

SUMMARY
The choroid plexus (ChP) epithelium is a source of secreted signaling factors in cerebrospinal fluid (CSF) and a key barrier between blood and brain. Here, we develop imaging tools to interrogate these functions in adult lateral ventricle ChP in whole-mount explants and in awake mice. By imaging epithelial cells in intact ChP explants, we observed calcium activity and secretory events that increased in frequency following delivery of serotonergic agonists. Using chronic two-photon imaging in awake mice, we observed spontaneous subcellular calcium events as well as strong agonist-evoked calcium activation and cytoplasmic secretion into CSF. Three-dimensional imaging of motility and mobility of multiple types of ChP immune cells at baseline and following immune challenge or focal injury revealed a range of surveillance and defensive behaviors. Together, these tools should help illuminate the diverse functions of this understudied body-brain interface.
Graphical Abstract
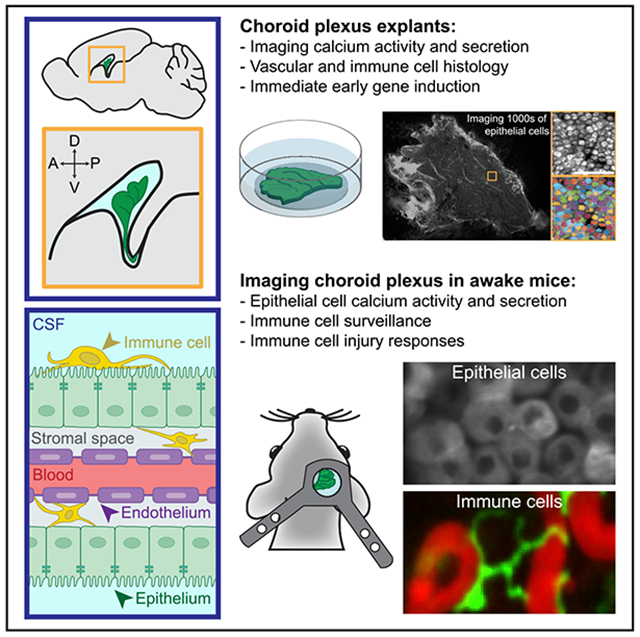
In Brief
The choroid plexus is a vital brain barrier and source of cerebrospinal fluid. Using two-photon imaging of choroid plexus epithelium in live explants and in awake mice, Shipley et al. establish a platform for investigating subcellular calcium activity and secretion in epithelial cells and diverse surveillance behaviors in immune cells.
INTRODUCTION
The choroid plexus (ChP) is a distinct, vital organ that extends into each ventricle in the brain. It is composed predominantly of epithelial cells that envelop a network of stromal cell types, including immune, mesenchymal, and vascular cells (Dani et al., 2019). The epithelial cells provide a source of cerebrospinal fluid (CSF) (Damkier et al., 2013) and associated growth-promoting factors for neural stem cells (Lehtinen et al., 2011; Fame and Lehtinen, 2020; Silva-Vargas et al., 2016). They also form a blood-CSF barrier that gates passage of nutrients, toxins, and immune cells from body to brain (Ghersi-Egea et al., 2018; Reboldi et al., 2009; Schwartz and Baruch, 2014; Shechter et al., 2013) and may regulate CSF composition via clearance of toxins and waste (Crossgrove et al., 2005). Thus, the sensing, secretory, and transcytotic functions of the ChP suggest diverse roles in regulating brain function. These roles may be disrupted in neurologic conditions ranging from hydrocephalus (Karimy et al., 2017) to Alzheimer’s disease (Balusu et al., 2016a; Marques et al., 2013). Furthermore, the ChP is an attractive target for enhancing drug delivery to the brain (Gonzalez et al., 2011; Haddad et al., 2013; Hudry and Vandenberghe, 2019).
Despite the importance of the ChP-CSF system, little is known about the behavior of mammalian ChP cell types in vivo. In vitro approaches exist for culturing ChP cell lines (Zheng and Zhao, 2002), dissociated ChP cells (Zheng et al., 1998), ChP epithelial cell sheets in transwell models (Strazielle and Ghersi-Egea, 1999), and ChP organoids (Pellegrini et al., 2020; Watanabe et al., 2012). Isolated ChP explants have also been used for analyzing secretion into conditioned medium (Gudeman et al., 1987, 1989; Lun et al., 2015a; Silva-Vargas et al., 2016) or for fixation and immunostaining (Dani et al., 2019; Lun et al., 2015a). Anatomical studies using light microscopy and electron microscopy (EM) have provided clues as to the cellular architecture of the ChP (e.g., Netsky and Shuangshoti, 1975). However, a major obstacle to progress in understanding the roles of ChP cells has been the lack of available tools for stable visualization and manipulation of specific ChP cell types in intact tissue in vitro and in vivo in a fluid environment deep within the brain.
Here, we adapted a suite of modern neuroscience tools to target the lateral ventricle (LV) ChP, providing optical access to this unexplored tissue in mice. We developed methods for volumetric two-photon imaging and non-rigid alignment of the ChP in acute explant preparations, as well as in awake mice across hours, days, and weeks. Dynamic cellular functions of other epithelia (e.g., in retina, lung, and salivary gland) are typically associated with changes in intracellular calcium (Ambudkar, 2016; Balaji et al., 2017; Concepcion and Feske, 2017; Narciso et al., 2017; Samanta and Parekh, 2016). For example, calcium signaling in salivary gland is important for on-demand secretion (Ambudkar, 2018, 2016). We found that ChP epithelial cells exhibited spontaneous subcellular calcium activity in vitro and in vivo. Serotonergic agonists evoked distributed increases in calcium activity, as well as secretory events measured using a sensor of exocytosis. We then visualized the motility and mobility of ChP immune cells in relation to ChP vasculature in awake mice at baseline and in response to peripheral immune stimulation and to laser-induced, focal ChP injury. Together, these methods provide a novel imaging platform for a wide range of studies imaging multiple genetically accessible ChP cell types in intact tissue at unprecedented spatial and temporal resolution.
RESULTS
Imaging ChP Explants
We first optimized adult LV ChP explant preparations (Dani et al., 2019; Lun et al., 2015a) to enable histological analyses (Figures 1A-1D) and stable live-cell imaging (Figures 1E, 1F, and S1A). Epithelial cells constitute the majority of adult ChP cells (Dani et al., 2019). In addition, the ChP contains immune cells (labeled by Cx3cr1+/GFP; Jung et al., 2000) consisting mostly of monocytes/macrophages but also including a smaller number of mast cells and dendritic cells (Dani et al., 2019; Van Hove et al., 2019). These immune cells evenly tiled the entire tissue under baseline conditions (Figures 1A-1C). The ChP could be divided into stereotyped zones defined by arterial and venous landmarks (Figures 1D, S1B, and S1C) (Dani et al., 2019). This vascular pattern strongly resembles that observed for human ChP (Hudson, 1960). As such, it provides an anatomical roadmap that allows specific subregions of the LV ChP to be identified and analyzed across mice within the same study, across studies from different labs, and across species.
Figure 1. Isolation, Immunostaining, and Calcium Imaging of Lateral Ventricle ChP Explants.

(A) Left: large leaf of LV ChP from a Cx3cr1+/GFP mouse immunostained with anti-GFP (green, immune cells) and PECAM (red, vasculature). Scale bar, 500 μm. Right: zoom-in of small dashed box. Scale bar, 100 μm. Cx3cr1+/GFP cells tile the ChP (confirmed in eight other mice).
(B) Positions of 1,781 Cx3cr1+/GFP cells from (A).
(C) Cumulative distribution of nearest-neighbor distances of each Cx3cr1+/GFP cell. Immune cells showed regular spacing (~30 μm) relative to random Poisson spacing (red trace; gray envelope, 1% acceptance interval).
(D) PECAM (red) and ACTA2 (green) immunostains demarcate stereotyped LV ChP regions (confirmed in three other mice). Purple arrowheads, arteries. Blue arrowheads, veins. Scale bar, 500 μm.
(E) Light path and setup for imaging LV ChP.
(F) Epifluorescence image containing a FoxJ1-Cre::Ai95D LV ChP explant expressing GCaMP6f in multiciliated ChP epithelial cells. Cells near stabilizing glue attachments at explant borders (asterisks) showed elevated GCaMP6f fluorescence (indicating unhealthy cells) and were excluded from subsequent analyses. Scale bar, 1 mm.
(G) Zoom-in of 122 epithelial cells (dashed box in F). Scale bar, 50 μm.
(H) Cell masks (see STAR Methods).
(I) Twenty labeled cells corresponding to traces in (K).
(J) Pink, traces surrounding each calcium transient with a fractional change in fluorescence, ΔF/F > 5σ (235 events across 122 cells from H). Red, mean calcium transient across traces.
(K) Five-minute time courses from cells in (I).
(L) Seventy-six percent of cells (93 of 122) in (H) exhibited calcium events.
(M) Average of all cross-correlations between binarized event time courses of all pairs of cells from (H) (computed at delays from −5 to +5 s), demonstrating that spontaneous events were uncorrelated across cells. We observed qualitatively similar results as in (G)–(M) in 25 other mice (not shown).
ChP explants were stabilized for acute in vitro imaging (Figure 1E). To visualize calcium activity in epithelial cells, we gently dissected and stabilized the entire LV ChP from one hemisphere. We expressed the calcium reporter GCaMP6f (using Ai95D mice; Madisen et al., 2015) in ChP epithelial cells (using FoxJ1-Cre mice that selectively target this cell population) (Figure 1F) (Lun et al., 2015a; Zhang et al., 2007). Using epifluorescence imaging, we could visualize spontaneous calcium activity across thousands of epithelial cells simultaneously (Figure 1F; Video S1). We focused on a subregion and performed activity-based cell segregation (STAR Methods), resulting in time courses of spontaneous activity in individual epithelial cells (Figures 1F-1K; Video S1). Most epithelial cells showed large, transient elevations in calcium activity lasting several seconds (Figures 1K and 1L). Such events were not synchronized across cells (Figure 1M). These findings suggest baseline regulation of calcium levels and calcium-dependent signal transduction in ChP epithelial cells.
Activation of Serotonin Receptors Stimulates Secretion via VAMP3-Mediated Exocytosis
Elevated calcium regulates many cellular processes, including gene transcription and secretion in other body epithelia, such as the salivary gland (Ambudkar, 2016). One factor previously shown to elevate calcium levels in ChP cell lines in culture is serotonin (5-HT [5-hydroxytryptamine]) (Esterle and Sanders-Bush, 1992; Sanders-Bush and Breeding, 1990). Metabolites of the 5-HT signaling pathway are present in the CSF (Toda et al., 2013). CSF-5-HT can originate from multiple sources, including direct release by dorsal raphe nucleus serotonergic neurons that course along the ventricles and in close proximity to the ChP (Narboux-Nême et al., 2008; Okaty et al., 2020; Tong et al., 2014), and via peripheral circulation, originating, for example, from 5-HT secretion in the gut (Stasi et al., 2019) or platelets (Cloutier et al., 2012). We found that 5-HT (Audhya et al., 2012; Toda et al., 2013) triggered coordinated waves of calcium activity that recruited increasing numbers of ChP epithelial cells across the explant with higher concentrations of 5-HT (Figures 2A, 2B, and S2E; Video S2).
Figure 2. Evoked Calcium Activity and Exocrine Secretion in ChP Epithelial Cells.

(A) Epifluorescence calcium imaging of ChP epithelial cells from FoxJ1-Cre::Ai95D LV ChP explant. Mean baseline fluorescence (left) and changes in fluorescence from baseline in response to 0, 5, 50, and 500 nM 5-HT. Scale bar, 100 μm.
(B) Time course of changes from baseline, averaged across the explant. Responses to at least one dose of 5-HT were observed in 18 of 19 mice and to all three doses in 10 of 19 mice (not shown).
(C) Htr2c expression in LV ChP (from Allen Brain Atlas; Lein et al., 2007). Scale bar, 500 μm.
(D) c-fos induction following injection of 5-HT2C agonist WAY-161503 (****p < 0.0001, t test, saline versus 3 mg/kg s.c.; left to right, n = 8, 2, 2, 2 and 8).
(E) Htr2cmRuby3 LV ChP labels 5-HT2C receptors in epithelial cells. Axial (left) and side-on (right; from dashed box at left) maximum projections show preferential apical (apposed to the CSF) versus basal (closer to vessels) localization. Scale bar, 10 μm.
(F) Two-photon imaging of FoxJ1-Cre::Ai95D explants. Higher concentrations of WAY-161503 activated more cells (green arrowheads), and cells activated at lower concentrations are not reactivated later. Responses were observed in 7 of 7 mice and to each dose in 5 of 7 mice (not shown). Scale bar, 10 μm.
(G) Confocal imaging of vesicle release from an example LV ChP epithelial cell following viral expression of VAMP3-pHluorin. Top left: maximum-intensity projection across baseline period shows fluorescent vesicle release (white punctae). Bottom left: similar projection following Hessian-based filtering. Middle panels: same as left but following application of WAY-161503 (500 nM). Right: vesicle release event masks segmented from the filtered movie. Scale bar, 5 μm.
(H) Cumulative number of VAMP3-pHluorin vesicle release events following application of WAY-161503 (red) or aCSF (blue).
The 5-HT2C serotonin receptor (Figure 2C) (Lein et al., 2007), a Gq/G11-coupled G protein-coupled receptor (GPCR), is the most highly expressed GPCR in ChP epithelial cells (Lun et al., 2015a). We found that subcutaneous (s.c.) injection of WAY-161503, a selective agonist of 5-HT2C (Rosenzweig-Lipson et al., 2006), drove robust immediate-early gene expression in ChP (Figures 2D, S2A, and S2B). Antibodies available for this receptor have typically shown low signal quality. Thus, we used genome editing to generate a Htr2cmRuby3 mouse line in which the fluorescent protein mRuby3 was inserted at the C-terminus of 5-HT2C (Figures S2C and S2D). 5-HT2C-mRuby3 was functional in these mice, as subcutaneous injection of WAY-161503 in Htr2cmRuby3 mice induced c-fos expression similar to that observed in wild-type mice (Figure 2D; mean ± SEM fold increase in c-fos mRNA expression in Htr2cmRuby3 mice receiving WAY-161503 [3 mg/kg] versus vehicle 80.6 ± 17.3, n = 4 heterozygous male mice; Htr2c expressed from X chromosome). Fluorescence of the mRuby3 tag revealed receptor localization throughout ChP epithelial cells, including at the apical and basal membranes (Figure 2E). This localization is consistent with the prediction that ChP epithelial cells can sense both central and peripheral sources of 5-HT (Figure S1C). Accordingly, using higher magnification two-photon calcium imaging, we obtained similar patterns of activation of an increasing number of cells with increasing concentrations of the 5-HT2C agonist WAY-161503 (Figures 2F and S2E; Video S3).
Application of 5-HT to dissociated ChP cells in culture can increase the transfer of water and protein secretion (Conn and Sanders-Bush, 1986; Esterle and Sanders-Bush, 1992; Watson et al., 1995). Our expression analyses confirmed that the secretory machinery commonly required for calcium-dependent gene induction, protein secretion, vesicle trafficking/release machinery, and/or homologs of proteins from other epithelia are expressed in ChP epithelial cells (e.g., Vamp3, Snap23, Stx12, Stxbp4), implicating vesicular exocytosis as a mechanism of protein secretion (Figures S2F-2SJ) (Dani et al., 2019; Lun et al., 2015a). In EM images of ChP epithelial cells, a high density of vesicles was observed near the apical membrane (Figure S2I, black arrows). VAMP3 showed the highest gene expression among vesicle proteins in the ChP (Figure S2G; RNA sequencing [RNA-seq] data from Lun et al., 2015a), and VAMP3 protein expression was confirmed by immunoblotting and immunostaining (Figures S2H and S2J). These data led us to investigate activity-dependent and VAMP3-mediated ChP exocytosis. Specifically, we used AAV-VAMP3-pHluorin (Urbina et al., 2018), a pH-sensitive variant of GFP, to visualize individual secretory events. pHluorin fluoresces upon plasma membrane fusion, when the lower pH (~5.6) inside intact exocytic vesicles changes to a pH of 7.4 upon exposure to the extracellular environment. The fluorescence signal disappears following endocytosis and re-acidification of the vesicles (Sankaranarayanan et al., 2000).
We first expressed VAMP3-pHluorin in the Z310 ChP epithelial cell line (Zheng and Zhao, 2002). Using total internal reflection fluorescence (TIRF) microscopy, a method with high signal-to-noise ratio, fast frame rate (2 frames/s), and narrow fluorescence excitation and emission ranges, we could capture numerous spontaneous vesicle fusion events (Figure S2K; Video S4). To evaluate ChP secretion in a more naturalistic setting, we transduced ChP in vivo with AAV-VAMP3-pHluorin, dissected ChP explants, and investigated vesicle fusion events in vitro. Because of the elaborate apical structure of ChP epithelial cells, including multiple microvilli and cilia (Figure S2I), these cells were not amenable to TIRF microscopy (axial resolution < 100 nm) without compressing the cells against a coverglass, a procedure that could compromise cellular integrity or induce cellular responses to mechanical distortion. Instead, we used Airyscan confocal microscopy (ZEISS LSM880) that afforded comparable signal-to-noise ratio and frame rates (1.59 frames/s). We observed spontaneous VAMP3-mediated exocytosis in individual epithelial cells in whole ChP explants (Video S5). To monitor localized secretion events, we first performed spatial filtering of each image (Figure 2G). We then defined regions with co-active pixels and extracted fluorescence time courses (Figures 2G, 2H, and S2L). Strikingly, 5-HT2C activation by WAY-161503 (delivered at levels similar to those that drove calcium activity in Figure 2F) drove an increase in the rate of VAMP3-mediated exocytosis (Figures 2H and S2L; Video S5). Our data using live imaging at subcellular resolution demonstrate that 5-HT stimulates ChP exocytosis via activation of 5-HT2C. More generally, our findings validate a platform for fluorescence imaging in ChP explants, enabling high-resolution studies of calcium activation, secretion, and other processes.
In Vivo Imaging of ChP in Awake Mice
Virtually nothing is known about the activity of ChP cell types in vivo. We developed a deep-brain cannula implantation strategy that enables acute and longitudinal imaging of the ChP over weeks and months in awake mice. A cannula and glass window were surgically implanted above the LV (Figures 3A-3D), similar to our recent approach for imaging in visual thalamus (Liang et al., 2018). At 2–3 weeks post-surgery, windows were typically translucent, allowing bright-field imaging of ChP (Figure 3E).
Figure 3. Imaging Lateral Ventricle ChP in Awake Mice.

(A and B) Schematic of cannula (gray cylinder) with glass bottom, implanted above the LV ChP (green).
(C) Headpost placement.
(D) Head-fixed mouse on a trackball. An immersion well attached to the headpost allowed imaging using a high numerical aperture objective.
(E) Bright-field image of ChP through the cannula 27 days post-surgery. Dotted line outlines ChP. Scale bar, 1 mm.
(F) Epifluorescence images of ChP (arrowheads) from FoxJ1-Cre::Ai95D mice, 42–56 days after surgery. Scale bar, 1 mm.
(G) Tracking the same ChP (arrowheads) through a clear window across many days following surgery (similar results observed in nine other mice, not shown). Scale bar, 1 mm.
Similar to brain surgery in the clinical setting, insertion of the imaging cannula is an invasive procedure. We performed additional control experiments to determine the extent of the injury response and to verify the health of the preparation several weeks following surgery, at the time of imaging. As anticipated, GFAP-positive astrocytes and Cx3cr1-positive immune cells were enriched in cerebral cortical tissue adjacent to the cannula (Figures S3A-S3G). The density of glial cells (GFAP positive) and immune cells (Cx3cr1-positive) dropped to baseline levels by ~100 μm from the edge of the cannula (Figures S3A-S3G). The ventricular lining of the LV below and lateral to the implant did not show accumulation of GFAP- or Cx3cr1-positive cells and retained characteristic S100β-positive ependymal cells (Figure S3B). Importantly, immune cells from the ChP tissue located below the implant exhibited a ramified, non-activated morphology with extended processes and a level of tiling of the ChP that was indistinguishable from observations in contralateral ChP and in ChP from control mice that did not undergo surgery (see Figures 1A-1C, S3H, and S3I). Elevated CSF cytokine levels that were evident in some mice 1 day following surgery also returned to undetectably low levels in all mice by 3 weeks post-surgery (Figure S3J). These data demonstrate that at the time imaging began several weeks post-surgery, our imaging preparation did not show signs of persistent inflammation.
Epifluorescence images of LV ChP from transgenic mice expressing GCaMP6f in ChP epithelial cells (FoxJ1-Cre::Ai95D; Figure 3F; Video S6) demonstrated consistently high image quality across mice. Notably, anatomical features of the ChP were stable upon repeated imaging across weeks and months, with no evidence of substantial remodeling of vasculature across imaging sessions beginning several weeks following surgery (Figure 3G). Although the location of the ChP in the LV showed moderate mouse-to-mouse variability following surgery (Figure 3F), identification of arterial and venous landmarks allowed longitudinal imaging of a similar anatomical region of the ChP across mice and within the same mouse across sessions (Figures 3 and S1B).
To maximize spatial resolution and minimize bleaching during cellular imaging, we performed two-photon imaging using a long-working distance, high-numerical aperture objective coupled to the imaging window (see STAR Methods). We targeted local regions of interest within previously acquired epifluorescence images (Figures 4A-4C). In contrast to other brain tissues that can be largely pressurized and stabilized for two-photon imaging (Goldey et al., 2014; Liang et al., 2018), the ChP is only anchored at one edge near the base of the LV and is otherwise suspended in CSF. Therefore, the ChP often exhibited large and non-rigid motion in three dimensions during changes in behavior such as locomotion or adjustment of body posture (Video S6). As described below, we used different imaging strategies and custom registration algorithms to overcome these technical challenges.
Figure 4. Two-Photon Calcium Imaging of Epithelial Cells in Awake Mice.

(A) Epifluorescence image of GCaMP6f-expressing ChP epithelial cells (diagonal vascularized sheet; FoxJ1-Cre::Ai95D mouse). Scale bar, 1 mm.
(B) Zoomed-in image (dashed red square in A). Scale bar, 100 μm.
(C) Maximum projection of two-photon imaging volume encompassing the ChP region in (B). Scale bar, 100 μm.
(D) Average of images at a single plane. Scale bar, 50 μm.
(E) Individual epithelial cell (red square in D), annotation of cell outline and nucleus, and division into 12 sectors.
(F) Annotation of all cell outlines and nuclei in (D).
(G) Time-lapse of a single subcellular calcium event.
(H) Kymograph of activity across all 12 sectors of cell in (E) and (G). Red arrowhead, event from (G).
(I) Time course of brightest-sector activity (black, maximum across sectors in H) and median activity (red). Asterisks, peaks of subcellular events exceeding 3 SDs (dashed blue line) above a running mean.
(J and K) Brightest-sector (J) and median-sector (K) activity surrounding peak (t = 0) of all events for cell in (E). Thicker lines, mean traces. Similar results were observed in three other mice (not shown).
(L) Images of cross-sections of two sheets of GCaMP6-expressing epithelial cells separated by stromal space, beginning 25 min after injection of WAY-161503 (3 mg/kg, s.c.; similar results observed in two other mice, not shown). Scale bar, 50 μm.
(M) Zoom-in of a single epithelial cell reveals release of subcellular plumes (arrowheads) of intracellular contents including GCaMP6f into CSF. The basal side of the epithelium remained intact, consistent with apocrine secretion. Scale bar, 10 μm. Similar events were observed in a second mouse (not shown). See also Figure S4; Videos S7, S8, S9, and S10.
(N) Scanning EM of ChP 15 min following WAY-161503 (3 mg/kg, s.c.) reveals apocrine blebs (arrowheads). Scale bar, 5 μm.
First, video-rate two-photon imaging of a single plane allowed precise and high-speed tracking of small numbers of cells following in-plane alignment, particularly during periods of minimal brain motion while the mouse was stationary. For these analyses, occasional large tissue movement could be stabilized or omitted from further analyses. Second, for longitudinal tracking across hours, for which larger non-rigid motion and drift of the tissue out of plane were often evident, we instead used a volumetric imaging strategy (0.25–0.5 volumes/s, 31–62 planes/volume, volume dimensions 170 × 170 × 350 or 355 × 230 × 100 μm3; see STAR Methods). This approach was important for achieving stable cell tracking following non-rigid alignment in three dimensions (see Figure 5).
Figure 5. Three-Dimensional Imaging and Registration of ChP in Awake Mice.

(A) Maximum projections across a time-averaged two-photon imaging volume of Cx3cr1+/GFP immune cells (green) and Texas red dextran-labeled vasculature (red; i.p. injection). Projections from two mice are shown (similar results in 13 other mice, not shown). Scale bar, 100 μm.
(B) Registration algorithm (see STAR Methods). Step 1: correct for depth-dependent magnification due to tunable lens. Step 2: intra-volume alignment of each plane to its neighbor. Step 3: 3D translation of each volume to a local target. Steps 4 and 5: Z projection and X-Y alignment.
(C) Mean Z projection of a single volume before versus after step 2. Scale bar, 50 μm.
(D) Estimated X and Y corrections for each plane of volume in (C).
(E) Z-profile time lapse of vasculature before and after 3D registration. Columns, 600 volumes spanning ~63 min; rows, average fluorescence in the white box in (C) at each Z plane. White trace, estimated Z correction.
(F) Index of motion artifact (sliding estimate of SD/mean vasculature fluorescence across volumes; see STAR Methods). Registration reduced both large, transient motion artifacts (peaks in orange trace) and persistent, higher frequency motion (see J).
(G–I) Cumulative distributions of X and Y displacements of planes within each volume (G) and XY displacements (H) and Z displacements (I) across consecutive volumes. Data in (G)–(J) are from 20 sessions from 13 mice.
(J) Mean motion artifact (see F) per session, pre- versus post-registration. ****p < 0.0001, paired t test.
Imaging Calcium Activity and Apocrine Secretion in ChP Epithelial Cells In Vivo
We imaged ChP epithelial cell calcium activity using a transgenic mouse expressing GCaMP6f, which provided similar expression levels across cells and stable expression across days (Figures 4A-4C; see also Figure 1F). We first performed single-plane two-photon calcium imaging (Figure 4D). High-speed imaging (33–41 frames/s) revealed spontaneous subcellular calcium events lasting ~200 ms (Figure 4G; Videos S7 and S8). To quantify this observation, we manually outlined the borders and nuclei of individual cells (Figures 4D-4F). A typical subcellular event from one example cell is shown in Figure 4G. We segmented each cell into 12 radial sectors extending from the center of the nucleus (Figure 4E, bottom) and “unwrapped” the sectors to create a kymograph of averaged subcellular activity across frames (Figure 4H). A maximum-intensity projection across sectors revealed large subcellular events (Figure 4I) of a consistent duration and characteristic exponential decay (Figure 4J). The consistent dynamics and correlated changes across nearby pixels for this and other cells (Figure S4) further suggested that these events were not due to photon noise or brain motion. In contrast to maximum-intensity projections across sectors, median projections showed no significant fluctuations (Figures 4K, S4A, and S4B), consistent with the subcellular nature of these events.
Our earlier findings demonstrated that application of 5-HT2C agonist WAY-161503 evoked robust calcium responses in ChP epithelial cells in vitro and induced immediate-early gene expression following peripheral injection in vivo (Figures 2D, 2F, S2A, S2B, and S2E; Video S3). Furthermore, signatures of apocrine secretion (Figures 4M and 4N; Video S9) have previously been reported to occur in ChP ex vivo (Agnew et al., 1980; Farkaš, 2015; Gudeman et al., 1989). We therefore sought to define the dynamics of ChP calcium activity and apocrine secretion upon WAY-161503 delivery in vivo. To obtain stable estimates of calcium transients across tens of minutes (see above), we used volumetric imaging (0.32 volumes/s, 93 planes/volume, 3.8 μm spacing between planes). Subcutaneous injection of WAY-161503 resulted in robust increases in calcium activity that progressed along the epithelium over tens of minutes (Figure 4L; Video S9). The large differences in timing of activation of various cells may relate to cell-to-cell differences in 5-HT2C expression (Figure 2E), to slow changes in the concentration of WAY-161503, or to sequential sensing of signals released from activated neighboring cells. Cellular increases in calcium activity culminated in apocrine secretion, reflected by a release of cytoplasmic protrusions from the apical surface of the cell and cellular release of cytoplasmic contents directly into the CSF (Figures 4L-4N; Video S10). The basal portion of the cells including the nucleus remained intact. Taken together, these findings highlight novel in vitro and in vivo approaches to test and visualize calcium activity and distinct modes of exocrine signaling by ChP epithelial cells. Furthermore, these data establish a platform for testing how exogenous signals such as serotonin can stimulate calcium activation, gene transcription, and exocrine secretion.
ChP Immune Cells at Baseline and in Response to Local or Peripheral Stimulation
The ChP is not only important for secretion of water and proteins into the CSF but is also an essential barrier that protects the brain from harmful blood-borne factors (Ghersi-Egea et al., 2018; Saunders et al., 2018) and is implicated as a site of immune cell entry into the brain (Fame and Lehtinen, 2020; Ghersi-Egea et al., 2018; Reboldi et al., 2009; Schwartz and Baruch, 2014; Shechter et al., 2013). However, the in vivo functions of ChP immune cells in physiological or pathological conditions remain largely unexplored (Kierdorf et al., 2019). Thus, we investigated ChP immune cells under homeostatic, immune-challenged, and injury conditions.
We repeated the surgical approach described above in transgenic mice expressing GFP in Cx3cr1-positive immune cells (Jung et al., 2000). Following surgical recovery, we performed intraperitoneal (i.p.) injection of Texas red-conjugated dextrans that rapidly filled the major vessels and fine capillary networks of the ChP. These large dextrans (70 kDa) did not immediately leak into the ChP stromal space. We then performed two-color imaging of ChP immune cells and vasculature (Figure 5A), focusing on regions of ChP that were oriented parallel to the imaging plane and thus amenable to time-lapse volumetric imaging across the thickness of the tissue (Video S11).
For tracking of fine immune cell processes across seconds, minutes, and hours in awake mice, it was critical to develop a procedure for accurate alignment of the three-dimensional (3D) imaging volumes (see Figure 5B and legend). It was useful to estimate shifts in ChP using the stable, bright red dextran signal and then apply these shifts to both the imaged vasculature (red) and immune cells (green). Given that the individual frames were acquired at 15.5 frames/s, there was minimal within-plane non-rigid motion. However, brain motion could result in X and Y shifts in successive imaging planes within a volume (Video S12). Thus, alignment of each Z plane to a reference plane within each volume was important (Figures 5C and 5D). We then performed rigid-body 3D alignment. Following these corrections, images of static objects (e.g., vasculature) could be effectively stabilized (Figures 5E and 5F). As a final step, we calculated the mean intensity across Z planes for each volume and ran a second translational alignment. Across all 20 sessions from 13 mice, estimated intra-volume and inter-volume shifts in X, Y, and Z could be quite large, reflecting ChP suspension in CSF (Figures 5G-5J).
We observed substantial exploratory movements of ChP immune cell bodies and/or distal processes. In each of 26 fields of view from 14 mice, we observed large numbers of GFP-positive immune cells. Some of the cells were located within the ChP stromal space, while others were located on the apical surface of the ChP, in contact with LV CSF (i.e., epiplexus or Kolmer cells) (Figures 6A-6G). Epiplexus cell bodies often exhibited substantial mobility. For example, the cell in Video S13 (top left) traveled 210 μm in 1 h. Some epiplexus cell bodies moved at a constant rate, while others displayed saltatory movements (Figure 6D; Video S13). In contrast, the majority of GFP-positive immune cells located within the stromal space showed minimal cell body mobility. However, these cells possessed highly dynamic processes that extended and contracted (Figures 6E-6G; Video S13), similar to microglia in other brain areas (Hierro-Bujalance et al., 2018). These processes appeared to serve a surveillance function, as they frequently contacted vessels within the stromal space and retracted upon contact with other processes from the same or neighboring immune cells (Figures 6E-6G; Video S13).
Figure 6. ChP Immune Cells Perform Local Surveillance and Housekeeping In Vivo.

(A) Cross-section of ChP. Epiplexus immune cells (orange arrowhead) are located on apical (CSF-sensing) surface of epithelium (green sheet). Stromal immune cells (blue arrowheads) are located in stromal space between vasculature (red with purple endothelial cells) and epithelium.
(B) Top: axial mean projection of Cx3cr1+/GFP cells in LV ChP explant ex vivo. Bottom: side-on view. Arrowheads indicate stromal (blue) and epiplexus (orange) immune cells. Scale bar, 100 μm.
(C–G) Similar to (B) but from in vivo two-photon imaging (see also Videos S11 and S13). Scale bar, 25 μm. (C) Example epiplexus cells from four mice. Side-on views (bottom) indicate locations outside vascular plane (likely outside the epithelium). (D) Example epiplexus cell pausing, then traveling across the ChP surface (colored dots, cell location at 1 min intervals). (E–G) Example stromal immune cells showed either stationary cell bodies with processes that survey nearby vessels (E and F) and that retract following upon contacting a different immune cell (F) or, occasionally, cell body movement constrained by surrounding vessels (G).
(H) Left, middle: i.p.-injected red dextran (70 kDa) fills the ChP vasculature. Right: 2 days later, dextran has leaked into stromal space and accumulated within immune cells. Scale bar, 50 μm.
(I) Snapshots of dextran punctae accumulating within immune cell processes (arrowheads) in blue dashed box in (H).
These surveillance-like behaviors were reminiscent of immune cells in other parts of the brain that play key roles in sensing environmental perturbations and protecting against injury (Hickman et al., 2018; Kierdorf et al., 2019; Li and Barres, 2018). Indeed, we found that immune cells in the ChP appear to partake in similar functions. First, we noted that the fluorescent dextrans used to label vasculature were cleared from circulation over several days. In these experiments, ChP immune cells in the stromal space, but not epiplexus cells, took up fluorescent dextrans 30 min following injection (Figures 6H and 6I; Videos S14 and S15), and dextran-labeled punctae could be observed even 26 days following injection (Figures S5A-S5C). These data demonstrate that ChP immune cells participate in uptake of foreign material from the peripheral circulation, consistent with the known housekeeping functions of immune cells in other parts of the body and brain.
The ChP contributes to blood-brain communication during peripheral inflammation (Balusu et al., 2016b), and the effects of immune challenges on the ChP have been implicated in several neurologic conditions. For example, genetic markers of immune function and inflammation are upregulated in ChP of schizophrenia patients (Kim et al., 2016). Thus, we next considered the effects of peripheral administration of the bacterial endotoxin lipopolysaccharide (LPS), which induces inflammatory responses in mouse ChP (Balusu et al., 2016b; Marques et al., 2009), on ChP immune cell morphology ex vivo and in vivo. As expected, LPS induced an inflammatory cytokine response in serum and CSF (Figures 7A and S6A). Using immunohistochemistry, we found that although peripheral LPS administration did not affect tiling of immune cells across the ChP (Figures 1A-1C, S3H, and S3I), it triggered a marked repositioning of GFP-positive immune cell bodies and processes to regions surrounding the vasculature within the ChP (Figures 7B, 7C, and S6B).
Figure 7. ChP Immune Cells Respond to Systemic and Local Insults.

(A) Higher CSF cytokine levels 1 h after i.p. injection of LPS versus saline (mean ± SEM; n = 3 samples, each consisting of 25 μL pooled across three to six mice; IL-1α, **p = 0.0017; TNF-α, **p = 0.0072; CCL2, *p = 0.0260; IL1β, *p = 0.0451; and IFN-β, *p = 0.0212 [unpaired t tests]).
(B–D) Following LPS, immune cells flatten along vessels. (B) LV ChP explants from Cx3cr1+/GFP mice that received i.p. saline (left) or LPS (middle). Segmentation of immune cells (right panel, green), and periluminal region surrounding vasculature (blue; Figures S6B and S6C; STAR Methods) allowed assessment of overlap (yellow). Scale bar, 50 μm. (C) Percentage of periluminal region occupied by immune cell processes following i.p. saline (n = 15 explants, nine mice) or LPS (n = 20 explants, ten mice). ****p < 0.0001, Welch’s t test. Mean ± SEM. (D) In vivo imaging of immune cells (green) and vasculature (red) pre-LPS (left) and 3 h following i.p. LPS (right). Scale bar, 25 μm. Arrowheads, transitions of cell bodies to splayed morphology (see Video S16).
(E) Segmentation of periluminal region (STAR Methods).
(F) Fractional change in immune cell fluorescence (ΔF/F) in periluminal region across 4 h, relative to pre-LPS baseline (red line).
(G) Schematic of focal injury via brief, high-power focusing of a laser on a small region of ChP during in vivo imaging.
(H) Maximum projections of immune cells and vasculature before, 6 min after, and 1 h after a local burn of the region within the white box. At 6 min, dextran leaks out of damaged vessels (see Video S17). Immune cell bodies then migrate to the injury site. Scale bar, 50 μm.
(I) Average pre- and post-injury velocity of immune cells toward (positive) or away from (negative) the injury site (n = 15 cells, three mice). **p = 0.0075, paired t test.
To define the morphological dynamics of individual immune cells in response to LPS, we performed in vivo two-photon imaging during peripheral delivery of LPS. Many GFP-positive ChP immune cell bodies and processes that were initially located distal to vessels prior to LPS moved toward and spread along nearby vessels within ~45–60 min of LPS delivery (Figure 7D; Video S16). Using a custom algorithm to segment vasculature and define periluminal regions (Figures 7E, 7F, and S6C; STAR Methods), we confirmed that immune cell fluorescence increased in periluminal regions (Figure 7F). Not all Cx3cr1-expressing cells responded to LPS, consistent with the multiplicity of Cx3cr1-expressing ChP immune cell types that likely exhibit distinct responses to peripheral stimuli (Dani et al., 2019; Van Hove et al., 2019). This repositioning of ChP immune cells along the periluminal region may provide an extra layer of brain protection from harmful blood-borne signals during peripheral inflammation (Mottahedin et al., 2019).
In addition to the robust response of ChP immune cells following peripheral inflammation, we found that these cells often move toward sites of local injury. We induced a focal injury by high-power two-photon heating of a small area in the center of the field of view (89 × 57 μm2; Figure 7G). This triggered rapid recruitment of immune cells to the injury site from nearby regions of the ChP. Immune cells initiated movement immediately following the laser injury, transitioned to an apparently more activated state (retracted processes, larger cell bodies), and continued moving until they stabilized in an aggregate surrounding the injury site (Figure 7H; Video S17). Across three mice, most but not all immune cells moved toward the injury site (Figure 7I). The majority of the cells that did move toward the injury site were confirmed to be epiplexus cells (Video S18). Together, these findings reflect diverse contributions of different types of resident ChP immune cells to host defense.
DISCUSSION
The scarcity of experimental tools for selectively targeting, monitoring, and manipulating ChP cells has hindered progress in understanding this essential and distinct organ located deep within the brain. Despite its principal roles in producing CSF, forming a brain barrier, and secreting important health and growth-promoting factors for the brain (Fame and Lehtinen, 2020; Ghersi-Egea et al., 2018; Lun et al., 2015b; Saunders et al., 2018), remarkably little is known regarding the functions of its cellular networks. Here, we developed imaging and analysis approaches for monitoring and pharmacological manipulation of multiple ChP cell types in live explants and in awake mice. Using a combination of epifluorescence, confocal, and two-photon microscopy in ChP explants, we observed spontaneous calcium activity as well as spontaneous exocytotic fusion events in individual epithelial cells. Both of these processes were enhanced by application of agonists of the 5-HT2C receptor, which is highly expressed in ChP epithelial cells. Epifluorescence and two-photon microscopy in awake mice revealed subcellular spontaneous calcium activity and 5-HT2C agonist-evoked calcium activity and apocrine-type exocrine release. By developing tools for volumetric, multi-color two-photon imaging of vasculature and immune cells within and on the surface of the ChP in vivo, we uncovered spontaneous surveillance behaviors of immune cells as well as profound immune cell activation and translocation following peripheral or local perturbations. We hope this ChP imaging toolkit will accelerate the pace of discoveries regarding the diverse functions of this vital deep brain tissue.
Imaging the ChP In Vitro and In Vivo
The above methods for imaging ChP explants should be of broad utility, as this approach is relatively simple and inexpensive and allows tracking of tissue prior to and following controlled delivery of multiple drugs to the apical surface of the ChP. Our description of vascular landmarks should also improve repeatability within and across studies. The LV ChP tissue is thin and delicate and not entirely flat (albeit much flatter than third and fourth ventricle ChP). Thus, in order to obtain high-quality data, it was important to carefully extract the explant, stably mount it, and adjust fluid flow and osmolarity to avoid undue stretch and pressure (Figure S1A).
In vivo methods enabled monitoring of ChP in a largely natural environment during systemic delivery of drugs or perturbations (Figures 4, 6, and 7). Although in vivo imaging using a cannula has been demonstrated in many deep brain areas (e.g., Dombeck et al., 2010; Liang et al., 2018), motion of ChP tissue posed a particularly challenging problem, as the ChP is anchored at the ventromedial aspect of the LV, far from the dorsal ChP regions that we imaged. This likely contributed to substantial non-rigid motion in three dimensions beyond what is observed in other brain tissues that are pressurized and anchored by the imaging window. Thus, although our use of a treadmill to minimize head torque applied by the limbs likely reduced motion artifacts to some extent (Dombeck et al., 2007), it was critical to additionally use several methods for two-dimensional (2D) and non-rigid 3D co-registration of imaging datasets in order to attain subcellular resolution (Figure 5). Another option to reduce coupling of body and brain motion could be to anesthetize mice prior to imaging. Although this may be particularly useful for structural imaging studies, anesthesia could significantly alter the functional properties of the ChP.
We did not observe sustained inflammation of ChP for time points at which imaging was performed, several weeks following implantation (Figure S3). Intracranial pressure also normalized to baseline levels following this recovery period (data not shown). This recovery period also improved imaging clarity in comparison with acute imaging immediately following surgery (not shown), consistent with deep imaging in other brain regions (Goldey et al., 2014; Liang et al., 2018). Nevertheless, additional improvements to our approach could further minimize the invasive nature of the cannula implant. For example, with improved red and infrared fluorescent indicators, window implants for two- and three-photon imaging of ChP can be placed well above the dorsal surface of the LV (e.g., Wang et al., 2018; Weisenburger et al., 2019). Alternatively, lower profile GRIN lenses may be used for intraventricular imaging, albeit with a much smaller field of view and range of imaging depths.
Spontaneous and Evoked Calcium Activity and Vesicle Fusion in ChP
We observed diverse rates of spontaneous calcium activity and diverse thresholds for evoked activity across nearby cells. These differences may relate to differences in activity states or to subtypes of epithelial cells. In the future, such functional characterizations of epithelial cells can be merged with single-cell transcriptomics (Dani et al., 2019) to better understand potential divisions of labor across cells.
Spontaneous calcium transients were restricted to subregions of a cell. Future studies can assess whether these subcellular events relate to the subcellular vesicle fusion events we observed in explants or to activation of a single protrusion among the many protrusions on the apical surface of each epithelial cell (evident in EM images in Figure S2I). These events were particularly fast (~200 ms) when measured in vivo, possibly because of calcium imaging at warmer ambient temperatures in vivo versus in vitro.
Application of a 5-HT2C receptor agonist drove strong increases in calcium activity and increased rates of vesicular fusion. This calcium sensitivity of epithelial tissue to serotonin and associated agonists is consistent with previous reports using cultured, dissociated ChP cells (Watson et al., 1995). Higher concentrations of 5-HT2C agonist evoked large, apocrine-type secretory events (Figures 4L-4N; Videos S9 and S10) that have been reported in ChP and other epithelia, including sweat and mammary glands (Farkaš, 2015). Although these secretory events involve massive release of internal contents from an epithelial cell, they do not imply that cell health is compromised. Rather, this process may represent an efficient and rapid means for activity-dependent secretion of large amounts of cargo in response to an external stimulus, possibly in conjunction with other rapid changes (e.g., rapid activation of water and ion channels). Our studies set the stage for more in-depth investigations of how the ChP dynamically regulates the molecular composition of the CSF that bathes the CNS.
Immune Surveillance at the ChP
Immune cells have been proposed to enter the brain via the ChP (Ghersi-Egea et al., 2018; Reboldi et al., 2009; Schwartz and Baruch, 2014; Shechter et al., 2013), but little is known about the functions of resident ChP immune cells during baseline conditions or in response to peripheral immune challenge or local injury. Tracking of Cx3cr1-expressing ChP immune cells together with vascular labeling in vivo revealed vascular surveillance by stromal immune cell processes, while cell bodies remained largely immobile. This surveillance points to active maintenance and phagocytic roles at the blood-CSF barrier. Notably, we found that these stromal immune cells still contained 70 kDa dextrans weeks after i.p. injection. In contrast, epiplexus cells on the apical surface of the ChP showed much greater cell body mobility but did not take up dextrans.
ChP immune cells also responded to systemic LPS delivery by spreading their cell bodies and processes along the periluminal region near blood vessels, a finding confirmed using immunohis-tochemistry. This cellular response may reflect a means of protection against peripheral insults, and differs from that of cortical microglia, which retract their processes and adopt an amoeboid “activated” shape during inflammation (Pozner et al., 2015). Without access to the time-lapse in vivo imaging, it would not have been possible to determine whether the same local immune cells change their morphology and location or whether new immune cells had entered the same region of ChP. Indeed, despite previous reports that immune cells cross at the ChP (Ghersi-Egea et al., 2018; Reboldi et al., 2009; Schwartz and Baruch, 2014; Shechter et al., 2013), our imaging sessions did not reveal arrival or departure of new immune cells from either the CSF or the vasculature during baseline conditions or following LPS. Future studies should examine deeper regions of the LV across a broader range of conditions to more fully assess potential subregions that mediate transit of immune cells to and from the brain.
We also noted rapid mobilization of nearby immune cells following deliberate heating of a focal region of the field of view using transient, high-magnification and high-power two-photon imaging. Many of these cells were epiplexus cells, which acted as “first responders” by accumulating at the injury site. This behavior is strikingly different from that of Cx3cr1-positive microglia in cortex and other brain regions (Davalos et al., 2005; Pozner et al., 2015), which extend their processes toward a laser-induced lesion to contain the injury while their cell bodies remain stationary (Davalos et al., 2005). Our findings can inform surgical procedures involving focal heating of ChP, such as during cauterization of ChP to treat hydrocephalus (Warf, 2005).
In the future, it should be possible to use the in vivo imaging approach described here to assess the role of changes in calcium and other intracellular signals in immune cells, epithelial cells, and other stromal cell types in the ChP during these and other immune challenges and brain injuries. A better understanding of the dynamic roles of multiple ChP cell types in various barrier functions in the intact brain should spark new ideas for penetrating this barrier for drug delivery to the brain, as well as for fortifying this barrier across the lifespan. More generally, given that repeated in vivo access and chronic imaging may be more amenable in the ChP than in most other body epithelia other than skin epithelium (Mesa et al., 2015; Rompolas et al., 2016), this platform may provide a unique window into the general functions of barrier epithelia in their natural environments.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria Lehtinen (maria.lehtinen@childrens.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The registration and vascular segmentation algorithms generated for this study are available at https://github.com/LehtinenLab/Shipley2020. Original data are available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committees of Beth Israel Deaconess Medical Center (Figures 3, 5, 6, and 7), Boston Children’s Hospital (Figures 1, 2, 3, 4, 5, 6, and 7), and Brown University (Figures 3 and 4). Mouse lines used include FoxJ1-Cre (Zhang et al., 2007), Ai95D (Jax# 024105; Madisen et al., 2015), Cx3cr1+/GFP (Jax# 005582; Jung et al., 2000), Htr2CmRuby3, CD-1, and C57BL/6 (Figures 1 and 2: male and female mice; Figures 3, 4, 5, 6, and 7 male mice). Htr2CmRuby3 mice were generated by the Gene Manipulation & Genome Editing Core, IDDRC, BCH. Best-ranked sgRNAs near the targeting region in Htr2C genome were picked (Doench et al., 2016) and synthesized (Alt-R® CRISPR-Cas9 crRNA, Integrated DNA Technologies). Donor plasmid was custom made at GeneScript, prepared with EndoFree Plasmid Maxi Kit (QIAGEN). Alt-R® S.p. HiFi Cas9 Nuclease (Integrated DNA Technologies) was used for the editing. A mixture of crRNA (0.61 μM), Cas9 protein (10 ng/μl), and donor (10ng/μl) was injected into 0.5 dpc embryos harvested from C57BL/6NHsd (Envigo) mating pairs. Embryos that survived the injection were implanted into recipient pseudopregnant females and allowed to reach term. Tail biopsies from pups were genotyped to identify founders. The line was maintained in C57BL/6J.
METHOD DETAILS
ChP explant preparation
Whole ChP from the lateral ventricle was harvested using #5 forceps and fine-dissection scissors. To collect the LV ChP, the hindbrain was separated from the mid- and forebrain structures using a scalpel, followed by a bilateral cut along the midline to separate the cortex into two hemispheres. Each hemisphere was stabilized with forceps and a third of the rostral end was cut off, the developing hippocampus was rolled out using the flat surface of a scalpel, and the attached LV ChP was gently separated from the hippocampus/fornix using forceps.
LV ChP was transferred onto round coverslips (15 mm, Warner Instruments, Cat. 64-0733) that had been prepared as follows: briefly, coverslips were lightly coated with Silicone (Kwik-sil, World Precision Instruments, Item. 600022), and while wet (1x aCSF: 119 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 11 mM glucose, with fresh 2.0 mM magnesium chloride and 2.8 mM calcium chloride), a polycarbonate membrane (Whatman, Nucleopore, 13 mm wide, 8.0 μm pore size, Cat. 110414) was placed on the coverslip. Edges of the polycarbonate membrane were attached to the coverslip using adhesive (3M, Vetbond). These glass coverslips were kept at room temperature and allowed to cure. The ChP was flattened onto the membrane and secured using 3M Vetbond. All samples were placed in a holding chamber with continuously oxygenated (95% O2 / 5% CO2) 1x a CSF.
In Vitro Epithelial Cell Experiments
In vitro epifluorescence calcium imaging
Epifluorescence calcium recordings were acquired from FoxJ1-Cre::Ai95D ChP explants (see above) using a 4x, 0.28 NA objective (Olympus). A halogen lamp and FITC filter set (Olympus) were used for excitation and emission filtering. Green fluorescence was collected using an sCMOS camera (Hamamatsu). Images (2048 × 2048 pixels, 3.30 × 3.30 mm2) were acquired at 10 frames/s using MicroManager (NIH).
Segmentation of cell masks
Epifluorescence calcium videos were cropped to a small region (161 × 161 μm2) near the center of the sample, for ease of processing. We obtained cell masks based on cell shape, as follows: first, a mean image, I, was generated, and was locally contrast-normalized using:
Where G is the 2D Gaussian operation. The normalized image was then binarized and watershed to separate cells that were joined together. Objects smaller than 5 pixels were considered noise and discarded. The convex hulls of remaining objects were used as cell masks. Neuropil masks were estimated as the annulus spanning the region between the cell perimeter and the perimeter obtained after dilating the cell by 5 pixels.
Trace extraction
A raw trace, F(t), was extracted from each cell mask by calculating mean intensity across pixels in each mask, and for each movie frame. A neuropil activity trace, Fneuropil(t), was calculated in the same way, using the corresponding neuropil mask. A neuropil-corrected signal, Fcorrected(t), was calculated by subtracting the neuropil trace from the raw trace, and adding back the mean of the signal:
The signal was further normalized by:
Where median(Fcorrected, 500) denotes a moving median filter with a window size of 500 frames (50 s) surrounding the time t.
Calcium events were defined as peaks in which ΔF/F > (5 × std(ΔF/F)). Cells with traces that never exceeded this threshold were considered “inactive.”
In vitro two-photon calcium imaging
Two-photon microscopy was used to record calcium activity in explants in which ChP epithelial cells express GCaMP6f (in FoxJ1-Cre::Ai95D mice, using an Olympus MPE-RS two-photon microscope; 30.0 frames/s; 512x512 pixels/frame). All imaging was performed with a 25x, 1.0 NA objective (Olympus) at 4.5x digital zoom (~113 × 113 μm2). Laser power measured below the objective at 940 nm was 55 mW using a Mai Tai DeepSee laser (Spectra-Physics). To perform 3D recordings, the settings above were used in conjunction with a nPFocus250 piezo microscope stage (nPoint) moving axially in a sawtooth pattern. 3D volume recordings were acquired at ~0.25 Hz to capture baseline activity, during which time aCSF flowed through the perfusion chamber (Warner; performed at room temperature) containing the ChP explant. Subsequently, increasing concentrations of 5-HT or the 5-HT2C selective agonist, WAY-161503 (Tocris), in aCSF were introduced for one minute per concentration, with ten minute aCSF washouts in between drug deliveries. To measure bulk tissue fluorescence, a mean volume projection along the axial (z) dimension was performed to flatten each 3D volume into a 2D image, resulting in a 2D video across time. Average fluorescence across the 10 minutes baseline period prior to the first drug delivery was used as a baseline image. A ΔF image stack was constructed by subtracting this baseline image from each frame in the video. A trace of ΔF activity was calculated as the mean pixel intensity of each frame of the image stack.
VAMP3-pHluorin imaging
Cultured cells
Z310 cells were cultured on glass coverslips and lipofectamine transfected with pAAV-VAMP3-pHluorin. After 3 days, the cells were imaged using TIRF microscopy (Cocucci et al., 2012) with a 100x objective (1.46 NA, Carl Zeiss) and a 2x magnification lens placed in front of the CCD camera (QuantEM, Photometrics). This arrangement provided a final pixel size of 80 nm. ChP explants: AAV2/5-VAMP3-pHluroin was injected in utero i.c.v. in E14.5 embryos. LV ChP explants were then harvested at P18-P24. Each ChP was attached directly onto an imaging dish using Vetbond and immersed with 1.8 mL of aCSF. WAY-161503 was added until the final bath concentration reached 500 nM. Individual epithelial cells from explants were imaged using a ZEISS LSM880 Airyscan confocal microscope. The chamber, imaging dish holder, and all buffers used were maintained at 37°C.
To detect secreted vesicles, each image frame was first smoothed with a two-pixel radius Gaussian filter (160 nm). We then further filtered each image by calculating the determinate of the Hessian matrix at every pixel, and this image stack was used to isolate VAMP3 fusion events from cell background. Masks of regions involving a fusion event were obtained by PCA/ICA segmentation (Mukamel et al., 2009). Fluorescent traces were extracted by averaging fluorescence of all pixels within each mask. Each fluorescence trace was normalized to peak fluorescence, and sorted the traces by the time at which this peak occurred, in order to generate a heatmap of time courses of vesicle release events.
In Vivo Imaging Experiments
Headpost and cranial window placement
Mice used for in vivo two-photon imaging (8-20 weeks) were outfitted with a headpost (titanium, 0.7 g, H.E. Parmer) and 3 mm cranial window using minor modifications of techniques previously described (Goldey et al., 2014; Liang et al., 2018). Briefly, each cranial window implant was first prepared by gluing a 3 mm x 2 mm (diameter x height) stainless steel cylindrical cannula (MicroGroup) to a 3 mm diameter glass coverslip (Warner) using a UV-cured optical adhesive (Norland, type 71). Approximately 3 hours prior to surgical implantation, dexamethasone sodium phosphate (4 mg/ml, intramuscular) was administered in order to reduce brain edema. Anesthesia was induced using isoflurane (1%–3% in 100% O2, with flow rate titrated to a respiratory rate of 1 breath per minute). Using standard aseptic techniques and a stereomicroscope, a 3-mm diameter craniotomy was performed over the left side of the skull, centered using stereotactic coordinates (2.0 mm lateral and 0.2 mm posterior to Bregma). Next, portions of neocortex, corpus callosum, and hippocampal tissue were carefully and slowly aspirated to expose the lateral ventricle, with the specific purpose of preventing undue increase in intracranial pressure. The ChP was visualized floating within the ventricle. Hemostasis was achieved with copious irrigation using sterile phosphate-buffered saline and occasional use of gelfoam. At this point, the cranial window implant was inserted through the craniotomy site and lowered to a depth of approximately 2.0 mm below the skull where it pressed lightly on the surface of the thalamus and preserved direct visualization of the intact ChP. The cannula was temporarily affixed to the skull with Vetbond (3M) followed by a permanent seal with C&B Metabond (Parkell). A custom two-pronged titanium headpost was then affixed to the skull and again sealed with C&B Metabond (the headpost implantation can also be performed prior to the craniotomy according to investigator preference).
To create a low-profile adaptor to accommodate the water-immersion objective and light shielding, a custom 3D-printed imaging well (outer diameter of the base, inner diameter, height: 20 mm, 10 mm, 4 mm, or 7.5 mm, 5 mm, 1 mm) was then positioned around the cannula and glued to the cement and headpost. Animals were given Meloxicam (5 mg/kg, s.c.), individually housed, and allowed at least 2 weeks to recover before live imaging. The estimated success rate in obtaining clear windows was ~80% for a trained surgeon. In the first post-operative week, the mice were undisturbed and, during the second week, the mice were habituated to the imaging environment. Each mouse was placed on a custom 3D-printed running wheel and the animal’s head was fixed using clamps (Thorlabs) that attach to each prong of the two-pronged titanium headpost. The running wheel and associated flexible hinges were useful for decreasing brain motion, by decreasing the degree to which hindlimb-related forces couple to brain motion. During two-photon imaging sessions, the low-profile imaging well was covered with blackout fabric (Thorlabs).
In Vivo Epithelial Cell Experiments
Epifluorescence imaging
To initially localize the ChP and assess stability and orientation of the ChP post-surgery, an epifluorescence video was recorded while scanning axially through the tissue. To account for lensing effects from changing z planes, planes were registered with scaled rotations to each other using the StackReg plugin in Fiji (NIH).
Two-photon imaging of spontaneous activity
To capture high-speed subcellular and cellular activity in epithelial cells in vivo, two-photon microscopy was used to record calcium activity in a ~25 cells. Imaging of GCaMP6f-expressing epithelial cells (in FoxJ1-Cre::Ai95D mice, see above) was performed using a resonant-scanning two-photon microscope (Olympus, 512x512 pixels/frame; Bruker, 490× 372 pixels/frame). Spontaneous activity was recorded at a single imaging plane (Olympus, 30.0 frames/s; Bruker 41.5 frames/s). All imaging was performed with a 25x, 1.0 NA objective (Olympus) at 4.5x digital zoom (~113 × 113 μm2). Laser power measured below the objective was 55 mW using a Mai Tai DeepSee laser at 940 nm (Newport Corp.).
Registration/preprocessing of spontaneous activity
To compensate for rapid ChP motion caused by mouse locomotion and changes in posture, each frame was registered to a target image created by the mean of the first 500 frames. Registration was performed by cross-correlating the Fourier transform of each image with this target image (i.e., rigid-body translation correction; Guizar-Sicairos and Fienup, 2008).
Cellular and subcellular segmentation – spontaneous activity
After registration, a mean image across the entire recording was generated. Cell outlines and outlines of cell nuclei were manually drawn for each cell in the field of view (~20-40 cells). Cytoplasm masks were generated from the difference between cell mask and nucleus mask. Cytoplasm masks were subdivided into 12 radially symmetric subsections from the center of the nucleus. The neuropil area was an annulus surrounding the cell, calculated by dilating the cytoplasm masks by 10 pixels, and excluding pixels in the original cell mask from this dilated cell mask.
Subcellular trace extraction – spontaneous activity
First, the aligned video was down-sampled by a factor of 4. For each cell, the raw sector activity (i.e., a pie slice of the cell), Fraw(ε, t), was calculated as mean activity across pixels inside each of the cytoplasm sectors (θ) for every time point, t. Neuropil activity, Fneuropil(t) was calculated as mean pixel activity in the neuropil mask defined above, at every time point, t. Neuropil activity was subtracted from raw activity to generate a neuropil-corrected time course:
To normalize for different baseline section brightness, a rolling median of 6.67 s was subtracted from neuropil-subtracted signal:
To find subcellular calcium events, the maximum signal across cell sectors was calculated, for each time t, by taking a maximum projection across sectors, and then subtracting the median across sectors:
This approach generated a single trace of the largest fluorescence deviation from median fluorescence across cell sectors at every time point.
To identify subcellular events, a peak detector was applied to the above trace using a threshold based on the trace of median activity across sectors (‘median trace’, Fmed). First, Fmed was calculated as:
Subcellular calcium events were defined as local peaks of epochs in which ΔFmax > (5 ×std(ΔFmed(t))).
In vivo 3D imaging of epithelial cell responses to delivery of a serotonin agonist
To perform 3D recordings, the same imaging settings described above for spontaneous in vivo calcium imaging were used, but with the addition of a nPFocus250 piezo microscope stage (nPoint) that moved the imaging plane axially in a sawtooth pattern (scanning of 93 planes per volume across 350 μm of depth with a scan rate of frame rate of 30.0 frames/s, 512x512 pixels/frame, resulting in volume scanning of a 170x170x350 μm3 volume at 0.32 volumes/s).
To register these volumes, we first averaged together every ten volumes in order to improve signal-to-noise ratio. Since the observed effects of WAY-161503 were slow and long lasting, this approach did not overly compromise temporal resolution. Each plane of these average volumes was registered to the center z plane (middle plane) of the volume using the StackReg plugin in Fiji (NIH), creating a rectified volume. The maximum intensity projection of each of these rectified volumes were used to correct for inter-volume motion X-Y motion. Using the first volume as an anchor point, each volume was registered to the previous volume. X-Y plane transverse shifts were calculated by cross-correlating the Fourier transformations of the maximum intensity projection of a given volume and of the previous volume (Guizar-Sicairos and Fienup, 2008).
In Vivo Immune Cell Experiments
Dextran injection
Mice received intraperitoneal injections of dextran conjugated with Texas Red (70 kDa, 0.2 mg/g i.p.). ThermoFisher Scientific), delivered 30 minutes before imaging. Presence of dextran in vasculature was confirmed by two-photon imaging.
Two-photon imaging
3D volume recording was necessary to robustly track the ChP across long timescales due to mouse motion, changes in posture, and occasional axial drift of ChP. Two-photon imaging of immune cells and vasculature was performed using a resonant-scanning two-photon microscope (experiments were performed on two different microscopes: Olympus; 12.8 frame/s; 512× 512 pixels/frame; 0.16 volumes/s, 81 planes/volume; volume size: 254× 254× 400 μm3. Neurolabware: 15.5 frames/s; 796x512 pixels/frame; 0.25-0.5 volumes/s, 31-62 planes/volumes; volume size: 355× 230× 100 μm3). Volume scanning on the Olympus was achieved by using a piezo microscope stage (nPFocus250). Volume scanning on the Neurolabware microscope was achieved using a tunable focus lens (Optotune). All imaging was performed with a 25x, 0.95 NA objective (Olympus) at 2x zoom (~254 × 254 μm2 (Olympus), ~360 × 230 μm2 (Neurolabware)). Laser power at 940-960 nm (Mai Tai DeepSee laser, Spectra Physics) measured below the objective was 30-40 mW. Immune cells were confirmed to be located within or on the outside of the ChP based on colocalization with the fluorescent dextran-labeled vasculature pattern.
3D registration
Due to the rapid motility of immune cells across seconds, 3D registration of individual volumes was necessary to properly account for ChP movement at these rapid timescales (see Figure 5). To account for optical deformation and warping caused by the focus-tunable lens, a counter-warping correction was calculated for each imaging session. The first 30 volumes were averaged together to create a mean distorted volume. The affine transformation was used to iteratively match each plane to its neighbor, beginning with the brightest plane of the volume and moving up and down until the ends of the volume. Since affine transformations are linear functions, the adjacent transformations could be combined by multiplication of the augmented transformation matrix to generate the warp-correction of every focus-tunable lens plane to the reference. These matrices are calculated using the MultiStackReg plugin in Fiji (NIH). Since these deformations were due to the optical system, not motion of the sample, these corrections were applied to every volume prior to subsequent motion correction (Figure 5B, “Step 1”). For the Olympus microscope that uses a piezo microscope stage to scan axially, there is no deformation, and this step is skipped.
Due to rapid motion caused by mouse movement, it was necessary to account for intra-volume changes. Using the brightest plane as a stationary anchor plane, each plane was registered to its neighbor, using Fourier cross-correlation to estimate the X and Y shifts. These neighboring X and Y shifts were summed cumulatively so that each plane is aligned with the anchor plane. (Figure 5B, “Step 2”) (Guizar-Sicairos and Fienup, 2008).
After intra-volume alignment, reference volumes were generated by averaging every 20 volumes. To account for inter-volume lateral and axial shifts, each volume was then registered to its respective reference volume by cross-correlating the 3D Fourier transformation of the two volumes to find the X, Y, and Z shifts. Each reference volume was registered to the first reference volume using the same method (Figure 5B, “Step 3”).
Axial projections, such as mean, median, and maximum projections, were then performed (Figure 5B, “Step 4”). Finally, the movie of these projected images was further stabilized in three successive steps: (i.) matching each frame to the average of the first 50 frames, (ii.) matching each frame of the resulting movie iteratively to its neighbor, (iii.) matching each frame of the resulting movie to the average of the first 50 frames (Figure 5B, “Step 5”).
To estimate the degree of brain motion of ChP in vivo, we quantified the two kinds of correction for brain motion that were applied (see above). The first correction involved intra-volume XY displacements for each plane (Figure 5B, “Step 2”). The second involved inter-volume displacements from 3D translational registration (Figure 5B, “Step 3”), together with additional XY displacements common to all planes and derived from the registration of the 2D image stack resulting from axial mean projections of each volume (Figure 5B, “Step 5”). The intra-volume XY displacement reflected faster frame-to-frame motion within a given Z-scan (12.8-31 frames per second; 31-81 frames per volume). We quantified the distribution of intra-volume XY displacements using the Euclidean distance of intra-volume shifts in X and Y. We also calculated inter-volume displacements between successive volumes (0.16-0.97 volumes per second) to estimate the level of motion observed at these somewhat slower timescales (using the using the Euclidean distance of X and Y displacements between consecutive volumes, ΔXY, and using the absolute value of the shift in Z between consecutive volumes). These distributions were then expressed as cumulative distribution functions for each recording (Figures 5G and 5H). Overall, the degree of brain motion was substantially larger than what is observed for recordings in other brain regions such as in the neocortex.
Inter-volume motion artifacts
To assess the efficacy of our 3D registration in removing motion artifacts, we considered sets of five consecutive volumes of the red channel (vasculature), which was expected to be stable (i.e., near-constant voxel intensity) in the absence of brain motion at this timescale. Thus, we used the metric of local standard deviation as a proxy for inter-volume motion artifacts. A rolling standard deviation across five neighboring volumes was calculated for each 3D voxel. These standard deviation contributions were averaged to obtain a global estimate of image stability. To account for global intensity changes within and across recordings, we normalized this mean standard deviation signal by the mean fluorescence to obtain an estimate of inter-volume motion artifacts over time.
Re-registration of single-cell regions
After 3D registration of the entire region, individual cell regions of interest (ROIs) were selected for local re-registration. XYZ regions were determined manually. The selected regions were then re-registered in XY with Fourier transformation-based cross-correlation, and individual plane affine registration, as described in the “3D registration” section, above.
Recording “physiological housekeeping” by immune cells
To assess the uptake of dextran by immune cells, a 3D recording of the ChP was acquired for 1 hour before the injection of 70 kDa red dextran. Immediately after this recording, without moving the recording field of view, the mouse was injected with 70 kDa red dextran, and recorded for 1 hour, as the dextran filled the vessels. Another 48 hours later, using local vascular features and tissue morphology, we imaged the same volume of ChP for an additional hour, without injecting more red dextran. All three recordings used the same acquisition parameters.
Response to LPS
To elicit a peripheral inflammatory response, 5 mg/kg lipopolysaccharide (LPS, Sigma) was delivered i.p. (Monje et al., 2003). An equal volume of saline was used as a control.
Quantifying in vitro “vessel coating” by immune cells
To quantify immune cell alignment with the periluminal region immediately adjacent to vessels, whole-mount LV ChP explants of LPS- and saline-injected Cx3cr1+/GFP mice were isolated and immunostained for GFP to label immune cells, and PECAM to label vasculature. Using a 500 pixel x 500 pixel ROI (225 × 225 μm2), the image pixel intensities were rescaled to the range 0-1 (20th percentile of pixel brightness rescaled to 0; 90th percentile of pixel brightness rescaled to 1). A first step to defining the periluminal region was to develop an automated algorithm to segment the vasculature. Segmenting vasculature involved identifying image regions that contain tube-like structures. Additionally, these tubes may be of different sizes (e.g., capillaries versus veins), and may also join together in junctions. Based on this structural description, a Jerman filter (Jerman et al., 2016) based on the local second-order derivative of the image (filter widths from 8 to 15 pixels; regularization factor τ = 1) was used to enhance pixels that were part of tube-like structures. The resulting image was then binarized to separate vasculature from the background.
To validate the automated vessel segmentation algorithm used as an initial step toward defining periluminal space surrounding vessels, the vasculature image was manually segmented as a ground truth comparison. Using the Selection Brush tool in Fiji, the vessels were hand traced and converted to a manual binary mask. The same region was automatically segmented, as above, to generate an automatic binary mask. The automated and binary masks were compared with the contour matching function, bfscore.m, in MATLAB. Briefly, the Boundary F1 score measures the how closely a predicted boundary matches a ground truth boundary. This algorithm was chosen over Dice or Jaccard similarity coefficients, as our principal goal involved defining the accuracy of the estimate of periluminal boundary.
To obtain an estimate of periluminal space, the edge of the binary image of vasculature (see above) was then dilated with a disk kernel with a width of 5 pixels (~1.6 μm). Pixels belonging to this dilated mask but not to the original vascular mask were considered to belong to the periluminal region.
Pixels containing immune cell bodies or processes were defined as follows, from the green emission image. First, the image pixel intensities were rescaled to the range 0-1 (10th percentile of pixel brightness rescaled to 0; 98th percentile of pixel brightness rescaled to 1). The image was then binarized, and dilated using a disk kernel with a width of 5 pixels. The degree of immune cell process occupancy within the periluminal region (“vessel coating”) was estimated as the proportion of vessel edge that overlapped with the binarized and dilated immune cells.
In vivo vessel coating
To quantify in vivo changes in immune cell occupancy of periluminal regions near vessels in response to LPS, a four-hour, four-dimensional (X Y x Z x T stack) dataset was mean-projected along the Z axis to produce an XYT image stack. This image stack was downsampled to 120 volumes (by averaging successive sets of 30 volumes), to improve the signal-to-noise ratio. This image stack was split into two channels: immune cells and vasculature. To define the periluminal region, vascular image stack intensity levels were rescaled to the range 0-1 (20th percentile of pixel brightness rescaled to 0; 98th percentile of pixel brightness rescaled to 1), and registered using the MultiStackReg function in Fiji, based on motion estimates from the immune cell image stack (NIH). A median projection of the vascular channel was computed, and pixels belonging to vessels were enhanced relative to the background using a Jerman filter (Jerman et al., 2016)(filter width from 8 to 10 pixels; regularization factor, τ = 1; see above). The gradient of the Jerman filtered image was calculated to find the vessel edges, and the gradient image was binarized, and morphologically closed (dilation, followed by erosion of a binary image, resulting in the filling of small holes; the structuring element was a disk with radius of 2 pixels), defining the periluminal regions of the image.
The immune cell image stack intensity levels were rescaled to the range 0-1 (10th percentile of pixel brightness levels, rescaled to 0; 98th percentile of pixel brightness levels, rescaled to 1). For each time frame in the image stack, t, the fluorescence in the periluminal region, F(t) was calculated as mean pixel intensity in the periluminal region defined above.
GFP fluorescence in the periluminal region, F(t), was measured at each frame in the image stack as the mean immune cell pixel intensity in the periluminal region defined above. The average fluorescence in the 1 hour prior to LPS injection was used as a baseline, F0. Change in fluorescence in response to LPS was expressed as: ΔF/F0.
Immune Cell Imaging During Acute Chp Injury
In this experiment, a local region of the ChP of an adult awake mouse was heated using the imaging laser, together with two-photon imaging of immune cells and vasculature prior to and following the heating. After one hour of imaging of 3D volumes of immune cells and vasculature (as detailed above) at 2x magnification and 30-40 mW power, the scan settings were changed to 8x magnification and ~150 mW for two minutes to induce focal heating of a local region of the tissue. Immediately following this tissue manipulation, the previous laser scanning settings were returned to observe immune cell activity in response to the focal damage.
Tracking immune cell acute response to laser burn injury
Immune cell body positions relative to the rectangular region targeted for focal laser heating were sampled every 5 minutes for 30 minutes before heating and in the one hour after heating. A distance transformation was used to calculate the closest distance from each cell to the rectangular injury site at each time point (using bwdist.m in MATLAB). Velocities of these cells toward the burn site (Δ distance /Δ time) were calculated for the 30-minute period before the injury, and separately for the 60-minute period after the injury. In cases where the cell entered the burn site prior to the end of the recording period, the velocity was calculated using the displacement that took place until the moment that the cell entered the burn site.
Other Supporting Experiments
Immunostaining and immunoblotting
ChP explants were dissected and fixed in 4% paraformaldehyde for 10 min at room temperature. Samples were incubated in primary antibodies overnight at 4°C and in secondary antibodies at room temperature for two hours. For ACTA2 staining, samples were permeabilized with 0.1% Triton X-100 in PBS prior to primary antibody incubation. For GFP and PECAM (CD31) staining, samples were blocked (0.3% Triton X-100, 5% goat serum in PBS) for 1 hour prior to primary antibody incubation. All samples were counterstained with Hoechst 33342 (Invitrogen, H3570, 1:10,000) and mounted onto slides using Fluoromount-G (SouthernBiotech). Standard protocols were used for immunoblotting.
Nearest-neighbor analysis
Locations of cell centers were selected using the Cell Counter plugin in Fiji. The tissue outline was drawn manually and made into a binary mask using Fiji. The nearest-neighbor distance was calculated for each individual cell, and the cumulative distribution function was plotted using the spatstat package in R (Baddedy et al., 2015).
Simulated distributions based on a 2D Poisson distribution were generated iteratively 100 times. The mean of these simulated distributions yielded the “Poisson” estimate, while their extrema yielded a p = 0.01 acceptance interval.
Cytokine FACS array
Pure CSF samples were collected from the cisterna magna. Blood samples were collected by tail-nick one hour following saline or LPS injection. The samples were coagulated, centrifuged and diluted five-fold. Post-surgical CSF samples were diluted two- or three-fold; post-LPS CSF samples were not diluted. For the 13-plex cytokine FACS-ELISA analysis, all samples were processed according to the manufacturer’s instructions. After resuspension, the beads were run on a FACS Celesta (BD Biosciences) and FACS results were analyzed by LegendPlex v7.1 software.
Quantitative RT-qPCR
RNA samples were prepared using either the RecoverAll Total Nucleic Acid Isolation Kit (Ambion) or the mirVana miRNA isolation kit, following the manufacturer’s specifications. Extracted RNA was quantified spectrophotometrically and 100 ng was reverse-transcribed into cDNA using the ImProm-II Reverse Transcription System (Promega) or ABI High Capacity cDNA Reverse Transcription Kit (4368813, Thermo Fisher). Primers were purchased from Thermo Fisher (Taqman Gene expression assays, informed by “Best Coverage”) and q-PCR reactions were conducted performed in duplicate using Taqman Fast Univ. PCR Master Mix. Cycling was executed using the StepOnePlus Real-Time PCR System (Invitrogen) and analysis of relative gene expression was performed using the 2−ΔΔCT method (Livak and Schmittgen, 2001). Gene expression readouts were normalized to eukaryotic 18S rRNA or Gapdh as internal controls.
Transmission and Scanning EM
Lateral ventricle ChP tissue from adult mouse brain was micro-dissected and processed for EM using standard methods (Coulter et al., 2018).
QUANTIFICATION AND STATISTICAL ANALYSIS
To achieve robust and unbiased results while minimizing animal use, whenever possible, we focused on within-mouse comparisons (e.g., changes in immune cell motility before and after tissue injury, Figure 7I), which affords greater sensitivity. It was not possible for experimenter to be not blinded to experimental conditions, except in analysis of LPS versus saline control (Figure 7C). We attempted to adhere to principles of Good Laboratory Practice (https://www.who.int/tdr/publications/documents/glp-handbook.pdf). Unbiased results were obtained by prospectively defining exclusion criteria (e.g., acquisition criteria [alignment, image quality], viral expression/localization). All descriptions of statistical significance, statistical tests used, and exact values and representations of n can be found in the figure legends. Software packages used for statistical tests can be found in the STAR Methods Key Resource Table under Software and Algorithms.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-ACTA2-Cy3 | Sigma-Aldrich | #C6198, RRID: AB_476856 |
| Mouse monoclonal anti-AQP1 | Santa Cruz | #sc-32737; RRID: AB_6266923 |
| Rat monoclonal anti-CD31 | BD Pharmingen | #550274, RRID: AB_393571 |
| Chicken polyclonal anti-GFP | Abcam | #ab13970; RRID: AB_300798 |
| Rabbit polyclonal anti-VAMP3 | Synaptic System | #104103; RRID:AB_887812 |
| Rabbit polyclonal anti-Iba1 | Wako | #019-19741; RRID:AB_839504 |
| Rabbit polyclonal anti-GFAP | Dako | #Z0334; RRID:AB_10013382 |
| Rabbit polyclonal anti-S100β | Dako | #Z0311; RRID:AB_10013383 |
| Bacterial and Virus Strains | ||
| pAAV-VAMP3-pHluorin | VAMP3-pHluorin coding sequence donated by Stephanie Gupton (Urbina et al., 2018) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| WAY-161503 hydrochloride | Tocris | #1801 |
| Serotonin hydrochloride | Sigma | #H9523 |
| Dextran, Texas Red, 70,000 MW Neutral | Life Technologies | #D1830 |
| Lipopolysaccharide | Sigma | #L3024 |
| Alt-R CRISPR Cas9 Nuclease V3 | Integrated DNA Technologies | #1081060 |
| HBSS | Fisher | #SH30031FS |
| Isoflurane | Patterson Veterinary | #07-893-2374 |
| Meloxicam (NSAID analgesic) | Patterson Veterinary | #07-893-5916 |
| Saline | N/A | |
| Critical Commercial Assays | ||
| TaqMan Fast Universal PCR Master Mix Gene Expression Assay | Thermo Fisher | #4366072 |
| LEGENDplex Mouse Inflammation Panel (13-plex) w/VbP | Biolegend | #740446 |
| EndoFree Plasmid Maxi Kit | QIAGEN | #12362 |
| miRNA isolation kit | mirVana | AM1560 |
| Experimental Models: Cell Lines | ||
| Z310 cells | (Zheng and Zhao, 2002) | N/A |
| Experimental Models: Organisms/Strains | ||
| FoxJ1-CRE | (Zhang et al., 2007) | N/A |
| Cx3cr1-GFP | Jackson Laboratory | 005582; RRID:IMSR_JAX:005582 |
| Ai95D(RCL-GCaMP6f)-D | Jackson Laboratory | 028865; RRID:IMSR_JAX:028865 |
| Htr2CmRuby3 | This paper | N/A |
| CD-1 | Charles River | N/A |
| C57BL/6 | Jackson Laboratory | 000664; RRID:IMSR_JAX:000664 |
| Oligonucleotides | ||
| c-fos; Mm00487425_m1 | Thermo Fisher | Mm00487425_m1; #4453320 |
| Snap23; Mm01330351_mH | Thermo Fisher | Mm01330351_mH; #4448892 |
| Stx12; Mm00505689_m1 | Thermo Fisher | Mm00505689_m1; #4448892 |
| Stxbp4; Mm00488497_m1 | Thermo Fisher | Mm00488497_m1; #4448892 |
| Vamp3; Mm01268442_g1 | Thermo Fisher | Mm01268442_g1; #4448892 |
| Vamp2; Mm01325243_m1 | Thermo Fisher | Mm01325243_m1; #4448892 |
| Vmat2; Mm00553058_m1 | Thermo Fisher | Mm00553058_m1; #4448892 |
| c-fos; Mm00487425_m1 | Thermo Fisher | Mm00487425_m1; #4453320 |
| 18S; Mm03928990_g1 | Thermo Fisher | Mm03928990_g1; #4331182 |
| crRNA Alt-R® CRISPR-Cas9 crRNA, 2 nmol | Integrated DNA Technologies | N/A |
| Software and Algorithms | ||
| MATLAB R2018b | Mathworks | https://www.mathworks.com/products/matlab.html |
| Custom 3D registration algorithm | This paper | https://github.com/LehtinenLab/Shipley2020 |
| Vascular segmentation algorithm | This paper | https://github.com/LehtinenLab/Shipley2020 |
| Fiji 1.52i | NIH | https://fiji.sc/ RRID:SCR_002285 |
| Scanbox | Neurolabware | https://scanbox.org/ |
| GraphPad Prism7 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| MicroManager | NIH | https://micro-manager.org |
| RStudio 1.2.5019 | RStudio, Inc | https://rstudio.com |
| Spatstat R package | (Baddedy et al., 2015) | https://spatstat.org/ |
| FV31S-SW - FluoView | Olympus | https://www.olympus-lifescience.com/en/support/downloads/ |
| LegendPlex v7.1 | Biolegend | https://www.biolegend.com/en-us/legendplex |
| Other | ||
| 25x, 0.95 NA, 8mm W.D. objective | Olympus | XLSLPLN25XSVMP2 |
| 25x, 1.00 NA, 4mm W.D. objective | Olympus | XLPLN25XSVMP2 |
| 4x, 0.28 NA objective | Olympus | XLFLUOR 4x/340 |
| 100x, 1.46 NA objective | Carl Zeiss Microscopy | 420792-9800-000 |
| Dissection microscope | Carl Zeiss Microscopy | 3715211/3715298 |
| Confocal microscope with Airyscan | Carl Zeiss Microscopy | LSM 880 |
| TIRF microscope | Intelligent Imaging Innovations, Inc | Marianas |
| Two-photon microscope | Neurolabware | http://neurolabware.com/ |
| IR cutoff filter – 775nm | Semrock | FF775-Di01 −25x36 |
| Red/green emission dichroic – 562 nm | Semrock | FF562-Di03-25x36 |
| Green emission filter – 510 nm center, 84 nm width | Semrock | FF01-510/84-50 |
| Red emission filter – 607 nm center, 70 nm width | Semrock | FF01-607/70 |
| Two-photon microscope | Bruker | https://www.bruker.com/ |
| Excitation dichroic mirror | Chroma | Z470/561/NIR-Trans |
| NIR blocker – block 458-473 nm, 561-568 nm | Chroma | Custom |
| Emission dichroic mirror – 560 nm | Chroma | T560lpxr |
| Blue/green emission dichroic mirror – 495 nm | Chroma | AT495lp |
| Green emission filter – 525 nm center, 50 nm width | Chroma | ET595/50 |
| Two-photon microscope | Olympus | FVMPE-RS |
| IR cutoff filter – 685nm | Olympus | BA685 |
| Excitation dichroic mirror | Olympus | FV30-SDM-M |
| Red/green Emission dichroic mirror – 570 nm | Olympus | SDM570 |
| Green emission filter – 518 nm center, 45 nm width | Olympus | BA575-645 |
| Red emission filter – 610 nm center, 70 nm band. | Olympus | BA495-540 |
| Nanopositioning Piezo Stage | nPoint | nPFocus250 |
| Tunable focus lens | Optotune | https://www.optotune.com/products/focus-tunable-lenses |
| CCD Camera | Photometrics | QuantEM:512SC |
| SCMOS Camera | Hamamatsu | OrcaFlash 4.0LT |
| CCD Camera | Andor | Andor iXon Ultra |
| Ti:Sapphire laser | Spectra-Physics | Mai Tai HP DeepSee |
| Blackout fabric | Thorlabs | Cat#BK5 |
| Coverglass 15 mm | Warner | SteREO Discovery.V8 |
| Ultra Quiet Imaging Chamber, high profile, wide bath (Model #JG-23W/HP) | Warner | #64-0703 |
| Heater controller single channel | Warner | #64-1487 |
| Tygon tubing E-3603 (1.59mm ID x 3.18mm OD x 50ft) | VWR | #64-0100 (TC-324B) |
| Luer connector kit | Harvard Apparatus | 89403-846 |
| Water bath for preheating solutions (2 Liter) | VWR | 72-1407 |
| Polycarbonate membrane Nucleopore; 13 mm wide; 8.0 μm pore size | Whatman | #110414 |
| Stereotaxic frame | KOPF | Model 940 |
| Heating pad | CWE | Model TC-1000 |
| Gelfoam | Moore Medical | #2928 |
| Titanium headpost | HE Parmer (Goldey et al., 2014) | Custom |
| Stainless steel cannula | MicroGroup | Custom |
| Round cover glass; 3 mm | Harvard Apparatus | Cat#64-0720 |
| UV-cured adhesive | Norland Products | Cat#7106 |
| Vetbond | 3M | Cat#1469SB |
| C&B Metabond | Parkell | Cat#S380 |
| Silicone (Kwik-sil) | WPI | #600022 |
Supplementary Material
Highlights.
Two-photon imaging of choroid plexus epithelium in live explants and in awake mice
Spontaneous subcellular calcium events in epithelial cells in awake mice
Serotonin boosts epithelial calcium activity and multiple forms of secretion
Distinct surveillance and defensive behaviors in stromal and epiplexus immune cells
ACKNOWLEDGMENTS
We thank members of the Lehtinen, Andermann, and Moore labs for helpful discussions; A. Lutas, K. Fernando, S. Marsh, M. Webb, C. Chen, O. Alturkistani, and the IDDRC Imaging Core for advice and technical assistance; M. Bhaumik, BCH Genome Editing Core, and G. Marsischky for Htr2cmRuby3 design; M. Ericsson, HMS EM Facility; W.H. Fowle, NEU EM Facility; C. Wu at the BCH Viral Core; S. Gupton for VAMP3 expression vector; and J. Zhang for Z310 cells. This work was supported by a National Science Foundation (NSF) Graduate Research Fellowship (F.B.S.); a Glenn Foundation/American Federation for Aging Research (AFAR) Postdoctoral Fellowship and a Reagan Sloane Shanley Research Internship (N.D.); a William Randolph Hearst Fellowship (N.D., J.C.); NIH grant T32 HL110852 (J.C., R.M.F.); an Office of Faculty Development (OFD)/Basic/Translational Research Executive Committee (BTREC)/Clinical and Translational Research Executive Committee (CTREC) Faculty Development Fellowship Award (R.M.F.); NIH National Institute of Allergy and Infectious Diseases (NIAID) grant R01-AI130591 (T Kishkovich, K.H.); National Heart, Lung, and Blood Institute (NHLBI) grant R35-HL145242 (M.J.H.); GM075252 (T Kirchhausen); a gift from the Korn family and NSF Next Generation Networks for Neuroscience (NeuroNex) grant 1707352 (C.I.M.); the David Mahoney Neuroimaging Grant Program – Dana Foundation, NIH grants DP2 DK105570, DP1 AT010971, R01 DK109930, R21 EY03043-02, the McKnight Foundation, the Pew Foundation, the Smith Family Foundation, the Klarman Family Foundation, and the AFAR (M.L.A.); the Pediatric Hydrocephalus Foundation, a Tommy Fuss Center Innovation Grant, Simons Foundation Autism Research Initiative [SFARI] award 610670, a Harvard Brain Science Initiative Bipolar Disorder Seed Grant, and NIH grant R01 NS088566 (M.K.L.); Human Frontier Science Program grant RGP0063/2018 (C.W., M.K.L.); NIH grant RF1DA048790 (C.I.M., M.K.L.); New York Stem Cell Foundation (NYSCF) grants NYSCF-R-NI39 (C.W.) and NYSCF-R-NI38 (M.K.L.); and Boston Children’s Hospital (BCH) Intellectual and Developmental Disabilities Research Center (IDDRC) grant 1U54HD090255. C.W. and M.K.L. are New York Stem Cell Foundation – Robertson Investigators. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.neuron.2020.08.024.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agnew WF, Yuen TG, and Achtyl TR (1980). Ultrastructural observations suggesting apocrine secretion in the choroid plexus: a comparative study. Neurol. Res 1, 313–332. [DOI] [PubMed] [Google Scholar]
- Ambudkar IS (2016). Calcium signalling in salivary gland physiology and dysfunction. J. Physiol 594, 2813–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar I (2018). Calcium signaling defects underlying salivary gland dysfunction. Biochim. Biophys. Acta Mol. Cell Res 1865 (11 Pt B), 1771–1777. [DOI] [PubMed] [Google Scholar]
- Audhya T, Adams JB, and Johansen L (2012). Correlation of serotonin levels in CSF, platelets, plasma, and urine. Biochim. Biophys. Acta 1820, 1496–1501. [DOI] [PubMed] [Google Scholar]
- Baddedy A, Rubak E, and Turner R (2015). Spatial Point Patterns: Methodology and Applications with R (Boca Raton, FL: Chapman and Hall/ CRC Press; ). [Google Scholar]
- Balaji R, Bielmeier C, Harz H, Bates J, Stadler C, Hildebrand A, and Classen A-K (2017). Calcium spikes, waves and oscillations in a large, patterned epithelial tissue. Sci. Rep 7, 42786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balusu S, Brkic M, Libert C, and Vandenbroucke RE (2016a). The choroid plexus-cerebrospinal fluid interface in Alzheimer’s disease: more than just a barrier. Neural Regen. Res 11, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, Brkic M, Demeestere D, Vanhooren V, Hendrix A, et al. (2016b). Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med 8, 1162–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier N, Paré A, Farndale RW, Schumacher HR, Nigrovic PA, Lacroix S, and Boilard E (2012). Platelets can enhance vascular permeability. Blood 120, 1334–1343. [DOI] [PubMed] [Google Scholar]
- Cocucci E, Aguet F, Boulant S, and Kirchhausen T (2012). The first five seconds in the life of a clathrin-coated pit. Cell 150, 495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion AR, and Feske S (2017). Regulation of epithelial ion transport in exocrine glands by store-operated Ca2+ entry. Cell Calcium 63, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, and Sanders-Bush E (1986). Agonist-induced phosphoinositide hydrolysis in choroid plexus. J. Neurochem 47, 1754–1760. [DOI] [PubMed] [Google Scholar]
- Coulter ME, Dorobantu CM, Lodewijk GA, Delalande F, Cianferani S, Ganesh VS, Smith RS, Lim ET, Xu CS, Pang S, et al. (2018). The ESCRT-III protein CHMP1A mediates secretion of sonic hedgehog on a distinctive subtype of extracellular vesicles. Cell Rep. 24, 973–986.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Li GJ, and Zheng W (2005). The choroid plexus removes beta-amyloid from brain cerebrospinal fluid. Exp. Biol. Med. (Maywood) 230, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damkier HH, Brown PD, and Praetorius J (2013). Cerebrospinal fluid secretion by the choroid plexus. Physiol. Rev 93, 1847–1892. [DOI] [PubMed] [Google Scholar]
- Dani N, Herbst RH, Habib N, Head J, Dionne D, Nguyen L, McCabe C, Cui J, Shipley FB, Jang A, et al. (2019). A cellular and spatial map of the choroid plexus across brain ventricles and ages. bioRxiv. 10.1101/627539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, and Gan W-B (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, and Tank DW (2007). Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron 56, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombeck DA, Harvey CD, Tian L, Looger LL, and Tank DW (2010). Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci 13, 1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterle TM, and Sanders-Bush E (1992). Serotonin agonists increase transferrin levels via activation of 5-HT1C receptors in choroid plexus epithelium. J. Neurosci 12, 4775–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, and Lehtinen MK (2020). Emergence and developmental roles of the cerebrospinal fluid system. Dev. Cell 52, 261–275. [DOI] [PubMed] [Google Scholar]
- Farkaš R (2015). Apocrine secretion: new insights into an old phenomenon. Biochim. Biophys. Acta 1850, 1740–1750. [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea J-F, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, and Engelhardt B (2018). Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 135, 337–361. [DOI] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nat. Protoc 9, 2515–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AM, Leadbeater WE, Burg M, Sims K, Terasaki T, Johanson CE, Stopa EG, Eliceiri BP, and Baird A (2011). Targeting choroid plexus epithelia and ventricular ependyma for drug delivery to the central nervous system. BMC Neurosci. 12, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudeman DM, Nelson SR, and Merisko EM (1987). Protein secretion by choroid plexus: isolated apical fragments synthesize protein in vitro. Tissue Cell 19, 101–109. [DOI] [PubMed] [Google Scholar]
- Gudeman DM, Brightman MW, Merisko EM, and Merril CR (1989). Release from live choroid plexus of apical fragments and electrophoretic characterization of their synthetic products. J. Neurosci. Res 24, 184–191. [DOI] [PubMed] [Google Scholar]
- Guizar-Sicairos M, and Fienup JR (2008). Direct image reconstruction from a Fourier intensity pattern using HERALDO. Opt. Lett 33, 2668–2670. [DOI] [PubMed] [Google Scholar]
- Haddad MR, Donsante A, Zerfas P, and Kaler SG (2013). Fetal brain-directed AAV gene therapy results in rapid, robust, and persistent transduction of mouse choroid plexus epithelia. Mol. Ther. Nucleic Acids 2, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J (2018). Microglia in neurodegeneration. Nat. Neurosci 21, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hierro-Bujalance C, Bacskai BJ, and Garcia-Alloza M (2018). In vivo imaging of microglia with multiphoton microscopy. Front. Aging Neurosci 10, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, and Vandenberghe LH (2019). Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron 101, 839–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson AJ (1960). The development of the vascular pattern of the choroid plexus of the lateral ventricles. J. Comp. Neurol 115, 171–186. [DOI] [PubMed] [Google Scholar]
- Jerman T, Pernus F, Likar B, and Spiclin Z (2016). Enhancement of vascular structures in 3D and 2D angiographic images. IEEE Trans. Med. Imaging 35, 2107–2118. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, and Littman DR (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimy JK, Zhang J, Kurland DB, Theriault BC, Duran D, Stokum JA, Furey CG, Zhou X, Mansuri MS, Montejo J, et al. (2017). Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat. Med 23, 997–1003. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Masuda T, Jordão MJC, and Prinz M (2019). Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat. Rev. Neurosci 20, 547–562. [DOI] [PubMed] [Google Scholar]
- Kim S, Hwang Y, Lee D, and Webster MJ (2016). Transcriptome sequencing of the choroid plexus in schizophrenia. Transl. Psychiatry 6, e964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. (2011). The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69, 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Li Q, and Barres BA (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol 18, 225–242. [DOI] [PubMed] [Google Scholar]
- Liang L, Fratzl A, Goldey G, Ramesh RN, Sugden AU, Morgan JL, Chen C, and Andermann ML (2018).A fine-scale functional logic to convergence from retina to thalamus. Cell 173, 1343–1355.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Δ Δ C(T)) method. Methods 25, 402–08. [DOI] [PubMed] [Google Scholar]
- Lun MP, Johnson MB, Broadbelt KG, Watanabe M, Kang YJ, Chau KF, Springel MW, Malesz A, Sousa AMM, Pletikos M, et al. (2015a). Spatially heterogeneous choroid plexus transcriptomes encode positional identity and contribute to regional CSF production. J. Neurosci 35, 4903–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Monuki ES, and Lehtinen MK (2015b). Development and functions of the choroid plexus-cerebrospinal fluid system. Nat. Rev. Neurosci 16, 445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, Sousa N, Correia-Neves M, and Palha JA (2009). Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J. Cereb. Blood Flow Metab 29, 921–932. [DOI] [PubMed] [Google Scholar]
- Marques F, Sousa JC, Sousa N, and Palha JA (2013). Blood-brain-barriers in aging and in Alzheimer’s disease. Mol. Neurodegener 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa KR, Rompolas P, Zito G, Myung P, Sun TY, Brown S, Gonzalez DG, Blagoev KB, Haberman AM, and Greco V (2015). Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature 522, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje ML, Toda H, and Palmer TD (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765. [DOI] [PubMed] [Google Scholar]
- Mottahedin A, Joakim Ek C, Truvé K, Hagberg H, and Mallard C (2019). Choroid plexus transcriptome and ultrastructure analysis reveals a TLR2-specific chemotaxis signature and cytoskeleton remodeling in leukocyte trafficking. Brain Behav. Immun 79, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narboux-Nême N, Pavone LM, Avallone L, Zhuang X, and Gaspar P (2008). Serotonin transporter transgenic (SERTcre) mouse line reveals developmental targets of serotonin specific reuptake inhibitors (SSRIs). Neuropharmacology 55, 994–1005. [DOI] [PubMed] [Google Scholar]
- Narciso CE, Contento NM, Storey TJ, Hoelzle DJ, and Zartman JJ (2017). Release of applied mechanical loading stimulates intercellular calcium waves in Drosophila wing discs. Biophys. J 113, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netsky M, and Shuangshoti S (1975). The Choroid Plexus in Health and Disease (New York: Elsevier; ). [Google Scholar]
- Okaty BW, Sturrock N, Escobedo Lozoya Y, Chang Y, Senft RA, Lyon KA, Alekseyenko OV, and Dymecki SM (2020). A single-cell transcriptomic and anatomic atlas of mouse dorsal raphe Pet1 neurons. eLife 9, e55523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, and Lancaster MA (2020). Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 369, eaaz5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozner A, Xu B, Palumbos S, Gee JM, Tvrdik P, and Capecchi MR (2015). Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC:G5-tdT reporter mouse. Front. Mol. Neurosci 8, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, and Sallusto F (2009). C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol 10, 514–523. [DOI] [PubMed] [Google Scholar]
- Rompolas P, Mesa KR, Kawaguchi K, Park S, Gonzalez D, Brown S, Boucher J, Klein AM, and Greco V (2016). Spatiotemporal coordination of stem cell commitment during epidermal homeostasis. Science 352, 1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig-Lipson S, Zhang J, Mazandarani H, Harrison BL, Sabb A, Sabalski J, Stack G, Welmaker G, Barrett JE, and Dunlop J (2006). Antiobesity-like effects of the 5-HT2C receptor agonist WAY-161503. Brain Res. 1073–1074, 240–251. [DOI] [PubMed] [Google Scholar]
- Samanta K, and Parekh AB (2016). Store-operated Ca2+ channels in airway epithelial cell function and implications for asthma. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders-Bush E, and Breeding M (1990). Serotonin1c receptor reserve in choroid plexus masks receptor subsensitivity. J. Pharmacol. Exp. Ther 252, 984–988. [PubMed] [Google Scholar]
- Sankaranarayanan S, De Angelis D, Rothman JE, and Ryan TA (2000). The use of pHluorins for optical measurements of presynaptic activity. Biophys. J 79, 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Dziegielewska KM, Møllgård K, and Habgood MD (2018). Physiology and molecular biology of barrier mechanisms in the fetal and neonatal brain. J. Physiol 596, 5723–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, and Baruch K (2014). The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J. 33, 7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, Kim K-W, Klein E, Kalchenko V, Bendel P, et al. (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity 38, 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Vargas V, Maldonado-Soto AR, Mizrak D, Codega P, and Doetsch F (2016). Age-dependent niche signals from the choroid plexus regulate adult neural stem cells. Cell Stem Cell 19, 643–652. [DOI] [PubMed] [Google Scholar]
- Stasi C, Sadalla S, and Milani S (2019). The relationship between the serotonin metabolism, gut-microbiota and the gut-brain axis. Curr. Drug Metab 20, 646–655. [DOI] [PubMed] [Google Scholar]
- Strazielle N, and Ghersi-Egea JF (1999). Demonstration of a coupled metabolism-efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J. Neurosci 19, 6275–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Homma D, Tokuoka H, Hayakawa I, Sugimoto Y, Ichinose H, and Kawasaki H (2013). Birth regulates the initiation of sensory map formation through serotonin signaling. Dev. Cell 27, 32–46. [DOI] [PubMed] [Google Scholar]
- Tong CK, Chen J, Cebrián-Silla A, Mirzadeh Z, Obernier K, Guinto CD, Tecott LH, García-Verdugo JM, Kriegstein A, and Alvarez-Buylla A (2014). Axonal control of the adult neural stem cell niche. Cell Stem Cell 14, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina FL, Gomez SM, and Gupton SL (2018). Spatiotemporal organization of exocytosis emerges during neuronal shape change. J. Cell Biol 217, 1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, Vandamme N, De Schepper S, Van Isterdael G, Scott CL, et al. (2019). A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci 22, 1021–1035. [DOI] [PubMed] [Google Scholar]
- Wang T, Ouzounov DG, Wu C, Horton NG, Zhang B, Wu CH, Zhang Y, Schnitzer MJ, and Xu C (2018). Three-photon imaging of mouse brain structure and function through the intact skull. Nat. Methods 15, 789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf BC (2005). Comparison of endoscopic third ventriculostomy alone and combined with choroid plexus cauterization in infants younger than 1 year of age: a prospective study in 550 African children. J. Neurosurg 103 (6, Suppl), 475–481. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Kang YJ, Davies LM, Meghpara S, Lau K, Chung CY, Kathiriya J, Hadjantonakis AK, and Monuki ES (2012). BMP4 sufficiency to induce choroid plexus epithelial fate from embryonic stem cell-derived neuroepithelial progenitors. J. Neurosci 32, 15934–15945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JA, Elliott AC, and Brown PD (1995). Serotonin elevates intracellular Ca2+ in rat choroid plexus epithelial cells by acting on 5-HT2C receptors. Cell Calcium 17, 120–128. [DOI] [PubMed] [Google Scholar]
- Weisenburger S, Tejera F, Demas J, Chen B, Manley J, Sparks FT, Martinez Traub F, Daigle T, Zeng H, Losonczy A, et al. (2019). Volumetric Ca(2+) imaging in the mouse brain using hybrid multiplexed sculpted light microscopy. Cell 177, 1050–1066.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang G, Shornick LP, Roswit WT, Shipley JM, Brody SL, and Holtzman MJ (2007).A transgenic FOXJ1-Cre system for gene inactivation in ciliated epithelial cells. Am. J. Respir. Cell Mol. Biol 36, 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, and Zhao Q (2002). Establishment and characterization of an immortalized Z310 choroidal epithelial cell line from murine choroid plexus. Brain Res. 958, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Zhao Q, and Graziano JH (1998). Primary culture of choroidal epithelial cells: characterization of an in vitro model of blood-CSF barrier. In Vitro Cell. Dev. Biol. Anim 34, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The registration and vascular segmentation algorithms generated for this study are available at https://github.com/LehtinenLab/Shipley2020. Original data are available from the Lead Contact upon request.