

ABSTRACT
Joubert syndrome is a rare brain malformation characterized by the absence or underdevelopment of the cerebellar vermis. Infants with Joubert syndrome usually present with hypotonia, developmental delay, oculomotor apraxia, and respiratory abnormalities. Seizures in Joubert syndrome are not uncommon. Infantile spasms as presentation are hitherto unreported. Here we present a rare case of an 8-month-old infant diagnosed as Joubert syndrome with ZNF423 mutation who presented with West syndrome. Early diagnosis and appropriate management of the child effectively reduced the spasms.
KEYWORDS: Epileptic spasms, Joubert syndrome, West syndrome
INTRODUCTION
Joubert syndrome (JS) is a rare autosomal recessive disorder, named after Marie Joubert in 1969.[1] It is characterized by abnormal respiratory pattern, abnormal eye movements, hypotonia, ataxia, and developmental delay with neuropathologic abnormalities of cerebellum and brainstem. As this syndrome is difficult to diagnose clinically, presence of typical molar tooth sign on axial magnetic resonance imaging (MRI) is diagnostic.[2] Though seizures may be seen in JS, epileptic spasms with EEG evidence of hypsarrhythmia as the primary presentation in children have not been reported so far. The case reported is an infant with JS who presented as West syndrome in whom a novel mutation in ZNF423 gene was detected.
CASE DESCRIPTION
An 8-month-old male infant with uneventful perinatal period presented with episodes of spasms of trunk along with stiffening of the arms and legs lasting for about 1–2sec associated with arching of the back. It was occurring more upon awakening and often occurred in multiple clusters with increased frequency over the previous 1 month. Clinical assessment revealed global developmental delay with a developmental age of 2–3 months. General examination revealed subtle facial dysmorphism. Epileptic flexor spasms were witnessed on examination. Vision and hearing were intact. Central nervous system examination showed mild central hypotonia with preserved reflexes. EEG showed features of classic hypsarrhythmia. Thus, a clinical diagnosis of West syndrome was made. However, the presence of typical molar tooth sign of midbrain [Figure 1], prominent cerebellar folia, small cerebellar vermis [Figure 2], and a prominent fourth ventricle on neuroimaging was diagnostic of JS. He was managed as per West syndrome protocol with prednisolone and valproate with which his spasms subsided, and there was gradual improvement in development. Repeat EEG showed a complete remission of the hypsarrhythmia. As the neuroimaging was diagnostic of JS and clinical condition was mimicking West syndrome, genetic analysis was performed. Clinical exome sequencing revealed a compound heterozygous mutation involving exon 4 (c.1192C>T)(p.Arg398Trp) and exon 5 (c.272C>T)(p.pro91Leu) in the ZNF423 gene consistent with JS type 19.
Figure 1.

MRI brain showing typical molar tooth sign of midbrain
Figure 2.
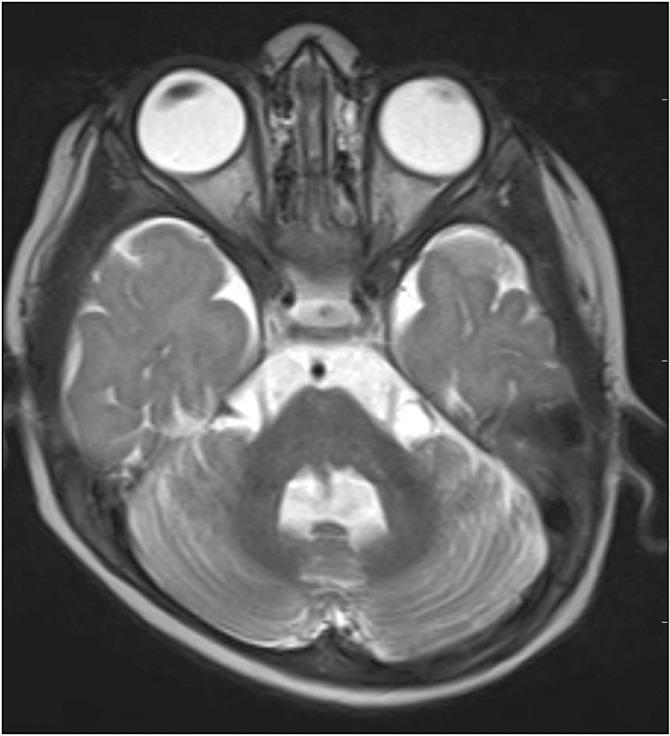
Prominent cerebellar folia and small cerebellar vermis
DISCUSSION
JS is a rare autosomal recessive disorder characterized by variable clinical presentation but with typical neuroradiological findings. Prevalence is around 1 in 100,000, and by 2009 only 200 cases have been reported all over the world.[3] Because of the nonspecific clinical presentation, diagnosis is often delayed.[4]
Usual presentation of JS includes hypotonia, developmental delay, irregular breathing patterns, and visual defects. But our child had only hypotonia and developmental delay with normal breathing and preserved vision. Epileptic seizures are not a frequent symptom (3%) in JS and if present are usually associated with cortical organization defects such as white matter cysts, corpus callosum dysgenesis, and dilation of the ventricular system, though our case had no such cortical malformations. Mugundhan et al.[5] have reported a case who had focal seizures since childhood, but the diagnosis of JS was made only in the third decade by classical findings in neuroimaging.
Abnormal EEG and/or seizures are present in some affected individuals with JS; however, the exact incidence is unknown.[6] A study by Fennell et al.[7] identified greater cognitive impairment in individuals with JS and abnormal EEG. Uncrossed epileptic seizures in an adult with JS have been reported by López Ruiz et al.[8] Although epilepsy is a rare feature of JS, abnormal EEG was reported in 45% in a case series by Elhassanien and Alghaiaty,[9] and 50% of them have focal seizures. Nonetheless, the association of epileptic spasms with JS like in our case has not been reported in literature so far.
In our case, genetic analysis revealed a compound heterozygous mutation involving exon 4 (c.1192C>T)(p.Arg398Trp) and exon 5 (c.272C>T)(p.pro91Leu) in the ZNF423 gene confirming JS 19. Two cases of ZNF423 homozygous mutation have been described by Chaki et al.[10] in 2012, who presented with nephronophthisis and cerebellar vermis hypoplasia but not epileptic seizures. Heterozygous mutation of ZNF423 with classical neuroimaging findings of JS 19 has not been described in literature so far. The heterozygous mutations caused a dominant-negative effect on protein function in cellular studies.[10] As there is no case report of heterozygous JS 19, the primary presentation of this entity remains unknown. Whether epileptic spasms could be the presentation needs to be confirmed with larger case series. Our case highlights the rare presentation of JS 19 with infantile spasms. This adds to the existing literature about the association between infantile spasms and JS.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
We express our sincere thanks to Dr. Saji James, Professor, Department of Paediatrics, Sri Ramachandra Institute of Higher Education and Research, Chennai, India.
REFERENCES
- 1.Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–25. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- 2.Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–21. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- 3.Choh SA, Choh NA, Bhat SA, Jehangir M. MRI findings in Joubert syndrome. Indian J Pediatr. 2009;76:231–5. doi: 10.1007/s12098-008-0232-1. [DOI] [PubMed] [Google Scholar]
- 4.Akcakus M, Gunes T, Kumandas S, Kurtoglu S, Coskun A. Joubert syndrome: report of a neonatal case. Paediatr Child Health. 2003;8:499–502. doi: 10.1093/pch/8.8.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mugundhan K, Mayan MCV, Nidhin PD, Loganathan G, Balamurugan N. Joubert syndrome associated with seizures. J Assoc Physicians India. 2017;65:96. [PubMed] [Google Scholar]
- 6.Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726–31. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 7.Fennell EB, Gitten JC, Dede DE, Maria BL. Cognition, behavior, and development in Joubert syndrome. J Child Neurol. 1999;14:592–6. doi: 10.1177/088307389901400907. [DOI] [PubMed] [Google Scholar]
- 8.López Ruiz P, García García ME, Dicapua Sacoto D, Marcos-Dolado A. Uncrossed epileptic seizures in Joubert syndrome. BMJ Case Rep. 2015;2015:bcr2014207719. doi: 10.1136/bcr-2014-207719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elhassanien AF, Alghaiaty HA. Joubert syndrome: clinical and radiological characteristics of nine patients. Ann Indian Acad Neurol. 2013;16:239–44. doi: 10.4103/0972-2327.112480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell. 2012;150:533–48. doi: 10.1016/j.cell.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]