

Abstract
Adropin is a nutritionally regulated peptide hormone, secreted primarily by the liver, which modulates metabolic homeostasis in a number of tissues. Growing evidence suggests that adropin is an important regulatory component in a number of cardiovascular pathologies, and may be central to the control of cardiac fuel metabolism and vascular function. In this mini-review, we examine the known facets of adropin biology, discuss open questions in the field, and speculate on the therapeutic potential of targeting adropin-related signaling pathways in cardiovascular diseases.
Keywords: adropin, cardiac metabolism, endothelial biology, energy homeostasis, vascular function
INTRODUCTION
Multiple classes of hormones regulate cellular function and homeostasis in the heart and vascular system. Each class is structurally distinct and exhibits different modes of action, such that peptide, catecholamine, and eicosanoid hormones act via plasma membrane receptors, whereas steroid, retinoid, and thyroid hormones act through nuclear receptors. Hormones in the cardiovascular system contribute to homeostasis by regulating hemodynamic effects such as systemic vascular resistance, heart rate, vascular tone, and cardiac output (1), and nonhemodynamic effects such as inflammation, reactive oxygen species production, and metabolic control (2). Adropin is a recently identified peptide hormone regulated by fasting and changes in dietary macronutrients and serves a physiological role in energy homeostasis and metabolic regulation (3). In particular, adropin demonstrates systemic effects on insulin sensitivity and glucose metabolism in the heart, liver, and skeletal muscle. A growing number of studies have shown that adropin has important cardiovascular functions, and our understanding of these regulatory effects have rapidly increased in the past three years. Adropin has been shown to regulate cardiac energy substrate flexibility (4, 5) and protect vascular endothelial cells from damage (6, 7). These studies suggest that stimulation of adropin signaling may be beneficial for overall cardiovascular function, and further work is underway to elucidate the molecular pathways underlying these outcomes. This review highlights the known cardiovascular effects of adropin and discusses its potential therapeutic role in disorders such as diabetic cardiomyopathy, atherosclerosis, and heart failure.
BACKGROUND ON ADROPIN BIOLOGY
Adropin is a highly conserved 76-amino acid peptide hormone encoded by the energy homeostasis-associated gene (Enho), which is primarily expressed in the liver and brain (3). Adropin was initially identified through microarray screening for differentially expressed genes in melanocortin-3 receptor-deficient (Mc3r−/−) mice with hypothalamic obesity. The authors of this original study determined that circulating adropin levels are regulated by energy balance states and dietary macronutrients (3). Hepatic Enho expression is downregulated in the livers of high-fat diet-induced obesity (DIO), Mc3r−/−, and leptin-deficient mice. Transgenic overexpression or systemic adropin treatment in DIO mice attenuated glucose intolerance, insulin resistance, and hepatosteatosis independent of body weight, energy expenditure, or food intake. It was also observed that liver Enho mRNA expression was regulated by LXRα, a nuclear receptor involved in cholesterol and triglyceride metabolism, linking adropin function to carbohydrate and lipid metabolism (3). Overall, the authors concluded that adropin plays an essential role in glucose homeostasis and lipid metabolism, protecting against hepatic steatosis and hyperinsulinemia associated with obesity.
Later studies determined that adropin acts as a secretory peptide, whose action is transmitted to peripheral tissues through extracellular receptors. Amino acids 1–33 are considered to encode the secretory signal peptide, with residues 34–76 being the biologically active secreted domain (3). Human, mouse, and rat adropin amino acid sequences are 100% identical (3), and three putative membrane receptors have been reported so far (Fig. 1). In studies to determine its signaling pathway, adropin was initially characterized as a plasma membrane-binding protein, which interacts with the Notch signaling pathway in neurons to modulate intracellular communication during nervous system development (8). In this study, it was shown that adropin interacts with the brain-specific, membrane-tethered, Notch1 ligand NB-3/Contactin 6. Adropin knockout mice displayed impaired motor coordination and synapse formation in the cerebellum, similar to the NB-3 knockout mice phenotype (8), suggesting that adropin regulates physical activity and motor coordination through NB-3-induced activation of Notch signaling (8). More recently, it was shown that adropin potentially interacts with an orphan G-protein-coupled receptor (GPR19) in the brain to inhibit water drinking (9). Using bioinformatics, mRNA screening, and a “deductive reasoning strategy,” (10) GPR19 was identified from the existing list (IUPHAR) of orphan G-protein-coupled receptors (9). In vivo Grp19 gene silencing using small interfering RNA (siRNA) in the brain attenuated the inhibitory effect of adropin on water deprivation-induced drinking, suggesting an effect on central physiological regulation.
Figure 1.

Adropin regulates various signaling pathways to enhance insulin sensitivity, glucose metabolism, motor coordination, and endothelial function. In neurons (A), adropin binds to the Notch ligand NB3 to activate Notch1 signaling and regulate brain development. In addition, adropin activates mitogen-activated protein kinase (MAPK) signaling in endothelial cells (B) through either the VEGF receptor 2 (VEGFR2) [to increase nitric oxide (NO) bioavailability via endothelial NO synthase (eNOS) upregulation], or in cardiomyocytes (C) through G-protein coupled receptor 19 (GPR19) [to regulate pyruvate dehydrogenase kinase 4 (PDK4) expression and increase pyruvate dehydrogenase (PDH) activity]. Note: In this figure, VEGFR2 is represented as a plasma membrane receptor, but it has not yet been determined whether it acts as an extracellular receptor, or as part of an endosomal pathway, in adropin signaling (6). Figure produced with license using Biorender software.
Subsequent studies have strengthened the case for adropin as an endogenous ligand for GPR19. Rao and Herr (11) investigated the pathophysiological relevance of GPR19 in breast cancer metastasis and discovered that GPR19 overexpression upregulates E-cadherin, an essential molecule for cell-cell adhesion. Cell-based assays showed that adropin activates GPR19 via MAP kinase (MAPK) signaling, causing phenotypic and functional changes such as decreased cell invasion (11). More recently, Thapa et al. (12) determined that adropin regulates pyruvate dehydrogenase in cardiac-derived H9c2 cells through a novel GPCR-MAPK-PDK4 signaling pathway. Adropin activates GPR19 to stimulate p44/42 phosphorylation, consequently decreasing pyruvate dehydrogenase kinase 4 (PDK4) expression and the inhibitory phosphorylation of pyruvate dehydrogenase (PDH), to regulate cardiac cell bioenergetics. Knockdown of GPR19 in vitro blocked the effects of adropin treatment on PDK4, suggesting that GPR19 acts as the putative cellular receptor for adropin in cardiac cells.
Lovren et al. (6) showed that VEGFR2 was required for adropin signaling in cardiac and noncardiac endothelial cells, thereby suggesting that this protein may act as a third receptor for adropin (Fig. 1). The endothelial effects of adropin signaling are discussed in the following sections.
REGULATION OF VASCULAR FUNCTION BY ADROPIN
Adropin was first linked to cardiovascular homeostasis as a novel regulator of endothelial function (6). This study determined that adropin is constitutively expressed in endothelial cells, and promotes critical functions such as proliferation, migration, and capillary tube formation while attenuating vascular permeability and TNF-α-induced apoptosis. Adropin exerts an endothelial protective effect by increasing nitric oxide (NO) synthesis through activation of endothelial NO synthase (eNOS). Specific inhibitors for Akt (LY294002) and Erk1/2 (PD98059) blocked eNOS activation by adropin, suggesting that adropin-driven NO production via eNOS is Akt- and Erk1/2-dependent. Consequently, it was determined that adropin modulates eNOS bioactivity through upstream activation of VEGFR2, an essential receptor that regulates endothelial cell function and angiogenesis, with resultant activation of PI3K-Akt and ERK1/2 signaling pathways. The authors did not determine whether VEGFR2-mediated adropin signaling was endosomal, or transduced through the plasma membrane, raising the question if VEGFR2 acts as a physiological receptor or pathway regulator. To further support the role of adropin in mediating endothelial function, in vivo studies demonstrated that adropin stimulates neovascularization in an eNOS-dependent manner (6).
As a regulator of endothelial function, adropin influences different hemodynamic variables such as blood flow, vascular dilation, and mean arterial pressure. Adropin improved limb perfusion following hindlimb ischemia in wild-type mice, but not eNOS knockout mice, suggesting that the resultant activation of eNOS by adropin increased skeletal muscle blood flow (6). However, Stein et al. (9) observed that there was no significant difference in mean arterial pressure upon the central administration of adropin to the brain via an indwelling lateral cerebroventricle cannula in conscious, unrestrained male rats. The relationship between adropin and blood pressure has been explored in various human studies with conflicting results. Altincik et al. (13) showed that serum adropin levels have no correlation to blood pressure variables in obese children, whereas a cross-sectional study demonstrated that plasma adropin levels are negatively correlated with blood pressure in hypertensive adult patients (14). In direct contrast, Celik et al. (15) observed that patients with hypertension exhibit high levels of adropin even after antihypertensive treatment. Together, these studies suggest that adropin influences hemodynamic parameters to potentially serve as an independent predictor of vascular endothelial dysfunction. However, further work that goes beyond correlative studies will be required to determine the exact function of adropin in human endothelial function.
Vascular stiffening is a hallmark of normal aging and an outcome of several cardiovascular pathologies such as systolic hypertension, stroke, atherosclerosis, and heart failure. Aging-induced arterial stiffening is reduced by aerobic exercise training, which is driven by elevated production of nitric oxide (16). The potential association between adropin and exercise training-induced changes in arterial stiffness was investigated by Fujie et al. (17). In an initial cross-sectional study, it was observed that serum adropin levels negatively correlated with carotid stiffness, and positively correlated with plasma nitric oxide levels and cardiorespiratory fitness in middle-aged and older adults. To examine the effect of aerobic exercise training, an interventional study was performed, which demonstrated that training induced increases in adropin that correlated with reduced arterial stiffness and increased plasma NOx levels. The data from these studies suggest that an increase in adropin induced by aerobic exercise training may participate in the upregulation of arterial eNOS, consequently alleviating arterial stiffness in middle-aged and older adults (17).
Atherosclerosis is a chronic inflammatory condition of the arteries, characterized by excessive fibrosis of the intima, hyperlipidemia, fatty plaques formation, infiltration of macrophages, proliferation of smooth muscle cells (SMC), accumulation of connective tissue components, and formation of thrombus (18). Sato et al. (7) demonstrated that adropin suppresses atherosclerosis by attenuating the inflammatory response of endothelial cells and monocyte-derived macrophages, monocyte-endothelial cell adhesion, and the migration and proliferation of vascular smooth muscle cells (VSMCs). Adropin regulates monocyte differentiation, specifically its anti-inflammatory phenotype, by the upregulation of PPAR-γ expression (7). However, it remains to be fully determined whether adropin directly exerts anti-inflammatory effects, or if these are mediated by the increase in endothelial NO production. Furthermore, Sato et al. (7) observed that adropin increased fibronectin and elastin expression in VSMCs via upregulation of the PI3K-Akt pathway, suggesting that adropin modulates plaque stability and vascular elasticity. This conclusion was further supported by in vivo studies, in which adropin infusion significantly reduced the development of atherosclerotic lesions in Apoe−/− mice, independent of metabolic defects and blood pressure (7).
SYSTEMIC PHYSIOLOGICAL EFFECTS OF ADROPIN
In the first study to address the role of adropin in energy metabolism outside of the liver, Gao et al. (19) demonstrated that adropin mediates substrate oxidation preference in skeletal muscle by regulating PDH activity. Indirect calorimetry showed that adropin knockout (AdrKO) mice preferentially oxidize fat over carbohydrates, whereas adropin transgenic (AdrTG) mice favor glucose oxidation over fatty acid oxidation (FAO). Mechanistically, these changes correlated with a decrease in carnitine palmitoyltransferase-1B (CPT1B) activity, a mitochondrial fatty acid translocase protein that is essential for mitochondrial FAO in muscle. Consistent with the preference for glucose oxidation in AdrTG mice, it was observed that adropin stimulates skeletal muscle pyruvate oxidation by reducing PDK4 levels (19). To further validate the observed effects of adropin, administration of synthetic adropin resulted in increased acetylation of PGC-1α, inhibited CPT1B activity, decreased PDK4 expression, and increased PDH activity (19).
In an elegant follow-up study by the same group, Gao et al. (20) investigated the therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obesity (DIO) with insulin resistance. The DIO state is characterized by dysregulation of glucose and fatty acid metabolism, and it was hypothesized that adropin treatment would exert therapeutic effects by regulating insulin actions to ameliorate metabolic inflexibility. Synthetic adropin peptide (34–76) improved glucose tolerance and alleviated whole-body insulin resistance in DIO mice. This was achieved by enhancing glucose oxidation and distal insulin signaling pathways in skeletal muscle. In addition, adropin treatment reduced metabolites from incomplete FAO (which arise from a diet-induced impairment of FAO activity), increased glycolytic flux through oxidative and nonoxidative pathways, and decreased muscle fatty acid uptake by downregulating the sarcolemmal fatty acid translocase, CD36. Interestingly, the authors further observed that adropin induces expression of HES1 in skeletal muscle, which consequently represses PGC-1α, a central regulator of energy metabolism. As HES1 is a target of Notch1 signaling, this finding may suggest a novel link between adropin function and the Notch pathway beyond the brain (8, 20). Overall, adropin therapy improves oxidative glucose uptake and utilization, while limiting fat oxidation and amplifying skeletal muscle insulin signaling actions (20), suggesting that adropin signaling may have a therapeutic benefit in glucose metabolism disorders.
REGULATION OF CARDIAC ENERGY METABOLISM BY ADROPIN
The comprehensive research conducted by Butler et al. (19, 20), mentioned above, raised questions of whether adropin could regulate energy metabolism in other striated muscle types, specifically cardiac muscle. Cardiomyocytes preferentially utilize fatty acids as a carbon source to sustain contractile performance (21). First, Thapa et al. (5) investigated whether adropin could regulate cardiac substrate metabolism following prolonged exposure to a high-fat diet in mice. It was observed that adropin treatment restores cardiac glucose oxidation in DIO mice by increasing PDH activity through reduced inhibitory lysine acetylation. Adropin reduced the expression of the mitochondrial acetyltransferase GCN5L1, resulting in decreased PDH acetylation and increased cardiac glucose utilization in DIO mice in vivo (5). In a contemporaneous study, Altamimi et al. (4) explored the influence of adropin on cardiac energy metabolism, insulin signaling, and cardiac efficiency ex vivo. It was shown that adropin treatment improved ex vivo cardiac function and efficiency, concomitant with enhanced inhibition of FAO by insulin. In addition, adropin stimulated downstream activation of cardiac insulin signaling via MAPK and FOXO1 signaling (4). The authors concluded that adropin acts on its putative receptor, GPR19, to reduce PDK4 protein levels and to activate MAPK signaling, consequently stimulating PDH activity to enhance glucose oxidation. This may occur in concert with the activation of downstream insulin signaling, thereby improving glucose uptake in addition to utilization in lean mice (4).
LOOKING INTO THE FUTURE: THERAPEUTIC POTENTIAL OF THE ADROPIN SIGNALING PATHWAY
A number of papers published recently have shown that circulating adropin levels correlate with several disease states in humans, and generally show that reduced adropin levels are linked with enhanced disease progression (e.g., 14, 17). These studies indicate that reduced adropin levels may be a promising biomarker for cardiovascular diseases, and this arena is likely to be the main focus of adropin translational studies for the foreseeable future. Although much more needs to be known about the biological pathways underlying adropin function, there are several possible avenues that may lead to a therapeutic role for adropin signaling in the long term.
Significantly reduced adropin levels are observed in obese and diabetic mice, suggesting that adropin levels may be linked to metabolic homeostasis (3). Recent work from our group has demonstrated that acute adropin treatment can restore cardiac glucose oxidation in prediabetic obese mice (5), suggesting that adropin may have a therapeutic role in treating cardiometabolic diseases. Diabetic cardiomyopathy (DCM) is a clinical condition of ventricular dysfunction that occurs in the absence of other cardiac risk factors (such as coronary artery disease, hypertension, and dyslipidemia) in individuals with diabetes. DCM is initially characterized by myocardial fibrosis, tissue remodeling, and associated diastolic dysfunction, which eventually evolves into decreased cardiac output and clinical heart failure (22). A myriad of cardiac metabolic abnormalities such as hyperglycemia, systemic insulin resistance, impaired cardiac insulin signaling, oxidative stress, and mitochondrial dysfunction have been implicated in the development and progression of DCM. Patients with DCM display increased free fatty acid (FFA) levels and decreased myocardial glucose uptake, which lead to the reliance on fatty acid oxidation (23). Increased FFA availability leads to the upregulation of CD36, elevated PPARα levels, and impaired glucose uptake through GLUT4 (23). Studies show that adropin reverses this phenotype in skeletal muscle of DIO mice (20) and ex vivo hearts from lean nonfasting mice (4), by increasing glycolytic substrate flux through upregulated insulin pathways. This indicates that adropin may improve metabolic flexibility by stimulating glucose oxidation in the diabetic heart, which could potentially mitigate the effects of DCM. Although this may be useful in DCM, it should be noted that it is unclear whether this would be a beneficial feature in the failing heart, whether studies have shown that insulin resistance can have a beneficial role in preventing cardiac glucotoxicity (24). Numerous studies have been conducted to address potential energy deficits in DCM by pharmacological inhibition of cardiomyocyte fatty acid oxidation; however, drugs developed for this purpose so far (25) have had limited efficacy as primary therapy, often due to off-target effects. The use of adropin to directly enhance glucose oxidation may be an alternative approach to treat DCM, with an endogenous peptide potentially helping to circumvent deleterious off-target effects. However, the therapeutic delivery of peptide hormones is restricted by their pharmacokinetics and potential for proteolytic degradation, so the development of small-molecule mimetics that activate the adropin signaling pathway may be required to circumvent these limitations.
Adropin has been shown to modulate endothelial function via upregulation of eNOS (6), thereby increasing the bioavailability of NO and contributing to decreased arterial stiffness (17). Studies on the correlation between adropin and the pathogenesis of cardiovascular diseases have mainly concentrated on the protection and functional regulation of endothelial cells. Sato et al. (7) showed that adropin exerts antiatherosclerotic effects by suppressing monocyte-endothelial cell adhesion and smooth muscle cell proliferation. These studies indicate that adropin may be a novel target to limit diseases characterized by endothelial dysfunction, in addition to its effects on metabolism. The pathophysiological mechanisms underlying various cardiovascular diseases involve crucial signaling molecules that are regulated by adropin, which subsequently modulates vascular inflammation, lipotoxicity, insulin resistance, oxidative stress, and substrate use flexibility (22). Harnessing the biological effects of adropin pathway stimulation could lead to the development of new vasoprotective therapeutic approaches.
Finally, the metabolic effects of adropin may be useful in the treatment of pulmonary hypertension. Pulmonary arterial hypertension (PAH) is a hemodynamic state, characterized by elevated pulmonary vascular resistance and proliferative vascular remodeling that obstructs the lumen of small pulmonary arteries (26). Inhibition of PDH is a key phenomenon underlying PAH, which is achieved through several mechanisms. These include induction of PDKs to phosphorylate and downregulate PDH activity, and the inhibitory acetylation of PDH via downregulation of the mitochondrial deacetylase enzyme SIRT3 (27, 28). Michelakis et al. (27) demonstrated that PDK expression is upregulated in the pulmonary arteries of human PAH lungs and is important for metabolic remodeling. Treatment with the PDK inhibitor dichloroacetate (DCA) ex vivo increased PDH activity and mitochondrial function, which is analogous to the effects of adropin treatment (4, 5, 20, 27) and indicates that adropin could theoretically serve as a potential alternative to DCA to improve hemodynamics and functional capacity in PAH patients. Interestingly, DCA treatment of PAH was less effective in patients with single-nucleotide polymorphism (SNP) mutations in SIRT3 (27), a factor that is common in patients with metabolic syndrome (29). The presence of these variants caused PDK-independent inhibition of PDH, which was not responsive to DCA. As adropin can increase the activity of PDH by decreasing its acetylation (5), adropin treatment may circumvent the resistance to DCA in SIRT3 mutant patients by preventing inhibitory PDH hyperacetylation (Fig. 2). Together, these studies suggest that adropin may serve as a novel alternative to DCA for PAH therapy.
Figure 2.
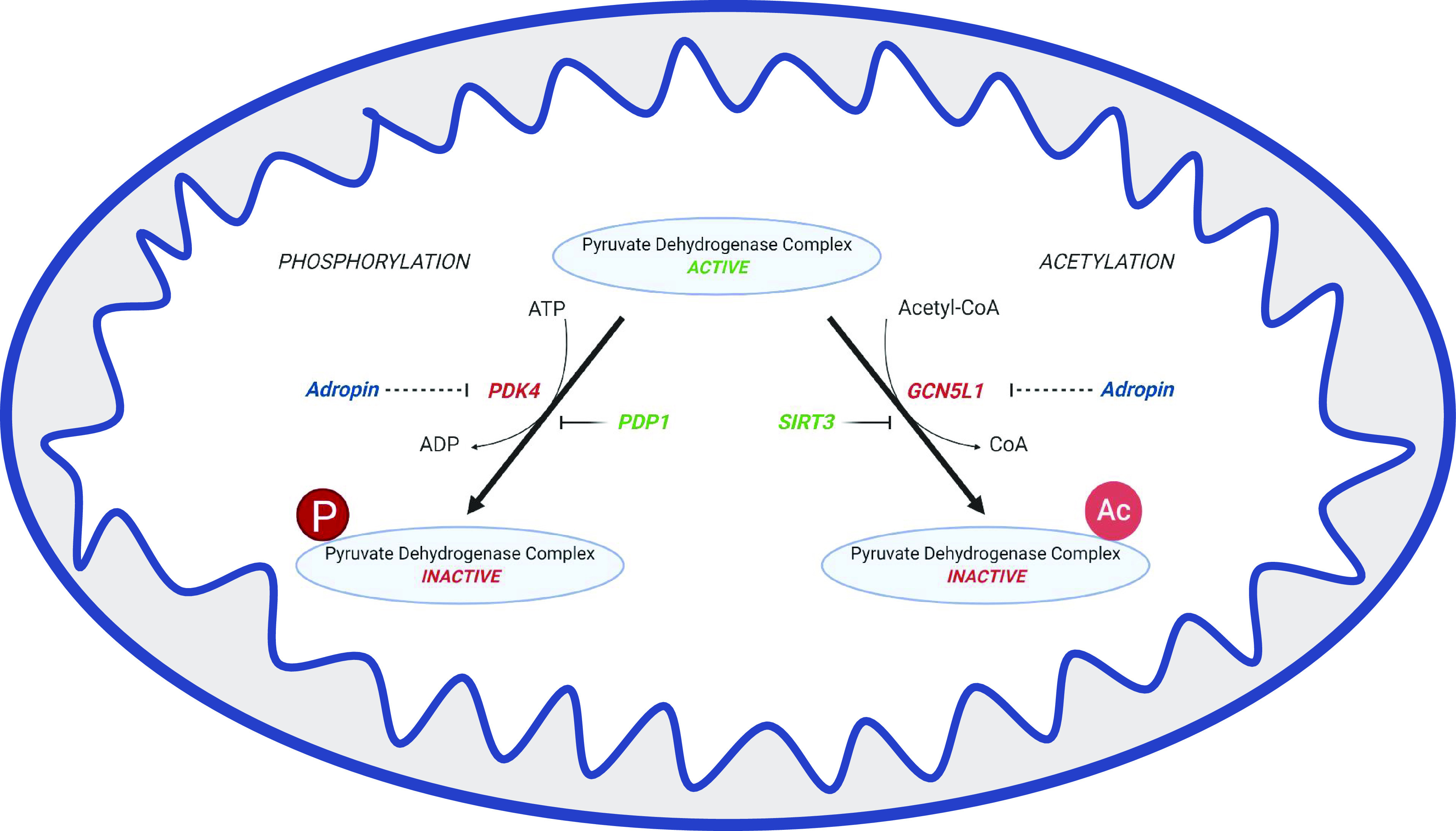
Phosphorylation and lysine acetylation are reversible posttranslational modifications that regulate pyruvate dehydrogenase (PDH) activity, thereby governing mitochondrial bioenergetic output. Pyruvate dehydrogenase kinase 4 (PDK4) inactivates PDH by phosphorylation, which is reversed by pyruvate dehyrogenase phosphotase 1 (PDP1), the sirtuin deacetylase SIRT3 deacetylates PDH to induce mitochondrial respiration, which is counteracted by the activity of the GCN5-like 1 (GCN5L1) acetyltransferase activity. Adropin reduces the expression of both PDK4 and GCN5L1 in different cardiac cell types, resulting in increased PDH activity via reduced inhibitory phosphorylation and/or acetylation.
CONCLUSIONS AND FUTURE DIRECTIONS
Adropin biology is an emerging field of biomedical research whose molecular, biochemical, and structural properties still need to be explored. Also, studies need to be conducted to fully elucidate the signaling mechanisms by which adropin acts, especially in different cardiovascular cell populations. Several studies show that adropin acts on different receptors in various tissues such as in brain tissue, vascular endothelial cells, cardiomyocytes, and fibroblasts to mediate metabolic homeostasis (8, 9, 12). Identification of these receptors in the vasculature, and their downstream signaling actions, will facilitate targeted exploitation of adropin biology for therapeutic purposes in cardiometabolic diseases.
A second open question is whether adropin biology may offer a direct link between liver disease and cardiovascular dysfunction. Exposure to a long-term HFD reduces adropin secretion from the liver (3), and adropin deficiency worsens HFD-induced metabolic defects such as adiposity, hyperglycemia, and insulin resistance (30). Insulin resistance triggers endothelial dysfunction through impaired eNOS activity (31) and is a major factor in both liver disease and cardiovascular dysfunction. Recent studies showed that patients with nonalcoholic fatty liver disease (NAFLD) displayed a marked eNOS dysfunction (32) and a decrease in serum adropin levels (33), which may contribute to a higher cardiovascular risk. Based on these studies, it may be concluded that liver dysfunction reduces adropin expression, which may result in endothelial defects and cardiovascular complications. Future studies to elucidate a causal relationship between adropin, liver disease, and cardiovascular pathology are warranted. In particular, it will be interesting to understand whether a decrease in adropin secretion by the liver acts only as a signaling event that promotes vascular dysfunction, or whether its effects on the liver have direct pleiotropic effects on the development of metabolism-related cardiovascular diseases.
GRANTS
This work was supported by NIH Grants R01HL147861 and R01HL132917 (to I. Scott).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
B.A.S.M. drafted manuscript; B.A.S.M. and I.S. edited and revised manuscript; B.A.S.M. and I.S. approved final version of manuscript.
REFERENCES
- 1.Montani JP, Liard JF, Schoun J, Möhring J. Hemodynamic effects of exogenous and endogenous vasopressin at low plasma concentrations in conscious dogs. Circ Res 47: 346–355, 1980. doi: 10.1161/01.RES.47.3.346. [DOI] [PubMed] [Google Scholar]
- 2.Cooper-DeHoff RM, Pepine CJ. Metabolic syndrome and cardiovascular disease: challenges and opportunities. Clin Cardiol 30: 593–597, 2007. doi: 10.1002/clc.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar KG, Trevaskis JL, Lam DD, Sutton GM, Koza RA, Chouljenko VN, Kousoulas KG, Rogers PM, Kesterson RA, Thearle M, Ferrante AW, Jr., Mynatt RL, Burris TP, Dong JZ, Halem HA, Culler MD, Heisler LK, Stephens JM, Butler AA. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab 8: 468–481, 2008. doi: 10.1016/j.cmet.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Altamimi TR, Gao S, Karwi QG, Fukushima A, Rawat S, Wagg CS, Zhang L, Lopaschuk GD. Adropin regulates cardiac energy metabolism and improves cardiac function and efficiency. Metabolism 98: 37–48, 2019. doi: 10.1016/j.metabol.2019.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Thapa D, Xie B, Zhang M, Stoner MW, Manning JR, Huckestein BR, Edmunds LR, Mullett SJ, McTiernan CF, Wendell SG, Jurczak MJ, Scott I. Adropin treatment restores cardiac glucose oxidation in pre-diabetic obese mice. J Mol Cell Cardiol 129: 174–178, 2019. doi: 10.1016/j.yjmcc.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lovren F, Pan Y, Quan A, Singh KK, Shukla PC, Gupta M, Al-Omran M, Teoh H, Verma S. Adropin is a novel regulator of endothelial function. Circulation 122: S185–S192, 2010. doi: 10.1161/CIRCULATIONAHA.109.931782. [DOI] [PubMed] [Google Scholar]
- 7.Sato K, Yamashita T, Shirai R, Shibata K, Okano T, Yamaguchi M, Mori Y, Hirano T, Watanabe T. Adropin contributes to anti-atherosclerosis by suppressing monocyte-endothelial cell adhesion and smooth muscle cell proliferation. Int J Mol Sci 19: 1293, 2018. doi: 10.3390/ijms19051293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong C-M, Wang Y, Lee JT, Huang Z, Wu D, Xu A, Lam KS. Adropin is a brain membrane-bound protein regulating physical activity via the NB-3/Notch signaling pathway in mice. J Biol Chem 289: 25976–25986, 2014. doi: 10.1074/jbc.M114.576058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein LM, Yosten GL, Samson WK. Adropin acts in brain to inhibit water drinking: potential interaction with the orphan G protein-coupled receptor, GPR19. Am J Physiol Regul Integr Comp Physiol 310: R476–R480, 2016. doi: 10.1152/ajpregu.00511.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yosten GL, Redlinger LJ, Samson WK. Evidence for an interaction of neuronostatin with the orphan G protein-coupled receptor, GPR107. Am J Physiol Regul Integr Comp Physiol 303: R941–R949, 2012. doi: 10.1152/ajpregu.00336.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao A, Herr DR. G protein-coupled receptor GPR19 regulates E-cadherin expression and invasion of breast cancer cells. Biochim Biophys Acta Mol Cell Res 1864: 1318–1327, 2017. doi: 10.1016/j.bbamcr.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 12.Thapa D, Stoner MW, Zhang M, Xie B, Manning JR, Guimaraes D, Shiva S, Jurczak MJ, Scott I. Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol 18: 25–32, 2018. doi: 10.1016/j.redox.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altincik A, Sayin O. Evaluation of the relationship between serum adropin levels and blood pressure in obese children. J Pediatr Endocrinol Metab 28: 1095–1100, 2015. doi: 10.1515/jpem-2015-0051. [DOI] [PubMed] [Google Scholar]
- 14.Gu X, Li H, Zhu X, Gu H, Chen J, Wang L, Harding P, Xu W. Inverse correlation between plasma adropin and ET-1 levels in essential hypertension: a cross-sectional study. Medicine (Baltimore) 94: e1712, 2015. doi: 10.1097/MD.0000000000001712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colik HT, Akkaya N, Erdamar H, Gok S, Kazanci F, Demircelik B, Cakmak M, Yigitoglu R. The effects of valsartan and amlodipine on the levels of irisin, adropin, and perilipin. Clin Lab 61: 1889–1895, 2015. doi: 10.7754/clin.lab.2015.150420. [DOI] [PubMed] [Google Scholar]
- 16.Zieman SJ, Melenovsky V, Kass DA. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol 25: 932–943, 2005. doi: 10.1161/01.ATV.0000160548.78317.29. [DOI] [PubMed] [Google Scholar]
- 17.Fujio S, Hasegawa N, Sato K, Fujita S, Sanada K, Hamaoka T, Iemitsu M. Aerobic exercise training-induced changes in serum adropin level are associated with reduced arterial stiffness in middle-aged and older adults. Am J Physiol Heart Circ Physiol 309: H1642–H1647, 2015. doi: 10.1152/ajpheart.00338.2015. [DOI] [PubMed] [Google Scholar]
- 18.Singh RB, Mengi SA, Xu Y-J, Arneja AS, Dhalla NS. Pathogenesis of atherosclerosis: a multifactorial process. Exp Clin Cardiol 7: 40–53, 2002. [PMC free article] [PubMed] [Google Scholar]
- 19.Gao S, McMillan RP, Jacas J, Zhu Q, Li X, Kumar GK, Casals N, Hegardt FG, Robbins PD, Lopaschuk GD, Hulver MW, Butler AA. Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes 63: 3242–3252, 2014. doi: 10.2337/db14-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao S, McMillan RP, Zhu Q, Lopaschuk GD, Hulver MW, Butler AA. Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol Metab 4: 310–324, 2015. doi: 10.1016/j.molmet.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 22.Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res 122: 624–638, 2018. doi: 10.1161/CIRCRESAHA.117.311586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isfort M, Stevens SC, Schaffer S, Jong CJ, Wold LE. Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail Rev 19: 35–48, 2014. doi: 10.1007/s10741-013-9377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol 305: H1693–1697, 2013. doi: 10.1152/ajpheart.00854.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fillmore N, Mori J, Lopaschuk GD. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br J Pharmacol 171: 2080–2090, 2014. doi: 10.1111/bph.12475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai Y-C, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res 115: 115–130, 2014. doi: 10.1161/CIRCRESAHA.115.301146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, O'Regan DP, Zhao L, Wharton J, Kiely DG, Kinnaird A, Boukouris AE, White C, Nagendran J, Freed DH, Wort SJ, Gibbs JS, Wilkins MR. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 9: eaao4583, 2017. doi: 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- 28.Ozden O, Park S-H, Wagner BA, Song HY, Zhu Y, Vassilopoulos A, Jung B, Buettner GR, Gius D. SIRT3 deacetylates and increases pyruvate dehydrogenase activity in cancer cells. Free Radic Biol Med 76: 163–172, 2014. doi: 10.1016/j.freeradbiomed.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stančáková A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV, Jr, Kahn CR, Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 44: 177–190, 2011. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen S, Zeng K, Liu Q-C, Guo Z, Zhang S, Chen X-R, Lin J-H, Wen J-P, Zhao C-F, Lin X-H, Gao F. Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis 8: e3008–e3008, 2017. doi: 10.1038/cddis.2017.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest 94: 1172–1179, 1994. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Persico M, Masarone M, Damato A, Ambrosio M, Federico A, Rosato V, Bucci T, Carrizzo A, Vecchione C. Non alcoholic fatty liver disease and eNOS dysfunction in humans. BMC Gastroenterol 17: 35, 2017. doi: 10.1186/s12876-017-0592-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kutlu O, Altun Ö, Dikker O, Aktaş Ş, Özsoy N, Arman Y, Çil EO, Özcan M, Yoldemir SA , Akarsu M, Toprak ID, Kırna K , Kutlu Y, Toprak Z , Eruzun H, Tükek T. Serum Adropin Levels Are Reduced in Adult Patients with Nonalcoholic Fatty Liver Disease. Med Princ Pract 28: 463–469, 2019. doi: 10.1159/000500106. [DOI] [PMC free article] [PubMed] [Google Scholar]