

Abstract
Identification of the metastatic potential represents one of the most important tasks for molecular imaging of cancer. While molecular imaging of metastases has witnessed substantial progress as an area of clinical inquiry, determining precisely what differentiates the metastatic phenotype has proven to be more elusive. In this study, we utilize both the morphological and molecular information provided by 3D optical diffraction tomography and Raman spectroscopy, respectively, to propose a label-free route for optical phenotyping of cancer cells at single-cell resolution. By using an isogenic panel of cell lines derived from MDA-MB-231 breast cancer cells that vary in their metastatic potential, we show that 3D refractive index tomograms can capture subtle morphological differences among the parental, circulating tumor cells, and lung metastatic cells. By leveraging its molecular specificity, we demonstrate that coarse Raman microscopy is capable of rapidly mapping a sufficient number of cells for training a random forest classifier that can accurately predict the metastatic potential of cells at a single-cell level. We also perform multivariate curve resolution alternating least squares decomposition of the spectral dataset to demarcate spectra from cytoplasm and nucleus, and test the feasibility of identifying metastatic phenotypes using the spectra only from the cytoplasmic and nuclear regions. Overall, our study provides a rationale for employing coarse Raman mapping to substantially reduce measurement time thereby enabling the acquisition of reasonably large training datasets that hold the key for label-free single-cell analysis and, consequently, for differentiation of indolent from aggressive phenotypes.
Keywords: Raman spectroscopy, optical diffraction tomography, breast cancer, metastasis, random forests, single-cell phenotyping
1. Introduction
Timely assessment of risk is critical for the detection and treatment of metastatic disease, which remains the main reason for cancer-related mortality (Chaffer and Weinberg, 2011). The current clinical standard for assessment of metastatic risk relies on pathologic examination of sentinel lymph nodes following biopsy. In addition to being an invasive procedure, identification of metastatic cells in lymph node biopsies can be challenging and lead to an increase in false negatives (Liu, C. et al., 2019). Early detection of metastasis requires tools that can recognize the metastatic potential of cancer cells derived from the primary tumor or liquid biopsies. As primary tumors grow, a small fraction of the cancer cells termed circulating tumor cells (CTC) undergo epithelial to mesenchymal transition (EMT), locally invade the surrounding stroma, intravasate and are shed into the bloodstream leveraging their enhanced motility (Chaffer et al., 2016). A few of these cells survive in the circulation to extravasate, locally invade and form premetastatic niches in secondary organs (e.g. lungs in breast cancer), and colonize through re-acquisition of epithelial characteristics via mesenchymal to epithelial transition (MET) (Chaffer et al., 2016). While our understanding of the processes involved in metastasis has improved substantially in recent years, detecting phenotypic subtypes with metastatic competence has proven to be elusive due in part to the substantial heterogeneity observed in these cell populations (Hsiao et al., 2010). Furthermore, our understanding of what imparts metastatic potential remains rudimentary, and biomarkers that can recognize such competence across different carcinomas are still lacking.
Early genomic analyses of tumors revealed additional organ-specific mutations in metastatic tumors despite sharing common ancestors (Brastianos et al., 2015). Similarly, transcriptional analyses of breast cancer metastasis to various organs including lungs and brain have also identified largely distinct signatures characteristic of organotropism (Bos et al., 2009, Minn et al., 2005). However, these population-based analyses require elaborate sample preparation and fail to capture the variations in phenotypes at a single-cell level. Recently, we and others have also investigated the physical properties associated with the differences in metastatic phenotypes in specialized microfluidic platforms (Liu, Z. et al., 2020, Yankaskas et al., 2019, Ma et al., 2018, Che et al., 2017, Sarioglu et al., 2015b, Sarioglu et al., 2015a, Jeon et al., 2015, Han et al., 2007). The pursuit of isolating CTC from blood to determine the course of metastatic disease has resulted in the proliferation of several cell labeling methods that leverage known epithelial markers for identification (Labib et al., 2018, Poudineh et al., 2017, Green et al., 2016, Ozkumur et al., 2013, Stott et al., 2010, Hu et al., 2005). However, the use of epithelial markers may not be sufficient to detect CTC that undergo EMT to acquire mesenchymal properties, particularly in triple-negative breast cancers (Gao et al., 2018). Also, the sensitivity of these methods is challenged by the small number of known markers of metastatic progression that can be targeted simultaneously.
To address these challenges, several techniques based on optical microscopy and imaging have attempted label-free phenotyping of cancer cells (Lam et al., 2019, Winnard Jr et al., 2017, Basu et al., 2014, Di et al., 2014, Chhetri et al., 2012). For example, phenotypic changes of 4T1 murine breast cancer cells in response to drug treatment were characterized in terms of morphological parameters extracted from fluorescence images in three-dimensional (3D) cultures (Di et al., 2014). Rohde and co-workers have developed an automated platform for morphological analysis of cellular phenotypes using transport-based morphometry (Basu et al., 2014), which we recently used to analyze the quantitative phase images of activated and naïve CD8+ T cells (Karandikar et al., 2019). Similar optical methods have also been leveraged for single-cell analysis of cancer phenotypes (Hai et al., 2019, Kim, M. G. et al., 2017), which often require large datasets for building robust prediction models.
In this study, we employed 3D optical diffraction tomography (ODT) and label-free Raman spectroscopy to quantitatively investigate both morphological and molecular differences between isogenic breast cancer cells of varying metastatic potential. We used a set of three isogenic cell lines composed of the parental MDA-MB-231 triple-negative breast cancer cell line (P231), circulating tumor cells (CTC), and lung metastatic cells (LM) where the latter two were derived respectively from the circulation and lungs of a mouse bearing parental P231 cells (Rizwan et al., 2018, Rizwan et al., 2015). Compared to the widely used qualitative phase imaging methods, such as Zernike phase contrast and differential interference contrast, quantitative phase imaging methods recover the phase delay caused by the sample, decoupled from absorption information. ODT is a form of quantitative phase imaging that allows morphological analysis of single cells based on their 3D refractive index (RI) profiles (Kim, K. et al., 2013, Sung et al., 2009, Choi, Wonshik et al., 2007). In addition to providing traditional measures of morphology such as area and aspect ratio, the RI information allows a label-free and non-contact route for the determination of cell dry mass and local thickness of specimens with nanometric sensitivity (Popescu et al., 2008). The additional morphological insights provided by optical diffraction tomography have been increasingly exploited for label-free and stain-free in vitro analysis of cells and tissues (Lee et al., 2017, Kim et al., 2013, Choi et al., 2007). Yet, most of the cellular studies have focused on either visualization of morphological dynamics in response to external stimuli such as drug exposure in single-cells or rapid identification of cells such as bacteria and white blood cells using deep learning by leveraging large datasets (Park et al., 2018, Jo et al., 2018). Its utility in assessment of phenotypic differences among closely related mammalian cancer cells, particularly in data-limited settings, remains largely unexplored.
Raman spectroscopy, on the other hand, provides a label-free route for assessment of biological specimens with exquisite molecular specificity (Paidi, Santosh K. et al., 2020, Kong et al., 2015). This optical technique, based on the inelastic scattering of light, probes vibrational modes of molecules and allows direct profiling of molecular composition of biological specimens including live cells and tissues in their native states (Paidi et al., 2020, Kong et al., 2015, Stone et al., 2002). The simple integration of Raman spectroscopy with optical microscopy facilitates seamless vibrational spectroscopic imaging at diffraction-limited spatial resolution with subcellular resolution. Several groups including our own laboratory have exploited the high resolution and rich molecular information afforded by Raman spectroscopic imaging to study the molecular progression of cancer (Paidi et al., 2020, Paidi, Santosh K. et al., 2019, Paidi, Santosh Kumar et al., 2017, Winnard Jr et al., 2017, Kann et al., 2015). Due to the low likelihood of spontaneous Raman scattering, most single-cell imaging studies have focused on employing nanoparticles for plasmonic enhancement of signals and selective tagging of subcellular regions of interest (Xu et al., 2018, Kang et al., 2015). Therefore, only a few studies have attempted label-free characterization of cells for studying biological processes associated with physiological changes, disease progression, and drug response (El-Mashtoly et al., 2015, Okada et al., 2012, Hamada et al., 2008). Our recent label-free Raman investigation in pellets of isogenic breast cancer cell lines that exhibit organotropism to brain, liver, lung, and spine revealed distinct metastatic organ-specific spectral signatures that were confirmed by metabolomics analysis (Winnard Jr et al., 2017). Due to the long acquisition time, label-free Raman spectroscopic studies have either exploited high-resolution single-cell maps for analysis of limited cells or bulk sampling of cell populations that permits the use of machine learning algorithms for classification problems by generating large datasets at the cost of spatial information. This tradeoff between obtaining higher spatial resolution maps and acquiring sufficiently large datasets amenable for machine learning has largely prevented the use of machine learning techniques to learn and predict cellular phenotypes from Raman images with single-cell analytical resolution.
Therefore, in this study, we sought to test whether morphological attributes encoded by ODT and biomolecular insights obtained using Raman spectroscopy can predict the phenotype of the closely related isogenic cell lines P231, CTC and LM of varying metastatic potentials with statistical confidence. A variety of data analysis and machine learning techniques have been leveraged to unravel latent information from optical spectroscopy and imaging datasets (Paidi et al., 2020). Here, we use random forest classifiers and multivariate curve resolution – alternating least squares (MCR-ALS) analysis, respectively, for developing classification routines and identifying spectra that are rich in contributions from specific subcellular regions. Random forests are ensemble classifiers that employ a collection of decision trees constructed by random sampling of instances and variables in each tree to yield fast and generalizable models that are void of dependence on specific features or training instances (Parmar et al., 2018). Due to these characteristics and the ability to parallelize the tree construction, random forest classifiers are gaining attention in image classification and vibrational spectroscopy (Mittal et al., 2018). MCR-ALS analysis allows decomposition of spectral datasets into pure component-like loading spectra and their scores under positivity constraints for each spectrum in the dataset (Felten et al., 2015).
Using the 3D RI profiles obtained from ODT of single cells, we compared the distributions of morphological parameters such as area, aspect ratio, and dry mass across the three classes, and used their combination to train and test random forest classifiers for automated identification. By leveraging coarse Raman sampling of single cancer cells to reduce the acquisition time and obtain spectral maps from a larger number of cells, we explored the intersection of the abovementioned resolution-sampling tradeoff to find a solution for identifying metastatic phenotype of cancer cells with single-cell analytical resolution. To show that coarse Raman maps capture sufficient information for achieving single-cell phenotyping, we used random forests to iteratively test spectral maps of individual cells against classifiers trained on the data from the remaining cells in the dataset. Furthermore, we used MCR-ALS analysis to identify the spectra from subcellular compartments and test the utility of random forest classification in predicting the metastatic phenotype when only spectra from either nucleus or cytoplasm are available. The ability to use specific subcellular regions of the cell for chemical imaging is expected to further reduce the spectral acquisition time and boost the number of cells in the training dataset to capture population heterogeneity. Such label-free identification of cells with high metastatic competence would not only have a profound impact on the prediction of a patient’s risk of developing metastasis but also inform the design of optimal, personalized therapeutic treatments.
2. Materials and methods
Cell culture
An isogenic panel of varying metastatic potential derived from the human breast cancer cell line MDA-MB-231 was used in this study. In addition to the td-Tomato expressing parental MDA-MB-231 cells (P231), the panel consisted of CTC and LM cells previously obtained after orthotopic implantation of the parental cells in the fourth right mammary fat pad of female athymic nu/nu female mouse (NCI) as detailed in our previous publications (Rizwan et al., 2018, Rizwan et al., 2015). The three cell lines were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin and maintained at 37 °C and 5% CO2 in a humidified incubator.
Optical diffraction tomography and data analysis
The three cell lines were seeded in glass coverslip-bottom Petri dishes for tomography. The morphological assessment of the cells was performed on an ODT system (HT-1H, Tomocube Inc., Republic of Korea) comprised of a 60X water-immersion objective (1.2 NA), an off-axis Mach-Zehnder interferometer with a 532 nm laser and a digital micromirror device (DMD) for tomographic scanning of each cell (Shin et al., 2015). The 3D RI distribution of the cells was reconstructed from the interferograms using the Fourier diffraction theorem as described previously (Kim, K., Yoon et al., 2013). TomoStuido (Tomocube Inc, Republic of Korea) was used to perform reconstruction and visualization of 3D RI maps and their 2D maximum intensity projections (MIP). The 2D MIP images were segmented using CellProfiler™ (v3.1.9) software to isolate single-cells using Otsu two-class thresholding and neglecting the partial cells at the boundaries of raw images (McQuin et al., 2018, Otsu, 1979). After segmentation, we obtained 57, 35, and 44 cells in P231, CTC, and LM classes respectively. The area of each cell was calculated by counting the number of non-zero pixels in their corresponding segmentation masks generated by the CellProfiler™ software (Wu et al., 2015). Similarly, the perimeter and aspect ratio were calculated respectively as the number of non-zero pixels at the edges of the masks and the ratio of major and minor axes lengths (Wu et al., 2015). The cell dry mass was calculated from the 3D RI profile (Phillips et al., 2012). The morphological parameters were used to train a random forest classifier with 100 trees using the MATLAB TreeBagger class and inspect the out-of-bag-error.
Raman spectroscopic imaging
The cells from three different passages (biological repeats) for each class were seeded on quartz slides (1 in × 1 in) coated with poly-lysine and incubated overnight to facilitate cell attachment for Raman imaging. The cells were fixed using 4% paraformaldehyde and washed prior to imaging in phosphate buffered saline (PBS) at room temperature. Five cells from each slide (technical repeats) were randomly selected for Raman mapping. The coarse single-cell Raman imaging experiments were performed on a HORIBA XploRA PLUS confocal Raman microscope. A 532 nm diode laser was used for excitation and delivered to the sample via a 60X water immersion objective (1.2 NA). The backscattered Raman light was dispersed using an 1800 lines/mm diffraction grating and imaged on a thermoelectrically cooled CCD coupled to the microscope. The spectra were acquired from the points on a coarse rectangular grid overlaid on each single cell to obtain spectra from various subcellular regions and capture intracellular spatial heterogeneity. Each spectrum in the fingerprint region (600–1950 cm−1) was acquired by exposing the sample to a laser power ca. 1 mW at each point for 2.5s (5 accumulations of 0.5s exposure). An average of 67 spectra were measured from each single cell in the dataset. Together with the extra spectra collected from points outside the cell on the rectangular grid, the average spectral acquisition time for each cell was under 4 minutes. The coarse Raman spectral acquisition, therefore, reduces the time of acquisition substantially as only a small number of spatial points are sampled within each cell as opposed to imaging the entire cell at a high resolution. For example, even imaging at a pixel size of 1μM with the same acquisition parameters would require about ca. 15 minutes (2.5s × 400 spectra) for each cell.
Raman data analysis
All the Raman spectral analysis was performed in MATLAB 2017b (Mathworks) environment. Spectroscopic imaging of each cell provided a hyperspectral dataset, where each pixel on the rectangular mapping grid corresponds to a Raman spectrum. The hyperspectral datasets from all the cells were unfolded (by preserving the spatial information and cell identity) and concatenated to form a combined spectral dataset for further analysis. The spectra in the fingerprint region were subjected to background subtraction using a fifth-order best-fit polynomial-based fluorescence removal method and cosmic ray removal using median filtering on the groups of spectra from each cell. Next, the points on the mapping grid exterior of the imaged cell were identified by Otsu thresholding on the 1452 cm−1 peak (CH2 bending mode of proteins) intensity and labeled separately for further analysis. The spectra were finally vector normalized to remove the variations in laser power across the experiments.
To identify spectra from specific subcellular regions, we performed MCR-ALS analysis for decomposing each spectrum into its constituents by iterative fitting under nonnegativity constraints on the obtained component spectra (loadings) and their contributions (scores) (Felten et al., 2015). The components were identified as rich in cytoplasm, nucleus, and background (quartz and water) characteristics and confirmed by re-constructing the score maps for each cell. Each spatial location in the cell is assigned either cytoplasm, nucleus, mixed or background based on the Otsu thresholding of the cytoplasm-like and nucleus-like component scores, which were both negatively correlated (Otsu, 1979). The scores corresponding to cytoplasm-like and nucleus-like constituents were compared across the three cell lines through violin plots with outlier suppression for clarity. The significance of differences between medians was determined using Wilcoxon rank-sum test with the conventional threshold.
Random forest classifiers (bootstrap-aggregated or bagged decision trees) were trained using the TreeBagger class in MATLAB to enable the identification of the metastatic phenotypes. We chose this implementation as it provides options to invoke Breiman’s original random forest algorithm and use parallel computing for improved training speeds on larger datasets in the future (Breiman, 2001). It is worth noting that similar performance can also be achieved using other available implementations of the algorithm. We used a leave-one-cell-out protocol by leaving one cell out each time as a test case and training random forest classifiers on spectra from the remaining cells. One hundred iterations (~4s per iteration) of training were performed for each test case by selecting randomized subsets of training data to ensure equal membership for all the three classes and to avoid overtraining for the class with high data availability. The spectra of the excluded cell were subjected as a test dataset and the class label for the entire cell was determined according to the following class assignment criterion. Since the test dataset (left-out cell) remained the same for all training iterations, the median of predicted labels for each spectrum was used for decision making at the cell level. For each cell, the majority class was assigned as the predicted class if its membership was at least 30% higher than the random chance prediction and 30% higher than the membership of the second majority class. If these conditions were not met by the majority class, the test cell was labeled unclassified. To verify the sufficiency of cytoplasm and nucleus spectra alone for the identification of metastatic phenotype, the random forest classifiers were separately run on the cytoplasm- and nucleus-rich spectra identified by the MCR-ALS analysis, in addition to running them on the entire spectral dataset.
3. Results and discussion
The availability of isogenic breast cancer cells of varying metastatic potential derived from the same MDA-MB-231 human breast cancer cell line (Fig. 1A) enabled us to investigate the utility of label-free optical imaging for the identification of metastatic phenotypes at the single-cell level. We used parental P231 cells along with their circulating (CTC) and lung metastatic (LM) variants to assess the efficacy of ODT (Fig. 1B) and Raman spectroscopy (Fig. 1C) for capturing the phenotypic differences in terms of their morphological and molecular attributes. Our previous characterization of these cell lines confirmed their distinct metastatic abilities commensurate with the stage and organ from which they were isolated (Rizwan et al., 2018). Our recent investigation of the biophysical properties of these cells revealed that the LM cells are most motile and least stiff, which bestow them with unique invasive capability (Liu et al., 2020).
Figure 1. Label-free identification of metastatic phenotypes.
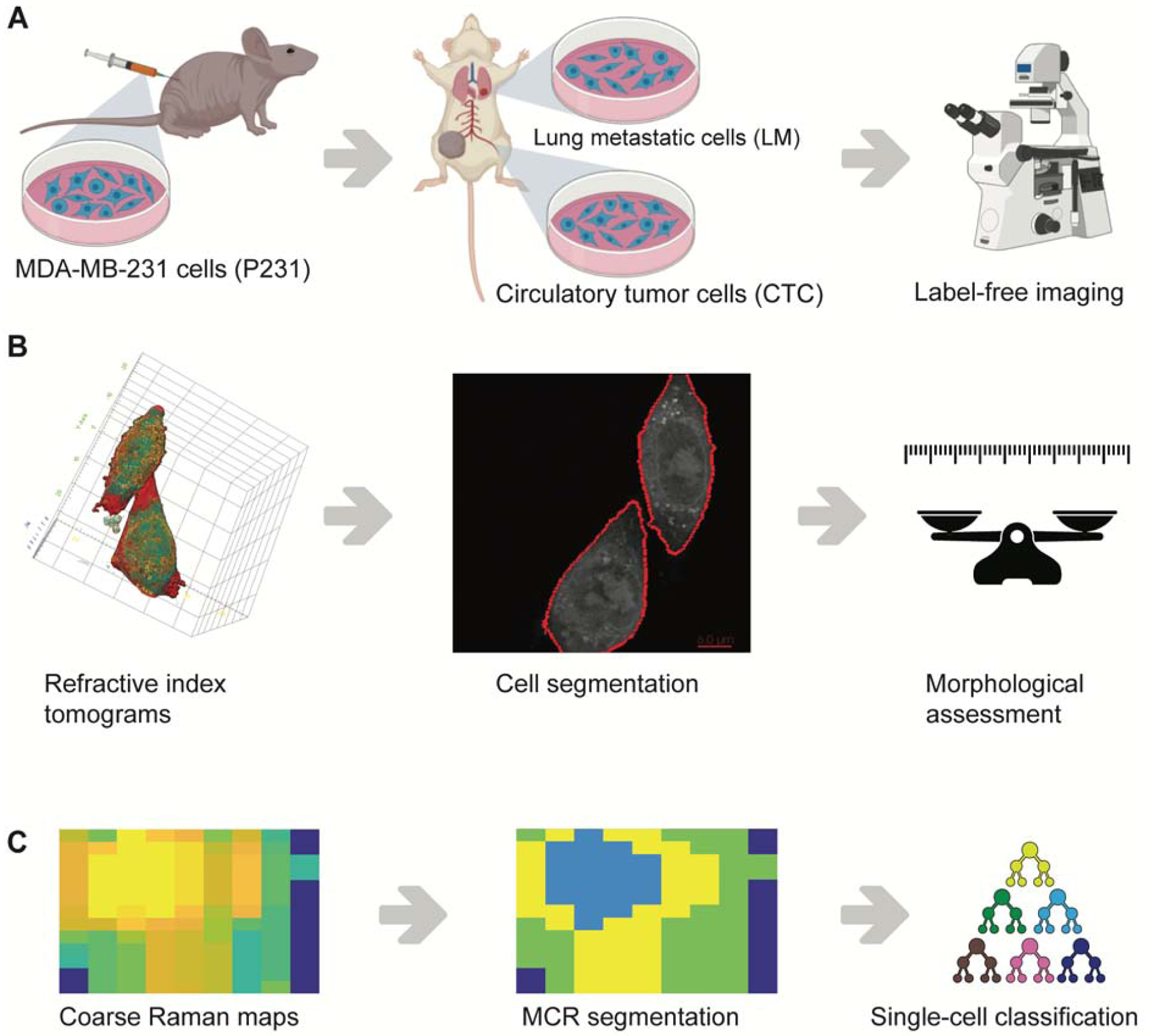
(A) Circulating tumor cells (CTC) and lung metastatic cells (LM) used in the study were isolated from the blood and lungs of mice bearing parental MDA-MB-231 (P231) tumor xenografts. (B) Refractive index tomograms were segmented to isolate single-cells for morphological assessment. (C) Coarse Raman maps of single-cells were subjected to MCR-ALS analysis to identify subcellular regions rich in cytoplasm and nucleus prior to the use of supervised classification using random forests.
To understand the morphological differences among the phenotypically distinct isogenic cell lines, we acquired 3D RI tomograms of single cancer cells belonging to each group. While the RI tomograms of cells from all three cell groups (Fig. 2A–C) show expected intracellular heterogeneity arising from RI variations across subcellular compartments, the intercellular differences are not apparent from gross visual inspection. Therefore, the maximum intensity projections of the RI tomograms (Fig. 2D–F) were subjected to further assessment using CellProfiler™ software to quantify morphological parameters such as area and aspect ratio. Also, we calculated the cell dry mass directly from the 3D RI tomograms to include an additional dimension in the morphological analysis that cannot be readily measured from brightfield or phase contrast microscopy. As seen in Fig. 2G, we observed that the area of the cells increased steadily with the increase in metastatic potential from P231 to LM. However, a significant increase in aspect ratio (Fig. 2H) was only observed for the CTC in comparison to the P231 and LM classes, while the differences between the latter were not statistically significant. These observations are consistent with the characteristics of EMT and MET processes in metastasis of P231 cells to lungs that respectively result in the acquisition of a spindle shape by the CTC for enhanced motility to reach the metastatic site and re-acquisition of epithelial shape for promoting the proliferation of LM cells to form metastatic tumors (Chaffer et al., 2016). We observed that the cell dry mass (Fig. 2I) increased with the metastatic potential of the isogenic cells, but the difference was statistically significant only for LM cells in comparison to P231 and CTC. The increase in the cell dry mass of the LM cells is consistent with the prior observation of an increased RI and cell dry mass of cancer cells in comparison with normal cells due to the higher accumulation of proteins associated with the higher proliferation of the former group (Choi, Woo June et al., 2010). Our observation expands this idea to the metastatic regime and provides a rationale to explore cell dry mass as a potential biomarker of invasiveness in future studies.
Figure 2. Morphological assessment of metastatic phenotypes.
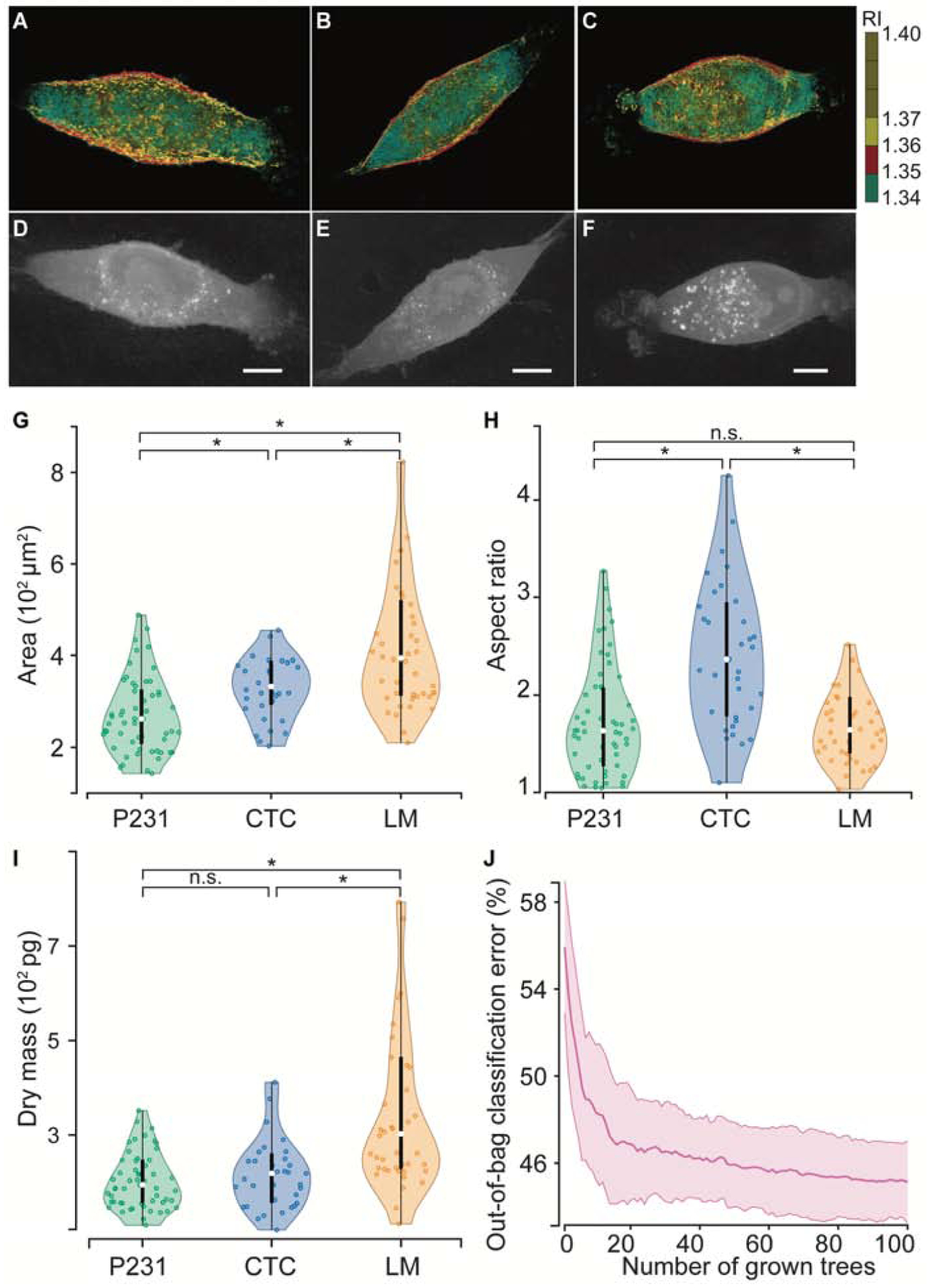
Representative 3D refractive tomograms of (A) P231, (B) CTC, and (C) LM cells show the intracellular variation of the refractive index. The maximum intensity projections of (D) P231, (E) CTC, and (F) LM were used for determining the 2D morphological parameters. The violin plots show the variations in (G) area, (H) aspect ratio, and (I) dry mass across the three classes. (J) The out-of-bag classification error plot, which illustrates the evolution of error as a function of number of decision trees included in the forest, shows that random forests built on the morphological parameters fail to accurately predict metastatic phenotypes. The scale bars represent 5 μm. * represents statistically significant differences at p < 0.05 threshold (Wilcoxon rank-sum test), whereas n.s. indicates a lack of statistically significant differences.
While the phenotypically distinct cell lines showed variable differences in individual morphological parameters, their utility for identifying phenotypes of single cancer cells is dependent on the existence of clear class boundaries between the three classes. The violin and box plots (Fig. 2G–I) show the appreciable overlap in the distribution of each morphological parameter across the metastatic potential thus making univariate analyses challenging for class separation. While prior studies have leveraged deep learning methods for cellular classification based on latent morphological features from the complete cell images, they have largely probed simpler systems such as bacteria, blood cells, immune cells, and anthrax spores compared to the current cohort of isogenic breast cancer cells (Jo et al., 2018, Park et al., 2018). Since our current study is focused on the detection of the cellular phenotypes within the constraints of small training datasets, we trained random forest classifier to test if supervised models leveraging these three morphological parameters can accurately predict the metastatic phenotype of test samples. The out-of-bag classification error rate (Fig. 2J), calculated for each training sample by testing them against the decision trees in the forest that did not use them for training, was found to asymptotically plateau around 46% (compared to 33.3% random chance). These results indicate that while phenotype differences among the isogenic cells show subtle but significant morphological differences, they are not sufficient for robust classification of closely related cells at a single-cell analytical resolution, particularly when the training data is relatively scarce.
Next, we sought to check if molecular information provided by vibrational spectroscopy can enable the identification of metastatic phenotypes at a single-cell analytical resolution. Therefore, to build a dataset large enough for training machine learning models, we performed coarse Raman microscopy of all three isogenic cell lines. The spectral dataset consisted of ca. 3000 spectra collected from 45 cells across the three cell types. The entirety of each cell was mapped coarsely (average of 67 pixels per cell) to capture intracellular heterogeneity. While these coarse Raman images (Fig. 3A) do not offer diffraction-limited spatial resolution, they capture enough information for the cell classification task and help significantly reduce the spectral acquisition time for each cell. The mean (+/− 1 s.d.) of the spectra from the three cell lines (Fig. 3B) show prominent peaks at 931 cm−1, 1003 cm−1, 1085 cm−1, 1303 cm−1, 1450 cm−1, 1658 cm−1 indicative of the common biological constituents of cells and tissues (Movasaghi et al., 2007). Since there are no discernible visible differences between the spectra of the three cell lines, we used MCR-ALS analysis to decompose the spectra into component spectra and their scores. MCR-ALS decomposition allows representation of each spectrum in the dataset as a weighted sum of iteratively generated pure component-like basis spectra, without requiring any composition estimates as inputs (Felten et al., 2015). In this study, a simple three-component MCR-ALS decomposition provided component loadings harboring features of cytoplasm, nucleus, and quartz background from the slide on which the cells were cultured (Fig. 3C). We identified MC1 and MC2 as loadings resembling cytoplasm and nucleus due to the prominence of cytoplasm features at 1003 cm−1 (C–C stretching vibration of the aromatic ring in the phenylalanine side chain) and 1639 cm−1 (amide I feature in proteins) in the former and nucleic acid features at 788 cm−1 (O-P-O stretching in DNA) and 1092 cm−1 (symmetric PO2− stretching in DNA) in the latter. The assignment is also justified by the strong negative correlation between the MC1 and MC2 scores for each cell, assessed by an average correlation coefficient of −0.98 over all the cells in the study.
Figure 3. MCR segmentation of single-cell Raman images.
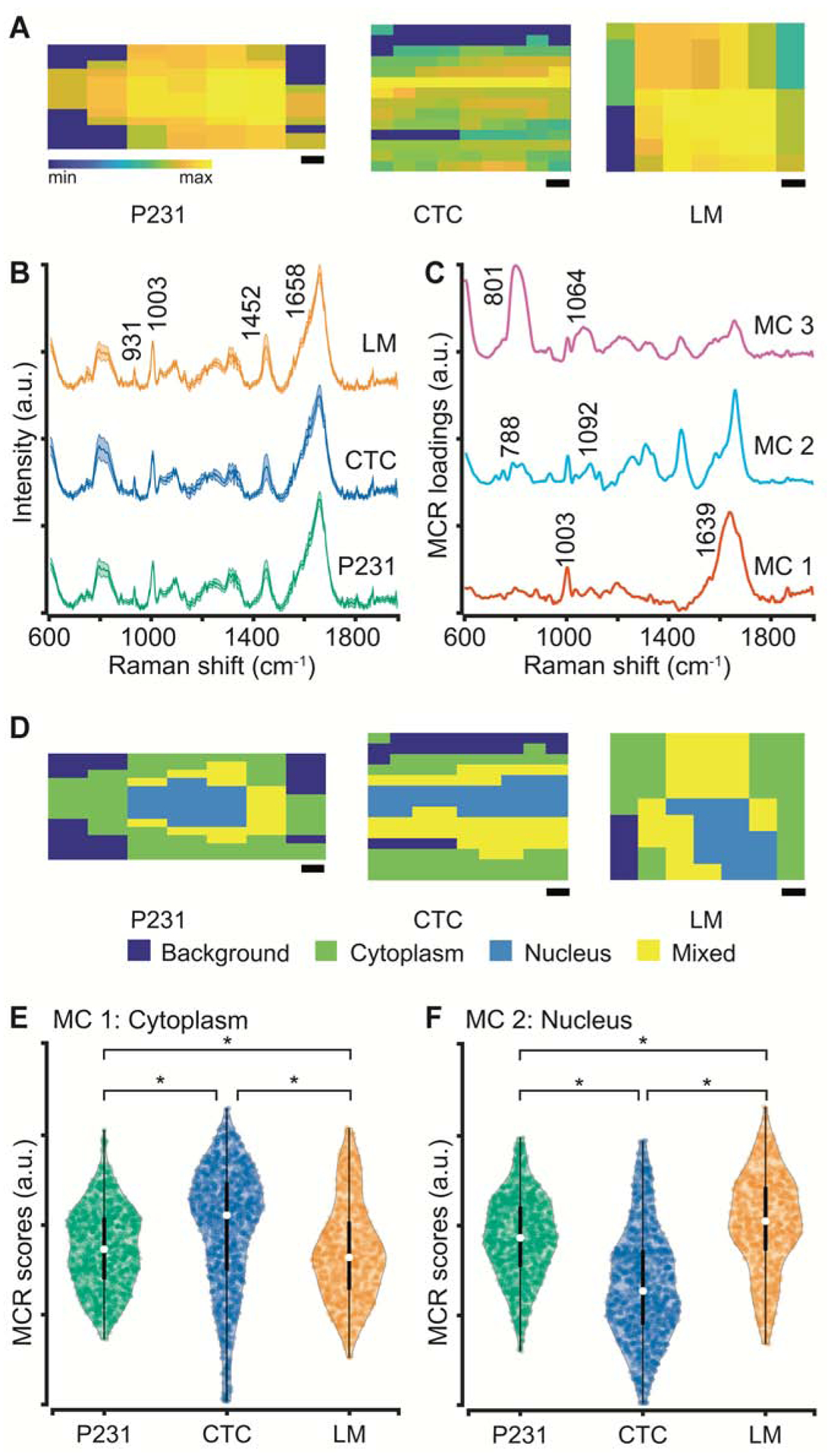
(A) Representative coarse Raman maps reconstructed using the 1452 cm−1 peak intensity shown for P231, CTC, and LM cells. (B) Mean Raman spectra (with the shadow representing 1 s.d. and vertical offset for clarity) are shown and some prominent biological peaks highlighted for the three isogenic cell lines used in the study. (C) The three MCR component loadings derived from the combined spectral dataset are shown. MC1, MC2, and MC3 respectively show cytoplasm-like, nucleus-like, and quartz background spectral features. (D) The segmentation maps constructed by thresholding on MCR component scores for the cells in panel A are shown. The violin plots with embedded box and whisker plots show the distribution of MCR scores for cytoplasm-like (E) and nucleus-like (F) loadings. The scale bars represent 2 μm. * represents statistically significant differences at p < 0.05 threshold (Wilcoxon rank-sum test).
We further verified the assignment by reconstructing the abundance maps for the scores of MC1 and MC2. We assigned each pixel as cytoplasm, nucleus, or mixed by thresholding on the MC1 and MC2 scores. Using this MCR-ALS decomposition of spectral dataset allows better visualization of the spatial demarcation between cytoplasm and nucleus, which was not apparent in the coarse Raman maps at individual wavenumbers. The identification of pixels as those rich in cytoplasm and nucleus allow us to dissect the heterogeneous single-cell Raman measurements into relatively homogenous subsets for identifying subcellular compartments that capture the information necessary for identification of metastatic phenotypes. The remaining loading MC3, showing features at 801 cm−1 and 1064 cm−1, captures the minor contributions of quartz substrate in the cell spectra. We compared the scores of the cytoplasm-like and nucleus-like components to understand the relative abundance of these components in the cells of varying metastatic potential. The violin plots of MC1 and MC2 show that while the median values for the cytoplasm scores are significantly higher for the CTC in comparison to the P231 cells, the median for the LM cells is significantly lower in comparison to both P231 and CTC groups. Since the nucleus scores are negatively correlated with the cytoplasm scores, their medians show an opposite trend. The similarity of P231 and LM scores and their deviation from CTC hint at the ability of Raman spectroscopy to identify the differences associated with the EMT and MET processes that make CTC dissimilar to the P231 and LM cells. These observations are consistent with our prior characterization of these cell lines that showed significant differences in the mRNA expression levels of osteopontin (OPN), CD44, and vimentin (VIM) – genes involved in EMT, cell migration, extracellular matrix organization, and cell adhesion – in CTC and LM cells compared to the P231 cells (Rizwan et al., 2018). Additionally,, the observed statistically significant differences in the MC component scores provide a rationale for exploring supervised classification techniques for the determination of metastatic phenotypes in the studied cancer cell lines.
We employed a multiclass random forest classifier to quantify our ability to classify P231, CTC, and LM cells based on the biochemical information encoded in their Raman spectra First, we subjected the entire spectral data consisting of spectra from all subcellular compartments (cytoplasm, nucleus, and mixed) to a leave-one-cell-out random forest classification task (as described in Methods). Briefly, we trained the classifier iteratively by leaving spectra from one cell at a time from the training dataset and subjecting it as a test dataset to the developed model. We observed satisfactory prediction performance across the three classes with only 4 misclassifications and 1 unclassification among 45 cells (Fig. 4A). The majority of misclassifications occurred between P231 and LM classes, while all CTC were classified accurately. This observation is in agreement with the similarity of both cytoplasm and nucleus MCR scores for P231 and LM classes and their deviation from CTC. The misclassifications of P231 cells can also be attributed to the presence of cells in the P231 group that have future propensity to intravasate into circulation and colonize lungs (i.e. future CTC and LM cells). Unlike most of the previous studies where the classification of spectra is done at a bulk level (Qiu et al., 2020, Winnard Jr et al., 2017, Marro et al., 2014), the leave-one-cell-out analysis allowed us to demonstrate not only the ability to identify metastatic potential at a single-cell level but also the robustness of such classification by completely excluding representation of the test data from the training dataset. Therefore, we believe that the classification accuracy of the leave-one-cell-out analysis can be further improved by mapping a larger number of cells to boost the training dataset and to better capture intercellular variability within and across classes.
Figure 4. Leave-one-cell-out random forest classification of Raman images at a single-cell level.
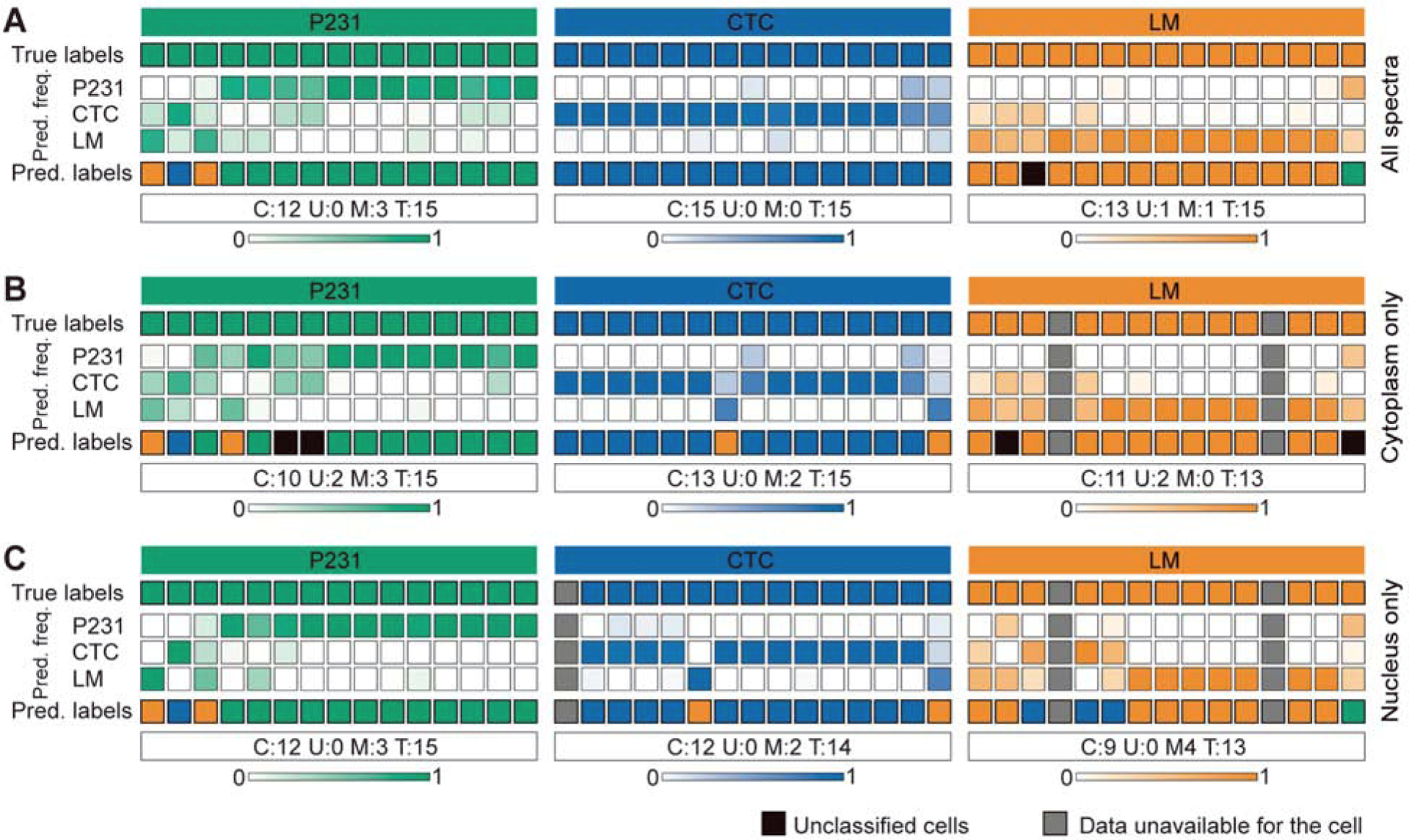
The leave-one-cell-out random forest predictions are shown for the multiclass classification task by including (A) all the spectra in the dataset, (B) spectra with high cytoplasm MC scores, and (C) spectra with high nucleus MC scores. Each column represents one unique cell, while the top and bottom rows respectively show the true and predicted class labels. The other rows show the normalized prediction frequencies (color bars at the bottom represent color scales) of spectra from each cell into the three classes. The classification results are summarized for each class to include the number of cells correctly classified (C), unclassified (U), and misclassified (M) out of the total (T) cells in the class.
While the leave-one-cell-out analysis provided an excellent prediction of phenotype for the cells in all the three classes, the use of the entire dataset comprised of spectra from different subcellular regions introduces substantial intra-class heterogeneity in the training dataset and may make prediction challenging. Such difficulty can be further exacerbated if the target phenotype changes are specifically guided by local molecular variations in particular regions, for example in the nucleus of cells treated with chemotherapeutic drugs. While there is no direct attribution of metastatic phenotypes observed in the isogenic panel employed in this study to specific compartments, we sought to train and test the random forest classifiers using subsets of the spectral dataset from the regions identified as cytoplasm and nucleus using MCR-ALS decomposition. The deviation of the observations from the baseline results obtained by subjecting the entire dataset will provide preliminary insights into specific localization of changes in subcellular regions that render the CTC and LM cells more metastatic in comparison to the parental P231 cells. First, we restricted our analysis to include only spectra that exhibit high scores for cytoplasm-like loading (MC1) in training and test datasets. The leave-one-cell-out analysis of the cytoplasm spectra (Fig. 4B) from the three classes yielded similar predictions with a slight improvement in the classification of LM cells, where a previously misclassified cell was now unclassified due to the relative increase of spectral classification into LM group. However, we found new unclassifications and misclassifications, respectively, in the P231 and CTC classes. Next, we performed the leave-one-cell-out analysis on the subset of dataset comprised only of spectra that show high scores of nucleus-like loading (MC2). We observed that the exclusion of cytoplasm spectra (Fig. 4C) resulted in the deterioration of performance in CTC and LM classes without affecting the P231 classification. Together, these results show that while the prediction of P231 and LM cells are primarily driven by the spectra acquired from the nucleus and cytoplasm respectively, the classification of CTC is more challenging and requires spectra from both regions. While these observations are preliminary and require further investigation in a larger cohort of cells, the results hint at the sufficiency of spectra from specific subcellular regions to predict subtle phenotypic differences associated with metastatic potential in closely related isogenic cells. In the present study, our experimental approach involved manual selection of randomly measured cells and the rectangular mapping grid for each cell. However, the true potential of the proposed coarse and selective subcellular mapping approaches can be realized through real-time multimodal integration of Raman microscopy with widefield imaging techniques such as ODT for identification of subcellular regions of interest and smart mapping for selective spectral acquisition from identified regions. Such developments will help reduce measurement time by allowing automatic stage translation between cells and avoiding spectral collection outside the cells.
4. Conclusions
In conclusion, our label-free optical study revealed morphological and molecular differences among isogenic breast cancer cells of progressively increasing metastatic potential. Using 3D RI tomograms, we showed that the parental P231, circulating CTC, and lung metastatic LM cells showed subtle yet significant variations in morphology as assessed by area, aspect ratio, and cell dry mass. The observations were consistent with prior evidence of EMT and MET processes that guide the metastatic progression of these MDA-MB-231 breast cancer cells. To uniquely predict the metastatic potential of these cells with single-cell analytical resolution, we used Raman spectroscopic imaging to capture their biomolecular composition along with the spatial details. The use of MCR-ALS decomposition allowed better visualization and demarcation of the nucleus and cytoplasm despite the low resolution of the coarse Raman images. Finally, our random forest classification models incorporating a leave-one-cell-out strategy provided a route identification of subtle metastatic phenotype of cells at a single-cell level based on the coarse Raman maps. Further classification using the spectra individually from cytoplasm and nucleus regions as identified by MCR-ALS decomposition showed that specific subsets were sufficient for the identification of metastatic phenotypes. Taken together, these studies show that optical imaging and spectroscopy are sensitive to the differences in the cellular states guided by biological processes. The ability to non-destructively probe single live cells and the lack of extensive sample preparation are the key advantages of the proposed technique over other approaches that may be used to study such subtle molecular differences in isogenic cells. We envision that coarse Raman imaging will be leveraged to build large spectral datasets from clinical samples that are amenable to machine learning analysis for determination of biomolecular phenotype/variant at a single-cell resolution to avoid loss of information associated with the population analyses. The developed approach can be readily integrated with emerging microfluidics platform to achieve simultaneous biophysical and biomolecular characterization of single cells and rapid identification of clinically relevant cellular phenotypes. The imaging protocol and leave-one-cell-out random forest routine can readily be extended to the investigation of a variety of phenomena such as drug response, stem cell differentiation, and immune cell activation.
Highlights.
Label-free morpho-molecular imaging, in combination with machine learning, allows phenotyping of isogenic breast cancer cells of varying metastatic potential.
Optical diffraction tomography captures subtle morphological differences consistent with the metastatic attributes of the panel of isogenic MDA-MB-231 cells.
Coarse Raman imaging provides sufficient biochemical information for predicting the metastatic phenotypes of single cancer cells using random forest classification.
MCR-ALS analysis allows a deeper understanding of the causal relationship with the sub-cellular components that determines the diagnostic power.
Acknowledgments
S.K.P. acknowledges the support of the SLAS Graduate Education Fellowship Grant. I.B. acknowledges the support from the National Cancer Institute (R01 CA238025), the National Institute of Biomedical Imaging and Bioengineering (2-P41-EB015871-31) and the National Institute of General Medical Sciences (DP2GM128198). K.G. acknowledges the support from the National Cancer Institute (R01 CA213428, R01 CA213492). The authors thank Tomocube Inc for use of the 3D ODT system. The schematic in Figure 1A was partially created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors disclose no potential conflicts of interest.
References
- Basu S, Kolouri S, Rohde GK, 2014. Proceedings of the National Academy of Sciences. 111, 3448–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver, Marc J, Gerald WL, Foekens JA, 2009. Nature. 459, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, 2015. Cancer discovery. 5, 1164–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L, 2001. Mach. Learning 45, 5–32. [Google Scholar]
- Chaffer CL, San Juan BP, Lim E, Weinberg RA, 2016. Cancer Metastasis Rev. 35, 645–654. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA, 2011. Science. 331, 1559–1564. [DOI] [PubMed] [Google Scholar]
- Che J, Yu V, Garon EB, Goldman JW, Di Carlo D, 2017. Lab on a Chip. 17, 1452–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhetri RK, Phillips ZF, Troester MA, Oldenburg AL, 2012. PLoS One. 7, e49148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Fang-Yen C, Badizadegan K, Oh S, Lue N, Dasari RR, Feld MS, 2007. Nature methods. 4, 717–719. [DOI] [PubMed] [Google Scholar]
- Choi WJ, Jeon DI, Ahn S, Yoon J, Kim S, Lee BH, 2010. Optics express. 18, 23285–23295. [DOI] [PubMed] [Google Scholar]
- Di Z, Klop MJ, Rogkoti V, Le Dévédec SE, van de Water B, Verbeek FJ, Price LS, Meerman JH, 2014. PloS one. 9, e109688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mashtoly SF, Yosef HK, Petersen D, Mavarani L, Maghnouj A, Hahn S, Kötting C, Gerwert K, 2015. Anal. Chem 87, 7297–7304. [DOI] [PubMed] [Google Scholar]
- Felten J, Hall H, Jaumot J, Tauler R, De Juan A, Gorzsás A, 2015. Nature protocols. 10, 217. [DOI] [PubMed] [Google Scholar]
- Gao D, Mittal V, Ban Y, Lourenco AR, Yomtoubian S, Lee S, 2018. Frontiers in biology. 13, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green BJ, Saberi Safaei T, Mepham A, Labib M, Mohamadi RM, Kelley SO, 2016. Angewandte Chemie International Edition. 55, 1252–1265. [DOI] [PubMed] [Google Scholar]
- Hai P, Imai T, Xu S, Zhang R, Aft RL, Zou J, Wang LV, 2019. Nature biomedical engineering. 3, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K, Fujita K, Smith NI, Kobayashi M, Inouye Y, Kawata S, 2008. J. Biomed. Opt 13, 044027. [DOI] [PubMed] [Google Scholar]
- Han A, Yang L, Frazier AB, 2007. Clin. Cancer Res 13, 139–143. [DOI] [PubMed] [Google Scholar]
- Hsiao Y, Chou M, Fowler C, Mason JT, Man Y, 2010. Journal of cancer. 1, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Bessette PH, Qian J, Meinhart CD, Daugherty PS, Soh HT, 2005. Proceedings of the national academy of sciences. 102, 15757–15761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD, 2015. Proceedings of the National Academy of Sciences. 112, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y, Cho H, Lee SY, Choi G, Kim G, Min H, Park Y, 2018. IEEE Journal of Selected Topics in Quantum Electronics. 25, 1–14. [Google Scholar]
- Kang JW, So PT, Dasari RR, Lim D, 2015. Nano letters. 15, 1766–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann B, Offerhaus HL, Windbergs M, Otto C, 2015. Adv. Drug Deliv. Rev 89, 71–90. [DOI] [PubMed] [Google Scholar]
- Karandikar SH, Zhang C, Meiyappan A, Barman I, Finck C, Srivastava PK, Pandey R, 2019. Anal. Chem 91, 3405–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Kim KS, Park H, Ye JC, Park Y, 2013. Optics express. 21, 32269–32278. [DOI] [PubMed] [Google Scholar]
- Kim K, Yoon H, Diez-Silva M, Dao M, Dasari RR, Park Y, 2013. J. Biomed. Opt 19, 011005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MG, Park J, Lim HG, Yoon S, Lee C, Chang JH, Shung KK, 2017. Scientific reports. 7, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong K, Kendall C, Stone N, Notingher I, 2015. Adv. Drug Deliv. Rev 89, 121–134. [DOI] [PubMed] [Google Scholar]
- Labib M, Mohamadi RM, Poudineh M, Ahmed SU, Ivanov I, Huang C, Moosavi M, Sargent EH, Kelley SO, 2018. Nature chemistry. 10, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam VK, Nguyen T, Phan T, Chung B, Nehmetallah G, Raub CB, 2019. Cytometry. 95, 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Park H, Kim K, Sohn Y, Jang S, Park Y, 2017. Scientific reports. 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ding J, Spuhler K, Gao Y, Serrano Sosa M, Moriarty M, Hussain S, He X, Liang C, Huang C, 2019. Journal of Magnetic Resonance Imaging. 49, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Lee SJ, Park S, Konstantopoulos K, Glunde K, Chen Y, Barman I, 2020. The FASEB Journal. 34, 9307–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YV, Middleton K, You L, Sun Y, 2018. Microsystems & Nanoengineering. 4, 1–13.31057891 [Google Scholar]
- Marro M, Nieva C, Sanz-Pamplona R, Sierra A, 2014. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 1843, 1785–1795. [DOI] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, 2018. PLoS biology. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J, 2005. Nature. 436, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, Yeh K, Leslie LS, Kenkel S, Kajdacsy-Balla A, Bhargava R, 2018. Proceedings of the National Academy of Sciences. 115, E5651–E5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movasaghi Z, Rehman S, Rehman IU, 2007. Applied Spectroscopy Reviews. 42, 493–541. [Google Scholar]
- Okada M, Smith NI, Palonpon AF, Endo H, Kawata S, Sodeoka M, Fujita K, 2012. Proceedings of the National Academy of Sciences. 109, 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu N, 1979. IEEE Trans. Syst. Man Cybern 9, 62–66. [DOI] [PubMed] [Google Scholar]
- Ozkumur E, Shah AM, Ciciliano JC, Emmink BL, Miyamoto DT, Brachtel E, Yu M, Chen P, Morgan B, Trautwein J, 2013. Science translational medicine. 5, 179ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidi SK, Pandey R, Barman I, 2020. Encyclopedia of Analytical Chemistry, 1–21. [Google Scholar]
- Paidi SK, Diaz PM, Dadgar S, Jenkins SV, Quick CM, Griffin RJ, Dings RP, Rajaram N, Barman I, 2019. Cancer Res. 79, 2054–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paidi SK, Rizwan A, Zheng C, Cheng M, Glunde K, Barman I, 2017. Cancer Res. 77, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Depeursinge C, Popescu G, 2018. Nature Photonics. 12, 578–589. [Google Scholar]
- Parmar A, Katariya R, Patel V, 2018, 758–763.
- Phillips KG, Jacques SL, McCarty OJT, 2012. Phys. Rev. Lett 109, 118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G, Park Y, Lue N, Best-Popescu C, Deflores L, Dasari RR, Feld MS, Badizadegan K, 2008. American Journal of Physiology-Cell Physiology. 295, C538–C544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudineh M, Aldridge PM, Ahmed S, Green BJ, Kermanshah L, Nguyen V, Tu C, Mohamadi RM, Nam RK, Hansen A, 2017. Nature nanotechnology. 12, 274–281. [DOI] [PubMed] [Google Scholar]
- Qiu S, Weng Y, Li Y, Chen Y, Pan Y, Liu J, Lin W, Chen X, Li M, Lin T, 2020. RSC Advances. 10, 14368–14373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizwan A, Bulte C, Kalaichelvan A, Cheng M, Krishnamachary B, Bhujwalla ZM, Jiang L, Glunde K, 2015. Scientific reports. 5, 10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizwan A, Paidi SK, Zheng C, Cheng M, Barman I, Glunde K, 2018. Scientific reports. 8, 11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, 2015a. Nature methods. 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, 2015b. Nature methods. 12, 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Kim K, Yoon J, Park Y, 2015. Opt. Lett 40, 5407–5410. [DOI] [PubMed] [Google Scholar]
- Stone N, Kendall C, Shepherd N, Crow P, Barr H, 2002. J. Raman Spectrosc 33, 564–573. [Google Scholar]
- Stott SL, Hsu C, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, 2010. Proceedings of the National Academy of Sciences. 107, 18392–18397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung Y, Choi W, Fang-Yen C, Badizadegan K, Dasari RR, Feld MS, 2009. Optics express. 17, 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnard PT Jr, Zhang C, Vesuna F, Kang JW, Garry J, Dasari RR, Barman I, Raman V, 2017. Oncotarget. 8, 20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Phillip JM, Khatau SB, Chen W, Stirman J, Rosseel S, Tschudi K, Van Patten J, Wong M, Gupta S, 2015. Scientific reports. 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Paidi SK, Qin Z, Huang Q, Yu C, Pagaduan JV, Buehler MJ, Barman I, Gracias DH, 2018. Nano letters. 19, 1409–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankaskas CL, Thompson KN, Paul CD, Vitolo MI, Mistriotis P, Mahendra A, Bajpai VK, Shea DJ, Manto KM, Chai AC, 2019. Nature biomedical engineering. 3, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]