

Abstract
Traumatic brain injury (TBI) is distinct from other neurological disorders because it is induced by a discrete event that applies extreme mechanical forces to the brain. This review describes how the brain senses, integrates, and responds to forces under both normal conditions and during injury. The response to forces is influenced by the unique mechanical properties of brain tissue, which differ by region, cell type, and subcellular structure. Elements such as the extracellular matrix, plasma membrane, transmembrane receptors, and cytoskeleton influences its properties. These same components also act as force-sensors, allowing neurons and glia to respond to their physical environment and maintain homeostasis. However, when applied forces become too large, as in TBI, these components may respond in an aberrant manner or structurally fail, resulting in unique pathological sequelae. This so-called “pathological mechanosensation” represents a spectrum of cellular responses, which vary depending on the overall biomechanical parameters of the injury and may be compounded by repetitive injuries. Such aberrant physical responses and/or damage to cells along with the resulting secondary injury cascades can ultimately lead to long-term cellular dysfunction and degeneration, often resulting in persistent deficits. Indeed, pathological mechanosensation not only directly initiates secondary injury cascades, but this post-physical damage environment provides the context in which these cascades unfold. Collectively, these points underscore the need to use experimental models that accurately replicate the biomechanics of TBI in humans. Understanding cellular responses in context with injury biomechanics may uncover therapeutic targets addressing various facets of trauma-specific sequelae.
Keywords: traumatic brain injury, biomechanics, force, mechanosensation, mechanotransduction, mechanobiology, acute, repetitive, extracellular matrix, plasma membrane, cytoskeleton
1. Epidemiology and Clinical Classifications
Traumatic brain injury (TBI) is unique from other neurological disorders or diseases in that it is caused by a mechanical insult—such as a blow or jolt to the head, or an object penetrating the skull—that results in brain dysfunction. Falls are the most common source of injury, though motor vehicle accidents, being struck by or against an object, and assault are also causes of harm (Centers for Disease Control and Prevention, 2019). The incidence of TBI varies with age and sex, with children ages 0–4 years of age, adolescents 15–19 years of age, adults over 75 years of age, and particularly males, comprising the most likely groups to experience a TBI-related hospital visit (Faul & Coronado, 2015). The likelihood of TBI exposure is also increased in athletes and military service members (Centers for Disease Control and Prevention et al., 2013; Theadom et al., 2014). TBI is a major cause of death and disability; in the United States alone there were approximately 2.87 million TBI-related emergency department visits, hospitalizations, and deaths in 2014, with global estimates of 27.08 million new cases in 2016 (Centers for Disease Control and Prevention, 2019; GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators, 2019). These numbers likely do not reflect the true incidence of TBI, as many mild injuries go unreported (Bell et al., 2017). In addition to the health burden, there is a significant economic burden as well, with estimated lifetime costs including direct costs like hospital care and indirect costs like lost productivity totaling $60.4 billion in US 2000 dollars (Finkelstein et al., 2006). With no effective therapeutic interventions, it is evident that further research on the mechanisms of injury are required.
Because there are numerous ways forces can be applied to the head to produce injury, TBI is a heterogeneous disorder. Symptoms can range from temporary, mild cognitive deficits to permanent, severely debilitating changes in motor function, emotion, and cognition (K. J. Dixon, 2017). Clinically, TBI is divided into three categories: mild, moderate, and severe (Table 1). These classifications are based in part on the Glasgow Coma Scale (GCS), which uses a numerical scale to quickly assess motor responsiveness, verbal performance, and eye opening as they relate to the depth and duration of impaired consciousness or coma (Teasdale & Jennett, 1974). GCS scores of 13–15 indicate a mild injury, scores of 9–12 indicate a moderate injury, and scores less than 9 indicate a severe injury. In addition to GCS scores shortly after injury, other symptoms are also used to classify injury severity. Mild injuries involve loss of consciousness for 0–30 minutes, altered consciousness or mental state for less than 24 hours, and posttraumatic amnesia of 0–1 days. Moderate injuries involve loss of consciousness for 30 minutes to 24 hours, altered consciousness or mental state for greater than 24 hours, and posttraumatic amnesia for 1–7 days. Severe injuries involve loss of consciousness and altered consciousness or mental state for greater than 24 hours, and posttraumatic amnesia for greater than 7 days (Department of Veterans Affairs & Department of Defense, 2016). Of note, these classifications are based on symptoms rather than the cause of injury or underlying pathology, although moderate and severe injuries can sometimes produce abnormalities on structural imaging scans (Department of Veterans Affairs & Department of Defense, 2016).
Table 1:
Classification of TBI severity
| (If a patient meets criteria in more than one category of severity, the higher severity level is assigned) | |||
|---|---|---|---|
| Criteria | Mild | Moderate | Severe |
| Structural imaging | Normal | Normal or abnormal | Normal or abnormal |
| Loss of consciousness (LOC) | 0–30 min | >30 min and <24 hours | >24 hours |
| Alterations of consciousness / mental state (AOC)* | Up to 24 hours | >24 hours; severity based on other criteria | |
| Posttraumatic amnesia (PTA) | 0–1 day | >1 day and <7 days | >7 days |
| Glasgow Coma Scale (GCS) (best available score in first 24 hours)** | 13–15 | 9–12 | <9 |
Alteration of mental status must be immediately related to the trauma to the head. Typical symptoms would be looking and feeling dazed and uncertain of what is happening, confusion, and difficulty thinking clearly or responding appropriately to mental status questions, and being unable to describe events immediately before or after the trauma event.
In April 2015, the DoD released a memorandum recommending against the use of GCS scores to diagnose TBI. See the memorandum for additional information.
Table reproduced from VA/DoD Clinical Practice Guideline for the Management of Concussion-Mild Traumatic Brain Injury.
Injury outcomes depend on both the type and severity of the initial event, termed the “primary injury,” as well as the multifaceted pathophysiological cascades—including inflammation, edema, hypoxia, metabolic deficits, reactive oxygen species activity, and excitotoxicity (K. J. Dixon, 2017; McIntosh et al., 1998)—that occur subsequently, termed the “secondary injury.” Collectively, damage from the primary insult and its evolving secondary injuries may initiate a continuum of developing neurodegenerative changes progressing for weeks, months, or even many years following injury, producing a wide range of outcomes (L. Wilson et al., 2017). Although the heterogeneity of TBI is gaining increased recognition (Saatman et al., 2008), its wide range of symptomatic outcomes, evolving pathology, and progressive degeneration remain poorly understood. This review will focus on damage caused by the primary injury, particularly how exactly outside forces are translated to the brain and its cells.
2. Brain Mechanics and Neural Mechanosensation
2.1. Basic Mechanics of Biological Materials
Because TBI is a physical injury, it is important to understand the mechanical properties of the brain as well as forces that act on it. Tissue mechanics of the central nervous system have recently been reviewed (Ayad et al., 2019; Meaney & Cullen, 2016). Briefly, mechanical properties encompass the physical attributes of a material as well as the behaviors that materials exhibit upon the application of forces. In engineering, the application of force is expressed in terms of stress, which is defined as the force applied over a cross-sectional area. There are five different types of stress: compression, tension, shear, torsion, and bending (Fig. 1). In response to stress, a material can change shape, or deform. The amount of deformation is measured as strain, which is a ratio of the dimensions of an object after the application of forces relative to the dimensions prior to the application of forces. The tendency of a material to deform in response to a force is termed Young’s modulus (E, also called the elastic modulus). Young’s modulus describes material under compression and tension (forces acting towards or away from a material surface, respectively), while the shear modulus (G) describes the response to shear stress (forces acting parallel to a material surface). However, most biological work uses the terms E, G, and stiffness interchangeably. The higher the elastic modulus, the stiffer the material and the greater the resistance to deformation. The amount of deformation (strain), together with stress, form a relationship that determine how a material behaves. For instance, many materials exhibit elastic behavior, meaning that when a force is applied it immediately deforms, and when that force is removed it instantly reverts back to its original shape. Other materials, including most biological materials, exhibit deformation and gradual recovery (hysteresis), with energy lost in this process. In addition, the behavior of most biological materials depends not only on the amount of stress, but also on the rate at which it is applied, where faster rates of loading elicit stiffer behavior and slower rates of loading elicit softer - or flowing - behavior. Because they display both the elastic properties of a solid which dominate the response upon high rate loading, and the viscous (resistance to flow) properties of a fluid which dominate the response for slow loading, these materials are termed viscoelastic.
Figure 1:
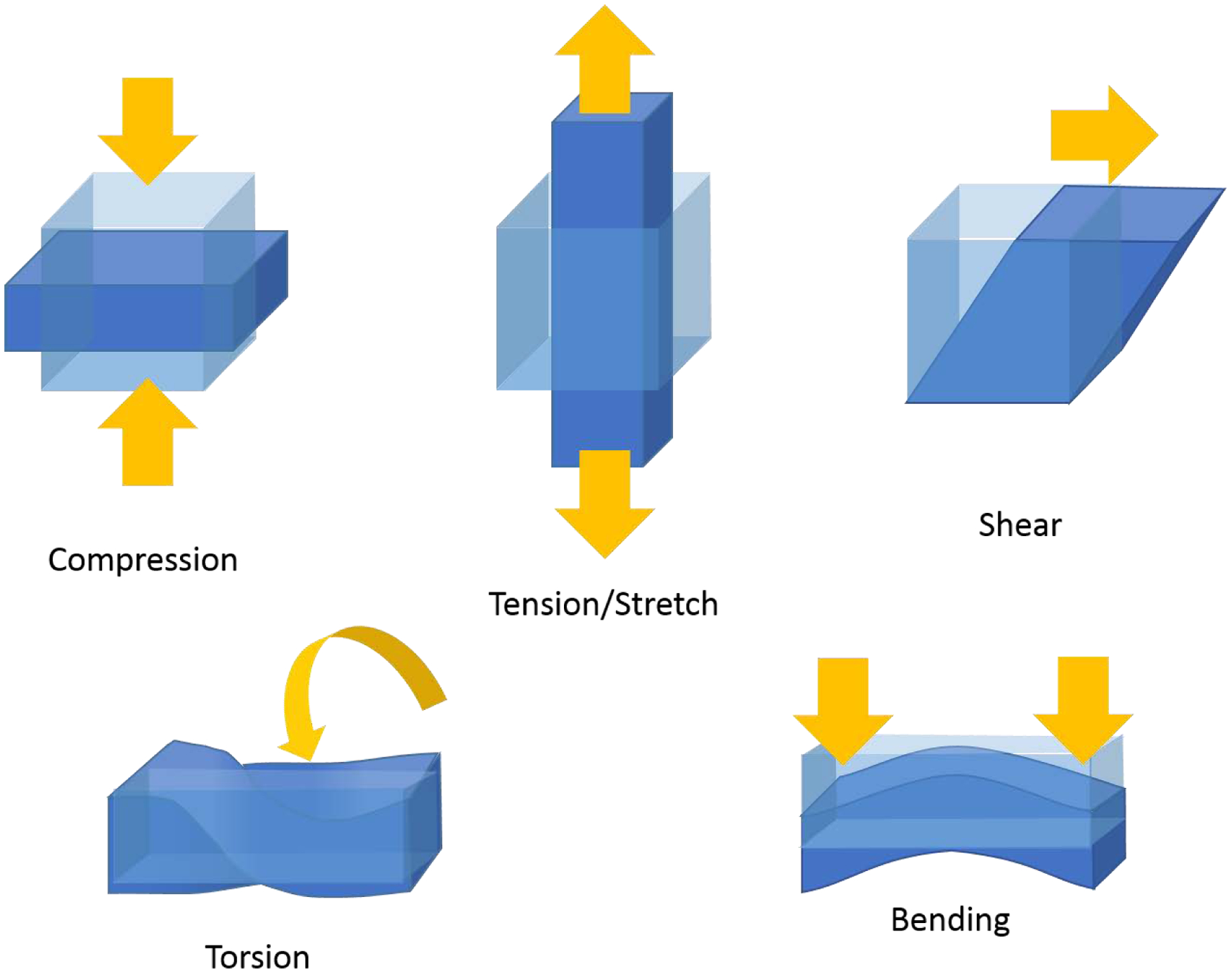
Types of stress. The five types of stress that deform materials.
2.2. Mechanical Properties of the Brain
The brain is a viscoelastic material and one of the softest tissues in the body (Moore et al., 2010). Although nearly incompressible, it is highly susceptible to tensile and shear forces (S. Cheng et al., 2008; Jin et al., 2013; Libertiaux et al., 2011). However, brain properties are not homogenous. At the macro scale, the brain displays differences in white matter and gray matter. White matter is stiffer than gray matter although it shows larger regional variation, is more viscous, responds less rapidly to stress, and its anisotropy means its response is highly dependent on the direction the stress is applied (Braun et al., 2014; Budday et al., 2015; Kruse et al., 2008; Prange & Margulies, 2002; van Dommelen et al., 2010). The stiffness of different brain regions is also heterogeneous; for example, the brainstem and other subcortical gray matter are stiffer than the cortex, while the cerebellum is much softer (C. L. Johnson et al., 2016; Murphy et al., 2013; J. Zhang et al., 2011). Age and disease also impact the mechanical properties, with the human brain softening in age and in many neurological diseases (Murphy et al., 2019; Sack et al., 2011). However, in addition to age and region, the species may also affect the mechanical properties (Elkin et al., 2011; MacManus et al., 2017b; Prange & Margulies, 2002). At the micro scale, different cell types exhibit diverse stiffness, and even within a cell different regions such as the soma and axon have varying stiffness and respond differently to mechanical stimulation (Gaub et al., 2020; Grevesse et al., 2015; Lu et al., 2006).
At the cellular level, several components confer the brain with its unique mechanical properties (Tyler, 2012). The plasma membranes surrounding all cells give rise to many of the viscoelastic properties of the brain as they are viscoelastic materials themselves (Crawford & Earnshaw, 1987). The phospholipid bilayer is sensitive to bending and compressive forces (Evans & Hochmuth, 1978). The integrity, stability, and elasticity of the plasma membrane are in part maintained by the cytoskeleton. Actin and spectrin, in addition to generating their own forces, are important for counteracting plasma membrane tension, particularly in the axon (Dubey et al., 2020; Galkin et al., 2012; Y. Zhang et al., 2017). Periodic actin-spectrin rings forming a lattice have been described in the axon and some dendrites, where they may stabilize or be stabilized by microtubules, as reviewed by Leite & Sousa (Leite & Sousa, 2016). Microtubules and neurofilaments provide key structural support to protect cells from compression forces (Ofek et al., 2009; Ouyang et al., 2013). Mechanical support is also provided by the extracellular matrix (ECM). In adults, the ECM occupies an estimated 20% of the brain and its composition is unique compared to the ECM of most other soft tissues (Nicholson & Syková, 1998). While the ECM of other organs is primarily made up of fibrous proteins like fibrillar collagens, brain ECM is almost entirely composed of non-fibrillar elements like glycosaminoglycans (e.g., hyaluronic acid), proteoglycans (e.g., lecticans), and glycoproteins (e.g., tenascins) (Ruoslahti, 1996; Zimmermann & Dours-Zimmermann, 2008). This relative lack of fibrillar proteins contributes to the low stiffness of the brain.
2.3. Physiological Mechanosensation in Neurons
Many of the components that give the brain its viscoelastic properties can also act as sensors of mechanical forces (Fig. 2A–E). Cells detect forces and convert them into biochemical signals in a process called mechanotransduction. It is important to note that brain cells are continuously subjected to forces even under physiological conditions. For instance, during development neural stem cell migration and differentiation is dependent on sensing ECM architecture and rigidity (Engler et al., 2006; Franco & Muller, 2011; Segel et al., 2019). In mature neurons, action potential propagation in axons is accompanied by membrane deformation (G. H. Kim et al., 2007), and dendritic spines “twitch” and rapidly contract in response to synaptic activity (Crick, 1982). The pulsing flow of blood through capillaries deforms the environment (Wagshul et al., 2011). Neurons must be able to detect, integrate, and respond to these signals.
Figure 2:
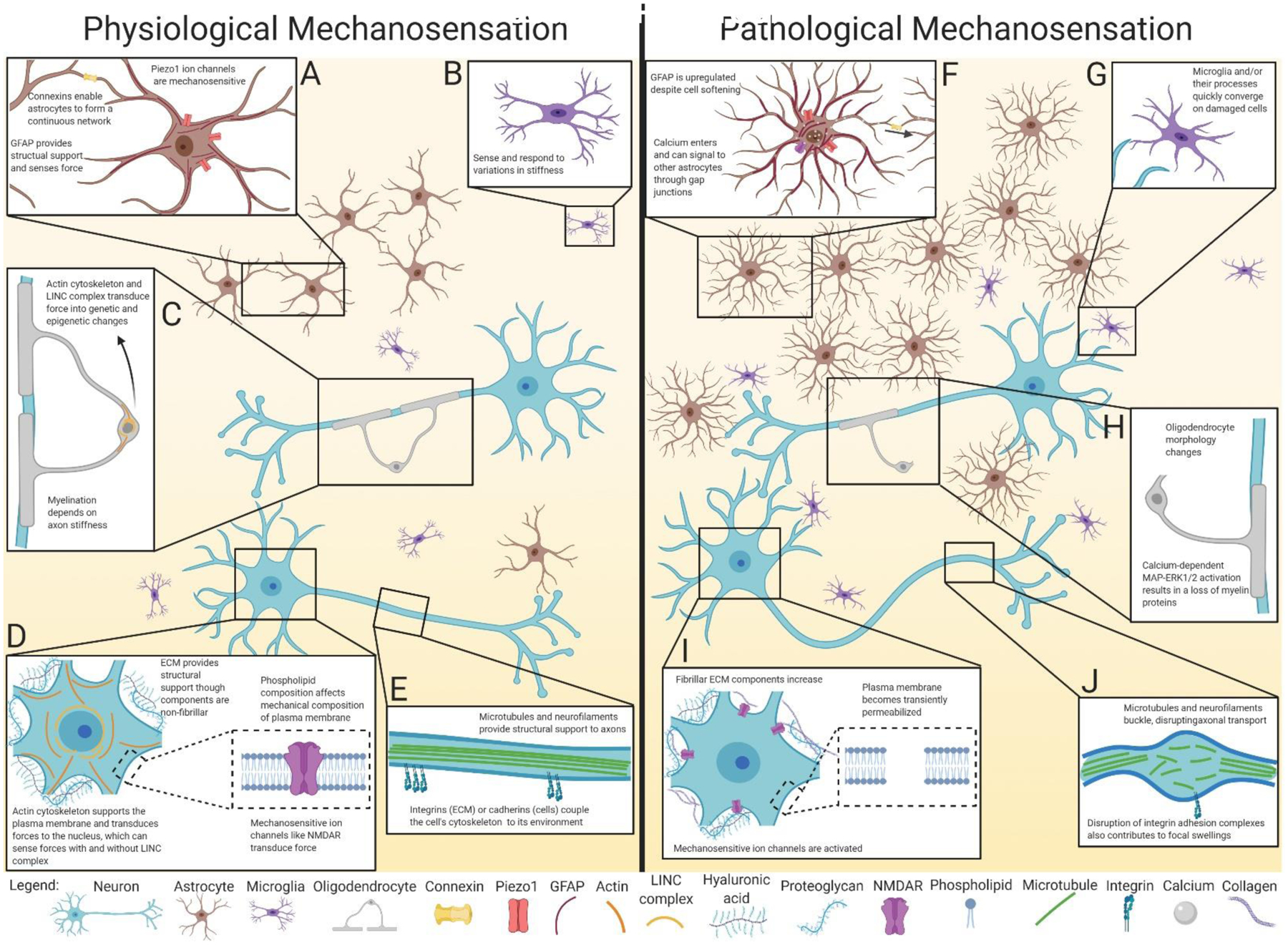
Physiological and pathological mechanosensation in neurons and glia. A: Astrocytes are linked together in a continuous network of gap junctions formed from connexins. This network may act as a scaffold to sense forces throughout the brain via mechanosensitive Piezo1 channels, along with cytoskeletal elements such as GFAP. B: Microglia can adapt their spread area, morphology, and cytoskeleton to changes in environmental stiffness. C: Oligodendrocytes sense force through their cytoskeleton and LINC complex, resulting in transcriptional changes to promote differentiation and maturation. Once mature, myelination itself depends on the ability to sense the stiffness and diameter of axons. D: Neuronal mechanical properties are affected by the composition of the ECM and plasma membrane. Applied forces are transduced through mechanosensitive components such as ion channels, cytoskeletal proteins, and the nucleus. E: In the axon, cytoskeletal proteins such as microtubules and neurofilaments provide structural support. The cytoskeleton is coupled to the environment via transmembrane proteins such as integrins and cadherins that transduce mechanical forces. F: In response to supra-threshold forces, astrocytes proliferate and become hypertrophic, upregulating components of the cytoskeleton like GFAP, despite becoming softer. The opening of mechanosensitive ion channels such as Piezo1 and NMDAR allow cations including calcium to enter the cell, which may signal to other connected astrocytes through gap junctions. G: Activated microglia and their processes converge on damaged cells and areas of increasing stiffness. H: Oligodendrocytes experiencing high strain display altered morphology and decreased myelin protein expression. Myelin organization can become disrupted, particularly in paranodal regions. I: Neuronal plasma membranes experience small tears, which along with activation of mechanosensitive ion channels, disrupt ion homeostasis. Supra-threshold forces also result in altered ECM composition, with a shift to stiffer substrates. J: Diffuse axonal injury results from the breakage of cytoskeletal components such as microtubules and neurofilaments, which along with disruption of adhesion complexes, results in focal swellings. Although beyond the scope of this graphic, note that mechanosensation pathways are also critical to brain vasculature homeostasis and pathophysiology, as reviewed in Monson et al. (2019), Baeyens et al. (2016), and Fang et al. (2020).
The first mechanosensitive molecules to be described were stretch-activated ion channels (Guharay & Sachs, 1984; Hudspeth, 1986; Martinac et al., 1987). Since then, a wide variety of mechanosensitive receptors have been found in both the peripheral and central nervous system, including members of the transient receptor potential (TRP) ion channel family, degenerin/epithelial sodium channel (DEG/ENaC) family including acid-sensing ion channels, two-pore potassium channel family, and Piezo family of cation channels (Bagriantsev et al., 2014; Ben-Shahar, 2011; Y.-R. Cheng et al., 2018; Choi et al., 2015; Dedman et al., 2009; Moran et al., 2004; Qiu et al., 2019). Other channels not classically thought of as mechanotransducers can also be affected by mechanical forces. For instance, it has long been known that the voltage-sensing mechanisms of many voltage-gated channels are sensitive to changes in the plasma membrane or other forms of mechanical disruption (Ben-Tabou et al., 1994; Boland & Drzewiecki, 2008; Piao et al., 2006; Shcherbatko et al., 1999).
More recently, in cultured rat cortical neurons, blocking extracellular calcium, voltage-gated sodium or calcium channels, or TRPV channels decreased or abolished the response to sub-traumatic forces (Gaub et al., 2020). The gating of NMDA receptors (NMDAR) is also mechanosensitive, possibly via interactions with the cytoskeleton and/or membrane lipids (L. R. Johnson et al., 2019; Maneshi et al., 2017). As for the mechanism of this mechanosensitivity, some ion channels are modulated by forces directly causing conformation changes to the protein. Others are activated indirectly by pressure, tension, stress, or stretch deformations at the plasma membrane that lead to conformational changes, as recently reviewed by Lim et al. (Lim et al., 2018). For instance, plasma membrane fatty acid composition fine-tunes the Piezo1 mechanical response, with saturated and unsaturated fatty acids differentially modulating channel activation or inactivation in the N2A neuronal cell line (Romero et al., 2019).
Transmembrane receptors that couple neurons to other cells (e.g., cadherins) or to the ECM (e.g., integrins) are also able to relay physical changes in the environment to the cell. Integrins, for example, link the actin cytoskeleton to ECM proteins like fibronectin. When integrins connected to actin experience a sufficient force, other integrins are recruited to form an integrin adhesion complex that grows into a signaling hub. Downstream signaling can control actin cytoskeletal dynamics which in turn can affect the localization of mechanosensitive transcription factors in addition to other downstream effectors, as recently reviewed by Chighizola et al. (Chighizola et al., 2019). While neurons do not have true focal adhesions, they do have adhesions termed focal points that contain molecules such as dispersed integrin and focal adhesion kinase (Armendáriz et al., 2014). Although much of the work on integrins has been performed on non-neuronal cells, there is evidence in both the peripheral and central nervous system that their signaling is important for neurite outgrowth, myelination, synaptic plasticity, and disease (Jaudon et al., 2020; Lin et al., 2012; Previtali et al., 2001; Y. Wang et al., 2019; X. Wu et al., 2016).
Physical changes in the environment do not only rely on signal transduction cascades from transmembrane receptors to alter gene expression. Gene expression can also be impacted by changes to nuclear architecture. The cytoskeleton is connected to the nucleus through the linker of the nucleoskeleton and cytoskeleton (LINC) complex, which transmits force to the nucleus and can result in spatial chromosome reorganization and subsequent transcriptional changes (Tajik et al., 2016). LINC-independent processes exist as well that directly deform the nucleus (Jahed & Mofrad, 2019). For instance, gating of the nuclear pore complex can be directly regulated by forces applied to the nucleus (Elosegui-Artola et al., 2017). Once again, although these studies were performed in non-neuronal cells, it is known that the nuclei of neurons can deform and move within cells, particularly during migration (Nakazawa & Kengaku, 2020), suggesting that these mechanisms may be at play in neurons as well.
2.4. Physiological Mechanosensation in Glia
Neurons are not the only mechanosensitive cells in the brain. Astrocytes can act as soft, compliant structures that surround neurons and may act to cushion them (Lu et al., 2006). These cells may act as a force-sensing scaffold across the brain as suggested by Ostrow & Sachs, as in vitro experiments have shown that mechanically stimulating one astrocyte produced calcium signaling in the entire interconnected network of astrocytes (Ostrow & Sachs, 2005). These cells have long been known to express stretch-activated and curvature-sensitive mechanotransducing ion channels, and more recent studies have shown that Piezo1 can regulate calcium oscillations and cytokine release (Bowman et al., 1992; Velasco-Estevez et al., 2020). Proteins involved with integrin adhesion complexes have been found to regulate glutamate transporter expression and astrocyte morphology (Avalos et al., 2004; Cho et al., 2018; Leyton et al., 2001). The cytoskeleton, including intermediate filaments like glial fibrillary acidic protein (GFAP), also plays a role in mechanotransduction in astrocytes. During migration, the astrocyte intermediate filament network of GFAP, vimentin, and nestin, together with the cytoskeletal linker plectin, promote the actin-driven treadmilling of adherens junctions (De Pascalis et al., 2018). This cytoskeletal remodeling during migration is also associated with changes in cell stiffness (Curry et al., 2017). Stiffness also increases with age and correlates with the complexity of actin, microtubules, and intermediate filaments (S.-M. Lee et al., 2015).
Cytoskeletal changes are also implicated in mechanosensitive oligodendrocyte differentiation and maturation. Mechanostimulation acting through the actin cytoskeleton and LINC complex mediate nuclear and epigenetic changes in oligodendrocyte progenitor cells that favor differentiation (M. Hernandez et al., 2016; Jagielska et al., 2017). Differentiation, maturation, and morphology are also associated with increased production of the microtubule network and are regulated by part of the focal adhesion complex, the mechanosensor p130Cas (Makhija et al., 2018; Shimizu et al., 2019). Once mature, myelination itself appears to be dependent on axon stiffness, among other properties (Espinosa-Hoyos et al., 2018).
Stiffness is an important property for microglia as well. When cultured on compliant substrates, microglia adapt their spread area, morphology, and actin cytoskeleton to the stiffness of their environment. When cultured on stiffness gradients, microglia migrate towards the stiffer regions in a process called durotaxis, which is amplified when the cells are activated (Bollmann et al., 2015). Furthermore, microglia (and astrocytes) respond to increasing stiffness with changes in morphology and upregulation of inflammatory genes and proteins (Moshayedi et al., 2014), demonstrating that cells not only sense but also react to mechanical properties.
2.5. Physiological Mechanosensation in Blood Vessels
In addition to neurons and glia, cells comprising the vasculature coursing through and around the brain can also detect changes in mechanical forces. As this topic has been reviewed elsewhere (Baeyens et al., 2016; Fang et al., 2020; Monson et al., 2019), a few major points are summarized here. Cerebral blood vessels are unique from the vasculature in other organs due in part to the meninges surrounding the brain. Within the subarachnoid space, the internal carotid and vertebral arteries gain a layer of pia-arachnoid tissue that remains with the arteries when they enter the brain parenchyma, forming the Virchow-Robin spaces. While in the subarachnoid space, the arteries are intermittently tethered to the pia and arachnoid by collagenous trabeculae that attach to an outer vascular coat (Monson et al., 2019). This tethering likely influences the stress-strain fields experienced across these regions.
The mechanical characteristics of blood vessels have been investigated for many years, but the properties of cerebral blood vessels specifically have received limited focus. Studies on large arteries leaving the Circle of Willis and their branches on the brain surface have shown that these cerebral arteries display nonlinear and anisotropic properties that are direction-dependent, similar to vessels elsewhere in the body. However, unlike other vessels, arteries from the brain are stiff in both the axial and circumferential directions. Unfortunately, the mechanical properties of smaller arteries and veins have not been characterized (Monson et al., 2019). On a cellular level, the mechanosensing abilities of endothelial cells lining the inside of vessels to detect and react to forces such as fluid shear stress from blood flow and circumferential cyclic expansion from heart propulsions has been covered extensively elsewhere, and the reader is pointed to recent reviews (Baeyens et al., 2016; Fang et al., 2020). Notably, many of the mechanosensors described for neurons and glia—ion channels, cell-cell and cell-ECM proteins, cytoskeleton, mitochondria, and nucleus—are also involved in endothelial cell mechanosensation.
By these numerous mechanisms, neurons and non-neuronal cells are able to detect and respond to physiological mechanical forces within their environment. Collectively, these capabilities allow the cells to sense, integrate, and respond to mechanical features in their environment across a micro- to macro- scale. These force-sensing and responding capabilities are not only necessary for homeostasis, but also for the brain - at a cellular level - to adapt and respond to changes in its physical environment. But how do individual cells and the brain as a whole respond when larger, non-homeostatic forces are applied?
3. Injury Biomechanics and Pathological Mechanosensation
3.1. Injury Biomechanics
Forces can be exerted, or loaded, on the head in several different ways, as described in detail by Meaney & Cullen (Meaney & Cullen, 2016). Forces applied relatively slowly (typically over timespans greater than 200 milliseconds) are termed static or quasi-static, while forces applied more rapidly (typically over less than 50 ms) are termed dynamic. Dynamic loading is the most common cause of injury, and can be divided into three types relevant to TBI: impact, impulsive, and blast overpressure (Fig. 3). In impact loading, the head strikes or is struck by an object. In contrast, the head does not strike an object during impulsive loading, and instead is set in motion due to another part of the body moving. In blast overpressure or shock wave loading, rapidly moving, very short (less than 5 ms) pressure waves travel through the brain. Shockwave propagation linked with blast exposure may induce complex, multi-faceted injury mechanisms associated with transmission of shear strain and stress waves through the brain (primary blast injury), impact loading (e.g., from shrapnel or hitting a larger object; termed secondary blast injury), and impulsive loading (head acceleration due to the body being thrown; termed tertiary blast injury). Further description of blast injury mechanisms and sequelae are beyond the scope of this article, but have recently been reviewed elsewhere (Chandra & Sundaramurthy, 2015; Magnuson et al., 2012). However, blast overpressure illustrates an important point: the physical injury event can initiate multiple loading mechanisms. For instance, an object striking the head often sets the head in motion, resulting in both impact and impulsive loading. Conversely, head motion initiated by rapid body movement often results in the head striking an object, leading to both impulsive and impact loading. Purely impulsive loading where the head does not strike an object after being set in motion, or purely impact loading to an immobilized head, are rare events.
Figure 3:
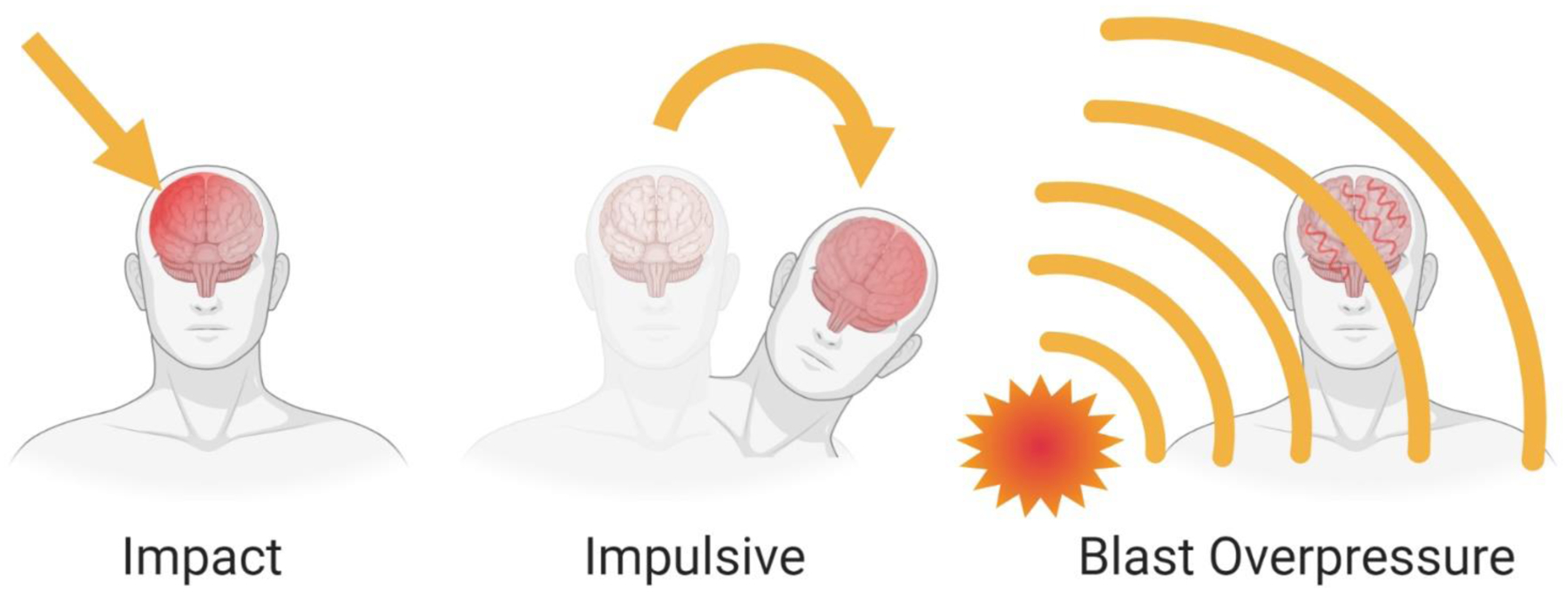
Types of dynamic loading in TBI. A: In impact loading, the head strikes or is struck by an object, producing contact forces at the site. B: In impulsive loading, the head is set in motion without direct impact to the head, for instance if an object strikes another part of the body or if inertial forces act on the body (e.g., a restrained occupant in a motor vehicle collision). This loading accelerates the head and causes rotation about the base of the neck and the craniocervical junction. C: In blast overpressure or shock wave loading, rapidly moving, very short pressure waves travel through the brain, producing a complex injury involving stress waves, with potential head impact and/or head acceleration.
Injuries resulting from dynamic loading can be further categorized as open- or closed-head. Open-head injuries occur when an object penetrates the brain. These relatively infrequent injuries can be devastating, but the majority of damage may be due to physical transection of tissue or cells from penetration of a foreign object and/or skull fragments, though shock wave forces and damage may also be generated from high-velocity projectiles (de Lanerolle et al., 2015). In contrast, non-penetrating closed-head injuries make up 88–97.5% of cases (Demetriades et al., 2004; Gennarelli et al., 1994; Maegele et al., 2007, 2019), and this review will focus on the mechanisms of this type of injury.
Within closed-head injuries, two broad categories of forces are at play: inertial and contact (Meaney & Cullen, 2016). Inertial forces are caused by impulsive loading due to head acceleration, while contact forces are caused by impact loading to the head. Although these forces may occur in isolation, as with impulsive loading in which only inertial forces are present, they are commonly seen in combination. For instance, in impact loading a blow to the head may cause a sudden head movement resulting in a secondary impulsive loading event, together generating both contact and inertial forces. Contact forces can have local effects at or near the impact site, and can have effects far from the impact area due to complex skull distortion or stress wave propagation. In either case, contact forces cause focal injuries as opposed to diffuse brain injury. Diffuse brain injury can be produced from inertial loading, which generally induces head acceleration. Head acceleration can produce differential movement of the skull and brain that can cause strain at the brain surface, and can also produce strain within the brain that can cause widespread damage. There are three types of head acceleration: translational, rotational, and angular (Fig. 4). Translational or linear acceleration occurs when the center of gravity of the brain moves in a straight line without rotation. This type of acceleration rarely occurs in isolation, and can produce focal injuries but not diffuse brain injuries. When the center of gravity of the brain (which is around the pineal region) does not move but the brain rotates around this region, rotational acceleration occurs. Again, this type of acceleration rarely occurs in isolation, but is capable of producing high levels of strain within the brain. When components of translational and rotational acceleration are combined so that the center of gravity of the brain moves in an angular manner, angular acceleration occurs. This type of acceleration is most common clinically and can produce almost every type of TBI. Because translational velocity increases farther outward from the axis of rotation, rotation produces differential displacements of tissue within the brain, resulting in tissue shearing (Ommaya et al., 2002; Ommaya & Gennarelli, 1974). In all cases, when loading deforms or strains the tissue beyond its functional or structural tolerance, injury occurs.
Figure 4:
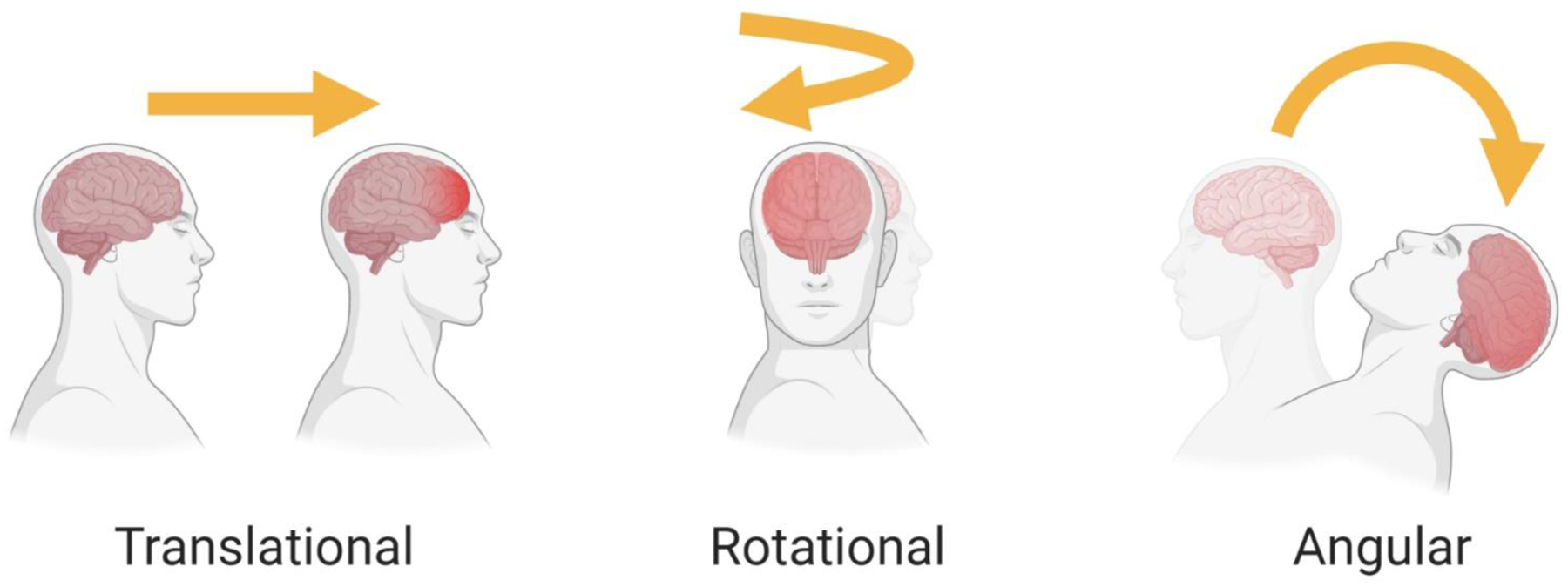
Types of head acceleration. A: Translational or linear acceleration occurs when the center of mass of the brain moves in the straight line without rotation, potentially producing a focal injury. B: Rotational acceleration occurs when the center of mass of the brain does not move while the head rotates around it, producing high levels of diffuse strain. C: Angular acceleration occurs when the center of mass of the brain moves in an angular manner, combining elements of translational and rotational acceleration.
Not only do the properties of the applied force, such as the direction, magnitude, and duration, influence the amount of damage that occurs, but so do the mechanical properties of the tissue itself. Though brain tissue is nearly incompressible, it is highly susceptible to tensile and shear forces (S. Cheng et al., 2008; Jin et al., 2013; Libertiaux et al., 2011). Measures of brain deformation in healthy human volunteers undergoing mild angular head acceleration have demonstrated patterns of radial-circumferential shear strain that are modified by internal membranes (such as the dura, flax cerebri, and tentorium) as well as brain region (Sabet et al., 2008). More specifically, shear strains are different among cortical gray matter, white matter, and deep gray matter (Chan et al., 2018). Compression in frontal regions and stretching in posterior regions has also been observed (Bayly et al., 2005). Injury can also change the mechanical properties of the tissue: a cortical impact study in mice showed a major decrease in shear stiffness of the impacted brain region immediately following injury that lasted for several days (Boulet et al., 2013). Studies in mice have also confirmed regional differences in mechanical responses to large-deformation dynamic micro-indentation, suggesting that distinct brain regions dissipate different amounts of energy for a given applied force, which may explain why some regions or cells are more vulnerable than others (MacManus et al., 2017a). Not surprisingly, at a cellular level, analyses of the biomechanics of single cortical neurons subjected to strains relevant to TBI found that the cell response exhibited hysteresis and time and rate dependencies emblematic of viscoelastic materials like the brain as a whole (Bernick et al., 2011). But how do these characteristics influence how cells respond to forces applied to the entire brain?
3.2. Modeling Injury Biomechanics Experimentally
Before examining the response of cells to injurious loading, it is important to first consider how to apply these forces in experimental models. Both the type of injury applied, as well as the characteristics of the substrate onto which it is applied, will influence the response. Therefore, the specific features of TBI that can be examined may not be generalizable to all cases, but rather will depend on the model used.
3.2.1. In Vitro Models
In vitro models of TBI typically involve growing neurons with or without additional cell types in a 2- or 3-dimensional matrix, and applying tensile, shear, or compressive forces to whole cells or specific cellular compartments. The most common 2D model is stretch injury, in which neurons are grown on elastic silicone membranes that are stretched by a rapid pressure pulse, resulting in two-dimensional strain applied to randomly oriented planar cultures non-uniformly (Cargill & Thibault, 1996; Tavalin et al., 1995) or equally in two directions (Geddes & Cargill, 2001). Stretch can be applied in one direction to isolated axonal projections in order to imitate injury in white matter tracts termed diffuse axonal injury, described further in the next section (Smith et al., 1999). In addition to tensile injuries, shear stress can also be applied to neurons grown in 2D by using fluid motion to deform cells (LaPlaca & Thibault, 1997). Of note, the type of injury applied influences the mechanical responses at both acute and delayed time points, emphasizing the need for caution in extrapolating specific injury mechanisms and responses to other scenarios (Geddes-Klein et al., 2006).
In addition to 2D configurations, 3D cultures and injury paradigms have also been created. These models may better represent loading mechanisms, thresholds, and consequences of injury than their 2D counterparts, as all strain fields in the brain are applied across three dimensions—an inescapable consequence of the 3D architecture of brain tissue. Neurons can be grown in 3D scaffolds with or without additional cells such as astrocytes. The presence of multiple cell types, increased complexity of neural cell morphology, and the increased cell-cell and cell-ECM contacts affect the transfer of strain to cells and better reflect the actual brain environment (Cullen & LaPlaca, 2006; LaPlaca et al., 2005). These 3D constructs can be subjected to shear or compressive forces, with rapid shear loading most closely replicating the inertial forces experienced in the nearly incompressible brain tissue (Cullen et al., 2011; LaPlaca et al., 2005).
Compared to animal models, the reduced complexity of these in vitro systems confers greater control over experimental conditions such as cell types, matrix constituents, and applied forces. It is also easy to measure responses immediately in real time or repeatedly in order to obtain cellular-level detail on injury mechanisms and outcomes. However, these models do not recapitulate the complexity of the intact brain, and their relevance to functional outcomes at the tissue and organism level such as behavior are unclear.
3.2.2. In Vivo Models
There are a variety of injury paradigms used to model TBI in vivo. Most methods have components of both focal and diffuse injury. The contribution of each element exists on a continuum, with some models incorporating more than one over the other.
One model that applies pure impact loading on an immobilized head to produce contact forces that result in mostly focal injuries is called controlled cortical impact (CCI), and was first developed in ferrets and later adapted for use in rats, mice, pigs, and primates (C. E. Dixon et al., 1991; King et al., 2010; Lighthall, 1988; Manley et al., 2006; Smith et al., 1995). In this method, a craniectomy is performed to expose the dura, after which a pneumatic impactor is used to rapidly drive a rod a precise depth into the brain tissue. Newer modifications to this protocol have used an impactor over a closed skull to produce a milder injury that can be administered repetitively (M L Prins et al., 2010).
In contrast, the fluid percussion injury (FPI) model produces mostly diffuse injuries along with a focal element. Initially developed in rabbits and cats, it was later adapted to rats and mice (Carbonell et al., 1998; C. E. Dixon et al., 1987; Lindgren & Rinder, 1966; McIntosh et al., 1989; Rinder, 1969; Sullivan et al., 1976). Instead of applying impact or impulse loading as would occur during TBI, FPI applies a sharp increase in intracranial pressure surrounding the brain. FPI involves using a pendulum to strike a tube of saline, creating a fluid pressure impulse that is delivered to the exposed dura of an animal attached to the other end of the tube. The dura can be exposed at the midline (central), lateral, or somewhere in between (parasagittal), and the position of this craniectomy affects injury outcomes (Floyd et al., 2002). Of note, work using a cut skull gel model to assess regional strain distribution revealed that the maximum site of strain was located in the lower brainstem while deformations were minor in other regions of the brain (Thibault et al., 1992). Although this model produces diffuse strain fields, they are not produced by the same forces that produce diffuse strain fields in inertial injuries experienced by humans. As such, though there may be some similarities in resulting pathologies, the mechanisms underlying those pathologies may vary depending on how forces are administered.
A third commonly used paradigm, the weight-drop model, can be mostly focal or mostly diffuse depending on how it is executed. Typically implemented as a closed-head injury in rats and mice, it involves dropping a weight from a defined distance onto the skull (Chen et al., 1996; Shapira et al., 1988). The severity of injury can be modulated by altering the weight or height from which it is dropped. When the head of the animal is fixed, a focal injury is produced. When the head is protected by a helmet or shield and allowed to rotate in response to injury in a version also called the impact-acceleration model, no focal contusion is observed and a diffuse injury is produced (Marmarou et al., 1994). However, for reasons explained below, it is not possible to reproduce the necessary acceleration to achieve a purely diffuse inertial injury in rodents. The diffuse injury produced in this model is due to impact loading and resultant contact forces.
To truly investigate diffuse injury caused by inertial forces, the head needs to rotate. Recent methods such as Closed-Head Impact Model of Engineered Rotational Acceleration (CHIMERA) have tried to incorporate head rotation into rodent models with more control than that achieved in weight-drop models (Namjoshi et al., 2014). However, measures of angular acceleration and velocity, which are responsible for producing high levels of shear strain, indicate that these levels do not reach scaled thresholds to produce concussion. Additionally, while there was diffuse pathology, the gradient emanated from the site of impact, suggesting that impact is likely the primary source behind observed pathology (Sauerbeck et al., 2018). The reason rodent brains cannot be used to mimic the inertial forces seen in TBI is that they are simply too small. Brain mass, in addition to rotational/angular acceleration, determines the amount of shearing force experienced. The smaller the brain mass, the greater the rotational acceleration needed to produce the same deformation fields observed in a larger brain. In rodents, the acceleration necessary to generate the forces seen in human injuries is unachievable (V. E. Johnson et al., 2015).
It is possible to scale the rotational/angular loading experienced by humans to animals with larger brains such as pigs and non-human primates (Cullen et al., 2016; Ommaya et al., 1967). Early work on non-human primates established that angular, not linear, acceleration was required to produce concussion and also recapitulated many pathologies seen in human injury (Adams et al., 1981; Gennarelli et al., 1982; Ommaya et al., 1966; Ommaya & Gennarelli, 1974; Yarnell & Ommaya, 1969). While research has moved away from using non-human primates, whose brains most closely resemble our own, pig brains also share many features with humans. In addition to both being gyrencephalic, pig and human brains have similar white matter to gray matter ratios (about 60:40); unlike lissencephalic rodent brains with a paucity of white matter (14:86 in rats and 10:90 in mice) (Bailey et al., 2009; Howells et al., 2010; K. Zhang & Sejnowski, 2000). These characteristics allow for pathology to develop in patterns and extents seen in humans (Cullen et al., 2016; McKee et al., 2009; Meaney et al., 1995).
This swine paradigm of angular head acceleration is the only model in use today capable of mechanistically recreating the biomechanical etiology of closed-head diffuse TBI. However, procuring and caring for pigs is expensive, and executing rotational injuries requires highly specialized equipment and personnel. While it is unwise to use rodents alone to study diffuse injuries, their ready availability and potential for transgenic manipulation make them vital for investigating other aspects of the heterogeneous injury spectrum characteristic of TBI, including but not limited to mechanisms of secondary injury such as cell death, neuroplasticity, inflammation, neurodegeneration, and accumulation of proteins such as amyloid beta and tau (W. H. Cheng et al., 2019; Edwards et al., 2020; Ertürk et al., 2016; Gold et al., 2018; Kokiko-Cochran et al., 2018; Muza et al., 2019; Schoch et al., 2012).
3.3. Pathological Mechanosensation in Neurons
Both in vivo and in vitro models have been used to explore how cells sense and react to injurious forces. This review will focus on the acute responses to supra-threshold loading, particularly those arising from diffuse strain fields (Fig. 2F–J, Fig. 5). For information on secondary injury cascades and longer-term responses, the reader is directed to a number of other review papers referenced at the end of this section.
Figure 5:
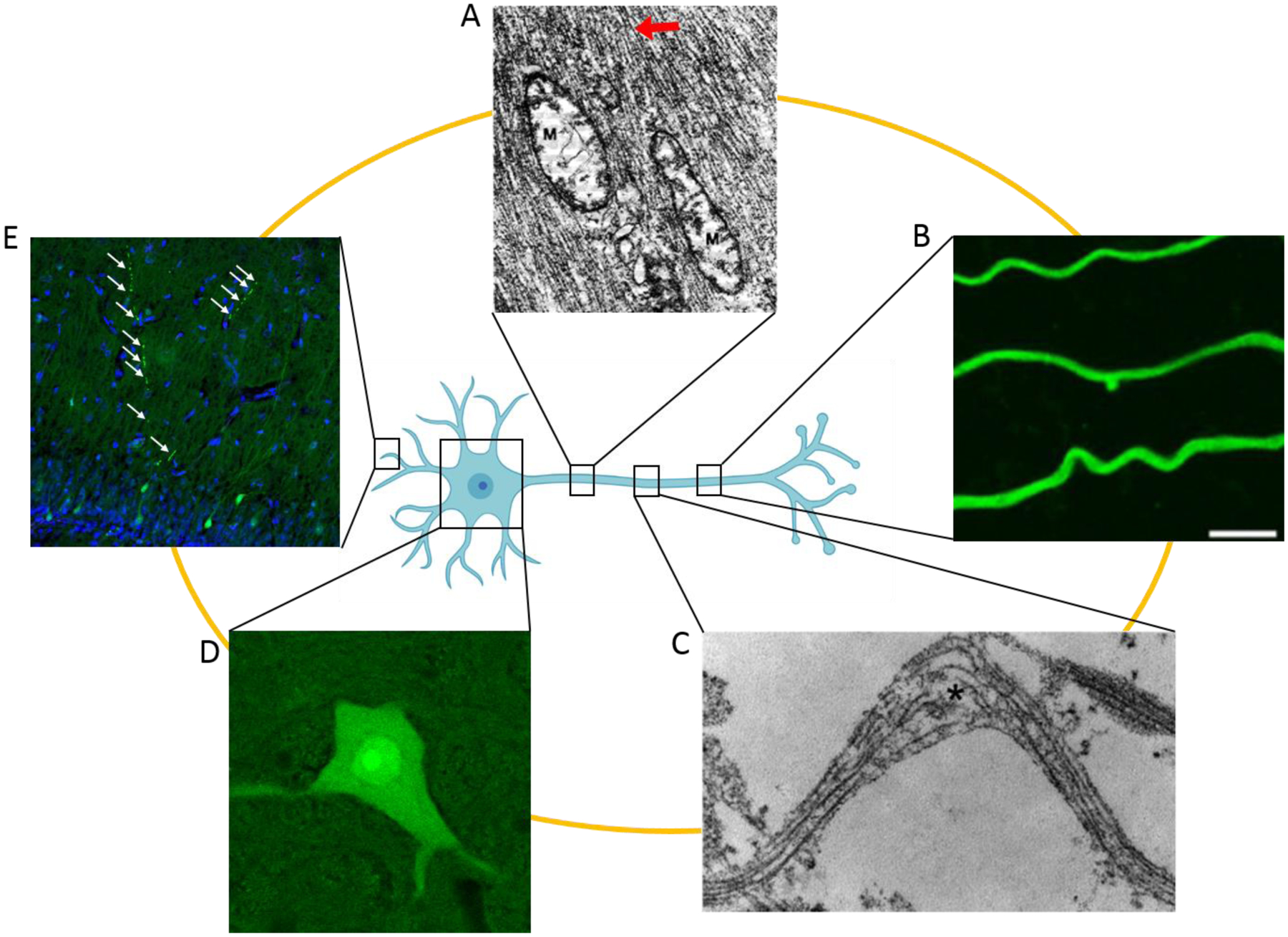
Examples of acute neuronal pathologies. A: Electron micrograph of permeabilized axon revealing dilated mitochondria with disrupted cristae (M) and abnormally tightly packed neurofilaments (arrow) within minutes after fluid percussion injury in cats (adapted with permission from Pettus, 1994). B: Fluorescence image of undulating axons labelled with βIII-tubulin 2 minutes after in vitro axonal stretch injury, scale bar 5 μm (reprinted with permission from Tang-Schomer 2012). C: Electron micrograph showing compaction and misalignment of axon microtubules, along with frayed broken ends (asterisk) minutes after in vitro axonal stretch injury (reprinted with permission from Tang-Schomer 2010). D: Fluorescence image of a permeabilized neuronal cell body in the superior colliculus flooded with a cell-impermeable dye 15 minutes after swine diffuse head acceleration injury. E: Fluorescence image of dendritic beading (arrows) in mechanoporated hippocampal dentate granule neurons labeled by a cell-impermeable dye 15 minutes after swine diffuse head acceleration injury. Cell-impermeable dye (Lucifer Yellow), green; nuclei (Hoechst), blue.
3.3.1. Cytoskeleton Abnormalities in Axons and Dendrites
Diffuse axonal injury (DAI) is one of the most common types of damage produced by TBI, and has been reviewed extensively elsewhere (V. E. Johnson, Stewart, & Smith, 2013; Smith & Meaney, 2000). Briefly, it occurs when inertial forces deform white matter, resulting in multifocal regions of swollen and disconnected axons. In severe cases, DAI is accompanied by tears in the white matter (primary axotomy) and intracranial hemorrhage, usually resulting in prolonged unconsciousness (coma) and poor outcomes (Gennarelli et al., 1982; Mata-Mbemba et al., 2015, 2018; O’Donnell et al., 2018; Smith et al., 2000). Whether severe or mild, DAI is the result of damage to the axonal cytoskeleton. In vitro stretch models have shown that breaks in microtubules along the axon produce discrete or periodic swellings called varicosities, leading to interruption of transport along the axon (Tang-Schomer et al., 2010, 2012). Whether or not microtubules break seems to depend on the microtubule-associated protein tau, as mathematical models show that the viscoelastic properties of the protein as well as binding interactions combine with strain rate to influence microtubule rupture (Ahmadzadeh et al., 2014, 2015). The involvement of the microtubules in DAI has also been confirmed in studies in guinea pig optic nerve, cat fluid percussion, and post-mortem human tissue, which show disruptions in neurofilaments as well (Christman et al., 1994; Maxwell & Graham, 1997; Pettus et al., 1994). While this secondary pathology can develop over the course of hours to days, there is also a more immediate response. Within minutes of injury from a variety of in vivo models, nodes of Ranvier and paranodal regions were noted to be particularly vulnerable to swelling (Greer et al., 2013; Maxwell, 1996; Maxwell et al., 1991). Within seconds of in vitro stretch injury, axons become undulated and misaligned before gradually returning to their pre-stretch orientation, indicative of loss of elasticity (Smith et al., 1999). Other in vitro work has demonstrated that acute mechanical perturbation of the neuronal integrin adhesion complexes that link the cytoskeleton to the ECM can induce focal swellings in neurites minutes after injury (although it should be noted that neuronal focal adhesion complexes are an in vitro epiphenomenon and do not exist in the brain, though perturbations to integrin likely do occur in vivo) (Hemphill et al., 2011).
Interestingly, injury to axons can result in dendritic alterations in the same neurons. Following in vitro axonal stretch, dendritic beading formed within minutes (Monnerie et al., 2010). Beading and dendritic spine loss seem to be dependent on sodium influx and NMDARs (Monnerie et al., 2010; Nagendran & Taylor, 2019). Even in acute hippocampal slices not subjected to mechanical injury but instead exposed transiently to NMDA, dendritic swelling and loss of microtubule associated protein 2 (MAP2) were observed immediately (Hoskison et al., 2007). Indeed, glutamate is known to modulate the dendritic cytoskeleton, as 8 hours of exposure caused a decrease in the number of microtubules in the distal region of retracting dendrites of cultured hippocampal neurons, and caused an increase in microtubule number in the dendritic shaft of both retracting and growing dendrites (M. T. Wilson & Keith, 1998). This cytoskeleton remodeling is likely to occur acutely after TBI, as neurotransmitter and ionic imbalances are an immediate consequence of injury (see below). In fact, in various rodent models of TBI, alterations in MAP2, neurofilaments, and/or dendritic morphology have been observed at time scales ranging from 10 minutes to 1 week after injury (Campbell et al., 2012; Gao et al., 2011; Hicks et al., 1995; Posmantur et al., 1996; Taft et al., 1992; Winston et al., 2013). The effects on dendritic spines also appear to be dependent on matrix metalloprotease 9, suggesting the involvement of the ECM as well (Pijet et al., 2019).
3.3.2. Mechanosensitive Ion Channels
Besides cytoskeletal disruptions, axonal injury has also been associated with increased levels of intracellular calcium. This increase is mediated by sodium influx through mechanosensitive sodium channels, which subsequently triggers the opening of voltage-gated calcium channels, as revealed in an in vitro stretch injury model (Wolf et al., 2001). The elevated calcium can lead to the proteolysis of the sodium channel α subunit 5–20 minutes after injury, resulting in persistent elevations in intracellular calcium (Iwata et al., 2004). Further work showed that stretch or bleb-inducing membrane damage caused leaking from voltage-gated sodium channel 1.6 (Nav1.6), which is present at axonal nodes of Ranvier (J. A. Wang et al., 2009).
Outside the context of DAI, other ion channels have been reported to respond to mechanical trauma. In vitro mechanical injuries have long been known to induce a persistent loss in the voltage-dependent magnesium block of NMDARs and potentiate their currents (LaPlaca & Thibault, 1998; L. Zhang et al., 1996). The immediate action on NMDAR initiates secondary signaling cascades that result in the enhancement of AMPA and GABAA currents, the former of which can last for days (Goforth et al., 2004; Kao et al., 2004; Spaethling et al., 2012). Outside of in vitro experiments, neocortical and hippocampal pyramidal cells in mouse brain slices express stretch-activated mechanically-gated channels capable of triggering action potentials (Nikolaev et al., 2015). In contrast, cerebellar Purkinje cells and locus coeruleus cells did not have any mechanically-gated currents and their robust rhythmic spike activity was resistant to mechanical modulation, suggesting they may be less vulnerable to TBI. Furthermore, rat FPI experiments have demonstrated that neuronal depolarization immediately after injury results in huge increases of extracellular potassium, which cause the release of neurotransmitters such as glutamate that result in further potassium flux (Katayama et al., 1990).
3.3.3. Plasma Membrane Permeability
Supra-threshold mechanical loading can alter ion homeostasis not only through changes in ion channels, but also by the temporary formation of micro- or nano-tears in neuronal plasma membrane. Acute increases in cytosolic calcium and efflux of lactate dehydrogenase dependent on loading rate have been reported since studies in the 1990s using in vitro membrane deformation by fluid shear stress (LaPlaca et al., 1997). Since then, other 2D or 3D in vitro models of stretch or shear injury have used variously sized cell-impermeable molecules to demonstrate that mechanical injury results in transient increases in plasma membrane permeability, also termed mechanoporation, that depend on strain rate and magnitude (Cullen et al., 2011; Geddes, Cargill, et al., 2003; Geddes, LaPlaca, et al., 2003; LaPlaca et al., 2006, 2009). Curiously, plasma membrane permeability has not been observed in unidirectional strain fields (Smith et al., 1999), suggesting that the complexity of the strain field is a major driver in inducing this mechanism of injury. The type of strain is also important, as compressive forces produce less or no permeability (Bar-Kochba et al., 2016; Cullen et al., 2011). The time course of these breaches in membrane integrity and subsequent repair can vary, as demonstrated by studies showing that neurons remain permeable to small molecules up to 5 minutes after severe stretch, but are only permeable to large molecules seconds after injury (Geddes, Cargill, et al., 2003). Additionally, neurons may undergo an initial round of membrane damage and repair, followed by a phase of secondary membrane degradation (Cullen et al., 2011). Although high strain rates and the ensuing structural compromise may lead to cell death in some cases (Cullen et al., 2011), in others the cells survive with functional deficits, such as electrophysiological disturbances (LaPlaca et al., 2006). The extent of the dysfunction may depend on the cell type, as cortical neurons were shown to exhibit longer increases in permeability than hippocampal neurons, but injury of hippocampal neurons produced larger increases in intracellular calcium and sustained ATP deficits, suggesting that secondary cascades and not primary membrane damage may underlie selective vulnerability of some neuron populations over others (Geddes, LaPlaca, et al., 2003).
Consistent with in vitro studies examining the relationship between strain and extent of permeability, comparing in vivo CCI injury in rats to stress/strain tissue patterns in a 3D finite elements simulation revealed that brain areas predicted to have the most stress/strain positively correlated with the number of cells with membrane damage (LaPlaca et al., 2019). Also similar to in vitro studies, weight drop injury in rats demonstrated that permeabilized neurons may experience membrane resealing, delayed permeability, or enduring membrane leakage (Farkas et al., 2006). Interestingly, central FPI in rats produced scattered somatic neuronal permeability that did not overlap with other markers of neuronal injury such as degeneration, stress, and axonal injury, indicating distinct injury phenotypes (Singleton & Povlishock, 2004). Some of these permeabilized neurons displayed signs of necrosis, while others looked ultrastructurally normal. This range of outcomes for permeabilized cells was also seen in a mouse CCI model (Whalen et al., 2008). On top of these primarily impact injuries, it has also been shown that neuronal permeability is produced in cortical and subcortical regions following diffuse head rotational (inertial) injuries in the pig, which more accurately reflect the biomechanics seen in most human injuries (Keating et al., 2020; Wofford et al., 2017).
In addition to somatic permeability, mechanoporation is also observed in axons and was actually first described in this compartment (Pettus et al., 1994). It has been debated whether axonal permeability triggers the disconnected axons that are a hallmark of DAI, with a rat impact acceleration model reporting axons with membrane damage and axons with impaired transport as distinct populations (Stone et al., 2004), while an in vitro fluid shear stress injury-induced mechanoporation did lead to axonal bead formation (Kilinc et al., 2008). Recently, studies using finite element simulations with or without molecule dynamics simulations showed that in a typical injury scenario, axons sustain deformations large enough to enable pore formation in the membrane bilayer, and support this mechanoporation as a more likely injury trigger than microtubule failure (Montanino et al., 2020; Montanino & Kleiven, 2018).
These simulations also illustrate that changes in permeability can be expected even below the threshold for pore formation (Montanino et al., 2020). This finding is not surprising, as many factors influence membrane permeability, including lipid composition. For instance, membrane permeability increases as the number of unsaturated fatty acids, either free or present in the hydrophobic tails of phospholipid molecules, increases (Ehringer et al., 1990). In contrast, sphingomyelin is suggested to increase resistance to water penetration in molecular dynamics simulations at equilibrium and under deformation, as well as increase bilayer stiffness (Saeedimasine et al., 2019). Cholesterol was also long thought to increase membrane bending rigidity, but recent evidence suggests that its effects are not universal but instead depend on the lipid composition of the membrane, as reviewed by Dimova (Dimova, 2014). Conversely, polyunsaturated fatty acids cause membranes to be more flexible, and are necessary for mechanosensation in peripheral touch receptor neurons (Vásquez et al., 2014). The ability of membrane lipid composition to alter properties such as bending rigidity may influence whether cells experience tears in response to mechanical loading.
3.3.4. Extracellular Matrix and Adhesions
The stiffness of a cell is also influenced by the local ECM composition (Viji Babu et al., 2019). The levels and distribution of different ECM components change in response to injury, as recently described in detail by George & Geller (George & Geller, 2018). To summarize, injury leads to the degradation of hyaluronic acid, accompanied by an increase in proteoglycans and fibrous ECM proteins. Although these changes are not acute, long-lasting alterations in ECM properties may influence how a neuron reacts to subsequent mechanical loading in the case of repetitive injuries. Furthermore, ECM composition is just part of the story: the number, density, and nature of cell-matrix adhesions (e.g., integrins) is also important. For instance, cortical neurons plated in a 3D matrix of varying concentrations of collagen demonstrated that neuronal viability after high-rate shear deformation decreased as the level of collagen increased, implicating ECM mechanical properties as well as cell-matrix adhesions in acute mechanotransduction events (Cullen, Lessing, et al., 2007). The number, density, and nature of cell-matrix and cell-cell (e.g., cadherins) adhesions likely contribute to the fidelity of strain transfer from the environment to the neuron and by extension affect the associated intracellular signaling cascades. They may also serve as fixed points preventing sliding between adjacent surfaces, which could concentrate stress and precipitate localized receptor or membrane failure. Clearly, the effects of supra-threshold forces on ECM, adhesions, plasma membrane, ion channels, and cytoskeleton are all interrelated and contribute to pathological mechanosensation in neurons.
3.3.5. Other Potential Mechanisms of Pathological Mechanosensation in Neurons
In addition to these known responses to the supra-threshold loading seen in TBI, there are likely other mechanisms of mechanotransduction. For instance, forces may affect receptor-ligand interactions. When two molecules are being pulled apart by tensile forces, greater force increases the likelihood that they separate in situations known as slip-bonds. In contrast, greater shear stress can actually promote adhesion in situations called catch-bonds (Sokurenko et al., 2008). Integrin, cadherin, and actin have all been shown to be capable of producing catch-bonds (Kong et al., 2009; C. Lee et al., 2013; Rakshit et al., 2012). However, above the optimum force for catch-bond formation, a receptor and ligand can be pulled apart like a slip-bond (C. Lee et al., 2013; Sokurenko et al., 2008). The high loading experienced during TBI could therefore disrupt receptor-ligand interactions, altering the cell-cell, cell-matrix, or cytoskeleton interactions.
While the cytoskeleton can transmit forces to mitochondria (Bartolák-Suki et al., 2017), the organelles may be able to directly sense forces themselves. In non-neuronal cells, mechanically deforming mitochondrial membranes led to mitochondrial fission that was not dependent on actin polymerization, but instead appeared to depend on the integral mitochondrial membrane adapter protein mitochondrial fission factor (MFF), which can detect and has a preference for mitochondrial tubules of smaller diameter. Deforming and constricting mitochondrial membranes reduces their diameter, causing MMF accumulation and recruitment of fission machinery like DRP1. Through fission, mitochondria may be able to resolve mechanical strain and avoid shearing or tearing (Carsten et al., 2017). Those outcomes may not be avoidable after supra-threshold forces such as those experienced during TBI. Indeed, in neurons mitochondria appear to be damaged acutely after injury. Within minutes of fluid percussion injury in cats, axonal mitochondria exhibited swelling and disruption of cristae that persisted for hours (Pettus et al., 1994). In cultures of neurons and astrocytes, stretch injury decreased neuronal mitochondria membrane potential 15 minutes after injury, which appeared to be at least partially linked to intracellular calcium elevation through NMDAR (Ahmed et al., 2000, 2002). This damage can have dire consequences for the cell (see section 3.5 Physiological and Functional Consequences of Pathological Loading). Given the effect of forces on mitochondria, it is likely that other organelles are affected by supra-threshold loading as well.
3.4. Pathological Mechanosensation in Glia
In addition to neurons, astrocytes also respond to biomechanical loading, though they seem to be less susceptible to strain-induced death than neurons (Cullen, Simon, et al., 2007). Mechanical injury causes astrocytes to release ATP extracellularly (Ahmed et al., 2000; Neary et al., 2003, 2005). Shortly after in vitro stretch injury, extracellular ATP can activate P2 purinergic receptors, which along with increases in intracellular calcium, resulted in ERK and Akt activation, which play a role in astrogliosis (Neary et al., 2003, 2005). While not necessarily an acute response to injury, ATP signaling through P2 receptors also resulted in an increase in astrocytic N-cadherin expression and cell surface localization 24 hours after stretch injury (Tran et al., 2008), suggesting that altered glia-glia or glia-axon adhesions may influence subsequent responses during repetitive injuries. Though extracellular ATP may increase, intracellular ATP concentrations decreased 15 minutes after stretch injury, which appeared to be caused by dysfunctional mitochondria with decreased membrane potentials (Ahmed et al., 2000). Interestingly, this injury in astrocytes seemed to influence mitochondrial function in neurons (Ahmed et al., 2000). Similar to neurons, mechanical injury resulted in an increase in intracellular sodium that was dependent on the severity of injury, and activated NMDAR (Floyd et al., 2005; Maneshi et al., 2017). Intracellular calcium also rises in response to shear stress. These increases occurred at areas of the cytoskeleton exhibiting the most strain, as shearing produced non-uniform distribution of forces across the cytoskeleton, particularly in cells having fewer stress fibers and lower tension before injury (Maneshi et al., 2018). Other proteins that comprise or interact with the cytoskeleton change as well, though these changes are not quite immediate. For instance, increased GFAP reactivity was seen at 48 hours in shear strain 3D culture system, along with hypertrophic process density, increased secretion of the ECM molecule chondroitin sulfate proteoglycan, and increased astrocyte density, depending on the strain rate (Cullen, Simon, et al., 2007). An increase in GFAP was correlated with a general softening in non-nuclear regions of astrocytes at the site of stretch injury and in the surrounding area (Miller et al., 2009). In fact, and contrary to long-held assumptions, in vivo the astrocytic glial scar is actually softer than surrounding tissue, and correlated with expression levels of cytoskeletal components GFAP and vimentin, as well as extracellular matrix components laminin and collagen IV (Moeendarbary et al., 2017). Even in injuries that do not produce a glial scar, astrocyte reactivity is a hallmark of TBI and impacts numerous processes over a variety of time scales (Burda et al., 2015).
While the biomechanical responses of astrocytes have received much attention, pathological mechanotransduction in other glial cells is less studied. It is clear that microglia can respond rapidly to injury, as in an intact living mouse brain, their processes have been observed to rapidly converge on the site of laser ablation injury without cell body movement in an ATP-dependent manner (Davalos et al., 2005). Microglia processes were also observed to contact diffusely injured axons 6 hours after a central FPI in pigs (Lafrenaye et al., 2015). The cell bodies themselves can also migrate rapidly, as illustrated by microglial localization to permeabilized neurons within 15 minutes of swine diffuse rotational injury (Wofford et al., 2017). As major players in the complex neuroinflammatory response, microglia impact many processes from secondary injury to repair across acute and chronic time points (Loane & Kumar, 2016).
Since axonal injury is a hallmark of diffuse brain injury, and as white matter damage has been detected with diffusion tensor imaging within hours to days after even mild injury (Bazarian et al., 2007; Herrera et al., 2017; Soni et al., 2020; Wilde et al., 2008; T. C. Wu et al., 2010), there has been interest in the mechanical responses of myelin and oligodendrocytes. Though the role of mechanical forces on immature oligodendrocyte differentiation and migration have been investigated (Makhija et al., 2020), only recently has the response of mature oligodendrocytes to supra-threshold forces been examined. High-magnitude tensile strain applied to cultured matured oligodendrocytes produced damage to cell morphology, partial cell loss, and decreased myelin protein expression (Chierto et al., 2019). This stretch-induced myelin protein loss was shown to be transient and reversible, and caused by calcium-dependent MAP-ERK1/2 activation (J. Kim et al., 2020). Interruption of the myelin sheath not only has implications for action potential conduction, but also for the mechanical response of the axon to loading. Spinal cord studies revealed that myelinated and demyelinated spinal cords displayed differences in stiffness and tensile stress, suggesting that myelin and axoglial junctions play a large role in determining the tensile response of the tissue (Shreiber et al., 2009). Indeed, unmyelinated axons are more susceptible to fluid percussion or in vitro stretch injury than their myelinated counterparts (Reeves et al., 2005, 2012; Staal & Vickers, 2011). Even in myelinated neurons, the mechanical response of the axon is dependent in part on the thickness of the myelin sheath (g-ratio) and the cholesterol concentration of the myelin (Zhu et al., 2016). In particular, the unmyelinated nodes of Ranvier experience larger strains that the rest of the axon (Zhu et al., 2016), and secondary injury abnormalities in paranodal myelin organization and adhesion have been observed days to weeks after injury (Marion et al., 2018; Reeves et al., 2010). Together, it is clear that the mechanical properties and responses of myelin and oligodendrocytes have significant roles in the progression of white matter pathology (Armstrong et al., 2016).
3.5. Pathological Mechanosensation in Blood Vessels
The biomechanics of cerebral blood vessels during TBI has recently been reviewed (Monson et al., 2019). To briefly summarize, blood vessels in the brain often tear and bleed as an immediate biophysical consequence of TBI. It is also possible that mechanical forces below the threshold for hemorrhage could contribute to vascular dysfunction including alterations in contractility through microstructural damage to vascular cells and ECM. However, little is known about the local loading conditions that blood vessels in the brain experience during TBI or how the vessels respond to these forces. It is likely that loading conditions vary depending on where the vessel is located in the brain (e.g., epidural, subarachnoid, parenchymal, etc.). Furthermore, the brain’s response to loading in a particular region may be influenced by the degree of vascularization. It is noteworthy that gray matter is more vascularized than white matter, and cerebral arteries are considerably stiffer than the surrounding tissue. Interestingly, branching segments of vessels appear to be more susceptible to mechanical failure than straight segments, suggesting that the resultant stress concentrations may be relevant to vessels experiencing uneven shear, such as the bridging veins extending from the surface of the brain to the relatively stiff sinuses. At the level of capillaries, the blood brain barrier is known to experience an immediate and biphasic (and in some cases, multiphasic) increase in permeability. Across vessel sizes, it is evident that application of mechanical forces during injury has an important role in vascular pathology, and further studies are needed to investigate the precise effects of different loading conditions.
3.6. Physiological and Functional Consequences of Pathological Loading
The failure of the cytoskeleton and plasma membrane, along with alterations in ion channels, ECM, and adhesions, in response to supra-threshold mechanical loading can lead to a wide range of consequences for the cell and the tissue (Loane et al., 2015). Deformation of various subcellular compartments may affect the diffusion of particles. Diffusion of substances in the cytoplasm is very complex, and depends on spatial restrictions due to the cytoskeleton, organelles, the physical geometry of the area or compartment, as well as other considerations (Luby-Phelps, 2000). The presence and concentration gradients of biomolecules and other signaling factors are often tightly regulated within subcellular compartments of the cytoplasm. In dendrites in particular, the regulation of calcium diffusion has been shown to depend on the presence of buffering proteins and crowding from large structures, as well as spine geometry (Biess et al., 2007, 2011; Cugno et al., 2019; Korkotian & Segal, 2006; Straube & Ridgway, 2009). When these compartments are deformed, diffusion rates can be expected to change.
Whether from altered diffusion or changes in ion channels and membrane permeability, acute loss of ionic homeostasis at the point of mechanical injury kicks off a variety of signaling cascades that contribute to secondary injury, including excitotoxicity and changes in energy metabolism (Giza & Hovda, 2001; McGinn & Povlishock, 2015). Mitochondrial dysfunction not only results in metabolic alterations, but also leads to increased reactive oxygen species (ROS), which further damage the cell (Hiebert et al., 2015). ROS, along with factors released by primary or secondary plasma membrane disruption and excitotoxic neuronal injury, drive early neuroinflammation, which can contribute to further secondary injury and persist chronically (Simon et al., 2017). These factors contribute to a variety of delayed cell death mechanisms in neurons that did not die as from the primary injury (Raghupathi, 2004; Stoica & Faden, 2010). Even neurons that survive all of these insults may display altered electrophysiological and network properties (Cohen et al., 2007; Goforth et al., 2011; Magou et al., 2015; Patel et al., 2012; Prado et al., 2005; Sharp et al., 2014; Wolf et al., 2017). Ultimately, long-term dysfunction and death may contribute to chronic neurodegenerative diseases (Smith et al., 2013).
4. Repetitive Brain Injuries
Although forces may only be transduced acutely, they can have lasting effects that can influence how the brain responds to a subsequent injury. At the macro level for instance, CCI in mice can alter the stiffness of the impacted brain region for several days (Boulet et al., 2013), which would change the response to loading if another injury were to occur within that time frame. At the micro level, changes to ECM composition or cell adhesions may also alter how subsequent forces are transferred to cells. Alterations to the cellular mechanosensation machinery can impact its ability to transduce signals, which along with an environment shaped by physical damage and secondary injury cascades, can make the cell more susceptible to further insults during windows of biomechanical vulnerability.
4.1. Temporal Windows of Biomechanical Vulnerability
The timing of repetitive injuries influences cellular vulnerability to further harm from another injury. Some studies suggest that a mild injury may actually be protective against a second injury by a process called preconditioning, which has been extensively studied as it relates to hypoxia and ischemia (Li et al., 2017). In TBI models, hippocampal cultures subjected to a low-level stretch administered 24 hours before a mild stretch resulted in less release of a common marker of damage (S100β) than mild injury alone (Slemmer & Weber, 2005). In vivo, rat weight drop models demonstrated that repetitive mild injuries prevented motor deficits that were seen in animals that received the single injuries (Allen et al., 2000; Mychasiuk et al., 2015). Weight-drop in adolescent mice produced changes in skull bone structure and integrity that reduced the percentage of animals experiencing skull fracture after another injury at the adult age compared to animals receiving adult injuries alone (McColl et al., 2018). These studies suggest that mild preconditioning injuries may strengthen the brain’s defenses under very specific conditions.
However, many studies support the notion that the brain is most vulnerable to increased damage during the first several days after the initial injury, after which time a second injury produces no more damage than a single injury. In mice, repetitive closed-skull CCI led to increased cognitive deficits and/or axonal injury only in animals injured 3 or 5 days apart, not 7 days apart (Longhi et al., 2005). Similarly, repetitive mild CCI in rats showed that the brain was most vulnerable to damage when injuries were delivered at 1 and 3, but not 7, day intervals (Huang et al., 2013). In pig models of diffuse rotational injury as well, repetitive injuries occurring within 3 days resulted in increased damage such as neuronal plasma membrane permeability, axonal damage, and cognitive dysfunction (Friess et al., 2009; Keating et al., 2020). Though Friess and colleagues also reported damage when injuries were separated by 7 days in pediatric animals, Keating et al. did not observe damage in adult animals receiving repetitive injuries separated by 7 days (Friess et al., 2009; Keating et al., 2020). Together these studies suggest that the brain is most vulnerable to a second injury within several days of receiving the first insult. It is important to note, however, that this increased vulnerability clearly depends on the severity of the injuries and likely the type of injury as well, both of which are related to the nature of the injury biomechanics. But how are cells sensitized to increased damage from repetitive injuries?
4.2. Mechanisms of Increased Vulnerability
4.2.1. Cytoskeleton Abnormalities in Neurites
An injury may not cause overt pathological changes in axons and dendrites initially, but can cause damage when administered repetitively. For instance, mice receiving a single closed-skull CCI displayed only small areas of axonal injury, while animals receiving another injury 24 hours later exhibited much greater axonal injury along with a loss of dendritic MAP2 staining (Laurer et al., 2001). Damaged dendrites with loss of MAP2 have also been observed in in vitro stretch injuries of organotypic hippocampal cultures injured 24 hours apart (Effgen & Morrison, 2017). Damage to the cytoskeleton itself during the first injury may prime further damage during a second injury, as cytoskeletal mislocalization has been observed in axonal growth cones after in vitro stretch injuries (Yap et al., 2017). Alternatively, the effects on neurites may be indirect. For instance, in vitro stretch injury of axons at strains of less than 5% did not cause DAI, but when a second identical insult was delivered 24 hours later, intracellular calcium concentrations increased to levels seen after a single high stretch of 20%, and the axons exhibited minor undulations and swellings (Yuen et al., 2009). Although no degeneration was observed after the first injury, it did lead to increased sodium channel expression after 24 hours, which was indirectly responsible for the increase in calcium. This channelopathy was suggested to lower the injury strain threshold and provide a mechanistic basis for increased vulnerability to a second injury (Yuen et al., 2009). Increases in intracellular calcium immediately activate the cytoskeleton-cleaving enzyme calpain whose levels remains elevated for several days, weakening the mechanical stability of the axon in particular (Büki et al., 1999; Ma, 2013; McGinn et al., 2009).
4.2.2. Plasma Membrane Permeability
Altered ion homeostasis can also result from plasma membrane disruptions. In addition to immediate tears, the plasma membrane can exhibit delayed permeability (Cullen et al., 2011; Farkas et al., 2006; M. L. Hernandez et al., 2019), which may impact the response to a repetitive injury. A first injury may prime cells for further damage. For instance, repetitive injuries separated by 15 minutes or 3 days resulted in increased plasma membrane permeability in subcortical oculomotor regions compared to single injuries or repetitive injuries separated by 7 days in a pig model of diffuse, closed-head acceleration (Keating et al., 2020).
Underlying this heightened susceptibility to damage from repetitive injuries could be alterations in plasma membrane composition. Evidence of lipid peroxidation, which damages membranes, occurs as early as 1 hour post-injury in a moderately severe mouse weight drop model (Hall et al., 2004). Mild CCI in rats results in hydrolysis of polyunsaturated free fatty acids and diacylglycerols by 30 minutes after injury (Homayoun et al., 2000). Mass spectrometry brain imaging after CCI or FPI revealed changes in most classes of lipids 3 days after injury, with some classes like ceramides or docosahexaenoic acid (DHA) exhibiting an increase as early as 1 day (Barbacci et al., 2017; Guo et al., 2017; Mallah et al., 2018; Roux et al., 2016). CCI also resulted in lipid dysregulation detectable in serum during the first week post-injury (Hogan et al., 2018). Altered lipid composition could change membrane properties such as stiffness, which would impact how cells respond to subsequent forces. Importantly, these lipid alterations can persist months to years after injury in animals and humans (Abdullah et al., 2014; Emmerich, Abdullah, Crynen, et al., 2016; Emmerich, Abdullah, Ojo, et al., 2016), highlighting the increased susceptibility and chronic effects of these injuries.
Another explanation for increased vulnerability to plasma membrane poration could involve faulty membrane repair mechanisms. Studies investigating plasma membrane permeability involve trapping a dye within a cell, indicating that mechanisms to repair the plasma membrane are rapidly deployed within minutes. These reparative mechanisms appear to include calcium-dependent vesicle exocytosis and cytoskeleton remodeling (Hendricks & Shi, 2014). However, repair may be energetically taxing for the neuron (Togo et al., 2000), and may be occurring at a time of metabolic dysfunction, as detailed below. Additionally, these mechanisms may be limited or unavailable as the secondary injury unfolds and calcium activates enzymes that digest the cytoskeleton and damage membranes (Deng et al., 2007; Homayoun et al., 1997, 2000; Mustafa et al., 2011). Together, these conditions may prevent the neuron from fully repairing its plasma membrane, resulting in secondary poration or increasing the vulnerability to damage from a second injury.
4.2.3. Mitochondria and Metabolic Changes
Mitochondria damage and subsequent alterations in cellular metabolism are acute responses to injury that may increase vulnerability to another insult. In a closed-skull impact model in mice, a second impact delivered 48 hours after the first resulted in cortical mitochondrial dysfunction 4 days after the final injury and increased markers of oxidative stress (Hubbard et al., 2019). A similar injury in rats caused decreases in cerebral glucose usage 24 hours after the first injury that recover by 3 days post injury, and administration of a second injury within this window caused the cerebral metabolic rate of glucose to become even more depressed and remained low for 3 days post-injury (Mayumi L. Prins et al., 2013). This pattern has also been observed in a rat FPI study (Selwyn et al., 2016). In a rat diffuse weight drop model, injuries repeated at 1, 2, 3, or 4, but not 5 days resulted in altered mitochondrial-related metabolism 48 hours after the last impact (Vagnozzi et al., 2007). A loss of mitochondrial function may inhibit the cell from having enough energy to adequately repair damage from primary and secondary injuries, rendering it more susceptible to subsequent damage. Mitochondrial dysfunction also results in ROS production and impaired calcium buffering capabilities, which together damage the plasma membrane, cytoskeleton, and other proteins (Hill et al., 2017) to potentially further lower the threshold for later injury.
4.2.4. Other Potential Mechanisms of Increased Vulnerability
Vessel disruption (hemorrhage) can expose neurons and glia to relatively unregulated whole blood, which can disrupt brain homeostasis. Even without an actual vessel tear, nervous system homeostasis may not be able to be maintained by vasculature that has been impaired by either mechanical forces directly or the biochemical environment of the injured brain. Similarly, the biphasic increase in BBB permeability may predispose the brain to damage from a subsequent injury (Monson et al., 2019). In addition, a single injury may prime cell death pathways that become fully activated upon a second injury. The expression or activation of pro-death proteins such as Bax and proteases such as calpains and caspases increase minutes to days after injury, while pro-survival proteins like Bcl-2 and Bcl-xL decrease (Raghupathi, 2004). Calpain-independent pathways involving overactive poly (ADP-ribose) polymerase (PARP-1) responding to DNA damage induced by oxidative stress can deplete the cell’s energy, also resulting in cell death (Barr & Conley, 2007). A subsequent injury resulting in more mitochondrial dysfunction and/or increases in intracellular calcium may trigger death in neurons that survived the first insult. And death is not just limited to neurons; oligodendrocytes also undergo apoptosis following TBI, leading to white matter loss (Flygt et al., 2013).
Cell death may be exacerbated by inflammation. Inflammatory cytokines are increased after rat FPI, followed by upregulation of apoptosis-related genes (Shojo et al., 2010). Inflammatory stimuli are also able to induce caspase activation, subtypes of which in addition to promoting apoptosis, can also mediate axonal degeneration (Kaushal et al., 2015). Furthermore, inflammatory mediators interact with various components of glutamate signaling to produce changes in receptors, transporters, and inflammatory genes (Simon et al., 2017). These interactions can trigger ROS generation and damage, which contribute to dendritic retraction, synaptic injury, damage to microtubules, and mitochondrial dysfunction in a process termed immunoexcitotoxicity (Blaylock & Maroon, 2011). Of note, inflammation has been observed a year after a single injury (V. E. Johnson, Stewart, Begbie, et al., 2013). Together, the inflammation, priming of cell death pathways, mitochondrial and metabolic changes, plasma membrane alterations, and cytoskeleton abnormalities initiated by a single injury can shift the vulnerability curve for pathological mechanosensation to render cells more susceptible to a second injury.
4.3. Consequences of Repetitive Injuries
Besides cell death, mechanically-induced changes in ion channels and ion homeostasis can impact the electrophysiological properties of neurons. Uniaxial stretching of squid giant axons at increasing rates depolarized the axonal membrane and the time needed to recover to baseline was nonlinear, with stretches of over 20% resulting incomplete recovery to the resting potential (Galbraith et al., 1993). Similarly, stretch injury in organotypic hippocampal slices resulted in altered electrophysiological function dependent on strain and strain rate in a complex manner (Kang & Morrison, 2015). When these organotypic cultures were stretched repetitively 24 or 72 hours apart, long term potentiation was lost (Effgen & Morrison, 2017). Thus, by altering ion homeostasis, repetitive injuries can have a long-term impact on the neuronal function. For instance, repetitive diffuse injuries have been shown to induce posttraumatic epilepsy, though this response can also be caused by single severe injuries (Reid et al., 2016; Shandra et al., 2019). Repetitive mild injuries in particular have been linked to chronic neurodegenerative diseases (Gardner et al., 2015; Gardner & Yaffe, 2015; McKee et al., 2009; Smith et al., 2013). Mechanisms involving excitotoxicity, increases in intracellular calcium, mitochondrial dysfunction, ROS-induced damage, and inflammation, may all contribute to the development of diseases such as chronic traumatic encephalitis (CTE), Alzheimer’s disease, Parkinson’s disease, and other dementias (Faden & Loane, 2015; Kulbe & Hall, 2017). It is clear that repetitive mechanical loading can cause primary and secondary damage with dire consequences.
5. Application of Biomechanics to Advance the Investigation and Treatment of TBI
The impacts of specific types of force application and of immediate physical damage itself are currently underappreciated in neurotrauma research. However, these acute mechanical responses either predispose or directly initiate mechanisms of secondary cellular dysfunction and/or death. Although these secondary mechanisms of damage are not unique to TBI, primary physical damage from force loading, along with the physically damaged environment in which secondary injury cascades unfold, is unique to trauma. Since this post-physical damage environment is the context in which the well-documented secondary processes dominate, there is a need to accurately represent the tissue- and cellular-level injury biomechanics in the development and application of reduced models of TBI.
Clearly, to fully describe the physical causation and physiological consequences of TBI, it is vital to establish relationships between defined cell- and tissue-level biomechanical inputs and acute structural or biophysical alterations. Understanding this relationship is important as the relative contributions of primary (physical) and secondary (pathophysiological) damage may vary according to the particular type of forces experienced or severity of injury. Therefore, consideration of injury biomechanics is critically important in utilizing pre-clinical models to accurately identify pathophysiology and develop targeted treatments specific to the types of forces producing injury. Ideally, animal models of TBI should reflect human injury with respect to biomechanical inputs (with appropriate scaling if necessary), immediate cellular/tissue biophysical responses, and neuropathology distribution and types. Moving forward, animal models should be vetted based on their ability to address all three of these areas, which will enable the opportunity to define and integrate the physical causation of injury as well as the biological consequences.
Information gained from pre-clinical work will be critical for the development of mechanistically based, targeted therapeutics for neuroprotection following TBI. It is evident that the response of the brain to supra-threshold loading depends on the types of forces that are applied. For example, pure impulse (inertial) loading produces different kinds of damage than pure impact (contact) loading. Likewise, the failure of certain structures but not others may differentially alter homeostasis. As such, different therapies might be expected to be more efficacious at treating one form of injury and its specific type of damage over another. Thus, pre-clinical studies of treatments that utilize biomechanically-relevant injury paradigms to target injury type- or causation-specific processes may result in useful therapies unique to a particular kind of loading. Similarly, clinical trials that include specific injury types or biomechanical causations as inclusion criteria for enrollment or subgroup analyses may demonstrate success in certain injury populations. Although determining the precise forces experienced by any given individual during TBI is likely not possible, in certain high-risk groups such as athletes or war fighters, wearable force sensors can provide this type of information (Rowson et al., 2018). Ultimately, clinical guidelines for treatment based on the type of injury and its biomechanical causation would be valuable
While more information is needed to know exactly how applying biomechanics would be implemented, one can envision an instance where a wearable force sensor on the helmet or in the mouth guard of an injured football player can be accessed by a doctor so the physician has a general sense of the loading experienced. Either the knowledge of the doctor or software could inform the physician which pathologies the athlete is (or is not) at risk for. This information could guide initial diagnostics—as well as compliment biomarker-based measurements—and potentially enable pre-symptomatic treatments for patients before actual pathology has blossomed. Patient-specific biomechanical loading could also influence guidelines for return to play, sub-specialists for follow-up care, and the time course and duration of monitoring. In this way, each patient could receive specialized care tailored to the type of damage likely to occur based on the particular biomechanical loading experienced.
6. Conclusions
Because TBI is a physical injury, it is important to recognize the mechanical properties of the brain and the forces that act on it in order to understand the brain’s response to injury. The brain’s properties vary at both the macro and micro scales, with various subcellular components including the extracellular matrix, plasma membrane, transmembrane receptors, and cytoskeleton all contributing to its properties and force-sensing abilities. Under physiological conditions, these elements are able to adequately transduce forces and allow neurons and glia to detect, integrate, and react to changes in their physical environment. However, when loading becomes too great, as in TBI, these mechanosensors break down, damaging the cell. Even if these components do not fail completely in response to an initial injury, the threshold for damage may be lowered and secondary injury cascades and/or repetitive injuries can induce failure.
The concept of immediate structural damage should be considered in context with evolving, secondary pathophysiology, and potential interrelationships should be addressed. Understanding cell and tissue injury biomechanics is important for establishing links between physical and physiological consequences, including how different types of supra-threshold loading affect specific mechanotransducers or brain regions. Proper consideration of TBI biomechanics is critical to moving the field forward, as injury models not representative of human injury will fail to replicate mechanisms of primary damage, acute reparative processes, and the underlying environment in which secondary damage ensues; thus having little or no hope of determining accurate mechanisms of pathology or establishing effective treatments capable of translating clinically. Models with sufficient biomechanical fidelity to human TBI are critical to advance our understanding of the cellular, tissue, and whole-organism responses to neurotrauma. Replicating the causes and consequences of human injury will facilitate the development of targeted therapeutics to address the damage inflicted upon cells by specific mechanisms of injury.
Highlights.
TBI is caused by extreme mechanical forces acting on the brain
Cytoskeleton, plasma membrane, and transmembrane proteins sense forces in the brain
Such biological force-sensors affect brain homeostasis and influence TBI responses
These components can structurally fail due to mechanical forces produced during TBI
Considering biomechanical parameters will advance the study and treatment of TBI
Acknowledgements
The authors thank John O’Donnell and Kathryn Wofford for constructive feedback. Financial support was provided by the Department of Veterans Affairs (BLR&D Merit Review I01-BX005017 & BLR&D Merit Review I01-BX003748), the National Institutes of Health (R03-NS116301), and the Department of Health of tsylvania (PA Consortium on TBI: PACT). Figures 2–5 were created fully or in part with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullah L, Evans JE, Ferguson S, Mouzon B, Montague H, Reed J, Crynen G, Emmerich T, Crocker M, Pelot R, Mullan M, & Crawford F (2014). Lipidomic analyses identify injury-specific phospholipid changes 3 mo after traumatic brain injury. FASEB Journal, 28(12), 5311–5321. 10.1096/fj.14-258228 [DOI] [PubMed] [Google Scholar]
- Adams JH, Graham DI, & Gennarelli TA (1981). Acceleration induced head injury in the monkey. II. Neuropathology. Acta Neuropathologica. Supplementum, 7, 26–28. 10.1007/978-3-642-81553-9_8 [DOI] [PubMed] [Google Scholar]
- Ahmadzadeh H, Smith DH, & Shenoy VB (2014). Viscoelasticity of tau proteins leads to strain rate-dependent breaking of microtubules during axonal stretch injury: predictions from a mathematical model. Biophysical Journal, 106(5), 1123–1133. 10.1016/j.bpj.2014.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh H, Smith DH, & Shenoy VB (2015). Mechanical Effects of Dynamic Binding between Tau Proteins on Microtubules during Axonal Injury. Biophysical Journal, 109(11), 2328–2337. 10.1016/j.bpj.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SM, Rzigalinski BA, Willoughby KA, Sitterding HA, & Ellis EF (2000). Stretch-Induced Injury Alters Mitochondrial Membrane Potential and Cellular ATP in Cultured Astrocytes and Neurons. [PubMed]
- Ahmed SM, Weber JT, Liang S, Willoughby KA, Sitterding HA, Rzigalinski BA, & Ellis EF (2002). NMDA receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. Journal of Neurotrauma, 19(12), 1619–1629. 10.1089/089771502762300274 [DOI] [PubMed] [Google Scholar]
- Allen GV, Gerami D, & Esser MJ (2000). Conditioning effects of repetitive mild neurotrauma on motor function in an animal model of focal brain injury. Neuroscience, 99(1), 93–105. 10.1016/s0306-4522(00)00185-8 [DOI] [PubMed] [Google Scholar]
- Armendáriz BG, Masdeu M. del M., Soriano E, Ureña JM, & Burgaya F (2014). The diverse roles and multiple forms of focal adhesion kinase in brain. The European Journal of Neuroscience, 40(11), 3573–3590. 10.1111/ejn.12737 [DOI] [PubMed] [Google Scholar]
- Armstrong RC, Mierzwa AJ, Sullivan GM, & Sanchez MA (2016). Myelin and oligodendrocyte lineage cells in white matter pathology and plasticity after traumatic brain injury. Neuropharmacology, 110(Pt B), 654–659. 10.1016/j.neuropharm.2015.04.029 [DOI] [PubMed] [Google Scholar]
- Avalos AM, Arthur WT, Schneider P, Quest AFG, Burridge K, & Leyton L (2004). Aggregation of integrins and RhoA activation are required for Thy-1-induced morphological changes in astrocytes. The Journal of Biological Chemistry, 279(37), 39139–39145. 10.1074/jbc.M403439200 [DOI] [PubMed] [Google Scholar]
- Ayad NME, Kaushik S, & Weaver VM (2019). Tissue mechanics, an important regulator of development and disease. Philosophical Transactions of the Royal Society B: Biological Sciences, 374(1779). 10.1098/rstb.2018.0215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens N, Schwartz MA, & Kozminski KG (2016). Biomechanics of vascular mechanosensation and remodeling. 27, 7–11. 10.1091/mbc.E14-11-1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Gracheva EO, & Gallagher PG (2014). Piezo proteins: regulators of mechanosensation and other cellular processes. The Journal of Biological Chemistry, 289(46), 31673–31681. 10.1074/jbc.R114.612697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey EL, McCulloch J, Sudlow C, & Wardlaw JM (2009). Potential animal models of lacunar stroke: a systematic review. Stroke, 40(6), e451–8. 10.1161/STROKEAHA.108.528430 [DOI] [PubMed] [Google Scholar]
- Bar-Kochba E, Scimone MT, Estrada JB, Franck C, Faul M, Xu L, Wald MM, Coronado V, Dellinger AM, Alexander MP, Inglese M, Johnson VE, Stewart W, Smith DH, Scheid R, Walther K, Guthke T, Preul C, Cramon D. Y. von, … Dingle Y-TL (2016). Strain and rate-dependent neuronal injury in a 3D in vitro compression model of traumatic brain injury. Scientific Reports, 6(April), 30550 10.1038/srep30550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacci DC, Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, Balaban CD, Schultz JA, Gouty S, Cox BM, & Woods AS (2017). Mass Spectrometric Imaging of Ceramide Biomarkers Tracks Therapeutic Response in Traumatic Brain Injury. ACS Chemical Neuroscience, 8(10), 2266–2274. 10.1021/acschemneuro.7b00189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TL, & Conley YP (2007). Poly(ADP-ribose) polymerase-1 and its clinical applications in brain injury. The Journal of Neuroscience Nursing : Journal of the American Association of Neuroscience Nurses, 39(5), 278–284. 10.1097/01376517-200710000-00004 [DOI] [PubMed] [Google Scholar]
- Bartolák-Suki E, Imsirovic J, Nishibori Y, & Krishnan R (2017). Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int J Mol Sci, 18(8), 1812 10.3390/ijms18081812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly PV, Cohen TS, Leister EP, Ajo D, Leuthardt EC, & Genin GM (2005). Deformation of the human brain induced by mild acceleration. Journal of Neurotrauma, 22(8), 845–856. 10.1089/neu.2005.22.845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V, & Peterson D (2007). Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. Journal of Neurotrauma, 24(9), 1447–1459. 10.1089/neu.2007.0241 [DOI] [PubMed] [Google Scholar]
- Bell JM, Breiding MJ, & DePadilla L (2017). CDC’s efforts to improve traumatic brain injury surveillance. Journal of Safety Research, 62, 253–256. 10.1016/j.jsr.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shahar Y (2011). Sensory functions for degenerin/epithelial sodium channels (DEG/ENaC). Advances in Genetics, 76, 1–26. 10.1016/B978-0-12-386481-9.00001-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tabou S, Keller E, & Nussinovitch I (1994). Mechanosensitivity of voltage-gated calcium currents in rat anterior pituitary cells. The Journal of Physiology, 476(1), 29–39. [PMC free article] [PubMed] [Google Scholar]
- Bernick KB, Prevost TP, Suresh S, & Socrate S (2011). Biomechanics of single cortical neurons. Acta Biomaterialia, 7(3), 1210–1219. 10.1016/j.actbio.2010.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biess A, Korkotian E, & Holcman D (2007). Diffusion in a dendritic spine: the role of geometry. Physical Review. E, Statistical, Nonlinear, and Soft Matter Physics, 76(2 Pt 1), 21922 10.1103/PhysRevE.76.021922 [DOI] [PubMed] [Google Scholar]
- Biess A, Korkotian E, & Holcman D (2011). Barriers to diffusion in dendrites and estimation of calcium spread following synaptic inputs. PLoS Computational Biology, 7(10), e1002182 10.1371/journal.pcbi.1002182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaylock RL, & Maroon J (2011). Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surgical Neurology International, 2, 107 10.4103/2152-7806.83391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland LM, & Drzewiecki MM (2008). Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochemistry and Biophysics, 52(2), 59–84. 10.1007/s12013-008-9027-2 [DOI] [PubMed] [Google Scholar]
- Bollmann L, Koser DE, Shahapure R, Gautier HOB, Holzapfel GA, Scarcelli G, Gather MC, Ulbricht E, & Franze K (2015). Microglia mechanics: Immune activation alters traction forces and durotaxis. Frontiers in Cellular Neuroscience, 9(September), 1–16. 10.3389/fncel.2015.00363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulet T, Kelso ML, & Othman SF (2013). Long-term in vivo imaging of viscoelastic properties of the mouse brain after controlled cortical impact. Journal of Neurotrauma, 30(17), 1512–1520. 10.1089/neu.2012.2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman CL, Ding JP, Sachs F, & Sokabe M (1992). Mechanotransducing ion channels in astrocytes. Brain Research, 584(1–2), 272–286. 10.1016/0006-8993(92)90906-p [DOI] [PubMed] [Google Scholar]
- Braun J, Guo J, Lützkendorf R, Stadler J, Papazoglou S, Hirsch S, Sack I, & Bernarding J (2014). NeuroImage High-resolution mechanical imaging of the human brain by three-dimensional multifrequency magnetic resonance elastography at 7T. NeuroImage, 90, 308–314. 10.1016/j.neuroimage.2013.12.032 [DOI] [PubMed] [Google Scholar]
- Budday S, Nay R, Rooij R. De, Steinmann P, Wyrobek T, Ovaert TC, & Kuhl E (2015). Mechanical properties of gray and white matter brain tissue by indentation. Journal of the Mechanical Behavior of Biomedical Materials, 46, 318–330. 10.1016/j.jmbbm.2015.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büki A, Siman R, Trojanowski JQ, & Povlishock JT (1999). The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. Journal of Neuropathology and Experimental Neurology, 58(4), 365–375. 10.1097/00005072-199904000-00007 [DOI] [PubMed] [Google Scholar]
- Burda JE, Bernstein AM, & Sofroniew MV (2015). Astrocyte roles in traumatic brain injury. Experimental Neurology. 10.1016/j.expneurol.2015.03.020 [DOI] [PMC free article] [PubMed]
- Campbell JN, Register D, & Churn SB (2012). Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. Journal of Neurotrauma, 29(2), 201–217. 10.1089/neu.2011.1761 [DOI] [PubMed] [Google Scholar]
- Carbonell WS, Maris DO, McCall T, & Grady MS (1998). Adaptation of the fluid percussion injury model to the mouse. Journal of Neurotrauma, 15(3), 217–229. 10.1089/neu.1998.15.217 [DOI] [PubMed] [Google Scholar]
- Cargill RS 2nd, & Thibault LE (1996). Acute alterations in [Ca2+]i in NG108–15 cells subjected to high strain rate deformation and chemical hypoxia: an in vitro model for neural trauma. Journal of Neurotrauma, 13(7), 395–407. 10.1089/neu.1996.13.395 [DOI] [PubMed] [Google Scholar]
- Carsten S, Helle J, Feng Q, Aebersold MJ, Vahid A, Sirianni A, Mostowy S, Hirt L, Gru RR, Idema T, Zambelli T, Snedeker JG, & An S (2017). Mechanical force induces mitochondrial fission. 1–26. [DOI] [PMC free article] [PubMed]
- Centers for Disease Control and Prevention. (2019). Surveillance Report of Traumatic Brain Injury-related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2014. Centers for Disease Control and Prevention, U.S. Department of Health and Human Services. [Google Scholar]
- Centers for Disease Control and Prevention, National Institutes of Health, Department of Defense, & VA Leadership Panel. (2013). Report to Congress on Traumatic Brain Injury in the United States: Understanding the Public Health Problem among Current and Former Military Personnel.
- Chan DD, Knutsen AK, Lu YC, Yang SH, Magrath E, Wang WT, Bayly PV, Butman JA, & Pham DL (2018). Statistical Characterization of Human Brain Deformation during Mild Angular Acceleration Measured in Vivo by Tagged Magnetic Resonance Imaging. Journal of Biomechanical Engineering, 140(10), 1–13. 10.1115/1.4040230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra N, & Sundaramurthy A (2015). Acute Pathophysiology of Blast Injury—From Biomechanics to Experiments and Computations: Implications on Head and Polytrauma. (Kobeissy FH (ed.)). [PubMed] [Google Scholar]
- Chen Y, Constantini S, Trembovler V, Weinstock M, & Shohami E (1996). An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. Journal of Neurotrauma, 13(10), 557–568. 10.1089/neu.1996.13.557 [DOI] [PubMed] [Google Scholar]
- Cheng S, Clarke EC, & Bilston LE (2008). Rheological properties of the tissues of the central nervous system: a review. Medical Engineering & Physics, 30(10), 1318–1337. 10.1016/j.medengphy.2008.06.003 [DOI] [PubMed] [Google Scholar]
- Cheng WH, Martens KM, Bashir A, Cheung H, Stukas S, Gibbs E, Namjoshi DR, Button EB, Wilkinson A, Barron CJ, Cashman NR, Cripton PA, & Wellington CL (2019). CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimer’s Research & Therapy, 11(1), 6 10.1186/s13195-018-0461-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y-R, Jiang B-Y, & Chen C-C (2018). Acid-sensing ion channels: dual function proteins for chemo-sensing and mechano-sensing. Journal of Biomedical Science, 25(1), 46 10.1186/s12929-018-0448-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chierto E, Simon A, Castoldi F, Meffre D, Cristinziano G, Sapone F, Carrete A, Borderie D, Etienne F, Rannou F, Morrison B, Massaad C, & Jafarian-Tehrani M (2019). Mechanical Stretch of High Magnitude Provokes Axonal Injury, Elongation of Paranodal Junctions, and Signaling Alterations in Oligodendrocytes. Molecular Neurobiology, 56(6), 4231–4248. 10.1007/s12035-018-1372-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chighizola M, Dini T, Lenardi C, Milani P, Podestà A, & Schulte C (2019). Mechanotransduction in neuronal cell development and functioning. Biophysical Reviews, 11(5), 701–720. 10.1007/s12551-019-00587-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Muthukumar AK, Stork T, Coutinho-Budd JC, & Freeman MR (2018). Focal adhesion molecules regulate astrocyte morphology and glutamate transporters to suppress seizure-like behavior. Proceedings of the National Academy of Sciences of the United States of America, 115(44), 11316–11321. 10.1073/pnas.1800830115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Sun D, & Jakobs TC (2015). Astrocytes in the optic nerve head express putative mechanosensitive channels. Molecular Vision, 21, 749–766. [PMC free article] [PubMed] [Google Scholar]
- Christman CW, Grady MS, Walker SA, Holloway KL, & Povlishock JT (1994). Ultrastructural studies of diffuse axonal injury in humans. Journal of Neurotrauma, 11(2), 173–186. 10.1089/neu.1994.11.173 [DOI] [PubMed] [Google Scholar]
- Cohen AS, Pfister BJ, Schwarzbach E, Grady MS, Goforth PB, & Satin LS (2007). Injury-induced alterations in CNS electrophysiology. Progress in Brain Research, 161, 143–169. 10.1016/S0079-6123(06)61010-8 [DOI] [PubMed] [Google Scholar]
- Crawford GE, & Earnshaw JC (1987). Viscoelastic relaxation of bilayer lipid membranes. Frequency-dependent tension and membrane viscosity. Biophysical Journal, 52(1), 87–94. 10.1016/S0006-3495(87)83191-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F (1982). Do dendritic spines twitch? Trends in Neurosciences, 5, 44–46. 10.1016/0166-2236(82)90020-0 [DOI] [Google Scholar]
- Cugno A, Bartol TM, Sejnowski TJ, Iyengar R, & Rangamani P (2019). Geometric principles of second messenger dynamics in dendritic spines. Scientific Reports, 9(1), 11676 10.1038/s41598-019-48028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Harris JP, Browne KD, Wolf JA, Duda JE, Meaney DF, Margulies SS, & Smith DH (2016). Chapter 17: A Porcine Model of Traumatic Brain Injury via Head Rotational Acceleration. In Injury Models of the Central Nervous System (Vol. 1462). 10.1007/978-1-4939-3816-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, & LaPlaca MC (2006). Neuronal response to high rate shear deformation depends on heterogeneity of the local strain field. Journal of Neurotrauma, 23(9), 1304–1319. 10.1089/neu.2006.23.1304 [DOI] [PubMed] [Google Scholar]
- Cullen DK, Lessing MC, & LaPlaca MC (2007). Collagen-dependent neurite outgrowth and response to dynamic deformation in three-dimensional neuronal cultures. Annals of Biomedical Engineering, 35(5), 835–846. 10.1007/s10439-007-9292-z [DOI] [PubMed] [Google Scholar]
- Cullen DK, Simon CM, & LaPlaca MC (2007). Strain rate-dependent induction of reactive astrogliosis and cell death in three-dimensional neuronal-astrocytic co-cultures. Brain Research, 1158, 103–115. 10.1016/j.brainres.2007.04.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Vernekar VN, & LaPlaca MC (2011). Trauma-Induced Plasmalemma Disruptions in Three-Dimensional Neural Cultures Are Dependent on Strain Modality and Rate. Journal of Neurotrauma, 28(November), 2219–2233. 10.1089/neu.2011.1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry N, Ghézali G, Kaminski Schierle GS, Rouach N, & Kaminski CF (2017). Correlative STED and Atomic Force Microscopy on Live Astrocytes Reveals Plasticity of Cytoskeletal Structure and Membrane Physical Properties during Polarized Migration. Frontiers in Cellular Neuroscience, 11, 104 10.3389/fncel.2017.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, & Gan W-B (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience, 8(6), 752–758. 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, & Bandak FA (2015). Neuropathology of traumatic brain injury: comparison of penetrating, nonpenetrating direct impact and explosive blast etiologies. Seminars in Neurology, 35(1), 12–19. 10.1055/s-0035-1544240 [DOI] [PubMed] [Google Scholar]
- De Pascalis C, Pérez-González C, Seetharaman S, Boëda B, Vianay B, Burute M, Leduc C, Borghi N, Trepat X, & Etienne-Manneville S (2018). Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. The Journal of Cell Biology, 217(9), 3031–3044. 10.1083/jcb.201801162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedman A, Sharif-Naeini R, Folgering JHA, Duprat F, Patel A, & Honoré E (2009). The mechano-gated K2P channel TREK-1. European Biophysics Journal, 38(3), 293–303. 10.1007/s00249-008-0318-8 [DOI] [PubMed] [Google Scholar]
- Demetriades D, Kuncir E, Murray J, Velmahos GC, Rhee P, & Chan L (2004). Mortality prediction of head Abbreviated Injury Score and Glasgow Coma Scale: analysis of 7,764 head injuries. Journal of the American College of Surgeons, 199(2), 216–222. 10.1016/j.jamcollsurg.2004.02.030 [DOI] [PubMed] [Google Scholar]
- Deng Y, Thompson BM, Gao X, & Hall ED (2007). Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Experimental Neurology, 205(1), 154–165. 10.1016/j.expneurol.2007.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Department of Veterans Affairs, & Department of Defense. (2016). VA / DoD CLINICAL PRACTICE GUIDELINE FOR THE MANAGEMENT OF CONCUSSION-MILD TRAUMATIC BRAIN INJURY.
- Dimova R (2014). Recent developments in the field of bending rigidity measurements on membranes. Advances in Colloid and Interface Science, 208, 225–234. 10.1016/j.cis.2014.03.003 [DOI] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, & Hayes RL (1991). A controlled cortical impact model of traumatic brain injury in the rat. Journal of Neuroscience Methods, 39(3), 253–262. 10.1016/0165-0270(91)90104-8 [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, & Hayes RL (1987). A fluid percussion model of experimental brain injury in the rat. Journal of Neurosurgery, 67(1), 110–119. 10.3171/jns.1987.67.1.0110 [DOI] [PubMed] [Google Scholar]
- Dixon KJ (2017). Pathophysiology of Traumatic Brain Injury. Physical Medicine and Rehabilitation Clinics of North America, 28(2), 215–225. 10.1016/j.pmr.2016.12.001 [DOI] [PubMed] [Google Scholar]
- Dubey S, Bhembre N, Bodas S, Veer S, Ghose A, Callan-Jones A, & Pullarkat P (2020). The axonal actin-spectrin lattice acts as a tension buffering shock absorber. ELife, 9 10.7554/eLife.51772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G 3rd, Zhao J, Dash PK, Soto C, & Moreno-Gonzalez I (2020). Traumatic Brain Injury Induces Tau Aggregation and Spreading. Journal of Neurotrauma, 37(1), 80–92. 10.1089/neu.2018.6348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Effgen GB, & Morrison B 3rd. (2017). Electrophysiological and Pathological Characterization of the Period of Heightened Vulnerability to Repetitive Injury in an in Vitro Stretch Model. Journal of Neurotrauma, 34(4), 914–924. 10.1089/neu.2016.4477 [DOI] [PubMed] [Google Scholar]
- Ehringer W, Belcher D, Wassal SR, & Stillwell W (1990). A comparison of the effects of linolenic (18 : 3 n 3) and docosahexaenoic (22 : 6 n 3) acids on phospholipid bilayers. Chemistry and Physics of Lipids, 54, 79–88. [DOI] [PubMed] [Google Scholar]
- Elkin BS, Ilankovan AI, & Iii BM (2011). A Detailed Viscoelastic Characterization of the P17 and Adult Rat Brain. 2244(November), 2235–2244. 10.1089/neu.2010.1604 [DOI] [PubMed] [Google Scholar]
- Elosegui-Artola A, Andreu I, Beedle AEM, Lezamiz A, Uroz M, Kosmalska AJ, Oria R, Kechagia JZ, Rico-Lastres P, Le Roux A-L, Shanahan CM, Trepat X, Navajas D, Garcia-Manyes S, & Roca-Cusachs P (2017). Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell, 171(6), 1397–1410.e14. 10.1016/j.cell.2017.10.008 [DOI] [PubMed] [Google Scholar]
- Emmerich T, Abdullah L, Crynen G, Dretsch M, Evans J, Ait-ghezala G, Reed J, Montague H, Chaytow H, Mathura V, Martin J, Pelot R, Ferguson S, Bishop A, Phillips J, Mullan M, & Crawford F (2016). Plasma Lipidomic Profiling in a Military Population of Mild Traumatic Brain Injury and Post-Traumatic Stress Disorder with Apolipoprotein E e 4 - Dependent Effect. 1348, 1331–1348. 10.1089/neu.2015.4061 [DOI] [PubMed] [Google Scholar]
- Emmerich T, Abdullah L, Ojo J, Mouzon B, Nguyen T, Laco GS, Crynen G, Evans JE, Reed J, Mullan M, & Crawford F (2016). Mild TBI Results in a Long-Term Decrease in Circulating Phospholipids in a Mouse Model of Injury. NeuroMolecular Medicine, 1–14. 10.1007/s12017-016-8436-4 [DOI] [PubMed]
- Engler AJ, Sen S, Sweeney HL, & Discher DE (2006). Matrix elasticity directs stem cell lineage specification. Cell, 126(4), 677–689. 10.1016/j.cell.2006.06.044 [DOI] [PubMed] [Google Scholar]
- Ertürk A, Mentz S, Stout EE, Hedehus M, Dominguez SL, Neumaier L, Krammer F, Llovera G, Srinivasan K, Hansen DV, Liesz A, Scearce-Levie KA, & Sheng M (2016). Interfering with the Chronic Immune Response Rescues Chronic Degeneration After Traumatic Brain Injury. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 36(38), 9962–9975. 10.1523/JNEUROSCI.1898-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Hoyos D, Jagielska A, Homan KA, Du H, Busbee T, Anderson DG, Fang NX, Lewis JA, & Van Vliet KJ (2018). Engineered 3D-printed artificial axons. Scientific Reports, 8(1), 478 10.1038/s41598-017-18744-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EA, & Hochmuth RM (1978). Mechanochemical Properties of Membranes In Bronner F & Kleinzeller A (Eds.), Membrane Properties: Mechanical Aspects, Receptors, Energetics and Calcium-Dependence of Transport (Vol. 10, pp. 1–64). Academic Press; 10.1016/S0070-2161(08)60833-3 [DOI] [Google Scholar]
- Faden AI, & Loane DJ (2015). Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics : The Journal of the American Society for Experimental NeuroTherapeutics, 12(1), 143–150. 10.1007/s13311-014-0319-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Wu D, & Birukov KG (2020). Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr Physiol, 9(2), 873–904. 10.1002/cphy.c180020.Mechanosensing [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas O, Lifshitz J, & Povlishock JT (2006). Mechanoporation Induced by Diffuse Traumatic Brain Injury: An Irreversible or Reversible Response to Injury? Journal of Neuroscience, 26(12), 3130–3140. 10.1523/JNEUROSCI.5119-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faul M, & Coronado V (2015). Epidemiology of traumatic brain injury. Handbook of Clinical Neurology, 127, 3–13. 10.1016/B978-0-444-52892-6.00001-5 [DOI] [PubMed] [Google Scholar]
- Finkelstein EA, Corso PS, & Miller TR (2006). The Incidence and Economic Burden of Injuries in the United States. Oxford University Press. [Google Scholar]
- Floyd CL, Golden KM, Black RT, Hamm RJ, & Lyeth BG (2002). Craniectomy position affects morris water maze performance and hippocampal cell loss after parasagittal fluid percussion. Journal of Neurotrauma, 19(3), 303–316. 10.1089/089771502753594873 [DOI] [PubMed] [Google Scholar]
- Floyd CL, Gorin FA, & Lyeth BG (2005). Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia, 51(1), 35–46. 10.1002/glia.20183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flygt J, Djupsjö A, Lenne F, & Marklund N (2013). Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. The European Journal of Neuroscience, 38(1), 2153–2165. 10.1111/ejn.12179 [DOI] [PubMed] [Google Scholar]
- Franco SJ, & Muller U (2011). Extracellular matrix functions during neuronal migration and lamination in the mammalian central nervous system. Developmental Neurobiology, 71(11), 889–900. 10.1002/dneu.20946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friess SH, Ichord RN, Ralston J, Ryall K, Helfaer MA, Smith C, & Margulies SS (2009). Repeated traumatic brain injury affects composite cognitive function in piglets. Journal of Neurotrauma, 1121(July), 090330061141047 10.1089/neu.2008-0845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith JA, Thibault LE, & Matteson DR (1993). Mechanical and electrical responses of the squid giant axon to simple elongation. Journal of Biomechanical Engineering, 115(1), 13–22. 10.1115/1.2895464 [DOI] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, & Egelman EH (2012). Actin filaments as tension sensors. Current Biology : CB, 22(3), R96–101. 10.1016/j.cub.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Deng P, Xu ZC, & Chen J (2011). Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS ONE, 6(9). 10.1371/journal.pone.0024566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, & Yaffe K (2015). Traumatic brain injury in later life increases risk for Parkinson disease. Annals of Neurology, 77(6), 987–995. 10.1002/ana.24396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, & Yaffe K (2015). Epidemiology of mild traumatic brain injury and neurodegenerative disease. Molecular and Cellular Neuroscience, 66(PB), 75–80. 10.1016/j.mcn.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub BM, Kasuba KC, Mace E, Strittmatter T, Laskowski PR, Geissler SA, Hierlemann A, Fussenegger M, Roska B, & Müller DJ (2020). Neurons differentiate magnitude and location of mechanical stimuli. Proceedings of the National Academy of Sciences of the United States of America, 117(2), 848–856. 10.1073/pnas.1909933117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators. (2019). Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet. Neurology, 18(1), 56–87. 10.1016/S1474-4422(18)30415-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes-Klein DM, Schiffman KB, & Meaney DF (2006). Mechanisms and consequences of neuronal stretch injury in vitro differ with the model of trauma. Journal of Neurotrauma, 23(2), 193–204. 10.1089/neu.2006.23.193 [DOI] [PubMed] [Google Scholar]
- Geddes DM, & Cargill RS 2nd. (2001). An in vitro model of neural trauma: device characterization and calcium response to mechanical stretch. Journal of Biomechanical Engineering, 123(3), 247–255. [DOI] [PubMed] [Google Scholar]
- Geddes DM, Cargill RS 2nd, & LaPlaca MC (2003). Mechanical Stretch to Neurons Results in a Strain Rate and Magnitude-Dependent Increase in Plasma Membrane Permeability. 20(10), 1039–1049. [DOI] [PubMed] [Google Scholar]
- Geddes DM, LaPlaca MC, & Cargill RS (2003). Susceptibility of hippocampal neurons to mechanically induced injury. Experimental Neurology, 184(1), 420–427. 10.1016/S0014-4886(03)00254-1 [DOI] [PubMed] [Google Scholar]
- Gennarelli TA, Champion HR, Copes WS, & Sacco WJ (1994). Comparison of mortality, morbidity, and severity of 59,713 head injured patients with 114,447 patients with extracranial injuries. The Journal of Trauma, 37(6), 962–968. 10.1097/00005373-199412000-00016 [DOI] [PubMed] [Google Scholar]
- Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, & Marcincin RP (1982). Diffuse axonal injury and traumatic coma in the primate. Annals of Neurology, 12(6), 564–574. 10.1002/ana.410120611 [DOI] [PubMed] [Google Scholar]
- George N, & Geller HM (2018). Extracellular matrix and traumatic brain injury. February 2017, 573–588. 10.1002/jnr.24151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza CC, & Hovda DA (2001). The Neurometabolic Cascade of Concussion. Journal of Athletic Training, 36(3), 228–235. 10.1227/NEU.0000000000000505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goforth PB, Ellis EF, & Satin LS (2004). Mechanical injury modulates AMPA receptor kinetics via an NMDA receptor-dependent pathway. Journal of Neurotrauma, 21(6), 719–732. 10.1089/0897715041269704 [DOI] [PubMed] [Google Scholar]
- Goforth PB, Ren J, Schwartz BS, & Satin LS (2011). Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. Journal of Neurophysiology, 105(5), 2350–2363. 10.1152/jn.00467.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold EM, Vasilevko V, Hasselmann J, Tiefenthaler C, Hoa D, Ranawaka K, Cribbs DH, & Cummings BJ (2018). Repeated Mild Closed Head Injuries Induce Long-Term White Matter Pathology and Neuronal Loss That Are Correlated With Behavioral Deficits. ASN Neuro, 10, 1759091418781921. 10.1177/1759091418781921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, Hånell A, McGinn MJ, & Povlishock JT (2013). Mild traumatic brain injury in the mouse induces axotomy primarily within the axon initial segment. Acta Neuropathologica, 126(1), 59–74. 10.1007/s00401-013-1119-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevesse T, Dabiri BE, Parker KK, & Gabriele S (2015). Opposite rheological properties of neuronal microcompartments predict axonal vulnerability in brain injury. Scientific Reports, 5, 1–10. 10.1038/srep09475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guharay F, & Sachs F (1984). Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. The Journal of Physiology, 352(1), 685–701. 10.1113/jphysiol.1984.sp015317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Zhou D, Zhang M, Li T, Liu Y, Xu Y, Chen T, & Li Z (2017). Monitoring changes of docosahexaenoic acid-containing lipids during the recovery process of traumatic brain injury in rat using mass spectrometry imaging. May, 1–9. 10.1038/s41598-017-05446-2 [DOI] [PMC free article] [PubMed]
- Hall ED, Detloff MR, Johnson K, Kupina NC, & Al HET (2004). Peroxynitrite-Mediated Protein Nitration and Lipid Peroxidation in a Mouse Model of Traumatic Brain Injury. Journal of Neurotrauma, 21(1), 9–20. [DOI] [PubMed] [Google Scholar]
- Hemphill MA, Dabiri BE, Gabriele S, Kerscher L, Franck C, Josue A, Alford PW, & Parker KK (2011). A Possible Role for Integrin Signaling in Diffuse Axonal Injury. 6(7). 10.1371/journal.pone.0022899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks BK, & Shi R (2014). Mechanisms of neuronal membrane sealing following mechanical trauma. 30(4), 627–644. 10.1007/s12264-013-1446-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez ML, Chatlos T, Gorse KM, & Lafrenaye AD (2019). Neuronal Membrane Disruption Occurs Late Following Diffuse Brain Trauma in Rats and Involves a Subpopulation of NeuN Negative Cortical Neurons. Frontiers in Neurology, 10(November), 1–13. 10.3389/fneur.2019.01238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez M, Patzig J, Mayoral SR, Costa KD, Chan JR, & Casaccia P (2016). Mechanostimulation Promotes Nuclear and Epigenetic Changes in Oligodendrocytes. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 36(3), 806–813. 10.1523/JNEUROSCI.2873-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera JJ, Bockhorst K, Kondraganti S, Stertz L, Quevedo J, & Narayana PA (2017). Acute White Matter Tract Damage after Frontal Mild Traumatic Brain Injury. Journal of Neurotrauma, 34(2), 291–299. 10.1089/neu.2016.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks RR, Smith DH, & McIntosh TK (1995). Temporal response and effects of excitatory amino acid antagonism on microtubule-associated protein 2 immunoreactivity following experimental brain injury in rats. Brain Research, 678(1–2), 151–160. 10.1016/0006-8993(95)00179-t [DOI] [PubMed] [Google Scholar]
- Hiebert JB, Shen Q, Thimmesch AR, & Pierce JD (2015). Traumatic brain injury and mitochondrial dysfunction. The American Journal of the Medical Sciences, 350(2), 132–138. 10.1097/MAJ.0000000000000506 [DOI] [PubMed] [Google Scholar]
- Hill RL, Singh IN, Wang JA, & Hall ED (2017). Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochemistry International, 111, 45–56. 10.1016/j.neuint.2017.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan SR, Phan JH, Alvarado-Velez M, Wang MD, Bellamkonda RV, Fernandez FM, & Laplaca MC (2018). Discovery of Lipidome Alterations Following Traumatic Brain Injury via High-Resolution Metabolomics. Journal of Proteome Research. 10.1021/acs.jproteome.8b00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun P, Parkins NE, Soblosky J, Carey ME, Rodriguez De Turco EB, & Bazan NG (2000). Cortical impact injury in rats promotes a rapid and sustained increase in polyunsaturated free fatty acids and diacylglycerols. Neurochemical Research, 25(2), 269–276. 10.1023/A:1007583806138 [DOI] [PubMed] [Google Scholar]
- Homayoun P, Rodriguez de Turco EB, Parkins NE, Lane DC, Soblosky J, Carey ME, & Bazan NG (1997). Delayed phospholipid degradation in rat brain after traumatic brain injury. Journal of Neurochemistry, 69(1), 199–205. 10.1046/j.1471-4159.1997.69010199.x [DOI] [PubMed] [Google Scholar]
- Hoskison MM, Yanagawa Y, Obata K, & Shuttleworth CW (2007). Calcium-dependent NMDA-induced dendritic injury and MAP2 loss in acute hippocampal slices. Neuroscience, 145(1), 66–79. 10.1016/j.neuroscience.2006.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Rewell SSJ, O’Collins V, Sena ES, van der Worp HB, Traystman RJ, & Macleod MR (2010). Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. Journal of Cerebral Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 30(8), 1412–1431. 10.1038/jcbfm.2010.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Coats JS, Mohd-Yusof A, Yin Y, Assaad S, Muellner MJ, Kamper JE, Hartman RE, Dulcich M, Donovan VM, Oyoyo U, & Obenaus A (2013). Tissue vulnerability is increased following repetitive mild traumatic brain injury in the rat. Brain Research, 1499, 109–120. 10.1016/j.brainres.2012.12.038 [DOI] [PubMed] [Google Scholar]
- Hubbard WB, Joseph B, Spry M, Vekaria HJ, Saatman KE, & Sullivan PG (2019). Acute Mitochondrial Impairment Underlies Prolonged Cellular Dysfunction after Repeated Mild Traumatic Brain Injuries. Journal of Neurotrauma, 36(8), 1252–1263. 10.1089/neu.2018.5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudspeth AJ (1986). The ionic channels of a vertebrate hair cell. Hearing Research, 22(1), 21–27. 10.1016/0378-5955(86)90070-5 [DOI] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen X-H, Taylor AG, Meaney DF, & Smith DH (2004). Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 24(19), 4605–4613. 10.1523/JNEUROSCI.0515-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagielska A, Lowe AL, Makhija E, Wroblewska L, Guck J, Franklin RJM, Shivashankar GV, & Van Vliet KJ (2017). Mechanical Strain Promotes Oligodendrocyte Differentiation by Global Changes of Gene Expression. Frontiers in Cellular Neuroscience, 11, 93 10.3389/fncel.2017.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahed Z, & Mofrad MR (2019). The nucleus feels the force, LINCed in or not! Current Opinion in Cell Biology, 58, 114–119. 10.1016/j.ceb.2019.02.012 [DOI] [PubMed] [Google Scholar]
- Jaudon F, Thalhammer A, & Cingolani LA (2020). Integrin adhesion in brain assembly: From molecular structure to neuropsychiatric disorders. The European Journal of Neuroscience. 10.1111/ejn.14859 [DOI] [PubMed]
- Jin X, Zhu F, Mao H, Shen M, & Yang KH (2013). A comprehensive experimental study on material properties of human brain tissue. Journal of Biomechanics, 46(16), 2795–2801. 10.1016/j.jbiomech.2013.09.001 [DOI] [PubMed] [Google Scholar]
- Johnson CL, Schwarb H, McGarry DJ, Anderson M, A. T, Huesmann GR, Sutton BP, & Cohen NJ (2016). Viscoelasticity of subcortical gray matter structures. Human Brain Mapping, 37(12), 4221–4233. 10.1002/hbm.23314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR, Battle AR, & Martinac B (2019). Remembering Mechanosensitivity of NMDA Receptors. Frontiers in Cellular Neuroscience, 13(December), 1–11. 10.3389/fncel.2019.00533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Meaney DF, Cullen DK, & Smith DH (2015). Animal models of traumatic brain injury. Handbook of Clinical Neurology, 127, 115–128. 10.1080/01425690701737481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, & Stewart W (2013). Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain : A Journal of Neurology, 136(Pt 1), 28–42. 10.1093/brain/aws322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, & Smith DH (2013). Axonal pathology in traumatic brain injury. Experimental Neurology, 246, 35–43. 10.1016/j.expneurol.2012.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang WH, & Morrison B (2015). Functional tolerance to mechanical deformation developed from organotypic hippocampal slice cultures. Biomechanics and Modeling in Mechanobiology, 14(3), 561–575. 10.1007/s10237-014-0622-4 [DOI] [PubMed] [Google Scholar]
- Kao C-Q, Goforth PB, Ellis EF, & Satin LS (2004). Potentiation of GABA(A) currents after mechanical injury of cortical neurons. Journal of Neurotrauma, 21(3), 259–270. 10.1089/089771504322972059 [DOI] [PubMed] [Google Scholar]
- Katayama Y, Becker DP, Tamura T, & Hovda DA (1990). Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. Journal of Neurosurgery, 73(6), 889–900. 10.3171/jns.1990.73.6.0889 [DOI] [PubMed] [Google Scholar]
- Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, & LeBlanc AC (2015). Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death and Differentiation, 22(10), 1676–1686. 10.1038/cdd.2015.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating CE, Browne KD, Duda JE, & Cullen DK (2020). Neurons in Subcortical Oculomotor Regions are Vulnerable to Plasma Membrane Damage after Repetitive Diffuse Traumatic Brain Injury in Swine. Journal of Neurotrauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilinc D, Gallo G, & Barbee KA (2008). Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Experimental Neurology, 212(2), 422–430. 10.1016/j.expneurol.2008.04.025 [DOI] [PubMed] [Google Scholar]
- Kim GH, Kosterin P, Obaid AL, & Salzberg BM (2007). A mechanical spike accompanies the action potential in Mammalian nerve terminals. Biophysical Journal, 92(9), 3122–3129. 10.1529/biophysj.106.103754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Adams AA, Gokina P, Zambrano B, Jayakumaran J, Dobrowolski R, Maurel P, Pfister BJ, & Kim HA (2020). Mechanical stretch induces myelin protein loss in oligodendrocytes by activating Erk1/2 in a calcium-dependent manner. Glia, March, 1–16. 10.1002/glia.23827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Robinson T, Dixon CE, Rao GR, Larnard D, & Nemoto CEM (2010). Brain temperature profiles during epidural cooling with the ChillerPad in a monkey model of traumatic brain injury. Journal of Neurotrauma, 27(10), 1895–1903. 10.1089/neu.2009.1178 [DOI] [PubMed] [Google Scholar]
- Kokiko-Cochran ON, Saber M, Puntambekar S, Bemiller SM, Katsumoto A, Lee Y-S, Bhaskar K, Ransohoff RM, & Lamb BT (2018). Traumatic Brain Injury in hTau Model Mice: Enhanced Acute Macrophage Response and Altered Long-Term Recovery. Journal of Neurotrauma, 35(1), 73–84. 10.1089/neu.2017.5203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, García AJ, Mould AP, Humphries MJ, & Zhu C (2009). Demonstration of catch bonds between an integrin and its ligand. The Journal of Cell Biology, 185(7), 1275–1284. 10.1083/jcb.200810002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkotian E, & Segal M (2006). Spatially confined diffusion of calcium in dendrites of hippocampal neurons revealed by flash photolysis of caged calcium. Cell Calcium, 40(5–6), 441–449. 10.1016/j.ceca.2006.08.008 [DOI] [PubMed] [Google Scholar]
- Kruse SA, Rose GH, Glaser KJ, Manduca A, Felmlee JP, Jr CRJ, & Ehman RL (2008). Magnetic resonance elastography of the brain. 39, 231–237. 10.1016/j.neuroimage.2007.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbe JR, & Hall ED (2017). Chronic traumatic encephalopathy-integration of canonical traumatic brain injury secondary injury mechanisms with tau pathology. Progress in Neurobiology, 158, 15–44. 10.1016/j.pneurobio.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenaye AD, Todani M, Walker SA, & Povlishock JT (2015). Microglia processes associate with diffusely injured axons following mild traumatic brain injury in the micro pig. Journal of Neuroinflammation, 12(1), 186 10.1186/s12974-015-0405-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlaca MC, Cullen DK, McLoughlin JJ, & Cargill RS 2nd. (2005). High rate shear strain of three-dimensional neural cell cultures: a new in vitro traumatic brain injury model. Journal of Biomechanics, 38(5), 1093–1105. 10.1016/j.jbiomech.2004.05.032 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Lee VM, & Thibault LE (1997). An in vitro model of traumatic neuronal injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. Journal of Neurotrauma, 14(6), 355–368. 10.1089/neu.1997.14.355 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Lessing MC, Prado GR, Zhou R, Tate CC, Geddes-Klein D, Meaney DF, & Zhang L (2019). Mechanoporation is a potential indicator of tissue strain and subsequent degeneration following experimental traumatic brain injury. Clinical Biomechanics, 64(April), 2–13. 10.1016/j.clinbiomech.2018.05.016 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Prado GR, Cullen DK, & Irons HR (2006). High rate shear insult delivered to cortical neurons produces heterogeneous membrane permeability alterations. Annual International Conference of the IEEE Engineering in Medicine and Biology - Proceedings, 2384–2387. 10.1109/IEMBS.2006.260633 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Prado GR, Cullen D, & Simon CM (2009). Plasma membrane damage as a marker of neuronal injury. Conference Proceedings : … Annual International Conference of the IEEE Engineering in Medicine and Biology Society. IEEE Engineering in Medicine and Biology Society. Conference, 2009, 1113–1116. 10.1109/IEMBS.2009.5334457 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, & Thibault LE (1997). An in vitro traumatic injury model to examine the response of neurons to a hydrodynamically-induced deformation. Annals of Biomedical Engineering, 25(4), 665–677. 10.1007/BF02684844 [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, & Thibault LE (1998). Dynamic mechanical deformation of neurons triggers an acute calcium response and cell injury involving the N-methyl-D-aspartate glutamate receptor. Journal of Neuroscience Research, 52(2), 220–229. [DOI] [PubMed] [Google Scholar]
- Laurer HL, Bareyre FM, Lee VMYC, Trojanowski JQ, Longhi L, Hoover R, Saatman KE, Raghupathi R, Hoshino S, Grady MS, & McIntosh TK (2001). Mild head injury increasing the brain’s vulnerability to a second concussive impact. Journal of Neurosurgery, 95(5), 859–870. 10.3171/jns.2001.95.5.0859 [DOI] [PubMed] [Google Scholar]
- Lee C, Lou J, Wen K, McKane M, Eskin SG, Ono S, Chien S, Rubenstein PA, Zhu C, & McIntire LV (2013). Actin depolymerization under force is governed by lysine 113:glutamic acid 195-mediated catch-slip bonds. Proceedings of the National Academy of Sciences of the United States of America, 110(13), 5022–5027. 10.1073/pnas.1218407110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-M, Nguyen T-H, Na K, Cho I-J, Woo DH, Oh J-E, Lee CJ, & Yoon E-S (2015). Nanomechanical measurement of astrocyte stiffness correlated with cytoskeletal maturation. Journal of Biomedical Materials Research. Part A, 103(1), 365–370. 10.1002/jbm.a.35174 [DOI] [PubMed] [Google Scholar]
- Leite SC, & Sousa MM (2016). The neuronal and actin commitment: Why do neurons need rings? Cytoskeleton, 73(9), 424–434. 10.1002/cm.21273 [DOI] [PubMed] [Google Scholar]
- Leyton L, Schneider P, Labra CV, Rüegg C, Hetz CA, Quest AF, & Bron C (2001). Thy-1 binds to integrin beta(3) on astrocytes and triggers formation of focal contact sites. Current Biology : CB, 11(13), 1028–1038. 10.1016/s0960-9822(01)00262-7 [DOI] [PubMed] [Google Scholar]
- Li S, Hafeez A, Noorulla F, Geng X, Shao G, Ren C, Lu G, Zhao H, Ding Y, & Ji X (2017). Preconditioning in neuroprotection: From hypoxia to ischemia. Progress in Neurobiology, 157, 79–91. 10.1016/j.pneurobio.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libertiaux V, Pascon F, & Cescotto S (2011). Experimental verification of brain tissue incompressibility using digital image correlation. Journal of the Mechanical Behavior of Biomedical Materials, 4(7), 1177–1185. 10.1016/j.jmbbm.2011.03.028 [DOI] [PubMed] [Google Scholar]
- Lighthall JW (1988). Controlled cortical impact: a new experimental brain injury model. Journal of Neurotrauma, 5(1), 1–15. 10.1089/neu.1988.5.1 [DOI] [PubMed] [Google Scholar]
- Lim CG, Jang J, & Kim C (2018). Cellular machinery for sensing mechanical force. BMB Reports, 51(12), 623–629. 10.5483/BMBRep.2018.51.12.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Frieboes LS, Forootan M, Palispis WA, Mozaffar T, Jafari M, Steward O, Gall CM, & Gupta R (2012). Biophysical stimulation induces demyelination via an integrin-dependent mechanism. Annals of Neurology, 72(1), 112–123. 10.1002/ana.23592 [DOI] [PubMed] [Google Scholar]
- Lindgren S, & Rinder L (1966). Experimental studies in head injury. II. Pressure propagation in “percussion concussion”. Biophysik, 3(2), 174–180. 10.1007/BF01191611 [DOI] [PubMed] [Google Scholar]
- Loane DJ, & Kumar A (2016). Microglia in the TBI brain: The good, the bad, and the dysregulated. Experimental Neurology, 275 Pt 3(0 3), 316–327. 10.1016/j.expneurol.2015.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loane DJ, Stoica BA, & Faden AI (2015). Neuroprotection for traumatic brain injury. Handbook of Clinical Neurology, 127, 343–366. 10.1016/B978-0-444-52892-6.00022-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhi L, Saatman KE, Fujimoto S, Raghupathi R, Meaney DF, Davis J, McMillan A, Conte V, Laurer HL, Stein S, Stocchetti N, & McIntosh TK (2005). Temporal window of vulnerability to repetitive experimental concussive brain injury. Neurosurgery, 56(2), 364–373. 10.1227/01.NEU.0000149008.73513.44 [DOI] [PubMed] [Google Scholar]
- Lu YB, Franze K, Seifert G, Steinhäuser C, Kirchhoff F, Wolburg H, Guck J, Janmey P, Wei EQ, Käs J, & Reichenbach A (2006). Viscoelastic properties of individual glial cells and neurons in the CNS. Proceedings of the National Academy of Sciences of the United States of America, 103(47), 17759–17764. 10.1073/pnas.0606150103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K (2000). Cytoarchitecture and physical properties of cytoplasm: volume, viscosity, diffusion, intracellular surface area. International Review of Cytology, 192, 189–221. 10.1016/s0074-7696(08)60527-6 [DOI] [PubMed] [Google Scholar]
- Ma M (2013). Role of calpains in the injury-induced dysfunction and degeneration of the mammalian axon. Neurobiology of Disease, 60, 61–79. 10.1016/j.nbd.2013.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacManus DB, Pierrat B, Murphy JG, & Gilchrist MD (2017a). A viscoelastic analysis of the P56 mouse brain under large-deformation dynamic indentation. Acta Biomaterialia, 48, 309–318. 10.1016/j.actbio.2016.10.029 [DOI] [PubMed] [Google Scholar]
- MacManus DB, Pierrat B, Murphy JG, & Gilchrist MD (2017b). Region and species dependent mechanical properties of adolescent and young adult brain tissue. Scientific Reports, 7(1), 1–12. 10.1038/s41598-017-13727-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegele M, Engel D, Bouillon B, Lefering R, Fach H, Raum M, Buchheister B, Schaefer U, Klug N, & Neugebauer E (2007). Incidence and outcome of traumatic brain injury in an urban area in Western Europe over 10 years. European Surgical Research., 39(6), 372–379. 10.1159/000107097 [DOI] [PubMed] [Google Scholar]
- Maegele M, Lefering R, Sakowitz O, Kopp MA, Schwab JM, Steudel W-I, Unterberg A, Hoffmann R, Uhl E, & Marzi I (2019). The Incidence and Management of Moderate to Severe Head Injury. Deutsches Arzteblatt International, 116(10), 167–173. 10.3238/arztebl.2019.0167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson J, Leonessa F, & Ling GSF (2012). Neuropathology of explosive blast traumatic brain injury. Current Neurology and Neuroscience Reports, 12(5), 570–579. 10.1007/s11910-012-0303-6 [DOI] [PubMed] [Google Scholar]
- Magou GC, Pfister BJ, & Berlin JR (2015). Effect of acute stretch injury on action potential and network activity of rat neocortical neurons in culture. Brain Research, 1624, 525–535. 10.1016/j.brainres.2015.07.056 [DOI] [PubMed] [Google Scholar]
- Makhija EP, Espinosa-Hoyos D, Jagielska A, & Van Vliet KJ (2020). Mechanical regulation of oligodendrocyte biology. Neuroscience Letters, 717(December 2019), 134673 10.1016/j.neulet.2019.134673 [DOI] [PubMed] [Google Scholar]
- Makhija EP, Jagielska A, Zhu L, Bost AC, Ong W, Chew SY, Shivashankar GV, & Van Vliet KJ (2018). Mechanical Strain Alters Cellular and Nuclear Dynamics at Early Stages of Oligodendrocyte Differentiation. Frontiers in Cellular Neuroscience, 12, 59 10.3389/fncel.2018.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallah K, Quanico J, Trede D, Kobeissy F, Zibara K, Salzet M, & Fournier I (2018). Lipid Changes Associated with Traumatic Brain Injury Revealed by 3D MALDI-MSI [Research-article]. Analytical Chemistry, 90(17), 10568–10576. 10.1021/acs.analchem.8b02682 [DOI] [PubMed] [Google Scholar]
- Maneshi MM, Maki B, Gnanasambandam R, Belin S, Popescu GK, Sachs F, & Hua SZ (2017). Mechanical stress activates NMDA receptors in the absence of agonists. Scientific Reports, 7(November 2016), 1–10. 10.1038/srep39610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneshi MM, Sachs F, & Hua SZ (2018). Heterogeneous cytoskeletal force distribution delineates the onset Ca2+ influx under fluid shear stress in astrocytes. Frontiers in Cellular Neuroscience, 12(March), 1–9. 10.3389/fncel.2018.00069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Rosenthal G, Lam M, Morabito D, Yan D, Derugin N, Bollen A, Knudson MM, & Panter SS (2006). Controlled cortical impact in swine: pathophysiology and biomechanics. Journal of Neurotrauma, 23(2), 128–139. 10.1089/neu.2006.23.128 [DOI] [PubMed] [Google Scholar]
- Marion CM, Radomski KL, Cramer NP, Galdzicki Z, & Armstrong RC (2018). Experimental Traumatic Brain Injury Identifies Distinct Early and Late Phase Axonal Conduction Deficits of White Matter Pathophysiology, and Reveals Intervening Recovery. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 38(41), 8723–8736. 10.1523/JNEUROSCI.0819-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmarou A, Foda MA, van den Brink W, Campbell J, Kita H, & Demetriadou K (1994). A new model of diffuse brain injury in rats. Part I: Pathophysiology and biomechanics. Journal of Neurosurgery, 80(2), 291–300. 10.3171/jns.1994.80.2.0291 [DOI] [PubMed] [Google Scholar]
- Martinac B, Buechner M, Delcour AH, Adler J, & Kung C (1987). Pressure-sensitive ion channel in Escherichia coli. Proceedings of the National Academy of Sciences, 84(8), 2297 LP–2301. 10.1073/pnas.84.8.2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Mbemba D, Mugikura S, Nakagawa A, Murata T, Ishii K, Kushimoto S, Tominaga T, Takahashi S, & Takase K (2018). Traumatic midline subarachnoid hemorrhage on initial computed tomography as a marker of severe diffuse axonal injury. Journal of Neurosurgery, 129(5), 1317–1324. 10.3171/2017.6.JNS17466 [DOI] [PubMed] [Google Scholar]
- Mata-Mbemba D, Mugikura S, Nakagawa A, Murata T, Kato Y, Tatewaki Y, Li L, Takase K, Ishii K, Kushimoto S, Tominaga T, & Takahashi S (2015). Intraventricular hemorrhage on initial computed tomography as marker of diffuse axonal injury after traumatic brain injury. Journal of Neurotrauma, 32(5), 359–365. 10.1089/neu.2014.3453 [DOI] [PubMed] [Google Scholar]
- Maxwell WL (1996). Histopathological changes at central nodes of Ranvier after stretch-injury. Microscopy Research and Technique, 34(6), 522–535. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, & Graham DI (1997). Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. Journal of Neurotrauma, 14(9), 603–614. 10.1089/neu.1997.14.603 [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Irvine A, Graham, Adams JH, Gennarelli TA, Tipperman R, & Sturatis M (1991). Focal axonal injury: the early axonal response to stretch. Journal of Neurocytology, 20(3), 157–164. 10.1007/BF01186989 [DOI] [PubMed] [Google Scholar]
- McColl TJ, Brady RD, Shultz SR, Lovick L, Webster KM, Sun M, McDonald SJ, O’Brien TJ, & Semple BD (2018). Mild Traumatic Brain Injury in Adolescent Mice Alters Skull Bone Properties to Influence a Subsequent Brain Impact at Adulthood: A Pilot Study. Frontiers in Neurology, 9, 372 10.3389/fneur.2018.00372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL, Wang KKW, & Povlishock JT (2009). Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. Journal of Neuropathology and Experimental Neurology, 68(3), 241–249. 10.1097/NEN.0b013e3181996bfe [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn MJ, & Povlishock JT (2015). Cellular and molecular mechanisms of injury and spontaneous recovery In Grafman J & Salazar AM (Eds.), Handbook of Clinical Neurology (Vol. 127). 10.1016/B978-0-444-52892-6.00005-2 [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Saatman KE, Raghupathi R, Graham DI, Smith DH, Lee VM, & Trojanowski JQ (1998). The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. In Neuropathology and applied neurobiology (Vol. 24, Issue 4, pp. 251–267). [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, & Faden AL (1989). Traumatic brain injury in the rat: characterization of a lateral fluid-percussion model. Neuroscience, 28(1), 233–244. 10.1016/0306-4522(89)90247-9 [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee H-S, Kubilus CA, & Stern RA (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. Journal of Neuropathology and Experimental Neurology, 68(7), 709–735. 10.1097/NEN.0b013e3181a9d503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney DF, & Cullen DK (2016). Biomechanical Basis of Traumatic Brain Injury In Winn HR (Ed.), Youmans and Winn Neurological Surgery (7th ed., pp. 2755–2764). Elsevier. [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, & Gennarelli TA (1995). Biomechanical analysis of experimental diffuse axonal injury. Journal of Neurotrauma, 12(4), 689–694. 10.1089/neu.1995.12.689 [DOI] [PubMed] [Google Scholar]
- Miller WJ, Leventhal I, Scarsella D, Haydon PG, Janmey P, & Meaney DF (2009). Mechanically induced reactive gliosis causes ATP-mediated alterations in astrocyte stiffness. Journal of Neurotrauma, 26(5), 789–797. 10.1089/neu.2008.0727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeendarbary E, Weber IP, Sheridan GK, Koser DE, Soleman S, Haenzi B, Bradbury EJ, Fawcett J, & Franze K (2017). The soft mechanical signature of glial scars in the central nervous system. Nature Communications, 8, 14787 10.1038/ncomms14787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnerie H, Tang-Schomer MD, Iwata A, Smith DH, Kim HA, & Le Roux PD (2010). Dendritic alterations after dynamic axonal stretch injury in vitro. Experimental Neurology, 224(2), 415–423. 10.1016/j.expneurol.2010.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monson KL, Converse MI, & Manley GT (2019). Cerebral blood vessel damage in traumatic brain injury. Clinical Biomechanics, 64(January 2018), 98–113. 10.1016/j.clinbiomech.2018.02.011 [DOI] [PubMed] [Google Scholar]
- Montanino A, & Kleiven S (2018). Utilizing a structural mechanics approach to assess the primary effects of injury loads onto the axon and its components. Frontiers in Neurology, 9(August), 1–12. 10.3389/fneur.2018.00643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanino A, Saeedimasine M, Villa A, & Kleiven S (2020). Localized Axolemma Deformations Suggest Mechanoporation as Axonal Injury Trigger. 11(January), 1–14. 10.3389/fneur.2020.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SW, Roca-Cusachs P, & Sheetz MP (2010). Stretchy proteins on stretchy substrates: the important elements of integrin-mediated rigidity sensing. Developmental Cell, 19(2), 194–206. 10.1016/j.devcel.2010.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MM, Xu H, & Clapham DE (2004). TRP ion channels in the nervous system. Current Opinion in Neurobiology, 14(3), 362–369. 10.1016/j.conb.2004.05.003 [DOI] [PubMed] [Google Scholar]
- Moshayedi P, Ng G, Kwok JCF, Yeo GSH, Bryant CE, Fawcett JW, Franze K, & Guck J (2014). The relationship between glial cell mechanosensitivity and foreign body reactions in the central nervous system. Biomaterials, 35(13), 3919–3925. 10.1016/j.biomaterials.2014.01.038 [DOI] [PubMed] [Google Scholar]
- Murphy MC, Huston J 3rd, & Ehman RL (2019). MR elastography of the brain and its application in neurological diseases. NeuroImage, 187, 176–183. 10.1016/j.neuroimage.2017.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MC Iii, J. H Jr, C. RJ, Glaser KJ, Senjem ML, Manduca A, Felmlee JP, & Ehman RL (2013). Measuring the Characteristic Topography of Brain Stiffness with Magnetic Resonance Elastography. 8(12), 1–14. 10.1371/journal.pone.0081668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AG, Wang JA, Carrico KM, & Hall ED (2011). Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. Journal of Neurochemistry, 117(3), 579–588. 10.1111/j.1471-4159.2011.07228.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza P, Bachmeier C, Mouzon B, Algamal M, Rafi NG, Lungmus C, Abdullah L, Evans JE, Ferguson S, Mullan M, Crawford F, & Ojo JO (2019). APOE Genotype Specific Effects on the Early Neurodegenerative Sequelae Following Chronic Repeated Mild Traumatic Brain Injury. Neuroscience, 404, 297–313. 10.1016/j.neuroscience.2019.01.049 [DOI] [PubMed] [Google Scholar]
- Mychasiuk R, Hehar H, Van Waes L, & Esser MJ (2015). Diet, age, and prior injury status differentially alter behavioral outcomes following concussion in rats. Neurobiology of Disease, 73, 1–11. 10.1016/j.nbd.2014.09.003 [DOI] [PubMed] [Google Scholar]
- Nagendran T, & Taylor AM (2019). Unique Axon-to-Soma Signaling Pathways Mediate Dendritic Spine Loss and Hyper-Excitability Post-axotomy. Frontiers in Cellular Neuroscience, 13, 431 10.3389/fncel.2019.00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa N, & Kengaku M (2020). Mechanical Regulation of Nuclear Translocation in Migratory Neurons. Frontiers in Cell and Developmental Biology, 8, 150 10.3389/fcell.2020.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA, & Wellington CL (2014). Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Molecular Neurodegeneration, 9, 55 10.1186/1750-1326-9-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Tran M, & Feld J (2005). Traumatic Injury Activates Protein Kinase B/Akt in Cultured Astrocytes: Role of Extracellular ATP and P2 Purinergic Receptors. Journal of Neurotrauma, 22(4), 491–500. [DOI] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Willoughby KA, & Ellis EF (2003). Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 23(6), 2348–2356. 10.1523/JNEUROSCI.23-06-02348.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, & Syková E (1998). Extracellular space structure revealed by diffusion analysis. Trends in Neurosciences, 21(5), 207–215. 10.1016/S0166-2236(98)01261-2 [DOI] [PubMed] [Google Scholar]
- Nikolaev YA, Dosen PJ, Laver DR, Van Helden DF, & Hamill OP (2015). Single mechanically-gated cation channel currents can trigger action potentials in neocortical and hippocampal pyramidal neurons. Brain Research, 1608, 1–13. 10.1016/j.brainres.2015.02.051 [DOI] [PubMed] [Google Scholar]
- O’Donnell JC, Browne KD, Kilbaugh TJ, Chen HI, Whyte J, & Cullen DK (2018). Challenges and Demand for Modeling Disorders of Consciousness following Traumatic Brain Injury. Neuroscience & Biobehavioral Reviews, December, 0–1. 10.1016/j.neubiorev.2018.12.015 [DOI] [PMC free article] [PubMed]
- Ofek G, Wiltz DC, & Athanasiou KA (2009). Contribution of the cytoskeleton to the compressive properties and recovery behavior of single cells. Biophysical Journal, 97(7), 1873–1882. 10.1016/j.bpj.2009.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ommaya AK, & Gennarelli TA (1974). Cerebral concussion and traumatic unconsciousness. Brain, 97(4), 633–654. [DOI] [PubMed] [Google Scholar]
- Ommaya AK, Goldsmith W, & Thibault L (2002). Biomechanics and neuropathology of adult and paediatric head injury. British Journal of Neurosurgery, 16(3), 220–242. 10.1080/0268869022014882 [DOI] [PubMed] [Google Scholar]
- Ommaya AK, Hirsch AE, Flamm ES, & Mahone RH (1966). Cerebral concussion in the monkey: an experimental model. Science (New York, N.Y.), 153(3732), 211–212. 10.1126/science.153.3732.211 [DOI] [PubMed] [Google Scholar]
- Ommaya AK, Yarnell P, Hirsch AE, & Harris EH (1967, February). Scaling of Experimental Data on Cerebral Concussion in Sub-Human Primates to Concussion Threshold for Man. 11th Stapp Car Crash Conference (1967). 10.4271/670906 [DOI]
- Ostrow LW, & Sachs F (2005). Mechanosensation and endothelin in astrocytes - Hypothetical roles in CNS pathophysiology. Brain Research Reviews, 48(3), 488–508. 10.1016/j.brainresrev.2004.09.005 [DOI] [PubMed] [Google Scholar]
- Ouyang H, Nauman E, & Shi R (2013). Contribution of cytoskeletal elements to the axonal mechanical properties. Journal of Biological Engineering, 7(1), 21 10.1186/1754-1611-7-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TP, Ventre SC, & Meaney DF (2012). Dynamic changes in neural circuit topology following mild mechanical injury in vitro. Annals of Biomedical Engineering, 40(1), 23–36. 10.1007/s10439-011-0390-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus EH, Christman CW, Giebel ML, & Povlishock JT (1994). Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. Journal of Neurotrauma, 11, 507–522. 10.1089/neu.1994.11.507 [DOI] [PubMed] [Google Scholar]
- Piao L, Lee H, Park C-K, Cho I-H, Piao ZG, Jung SJ, Choi S-Y, Lee SJ, Park K, Kim J-S, & Oh SB (2006). Mechanosensitivity of voltage-gated K+ currents in rat trigeminal ganglion neurons. Journal of Neuroscience Research, 83(7), 1373–1380. 10.1002/jnr.20810 [DOI] [PubMed] [Google Scholar]
- Pijet B, Stefaniuk M, & Kaczmarek L (2019). MMP-9 Contributes to Dendritic Spine Remodeling Following Traumatic Brain Injury. Neural Plasticity, 2019, 3259295 10.1155/2019/3259295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Liu SJ, Heck K, Taft WC, Clifton GL, & Hayes RL (1996). Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. Journal of Neuropathology and Experimental Neurology, 55(1), 68–80. 10.1097/00005072-199601000-00007 [DOI] [PubMed] [Google Scholar]
- Prado GR, Ross JD, DeWeerth SP, & LaPlaca MC (2005). Mechanical trauma induces immediate changes in neuronal network activity. Journal of Neural Engineering, 2(4), 148–158. 10.1088/1741-2560/2/4/011 [DOI] [PubMed] [Google Scholar]
- Prange MT, & Margulies SS (2002). Age-Dependent Properties of the Brain Undergoing Large Deformation. Journal of Biomechanical Engineering, 124(April), 244–252. 10.1115/1.1449907 [DOI] [PubMed] [Google Scholar]
- Previtali SC, Feltri ML, Archelos JJ, Quattrini A, Wrabetz L, & Hartung H (2001). Role of integrins in the peripheral nervous system. Progress in Neurobiology, 64(1), 35–49. 10.1016/s0301-0082(00)00045-9 [DOI] [PubMed] [Google Scholar]
- Prins ML, Hales A, Reger M, Giza CC, & Hovda DA (2010). Repeat traumatic brain injury in the juvenile rat is associated with increased axonal injury and cognitive impairments. Developmental Neuroscience, 32(5–6), 510–518. 10.1159/000316800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins Mayumi L., Alexander D, Giza CC, & Hovda DA (2013). Repeated mild traumatic brain injury: mechanisms of cerebral vulnerability. Journal of Neurotrauma, 30(1), 30–38. 10.1089/neu.2012.2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Guo J, Kala S, Zhu J, Xian Q, Qiu W, Li G, Zhu T, Meng L, Zhang R, Chan HC, Zheng H, & Sun L (2019). The Mechanosensitive Ion Channel Piezo1 Significantly Mediates In Vitro Ultrasonic Stimulation of Neurons. IScience, 21, 448–457. 10.1016/j.isci.2019.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R (2004). Cell death mechanisms following traumatic brain injury. Brain Pathology (Zurich, Switzerland), 14(2), 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakshit S, Zhang Y, Manibog K, Shafraz O, & Sivasankar S (2012). Ideal, catch, and slip bonds in cadherin adhesion. Proceedings of the National Academy of Sciences of the United States of America, 109(46), 18815–18820. 10.1073/pnas.1208349109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Greer JE, Vanderveer AS, & Phillips LL (2010). Proteolysis of submembrane cytoskeletal proteins ankyrin-G and αII-spectrin following diffuse brain injury: a role in white matter vulnerability at Nodes of Ranvier. Brain Pathology (Zurich, Switzerland), 20(6), 1055–1068. 10.1111/j.1750-3639.2010.00412.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Phillips LL, & Povlishock JT (2005). Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Experimental Neurology, 196(1), 126–137. 10.1016/j.expneurol.2005.07.014 [DOI] [PubMed] [Google Scholar]
- Reeves TM, Smith TL, Williamson JC, & Phillips LL (2012). Unmyelinated axons show selective rostrocaudal pathology in the corpus callosum after traumatic brain injury. Journal of Neuropathology and Experimental Neurology, 71(3), 198–210. 10.1097/NEN.0b013e3182482590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid AY, Bragin A, Giza CC, Staba RJ, & Engel JJ (2016). The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia, 57(10), 1558–1567. 10.1111/epi.13486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinder L (1969). “Concussive response” and intracranial pressure changes at sudden extradural fluid volume input in rabbits. Acta Physiologica Scandinavica, 76(3), 352–360. 10.1111/j.1748-1716.1969.tb04478.x [DOI] [PubMed] [Google Scholar]
- Romero LO, Massey AE, Mata-Daboin AD, Sierra-Valdez FJ, Chauhan SC, Cordero-Morales JF, & Vásquez V (2019). Dietary fatty acids fine-tune Piezo1 mechanical response. Nature Communications, 10(1), 1–14. 10.1038/s41467-019-09055-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, Balaban CD, Barbacci D, Schultz JA, Gouty S, Cox BM, & Woods AS (2016). Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. Journal of Neuroscience Methods, 272(2015), 1–14. 10.1016/j.jneumeth.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowson B, Tyson A, Rowson S, & Duma S (2018). Measuring head impacts: accelerometers and other sensors. Handbook of Clinical Neurology, 158, 235–243. 10.1016/B978-0-444-63954-7.00023-9 [DOI] [PubMed] [Google Scholar]
- Ruoslahti E (1996). Brain extracellular matrix. Glycobiology, 6(5), 489–492. 10.1093/glycob/6.5.489 [DOI] [PubMed] [Google Scholar]
- Saatman KE, Duhaime A-C, Bullock R, Maas AIR, Valadka A, & Manley GT (2008). Classification of traumatic brain injury for targeted therapies. Journal of Neurotrauma, 25(7), 719–738. 10.1089/neu.2008.0586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabet AA, Christoforou E, Zatlin B, Genin GM, & Bayly PV (2008). Deformation of the human brain induced by mild angular head acceleration. Journal of Biomechanics, 41(2), 307–315. 10.1016/j.jbiomech.2007.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack I, Streitberger K-J, Krefting D, Paul F, & Braun J (2011). The influence of physiological aging and atrophy on brain viscoelastic properties in humans. PloS One, 6(9), e23451 10.1371/journal.pone.0023451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeedimasine M, Montanino A, Kleiven S, & Villa A (2019). Role of lipid composition on the structural and mechanical features of axonal membranes : a molecular simulation study. Scientific Reports, May, 1–12. 10.1038/s41598-019-44318-9 [DOI] [PMC free article] [PubMed]
- Sauerbeck AD, Fanizzi C, Kim JH, Gangolli M, Bayly PV, Wellington CL, Brody DL, & Kummer TT (2018). ModCHIMERA: A novel murine closed-head model of moderate traumatic brain injury. Scientific Reports, 8(1), 1–17. 10.1038/s41598-018-25737-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch KM, Madathil SK, & Saatman KE (2012). Genetic manipulation of cell death and neuroplasticity pathways in traumatic brain injury. Neurotherapeutics : The Journal of the American Society for Experimental NeuroTherapeutics, 9(2), 323–337. 10.1007/s13311-012-0107-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel M, Neumann B, Hill MFE, Weber IP, Viscomi C, Zhao C, Young A, Agley CC, Thompson AJ, Gonzalez GA, Sharma A, Holmqvist S, Rowitch DH, Franze K, Franklin RJM, & Chalut KJ (2019). Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature, 573(7772), 130–134. 10.1038/s41586-019-1484-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwyn RG, Cooney SJ, Khayrullina G, Hockenbury N, Wilson CM, Jaiswal S, Bermudez S, Armstrong RC, & Byrnes KR (2016). Outcome after Repetitive Mild Traumatic Brain Injury Is Temporally Related to Glucose Uptake Profile at Time of Second Injury. Journal of Neurotrauma, 33(16), 1479–1491. 10.1089/neu.2015.4129 [DOI] [PubMed] [Google Scholar]
- Shandra O, Winemiller AR, Heithoff BP, Munoz-Ballester C, George KK, Benko MJ, Zuidhoek IA, Besser MN, Curley DE, Edwards GF 3rd, Mey A, Harrington AN, Kitchen JP, & Robel S (2019). Repetitive Diffuse Mild Traumatic Brain Injury Causes an Atypical Astrocyte Response and Spontaneous Recurrent Seizures. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 39(10), 1944–1963. 10.1523/JNEUROSCI.1067-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira Y, Shohami E, Sidi A, Soffer D, Freeman S, & Cotev S (1988). Experimental closed head injury in rats: mechanical, pathophysiologic, and neurologic properties. Critical Care Medicine, 16(3), 258–265. 10.1097/00003246-198803000-00010 [DOI] [PubMed] [Google Scholar]
- Sharp DJ, Scott G, & Leech R (2014). Network dysfunction after traumatic brain injury. Nature Reviews. Neurology, 10(3), 156–166. 10.1038/nrneurol.2014.15 [DOI] [PubMed] [Google Scholar]
- Shcherbatko A, Ono F, Mandel G, & Brehm P (1999). Voltage-dependent sodium channel function is regulated through membrane mechanics. Biophysical Journal, 77(4), 1945–1959. 10.1016/S0006-3495(99)77036-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Osanai Y, Tanaka KF, Thai TQ, Abe M, Natsume R, Sakimura K, & Ikenaka K (2019). Mechanical regulation of oligodendrocyte morphology and maturation by the mechanosensor p130Cas. Journal of Neurochemistry, 150(2), 158–172. 10.1111/jnc.14657 [DOI] [PubMed] [Google Scholar]
- Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, & Borlongan CV (2010). Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience, 171(4), 1273–1282. 10.1016/j.neuroscience.2010.10.018 [DOI] [PubMed] [Google Scholar]
- Shreiber DI, Hao H, & Elias RAI (2009). Probing the influence of myelin and glia on the tensile properties of the spinal cord. Biomechanics and Modeling in Mechanobiology, 8(4), 311–321. 10.1007/s10237-008-0137-y [DOI] [PubMed] [Google Scholar]
- Simon DW, McGeachy MJ, Bayır H, Clark RSB, Loane DJ, & Kochanek PM (2017). The far-reaching scope of neuroinflammation after traumatic brain injury. Nature Reviews Neurology. 10.1038/nrneurol.2017.13 [DOI] [PMC free article] [PubMed]
- Singleton RH, & Povlishock JT (2004). Identification and Characterization of Heterogeneous Neuronal Injury and Death in Regions of Diffuse Brain Injury: Evidence for Multiple Independent Injury Phenotypes. Journal of Neuroscience, 24(14), 3543–3553. 10.1523/JNEUROSCI.5048-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slemmer JE, & Weber JT (2005). The extent of damage following repeated injury to cultured hippocampal cells is dependent on the severity of insult and inter-injury interval. Neurobiology of Disease, 18(3), 421–431. 10.1016/j.nbd.2004.09.022 [DOI] [PubMed] [Google Scholar]
- Smith DH, Johnson VE, & Stewart W (2013). Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nature Reviews. Neurology, 9(4), 211–221. 10.1038/nrneurol.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, & Meaney DF (2000). Axonal Damage in Traumatic Brain Injury. The Neuroscientist, 6(6), 483–495. 10.1177/107385840000600611 [DOI] [Google Scholar]
- Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, & Meaney DF (2000). Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. Journal of Neurosurgery, 93(2), 315–322. 10.3171/jns.2000.93.2.0315 [DOI] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, & McIntosh TK (1995). A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. Journal of Neurotrauma, 12(2), 169–178. 10.1089/neu.1995.12.169 [DOI] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, & Meaney DF (1999). High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 19(11), 4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokurenko EV, Vogel V, & Thomas WE (2008). Catch-bond mechanism of force-enhanced adhesion: counterintuitive, elusive, but … widespread? Cell Host & Microbe, 4(4), 314–323. 10.1016/j.chom.2008.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni N, Vegh V, To XV, Mohamed AZ, Borges K, & Nasrallah FA (2020). Combined Diffusion Tensor Imaging and Quantitative Susceptibility Mapping Discern Discrete Facets of White Matter Pathology Post-injury in the Rodent Brain. Frontiers in Neurology, 11, 153 10.3389/fneur.2020.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaethling J, Le L, & Meaney DF (2012). NMDA receptor mediated phosphorylation of GluR1 subunits contributes to the appearance of calcium-permeable AMPA receptors after mechanical stretch injury. Neurobiology of Disease, 46(3), 646–654. 10.1016/j.nbd.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal JA, & Vickers JC (2011). Selective vulnerability of non-myelinated axons to stretch injury in an in vitro co-culture system. Journal of Neurotrauma, 28(5), 841–847. 10.1089/neu.2010.1658 [DOI] [PubMed] [Google Scholar]
- Stoica BA, & Faden AI (2010). Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics : The Journal of the American Society for Experimental NeuroTherapeutics, 7(1), 3–12. 10.1016/j.nurt.2009.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DO, Dialo AO, Rubin DG, Mutlu LK, Povlishock JT, & Helm GA (2004). Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Experimental Neurology, 190(1), 59–69. 10.1016/j.expneurol.2004.05.022 [DOI] [PubMed] [Google Scholar]
- Straube R, & Ridgway D (2009). Investigating the effects of molecular crowding on Ca2+ diffusion using a particle-based simulation model. Chaos (Woodbury, N.Y.), 19(3), 37110 10.1063/1.3207820 [DOI] [PubMed] [Google Scholar]
- Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, & Wist AO (1976). Fluid-percussion model of mechanical brain injury in the cat. Journal of Neurosurgery, 45(5), 521–534. [PubMed] [Google Scholar]
- Taft WC, Yang K, Dixon CE, & Hayes RL (1992). Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. Journal of Neurotrauma, 9(3), 281–290. 10.1089/neu.1992.9.281 [DOI] [PubMed] [Google Scholar]
- Tajik A, Zhang Y, Wei F, Sun J, Jia Q, Zhou W, Singh R, Khanna N, Belmont AS, & Wang N (2016). Transcription upregulation via force-induced direct stretching of chromatin. Nature Materials, 15(12), 1287–1296. 10.1038/nmat4729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, & Smith DH (2012). Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Experimental Neurology, 233(1), 364–372. 10.1016/j.expneurol.2011.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, & Smith DH (2010). Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 24(5), 1401–1410. 10.1096/fj.09-142844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalin SJ, Ellis EF, & Satin LS (1995). Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. Journal of Neurophysiology, 74(6), 2767–2773. 10.1152/jn.1995.74.6.2767 [DOI] [PubMed] [Google Scholar]
- Teasdale G, & Jennett B (1974). ASSESSMENT OF COMA AND IMPAIRED CONSCIOUSNESS: A Practical Scale. The Lancet, 304(7872), 81–84. 10.1016/S0140-6736(74)91639-0 [DOI] [PubMed] [Google Scholar]
- Theadom A, Starkey NJ, Dowell T, Hume PA, Kahan M, McPherson K, & Feigin V (2014). Sports-related brain injury in the general population: an epidemiological study. Journal of Science and Medicine in Sport, 17(6), 591–596. 10.1016/j.jsams.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Thibault LE, Meaney DF, Anderson BJ, & Marmarou A (1992). Biomechanical aspects of a fluid percussion model of brain injury. Journal of Neurotrauma, 9(4), 311–322. 10.1089/neu.1992.9.311 [DOI] [PubMed] [Google Scholar]
- Togo T, Alderton JM, & Steinhardt RA (2000). The mechanism of cell membrane repair. Zygote (Cambridge, England), 8 Suppl 1, S31–2. [PubMed] [Google Scholar]
- Tran MD, Wanner IB, & Neary JT (2008). Purinergic receptor signaling regulates N-cadherin expression in primary astrocyte cultures. Journal of Neurochemistry, 105(1), 272–286. 10.1111/j.1471-4159.2008.05214.x [DOI] [PubMed] [Google Scholar]
- Tyler WJ (2012). The mechanobiology of brain function. Nature Reviews Neuroscience, 13(12), 867–878. 10.1038/nrn3383 [DOI] [PubMed] [Google Scholar]
- Vagnozzi R, Tavazzi B, Signoretti S, Amorini AM, Belli A, Cimatti M, Delfini R, Di Pietro V, Finocchiaro A, & Lazzarino G (2007). Temporal window of metabolic brain vulnerability to concussions: mitochondrial-related impairment--part I. Neurosurgery, 61(2), 379 10.1227/01.NEU.0000280002.41696.D8 [DOI] [PubMed] [Google Scholar]
- van Dommelen JAW, van der Sande TPJ, Hrapko M, & Peters GWM (2010). Mechanical properties of brain tissue by indentation : Interregional variation. Journal of the Mechanical Behavior of Biomedical Materials, 3(2), 158–166. 10.1016/j.jmbbm.2009.09.001 [DOI] [PubMed] [Google Scholar]
- Vásquez V, Krieg M, Lockhead D, & Goodman MB (2014). Phospholipids that contain polyunsaturated fatty acids enhance neuronal cell mechanics and touch sensation. Cell Reports, 6(1), 70–80. 10.1016/j.celrep.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Estevez M, Rolle SO, Mampay M, Dev KK, & Sheridan GK (2020). Piezo1 regulates calcium oscillations and cytokine release from astrocytes. Glia, 68(1), 145–160. 10.1002/glia.23709 [DOI] [PubMed] [Google Scholar]
- Viji Babu PK, Rianna C, Mirastschijski U, & Radmacher M (2019). Nano-mechanical mapping of interdependent cell and ECM mechanics by AFM force spectroscopy. Scientific Reports, 9(1), 12317 10.1038/s41598-019-48566-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagshul ME, Eide PK, & Madsen JR (2011). The pulsating brain: A review of experimental and clinical studies of intracranial pulsatility. Fluids and Barriers of the CNS, 8(1), 5 10.1186/2045-8118-8-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JA, Lin W, Morris T, Banderali U, Juranka PF, & Morris CE (2009). Membrane trauma and Na+ leak from Nav1 .6 channels. AM J Phyiol Cell Physiol, 294, 823–834. 10.1152/ajpcell.00505.2008. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang X, Tian J, Shan J, Hu Y, Zhai Y, & Guo J (2019). Talin promotes integrin activation accompanied by generation of tension in talin and an increase in osmotic pressure in neurite outgrowth. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 33(5), 6311–6326. 10.1096/fj.201801949RR [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Dalkara T, You Z, Qiu J, Bermpohl D, Mehta N, Suter B, Bhide PG, Lo EH, Ericsson M, & Moskowitz M. a. (2008). Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. Journal of Cerebral Blood Flow and Metabolism : Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 28, 490–505. 10.1038/sj.jcbfm.9600544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde EA, McCauley SR, Hunter JV, Bigler ED, Chu Z, Wang ZJ, Hanten GR, Troyanskaya M, Yallampalli R, Li X, Chia J, & Levin HS (2008). Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology, 70(12), 948–955. 10.1212/01.wnl.0000305961.68029.54 [DOI] [PubMed] [Google Scholar]
- Wilson L, Stewart W, Dams-O’Connor K, Diaz-Arrastia R, Horton L, Menon DK, & Polinder S (2017). The chronic and evolving neurological consequences of traumatic brain injury. The Lancet. Neurology, 16(10), 813–825. 10.1016/S1474-4422(17)30279-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MT, & Keith CH (1998). Glutamate modulation of dendrite outgrowth: alterations in the distribution of dendritic microtubules. Journal of Neuroscience Research, 52(5), 599–611. [DOI] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, & Burns MP (2013). Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. Journal of Neurotrauma, 30(23), 1966–1972. 10.1089/neu.2013.2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofford KL, Harris JP, Browne KD, Brown DP, Grovola MR, Mietus CJ, Wolf JA, Duda JE, Putt ME, Spiller KL, & Kacy Cullen D (2017). Rapid Neuroinflammatory Response Localized to Injured Neurons After Diffuse Traumatic Brain Injury in Swine. Experimental Neurology, 290, 85–94. 10.1016/j.expneurol.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Johnson BN, Johnson VE, Putt ME, Browne KD, Mietus CJ, Brown DP, Wofford KL, Smith DH, Grady MS, Cohen AS, & Cullen DK (2017). Concussion Induces Hippocampal Circuitry Disruption in Swine. Journal of Neurotrauma, 34(14), 2303–2314. 10.1089/neu.2016.4848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, & Smith DH (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 21(6), 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TC, Wilde EA, Bigler ED, Yallampalli R, McCauley SR, Troyanskaya M, Chu Z, Li X, Hanten G, Hunter JV, & Levin HS (2010). Evaluating the relationship between memory functioning and cingulum bundles in acute mild traumatic brain injury using diffusion tensor imaging. Journal of Neurotrauma, 27(2), 303–307. 10.1089/neu.2009.1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Muthuchamy M, & Reddy DS (2016). Atomic Force Microscopy Protocol for Measurement of Membrane Plasticity and Extracellular Interactions in Single Neurons in Epilepsy. Frontiers in Aging Neuroscience, 8, 88 10.3389/fnagi.2016.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap YC, King AE, Guijt RM, Jiang T, Blizzard A, Breadmore MC, & Dickson TC (2017). Mild and repetitive very mild axonal stretch injury triggers cystoskeletal mislocalization and growth cone collapse. 1–19. [DOI] [PMC free article] [PubMed]
- Yarnell P, & Ommaya AK (1969). Experimental Cerebral Concussion in the Rhesus Monkey. Bull NY Acad Med, 45(1), 39–45. [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Browne KD, Iwata A, & Smith DH (2009). Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. Journal of Neuroscience Research, 87(16), 3620–3625. 10.1002/jnr.22161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Green MA, Sinkus R, & Bilston LE (2011). Viscoelastic properties of human cerebellum using magnetic resonance elastography. Journal of Biomechanics, 44(10), 1909–1913. 10.1016/j.jbiomech.2011.04.034 [DOI] [PubMed] [Google Scholar]
- Zhang K, & Sejnowski TJ (2000). A universal scaling law between gray matter and white matter of cerebral cortex. Proceedings of the National Academy of Sciences of the United States of America, 97(10), 5621–5626. 10.1073/pnas.090504197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, & Satin LS (1996). Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science (New York, N.Y.), 274(5294), 1921–1923. 10.1126/science.274.5294.1921 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Abiraman K, Li H, Pierce DM, Tzingounis AV, & Lykotrafitis G (2017). Modeling of the axon membrane skeleton structure and implications for its mechanical properties. PLoS Computational Biology, 13(2), e1005407 10.1371/journal.pcbi.1005407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Gatti DL, & Yang KH (2016). Nodal versus Total Axonal Strain and the Role of Cholesterol in Traumatic Brain Injury. Journal of Neurotrauma, 33(9), 859–870. 10.1089/neu.2015.4007 [DOI] [PubMed] [Google Scholar]
- Zimmermann DR, & Dours-Zimmermann MT (2008). Extracellular matrix of the central nervous system: from neglect to challenge. Histochemistry and Cell Biology, 130(4), 635–653. 10.1007/s00418-008-0485-9 [DOI] [PubMed] [Google Scholar]