

Abstract
Our laboratory recently demonstrated that seizures activate the hypothalamic-piuitary-adrenal (HPA) axis, increasing circulating levels of corticosterone [1]. Given the well-established proconvulsant actions of corticosterone, we hypothesized that seizure-induced activation of the HPA axis may contribute to future seizure susceptibility. Further, since hypercortisolism is associated with depression, we propose that seizure-induced activation of the HPA axis may contribute to comorbid depression and epilepsy. To test this hypothesis, we generated mice lacking the GABAAR δ subunit specifically in corticotropin-releasing hormone (CRH) neurons (Gabrd/Crh mice), which exhibit hyporeactivity of the HPA axis [2]. Gabrd/Crh mice exhibit blunted seizure-induced elevations in corticosterone, establishing a useful tool to investigate the contribution of HPA axis dysfunction on epilepsy and associated comorbidities. Interestingly, Gabrd/Crh mice exhibit decreased acute seizure susceptibility following kainic acid administration. Furthermore, chronically epileptic Gabrd/Crh mice exhibit a decrease in both spontaneous seizure frequency and depression-like behaviors compared to chronically epileptic Cre−/− littermates. Seizure susceptibility and associated depression-like behaviors can be restored to wild type levels by treating Gabrd/Crh mice with exogenous corticosterone. Similarly, chemogenetic activation of CRH neurons in the PVN is sufficient to increase seizure susceptibility; whereas, chemogenetic inhibition of CRH neurons in the paraventricular nucleus of the hypothalamus (PVN) is sufficient to decrease seizure susceptibility and depression-like behaviors in chronically epileptic mice. These data suggest that seizure-induced activation of the HPA axis promotes seizure susceptibility and comorbid depression-like behaviors, suggesting that the HPA axis may be a novel target for seizure control.
Keywords: epilepsy, depression, seizures, stress, HPA axis, corticosterone, GABA
1. Introduction
Stress is the most commonly reported precipitating factor for seizures [3–9] (for review see [10, 11]). The increased seizure susceptibility associated with stress is thought to be mediated by stress hormones, which have been shown to exhibit proconvulsant actions in animal models. Chronic stress has been demonstrated to increase seizure susceptibility [4, 11, 12] and accelerate epileptogenesis [13] in a variety of experimental epilepsy models. Further, administration of exogenous stress hormones, including both corticosterone and CRH, have been shown to exert proconvulsant actions in numerous experimental models of epilepsy (for review see [14]) and corticosterone treatment designed to mimic chronic stress has been shown to increase the frequency of epileptiform events in chronically epileptic mice [15].
Interestingly, basal levels of corticosterone are elevated in patients with epilepsy and are further increased following seizures [16–18], suggesting that the regulation of the HPA axis may be fundamentally altered in patients with epilepsy (for review see [19]). Consistent with this idea, our laboratory recently demonstrated that seizures activate the HPA axis in multiple epilepsy models [1] and it was recently demonstrated that the glucocorticoid antagonist, RU486, decreases neuropathological changes following status epilepticus [20]. These findings force us to reevaluate the relationship between stress and epilepsy and suggest that the HPA axis may impact epilepsy outcomes independent of stress. Given the proconvulsant actions of stress hormones, these findings suggest that seizure-induced elevations in corticosterone may influence future seizure susceptibility. Further, hypercortisolism is also associated with major depression [21–24], leading us to hypothesize that seizure-induced activation of the HPA axis may also contribute to the comorbidity of depression and epilepsy. Despite the report that 55% of patients with temporal lobe epilepsy suffer from depression [25], the 25-fold increase in suicide rate [26], and significant impact on the quality of life of these patients [27], the pathological mechanisms underlying the comorbidity between depression and epilepsy remains unclear. Stress is a trigger for both epilepsy and depression and significant accumulating evidence, from both human and animal studies, suggests that the HPA axis may play a role in the comorbidity of these two disorders [28, 29] (for review see [30]). Dysfunction of the HPA axis has been implicated in the pathogenesis of depression (for review see [31]) and epilepsy is associated with a dysregulation in the control of the HPA axis [28, 29], leading us to the hypothesis that seizure-induced activation of the HPA axis may play a role in the comorbidity of these disorders. Interestingly, previous studies demonstrated elevations in corticosterone levels in chronically epileptic mice [28, 29], which correlated with increased depression-like behaviors. A recent study also demonstrated that in vulnerable animals, subjection to social defeat stress decreased the threshold to induce status epilepticus, accelerated epileptogenesis, and increased the comorbidity with depression in chronically epileptic mice [32].
Here we utilized a mouse model generated by our laboratory, which exhibits hyporeactivity of the HPA axis [2] to examine the relationship between seizure-induced activation of the HPA axis, seizure susceptibility, and the comorbidity with depression. Our laboratory previously demonstrated a role for the GABAA receptor (GABAAR) δ subunit [33] and excitatory actions of GABA on CRH neurons in the stress-induced [33] and seizure-induced [1] activation of the HPA axis. Thus, loss of the GABAAR δ subunit specifically in CRH neurons in Gabrd/Crh mice results in a suppression of stress-induced elevations in corticosterone [2]. Here we demonstrate that seizure-induced elevations in corticosterone are also blunted in Gabrd/Crh mice which is associated with decreased seizure susceptibility and decreased depression-like behaviors in chronically epileptic mice. Exogenous corticosterone administration or chemogenetic activation of CRH neurons in the PVN is sufficient to increase seizure susceptibility and chemogenetic inhibition of CRH neurons in the PVN decreases seizure frequency and depression-like behaviors in chronically epileptic mice.
2. Materials and Methods
2.1. Animals
Adult (>P60) male Gabrd/Crh mice and Cre−/− littermates were bred and housed at Tufts University School of Medicine’s Division of Laboratory Animal Medicine facility. Animals had ad libitum access to food and water. All procedures were approved by the Tufts University Institutional Animal Care and Use Committee.
The generation and characterization of the Gabrd/Crh mouse model was previously published by our laboratory [2]. The CRH-Cre line used in the current study was also previously characterized by our laboratory [33].
Both mouse lines were generated on a 129/Sv background and backcrossed to a C57Bl/6 background for over 12 generations. Genotyping of the Gabrd/Crh mice was carried out in-house using the following primers:
| Gabrd | 5’: AGCAACTTTGCTTGCGCTG |
| 3’: TTGATAGCTGAAGCCCGTGG | |
| CRH-Cre | 5’: CTGTCTTGTCTGTGGGTGTCCGAT |
| 3’: CGGCAAACGGACAGAAGCATT |
The expected PCR product size is 449 bp for wild type and 543 bp for floxed Gabrd mice and the product size is 310 bp for CRH-Cre.
The Gabrd/Crh mice are homozygous for the floxed Gabrd allele and express the CRH-Cre transgene. Zygosity for the Cre transgene was not determined since one allele is sufficient for Cre-mediated excision of floxed alleles. The Cre−/− mice used in this study are homozygous for the floxed Gabrd gene, but do not express the CRH-Cre transgene.
2.2. Corticosterone Measurements
Trunk blood was collected in CAPIJECT® (T-MG) tubes at baseline, 2 hrs following kainic acid-induced seizures, or in chronically epileptic mice (60 days post pilocarpine-induced status epilepticus). Trunk blood was centrifuged at 14K rpm and serum collected and stored at −20°C prior to subjection to the ELISA analysis. Corticosterone was measured using an enzyme immunoassay kit, according to the manufacturer’s instructions (Enzo Pharmaceuticals, New Jersey). Briefly, samples were run in duplicate and compared to a standard curve of known corticosterone concentrations. Samples from all experimental groups were run in parallel.
2.3. Electroencephalogram (EEG) recording
For all EEG recordings, mice were implanted with chronic EEG headmounts. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine and lengthwise incision was made along the scalp and a pre-fabricated headmount (Pinnacle Technology, part #8201) was fixed to the skull with four screws, two which serve as EEG leads in the somatosensory and motor cortices of the left hemisphere, one a reference ground, and one an animal ground. The site was closed with dental cement and the mice were allowed to recover for a minimum of 5 days prior to experimentation. Electroencephalogram recordings were collected using a 100x gain preamplifier high pass filtered at 1.0 Hz (Pinnacle Technology, part #8202-SE) and tethered turnkey system (Pinnacle Technology, part #8200).
EEG recordings were performed in awake, behaving animals in response to acute treatment with the chemoconvulsant kainic acid (KA, 20mg/kg, i.p.) or in chronically epileptic mice following pilocarpine-induced status epilepticus (340mg/kg, i.p.). For the kainic acid experiments, electrographic epileptiform activity was measured for 2 hrs following KA administration and the latency to the first seizure event and total time exhibiting abnormal electrographic epileptiform activity was measured. Abnormal epileptiform activity was detected as paroxysmal activity having a sudden onset and an amplitude at least 2.5x the standard deviation of the baseline and a consistent change in the Power of the fast Fourier transform of the EEG, including a change in the Power and the frequency of activity over the course of the event. These parameters have been used previously by our laboratory to detect epileptiform activity [1, 34–36]. Abnormal electrographic activity, including ictal activity and rhythmic spiking lasting longer than 30 s were collectively defined as “epileptiform activity”. Latency to the first seizures was defined as the time elapsed from the injection of kainic acid to the start of the first identified electrographic seizure. The amount of time exhibiting epileptiform activity divided by the length of the total recording period (120 mins) × 100 was calculated as the “% time epileptiform activity” and compared between experimental groups. For assessment of seizure frequency in chronic epileptic mice, we employed the pilocarpine model. Mice were treated with 1mg/kg scopolamine 30 minutes prior to treatment with 340mg/kg pilocarpine. Mice were continually observed until they reached status epilepticus. An additional 50mg/kg pilocarpine was administered if necessary for the mice to reach status epilepticus. After 3 hrs of continuous status epilepticus, mice were given 5mg/kg diazepam. The percent change in body weight was determined immediately after diazepam treatment and survival rate was determined 48 hours post-status epilepticus. After 4 weeks of recovery/latent period, chronic video/EEG (24/7) was performed over a continuous 21-day period. In a subset of Gabrd/Crh mice, a 10mg, 21-day, slow-release corticosterone pellet was implanted (Innovative Research of America) immediately prior to chronic EEG recording to assess the impact on seizure frequency in chronically epileptic Gabrd/Crh mice. These animals were exposed to elevated corticosterone, measured previously to reach stress-induced levels [2], throughout the entire chronic EEG recording period. Epileptiform activity was initially identified using DClamp [37] and by manual observation. DClamp was able to detect periods abnormal of rhythmic spiking as well as ictal events. The epileptiform activity was subsequently verified based on visual inspection of the electrographic activity and ictal events were also verified by video analysis of the behavioral seizures. The behavioral seizures were scored using a modified Racine scale in which 1 = uncontrollable whisker movement, 2 = head bobbing, 3 = forelimb clonus, 4 = abnormal rearing, and 5 = rearing and falling/tonic clonic seizures. Epileptiform activity was again identified as paroxysmal activity 2.5x the standard deviation of the baseline. High frequency epileptiform activity with a sudden onset and a behavioral seizure component were classified as “ictal events”. The number of ictal events observed per day was measured. The average number of seizures/day was calculated per animal and then averaged between animals. LabChart Pro software (ADInstruments) was used for data acquisition and analysis in addition to DClamp.
2.4. Immunohistochemistry
Immunohistochemistry was performed as previously described [33, 38, 39]. To evaluate the activation of neurons in the PVN, mice were sacrified 2 hours following KA administration. Mice were sacrificed 60 days following pilocarpine administration to evaluate the number of neurons in the PVN activated in the PVN of chronically epileptic mice. Mice were deeply anesthetized with isoflurane, euthanized by decapitation, and the brains were rapidly removed, fixed by immersion fixation in 4% paraformaldehyde overnight at 4°C, cryoprotected in 10–30% sucrose, and frozen at −80°C until use. Coronal sections (40 μm) were prepared using a Leica cryostat and an equal number of sections per experimental group were processed in parallel to ensure equivalent treatment. For the c-fos staining, sections were blocked with 10% normal goat serum for 1 h, and probed with a primary polyclonal anti-c-fos antibody (1:10,000, Sigma) overnight at 4°C and a secondary anti-rabbit Alexa 488 antibody (Invitrogen) for 2 hrs at room temperature. For CRH immunostaining, mice were infused with colchine (50μg in a 2μl volume) unilaterally into the lateral ventricle (0.5mm posterior, 1mm lateral, 2mm depth) 24 hrs prior to sacrifice. The sections were incubated a rabbit anti-CRH antibody (kindly provided by Dr. Paul Sawchenko on behalf of Dr. Wylie Vale, Salk Institute for Biological Studies, 1:10,000) for 4 days at 4°C, followed by a biotinylated anti-rabbit secondary antibody (VectaStain Elite ABC Kit, Vector Labs, 1:200) for 2 hours at room temperature (RT), and a streptavidin conjugated AlexaFluor 488 antibody (Molecular probes, 1:1,000) for 2 hours at RT. Sections were mounted with a mounting medium containing DAPI (Vector Laboratories) which enabled localization of the region of interest, the PVN, based on anatomical structures, cell density, and cell distribution. Fluorescence was visualized using a Nikon A1R confocal and the number of c-fos-positive neurons in the PVN was measured in an equal number of matched sections using NIH ImageJ software. For the current study, the extent of c-fos activation was only quantified in the PVN. Ongoing studies in the laboratory are investigating differential patterns of activation in other brain regions.
2.5. Electrophysiology
Electrophysiological recordings were performed as previously described [33, 39]. Adult Gabrd/Crh mice and Cre−/− littermates were sacrificed by decapitation under anesthesia with isoflurane two hours following vehicle or KA administration and the brain was rapidly dissected out and placed immediately in ice-cold, oxygenated normal artificial cerebral spinal fluid (nACSF) (containing 126 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 10 mM dextrose; 300–310 mOsm) containing 3 mM kynurenic acid. Acute sections (350 μm), containing the PVN, were prepared using a Leica VT1000S vibratome and stored in oxygenated nACSF at 33°C for at least 1 h before recording. Slices were placed into a recording chamber maintained at 33°C (in-line heater, Warner Instruments) and perfused at a high flow rate (≥ 4 ml/min) with 95% O2 / 5% CO2 to maintain adequate O2 tension and physiological pH (7.3–7.4). CRH neurons were visually identified (Sarkar et al., 2011) and the identity confirmed using morphological and electrophysiological methods [40].
Current clamp recordings were carried out in the I = 0 configuration using electrodes with DC resistance of 5–8 MΩ and an intracellular solution containing 130 mM K-gluconate, 10 mM KCl, 4 mM NaCl, 10 mM HEPES, 0.1 mM EGTA, 2 mM Mg-ATP, and 0.3 mM Na-GTP (pH 7.25; 280–290 mOsm). After stabilization of the holding current and membrane properties, the spontaneous firing rate of CRH neurons was measured over a 5 min period. Whole cell capacitance and series resistance was measured throughout the course of the experiment and data was eliminated from analysis if there was >20% change. Data was collected and analyzed using Powerlab software (ADInstruments).
2.6. Behavioral Tests
Behavioral tests were conducted on chronically epileptic mice 60 days following pilocarpine-induced status epilepticus. A subset of Gabrd/Crh mice were implanted with a 10mg, 21-day, slow-release corticosterone pellet (Innovative Research of America) 14 days prior to behavioral testing. Mice were anesthetized with ketamine/xylazine, a small 1 cm incision was made on the back of the neck, and a small slow-release pellet (or nothing for sham) was placed underneath the skin. The incision was closed and the animals were allowed to recover for 14 days until experimentation, throughout which time the animals were exposed to elevated levels of corticosterone.
Forced Swim Test
Mice were placed into a 5 L plastic beaker (20cm in diameter) containing 3 L of water (23–25°C) for 6 minutes. A video camera was used to record each session. The latency to the first bout of immobility and total time spent immobile during the 6 minute test was measured.
2.7. DREADD experiments
To enhance the seizure-induced activation of the HPA axis in Gabrd/Crh mice which have HPA axis hypofunction, Gabrd/Crh mice were stereotaxically injected bilaterally with 250nl of AAV-hSyn-DIO-hM3D(Gq)-mCherry (Gq DREADD) into the PVN using the following stereotaxic coordinates (0.45mm posterior, ±0.05mm lateral, and 5.0mm depth). Similarly, to inhibit the seizure-induced activation of the HPA axis, CRH-Cre mice were injected with 250nl of AAV-hSyn-DIO-hM4D(Gi)-mCherry (Gi DREADD). Mice were also injected with 250nl of AAV-GFP (Vector Biolabs, Serotype 2) to serve as controls. Injections were performed 28 days prior to experimentation to allow for optimal expression. Accurate targeting and specific expression of the DREADD viruses in CRH neurons was confirmed by post-hoc analysis using immunohistochemistry to confirm colocalization with the endogenous CRH peptide. Thirty minutes prior to the behavioral experiments, all experimental animals were injected with 3 mg/kg clozapine-N-oxide (CNO) to control for potential off-target effects [41]. To assess the impact of silencing or activating CRH neurons in the PVN on seizure frequency, mice were treated with CNO in the drinking water (5mg/200ml, a protocol provided on Bryan Roth’s website). Mice were treated with 7 days of normal drinking water followed by 7 days with CNO, a treatment protocol which was then repeated twice and counterbalanced between experiments. We observed a similar effect of CNO treatment over the two separate weeks of treatment. Therefore, the average seizure frequency was collapsed over the weeks of either vehicle or CNO treatment and represented as “average # seizures/day”. Seizure frequency was determined during each week of treatment and averaged for water or CNO treatment for each animal and then averaged between animals.
2.8. Statistical Analyses
Statistical significance between Gabrd/Crh mice and Cre−/− littermates was determined using a Student’s t-test. Statistical significance between the data sets of Cre−/− littermates, Gabrd/Crh mice, and Gabrd/Crh mice treated with corticosterone was conducted using a one-way ANOVA. A two-way ANOVA was used to compared between genotypes (Cre−/− vs. Gabrd/Crh) and treatment (vehicle vs. KA or pilocarpine). A Tukey’s multiple comparisons test was used to determine statistical significance. All data are represented as the average ± SEM.
3. Results
3.1. Suppression of the seizure-induced activation of the HPA axis in Gabrd/Crh mice.
Previously, our laboratory demonstrated seizure-induced activation of the HPA axis, resulting in increased corticosterone levels, following seizures induced with both kainic acid (KA) and pilocarpine [1]. Due to the evidence for a role of GABAAR δ subunit-containing receptors in the regulation of CRH neurons in the PVN and, thus, in the regulation of the HPA axis [33], we generated mice lacking the GABAAR δ subunit specifically in CRH neurons (Gabrd/Crh mice), which has been previously characterized by our laboratory [2]. Due to the excitatory actions of GABA, which plays a role in mounting the physiological response to stress [33], the loss of the GABAAR δ subunit in CRH neurons (Gabrd/Crh mice) paradoxically results in a decrease in stress-induced elevations in corticosterone [2]. The loss of the GABAAR δ subunit in the PVN was previously verified in Gabrd/Crh mice [2]. Here we investigate whether the seizure-induced activation of the HPA axis is also altered in Gabrd/Crh mice. Circulating corticosterone levels were measured in control Gabrd/Crh mice and Cre−/− littermates and two hrs following seizures induced with KA (20mg/kg, i.p.). Corticosterone levels are increased in Cre−/− mice two hrs following KA-induced seizures (470.0 ± 99.9 ng/ml) compared to vehicle-treated controls (65.7 ± 24.6 ng/ml) (Figure 1a). The seizure-induced elevation in corticosterone is suppressed in Gabrd/Crh mice (57.6 ± 24.3 ng/ml) compared to Cre−/− littermates (470.0 ± 99.9 ng/ml) (Figure 1). However, the baseline levels of corticosterone are not different between Gabrd/Crh mice (69.9 ± 15.6 ng/ml) and Cre−/− littermates (63.1 ± 18.2 ng/ml) (Figure 1). Administration of exogenous corticosterone increases circulating corticosterone in Gabrd/Crh mice (302.1 ± 105.3 ng/ml) to levels comparable to wild type (Cre−/−: 470.0 ± 99.9 ng/ml) (Figure 1; n = 5 – 12 mice per experimental group; * denotes p<0.05 using a two-way ANOVA).
Figure 1: Blunted acute seizure-induced elevations in corticosterone in Gabrd/Crh mice.
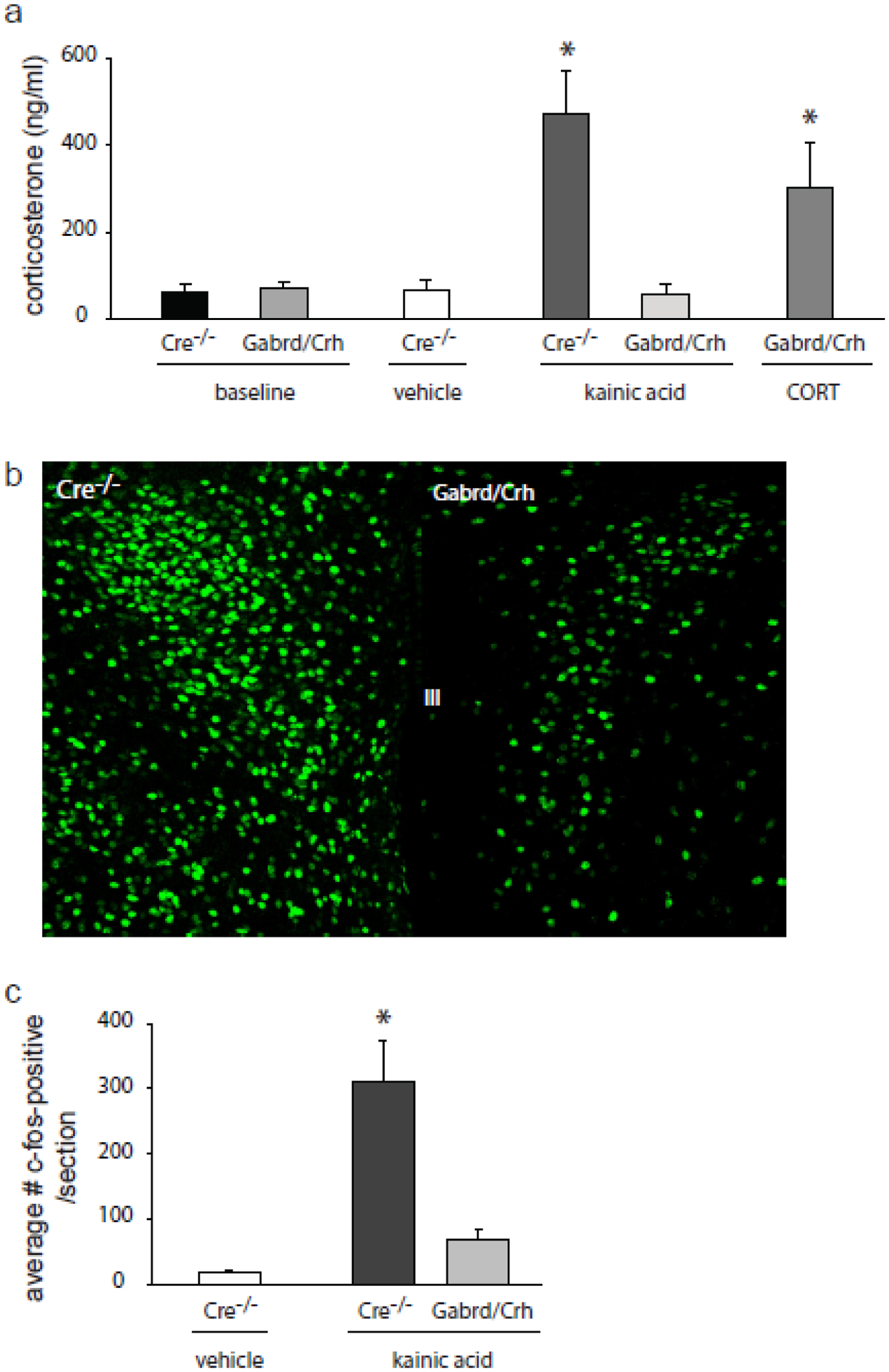
a, Average circulating corticosterone levels in Gabrd/Crh mice and Cre−/− littermates at baseline and 2 hrs following seizures induced with kainic acid (20mg/kg, i.p.) compared to vehicle-treated Cre−/− littermates and Gabrd/Crh mice treated with exogenous corticosterone (21-day slow-release corticosterone pellet). n = 5 – 12 mice per experimental group; * denotes p<0.05 using a two-way ANOVA. b, Representative images of c-fos expression in the PVN of Gabrd/Crh mice and Cre−/− littermates 2 hrs following seizures induced with kainic acid. c, The average number of c-fos-positive neurons in the PVN of Gabrd/Crh miceand Cre−/− littermates following kainic acid-induced seizures or in vehicle-treated controls. n = 6 sections/mouse, 3 mice per experimental group. * denotes p<0.05 using a one-way ANOVA.
Consistent with the suppression of the seizure-induced activation of the HPA axis in Gabrd/Crh mice, c-fos activation in the PVN two hrs following KA-induced seizures is decreased in Gabrd/Crh mice compared to Cre−/− controls (Figure 1b,c). The number of c-fos positive neurons in the PVN 2 hrs following seizures induced with KA is decreased in Gabrd/Crh mice (68.0 ± 15.5) and vehicle-treated mice (16.3 ± 3.1) compared to KA-treated Cre−/− mice (311.8 ± 62.3) (Figure 1c; n = 6 sections per mouse, 3 – 6 mice per experimental group; * denotes p<0.05 using a two-way ANOVA). Together, these data demonstrate suppression of the seizure-induced activation of the HPA axis in Gabrd/Crh mice, similar to our observations that the stress-induced activation of the HPA axis is blunted in these mice [1].
3.2. Suppression of the seizure-induced activation of the HPA axis is associated with decreased acute seizure susceptibility.
To explore the relationship between seizure-induced activation of the HPA axis and acute seizure susceptibility, electrographic epileptiform activity following KA administration was measured in Gabrd/Crh mice and Cre−/− littermates. The latency to the first seizure was not different between Gabrd/Crh mice (450.7 ± 171.3 s) and Cre−/− littermates (625.6 ± 170.8 s), which is understandable since the first seizure is required to activate the HPA axis. However, the total epileptiform activity measured during the two hrs following KA administration is decreased in Gabrd/Crh mice (5.0 ± 1.4 %) compared to Cre−/− mice (67.8 ± 6.0 %) (Figure 2). Treatment of Gabrd/Crh mice with exogenous corticosterone did not significantly alter the latency to the first seizure (341.4 ± 121.2 s) but did increase the percent time exhibiting epileptiform activity (62.5 ± 11.7 %) compared to untreated Gabrd/Crh mice (latency: 450.7 ± 171.3 s; percent time: 5.0 ± 1.4 %) (Figure 2; n = 7 – 8 mice per experimental group; * denotes p<0.05 using a one-way ANOVA). These data demonstrate that suppression of the seizure-induced activation of the HPA axis is associated with decreased acute seizure susceptibility.
Figure 2: Decreased seizure susceptibility in Gabrd/Crh mice.
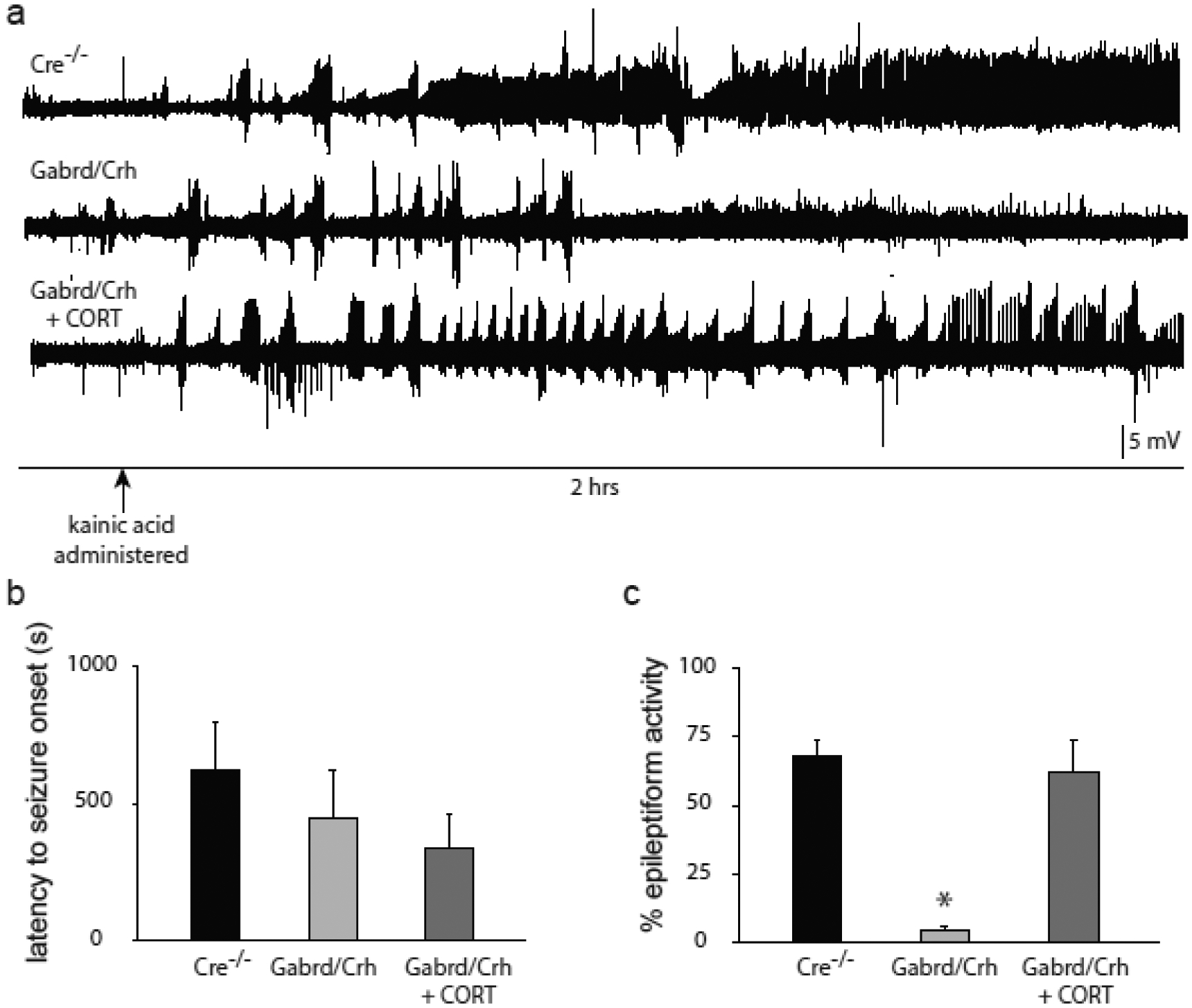
a) Representative electrographic epileptiform activity over the 2 hr recording period in Gabrd/Crh mice and Cre−/− littermates and Gabrd/Crh mice treated with exogenous corticosterone. b, The average latency to the onset of the first electrographic seizure in Gabrd/Crh mice, Cre−/− littermates, and Gabrd/Crh mice treated with exogenous corticosterone. c, The average percent time exhibiting epileptiform activity in Gabrd/Crh mice, Cre−/− littermates, and Gabrd/Crh mice treated with exogenous corticosterone. n = 7 – 8 mice per experimental group; * denotes p<0.05 using a one-way ANOVA.
3.3. The seizure-induced activation of the HPA axis is blunted in chronically epileptic Gabrd/Crh mice.
The experiments presented above demonstrate seizure-induced activation of the HPA axis in response to substantial seizure activity induced by acute administration of a chemoconvulsant. To determine whether the HPA axis is activated by episodic seizures in chronically epileptic mice, we performed similar experiments using the pilocarpine model. Circulating corticosterone levels were measured in control and chronically epileptic Gabrd/Crh mice and Cre−/− littermates 60 days following status epilepticus induced with pilocarpine, at which time the mice are in the chronic epilepsy period, confirmed with 24/7 video, EEG recording. Corticosterone levels are increased in chronically epileptic Cre−/− mice (104.9 ± 30.1 ng/ml) compared to vehicle-treated controls (27.7 ± 5.4 ng/ml) (Figure 3). In contrast, corticosterone levels are not significantly increased in chronically epileptic Gabrd/Crh mice (25.0 ± 7.4 ng/ml) compared to Gabrd/Crh controls (31.6 ± 6.3 ng/ml) (Figure 3). Treatment with exogenous corticosterone increases circulating corticosterone in chronically-epileptic Gabrd/Crh mice (211.5 ± 56.4 ng/ml) (Figure 3; n = 9 – 19 mice per experimental group; * denotes p<0.05 using a one-way ANOVA).
Figure 3: Blunted corticosterone levels in chronically epileptic Gabrd/Crh mice.
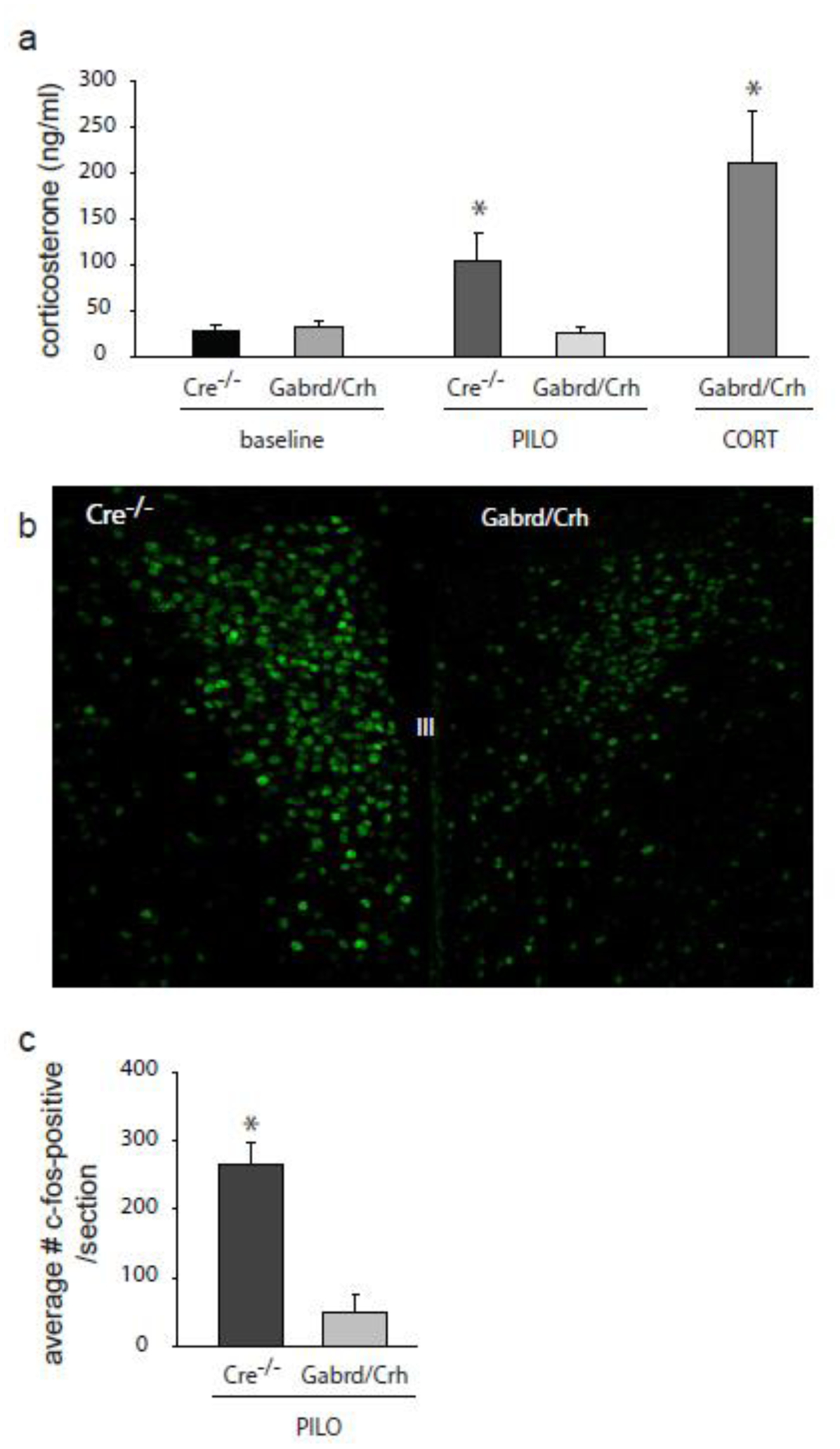
a, Average circulating corticosterone levels in Gabrd/Crh mice and Cre−/− littermates at baseline and in chronically epileptic mice 60 days following pilocarpine-induced status epilepticus. The blunted corticosterone levels in chronically epileptic Gabrd/Crh mice are increased with exogenous corticosterone treatment (21-day slow-release corticosterone pellet). n = 9 – 19 mice per experimental group; * denotes p<0.05 using a one-way ANOVA. b, Representative images of c-fos expression in the PVN of Gabrd/Crh mice and Cre−/− littermates 60 days following pilocarpine-induced status epilepticus. c, The average number of c-fos-positive neurons in the PVN of Gabrd/Crh mice and Cre−/− littermates 60 days following pilocarpine-induced status epilepticus or in vehicle-treated controls. n = 6 sections/mouse, 3 mice per experimental group. * denotes p<0.05 using a two-way ANOVA.
Suppressed activation of the HPA axis in chronically epileptic Gabrd/Crh mice is also suggested based on the decreased c-fos expression in the PVN of Gabrd/Crh mice compared to Cre−/− controls. The number of c-fos positive neurons in the PVN of chronically epileptic mice is decreased at 60 days post-status epilepticus in Gabrd/Crh mice (48.8 ± 27.3) and vehicle-treated controls (16.3 ± 3.1) compared to chronically epileptic Cre−/− mice (265.6 ± 32.9) (Figure 3; n = 6 sections per mouse, 3 – 6 mice per experimental group; * denotes p<0.05 using a two-way ANOVA). These data demonstrate that the HPA axis is activated in chronically epileptic mice, which we propose may contribute to seizure susceptibility and associated comorbidities, such as depression-like behaviors.
Consistent with blunted seizure-induced activation of CRH neurons in Gabrd/Crh mice, the firing rate of CRH neurons in the PVN is decreased in Gabrd/Crh mice following KA-induced seizures. Two hrs following seizures induced with KA, the firing rate of CRH neurons is increased in Cre−/− mice (8.9 ± 1.9 Hz) compared to vehicle-treated controls (4.3 ± 0.9 Hz) (Figure 4), similar to previous reports [1]. However, the firing rate of CRH neurons in the PVN is not increased in Gabrd/Crh mice (3.8 ± 0.6 Hz) following seizures induced with KA (Figure 4) (n = 15–16 mice, 4–5 mice per experimental group; * denotes p<0.05 using a two-way ANOVA).
Figure 4: Decreased firing rate of CRH neurons following seizures in Gabrd/Crh mice.
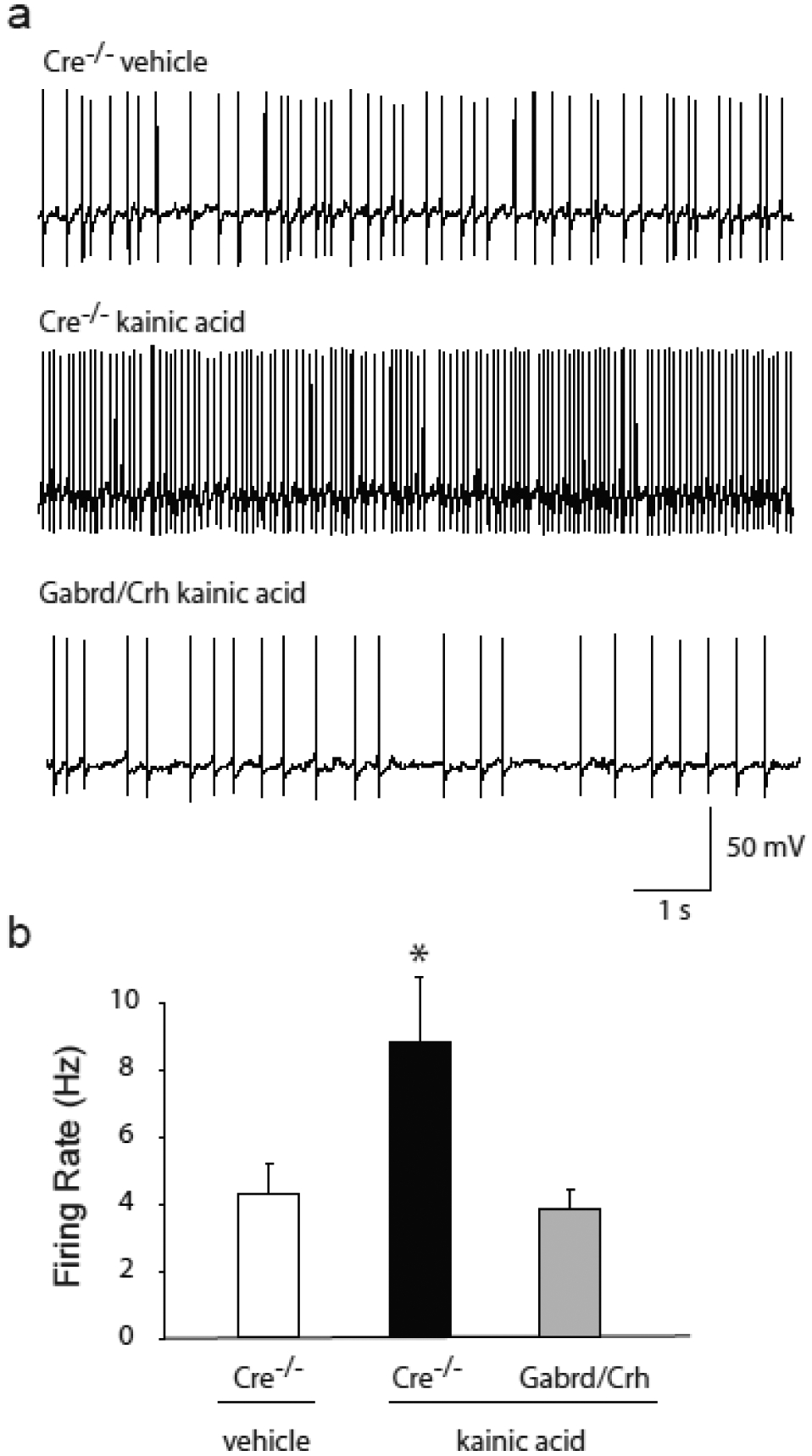
a, Representative traces of the spontaneous firing rate of CRH neurons 2 hrs following kainic acid-induced seizures in Gabrd/Crh mice and Cre−/− littermates, and in vehicle treated controls. b, The average firing rate of CRH neurons 2 hrs following kainic acid-induced seizures in Gabrd/Crh mice and Cre−/− littermates, and in vehicle treated controls. n = 15 – 16 slices, 4 – 5 mice per experimental group; * denotes p<0.05 using a two-way ANOVA
3.4. Seizure frequency is decreased chronically epileptic Gabrd/Crh mice.
To determine whether seizure-induced activation of the HPA axis contributes to seizure susceptibility in chronically epileptic mice, the daily seizure frequency was measured in Gabrd/Crh mice and Cre−/− littermates for 21 consecutive days beginning 4 weeks post pilocarpine-induced status epilepticus (Figure 5a). There was no difference in the survival rate (Cre−/−: 63.4 ± 7.6 %; Gabrd/Crh: 64.1 ± 7.8 %) or the percent change in body weight (Cre−/−: 6.6 ± 0.4 %; Gabrd/Crh: 7.3 ± 0.4 %) following pilocarpine-induced status epilepticus between Cre−/− and Gabrd/Crh mice (Figure 5b,c), suggesting that the initial epileptogenic insult was similar between experimental groups. However, the number of seizures per day was reduced in chronically-epileptic Gabrd/Crh mice (2.1 ± 0.7) compared to Cre−/− littermates (5.5 ± 1.2) (Figure 6b). Treatment of both Cre−/− littermates and Gabrd/Crh mice with exogenous corticosterone increased the number of seizures/day (Cre−/−+CORT: 7.3 ± 1.6; Gabrd/Crh+CORT: 5.9 ± 1.0) (Figure 6b), similar to previous reports [15]. Corticosterone increased seizure frequency to a greater extent in Gabrd/Crh mice (273.1% increase) compared to Cre−/− littermates (133.4 % increase) (n = 6 – 7 mice per experimental group; * denotes p<0.05 using a one-way ANOVA). These data suggest seizure-induced activation of the HPA axis contributes to seizure frequency in chronically epileptic mice.
Figure 5: Pilocarpine-induced status epilepticus is equivalent in Gabrd/Crh mice and Cre−/− littermates.
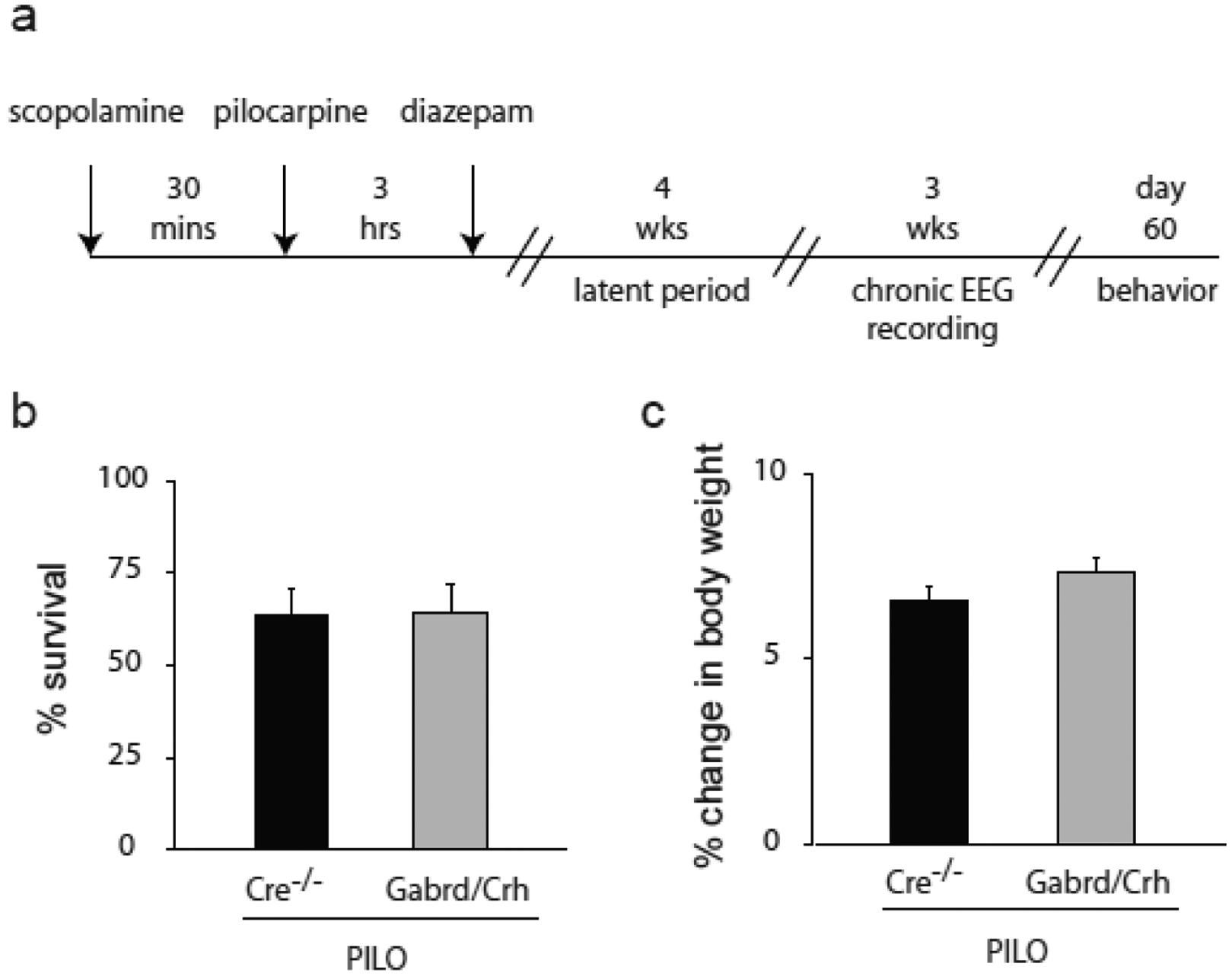
a, A diagram of the paradigm used to induce status epilepticus and assess spontaneous recurrent seizures and comorbid depression-like behaviors in chronically epileptic mice. b, The average survival rate following pilocarpine-induced status epilepticus in Gabrd/Crh mice and Cre−/− littermates. c, The percent body weight lost during the 3 hr period of pilocarpine-induced status epilepticus.
Figure 6: Decreased seizure frequency in chronically-epileptic Gabrd/Crh mice.
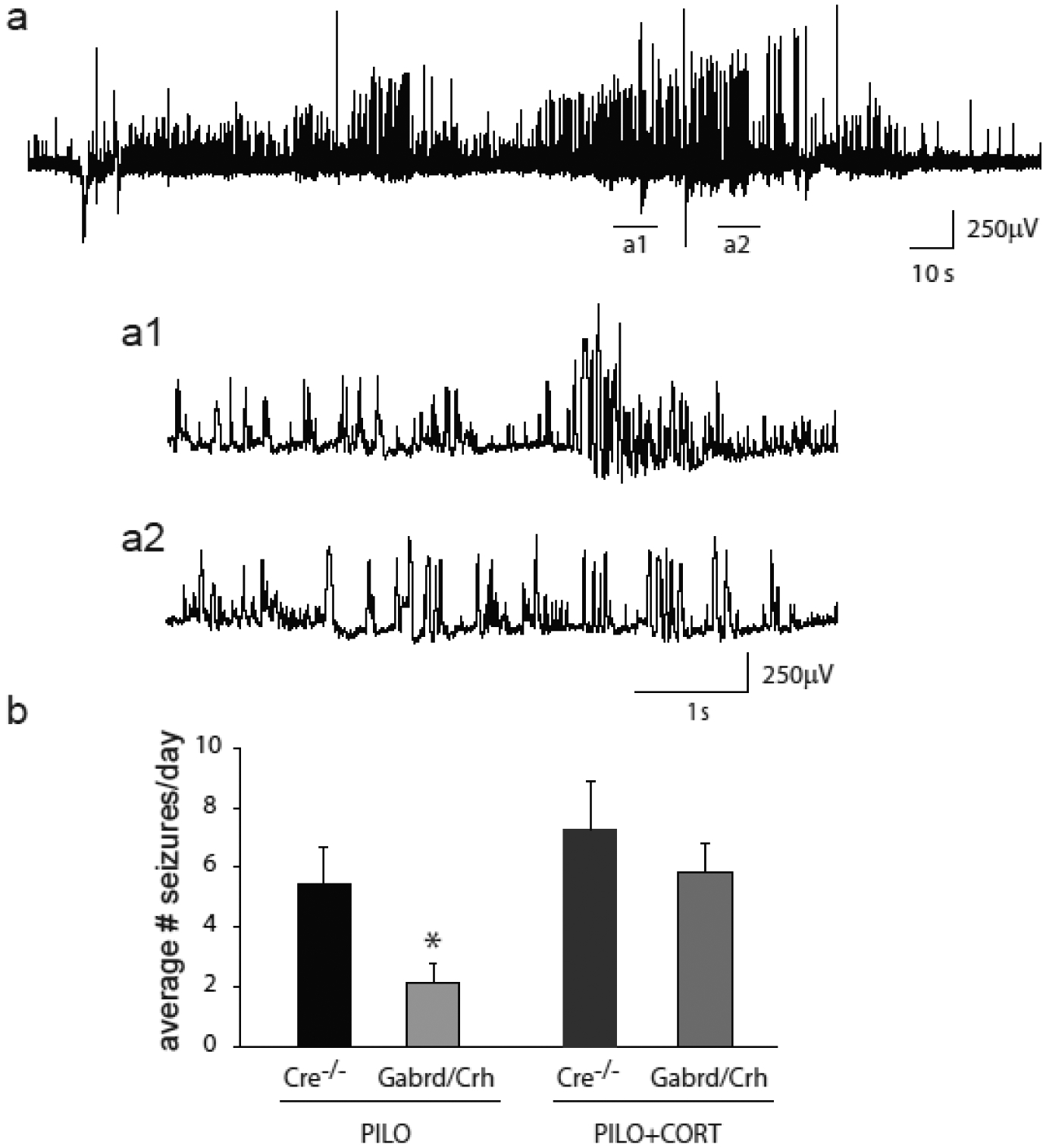
a, A representative electrographic spontaneous recurrent seizure in chronically epileptic mice following pilocarpine-induced status epilepticus. The underlined regions are shown below on a shorter time scale (a1, a2). b, The average number of seizures per day in vehicle-treated and corticosterone-treated chronically epileptic Gabrd/Crh mice and Cre−/− littermates. n = 6 – 7 mice per experimental group; * denotes p<0.05 using a one-way ANOVA.
3.5. Depression-like behaviors are decreased in chronically epileptic Gabrd/Crh mice.
Depression-like behavior was assessed in chronically epileptic Gabrd/Crh mice and Cre−/− littermates using the forced swim test. Chronically epileptic Cre−/− littermates exhibit an increase in depression-like behaviors compared to vehicle-treated controls, consistent with previous reports (for review see [42, 43]). Chronically epileptic Cre−/− littermates exhibit a decrease in the latency to the first bout of immobility in the forced swim test (66.5 ± 5.5 s) and an increase in the total time spent immobile (232.8 ± 5.3 s) compared to vehicle-treated, non-epileptic controls (latency: 87.3 ± 5.1 s; total time: 163.8 ± 8.2 s) (Figure 7a,b). Consistent with a role for seizure-induced activation of the HPA axis in comorbid depression in epilepsy, chronically epileptic Gabrd/Crh mice exhibit an increase in the latency to the first bout of immobility in the forced swim test (114.7 ± 17.6 s) and a decrease in the total time spent immobile (116.4 ± 15.4 s) compared to chronically epileptic Cre−/− mice (latency: 66.5 ± 5.5 s; total time: 232.8 ± 5.3 s) (Figure 7a,b). Further, treatment of Gabrd/Crh mice with exogenous corticosterone decreased the latency to the first bout of immobility (39.0 ± 5.2 s) and increased the total time spent immobile in the forced swim test (254.1 ± 11.0 s) (Figure 7a,b; n = 15 – 17 mice per experimental group; * denotes p<0.05 using a two-way ANOVA).
Figure 7: Decreased comorbid depression-like behaviors in chronically epileptic Gabrd/Crh mice.
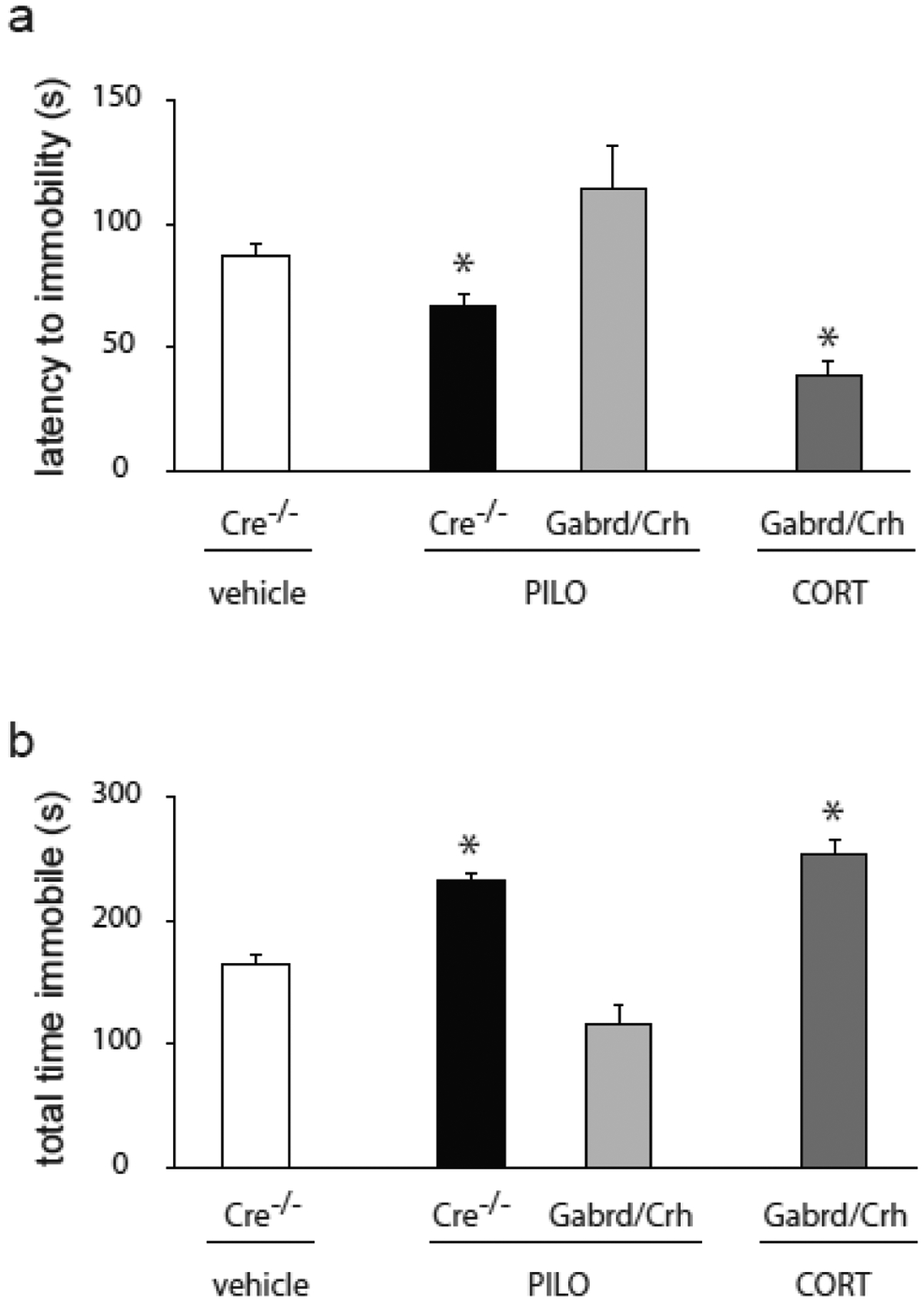
a, The average latency to the first bout of immobility in the forced swim test in chronically epileptic Gabrd/Crh mice and Cre−/− littermates, vehicle-treated controls, and corticosterone treated chronically epileptic Gabrd/Crh mice. b, The average total time spent immobile in the forced swim test in chronically epileptic Gabrd/Crh mice and Cre−/− littermates, vehicle-treated controls, and corticosterone treated chronically epileptic Gabrd/Crh mice. n = 15 – 17 mice per experimental group; * denotes p<0.05 using a two-way ANOVA.
3.6. Specifically activating or inhibiting CRH neurons in the PVN using DREADDs alters seizure frequency and comorbid depression-like behaviors
To specifically investigate the role of CRH neurons within the PVN, which govern the activity of the HPA axis, in seizure susceptibility and comorbid depression, we utilized a DREADD approach to specifically activate or inhibit these neurons. Activation of CRH neurons in the PVN with CNO (Gq DREADD) increased the number of seizures per day (9.6 ± 1.3) compared to seizure frequency in the same animals in the absence of CNO treatment (7.3 ± 1.4) (Figure 8a,b). In contrast, inhibiting CRH neurons in the PVN (Gi DREADD) decreased the number of seizures per day (3.0 ± 0.7) compared to seizure frequency in the same animals in the absence of CNO treatment (6.7 ± 1.4) (Figure 8a,b; n = 5 mice per experimental group; * denotes p<0.05 using a paired Student’s t-test).
Figure 8: Chemogenetic modulation of PVN CRH neurons alters seizure frequency and depression-like behaviors in chronically epileptic mice.
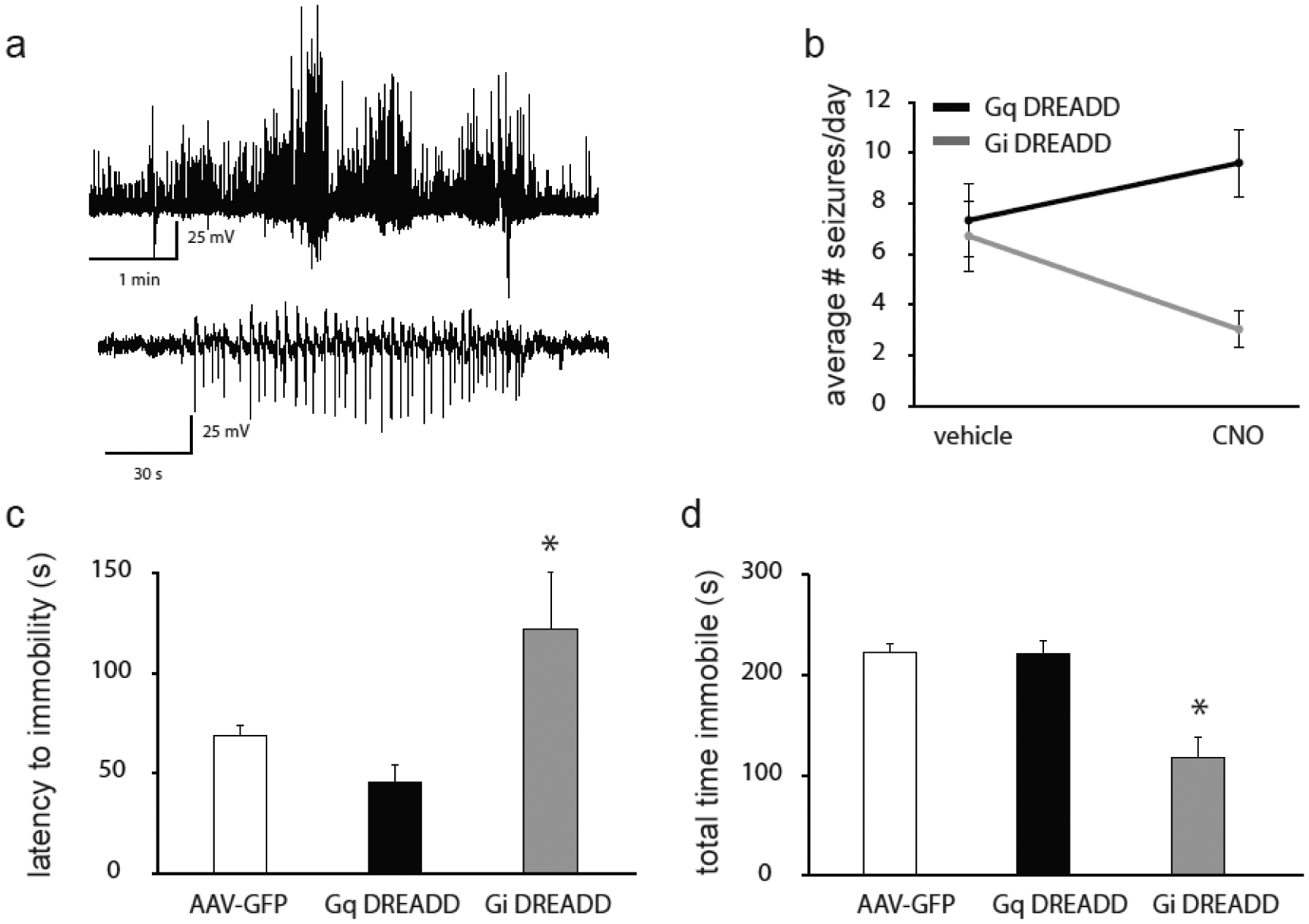
a, Representative electrographic seizures in chronically epileptic DREADD mice. a1, Representative electrographic activity corresponding to a tonic-clonic, Stage 5 behavioral seizure. a2, Representative electrographic activity corresponding to a Stage 3 behavioral seizure. b, The average number of seizures per day in Gq and Gi DREADD mice in the presence and absence of CNO. n = 5 mice per experimental group; * denotes p<0.05 using a paired Student’s t-test. The average latency to immobility (c) and the total time spent immobile (d) in the forced swim test in Gq and Gi DREADD mice and AAV-GFP controls. n = 10 – 20 mice per experimental group; * denotes p<0.05 using a one-way ANOVA.
Modulation of CRH neurons in the PVN using DREADDs also alters depression-like behaviors in chronically epileptic mice. Activation of CRH neurons in the PVN does not alter depression-like behaviors in the forced swim test in chronically epileptic Gq DREADD mice (latency to immobility: 45.7 ± 219.6 s; total time immobile: 219.6 ± 13.3 s) compared to chronically epileptic AAV-GFP mice (latency to immobility: 68.8 ± 4.8 s; total time immobile: 222.4 ± 7.6 s) (Figure 8c,d; n = 10 – 20 mice per experimental group; * denotes p<0.05 using a one-way ANOVA), which may be due to a ceiling effect. However, inhibiting CRH neurons in chronically epileptic mice decreases depression-like behaviors (Gi DREADD mice - latency to immobility: 122.0 ± 28.3 s; total time immobile: 116.4 ± 21.4 s) compared to chronically epileptic AAV-GFP mice (latency to immobility: 68.8 ± 4.8 s; total time immobile: 222.4 ± 7.6 s) (Figure 8c,d; n = 10 – 20 mice per experimental group; * denotes p<0.05 using a one-way ANOVA). These data suggest that seizure-induced activation of the HPA axis contributes to an increase in seizure frequency and associated comorbidities, consistent with the notion that dysregulation of the HPA axis negatively impacts epilepsy outcomes.
4. Discussion
This study utilizes genetic and chemogenetic approaches to modulate HPA axis function to investigate the relationship between seizure-induced activation of the HPA axis, seizure susceptibility, and associated comorbidities. Much of the research into the role of the HPA axis in epilepsy has focused on the role of stress on seizure susceptibility, given the evidence that stress is a major trigger for seizures in patients with epilepsy [11]. In addition to the impact of stress, recent studies have begun to investigate the impact of HPA axis dysfunction on epilepsy outcomes independent of stress [1, 20]. Blocking glucocorticoid receptors with RU486 has been shown to mitigate some of the neuropathological features of chronic epilepsy in mice [20]. The present study goes even further to demonstrate that genetic or chemogenetic suppression of the HPA axis decreases seizure susceptibility and comorbid depression-like behaviors.
The fact that suppression of the seizure-induced activation of the HPA axis alters seizure susceptibility in the kainic acid model suggests that seizure-induced activation of the HPA axis is capable of influencing even acute seizure susceptibility. This suggests that seizure- induced activation of the HPA axis is not due solely to long-term remodeling by episodic seizure activity, but rather acute activation of the HPA axis is sufficient to influence seizure susceptibility and severity. In contrast, the evidence that suppression of the seizure-induced activation of the HPA axis alters seizure frequency in chronically epileptic mice suggests acute intense seizure activity is not required for the seizure-induced activation of the HPA axis, but rather episodic seizures are also sufficient to activate the HPA axis and impact seizure activity. However, it is interesting to note that corticosterone levels are elevated in chronically epileptic mice (Figure 3), suggesting that there may also be HPA axis remodeling following chronic seizure activity.
Collectively, these findings force us to reevaluate the relationship between the HPA axis, epilepsy and associated comorbidities. Independent of stress, seizures activate the HPA axis, negatively impacting epilepsy outcomes. However, it is possible that stress itself may also impact the extent of seizure-induced activation of the HPA axis. It has been well established that early life stress can impact future stress reactivity [44]. Interestingly, previous studies also implicate early life stress in seizure susceptibility [45, 46]. A recent study demonstrated that early life stress decreased the threshold for status epilepticus, accelerated epileptogenesis, and increased comorbid depression-like behaviors [32]. The same study demonstrated that the animals vulnerable to increased seizure susceptibility and comorbid depression also exhibited increased corticosterone levels and increased adrenal weight [32], suggesting altered HPA axis function. Thus, the impact of stress may go beyond triggering seizures and may actually alter HPA axis function in a way that worsens seizures and associated comorbidities.
We propose that the seizure-induced activation of the HPA axis creates a proconvulsant environment, which facilitates future seizure susceptibility. The pathophysiological consequences of seizure-induced activation of the HPA axis has not been comprehensively explored. It is quite possible that seizure-induced HPA axis dysfunction may impact other aspects of epilepsy and associated comorbidities. For instance, seizure-induced activation of the HPA axis may influence seizure clustering, whereby seizure-induced elevations in stress hormones create a proconvulsant environment favoring seizure clustering. Unfortunately, we were unable to comprehensively explore this hypothesis in the current study due to a relatively limited assessment of seizure frequency in the chronically epileptic period. In order to definitively establish whether there are changes in seizure clustering, one would need to record seizure frequency over a longer time period. It is also possible that seizure-induced activation of the HPA axis may impact additional comorbidities associated with epilepsy, such as cognitive impairments and cardiovascular irregularities potentially associated with sudden unexpected death in epilepsy (SUDEP).
5. Conclusions
The data presented here demonstrate that seizure-induced activation of the HPA axis has a significant impact on epilepsy outcomes. These findings implicate HPA axis dysfunction in epilepsy beyond the largely explored impact of stress. It remains to be determined whether previous stress experience alters seizure-induced activation of the HPA axis, adding another level of complexity to the relationship between stress, the HPA axis, epilepsy, and associated comorbidities.
Highlights.
Suppression of the seizure-induced activation of the HPA axis decreases acute seizure susceptibility
Blunted seizure-induced elevation in corticosterone is associated with reduced seizure frequency in chronically epileptic mice
Suppression of the seizure-induced activation of the HPA axis decreases depression-like behaviors in chronically epileptic mice
Activation of CRH neurons in the PVN is sufficient to increase seizure susceptibility
Inhibition of CRH neurons in the PVN is sufficient to decrease depression-like behaviors in chronically epilepsy mice
Acknowledgments
J.M. was supported by NIH-NINDS grant R01NS073574 (J.M.) and R01NS102937 (J.M.). The behavioral and imaging studies were conducted in the Tufts Center for Neuroscience Research, P30 NS047243. A.H. was supported by a predoctoral fellowship from the Epilepsy Foundation and the Dean’s Award from Tufts University School of Medicine.
Financial Disclosures
Dr. Maguire receives funding from SAGE Therapeutics to perform preclinical studies, which are not directly related to the current manuscript. Dr. Maguire also serves as a member of the SAGE Therapeutics Scientific Advisory Board, a relationship that is approved and regulated by Tufts University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].O’Toole KK, Hooper A, Wakefield S, Maguire J. Seizure-induced disinhibition of the HPA axis increases seizure susceptibility. Epilepsy Research 2013;108(1):29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lee V, Sarkar J, Maguire J. Loss of Gabrd in CRH neurons blunts the corticosterone response to stress and diminishes stress-related behaviors. Psychoneuroendocrinology 2014;41:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nakken KO, Solaas MH, Kjeldsen MJ, Friis ML, Pellock JM, Corey LA. Which seizure-precipitating factors do patients with epilepsy most frequently report? Epilepsy Behav 2005;6:85–9. [DOI] [PubMed] [Google Scholar]
- [4].Sawyer NT, Escayg A. Stress and epilepsy: multiple models, multiple outcomes. J Clin Neurophysiol 2010;27:445–52. [DOI] [PubMed] [Google Scholar]
- [5].Sperling MR, Schilling CA, Glosser D, Tracy JI, Asadi-Pooya AA. Self-perception of seizure precipitants and their relation to anxiety level, depression, and health locus of control in epilepsy. Seizure 2008;17:302–7. [DOI] [PubMed] [Google Scholar]
- [6].Neugebauer R, Paik M, Hauser WA, Nadel E, Leppik I, Susser M. Stressful life events and seizure frequency in patients with epilepsy. Epilepsia 1994;35:336–43. [DOI] [PubMed] [Google Scholar]
- [7].Frucht MM, Quigg M, Schwaner C, Fountain NB. Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 2000;41:1534–9. [DOI] [PubMed] [Google Scholar]
- [8].Haut SR, Vouyiouklis M, Shinnar S. Stress and epilepsy: a patient perception survey. Epilepsy Behav 2003;4:511–4. [DOI] [PubMed] [Google Scholar]
- [9].Haut SR, Hall CB, Masur J, Lipton RB. Seizure occurrence: precipitants and prediction. Neurology 2007;69:1905–10. [DOI] [PubMed] [Google Scholar]
- [10].Lai CW, Trimble MR. Stress and epilepsy. Journal of Epilepsy 1997;10:177–86. [Google Scholar]
- [11].Maguire J, Salpekar JA. Stress, seizures, and hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy. Epilepsy Behav 2013;26:352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mackenzie G, Maguire J. Chronic stress compromises GABAergic inhibition in the hippocampus and increases seizure susceptibility. Journal of Neurophysiology. 2014. [Google Scholar]
- [13].Koe AS, Salzberg MR, Morris MJ, O’Brien TJ, Jones NC. Early life maternal separation stress augmentation of limbic epileptogenesis: the role of corticosterone and HPA axis programming. Psychoneuroendocrinology 2014;42:124–33. doi: 10.1016/j.psyneuen.2014.01.009. Epub;%2014 Jan 21.:124–33. [DOI] [PubMed] [Google Scholar]
- [14].Stress Joels M., the hippocampus, and epilepsy. Epilepsia 2009;50:586–97. [DOI] [PubMed] [Google Scholar]
- [15].Castro OW, Santos VR, Pun RYK, McKlveen JM, Batie M, Holland KD et al. Impact of Corticosterone Treatment on Spontaneous Seizure Frequency and Epileptiform Activity in Mice with Chronic Epilepsy. PLoS ONE 2012;7:e46044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Culebras A, Miller M, Bertram L, Koch J. Differential response of growth hormone, cortisol, and prolactin to seizures and to stress. Epilepsia 1987;28:564–70. [DOI] [PubMed] [Google Scholar]
- [17].Abbott RJ, Browning MC, Davidson DL. Serum prolactin and cortisol concentrations after grand mal seizures. Journal of Neurology, Neurosurgery & Psychiatry 1980;43:163–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pritchard PB, Wannamaker BB, Sagel J, Daniel CM. Serum prolactin and cortisol levels in evaluation of pseudoepileptic seizures. Ann Neurol 1985;18:87–9. [DOI] [PubMed] [Google Scholar]
- [19].Wulsin AC, Solomon MB, Privitera MD, Danzer SC, Herman JP. Hypothalamic-pituitary-adrenocortical axis dysfunction in epilepsy. Physiology & Behavior 2016;166:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wulsin AC, Herman JP, Danzer SC. RU486 Mitigates Hippocampal Pathology Following Status Epilepticus. Frontiers in Neurology 2016;7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].de Kloet ER. Hormones, brain and stress. Endocr Regul 2003;37:51–68. [PubMed] [Google Scholar]
- [22].de Kloet ER. Hormones and the stressed brain. Ann N Y Acad Sci 2004;1018:1–15.: 1–15. [DOI] [PubMed] [Google Scholar]
- [23].Steiner M, Dunn E, Born L. Hormones and mood: from menarche to menopause and beyond. J Affect Disord 2003;74:67–83. [DOI] [PubMed] [Google Scholar]
- [24].Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 2005;4:141–94. [DOI] [PubMed] [Google Scholar]
- [25].Lambert MV, Robertson MM. Depression in Epilepsy: Etiology, Phenomenology, and Treatment. Epilepsia 1999;40:s21–s47. [DOI] [PubMed] [Google Scholar]
- [26].Mazza M, Orsucci F, De RS, Bria P, Mazza S. Epilepsy and depression: risk factors for suicide? Clin Ter 2004;155:425–7. [PubMed] [Google Scholar]
- [27].Boylan LS, Flint LA, Labovitz DL, Jackson SC, Starner K, Devinsky O. Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology 2004;62:258–61. [DOI] [PubMed] [Google Scholar]
- [28].Zobel A, Wellmer J, Schulze-Rauschenbach S, Pfeiffer U, Schnell S, Elger C et al. Impairment of inhibitory control of the hypothalamic pituitary adrenocortical system in epilepsy. Eur Arch Psychiatry Clin Neurosci 2004;254:303–11. [DOI] [PubMed] [Google Scholar]
- [29].Mazarati AM, Shin D, Kwon YS, Bragin A, Pineda E, Tio D et al. Elevated plasma corticosterone level and depressive behavior in experimental temporal lobe epilepsy. Neurobiol Dis 2009;34:457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kanner AM. Depression in epilepsy: prevalence, clinical semiology, pathogenic mechanisms, and treatment. Biol Psychiatry 2003;54:388–98. [DOI] [PubMed] [Google Scholar]
- [31].Carroll BJ, Cassidy F, Naftolowitz D, Tatham NE, Wilson WH, Iranmanesh A et al. Pathophysiology of hypercortisolism in depression. Acta Psychiatr Scand Suppl 2007:90–103. [DOI] [PubMed] [Google Scholar]
- [32].Becker C, Bouvier E, Ghestem A, Siyoucef S, Claverie D, Camus F et al. Predicting and treating stress-Induced vulnerability to epilepsy and depression. Ann Neurol 2015;78:128–36. [DOI] [PubMed] [Google Scholar]
- [33].Sarkar J, Wakefield S, Mackenzie G, Moss SJ, Maguire J. Neurosteroidogenesis Is Required for the Physiological Response to Stress: Role of Neurosteroid-Sensitive GABAA Receptors. J Neurosci 2011;31:18198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacKenzie G, Maguire J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Research 2015;109:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci 2005;8:797–804. [DOI] [PubMed] [Google Scholar]
- [36].Sivakumaran S, Maguire J. Bumetanide reduces seizure progression and the development of pharmacoresistant status epilepticus. Epilepsia 2016;57:222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].White AM, Williams PA, Ferraro DJ, Clark S, Kadam SD, Dudek FE et al. Efficient unsupervised algorithms for the detection of seizures in continuous EEG recordings from rats after brain injury. J Neurosci Methods 2006;152:255–66. [DOI] [PubMed] [Google Scholar]
- [38].Maguire J, Ferando I, Simonsen C, Mody I. Excitability changes related to GABAA receptor plasticity during pregnancy. J Neurosci 2009;29:9592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee V, Maguire J. Impact of inhibitory constraint of interneurons on neuronal excitability. J Neurophysiol 2013;110:2520–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG. Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol 2002;14:929–32. [DOI] [PubMed] [Google Scholar]
- [41].Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017;357:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Danzer SC. Depression, stress, epilepsy and adult neurogenesis. Exp Neurol 2012;233:22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pineda E, Shin D, Sankar R, Mazarati AM. Comorbidity between epilepsy and depression: experimental evidence for the involvement of serotonergic, glucocorticoid, and neuroinflammatory mechanisms. Epilepsia 2010;51 Suppl 3:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].van Bodegom M, Homberg JR, Henckens MJAG. Modulation of the Hypothalamic-Pituitary-Adrenal Axis by Early Life Stress Exposure. Frontiers in Cellular Neuroscience 2017;11:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dube CM, Molet J, Singh-Taylor A, Ivy A, Maras PM, Baram TZ. Hyper-excitability and epilepsy generated by chronic early-life stress. Neurobiology of Stress 2015;2:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koe AS, Jones NC, Salzberg MR. Early Life Stress as an Influence on Limbic Epilepsy: An Hypothesis Whose Time has Come? Frontiers in Behavioral Neuroscience 2009;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]