoraxjnl-2018-212630. [DOI] [PubMed] [Google Scholar]
- 43.Nishida K, Brune KA, Putcha N, Mandke P, O'Neal WK, Shade D, Srivastava V, Wang M, Lam H, An SS, Drummond MB, Hansel NN, Robinson DN, Sidhaye VK. Cigarette smoke disrupts monolayer integrity by altering epithelial cell-cell adhesion and cortical tension. Am J Physiol Lung Cell Mol Physiol 313: L581–L591, 2017. doi: 10.1152/ajplung.00074.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel NR, Cunoosamy DM, Fagerås M, Taib Z, Asimus S, Hegelund-Myrbäck T, Lundin S, Pardali K, Kurian N, Ersdal E, Kristensson C, Korsback K, Palmér R, Brown MN, Greenaway S, Siew L, Clarke GW, Rennard SI, Make BJ, Wise RA, Jansson P. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. COPD 13: 1009–1019, 2018. doi: 10.2147/COPD.S150576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phillips JE Inhaled phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front Pharmacol 11: 259, 2020. doi: 10.3389/fphar.2020.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Posso SV, Quesnot N, Moraes JA, Brito-Gitirana L, Kennedy-Feitosa E, Barroso MV, Porto LC, Lanzetti M, Valença SS. AT-RVD1 repairs mouse lung after cigarette smoke-induced emphysema via downregulation of oxidative stress by NRF2/KEAP1 pathway. Int Immunopharmacol 56: 330–338, 2018. doi: 10.1016/j.intimp.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 47.Prange R, Thiedmann M, Bhandari A, Mishra N, Sinha A, Häsler R, Rosenstiel P, Uliczka K, Wagner C, Yildirim AÖ, Fink C, Roeder T. A Drosophila model of cigarette smoke induced COPD identifies Nrf2 signaling as an expedient target for intervention. Aging (Albany NY) 10: 2122–2135, 2018. doi: 10.18632/aging.101536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakkas LI, Mavropoulos A, Bogdanos DP. Phosphodiesterase 4 inhibitors in immune-mediated diseases: mode of action, clinical applications, current and future perspectives. Curr Med Chem 24: 3054–3067, 2017. doi: 10.2174/0929867324666170530093902. doi: 10.2174/0929867324666170530093902. [DOI] [PubMed] [Google Scholar]
- 49.Sekine T, Hirata T, Ishikawa S, Ito S, Ishimori K, Matsumura K, Muraki K. Regulation of NRF2, AP-1 and NF-κB by cigarette smoke exposure in three-dimensional human bronchial epithelial cells. J Appl Toxicol 39: 717–725, 2019. doi: 10.1002/jat.3761. [DOI] [PubMed] [Google Scholar]
- 50.Shibata S, Miyake K, Tateishi T, Yoshikawa S, Yamanishi Y, Miyazaki Y, Inase N, Karasuyama H. Basophils trigger emphysema development in a murine model of COPD through IL-4-mediated generation of MMP-12-producing macrophages. Proc Natl Acad Sci USA 115: 13057–13062, 2018. doi: 10.1073/pnas.1813927115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shrestha J, Bazaz SR, Es HA, Azari DY, Thierry B, Warkiani ME, Ghadiri M. Lung-on-a-chip: the future of respiratory disease models and pharmacological studies. Crit Rev Biotechnol 40: 213–230, 2020. doi: 10.1080/07388551.2019.1710458. [DOI] [PubMed] [Google Scholar]
- 52.Sidhaye VK, Holbrook JT, Burke A, Sudini KR, Sethi S, Criner GJ, Fahey JW, Berenson CS, Jacobs MR, Thimmulappa R, Wise RA, Biswal S. Compartmentalization of anti-oxidant and anti-inflammatory gene expression in current and former smokers with COPD. Respir Res 20: 190, 2019. doi: 10.1186/s12931-019-1164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sidhaye VK, Nishida K, Martinez FJ. Precision medicine in COPD: where are we and where do we need to go? Eur Respir Rev 27: 180022, 2018. doi: 10.1183/16000617.0022-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh D, Abbott-Banner K, Bengtsson T, Newman K. The short-term bronchodilator effects of the dual phosphodiesterase 3 and 4 inhibitor RPL554 in COPD. Eur Respir J 52: 1801074, 2018. doi: 10.1183/13993003.01074-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singh D, Beeh KM, Colgan B, Kornmann O, Leaker B, Watz H, Lucci G, Geraci S, Emirova A, Govoni M, Nandeuil MA. Effect of the inhaled PDE4 inhibitor CHF6001 on biomarkers of inflammation in COPD. Respir Res 20: 180, 2019. doi: 10.1186/s12931-019-1142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M, Novellino E, Brusselle GG, Wagner DE, Yildirim AÖ, Königshoff M. Reduced frizzled receptor 4 expression prevents WNT/β-catenin–driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 196: 172–185, 2017. doi: 10.1164/rccm.201605-0904OC. [DOI] [PubMed] [Google Scholar]
- 57.Sun Z, Li F, Zhou X, Chung KF, Wang W, Wang J. Stem cell therapies for chronic obstructive pulmonary disease: current status of pre-clinical studies and clinical trials. J Thorac Dis 10: 1084–1098, 2018. doi: 10.21037/jtd.2018.01.46. doi: 10.21037/jtd.2018.01.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sundar IK, Maremanda KP, Rahman I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol Lett 317: 92–101, 2019. doi: 10.1016/j.toxlet.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Surcel A, Ng WP, West-Foyle H, Zhu Q, Ren Y, Avery LB, Krenc AK, Meyers DJ, Rock RS, Anders RA, Meyers CLF, Robinson DN. Pharmacological activation of myosin II paralogs to correct cell mechanics defects. Proc Natl Acad Sci U S A 112: 1428–1433, 2015. doi: 10.1073/pnas.1412592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 122: 2749–2755, 2012. doi: 10.1172/JCI60324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wise RA, Holbrook JT, Criner G, Sethi S, Rayapudi S, Sudini KR, Sugar EA, Burke A, Thimmulappa R, Singh A, Talalay P, Fahey JW, Berenson CS, Jacobs MR, Biswal S; Broccoli Sprout Extract Trial Research Group. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: a randomized, double-blind, placebo controlled trial. PLoS ONE 11: e0163716, 2016. doi: 10.1371/journal.pone.0163716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu J, Murphy SL, Kochanek KD, Arias E. Mortality in the United States, 2018. CDC; https://www.cdc.gov/nchs/data/databriefs/db355-h.pdf [20 July 2020]. [PubMed] [Google Scholar]
- 63.Yanagisawa H, Hashimoto M, Minagawa S, Takasaka N, Ma R, Moermans C, Ito S, Araya J, Budelsky A, Goodsell A, Baron JL, Nishimura SL. Role of IL-17A in murine models of COPD airway disease. Am J Physiol Lung Cell Mol Physiol 312: L122–L130, 2017. doi: 10.1152/ajplung.00301.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang M, Shi R, Zhang Y, Shan H, Zhang Q, Yang X, Li Y, Zhang J. Nix/BNIP3L-dependent mitophagy accounts for airway epithelial cell injury induced by cigarette smoke. J Cell Physiol 234: 14210–14220, 2019. doi: 10.1002/jcp.28117. [DOI] [PubMed] [Google Scholar]
- 65.Zhang M, Tang J, Shan H, Zhang Q, Yang X, Zhang J, Li Y. p66Shc mediates mitochondrial dysfunction dependent on PKC activation in airway epithelial cells induced by cigarette smoke. Oxid Med Cell Longev 2018: 5837123, 2018., doi: 10.1155/2018/5837123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zou S-C, Pang L-L, Mao Q-S, Wu S-Y, Xiao Q-F. IL-9 exacerbates the development of chronic obstructive pulmonary disease through oxidative stress. Eur Rev Med Pharmacol Sci 22: 8877–8884, 2018. doi: 10.26355/eurrev_201812_16656. doi: 10.26355/eurrev_201812_16656. [DOI] [PubMed] [Google Scholar]
- 67.Zscheppang K, Berg J, Hedtrich S, Verheyen L, Wagner DE, Suttorp N, Hippenstiel S, Hocke AC. Human pulmonary 3D models for translational research. Biotechnol J 13: 1700341, 2018. doi: 10.1002/biot.201700341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zuo H, Faiz A, van den Berge M, Mudiyanselage SNHR, Borghuis T, Timens W, Nikolaev VO, Burgess JK, Schmidt M. Cigarette smoke exposure alters phosphodiesterases in human structural lung cells. Am J Physiol Lung Cell Mol Physiol 318: L59–L64, 2020. doi: 10.1152/ajplung.00319.2019. [DOI] [PubMed] [Google Scholar]
- 69.Zuo H, Han B, Poppinga WJ, Ringnalda L, Kistemaker LEM, Halayko AJ, Gosens R, Nikolaev VO, Schmidt M. Cigarette smoke up-regulates PDE3 and PDE4 to decrease cAMP in airway cells. Br J Pharmacol 175: 2988–3006, 2018. doi: 10.1111/bph.14347. [DOI] [PMC free article] [PubMed] [Google Scholar]