

Abstract
With the widespread development of new drugs to treat chronic liver diseases (CLDs), including viral hepatitis and nonalcoholic steatohepatitis (NASH), more patients are entering trials with abnormal baseline liver tests and with advanced liver injury, including cirrhosis. The current regulatory guidelines addressing the monitoring, diagnosis, and management of suspected drug-induced liver injury (DILI) during clinical trials primarily address individuals entering with normal baseline liver tests. Using the same laboratory criteria cited as signals of potential DILI in studies involving patients with no underlying liver disease and normal baseline liver tests may result in premature and unnecessary cessation of a study drug in a clinical trial population whose abnormal and fluctuating liver tests are actually due to their underlying CLD. This position paper focuses on defining best practices for the detection, monitoring, diagnosis, and management of suspected acute DILI during clinical trials in patients with CLD, including hepatitis C virus (HCV) and hepatitis B virus (HBV), both with and without cirrhosis and NASH with cirrhosis. This is one of several position papers developed by the IQ DILI Initiative, comprising members from 16 pharmaceutical companies in collaboration with DILI experts from academia and regulatory agencies. It is based on an extensive literature review and discussions between industry members and experts from outside industry to achieve consensus regarding the recommendations. Key conclusions and recommendations include (1) the importance of establishing laboratory criteria that signal potential DILI events and that fit the disease indication being studied in the clinical trial based on knowledge of the natural history of test fluctuations in that disease; (2) establishing a pretreatment value that is based on more than one screening determination, and revising that baseline during the trial if a new nadir is achieved during treatment; (3) basing rules for increased monitoring and for stopping drug for potential DILI on multiples of baseline liver test values and/or a threshold value rather than multiples of the upper limit of normal (ULN) for that test; (4) making use of more sensitive tests of liver function, including direct bilirubin (DB) or combined parameters such as aspartate transaminase:alanine transaminase (AST:ALT) ratio or model for end-stage liver disease (MELD) to signal potential DILI, especially in studies of patients with cirrhosis; and (5) being aware of potential confounders related to complications of the disease being studied that may masquerade as DILI events.
Electronic supplementary material
The online version of this article (10.1007/s40264-020-01014-2) contains supplementary material, which is available to authorized users.
Key Points
| Monitoring and stopping rules for potential drug-induced liver injury (DILI) events in patients with chronic liver diseases who enter clinical trials with abnormal baseline liver tests should be based on a knowledge of the expected test fluctuations that reflect the natural history of the disease and are specific to the disease being studied. |
| After establishing a potentially elevated baseline value for alanine transaminase (ALT), aspartate transaminase (AST), total bilirubin, or alkaline phosphatase derived from the mean of at least two pretreatment values, the criteria for further elevations that trigger increased monitoring and holding or stopping drug should be based on multiples of the baseline value or a specific threshold value, whichever comes first, and not solely on multiples of the upper limit of normal (ULN). |
| For clinical trials in patients with cirrhosis from hepatitis C virus (HCV), hepatitis B virus (HBV), or nonalcoholic steatohepatitis (NASH), lesser elevations of ALT, AST; elevations of direct bilirubin and alkaline phosphatase even without significant elevations of aminotransferases; changes in the AST:ALT ratio; and changes in the international normalized ratio (INR) or model for end-stage liver disease (MELD) score may all be more sensitive measures of potential DILI events than traditional criteria of multiples of ULN of ALT, AST, total bilirubin, or alkaline phosphatase. |
Introduction
With the development of new drugs for chronic hepatitis C virus (HCV), hepatitis B virus (HBV), nonalcoholic steatohepatitis (NASH), alcoholic liver disease (ALD), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC), increasing numbers of patients are entering clinical trials with abnormal liver tests at baseline. In addition, given the increasing prevalence of the metabolic syndrome and resultant nonalcoholic fatty liver disease (NAFLD) and NASH in the general population, many more patients with underlying chronic liver disease (CLD) and elevated aminotransferases will be entering clinical trials for nonhepatic conditions such as type 2 diabetes mellitus, obesity, hyperlipidemia, gout, hypertension, and others. Abnormal liver tests at entry confound the recognition and risk assessment of potential drug-induced liver injury (DILI) during clinical trials based on multiples of the upper limit of normal (ULN). Although most experts agree with Zimmerman’s [1] seminal observation that CLD does not pose an increased risk for developing DILI for most drugs, recent data confirmed his warning that the outcome of a DILI event may be more serious in those with advanced CLD, including a higher risk of mortality [2–4]. These published reports highlight the importance of defining best practices for monitoring, detecting, and managing DILI in clinical trials in patients with CLD.
Recommendations in current regulatory guidelines [5, 6] for triggering investigation of potential DILI or for stopping an investigational drug in a clinical trial are generally based on multiples of the ULN defined by a reference laboratory for alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin (TBL), and alkaline phosphatase (ALP) with or without accompanying symptoms, in a subject who entered the trial with no known underlying liver disease and baseline values within the normal range. These guidelines recommend that an increase of serum ALT or AST to > 3 × ULN in subjects taking a study drug should trigger closer monitoring [5, 6]. Recent analyses have shown that most approved drugs that resulted in severe DILI detected during postmarketing pharmacovigilance exhibited signs of less severe liver damage such as isolated elevation in ALT > 3 × ULN (i.e., without elevated TBL or symptoms) during preapproval clinical studies [7, 8]. Although an imbalance in the proportion of subjects in the interventional arm with ALT and AST elevations > 3 × ULN compared with those in the control arm may be a sensitive marker of the risk of DILI, this finding by itself in the absence of study subjects in clinical trials with more severe forms of hepatotoxicity can be found even without actual cases of DILI. This may in part be due to the phenomenon of adaptation (or drug tolerance), defined as mild liver injury followed by a period in which the liver “adapts,” and the injury subsides, despite continuing treatment with the causative agent [9, 10].
In addition, minor elevations of ALT and AST can also be observed in placebo-treated or healthy individuals in clinical trials because of the effects of physical exercise or diets [11, 12]. This has led an international DILI expert working group to suggest that isolated increases of ALT > 5 × ULN are a more appropriate threshold for suspected DILI in subjects who enter clinical trials with normal baseline liver tests [13, 14]. Nonetheless, some cases marked by such drug-induced rises of aminotransferases may also resolve through a process of adaptation.
Current published guidelines on monitoring for DILI may overestimate the risk of acute DILI in patients who enter clinical trials with elevated baseline liver tests. Applying the same thresholds to patients with CLD as are used in “normal” patients may lead to unnecessary cessation of study drug, the clinical trial, or even the entire drug development program. Spontaneous fluctuations of liver enzymes during the trial that are actually due to the underlying CLD may be confused with DILI. Hy’s law is based on Dr. Hyman Zimmerman’s [1] clinical observation that a patient who presents with jaundice as a result of hepatocellular DILI has at least a 10% chance of dying (or needing a liver transplant) from acute liver failure. This maxim has been borne out in a number of studies and is the foundation of a widely recognized benchmark available to the pharmaceutical industry and regulatory agencies for assessing a drug’s potential to cause severe DILI. According to the current regulatory guidelines, “a Hy’s law case” is defined by (1) ALT elevation of > 3 × ULN; (2) TBL of > 2 × ULN; (3) absence of initial cholestasis (ALP < 2 × ULN); and (4) no other cause to explain the elevated ALT and TBL has been identified upon completion of a comprehensive diagnostic evaluation. Calculating an R value based on the multiple of ALT above the ULN divided by the multiple of ALP above the ULN may signal hepatocellular DILI (R value > 5) or cholestatic or mixed DILI (R value ≤5). If the R value is < 5 or = 5 because of an elevated ALP, then the prognostic significance of an ALT > 3 × ULN and a TBL > 2 × ULN without another cause is not as clear. However, the validity of these biochemical threshold values to define Hy’s law in a clinical trial as a specific tool for assessing a drug’s potential to cause serious DILI is questionable in patients with preexisting liver disease who enter the trial with elevated ALT, AST, and/or TBL. Moreover, invoking Hy’s law with these fold increases above the ULN when the ALT, AST, and TBL elevations are already present at baseline could result in an artificially lower threshold for discontinuing the study drug because of trivial fluctuations during the treatment phase.
Currently, there is a lack of consensus in the pharmaceutical industry and regulatory agencies about how to monitor and manage potential DILI in subjects who have different underlying CLDs. Given the variability of baseline values, inclusion/exclusion criteria, disease-specific fluctuations of liver tests, rates of progression to advanced stages of liver disease, treatment responses, and risk-benefit assessments, it is reasonable to consider different criteria for monitoring, detecting, and managing DILI in clinical trials for each specific disease population. In response to this and other issues surrounding DILI, the IQ DILI Initiative was launched in June 2016 within the International Consortium for Innovation and Quality in Pharmaceutical Development (also known as the IQ Consortium). The IQ Consortium is a leading science-focused, not-for-profit organization addressing scientific and technical aspects of drug development and comprises 38 pharmaceutical and biotechnology companies. The IQ DILI Initiative is an affiliate of the IQ Consortium, comprising 16 IQ member companies, focused on establishing best practices for monitoring, diagnosing, managing, and preventing DILI. Working groups within IQ DILI comprise clinical and nonclinical experts in DILI from the companies, with consistent participation from nonindustry DILI experts.
This consensus paper reviews the challenges of detecting DILI in clinical trials of subjects with HCV infection, HBV infection, and cirrhosis as a result of NASH, HCV, and HBV. Based on an extensive literature review, a survey of IQ DILI Initiative member pharmaceutical companies, as well as carefully structured discussions between IQ DILI members and academic and regulatory experts, consensus opinions were formulated and recommendations proposed. These are primarily focused on disease-specific thresholds of standard tests, including ALT, AST, TBL, direct bilirubin (DB) and ALP, that should trigger stepped-up monitoring and a comprehensive diagnostic evaluation to exclude alternative causes of liver injury during these clinical trials. In this position paper, we primarily focus the discussion on hepatocellular DILI, the most common but not the sole histopathological pattern or clinicopathological phenotype of hepatotoxicity [15], in patients with the aforementioned CLDs. Because of the underlying CLDs being considered, a categorical separation in all cases between hepatocellular and other forms of DILI may be more difficult. Other papers from our consortium have specifically addressed DILI in clinical trials of noncirrhotic NASH and cholestatic liver diseases (primarily PBC, PSC) [16, 17]. Future papers will address DILI in clinical trials of ALD. The recommendations in this paper are based on the opinions of the authors and do not imply a regulatory mandate.
General Considerations
Determination of Baseline Liver Test Values
In subjects with underlying CLD, liver tests are often, although not always, abnormal prior to entry, and values often fluctuate over time during the study. Fluctuating levels of ALT are characteristic of HCV [18, 19]; and a flare of ALT levels in HBV can be associated with HBV reactivation with or without viral clearance [20–23]. Therefore, a single ALT determination done several weeks or even months before the first dose of study drug may not be an accurate reflection of a patient’s status when entering a trial. For the purposes of this discussion, we define the baseline liver tests as those done immediately (generally within 24 h) before the administration of the first dose of study drug. Screening liver tests are those done more in advance (usually days or weeks) of the first dose, during the screening period after an informed consent is signed and an individual has been enrolled in the trial.
Published proceedings of previous workshops convening academic, industry, and regulatory experts have suggested that samples should be obtained at two or more time points during the pretreatment screening phase to determine whether liver tests are stable or subject to fluctuation [15, 24–26]. Results of a recent survey conducted by the IQ DILI initiative revealed that 3 of 12 (25%) companies were using more than one determination of aminotransferase, TBL, and ALP during screening before baseline serum testing to generate mean baseline values available to the clinical investigators prior to administration of the first dose of study drug (unpublished data). In the proceedings of previous meetings, recommendations were made to obtain at least two determinations during the screening period separated by not less than 2 weeks and not more than 2 months prior to time of the initial dose of the study drug, with the mean value chosen as the screening value to meet the predefined inclusion/exclusion criteria [25]. Some experts have also advocated that if the second screening value exceeds the exclusion criteria for that liver test, or if the second value is more than 50% (1.5×) higher than the first value, then enrollment should be delayed and a third value obtained to aid with judgments about whether the subject’s underlying CLD is progressing and may limit eligibility [25].
With the use of a central laboratory in clinical trials, the results of baseline liver tests are often not available at the time the first dose of study drug is administered but are available soon after. To avoid prolonging the screening period and delaying dosing, some experts suggest that the screening tests should be done as close as possible to the first dose of study drug to qualify the patient for inclusion; a second measurement at baseline (within 24 h before the first administration of study drug) can be obtained and averaged with the previous screening value. Initial increased monitoring for changes in the underlying CLD and for DILI may be necessary in the event of a > 50% elevation of the baseline over the screening value in a subject who already has received the first dose of study drug.
Consensus Recommendations
In patients with underlying CLD, a single value of ALT, AST, TBL, ALP to establish the baseline is not appropriate, and two values should be obtained during the screening prior to the first dose of study drug to determine a mean value that will qualify the patient for inclusion into the trial and be used as a reference value for future changes.
While the gap between these screening values and the first dose of study drug could be 2 weeks to 2 months in CLDs such as HCV, HBV, and NASH; in CLDs with the possibility of more dynamic patterns of worsening or improving liver tests (such as any decompensating cirrhosis or acute-on-chronic liver failure [ACLF]), the screening values, including aminotransferases and bilirubin, should be within a week of the first dose of study drug, and combined with the baseline value (≤ 24 h prior to the first dose of study drug).
A second screening value that is > 50% higher than the first value should prompt a delay and re-evaluation of the severity of underlying liver disease and eligibility for the trial.
Baseline values of liver tests should also be obtained immediately prior (≤ 24 h) to administration of the first dose of study drug. In some protocols, if the results of these tests are available prior to administration of the first dose of study drug, they can be used as the second screening value to calculate a mean value and determine eligibility for the trial. If the tests are sent to a central laboratory and results are not immediately available at the time the first dose is administered, then those baseline values should be included in calculating the threshold values from which to assess changes during the trial possibly indicative of DILI, other intervening causes of liver injury, or fluctuations of the underlying CLD.
If only one biochemical assessment is done during screening in a clinical trial of a preexisting liver disease that is not prone to rapid prominent fluctuations of liver test measurements (e.g., NASH, chronic HCV), it should be done within 2 weeks of the planned first administration of study drug to determine whether the subject meets the inclusion/exclusion criteria.
If the baseline determinations are reported after the first dose of study drug is administered and are > 50% higher than the previous screening value(s) that allowed entry into the trial, a decision about holding or discontinuing study drug needs to be made based on the agreed inclusion/exclusion criteria for baseline liver tests. If continuing study drug, a schedule of increased monitoring should be put into place during the initial phase of the trial for that subject.
Normal Range of Alanine Transaminase
A major source of variation in normal reference ranges for ALT is the incomplete characterization of the reference populations from which the ULN of ALT is derived. The apparently healthy individuals whose values defined the “normal range” may have occult liver disease [27–30]. A number of studies have redefined the ULN for ALT in a healthy population, but these new suggested levels are often not utilized by commercial laboratories. These studies have included populations from different geographic areas [30–36], but comparisons between studies are limited by the heterogeneous criteria employed to exclude patients with underlying liver disease. Results have shown that the ULN of ALT in prospectively studied healthy populations without identifiable risk factors for liver disease (including NAFLD, NASH) ranges from 29 to 33 IU/l for males and from 19 to 25 IU/l for females [37]. These ranges are considered the true normal values by the American College of Gastroenterology (ACG) and the American Association for the Study of Liver Disease (AASLD). The current challenge (even in NASH clinical trials) is that the central laboratories utilized to analyze ALT do not define reference ranges based on these new lower normal levels.
An important question is, what impact will re-defining the ALT ULN have on the diagnosis of DILI. A number of drugs that were removed from the market because of hepatotoxicity, including bromfenac, ximelagatran, trovafloxacin, and troglitazone, all reported subjects in whom the ALT elevations were markedly elevated (> 10 × ULN in most cases); and a reduced ULN of ALT would not have altered the diagnosis of DILI [38–42]. Lowering the ULN of ALT currently used by commercial laboratories would likely help in identifying patients with unsuspected CLD, but it does not appear that defining a lower ULN for ALT will significantly impact identification of cases that trigger concern for serious DILI. On the other hand, low-grade elevations of ALT persisting during clinical trials may be an important signal in drugs administered chronically and may reflect potential subacute forms of DILI.
Consensus Recommendations
-
7.
Until or unless there is agreement on a standard reference range across all laboratories, we suggest the use of the ULN of ALT and AST as defined by the central laboratory participating in the clinical trial and a definition in the protocol of the reference ranges being used for normal ALT, AST, TBL, DB, and ALP.
Defined Multiples of Upper Limit of Normal versus Multiples of Baseline Liver Tests Combined with Threshold Values of Serum Liver Enzyme Activities as Triggers for Increased Liver Monitoring, Diagnostic Evaluation, and Stopping Rules
Traditionally in clinical trials, multiples of the ULN of ALT, AST, ALP, and TBL alone or in combination have been used as criteria to trigger interruption or stoppage of the drug, increased monitoring for DILI, investigations for causality assessment [43] and assignment as a potential Hy’s law case. An alternative strategy is to start with each subject’s liver tests prior to first dose of study drug (mean of screening and baseline values), and then use multiples of those values during the trial to trigger the above actions in that subject [43–45]. In particular, when subjects enter clinical trials with elevated baseline ALT values, it may not be appropriate to trigger the standard responses to evaluate a case of concern when the ALT rises to 3 × ULN, as this may reflect only a minor increase above the subject’s baseline and may be due to normal variability of the ALT fluctuations in the disease being studied. Focusing on an imbalance between the number of individuals in the treatment arm compared with the placebo arm who develop multiples of their own baseline ALT during the clinical trial may be more informative [6, 9, 24, 44, 46]. As previously recommended in the US FDA guidance on DILI, the recent European Association for the Study of the Liver (EASL) clinical practice guidelines on DILI suggested that a doubling of a subject’s baseline values may be considered a threshold increase warranting close observation for potential DILI during clinical trials [47]. The survey of 13 IQ DILI companies recently conducted by the IQ DILI Consortium found that 77% of the companies currently use multiples of a subject’s baseline aminotransferases to trigger increased monitoring for potential DILI or cessation of study drug in clinical trials where subjects enter with abnormal baseline values, generally defined as > 1.5 × ULN.
However, reliance solely on the subject’s baseline aminotransferases and multiples of those values as the basis for predicting potential DILI cases poses some additional dilemmas. A multiple of a high baseline value may allow a subject’s ALT to rise to very high levels before reaching criteria that would trigger increased monitoring for DILI. This has led some experts to propose a hybrid approach that combines using a multiple of a subject’s baseline ALT with a threshold value of ALT activity that serves as a “guardrail,” and allowing whichever comes first to provoke either holding or stopping the study drug [44, 48]. For example, such a hybrid threshold previously proposed for detecting hepatotoxicity and stopping rules in NASH trials included a serum AST and/or ALT > 3 × the baseline value or > 500 IU/L, whichever came first [45]. In general, such threshold values should exceed the usual range of fluctuations of aminotransferases and bilirubin commonly seen in the CLD being studied but should be lower if there is a known risk of DILI based on nonclinical data or data from other drugs in the same class. Consensus recommendations for hybrid thresholds for the specific CLDs that are discussed are given in the following sections. Concurrent elevations of both ALT and TBL should always be viewed as a more specific signal of functional liver impairment and a harbinger of severe DILI and should lead to earlier discontinuation, even when using criteria linked to baseline values.
Table 1 summarizes available data on the expected ranges and fluctuations of ALT and AST in patients entering clinical trials with several types of CLD that are the focus of this paper.
Table 1.
Magnitude of baseline liver test elevations and fluctuations in individuals with chronic liver diseases currently the focus of new drug development programs
| Underlying disease | Disease prevalence | Frequency of baseline liver test elevations | Magnitude of baseline liver test elevations | Pattern of liver test elevations over disease course |
|---|---|---|---|---|
| Hepatitis B |
HBsAg + global: 3.5–5% [55, 56] USA: 0.27% (higher among certain ethnic groups) |
ALT and AST elevated in most patients (> 80%) [57] with chronic active untreated infection with viremia; chronic carriers often have normal ALT. In those treated with neocleos(t)ide analogs, ALT often normal or near/normal |
Flare > 5 × ULN seen with successful antiviral therapy or with reactivation [62] |
Moderately elevated with intermittent flares. May be normal during immune tolerant and inactive carrier phases [52] |
| Hepatitis C |
Global: 0.8–1.1% [49] |
55–75% [53] |
Of those with ALT elevations [18], < 3 × ULN in ~ 65% 3–7 × ULN in ~ 30% > 7 × ULN in ~ 5% |
Mildly elevated with variable episodic fluctuations during acute exacerbations [54] |
| Cirrhosis | 0.076–0.27% [63, 64] | 40–70% (in decompensated cirrhosis) [65]. Specific rates unavailable for compensated cirrhosis. Patients may have normal laboratory values in both compensated and decompensated cirrhosis |
HBV (ALT 2–4 × ULN in compensated and 2–3 × ULN in decompensated cirrhosis [66, 67]) HCV (ALT 2–3 × ULN in compensated cirrhosis [68]) NASH (ALT < 2 × ULN in compensated cirrhosis [69, 70]) ALP < 2 × ULN in cirrhosis (underlying cause unspecified) [71] |
Mildly elevated and may decrease over time. AST:ALT ratio > 1 increases with progressive cirrhosis [72] |
The values expressing increased baseline levels as multiples of ULN are based on values of ULN used by laboratories participating in clinical trials that predate the updated reference range guidelines for liver chemistry tests now accepted by both the AASLD and the ACG and published in the American Journal of Gastroenterology online in 2016 [37]
AASLD American Association for the Study of Liver Diseases, ACG American College of Gastroenterology, ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, HBsAg HBV surface antigen, HBV hepatitis B virus, HCV hepatitis C virus, NASH nonalcoholic steatohepatitis, ULN upper limit of normal
Consensus Recommendations
-
8.
If baseline ALT or AST exceed 1.5 × ULN, use multiples of the subject’s baseline value as the trigger for increased monitoring for DILI or for holding or stopping drug.
-
9.
In addition to a multiple of the baseline value as the trigger for drug cessation to mitigate risk of serious DILI outcomes in study subjects, add a maximum threshold value of ALT or AST that can also trigger stopping the drug if it is reached prior to a defined multiple of a subject’s baseline values. This threshold value should be based on a knowledge of the usual fluctuations of aminotransferases and bilirubin in each specific chronic disease (see Table 1 and individual following sections); the potential risk of DILI with the study drug based on the mechanism of action of the drug and preclinical data or data from other drugs in the same class; and whether study subjects have advanced liver disease or cirrhosis.
Assessment of Drug-Induced Liver Injury (DILI) in Clinical Trials of Adults with Chronic Hepatitis C Virus (HCV) Infection
Introduction
Clinical development programs for HCV over the past decade have resulted in the approval of multiple oral direct-acting antiviral (DAA) regimens and issuance of a recent FDA guidance for developing DAAs [73]. DAAs have revolutionized the treatment of chronic HCV, achieving cure rates (also known as sustained viral response [SVR] rates) in the vast majority of patients with just 8–12 weeks of therapy [74].
While we acknowledge that further trials with new DAAs may not be forthcoming given the high rates of cure already achieved with the existing agents, the heterogeneous natural history of HCV, characterized by fluctuations of aminotransferases, could make monitoring and detecting DILI in future clinical trials challenging. Approximately 30% of patients with chronic HCV infection have persistently normal ALT levels [53] but, in a 22-month study period, changes of serum ALT levels more than threefold were observed in 27.5% of untreated patients with HCV [75]. Instances of severe hepatic events in HCV clinical trials with DAAs have been rare, with most occurring with protease inhibitor-containing regimens in patients with decompensated cirrhosis [74]. While fluctuations in aminotransferases were seen in untreated patients over time, most patients normalized their aminotransferases rapidly during DAA treatment [76]. Even though significant drug toxicity has not been seen with many DAAs in the context of clinical trials, strategies developed for assessing and monitoring for DILI during these trials are potentially applicable to drug development programs for other CLDs. In this section, we focus on patients with compensated liver disease as defined per HCV protocol inclusion/exclusion criteria. For patients with HCV and decompensated cirrhosis, recent FDA guidance recommended that specific hepatic safety monitoring and treatment discontinuation criteria should be discussed with the Division of Antiviral Products during the protocol development phase to incorporate case selection criteria and laboratory cutoff values specific to the population [73]. As such, specific recommendations for future trials in patients with HCV with decompensated cirrhosis are not further discussed in this section of the paper but are considered in the following section on cirrhosis.
Baseline Hepatic Inclusion/Exclusion Criteria
An extensive literature review of available protocols studying DAAs for HCV provided information on hepatic inclusion/exclusion criteria and laboratory test monitoring [77–82]. In trials that included patients with cirrhosis, the majority of patients did not have cirrhosis (55–80%). Since cirrhosis has been demonstrated to be a significant factor affecting treatment outcomes, determining whether enrollees into clinical trials have cirrhosis remains critical (see cirrhosis section). In the most recent HCV phase III trials, cirrhosis was defined as any one of the following: liver biopsy showing cirrhosis, FibroTest® score > 0.75, an AST:platelet ratio index (APRI) > 2, or a Fibroscan with a result of > 12.5 kPa. For inclusion in these trials, platelets must have been above 50,000, DB ≤ 1.5 ULN; international normalized ratio (INR) ≤ 1.5 × ULN; and albumin > 3 g/dl [77–83]. Patients with decompensated cirrhosis, defined as the presence of ascites, encephalopathy, or variceal hemorrhage, were typically excluded.
Prior to treatment, aminotransferases were measured twice: at screening (day − 28 to day 0) and at baseline prior to the first dose of the study drug. These values were required to be ≤ 10 × ULN with DB ≤ 1.5 × ULN in most of the cases, and ≤ 5 × ULN with DB ≤ ULN in some more conservative protocols [77–82]. While there are recommendations in the literature for doing multiple screening assessments in patients with abnormal aminotransferases [84], based on the IQ DILI survey results (unpublished), it appears that many companies relied on a limited assessment (one screening value) for inclusion of patients with HCV in DAA trials, and the measured value at baseline was used for comparison to subsequent values.
Consensus Recommendations
Patients with HCV with baseline aminotransferases > 10 × ULN and DB > 1.5 × ULN should be excluded from investigative drug studies and assessed for other causes of liver disease.
Exclusion criteria to limit study subjects to compensated cirrhosis should include any history of decompensating events (variceal bleeding, ascites, hepatic encephalopathy), and platelets < 50,000/mm3, albumin < 3 g/dl, INR > 1.5 × ULN, and DB > 1.5 × ULN.
Eligibility criteria for clinical trials of patients with HCV with decompensated cirrhosis should be established in a protocol-specific fashion with input from regulatory authorities.
Baseline aminotransferase levels used for follow-up comparisons during the trial should be determined by averaging the screening and baseline values unless the difference is > 50% and the baseline value is now > 1.5 × ULN. In the case of such a large difference, a third sample should be collected. If this third sample is collected after treatment for HCV has been administered for more than 3 days, then this most recent value is expected to be equal to or lower and should be used as the baseline value.
Liver Tests Driving Close Monitoring
Because it is important to distinguish drug hepatotoxicity from the underlying chronic HCV as the source of any aminotransferase abnormalities during clinical trials, typical monitoring frequency in HCV trials during a 12-week treatment duration were aminotransferase, ALP, and TBL monitoring performed at baseline, weeks 1, 2, 4, 6, 8, 10, and 12, and 4 weeks after the conclusion of treatment [77–82]. The pattern of virologic response during treatment is very similar across regimens and leads to a rapid undetectable viral load at week 4 in most cases. For example, after initiating treatment with ledipasvir–sofosbuvir, an HCV viral load < 25 IU/ml was observed in 27%, 87%, and 100% of patients at weeks 1, 2, and 4, respectively [77]. Biochemical responses were also very rapid. In patients with abnormal ALT at baseline treated with ledipasvir–sofosbuvir, 82%, 91%, and 94% had normalized ALT after weeks 1, 2, and 4, respectively [77, 85]. The concept of a post-treatment nadir value as the new reference baseline for subsequent hepatotoxicity evaluation was proposed following a workshop conducted on 9 November 2012 with regulatory experts across the globe and representatives from industry and academia (Fig. 1) [25, 48].
Fig. 1.
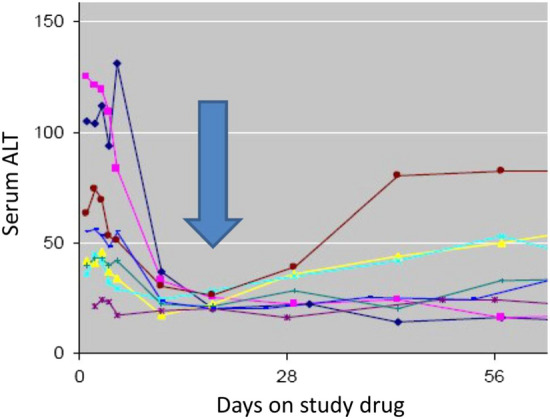
Biochemical response. Early fall in serum alanine aminotransferase (ALT) during effective treatment of viral hepatitis C. The colored curves represent the ALT pattern of response from eight different patients.
Reproduced from Kullak-Ublick et al. [48]
Consensus Recommendations
-
5.
Recommended key time points for assessing for DILI and measuring HCV RNA depend on the drug regimen and patient population. On-treatment measurements should include weeks 1, 2, 4, 8, 12, and 24 or at the end of therapy [73].
-
6.
A subject’s week 1 or week 2 aminotransferase measurements or any lower value captured (nadir) in response to the start of therapy should become the new reference baseline for further DILI assessment during the trial.
-
7.
Once HCV viral load becomes undetectable and ALT is normalized, subsequent ALT flares in the absence of new-onset HCV resistance with a rising viral load should be considered suspect for DILI and assessed as per regulatory guidance [5, 6].
-
8.
In the clinical trial setting, pretreatment and on-treatment blood samples should be stored as long as possible to facilitate retrospective assessment of DILI and allow monitoring of ALT concomitantly with viral load.
Liver Tests and Drug Stopping Rules
Stopping rules varied among different HCV treatment protocols depending on the initial reference value used as a starting point (baseline, nadir, or ULN). In some studies, the study drug was discontinued for an elevation of ALT and/or AST > 5 × baseline or nadir, or for an elevation of ALT > 3 × baseline and TBL > 2 × ULN confirmed by immediate repeat testing [77–79, 81]. In other HCV protocols, the study drug was to be discontinued if ALT ≥ 10 × ULN; if ALT ≥ 5 × ULN with symptoms and signs of hepatitis developing [80]; or if ALT > 2 × baseline or > 5 × ULN and either TBL > 2 × ULN or INR > 2 [82]. Without predicting what could occur with potential new drugs in the pipeline, the overall reported rates of serious adverse events have been < 10% in most published HCV studies, and treatment discontinuations were well below 5%, even in patients with comorbid conditions, such as human immunodeficiency virus (HIV) infection and cirrhosis [74]. No significant drug hepatotoxicity was seen with these DAA agents used in phase III clinical trials exploring short treatment durations (8, 12, and 24 weeks) [74]. Should there be a potential DILI event with elevated aminotransferases, either alone or in conjunction with TBL (not provoked by Gilbert’s syndrome, hemolysis, or a DAA effect on transporters of unconjugated bilirubin), and/or symptoms that meet the criteria recommended below and provoke stopping drug (Fig. 2), a proposed algorithm for initial and expanded testing to exclude other causes of hepatic injury other than DILI is outlined in the supplementary Causality Assessment Table (Table 1 in the electronic supplementary material [ESM]) and further detailed in a paper on best practices for causality assessment in preparation by the IQ DILI Consortium.
Fig. 2.

Algorithm for monitoring and management of potential DILI signals in phase II–III clinical trials in patients with HCV with normal or elevated baseline ALT. aBaseline ALT is derived from an average of two pretreatment ALT measurements 2 weeks apart. Elevated baseline is defined as ALT ≥ 1.5 × ULN. bSymptoms may be liver related (e.g., severe fatigue, nausea, vomiting, right upper quadrant pain) or immunologic reaction (e.g., rash, > 5% eosinophilia). cFor patients with Gilbert’s syndrome or hemolysis. dIn patients with a sizable stable early decrease in ALT during treatment (> 50% of baseline value), a new baseline, corresponding to the ALT nadir, should be established on an individual basis for subsequent determination of a DILI signal. eThe specific interval between the tests should be determined based on the patient’s clinical condition. ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, DB direct bilirubin, DILI drug-induced liver injury, HBcAb+ hepatitis B core antibody positive, HBV hepatitis B virus, HCV hepatitis C virus, TBL total bilirubin, ULN upper limit of normal
Consensus Recommendations
-
9.
Treatment should be stopped in patients who enter with normal baseline ALT and develop ALT ≥ 10 × ULN during the trial (even with normal TBL); and in those who enter with abnormal baseline ALT and develop confirmed ALT ≥ 5 × baseline or nadir value or ALT ≥ 500 U/l, whichever comes first (even with normal TBL); or for elevation of ALT > 3 × baseline and TBL > 2 × ULN (excluding those with Gilbert’s syndrome).
-
10.
In subjects who enter with normal baseline ALT, treatment should be stopped for elevation of ALT ≥ 3 × ULN with elevation of TBL ≥ 2 × ULN; and in those entering with abnormal baseline ALT, treatment should be stopped with elevation of ALT ≥2 × baseline or new nadir value or ALT ≥ 300 U/l (whichever comes first) with TBL ≥ 2 × ULN.
-
11.
Appearance or worsening of clinical symptoms (e.g., weakness, nausea, vomiting, jaundice) suggesting signs of clinical liver injury, in the setting of abnormal aminotransferases (at least 3 × ULN or 2 × baseline) or bilirubin, should also prompt discontinuation of therapy.
Lessons Learned from Postmarketing Experience with Direct-Acting Antivirals for Chronic HCV Applicable to Future Trials
HCV DAA agents have been demonstrated to be effective and safe in clinical studies that excluded subjects with concomitant chronic HBV infection, but safety data from clinical trials may not fully represent more diverse patient experiences in clinical practice [74]. Examination of several real-world HCV cohorts suggested the efficacy and safety of DAAs was similar to that in clinical trials (TARGET, TRIO) [86, 87], but the primary outcome was virological response, and some analyses were done retrospectively. In real-life experience, toxicities and drug–drug interactions (DDIs) are emerging, especially in specific populations who were often excluded from the clinical trials, such as HBV/HCV co-infected patients, patients with Child–Turcotte–Pugh (CTP) B or C cirrhosis, patients on transplant waiting lists, and transplant recipients [88].
After marketing authorization, the FDA issued a warning about the risk of serious liver injury in patients with underlying advanced liver disease treated with Viekira Pak®, Viekira XR® (ombitasvir, paritaprevir, ritonavir, dasabuvir) (AbbVie, IL, USA) and Technivie® (ombitasvir, paritaprevir, ritonavir) (AbbVie) [89] and more recently with the use of Mavyret® (glecaprevir, pibrentasvir) (AbbVie), Zepatier® (elbasvir, grazoprevir) (AbbVie), or Vosevi® (sofosbuvir, velpatasvir, voxilaprevir) (Gilead Sciences, CA, USA), all of which contain an HCV protease inhibitor [90]. This led to several label updates and contraindications to the use of certain protease inhibitor-containing regimens in patients with CTP B and C cirrhosis, many of whom were inappropriately classified as CTP A [91].
Patients co-infected with chronic HBV were excluded from initial HCV clinical trials. Postmarketing data indicate that treatment of HCV with DAAs may cause reactivation of HBV. In 2016, the FDA warned about the risk of HBV reactivation after receiving several reports in patients treated with DAAs [92]. This led to a label update and a boxed warning for all DAA drugs. A recent prospective study conducted in Taiwan in 111 patients with HCV and HBV infection (positive HBV surface antigen [HBsAg]) treated with ledipasvir and sofosbuvir showed HBV DNA increases in 53% (39/74) of those who entered the trial with detectable HBV DNA > 20 IU/L and in 84% (31/37) of those who were HBV DNA negative when entering the trial [93]. Overall, five patients (6.8%) had increased levels of HBV DNA with concomitant ALT increases > 2 × ULN through post-treatment week 12. The ALT elevations occurred as soon as week 4 in two patients. The underlying mechanism of HBV reactivation is not clear, but HCV/HBV co-infected individuals usually have low or undetectable HBV DNA levels, which may be due to induction of type I and III interferons by HCV [94]. After starting DAA treatment, a rapid HCV viral suppression leads to reduced activation of the interferon cascade, allowing for faster HBV replication [95, 96]. Outside of clinical trials, consensus guidelines for mitigation of risk (e.g., use of prophylaxis with HBV nucleot(s)ide analogs) and management of HBV re-emergence might be different, and monitoring should be adapted to the specific patient population and the regimen used [97–99].
Consensus Recommendations
-
12.
Evaluate all patients with cirrhosis carefully to avoid the use of protease-containing DAA regimens in those with decompensated disease (CTP class B or C).
-
13.
Evaluate potential DDIs in patients with HCV taking other medications and receiving DAAs.
-
14.
Assess the need for DAA pharmacokinetic measurements when evaluating potential DILI in subjects with advanced liver disease.
-
15.
Screen all patients with HCV for evidence of current or prior HBV infection before starting treatment with DAAs to avoid reactivation of HBV.
-
16.
Patients fulfilling the standard criteria for HBV treatment should receive nucleot(s)ide analog treatment in alignment with recent clinical guidelines.
Summary
The large clinical trial experience obtained with DAAs in the treatment of patients with chronic HCV has helped define the new baseline reference point (nadir) for aminotransferases, resetting the criteria by which to evaluate any future elevations, and prompting the appropriate diagnostic assessments to determine whether DILI might be present. As more experience is gained, it will be important to determine whether this strategy of defining a subject’s new on-treatment baseline for aminotransferases during a clinical trial might also be applicable for new drugs treating other CLDs in which response to treatment results in decrease or normalization of aminotransferases.
Assessment of DILI in Clinical Trials of Adults with Chronic Hepatitis B Virus (HBV) Infection
Introduction
It is estimated that approximately 250 million people worldwide are chronically infected with the HBV [56, 100], although a recent study utilizing modeling estimated that this number may be as high as 356 million [101]. Between 15 and 40% of patients develop serious sequelae of infection, and as many as 650,000 people per year die due to chronic hepatitis B (CHB)-related disease [23, 55]. Over the last two decades, significant advances have been made in the treatment of HBV, with most patients now achieving long-term viral suppression accompanied by a low risk of antiviral resistance on long-term oral therapy. However, unlike the cure rates experienced with DAA therapy for HCV, current antivirals for HBV have not achieved sustained viral eradication because HBV DNA remains in hepatocytes in the form of covalently closed circular (ccc) DNA [102]. This has led to numerous ongoing trials of medications and combinations of medications aimed at curing HBV [103–111].
Baseline Liver Test Inclusion/Exclusion Criteria
The natural history of chronic HBV infection is characterized by hepatitis flares and remissions and can be divided into four phases: immune tolerant, hepatitis B e antigen (HBeAg)-positive immune active, inactive, and HBeAg-negative immune reactivation phases [99, 112–115]. See Table 2.
Table 2.
| Phase | EASL proposed phase [99] | ALT | HBsAg | HBeAg | HBV DNA | Histology | |
|---|---|---|---|---|---|---|---|
| inflammation | Fibrosis | ||||||
| Immune tolerant |
Phase 1 Chronic infection |
Normal | Positive | Positive | Markedly elevated (typically > 10 million IU/ml) | Minimal | Minimal |
| Immune active |
Phase 2 Chronic hepatitis |
Elevated > 2 × but < 5 × ULN. Levels fluctuate | Positive | Positive | Elevated (≥ 20,000 IU/ml). Levels may fluctuate | Moderate–severe often with necrosis | Moderate–severe, variable |
| Inactive carrier |
Phase 3 Chronic infection |
Normal | Positive | Negative | Low or undetectable (< 2000 IU/ml) | Minimal | Mild–severe |
| Immune reactivation |
Phase 4 Chronic hepatitis |
Elevated > 2 × but < 5 × ULN. Levels fluctuate | Positive | Negative | Elevated (≥ 2000 IU/ml) | Moderate–severe often with necrosis | Moderate–severe |
| Occult HBV infection |
Phase 5 Resolved HBV infection |
Normal | Negative | Negative | Usually but not always undetectable | Variable HBV DNA (cccDNA) often detected in the liver | Variable |
ALT alanine aminotransferase, cccDNA covalently closed circular DNA, EASL European Association for the Study of the Liver, HBeAg hepatitis B e antigen, HBsAg HBV surface antigen, HBV hepatitis B virus, ULN upper limit of normal
Characterization of these phases has recently been updated by the EASL [99]. In addition to HBV DNA levels, HBeAg status, and liver histology, each phase is characterized by either an elevated or a normal ALT. However, these phases are not static and do not necessarily move from one stage to another in a directional or sequential manner. Thus, it is recommended to obtain serial ALT and HBV DNA levels to accurately characterize the phase of chronic infection [23, 99, 115]. A single ALT level is likely to be insufficient when evaluating a patient for enrollment in a clinical trial for chronic HBV therapy, or when designing liver-related monitoring and stopping rules. In spite of these caveats, a review of published studies of currently approved and marketed HBV treatments showed that most mean baseline ALT levels at initiation of clinical trials appeared to be obtained from a single value, and ranged from 114 to 199 U/l [58–61, 116–119]. The inclusion criterion for most trials was consistent with AASLD guideline recommendations of a requirement for an ALT at least > 2 × ULN.
Exclusion criteria for ALT upper limit level in some HBV clinical trials included > 10 × ULN or specific upper limit cutoff values such as ≤ 400 U/l for men and ≤ 300 U/l for women [120]. However, other trials [121–123] did not specify an upper limit. Unless a clinical trial was specifically targeting those with normal ALT values [124–126], defining an ALT upper limit is important as the assessment of response to treatment may be difficult if enrollment included patients undergoing a spontaneous reactivation or those with acute HBV. It is important to note that guidelines on HBV specifically define normal ALT as < 19 U/l for females and < 30 U/l for males [36, 115]. Since these values have not been incorporated into the normal reference ranges of central laboratories that are typically used in clinical trials, it is important to establish the definition of normal ALT prior to HBV trial initiation and prior to designing liver-related monitoring and stopping criteria for HBV trials. When designing trial eligibility and monitoring and stopping rules, differentiation should be made between patients entering a trial on nucleos(t)ide analogs versus those naïve to therapy or nonresponders to nucleos(t)ide analogs.
Patients with other liver diseases, hepatocellular carcinoma (HCC), and HIV infection, are typically excluded from HBV clinical trials. Specific inclusion and exclusion criteria may also depend on the mechanism of action of drug, pharmacokinetics/pharmacodynamics (PK/PD), nonclinical data, phase of the clinical trial, and target patient population.
Consensus Recommendations
The average of at least two consecutive ALT levels obtained prior to enrollment 2–4 weeks apart should be used to determine the baseline ALT level in HBV trials. These levels can be obtained during the screening period and at the baseline visit.
If there is a difference in ALT of > 50% between the two measurements, it is advisable to obtain a third value and to avoid enrollment of patients with an alternative diagnosis or those undergoing an HBV flare.
If continued ALT elevations occur, enrollment should be held until the underlying cause is identified or ALT levels stabilize.
Specific eligibility criteria may also depend on the mechanism of action of drug, PK/PD, nonclinical data, phase of the clinical trial, and target patient population.
Exclusion criteria for ALT level for patients on nucleos(t)ide analogs should be > 2 × ULN, since, in the absence of a flare, most subjects on this treatment will maintain ALT levels below this value.
Exclusion criteria for ALT level for patients naïve to therapy or who are nonresponders to nucleos(t)ide analogs should be > 7 × ULN or > 300 U/l, whichever comes first.
The definition of the normal range for ALT should be established prior to trial initiation and prior to designing liver-related monitoring and stopping criteria.
Patients with other liver diseases, including hepatitis delta virus (HDV) infection, patients with HCC, and patients with HIV should be excluded from clinical trials for HBV, especially in early drug development (phase I and II). If included in later-stage trials (phase III and IV), these patients should be studied as separate cohorts or subpopulations. Alternatively, these patients may be studied in stand-alone clinical trials (e.g., HBV/HIV co-infection or HCC due to HBV).
Differentiating an HBV Flare from DILI
HBV flares may occur spontaneously or be treatment induced during or after HBV therapy as well as in the setting of DAA therapy for HCV, drug-induced immunosuppression, and/or chemotherapy. The clinical spectrum can range from asymptomatic to hepatic decompensation with jaundice and coagulopathy and is characterized by an abrupt ALT elevation. While many definitions exist, an HBV flare most commonly presents with an abrupt rise of ALT levels to > 5 × ULN (accompanied by a rise in HBV DNA) in a person with underlying chronic HBV infection [62]. Flares occurring during HBV clinical trials have also been defined biochemically as ALT levels > 2 × baseline or > 10 × ULN and often signal a response to therapy [60, 127, 128]. In the clinical trial leading to marketing authorization for tenofovir [60], all flares were characterized by ALT > 10 × ULN and 2 × baseline; occurred during the first 8 weeks of initiating medication; and resolved within 4–8 weeks without interruption of therapy. However, since a flare may mimic acute DILI, detection and causality assignment in patients with HBV can be challenging. When DILI is suspected, clinical assessments should include serial routine quantitative measurements of HBV DNA (and HBsAg if available) as elevations of both typically precede the rapid rise of ALT characteristic of an HBV flare. HBsAg quantification can be especially useful in these cases when a baseline value has been obtained. The finding of stable HBsAg and HBV DNA levels at the time of an abrupt rise in ALT is more consistent with potential DILI and should prompt a thorough evaluation. In addition, in trials of patients with HBeAg-positive disease, e antigen loss and seroconversion to HBeAb can occur, signaled by a flare in ALT, but would not be expected during an episode related to DILI. Of note is the fact that the peak ALT elevations for anti-HBe(+) patients during a flare are considerably lower than those seen in HBeAg(+) patients [129].
HBV drug development has highlighted the fact that preclinical animal studies may not predict DILI events in humans. In a phase II HBV clinical trial evaluating fialuridine, an investigational nucleoside analog that did not demonstrate hepatotoxicity in preclinical animal studies, five participants experienced fatal hepatotoxicity associated with pancreatitis and lactic acidosis [130]. The pattern of liver test elevations was notable initially for increased bilirubin associated with mild elevations of ALT levels and lactic acidemia. Likely mechanisms of action for fialuridine hepatotoxicity include mitochondrial injury and inhibition of pyruvate oxidation [131]. Thus, in clinical trials studying drugs with potential mitochondrial toxicity, there should be an awareness that the development of jaundice with minimal ALT elevations may suggest DILI rather than a spontaneous HBV flare.
While HDV should be excluded during the screening period (unless being studied as a separate cohort in later phase III and IV trials), patients with chronic HBV are at continued risk for HDV superinfection [132]. HDV superinfection can present as acute hepatitis with increasing ALT levels as well as worsening liver disease [132], scenarios that can resemble DILI. Therefore, testing for HDV immunoglobulin G (IgG) and HDV IgM should be obtained during the evaluation of any new onset of ALT elevations as part of the causality assessment for DILI. Superinfections with acute hepatitis A virus (HAV), as well as other viruses (hepatitis E virus [HEV], Epstein Barr virus [EBV], cytomegalovirus [CMV]) can also mimic an HBV flare, and these viruses should be excluded by the appropriate serologic studies (see Table 1 in the ESM).
Consensus Recommendations
-
9.
Baseline HBsAg and HBV DNA quantification should be established prior to the start of the study.
-
10.
Evaluation of rapidly rising ALT levels should include blood tests for levels of HBV DNA and HBsAg.
-
11.
The finding of stable HBsAg and HBV DNA levels can assist in differentiating an HBV flare from DILI.
-
12.
In clinical trials studying drugs with potential mitochondrial toxicity, the development of jaundice with minimal ALT elevations may suggest DILI rather than a spontaneous HBV flare.
-
13.
HDV, HAV, and other viral superinfections (HEV, EBV, CMV) should be considered during the evaluation of the new onset of ALT elevations or worsening of liver disease with the appropriate serologic testing.
Monitoring and Stopping Criteria
Lessons learned during the extensive HCV drug development over the last decade should be applied to drug development for new HBV treatments. In particular, since it was noted that normalization or significant reductions of ALT values occurred within the first few weeks of DAA therapy for HCV [116], it was suggested that this new ALT nadir value be incorporated into DILI monitoring and stopping rules for HCV clinical trials [15, 25, 48].
Normalization or significant reductions of ALT values also may occur in some patients in response to HBV therapy, typically by week 12 [133], which is not as early as the ALT reduction seen with DAA therapy for HCV [116]. In published studies on drugs in development for HBV in which stopping rules were available for review, new ALT nadir values in response to therapy were not used for monitoring and discontinuation [134]. However, utilization of a new in-study nadir value is advisable in patients with HBV who experience a significant improvement in ALT level in response to HBV therapy. In addition, establishing liver-related monitoring and stopping criteria based solely on multiples of ULN may result in inconsistent and/or incorrect evaluation of the hepatotoxicity of the candidate drug. Thus, using the baseline ALT value or new nadir level combined with a gatekeeper ALT level threshold (whichever comes first) for monitoring and interruption/discontinuation of therapy may lead to a more accurate assessment (Figs. 3, 4). Any occurrences of liver decompensation that are considered secondary to DILI should trigger permanent discontinuation.
Fig. 3.

Algorithm for monitoring and management of potential DILI signals in phase II–III chronic HBV clinical trials in patients with HBV with normal or elevated baseline ALT who are nucleos(t)ide suppressedf. fThese levels pertain to subjects who are nucleos(t)ide suppressed when entering trials, and values may differ in subjects who are not nucleos(t)ide suppressed. gBaseline ALT is derived from an average of two pretreatment ALT measurements 2 weeks apart. hFor patients with Gilbert’s syndrome or hemolysis. iSymptoms may be liver related (e.g., severe fatigue, nausea, vomiting, right upper quadrant pain) or an immunologic reaction (e.g., rash, > 5% eosinophilia). jElevated baseline is defined as ALT ≥1.5 × ULN. kIn patients with a sizable stable early decrease in ALT during treatment (> 50% of baseline value), a new baseline, corresponding to the ALT nadir, should be established on an individual basis for subsequent determination of a DILI signal. lThe specific interval between the tests should be determined based on the patient’s clinical condition. ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, DB direct bilirubin, TBL total bilirubin, ULN upper limit of normal
Fig. 4.

Algorithm for monitoring and management of potential DILI signals in phase II and III clinical trials for new agents to treat HBV in naïve or non-nucleos(t)ide-suppressed patients with normal or elevated baseline ALT. mBaseline ALT is derived from an average of two pretreatment ALT measurements 2 weeks apart. nFor patients with Gilbert’s syndrome or hemolysis. oSymptoms may be liver related (e.g., severe fatigue, nausea, vomiting, right upper quadrant pain) or immunologic reaction (e.g., rash, > 5% eosinophilia). pElevated baseline is defined as ALT ≥1.5 × ULN. qIn patients with a sizable stable early decrease in ALT during treatment (> 50% of baseline value), a new baseline, corresponding to the ALT nadir, should be established on an individual basis for subsequent determination of a DILI signal. rThe specific interval between the tests should be determined based on the patient’s clinical condition. ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, DB direct bilirubin, TBL total bilirubin, ULN upper limit of normal
Finally, when designing clinical trial monitoring and stopping rules, differentiation should be made between patients entering a trial on nucleos(t)ide analogs versus those naïve to therapy or nonresponders to nucleos(t)ide analogs (Figs. 3, 4) [135].
Consensus Recommendations
-
14.
When a significant improvement in ALT level or new nadir in response to HBV therapy is achieved (e.g., a decrease of > 50% of the original ALT baseline to a new stable level during the trial), this ALT value should subsequently be utilized as the subject’s new baseline value during the trial to determine DILI monitoring and stopping rules.
-
15.
A combination of multiples of baseline ALT values, or multiples of a new nadir, as well as a threshold value (whichever comes first) should be used when assessing the hepatotoxicity of the candidate HBV drug (Figs. 3, 4).
-
16.
Consideration should be given to convening an ad hoc panel of external hepatology experts at the onset of the trial to be available if cases of suspected DILI occur in a clinical trial with no alternative causal explanation and to perform an unblinded safety assessment and consider a temporary pause of the trial.
-
17.
An episode of DILI resulting in hepatic decompensation should trigger permanent drug discontinuation.
-
18.
When designing clinical monitoring and stopping rules for liver safety signals, differentiation should be made between patients entering a trial on nucleos(t)ide analogs versus those naïve to therapy or nonresponders to nucleos(t)ide analogs (see Figs. 3, 4).
Inclusion of Patients with HBV in Clinical Trials for Other Indications and Implications for Causality Assessment of Potential DILI Cases
Patients with HBsAg positivity are typically excluded from non-HBV clinical trials. This is because HBV reactivation can occur, leading to increased levels of ALT and other liver-related blood tests, making differentiation from DILI a challenge. However, even individuals with serological markers of resolved infection (HBsAg negative, undetectable HBV DNA, HBcAb positive, with or without HBsAb) may still harbor cccDNA and integrated HBV DNA [115]. Thus, HBV could be reactivated when the immune system is suppressed by chemotherapy for cancer treatment, immunosuppression for transplantation, monoclonal antibody (e.g., rituximab) for the treatment of hematologic malignancies, or DAA therapy for the treatment of hepatitis C [136–139]. Therefore, when evaluating the etiology of liver test abnormalities in patients participating in these trials, reactivation of HBV as signaled by reappearance of circulating HBV DNA should be included as part of the causality assessment for potential DILI. Inclusion of individuals with isolated HBcAb (without elevated aminotransferases, HBV DNA, or other serologic markers, including HBsAb) into trials not involving immunosuppressive therapies should be considered on a case-by-case basis, dependent upon the mechanism of action of the investigational product.
Consensus Recommendations
-
19.
The eligibility of patients with HBV to participate in non-HBV clinical trials is dependent upon multiple factors, including the class of the candidate drug being evaluated and the status of the patient’s immune system.
-
20.
When subjects with HBV are included in clinical trials for other indications, complete HBV virological assessment should be established at baseline (including full serology and HBV DNA levels).
-
21.
Individuals with serological markers of resolved infection (HBsAg negative, undetectable HBV DNA, HBcAb positive with or without HBsAb) can still harbor cccDNA and may be at risk for reactivation when exposed to immunomodulatory medications. Their participation should be considered on a case-by-case basis.
-
22.
Evaluation of unexplained ALT elevations should include HBV DNA testing, even in subjects entering trials with isolated HBcAb positivity.
Summary
Numerous ongoing clinical trials of new HBV therapies are aimed not only at viral suppression but more recently at HBV cure. However, many gaps still exist in our knowledge that need to be resolved before we can improve on best practices for trial design and the criteria for monitoring and stopping rules in these patients. Publication of clinical study protocols has been a step in the right direction, but this is not done for all published trials. Furthermore, clarification on how eligibility criteria related to liver-related blood tests were chosen, as well as how monitoring and stopping rules were determined, would be beneficial for the field, since these criteria vary amongst HBV clinical trials. When designing trial eligibility and stopping rules, differentiation should be made between patients who are nucleos(t)ide suppressed and those who are naïve to therapy or nonresponders to nucleos(t)ide analogs. Lessons learned from HCV drug development, as well as from our expanding knowledge and research in the field of DILI, will likely result in improved assessment of the hepatotoxic potential of a candidate drug, improved causality assessment, and greater consistency across industry in clinical trial design and liver-related monitoring and stopping rules.
Assessment of DILI in Clinical Trials of Adults with Cirrhosis Associated with HCV, HBV, or Nonalcoholic Steatohepatitis
Introduction
The most advanced form of CLD is cirrhosis, the clinicopathological entity marked by advanced fibrosis and hepatic dysfunction. Cirrhosis is due primarily to NASH, HCV, HBV, ALD, PSC, and PBC and occurs in about 0.27% (approximately 1:400) of the US population [64]. With the advent of suppressive antiviral therapy for HBV and DAAs that cure HCV, multiple trials have been conducted recently in patients with advanced fibrosis and cirrhosis secondary to both HBV and HCV. There is evidence that long-term anti-HBV therapy with suppression of HBV DNA will result in fibrosis improvement and even reversal in those with bridging fibrosis and compensated cirrhosis [129, 140]. The successful use of DAA treatment for patients with HCV with cirrhosis has demonstrated a regression of hepatic fibrosis as assessed by histology and/or transient elastography after at least 6 months of follow-up off medication [141–143]. Regression of fibrosis with new drugs is also currently being investigated in the NASH population with moderate fibrosis and in those with compensated cirrhosis [126, 144–149]. There is also interest in new drugs for treating decompensated cirrhosis and its complications, including those focused on reversing fibrosis as assessed by reducing the hepatic venous portal vein pressure gradient (HVPG) and/or model for end-stage liver disease (MELD) score and on treating hepatic encephalopathy, hepatorenal syndrome, variceal bleeding, and ACLF [150–155].
Monitoring for and diagnosing DILI in clinical trials of subjects with cirrhosis is challenging because of the normal fluctuation of liver tests, possible progression of the underlying liver disease, varying compromised clearance rates of drugs and their metabolites by the liver, and the fact that most patients are taking numerous concomitant medications. These challenges highlight the need for increased vigilance in clinical trials in subjects with cirrhosis. This section covers monitoring for potential DILI in subjects with cirrhosis participating in clinical trials of treatments for HCV, HBV, and NASH. Trials in subjects with cirrhosis due to PSC and PBC have been covered in a recently published companion paper from the IQ DILI Consortium with a focus on underlying cholestatic liver disease [16]. Since sufficient data are not yet available from all subjects with cirrhosis participating in clinical trials (e.g., NASH cirrhosis), some recommendations are the result of consensus opinion from academic, regulatory, and industry experts in the field.
Although no data are yet available, the efficacy of new drugs in development may vary depending on the severity of cirrhosis (e.g., compensated or decompensated) caused by the underlying CLD. Compromise of hepatic function that alters the clearance of a study drug varies among patients with cirrhosis [156]. For drugs that are metabolized primarily in the liver, functional changes that affect the pharmacokinetic profile of a study drug may be due to diminished first-pass metabolism due to portal hypertension and shunting; reduced drug binding protein levels; primary changes to hepatocyte drug metabolism and transport; and changes in biliary excretion. A recognition of these functional changes has led regulatory agencies to recommend that hepatic impairment studies be done early in drug development for drugs intended for the treatment of populations with underlying liver disease in order to assess the risk of excessive drug exposure and potential hepatotoxicity [144, 156]. This is especially important when hepatic metabolism and/or excretion accounts for more than 20% of drug elimination, when the drug has a narrow therapeutic range, or when the drug metabolism and elimination is unknown. Currently, the CTP score is used to define mild, moderate, and severe hepatic impairment, but it is recognized that this score was not originally intended to be a guide for potential dose modification in patients with hepatic impairment [157].
Recent publications have illustrated the role of studying pharmacokinetic parameters in subjects with advanced liver disease and employing population pharmacokinetic modeling and physiologically based pharmacokinetic modeling to predicting the potential need for dose adjustments in these populations [158, 159]. In addition, assessment of functional hepatocyte metabolism and transport, and first-pass clearance reflecting portosystemic shunting, might better reflect the correlation of hepatic impairment with excessive drug exposure and the risk of DILI than correlation with the CTP score. A full discussion of this important topic is beyond the scope of this paper and will be the subject of another paper from the IQ Consortium.
Consensus Recommendations
Hepatic impairment studies need to be done early in drug development in all programs targeting patients with underlying liver disease, especially when studying the cirrhotic population.
Further research is needed in defining new tools to assess hepatic impairment that are potentially more directly correlated with risks of excessive drug exposure; and consideration should be given to employing these tests in clinical trials of subjects with cirrhosis in comparison with the traditional classification of hepatic impairment by CTP score.
Prior to conducting studies in subjects with compensated cirrhosis, studies in small cohorts of patients with varying degrees of hepatic impairment may be useful, with the modeling of pharmacokinetic data to simulate the systemic drug exposure profiles for a range of drug doses. These studies may predict the need for dose adjustments in this population.
Inclusion/Exclusion Criteria in Studies of Compensated Cirrhosis
Cirrhosis encompasses a broad clinical spectrum of disease. Histologic evidence of cirrhosis (generally classified by Ishak score > 4 or Metavir score > 3) without clinical evidence of disease and without varices, associated with a modest increase in HVPG of > 6–10 mmHg, is sometimes called stage 1 compensated cirrhosis, often corresponding to CTP class A disease [160, 161].
Stage 2 compensated cirrhosis is characterized by the absence of symptoms but the presence of portal hypertension with varices, and HVPG in the > 10–12 mmHg range. Decompensated cirrhosis is defined by the presence of symptoms and a history of “decompensating events,” including variceal bleeding, hepatic encephalopathy, and development of ascites and often includes physical and laboratory-based indications of portal hypertension and significant hepatic functional impairment, including splenomegaly, low platelet count, direct hyperbilirubinemia, and elevated INR [162].
Detecting early stage 1 compensated cirrhosis (stage F4) in patients with CLD entering clinical trials and differentiating it from bridging fibrosis (stage F3) may be challenging, as baseline liver tests may not be markedly abnormal. It should be noted that although abnormal aminotransferases are common in CLD, they may be normal in some patients with advanced fibrosis or cirrhosis [163, 164]. In a large cohort of patients with biopsy-proven NASH, the mean ALT level in those with cirrhosis (stage F4) was lower than in those with fibrosis stages 1–3 (46 IU/l vs. 70–78 IU/l, respectively), although mean ALP was higher (100 U/l vs. 79–89 U/l, respectively) [165]. AST levels are also relatively low in patients with compensated cirrhosis, with a mean of 57–67 U/l [166]. Compared with absolute ALT values, the AST/ALT ratio appears to be a more sensitive indicator of cirrhosis [167, 168]. In patients with HCV, an AST/ALT ratio of > 1 was present in only 4% of all patients without cirrhosis but in 79% of those with documented cirrhosis [169]. In a recent meta-analysis of 86 studies that evaluated the accuracy of clinical findings for identifying biopsy-proven cirrhosis from multiple causes, a platelet count < 160,000 × 109/l, a prolonged prothrombin time, or a serum albumin < 3.5 g/dl were more discriminating with a higher likelihood ratio for cirrhosis than an increased ALT or bilirubin [170]. A full discussion of noninvasive methods to diagnose cirrhosis is beyond the scope of this paper, but the APRI, the Fibrosis-4 (FIB-4) score, and the use of transient elastography imaging all may aid in characterizing the population entering screening for trials in CLD with cirrhosis [171–173].
In previous studies of subjects with compensated cirrhosis due to HCV, exclusion criteria have included another known underlying CLD (e.g., HBV), excessive alcohol intake, or exposure to any concomitant administration of herbal or dietary supplements linked to hepatotoxicity. A recent report by the US Drug-Induced Liver Injury Network (DILIN) listed the top ten individual drugs identified as causing DILI in clinical practice in their prospective long-term study [3]. These included primarily antibiotics/antimicrobials (ciprofloxacin, levofloxacin, azithromycin, amoxicillin–clavulanate, cefazolin, minocycline, nitrofurantoin, sulfamethoxazole–trimethoprim, and isoniazid) as well as diclofenac. The NIH-sponsored database LiverTox [174] provides up-to-date information on the hepatotoxicity of more than 500 drugs in the current database of more than 1200 prescription and nonprescription compounds. A classification of the hepatotoxic potential of marketed medications based on case reports of DILI has also recently been published by others [175–177]. Although an increased risk of DILI has not been confirmed in prospective studies, avoidance of these higher risk hepatotoxic medications while searching for a potentially safer alternative is recommended in subjects entering clinical trials in cirrhosis [178]. In addition, drugs known to potentially precipitate hepatic encephalopathy, gastrointestinal bleeding, or renal failure in patients with cirrhosis are generally avoided [179].
Information on baseline values of ALT, AST, and TBL in subjects entering clinical trials with compensated cirrhosis is provided in Table 1. Previously conducted studies of DAAs for treatment of HCV in patients with compensated cirrhosis allowed subjects to enroll with aminotransferases up to 5–10 × ULN, TBL up to 1.5–3 × ULN, INR prolonged to 2.2, albumin as low as 2.8–3.5 g/dl, and platelet counts as low as 50,000 × 109/l [161, 180–184]. In these studies, CTP scores were confined to < 7, and the mean baseline ALT and AST levels prior to receiving study drug(s) were as high as 102 IU/l and 101 IU/l in treatment naïve and treatment experienced subjects, respectively, with a range of up to 326 and 231 IU/l, respectively [68]. The upper limit of ALT and AST in patients with HCV and compensated cirrhosis may in fact be higher because some of these studies excluded subjects with baseline aminotransferase > 5 × ULN, and other studies excluded those with > 10 × ULN.
Studies in compensated cirrhosis due to HBV treated with pegylated interferon, nucleos(t)ide analogs, or a combination of nucleos(t)ide and thymosin generally allowed subjects to enroll with ALT levels up to 10 × ULN, TBL up to 2 × ULN, INR prolonged to 1.3, albumin as low as 2.8 gm/dl, and platelet counts as low as 50,000 × 109/l. In these studies, the mean baseline ALT levels prior to receiving study drug(s) varied between 62 and 160 IU/l. Median ALT was between 50 and 70, and—in some studies—as many as 22% of subjects had ALT values within normal limits. CTP scores were mostly 5–6 and generally < 7, but as high as 9 in one study. Exclusion criteria were any decompensating events; HCV, HIV, or HDV infection; renal insufficiency; or excessive alcohol consumption [185–191].
Studies are currently ongoing or have been completed with new drugs for NASH with compensated cirrhosis including studies with emricasan, the galectin-3 inhibitor GR-MD-02, and selonsertib [126, 146, 148]. Inclusion criteria in these studies included the absence of any decompensating events and allowed subjects to enroll with ALT < 8–10 × ULN, AST < 10 × ULN, albumin ≥2.8 g/dl, platelets > 60,000 × 109/l, DB < 2.0 mg/dl, and INR < 1.7. A recent draft guidance from the FDA on conducting clinical trials in NASH with compensated cirrhosis recommended that patients with ALT or AST elevation > 5 × ULN or TBL above the ULN at screening should not be enrolled and should be investigated for the possibility of concomitant liver disease (e.g., alcohol-associated liver disease or viral or autoimmune hepatitis) [162]. The presence of medium or large varices on endoscopy (even without a history of gastrointestinal bleeding) or an HVPG measured at ≥12 mmHg have been used to define decompensated cirrhosis or “late-stage” compensated cirrhosis with a high risk of decompensation and would serve to exclude subjects from compensated cirrhosis trials [192–194].
CTP scores based on the presence or absence of encephalopathy and ascites, and the laboratory parameters of TBL, albumin, and INR are usually < 7 in the Class A category (which defines compensated cirrhotic patients). The compensated NASH cirrhosis studies cited above allow subjects to enroll with CTP scores ≤7, and with MELD scores up to 12 in one study and 15 in another [146, 148]. In previous studies of treated patients with HCV-associated cirrhosis, the baseline MELD score was independently associated (multivariate analysis) with the risk of hepatic decompensation during a 72-week post-treatment observation period of 22%, 59%, and 83% in patients with baseline MELD scores of 6–9, 10–13, and > 14, respectively [195]. These data suggest that baseline MELD scores > 10 expose a subject with cirrhosis to a higher risk of hepatic decompensation during the course of a phase IIb/III clinical trial. In the survey of IQ DILI companies conducted by the consortium (unpublished data), 8 of 11 companies that had conducted trials that included patients with compensated cirrhosis used the CTP score as an exclusion criterion, 5 of 11 used the MELD score, and 3 of 11 used any indication or history of decompensation (e.g., variceal bleeding, peritonitis, hepatorenal syndrome, hepatic encephalopathy).
As recommended for clinical trials in noncirrhotic cohorts with HCV, HBV, and NASH, at least two values for biochemical liver tests are recommended during screening to establish baseline values in subjects with cirrhosis; these tests should also include albumin, INR, DB, and creatinine as assessments of liver and other organ dysfunction that can accompany progression to decompensation in subjects with cirrhosis. Because a rise in aminotransferases may not be as sensitive to increasing liver injury in subjects with cirrhosis compared with those without, requiring an increase of 50% of the second value over the initial value to trigger re-evaluation of the subject for inclusion (as discussed) may not be appropriate in clinical trials of cirrhosis. For these subjects, a smaller increase of 33% over the initial value in aminotransferases in the second determination may still signal significantly worsening disease and decompensation and warrant a repeat determination and delay in enrolling the subject in the trial [196].
Consensus Recommendations
-
4.
Consideration should be given to continuing some measurements of pharmacokinetic parameters during phase II and III studies in subjects with cirrhosis when studying drugs that undergo significant hepatic metabolism and excretion and/or demonstrate a narrow therapeutic range. Review of unblinded data by an independent data monitoring committee (DMC) to monitor for excessive exposure and potential increased risk for DILI and to differentiate this from progression of the underlying disease is recommended.
-
5.
Inclusion criteria for clinical trials in subjects with compensated cirrhosis must clearly delineate accepted methods for identifying compensated (and excluding decompensated) cirrhosis specific to the disease being studied based on clinical, imaging, biochemical evidence, and/or histologic scoring systems.
-
6.
In studies in patients with compensated cirrhosis, it is preferable to limit concomitant medications whenever clinically feasible utilizing such resources as LiverTox (https://livertox.nih.gov). Unsupervised complementary and alternative medicine use (including herbal and dietary supplements) should be strongly discouraged. DDIs known to affect potentially impaired hepatic transport and metabolism pathways of study drug in patients with cirrhosis (by demonstrated or modeled evidence) should also be considered and avoided.
-
7.
Decompensating events include the following: documented variceal hemorrhage, documented ascites (by imaging or examination), spontaneous bacterial peritonitis, and documented hepatic encephalopathy (with examination by an experienced clinician). Even in the absence of ascites or encephalopathy, the presence of any of the following abnormal laboratory values, including an albumin < 2.8 g/dL, INR > 1.7, or TBL > 2.0 mg/dl with a DB > 50% of the TBL have been used to exclude subjects from trials of compensated cirrhosis in patients with HBV, HCV, and NASH, since they would result in a CTP score ≥ 7. Patients with any of these clinical and laboratory features should be considered to have decompensated cirrhosis and be excluded from clinical trials of compensated cirrhosis.
-
8.
Inclusion of subjects with stage 2 compensated cirrhosis with portal hypertension, low platelet count, medium to large varices, and if available an HVPG ≥ 12 mmHg (even without any history of bleeding) in clinical trials of new treatments should be based on the population expected to use the intended treatment in a real-world setting, the population being recruited, and the study goals. Small proof-of-concept trials to support an expectation of adequate safety and efficacy should be performed prior to including these patients in larger clinical trials.
-
9.
Baseline CTP and MELD scores should be calculated, and patients with CTP ≥ 7 and those with MELD ≥ 10 should be excluded in trials of patients with compensated cirrhosis.
-
10.
It is advisable to obtain two screening determinations of hepatic biochemical tests 2–4 weeks apart (with the second as close as possible to the first dose of study drug) to calculate a mean value as the subject’s reference baseline. If there is any significant increase in TBL, DB, or INR, or more than a 33% rise in aminotransferases from the first to the second determination, a third measurement should be obtained to determine whether the subject’s underlying liver disease is worsening prior to initiation of study drug.
Changes in Liver Tests or Symptoms that Trigger Increased Monitoring in Patients with Cirrhosis
Large natural history studies in patients with hepatitis C and compensated cirrhosis who were followed at least 24 months off treatment (having failed to achieve SVR when treated with interferon and ribavirin) demonstrated a pattern of baseline laboratory values associated with a higher risk of progression to decompensated cirrhosis, liver transplant, and liver-related death. A model that included platelet count < 150,000 mm3, AST/ALT ratio > 0.8, TBL > 0.7 mg/dl and albumin ≤ 3.9 g/dl was the best predictor of clinical decompensation during follow-up [123]. In addition, a decrease in platelet count of > 15%, an increase in AST/ALT ratio of > 15%, an increase in TBL of > 15% compared with baseline, and a decrease in albumin of > 15% were correlated with a significant increase in clinical decompensation [197]. It is not known whether this degree of change or the time course for predicting clinical decompensation can be generalized to other causes of cirrhosis, but these data have led experts to recommend that subjects entering clinical trials with compensated cirrhosis need to be monitored more closely from the outset of the trial for changes that may be due to either DILI or disease progression [44]. A specific schedule for “close monitoring” has yet to be clearly defined [25]. If any data on peak time to onset of DILI have been previously obtained in the study drug in different patient populations or in drugs in the same class with the same mechanism of action, then these data should influence the enhanced “close monitoring” schedule put into place.
In patients with compensated cirrhosis, even mild elevations in ALT or AST during acute superimposed liver injury due to a suspect drug may represent extensive hepatocyte injury. In these patients, the hallmark of hepatocellular DILI may be decompensating events, such as the development of hepatic encephalopathy, ascites, or variceal bleeding, or laboratory changes reflecting decreased hepatic functional reserve, including increasing TBL, increased DB, coagulopathy, and reduced albumin [198]. This is illustrated by findings in a large series of > 1300 patients with compensated cirrhosis with precipitating events that caused either acute decompensation (AD) without organ failure, or decompensation with organ failure (ACLF) [199]. In those with AD, the mean ALT on admission to the hospital was only approximately 1.4 × ULN, whereas the mean TBL had risen to 5 × ULN and the mean INR was elevated to 1.5. In those with ACLF, mean ALT was still only 1.6 × ULN, but the mean TBL was 13 × ULN and the mean INR was 2.1. INR also increased with increasing grades of ACLF [152]. Because of the extent of hepatocyte damage at baseline and the potential limited capacity of ALT and AST elevations to reflect further acute DILI-related damage, any increases in the baseline values of tests reflecting liver functional capacity (INR, bilirubin, albumin) as well as defined multiples of baseline aminotransferases (rather than multiples of the ULN) have been recommended for prompting increased DILI monitoring [200]. Together with these flags, prespecified enzyme activity thresholds that are exceeded based on the ranges of ALT and AST seen during screening in each of these CLD (see Table 1) can serve as “guardrails” to trigger increased monitoring. In the previous two sections on clinical trials in HCV and HBV, alterations in aminotransferases were primarily focused on changes in ALT and not AST. This reflects the greater specificity of ALT for the liver and not muscle or red blood cells. With the advent of increasing fibrosis and cirrhosis, a rising AST and an increase in the AST:ALT ratio to near or > 1.0 can signal this progression in patients with NASH, HBV, and HCV. An abrupt change in that ratio with a relatively greater rise in ALT versus AST may signify DILI or some other cause of acute liver injury in patients with cirrhosis rather than a progression of the underlying disease [201].
Elevations in DB are considered more reflective of decreased liver function than are isolated elevations in unconjugated bilirubin (which are often due to Gilbert’s syndrome or hemolysis). Increases in ALP and TBL with only mild or no increases in aminotransferases may suggest biliary obstruction, making causality assessment more challenging. But in this population, where increases in ALT and AST may not correlate with the degree of hepatocyte damage from DILI, an unexplained rise in DB over baseline, even in the absence of significant elevations in aminotransferases, may also signal hepatocellular DILI requiring case-level evaluation.
Consensus Recommendations
-
11.
Baseline and monitored liver tests should include not only standard AST, ALT, ALP, and TBL but also readily available tests that reflect liver function, including DB, INR, and albumin.
-
12.
In phase I and IIa trials involving patients with cirrhosis, monitoring should be weekly or biweekly until an understanding of the hepatic safety profile, pharmacokinetic characteristics, and concentrations of the drug are obtained. Even in phase IIb and III trials, more frequent monitoring for hepatotoxicity over a longer duration is advised when compared with trials not including patients with cirrhosis.
-
13.
Because patients can decompensate quickly, consensus expert opinion supports increased monitoring during phase III trials tailored to the risk of hepatotoxicity of study drug in the population being studied. This could call for a schedule of monitoring as frequent as every week during the first month of the trial; biweekly for the next 2 months; at least once a month through 6 months, and a minimum of every 2 months for the duration of the trial.
-
14.
Multiples of baseline ALT, AST, ALP, and TBL elevations rather than multiples of ULN should be used as a reference for detection of DILI. Any increases in TBL, DB, or INR and doubling of the baseline value of ALT, AST, and ALP (provided that the values are also > ULN) should prompt a more vigilant approach to monitoring for potential DILI. Even increases limited to doubling of ALP and DB over baseline, even without concomitant significant elevations of aminotransferases but without evidence of biliary tract obstruction, should prompt increased monitoring for DILI.
-
15.
In trials of cirrhosis due to HCV and NASH, elevations of ALT out of proportion to AST resulting in a decreased AST/ALT ratio (so that ALT now exceeds AST) compared with a baseline ratio > 1.0 may suggest potential DILI or some other cause of acute hepatotoxicity rather than progression of the underlying cirrhotic liver disease (see Table 1 in the ESM).
Liver Test Thresholds or Symptoms that Trigger Stopping Study Drug in Patients with Compensated Cirrhosis
A wide range of “stopping rules” for individual study subjects have been used in clinical trials of new drugs for cirrhosis associated with HCV [180–184, 196, 200, 202]. In trials of DAAs in HCV-induced compensated cirrhosis, individuals meeting any of the following criteria were required to stop study drug: A confirmed elevation of ALT or AST > 5 × baseline or nadir value, a confirmed elevation of ALT > 3 × baseline and TBL > 2 × ULN, and any confirmed elevation of ALT > 15 × ULN without an alternate etiology [203]. An increase in DB > 1 mg/dl over the baseline value, or an INR > 1.5 if normal at baseline and increased by > 0.2 if 1.5–1.7 at baseline were also reasons to stop study drug in some studies [200, 203]. Using these criteria, no on-treatment cases of serious hepatotoxicity were observed and no Hy’s law cases were identified [203]. No individual met the prespecified liver-related stopping rules during the on-treatment phase, and ALT or AST increases > 5 × ULN were infrequent and generally transient. Because of this experience and given the concern that elevations in aminotransferases in response to DILI may be relatively constrained in patients with cirrhosis, more conservative criteria for triggering stopping drug in trials of patients with cirrhosis have been proposed [178].
In NASH trials with bridging fibrosis (stage F3) [204], ALT or AST > 2 × baseline and 5 × ULN with TBL > 2 × ULN or INR > 1.5 provoked stopping, as did an ALT or AST > 5 × baseline or 10–20 × ULN without an alternative etiology [200]. Stopping rules for NASH trials now being run in patients with compensated cirrhosis are currently not available [126, 145, 146, 148]. Owing to the differences in the underlying liver disease causing cirrhosis and baseline liver function at enrollment, optimal discontinuation criteria may need to be established for each clinical trial based on the particular study population, the specific disease being studied, and the risk profile of the investigational drug. Expert input from an external hepatic adjudication committee (HAC) comprising individuals with clinical expertise in the field of liver disease and DILI may be required to set optimal criteria for drug discontinuation prior to beginning complex trials in subjects with CLD and cirrhosis. When potential cases of DILI are identified in a clinical trial program, case-level assessments of causality as well as a comprehensive evaluation of risk for DILI associated with the study drug should be performed by an HAC with results made available to the unblinded DMC monitoring the trial. Blinded case-level assessments by the HAC are warranted for trials in which background disease progression effects are difficult to distinguish from potential adverse effects of the study drug. In the IQ DILI Consortium survey, 9 of 13 (69%) companies used an HAC for evaluating complex cases of potential DILI.
Consensus Recommendations
-
16.
Owing to the complexity of potential cases of DILI in trials including subjects with underlying cirrhosis, it is highly recommended to identify a HAC composed of experts in clinical liver disease and DILI for setting discontinuation criteria prior to study onset and for causality assessment of potential DILI cases during the study.
-
17.Suggested criteria for the discontinuation of study drug for subjects with compensated cirrhosis with HCV, HBV, or NASH with abnormal baseline aminotransferases, ALP, or bilirubin include the following:
- AST, ALT, or ALP > 2 × baseline or 5 × ULN (whichever comes first), along with TBL > 2 × ULN or INR > 1.5 or increased by 0.2 if starting with baseline INR ≥1.5
- AST or ALT > 3 × baseline or > 8 × ULN, whichever comes first
- Development of decompensated cirrhosis (variceal hemorrhage, hepatic encephalopathy, ascites)
- Increase in ALP > 2 × baseline and DB by > 1 mg/dl from baseline value without an alternative etiology.
-
18.Suggested criteria for discontinuation of study drug for subjects with cirrhosis with normal baseline ALT, AST, ALP, or bilirubin are as follows:
- AST, ALT, or ALP > 2 × baseline or 3 × ULN (whichever comes first), along with TBL > 2 × ULN or INR > 1.5 or increased by 0.2 if starting with baseline INR ≥1.5
- ALT or AST > 5 × ULN
- Development of decompensated cirrhosis
- Increase in ALP > 3 × ULN and DB by > 1 mg/dl from baseline value without an alternative etiology.
-
19.
Obtaining pharmacokinetics at the time of stopping study drug for suspected DILI should be strongly considered, with the sample to be analyzed later for quantitative levels of study drug and major drug metabolites in the circulation. Using this information, together with concomitant clinical and laboratory test results, a comprehensive analysis of the key determinants of systemic exposure of the study drug in patients with cirrhosis should be performed. This analysis may inform a need for optimal dose adjustments in subsequent studies of subjects with different levels of hepatic impairment.
The following table presents an algorithm that summarizes the above recommendations on increased monitoring for potential DILI events and on criteria for holding or stopping study drugs in clinical trials involving patients with compensated cirrhosis (see Table 3).
Table 3.
Algorithm for monitoring and management of possible DILI in clinical trials in patients with HCV, HBV, NASH, and compensated cirrhosis with normal or elevated baseline liver testsa
| Baseline evaluation | Treatment-emergent ALT, AST | Treatment-emergent AST:ALT | Treatment-emergent TBL, DB, ALP | Treatment-emergent INR | Decompensating events (VH, HE, ascites) | Action |
|---|---|---|---|---|---|---|
| Normal baseline < 1.5 × ULN of ALT, AST (with normal TBL, DB, ALP, INR) | ALT or AST ≥2 × baselineb | Rise in ALT out of proportion to AST, resulting in decreased ratio | Any increase ≥2 × baseline TBL, ALP (with ≥50% DB) even without elevations of AT | Any increase | None | Any of these changes should trigger increased monitoring with repeat testing within 1 week |
| ALT, AST ≥2 × baseline or 3 × ULN (whichever comes first) | TBL ≥2 × ULN | INR ≥1.5 | None | Combination of changes in AT and TBL or AT and INR: Stop study drug and proceed with causality assessmentc | ||
| ALT or AST ≥5 × ULN | No change | No change | None | Stop study drug and proceed with causality assessmentc | ||
| No change | ALP ≥3 × ULN and DB ≥1 mg/dl over baselined | No change | None | Stop study drug and proceed with causality assessmentc | ||
| Change or no change | Change or no change | Change or no change | Development of VH, HE, or ascites | Stop study drug and proceed with causality assessmentc | ||
| Abnormal Baseline | ALT or AST ≥2 × baseline or 5 × ULN (whichever comes first) |
TBL ≥2 × ULN if normal at baseline (with ≥50% DB) TBL ≥1.5 × baseline if abnormal at baseline, or DB ≥1 mg/dl over baseline |
INR ≥1.5 or increased by 0.2 if baseline INR ≥1.5 | None | Combination of changes in AT and TBL or AT and INR: Stop study drug and proceed with causality assessment | |
| ALT or AST ≥3 × baseline or 8 × ULN, whichever comes first | No change | No change | None | Stop study drug and proceed with causality assessmentc | ||
| No change | ALP ≥2 × baseline and DB ≥1 mg/dl over baseline | No change | None | Stop study drug and proceed with causality assessmentc | ||
| Change or no change | Change or no change | Change or no change | Development of VH, HE, or ascites | Stop study drug and proceed with causality assessmentc |
ALP alkaline phosphatase, ALT alanine aminotransferase , AST aspartate aminotransferase , AT aminotransferase, DB direct bilirubin , DILI drug-induced liver injury, HBV hepatitis B virus, HCV hepatitis C virus, HE hepatic encephalopathy, INR international normalized ratio, NASH nonalcoholic steatohepatitis , TBL total bilirubin , ULN upper limit of normal range, VH variceal hemorrhage
aVariations on criteria for holding and stopping drug in clinical trials of decompensated cirrhosis in HCV, HBV, and NASH are contained in the text
bProvided the values are also > ULN
cCausality assessment should include obtaining a PK sample at time of potential DILI event for later analysis of excessive exposure
dWithout an alternative etiology
Clinical Trials in Decompensated Cirrhosis
The cumulative proportion of patients with compensated cirrhosis who undergo progression to decompensated disease is thought to be approximately 10% per year and approximately 50% over 5 years [198, 205]. Thus, careful evaluation of patients entering clinical trials is important to ensure they meet the inclusion/exclusion criteria if the trial is limited to compensated cirrhosis.
A recent analysis of data from a placebo controlled clinical trial in patients with multi-etiology decompensated cirrhosis (CTP > 7; MELD < 25 [median 12]) undergoing treatment with glycerol phenylbutyrate added to standard-of-care therapy for hepatic encephalopathy illustrated the problem of applying conventional guidance to monitor for and diagnose DILI in clinical trials in this patient population when it is the intended group for treatment with a study drug [65, 154]. Baseline ALT values were abnormal in 42% of 178 subjects, AST in 71%, TBL in 67%, and INR in 62%. Both screening and baseline values just prior to first dose of study drug, and those collected during the trial, showed fluctuations with substantial variation over time, with or without exposure to study drug. During the trial, 23% of patients on study drug and 16% of those on placebo developed liver test abnormalities that otherwise would typically prompt treatment discontinuation and the undertaking of a comprehensive causality assessment (including 88% of these with concomitant ALT > 3 × ULN and TBL > 2.0 × ULN or INR > 1.5). The study subjects who surpassed these threshold liver test abnormalities included equal numbers with normal as well as abnormal baseline values. These data underscore the fact that the current FDA guidance for increased monitoring for DILI and stopping rules is not applicable for clinical trials of new drugs in individuals with decompensated cirrhosis.
The study of antiviral drugs in patients with cirrhosis with HBV and HCV provides substantial information about baseline liver tests in patients with decompensated cirrhosis. In these studies, there was a wide variation in the baseline tests and inclusion/exclusion criteria, and some included combinations of patients with both decompensated and compensated cirrhosis. Subjects with both HBV and HCV were included with CTP classes B and C (median scores 7–11 at baseline but up to 12 in one study in HBV and generally no upper limit) and median MELD scores of 10–16 (mean 15) [206–211]. In some studies of decompensated cirrhosis with HBV, inclusion required ALT > 1.5 × ULN or as high as 2.0 × ULN. Mean baseline ALT in HBV studies varied from 98 to 169 U/l, with a range up to approximately 360 U/l [212]. Mean baseline TBL ranged from about 2.0 to 4.5 mg/dl and mean baseline albumin from 2.8 to 3.0 g/dl. Fluctuations in bilirubin, prothrombin time (PT)-INR, and albumin during screening resulted in changes in the assignment of CTP classes in up to 10% of subjects between the time of initial screening and the time of first dose of study drug. Exclusion criteria in these studies in HBV have included HIV, HCV or other known viral infections, alcohol abuse, presence of hepatocellular carcinoma, and fulminant hepatic failure. Criteria for exclusion have also been ALT > 10 × or ≥15 × ULN, grade 3 or higher hepatic encephalopathy, MELD score > 25, serum creatinine > 2.0–2.5 mg/dl or creatinine clearance < 50 ml/min, serum sodium < 125 mEq/l, and platelet count < 35,000 × 109/l [206, 209–211].
In a study of subjects with NASH and decompensated cirrhosis (defined as a previous history of variceal hemorrhage or ascites), subjects with MELD scores ≥ 12 and ≤ 20 were included and those with ALT > 3 × ULN, AST > 5 × ULN, and alpha-fetoprotein (αAFP) > 50 μg/mL were excluded [153]. In another study of patients with NASH cirrhosis with elevated HVPG and small varices but no previous history or overt signs of decompensation, exclusion criteria were aminotransferase > 10 × ULN, platelets < 60,000 × 109/l, albumin < 2.8 g/dl, INR > 1.5, DB > 2.0 mg/dl, MELD > 15, and αAFP > 200 μg/ml [148].
Based on the data summarized above, conservative criteria based on expert opinion used to interrupt and potentially stop a study drug in this population and perform a causality assessment for potential DILI include any of the following: (1) ALT or AST > 2 × baseline or > 3 × post-baseline on-treatment nadir values; (2) TBL > 1.5 × baseline with DB ≥ 50% of the total, or any increase in DB ≥ 1 mg/dl over baseline; (3) any increase in INR to > 1.5 if starting with a normal INR at baseline or any increase of 0.2 if starting with an INR of ≥ 1.5; or (4) further decompensation events [196, 200, 201].
Decompensated Cirrhosis and the Risk of Excessive Study Drug Exposure Leading to DILI
Recently, the FDA has issued warnings about liver failure occurring after the misuse of protease inhibitor-containing regimens of DAAs used to treat HCV in patients with decompensated cirrhosis (CTP B and C), portal hypertension, and other preexisting risk factors such as liver cancer or alcohol abuse. These warnings have previously included Viekera Pak® (ombitasvir, paritaprevir, ritonavir, dasabuvir) (AbbVie, IL, USA) [88] and more recently Mavyret® (glecaprevir, pibrentasvir) (AbbVie), Zepatier® (elbasvir, grazoprevir) (AbbVie), and Vosevi® (sofosbuvir, velpatasvir, voxilaprevir) (Gilead Sciences, CA, USA) [90]. In some instances, liver injury and worsening liver function were associated with markedly increased exposure to some components of these medicines. A significant relationship between exposure to paritaprevir (a component of Viekira Pak®) and DILI risk was evident in CTP B subjects where the exposure was increased by 62% [213]. In subjects with more severe hepatic impairment, it was increased as much as 945%. A similar phenomenon was evident with Zepatier® (a combination of elbasvir and grazoprevir) where severe hepatic impairment was associated with a 12-fold increase in exposure to grazoprevir. Importantly, the FDA pointed out that many of the patients who decompensated were inappropriately categorized as CTP class A when they were actually CTP B [89].
There are also other instances where DILI appears to be linked to dose-related elevations in drug concentrations and overexposure in patients with moderate to severe liver impairment, including overt decompensation events from primary liver disease [214]. Since the marketing approval of obeticholic acid for PBC in May 2016, the FDA Adverse Event Reporting System had received reports of 19 deaths and 11 cases of serious liver injury in patients receiving this treatment [215]. Higher than recommended doses (5 mg daily instead of 10 mg twice weekly) were prescribed to these patients with advanced cirrhosis. Moderate and severe hepatic impairment studies in subjects with CTP class B and C of a single dose of obeticholic acid 10 mg resulted in total plasma drug exposures of 4- and 17-fold increases, respectively, over subjects with normal liver function [214]. Actual exposures in the livers of these patients are difficult to predict and may be significantly increased or decreased according to altered liver functional capacity and portosystemic shunting.
These examples highlight the essential need for full hepatic impairment studies (including CTP B and C) in preapproval drug development programs involving subjects with decompensated cirrhosis [216] and emphasize the recommendation that the determination of study drug concentration be part of the causality assessment when investigating potential DILI cases that have a potential drug exposure-related hepatotoxicity component [196]. In some instances, data from such studies may also justify protocol instructions to modify study drug dosing during the treatment phase in the presence of worsening cirrhosis.
Use of Model for End-Stage Liver Disease Score as a Criteria for Stopping Drug for Potential DILI
Alternatives to standard liver tests to monitor for DILI and assess severity may be called for in clinical trials in patients with decompensated cirrhosis. The MELD score (predicting 90-day survival without liver transplant based on measurements of bilirubin, INR, creatinine, and serum sodium) has been suggested at baseline and during treatment to help monitor for DILI in such patients and assess prognosis of DILI events [15, 25, 44]. Several studies of new therapies in patients with HCV and decompensated cirrhosis have resulted in improved MELD scores during the trial in patients who achieved SVR, mostly mediated by a reduction in TBL levels [209, 211]. This suggests that a worsening MELD score during such a trial may be a signal of potential DILI in a subject with an undetectable HCV RNA level during the acute deterioration of liver function. The MELD score has been shown to be a good predictor of survival in DILI-related acute liver failure in previous studies [217, 218]. An increase in the MELD score over the baseline score of > 10 points in a patient with decompensated cirrhosis is indicative of an absolute increase in mortality of 10%. An increase of 5 points in the MELD score has been used previously in trials of subjects with decompensated cirrhosis as a criteria to stop study drug; if the baseline MELD score is > 10, an increase of 3 points has been proposed as a stopping criteria [201]. As with other scoring systems, the MELD score may rise as a result of further decompensation of the underlying CLD, confounding the adjudication of potential DILI. Particularly in trials in decompensated cirrhosis (but even in compensated cirrhosis), it is not until a full comparison between study drug and placebo arms is conducted that it may be possible to discriminate between DILI and progression of the underlying liver disease.
Consensus Recommendations
-
20.
As part of the early-phase clinical development program, hepatic impairment studies, preferably with multiple dose strategies, should be conducted to quantify differences in drug clearance in these patients versus those with compensated cirrhosis and normal patients without CLD. In general, the extent of enrollment for these studies should include subjects with CTP classes A, B, and C to cover the postmarket treatment population of patients with decompensated cirrhosis.
-
21.
Studies that measure study drug hepatic metabolism and portosystemic shunting may be useful in predicting drug clearance changes that might result in either excessive (potentially hepatotoxic) or diminished (potentially inadequate) exposure to the agent.
-
22.Studies of new drugs in populations with decompensated cirrhosis directed at the underlying HBV, HCV, or NASH with the therapeutic goal of regression of fibrosis and/or reduction of portal hypertension should be discussed with the regulatory agencies prior to protocol development. In order to facilitate recognition of potential DILI events during the clinical trial, enrollment should be limited to a population characterized by the following:
- No criteria of ACLF.
- Stable decompensated cirrhosis and no active gastrointestinal bleeding, hepatic encephalopathy grade 3 or higher, refractory ascites, or spontaneous bacterial peritonitis or sepsis.
- CTP 7–12; MELD 10–24.
- At baseline, ALT < 150 U/l; TBL ≤ 3.0 mg/dl; albumin > 2.8 g/dl, creatinine ≤ 1.5 mg/dl.
-
23.
Due to fluctuating aminotransferase and biomarkers of liver function (TBL, DB, INR, albumin), as well as laboratory parameters and clinical assessments for MELD and CTP scores, baseline values and composite scores should be obtained as close as possible to the first dose of study drug. Vigorous monitoring as described for trials in compensated cirrhosis should also apply in this population.
-
24.Suggested criteria for temporarily holding or permanently stopping a study drug include the following:
- In subjects with elevated baseline ALT (defined as > 1.5 × ULN), an ALT > 2 × baseline or > 200 IU/l, whichever comes first; or 3 × post-baseline on-treatment nadir values
- In subjects with elevated baseline AST, an AST > 3 × baseline or > 300 IU/l, whichever comes first
- TBL > 1.5 × baseline with a DB of ≥ 50% of the total; or a DB increased ≥1 mg/dl over the baseline. Elevations of ALP and bilirubin without evidence of biliary obstruction, even without elevations in aminotransferase should prompt consideration of stopping in this population
- INR increase to > 1.5 if normal at baseline, or increased by 0.2 if baseline INR ≥ 1.5
- MELD score increased by 5 points over baseline (if ≤ 12 at baseline) or by 3 points over baseline (if > 12 at baseline).
-
25.
Causality assessment for potential DILI events in clinical trials of subjects with decompensated cirrhosis should include pharmacokinetic samples to measure drug exposure in patients whose deterioration merits stopping drug and withdrawal from the trial. Pharmacokinetic data should be provided to the HAC in consideration of recommendations to stop the trial due to excessive drug exposure or to adjust the dose in subjects with moderate and/or severe hepatic impairment. In assessing worsening liver function in such study subjects as to whether a causal association with the study drug exists, it is important to exclude other causes, including infection, exposure to other drugs, and gastrointestinal bleeding.
Summary
It is critical that clear inclusion/exclusion criteria that define and control enrollment be applied in clinical trials in patients with cirrhosis from HCV, HBV, or NASH to ensure the population conforms to compensated or decompensated cirrhosis. Studies to detect changes in drug clearance due to hepatic drug metabolism, portal shunting, or other factors should precede longer-term safety and efficacy studies. Several underlying features of HCV, HBV, and NASH with cirrhosis dictate changes in best practices for monitoring for and detecting potential DILI events in clinical trials of interventional drugs for these disease indications. Such changes include recognizing that extensive chronic liver injury may mask the usual correlation between further acute injury from hepatocellular DILI and significant rises in aminotransferases. Thus, using multiples of baseline and/or threshold values of aminotransferases (and not multiples of ULN) should be the basis for criteria signaling potential DILI. Also, more modest elevations of aminotransferases over baseline may still signal a DILI event. Tests of liver function, including total and DB or INR, even in the presence of only slight elevations of aminotransferases can also signal cholestatic or even hepatocellular DILI in these populations. Utilizing changes in AST:ALT ratios may also differentiate a potential DILI event during a clinical trial from an exacerbation of the underlying chronic disease that caused the cirrhosis. In clinical trials in patients with decompensated cirrhosis, rising MELD scores are a useful indicator of potential DILI. Because of the potential for worsening of the underlying liver function during these trials, even in patients entering with compensated cirrhosis, it is important to consider pharmacokinetic sampling at the time of a potential DILI event to detect unanticipated overexposure to the drug.
Gaps in the Recommendations and Opportunities to Address these Gaps
The increasing frequency of clinical trials involving patients with CLD with cirrhosis, and the incidental inclusion of patients with cirrhosis in clinical trials for other indications (e.g., diabetes, hypertension, hyperlipidemia) offers a unique opportunity to study the range of baseline liver tests that characterize each of these disease populations. Careful monitoring of the fluctuations of these parameters during the clinical trial in the placebo group (on standard of care) will no doubt improve our understanding of the expected variability over time and the natural history associated with each disease indication. This will require cooperation and collaboration of multiple sponsors conducting clinical trials in the same indications, and preferably using the same agreed upon criteria for definitions of subgroups of compensated and decompensated cirrhosis, and inclusion and exclusion criteria in each subgroup. This will allow the establishment of aggregated large databases within each disease population that will inform rational choices about clinical criteria that will mandate increased monitoring for DILI and holding and stopping rules and adequate investigations for causality assessment. It is also imperative that any cases of possible, probable, or likely DILI in clinical trials be thoroughly evaluated and adjudicated by experts. Such cases need to be followed closely to discern patterns and trends that illuminate early warning signals in patients with cirrhosis. They also need to be followed for clinical and laboratory signatures of reversal of drug-related worsening upon study drug discontinuation or dose modification that may differ based on the underlying liver disease and that potentially differ from the noncirrhotic population.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Dr. Poonam Mishra, Deputy Director for Safety, Division of Antiviral Products, Center for Drug Evaluation and Research, US Food and Drug Administration; Dr. Ben Shneider, Baylor College of Medicine; Dr. Hans Tillmann, East Carolina University School of Medicine; and all members of the IQ Abnormal Baselines Working Group for their helpful comments and suggestions about this paper and the members of the IQ Secretariat, who were instrumental in organizing and producing this paper, including Ann Marie Stanley, Alexis Myers, Fatou Sarr, and Maureen Hardwick.
Declarations
Funding
Not applicable. All work on this paper was done voluntarily and no compensation was paid to any of the authors.
Conflict of interest
Drs. Chalasani, Avigan, Dimick-Santos, and Rockey have no conflicts of interest that are directly relevant to the content of this paper. Dr. Chalasani has ongoing consulting activities (or had in the preceding 12 months) with NuSirt, AbbVie, Eli Lilly, Afimmune (DS Biopharma), Allergan (Tobira), Madrigal, Shire, Axovant, Coherus, Pronova (BASF), Siemens, and Genentech. These consulting activities are generally in the areas of NAFLD and drug hepatotoxicity. Dr. Chalasani receives research grant support to his institution from Intercept, Lilly, Galectin Therapeutics, and Cumberland. Over the last decade, Dr. Chalasani has served as a paid consultant to more than 30 pharmaceutical companies, and his outside activities have regularly been disclosed to his institutional authorities. Drs. Treem, Lonjon-Domanec, Seekins, Dash, Regev, Marcinak, and Patwardhan are full-time employees of Takeda,ssen Pharmaceutica NV, Bristol Myers Squibb, Genentech, Eli Lilly, Pfizer, and AbbVie, respectively. Dr. Palmer was a full-time employee of Takeda (formerly Shire) during the drafting of this paper and is now at Liver Consulting LLC. Drs. Palmer, Lewis, and Freston serve as consultants to several pharmaceutical companies for activities related to NAFLD and DILI but have not derived any financial or other compensation from activities related to developing this best practices document. Dr. Rockey receives ongoing research grant support to his institution from Galectin Therapeutics, Genfit, Gilead Sciences, Intercept Pharmaceuticals, Mallinckrodt Pharmaceuticals, Salix Pharmaceuticals, and Sequana Pharmaceuticals. Dr. Maller was a full-time employee of Pfizer during the drafting of this paper and is now at MEMS Biopharma Consulting, LLC. Dr. Di Bisceglie serves on adjudication committees for AbbVie and Daichi Sankyo, on a data safety monitoring committee for Eiger, and as a consultant and chief medical officer to HighTide Therapeutics, which is developing a therapy for fatty liver disease. He has not derived any financial or other compensation from activities related to developing this best practices document. Dr. Andrade serves as a consultant to several pharmaceutical companies for activities related to NAFLD and DILI but has not derived any financial or other compensation from activities related to developing this best practices document.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
All data reviewed and analyzed in this paper are already in the public domain in previous publications, abstracts, conference presentations, and on ClinicalTrials.gov.
Code availability
Not applicable.
IQ DILI Abnormal Baselines Working Group Members
Ajit Dash, M.D., Ph.D., MBBS, Genentech, Inc.; Isabelle Lonjon-Domanec, M.D,ssen Pharmaceutica NV; Eric Maller, M.D., Pfizer; John F. Marcinak, M.D., AbbVie; Melissa Palmer, M.D., Liver Consulting LLC (formerly with Takeda); Niti Patel, Ph.D., DABT, Takeda; Meenal Patwardhan, M.D., MHSA, AbbVie Inc.; Ritu Raheja, MPH/MBA, AbbVie Inc.; Arie Regev, M.D., Eli Lilly and Company; Daniel Seekins, M.D., Bristol Myers Squibb; William R. Treem, M.D., Takeda; Hui-Talia Zhang, M.D., Ph.D, Bayer HealthCare Pharmaceuticals.
Author contributions
All industry, academic, and regulatory-based authors contributed to the paper’s conception, design, and development through a series of in-person and virtual working sessions. Material preparation, data collection, and analysis were performed primarily by Will Treem, Melissa Palmer, Isabelle Lonjon-Domanec, Daniel Seekins, Jon Marcinak, and Ajit Dash. The first draft of the manuscript was written by the same six authors, and all authors subsequently commented on and edited all versions of the manuscript. All authors participated in the development and agreed upon the consensus recommendations in the paper and read and approved the final manuscript.
Footnotes
The views expressed are those of the authors and do not necessarily represent the position of, nor imply endorsement from, the US Food and Drug Administration or the US Government.
Contributor Information
William R. Treem, Email: William.Treem@takeda.com
Naga Chalasani, Email: nchalasa@iu.edu.
References
- 1.Zimmerman HJ. Hepatotoxicity: The adverse effects of drugs and other chemicals on the liver. 2. Philadelphia: Lippincott, Williams & Wilkins; 1999. [Google Scholar]
- 2.Chalasani N, Bjornsson E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology. 2010;138(7):2246–2259. doi: 10.1053/j.gastro.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, et al. Features and outcomes of 899 patients with drug-induced liver injury: The DILIN prospective study. Gastroenterology. 2015;148(7):1340–52 e7. [DOI] [PMC free article] [PubMed]
- 4.Russo MW, Watkins PB. Are patients with elevated liver tests at increased risk of drug-induced liver injury? Gastroenterology. 2004;126(5):1477–1480. doi: 10.1053/j.gastro.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 5.Health Canada. Guidance document: pre-market evaluation of hepatotoxicity in health products. https://www.canada.ca/content/dam/hc-sc/migration/hc-sc/dhp-mps/alt_formats/pdf/prodpharma/applic-demande/guide-ld/hepatotox_guide_ld-eng.pdf.
- 6.U.S. Food & Drug Administration (FDA). Guidance for industry drug-induced liver injury: premarketing clinical evaluation. https://www.fda.gov/downloads/Guidances/UCM174090.pdf. 2017.
- 7.Llanos L, Moreu R, Ortin T, Peiro AM, Pascual S, Bellot P, et al. The existence of a relationship between increased serum alanine aminotransferase levels detected in premarketing clinical trials and postmarketing published hepatotoxicity case reports. Aliment Pharmacol Ther. 2010;31(12):1337–1345. doi: 10.1111/j.1365-2036.2010.04298.x. [DOI] [PubMed] [Google Scholar]
- 8.Moylan CA, Suzuki A, Papay JI, Yuen NA, Ames M, Hunt CM. A pre-marketing ALT signal predicts post-marketing liver safety. Regul Toxicol Pharmacol. 2012;63(3):433–439. doi: 10.1016/j.yrtph.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 9.Watkins PB, Seligman PJ, Pears JS, Avigan MI, Senior JR. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48(5):1680–1689. doi: 10.1002/hep.22633. [DOI] [PubMed] [Google Scholar]
- 10.Dara L, Liu Z-X, Kaplowitz N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2016;36(2):158–165. doi: 10.1111/liv.12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenzweig P, Miget N, Brohier S. Transaminase elevation on placebo during phase I trials: Prevalence and significance. Br J Clin Pharmacol. 1999;48(1):19–23. doi: 10.1046/j.1365-2125.1999.00952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purkins L, Love ER, Eve MD, Wooldridge CL, Cowan C, Smart TS, et al. The influence of diet upon liver function tests and serum lipids in healthy male volunteers resident in a Phase I unit. Br J Clin Pharmacol. 2004;57(2):199–208. doi: 10.1046/j.1365-2125.2003.01969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming KM, West J, Aithal GP, Fletcher AE. Abnormal liver tests in people aged 75 and above: prevalence and association with mortality. Aliment Pharmacol Ther. 2011;34(3):324–334. doi: 10.1111/j.1365-2036.2011.04718.x. [DOI] [PubMed] [Google Scholar]
- 14.Makar GA, Weiner MG, Kimmel SE, Bennett D, Burke A, Yang YX, et al. Incidence and prevalence of abnormal liver associated enzymes in patients with atrial fibrillation in a routine clinical care population. Pharmacoepidemiol Drug Saf. 2008;17(1):43–51. doi: 10.1002/pds.1514. [DOI] [PubMed] [Google Scholar]
- 15.Avigan MI, Bjornsson ES, Pasanen M, Cooper C, Andrade RJ, Watkins PB, et al. Liver safety assessment: required data elements and best practices for data collection and standardization in clinical trials. Drug Saf. 2014;37(Suppl 1):S19–31. doi: 10.1007/s40264-014-0183-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer M, Regev A, Lindor K, Avigan MI, Dimick-Santos L, Treem W, et al. Consensus guidelines: best practices for detection, assessment and management of suspected acute drug-induced liver injury occurring during clinical trials in adults with chronic cholestatic liver disease. Aliment Pharmacol Ther. 2020;51(1):90–109. doi: 10.1111/apt.15579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Regev A, Palmer M, Avigan MI, Dimick-Santos L, Treem WR, Marcinak JF, et al. Consensus: guidelines: best practices for detection, assessment and management of suspected acute drug-induced liver injury during clinical trials in patients with nonalcoholic steatohepatitis. Aliment Pharmacol Ther. 2019;49(6):702–713. doi: 10.1111/apt.15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Jr, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 19.Hadziyannis SJ, Sette H, Jr, Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140(5):346–355. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 20.Flink HJ, Sprengers D, Hansen BE, van Zonneveld M, de Man RA, Schalm SW, et al. Flares in chronic hepatitis B patients induced by the host or the virus? Relation to treatment response during peg-interferon {alpha}-2b therapy. Gut. 2005;54(11):1604–1609. doi: 10.1136/gut.2004.062208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, et al. Chronic hepatitis B: virology, natural history, current management and a glimpse at future opportunities. Antiviral Res. 2015;121:47–58. doi: 10.1016/j.antiviral.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Lok AS, Lai CL, Leung N, Yao GB, Cui ZY, Schiff ER, et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology. 2003;125(6):1714–1722. doi: 10.1053/j.gastro.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 23.Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50(3):661–662. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- 24.Cai Z, Christianson AM, Stahle L, Keisu M. Reexamining transaminase elevation in phase I clinical trials: The importance of baseline and change from baseline. Eur J Clin Pharmacol. 2009;65(10):1025–1035. doi: 10.1007/s00228-009-0684-x. [DOI] [PubMed] [Google Scholar]
- 25.Merz M, Lee KR, Kullak-Ublick GA, Brueckner A, Watkins PB. Methodology to assess clinical liver safety data. Drug Saf. 2014;37(Suppl 1):S33–45. doi: 10.1007/s40264-014-0184-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sibille M, Deigat N, Durieu I, Guillaumont M, Morel D, Bienvenu J, et al. Laboratory data in healthy volunteers: reference values, reference changes, screening and laboratory adverse event limits in phase I clinical trials. Eur J Clin Pharmacol. 1999;55(1):13–19. doi: 10.1007/s002280050586. [DOI] [PubMed] [Google Scholar]
- 27.Dufour DR. Alanine aminotransferase: is it healthy to be "normal"? Hepatology. 2009;50(6):1699–1701. doi: 10.1002/hep.23358. [DOI] [PubMed] [Google Scholar]
- 28.Neuschwander-Tetri BA, Unalp A, Creer MH, Nonalcoholic Steatohepatitis Clinical Research N Influence of local reference populations on upper limits of normal for serum alanine aminotransferase levels. Arch Intern Med. 2008;168(6):663–666. doi: 10.1001/archinternmed.2007.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pacifico L, Ferraro F, Bonci E, Anania C, Romaggioli S, Chiesa C. Upper limit of normal for alanine aminotransferase: quo vadis? Clin Chim Acta. 2013;25(422):29–39. doi: 10.1016/j.cca.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 30.Ruhl CE, Everhart JE. Upper limits of normal for alanine aminotransferase activity in the United States population. Hepatology. 2012;55(2):447–454. doi: 10.1002/hep.24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabir A, Pourshams A, Khoshnia M, Malekzadeh F. Normal limit for serum alanine aminotransferase level and distribution of metabolic factors in old population of Kalaleh, Iran. Hepat Mon. 2013;13(10):e10640. doi: 10.5812/hepatmon.10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang HS, Um SH, Seo YS, An H, Lee KG, Hyun JJ, et al. Healthy range for serum ALT and the clinical significance of "unhealthy" normal ALT levels in the Korean population. J Gastroenterol Hepatol. 2011;26(2):292–299. doi: 10.1111/j.1440-1746.2010.06481.x. [DOI] [PubMed] [Google Scholar]
- 33.Kariv R, Leshno M, Beth-Or A, Strul H, Blendis L, Kokia E, et al. Re-evaluation of serum alanine aminotransferase upper normal limit and its modulating factors in a large-scale population study. Liver Int. 2006;26(4):445–450. doi: 10.1111/j.1478-3231.2006.01197.x. [DOI] [PubMed] [Google Scholar]
- 34.Kim HY, Kim CW, Lee CD, Choi JY, Park CH, Bae SH, et al. Can "healthy" normal alanine aminotransferase levels identify the metabolically obese phenotype? Findings from the Korea national health and nutrition examination survey 2008–2010. Dig Dis Sci. 2014;59(6):1330–1337. doi: 10.1007/s10620-013-2995-0. [DOI] [PubMed] [Google Scholar]
- 35.Lee JK, Shim JH, Lee HC, Lee SH, Kim KM, Lim YS, et al. Estimation of the healthy upper limits for serum alanine aminotransferase in Asian populations with normal liver histology. Hepatology. 2010;51(5):1577–1583. doi: 10.1002/hep.23505. [DOI] [PubMed] [Google Scholar]
- 36.Prati D, Taioli E, Zanella A, Della Torre E, Butelli S, Del Vecchio E, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med. 2002;137(1):1–10. doi: 10.7326/0003-4819-137-1-200207020-00006. [DOI] [PubMed] [Google Scholar]
- 37.Kwo PY, Cohen SM, Lim JK. ACG clinical guideline: Evaluation of abnormal liver chemistries. Am J Gastroenterol. 2017;112(1):18–35. doi: 10.1038/ajg.2016.517. [DOI] [PubMed] [Google Scholar]
- 38.Misbin RI. Subject: NDA 20-720 rezulin triple Therapy. Troglitazone efficacy supplement. https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20720S12S14_Rezulin.cfm. 2017.
- 39.Goldkind L, Laine L. A systematic review of NSAIDs withdrawn from the market due to hepatotoxicity: lessons learned from the bromfenac experience. Pharmacoepidemiol Drug Saf. 2006;15(4):213–220. doi: 10.1002/pds.1207. [DOI] [PubMed] [Google Scholar]
- 40.Senior JR. Evolution of the food and drug administration approach to liver safety assessment for new drugs: Current status and challenges. Drug Saf. 2014;37(Suppl 1):S9–17. doi: 10.1007/s40264-014-0182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucena M, Andrade R, Rodrigo L, Salmerón J, Alvarez A, Lopez-Garrido JM, et al. Trovafloxacin-induced acute hepatitis. Clin Infect Dis. 2000;30:400–401. doi: 10.1086/313680. [DOI] [PubMed] [Google Scholar]
- 42.Lee WM, Larrey D, Olsson R, Lewis JH, Keisu M, Auclert L, et al. Hepatic findings in long-term clinical trials of ximelagatran. Drug Saf. 2005;28(4):351–370. doi: 10.2165/00002018-200528040-00006. [DOI] [PubMed] [Google Scholar]
- 43.Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, et al. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89(6):806–815. doi: 10.1038/clpt.2011.58. [DOI] [PubMed] [Google Scholar]
- 44.Chalasani N, Regev A. Drug-induced liver injury in patients with preexisting chronic liver disease in drug development: how to identify and manage? Gastroenterology. 2016;151(6):1046–1051. doi: 10.1053/j.gastro.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 45.Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, et al. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology. 2011;54(1):344–353. doi: 10.1002/hep.24376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teschke R, Danan G. Diagnosis and management of drug-induced liver injury (DILI) in patients with pre-existing liver disease. Drug Saf. 2016;39(8):729–744. doi: 10.1007/s40264-016-0423-z. [DOI] [PubMed] [Google Scholar]
- 47.Andrade RJ, Aithal GP, Björnsson ES, Kaplowitz N, Kullak-Ublick GA, Larrey D, et al. EASL clinical practice guidelines: drug-induced liver injury. J Hepatol. 2019;70(6):1222–1261. doi: 10.1016/j.jhep.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 48.Kullak-Ublick GA, Merz M, Griffel L, Kaplowitz N, Watkins PB. Liver safety assessment in special populations (hepatitis B, C, and oncology trials) Drug Saf. 2014;37(Suppl 1):S57–62. doi: 10.1007/s40264-014-0186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polaris Observatory HCV Collaborators Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol Hepatol. 2017;2(3):161–176. doi: 10.1016/S2468-1253(16)30181-9. [DOI] [PubMed] [Google Scholar]
- 50.Chak E, Talal AH, Sherman KE, Schiff ER, Saab S. Hepatitis C virus infection in USA: an estimate of true prevalence. Liver Int. 2011;31(8):1090–1101. doi: 10.1111/j.1478-3231.2011.02494.x. [DOI] [PubMed] [Google Scholar]
- 51.Alter MJ. Epidemiology of hepatitis C. Hepatology. 1997;26(3 Suppl 1):62s–s65. doi: 10.1002/hep.510260711. [DOI] [PubMed] [Google Scholar]
- 52.Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, Swan T. Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology. 2015;62(5):1353–1363. doi: 10.1002/hep.27978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puoti C, Guarisco R, Spilabotti L, Bellis L, Mitidieri Costanza O, Dell' Unto O, et al. Should we treat HCV carriers with normal ALT levels? The '5Ws' dilemma. J Viral Hepat. 2012;19(4):229–235. doi: 10.1111/j.1365-2893.2011.01485.x. [DOI] [PubMed] [Google Scholar]
- 54.Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexander WJ, et al. The natural history of community-acquired hepatitis C in the United States. The Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. N Engl J Med. 1992;327(27):1899–905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 55.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386(10003):1546–1555. doi: 10.1016/S0140-6736(15)61412-X. [DOI] [PubMed] [Google Scholar]
- 57.Kumar M, Sarin SK, Hissar S, Pande C, Sakhuja P, Sharma BC, et al. Virologic and histologic features of chronic hepatitis B virus-infected asymptomatic patients with persistently normal ALT. Gastroenterology. 2008;134(5):1376–1384. doi: 10.1053/j.gastro.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 58.Heathcote EJ, Marcellin P, Buti M, Gane E, De Man RA, Krastev Z, et al. Three-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B. Gastroenterology. 2011;140(1):132–143. doi: 10.1053/j.gastro.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 59.Liaw YF, Gane E, Leung N, Zeuzem S, Wang Y, Lai CL, et al. 2-Year GLOBE trial results: telbivudine is superior to lamivudine in patients with chronic hepatitis B. Gastroenterology. 2009;136(2):486–495. doi: 10.1053/j.gastro.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 60.Marcellin P, Heathcote EJ, Buti M, Gane E, de Man RA, Krastev Z, et al. Tenofovir disoproxil fumarate versus adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359(23):2442–2455. doi: 10.1056/NEJMoa0802878. [DOI] [PubMed] [Google Scholar]
- 61.Pol S, Lampertico P. First-line treatment of chronic hepatitis B with entecavir or tenofovir in ‘real-life’ settings: from clinical trials to clinical practice. J Viral Hepat. 2012;19(6):377–386. doi: 10.1111/j.1365-2893.2012.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang M, Liaw Y-F. Hepatitis B flares in chronic hepatitis B: pathogenesis, natural course, and management. J Hepatol. 2014;61(6):1407–17. doi: 10.1016/j.jhep.2014.08.033. [DOI] [PubMed] [Google Scholar]
- 63.Fleming KM, Aithal GP, Solaymani-Dodaran M, Card TR, West J. Incidence and prevalence of cirrhosis in the United Kingdom, 1992–2001: a general population-based study. J Hepatol. 2008;49(5):732–738. doi: 10.1016/j.jhep.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 64.Scaglione S, Kliethermes S, Cao G, Shoham D, Durazo R, Luke A, et al. The epidemiology of cirrhosis in the United States: a population-based study. J Clin Gastroenterol. 2015;49(8):690–696. doi: 10.1097/MCG.0000000000000208. [DOI] [PubMed] [Google Scholar]
- 65.Jurek M, Mokhtarani M, Vierling JM, Coakley DF, Ganju J, Rowell R, et al. Variation in liver biochemistries in patients with decompensated cirrhosis: implications for assessing drug-induced liver injury in clinical trials. Pharm Med. 2016;30:95–101. [Google Scholar]
- 66.Gai XD, Wu WF. Effect of entecavir in the treatment of patients with hepatitis B virus-related compensated and decompensated cirrhosis. Exp Ther Med. 2017;14(4):3908–3914. doi: 10.3892/etm.2017.4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang X, Li J, Zhou L, Liu J, Wang J, Lu W. Comparison of telbivudine efficacy in treatment-naive patients with hepatitis B virus-related compensated and decompensated cirrhosis in 96 weeks. Eur J Gastroenterol Hepatol. 2014;26(4):396–403. doi: 10.1097/MEG.0000000000000062. [DOI] [PubMed] [Google Scholar]
- 68.Jacobson IM, Lawitz E, Kwo PY, Hezode C, Peng CY, Howe AYM, et al. Safety and efficacy of elbasvir/grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology. 2017;152(6):1372–82.e2. doi: 10.1053/j.gastro.2017.01.050. [DOI] [PubMed] [Google Scholar]
- 69.Sanyal AJ, Harrison SA, Ratziu V, Abdelmalek MF, Diehl AM, Caldwell S, et al. The natural history of advanced fibrosis due to nonalcoholic steatohepatitis: data from the simtuzumab trials. Hepatology. 2019;70(6):1913–1927. doi: 10.1002/hep.30664. [DOI] [PubMed] [Google Scholar]
- 70.Younossi ZM, Stepanova M, Anstee QM, Lawitz EJ, Wai-Sun Wong V, Romero-Gomez M, et al. Reduced patient-reported outcome scores associate with level of fibrosis in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2019;17(12):2552–2560. doi: 10.1016/j.cgh.2019.02.024. [DOI] [PubMed] [Google Scholar]
- 71.Mohieldein A, Azam Hyder M, Hasan M. Comparative levels of ALT, AST, ALP and GGT in liver associated diseases. European J Exp Biol. 2013;3(2):280–284. [Google Scholar]
- 72.Williams AL, Hoofnagle JH. Ratio of serum aspartate to alanine aminotransferase in chronic hepatitis. Relationship to cirrhosis. Gastroenterology. 1988;95(3):734–739. doi: 10.1016/s0016-5085(88)80022-2. [DOI] [PubMed] [Google Scholar]
- 73.U.S. Food & Drug Administration (FDA). Chronic hepatitis C virus infection: developing direct-acting antiviral drugs for treatment guidance for industry. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm225333.pdf.
- 74.Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS. Oral direct-acting agent therapy for hepatitis c virus infection: a systematic review. Ann Intern Med. 2017;166(9):637–648. doi: 10.7326/M16-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ito H, Yoshioka K, Ukai K, Watanabe K, Yano M, Ishigami M, et al. The fluctuations of viral load and serum alanine aminotransferase levels in chronic hepatitis C. Hepatol Res. 2004;30(1):11–17. doi: 10.1016/j.hepres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 76.Khan ST, McGuinty M, Corsi DJ, Cooper CL. Liver enzyme normalization predicts success of hepatitis C oral direct-acting antiviral treatment. Clin Invest Med. 2017;40(2):E73–e80. doi: 10.25011/cim.v40i2.28198. [DOI] [PubMed] [Google Scholar]
- 77.Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. Acad Nurse. 2014;370(16):1483–1493. doi: 10.1056/NEJMoa1316366. [DOI] [PubMed] [Google Scholar]
- 78.Bourlière M, Gordon SC, Flamm SL, Cooper CL, Ramji A, Tong M, et al. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N Engl J Med. 2017;376(22):2134–2146. doi: 10.1056/NEJMoa1613512. [DOI] [PubMed] [Google Scholar]
- 79.Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373(27):2599–2607. doi: 10.1056/NEJMoa1512610. [DOI] [PubMed] [Google Scholar]
- 80.Ferenci P, Bernstein D, Lalezari J, Cohen D, Luo Y, Cooper C, et al. ABT-450/r–ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983–1992. doi: 10.1056/NEJMoa1402338. [DOI] [PubMed] [Google Scholar]
- 81.Foster GR, Afdhal N, Roberts SK, Bräu N, Gane EJ, Pianko S, et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med. 2015;373(27):2608–2617. doi: 10.1056/NEJMoa1512612. [DOI] [PubMed] [Google Scholar]
- 82.Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med. 2014;370(3):211–21. doi: 10.1056/NEJMoa1306218. [DOI] [PubMed] [Google Scholar]
- 83.Naggie S, Cooper C, Saag M, Workowski K, Ruane P, Towner WJ, et al. Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373(8):705–713. doi: 10.1056/NEJMoa1501315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Regev A, Seeff LB, Merz M, Ormarsdottir S, Aithal GP, Gallivan J, et al. Causality assessment for suspected DILI during clinical phases of drug development. Drug Saf. 2014;37(1):47–56. doi: 10.1007/s40264-014-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–1898. doi: 10.1056/NEJMoa1402454. [DOI] [PubMed] [Google Scholar]
- 86.Welzel TM, Nelson DR, Morelli G, Di Bisceglie A, Reddy RK, Kuo A, et al. Effectiveness and safety of sofosbuvir plus ribavirin for the treatment of HCV genotype 2 infection: results of the real-world, clinical practice HCV-target study. Gut. 2017;66(10):1844–1852. doi: 10.1136/gutjnl-2016-311609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tapper EB, Bacon BR, Curry MP, Dieterich DT, Flamm SL, Guest LE, et al. Real-world effectiveness for 12 weeks of ledipasvir-sofosbuvir for genotype 1 hepatitis C: the trio health study. J Viral Hepat. 2017;24(1):22–27. doi: 10.1111/jvh.12611. [DOI] [PubMed] [Google Scholar]
- 88.Binda C, Tortora A, Garcovich M, Annicchiarico BE, Siciliano M. Toxicity and risks from drug-to-drug interactions of new antivirals for chronic hepatitis C. Eur Rev Med Pharmacol Sci. 2017;21(1 Suppl):102–111. [PubMed] [Google Scholar]
- 89.U.S. Food & Drug Administration (FDA). FDA Drug Safety Communication: FDA warns of serious liver injury risk with hepatitis C treatments Viekira Pak and Technivie. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-fda-warns-serious-liver-injury-risk-hepatitis-c-treatments-viekira-pak.
- 90.U.S. Food & Drug Administration (FDA). FDA warns about rare occurrence of serious liver injury with use of hepatitis C medicines Mavyret, Zepatier, and Vosevi in some patients with advanced liver disease. https://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-rare-occurrence-serious-liver-injury-use-hepatitis-c-medicines-mavyret-zepatier-and.
- 91.American Association for the Study of Liver Diseases (AASLD). HCV guidance: recommendations for testing, managing, and treating hepatitis C: Patients with decompensated cirrhosis. https://www.hcvguidelines.org/unique-populations/decompensated-cirrhosis.
- 92.Ma AT, Feld JJ. Hepatitis B reactivation with hepatitis C treatment: bringing some clarity to the black box. Gastroenterology. 2018;154(4):795–798. doi: 10.1053/j.gastro.2018.02.005. [DOI] [PubMed] [Google Scholar]
- 93.Liu CJ, Chuang WL, Sheen IS, Wang HY, Chen CY, Tseng KC, et al. Efficacy of ledipasvir and sofosbuvir treatment of HCV infection in patients coinfected with HBV. Gastroenterology. 2018;154(4):989–997. doi: 10.1053/j.gastro.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 94.Chene X, Uchida T, Xia Y, Umarova R, Liu C, Chen P. Diminished hepatic IFN response following HCV clearance triggers HBV reactivation in coinfection. J Clin Invest. 2020;130(6):3205–20. doi: 10.1172/JCI135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen G, Wang C, Chen J, Ji D, Wang Y, Wu V, et al. Hepatitis B reactivation in hepatitis B and C coinfected patients treated with antiviral agents: a systematic review and meta-analysis. Hepatology. 2017;66(1):13–26. doi: 10.1002/hep.29109. [DOI] [PubMed] [Google Scholar]
- 96.Meissner EG, Wu D, Osinusi A, Bon D, Virtaneva K, Sturdevant D, et al. Endogenous intrahepatic IFNs and association with IFN-free HCV treatment outcome. J Clin Invest. 2014;124(8):3352–3363. doi: 10.1172/JCI75938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sarin SK, Kumar M, Lau GK, Abbas Z, Chan HL, Chen CJ, et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int. 2016;10(1):1–98. doi: 10.1007/s12072-015-9675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560–1599. doi: 10.1002/hep.29800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.European Association for the Study of the Liver (EASL). EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370–98. [DOI] [PubMed]
- 100.World Health Organization (WHO). Global hepatitis report 2017. https://apps.who.int/iris/bitstream/handle/10665/255017/WHO-HIV-2017.06-eng.pdf;jsessionid=4F161916938B5451BA25A4368BD27CE7?sequence=1.
- 101.Razavi-Shearer D, Gamkrelidze I, Nguyen MH, Chen D-S, Van Damme P, Abbas Z, et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: a modelling study. Lancet Gastroenterol Hepatol. 2018;3(6):383–403. doi: 10.1016/S2468-1253(18)30056-6. [DOI] [PubMed] [Google Scholar]
- 102.Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64(12):1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- 103.Soriano V, Barreiro P, Benitez L, Peña JM, de Mendoza C. New antivirals for the treatment of chronic hepatitis B. Expert Opin Investig Drugs. 2017;26(7):843–51. doi: 10.1080/13543784.2017.1333105. [DOI] [PubMed] [Google Scholar]
- 104.National Institutes of Health (NIH). A study of different combination regimens including JNJ-73763989 and/or JNJ-56136379 for the treatment of chronic hepatitis B virus infection. 2019. https://ClinicalTrials.gov/show/NCT03982186.
- 105.National Institutes of Health (NIH). A study of ABI-H0731 + Nucleos(t)Ide as finite treatment for chronic hepatitis B patients. 2018. https://ClinicalTrials.gov/show/NCT03780543.
- 106.National Institutes of Health (NIH). A study evaluating ABI-H0731+ NUC vs NUC alone for the treatment of viremic HBeAg-positive, CHB Patients. 2018. https://ClinicalTrials.gov/show/NCT03577171.
- 107.National Institutes of Health (NIH). A study evaluating ABI-H0731 as adjunctive therapy in patients with chronic hepatitis B. 2018. https://ClinicalTrials.gov/show/NCT03576066.
- 108.National Institutes of Health (NIH). Myrcludex B vs entecavir in patients with HBeAg negative chronic hepatitis B. 2016. https://ClinicalTrials.gov/show/NCT02881008.
- 109.National Institutes of Health (NIH). Study of ARC-520 With or without other drugs used in the treatment of chronic chronic hepatitis B virus (HBV). 2015. https://ClinicalTrials.gov/show/NCT02577029.
- 110.National Institutes of Health (NIH). REP 2139-Mg and REP 2165-Mg Combination Therapy in Chronic Hepatitis B Infection. 2015. https://ClinicalTrials.gov/show/NCT02565719.
- 111.National Institutes of Health (NIH). A multicenter study to determine the depth and duration of hepatitis B surface antigen (HBsAg) reduction after single or multiple doses of ARC-520, in combination with entecavir in patients with chronic hepatitis B virus (HBV) infection. 2014. https://ClinicalTrials.gov/show/NCT02065336.
- 112.Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology. 2007;45(2):507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 113.Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology. 2006;43(2 Suppl 1):S173–S181. doi: 10.1002/hep.20956. [DOI] [PubMed] [Google Scholar]
- 114.Croagh CMN, Lubel JS. Natural history of chronic hepatitis B: phases in a complex relationship. World J Gastroenterol. 2014;20(30):10395–10404. doi: 10.3748/wjg.v20.i30.10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63(1):261–283. doi: 10.1002/hep.28156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chan HL, Heathcote EJ, Marcellin P, Lai CL, Cho M, Moon YM, et al. Treatment of hepatitis B e antigen positive chronic hepatitis with telbivudine or adefovir: a randomized trial. Ann Intern Med. 2007;147(11):745–754. doi: 10.7326/0003-4819-147-11-200712040-00183. [DOI] [PubMed] [Google Scholar]
- 117.Lau GK, Piratvisuth T, Luo KX, Marcellin P, Thongsawat S, Cooksley G, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352(26):2682–2695. doi: 10.1056/NEJMoa043470. [DOI] [PubMed] [Google Scholar]
- 118.U.S. Food & Drug Administration (FDA). Application number: 21-449 (adefovir dipivoxil). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/21-449_Hepsera_medr_P1.pdf.
- 119.U.S. Food & Drug Administration (FDA). Application number: 21-797, 21-798 (entacavir). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/21797_BARACLUDE_medr.PDF.
- 120.National Institutes of Health (NIH). Efficacy & safety of tenofovir disoproxil fumarate (TDF) plus peginterferon α-2a (Peg-IFN) versus TDF or peg-IFN monotherapy in chronic hepatitis B. 2011. https://clinicaltrials.gov/ct2/show/NCT01277601?term=gilead&cond=Hepatitis+B%2C+Chronic.
- 121.U.S. National Institute of Health (NIH). Evaluation of antiviral indications on chronic HBV infection with different transaminase levels. 2018. https://clinicaltrials.gov/ct2/show/NCT03740789.
- 122.Clinical Research Protocol National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Tenofovir disoproxil fumarate alone versus its combination with emtricitabine for treatment of chronic hepatitis B. https://clinicaltrials.gov/ProvidedDocs/73/NCT00524173/Prot_SAP_000.pdf.
- 123.Ghany MG, Kim HY, Stoddard A, Wright EC, Seeff LB, Lok AS. Predicting clinical outcomes using baseline and follow-up laboratory data from the hepatitis C long-term treatment against cirrhosis trial. Hepatology. 2011;54(5):1527–1537. doi: 10.1002/hep.24550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.National Institutes of Health (NIH). A study to compare tenofovir disoproxil fumarate versus adefovir dipivoxil for the treatment of HBeAg-positive chronic hepatitis B. 2005. https://clinicaltrials.gov/ct2/show/NCT00116805?term=gilead&cond=Hepatitis+B%2C+Chronic.
- 125.National Institutes of Health (NIH). Efficacy and safety study of tenofovir disoproxil fumarate (TDF) in chinese chronic hepatitis B (CHB) subjects with advanced fibrosis & compensated cirrhosis. 2014 February 28, 2018. https://clinicaltrials.gov/ct2/show/NCT02224456?term=gilead&cond=Hepatitis+B%2C+Chronic.
- 126.National Institutes of Health (NIH). Safety and efficacy of selonsertib in adults with compensated cirrhosis due to nonalcoholic steatohepatitis (NASH) (STELLAR 4). 2017. https://clinicaltrials.gov/ct2/show/NCT03053063.
- 127.Teschke R, Danan G. Drug-induced liver injury: is chronic liver disease a risk factor and a clinical issue? Expert Opin Drug Metab Toxicol. 2017;13(4):425–438. doi: 10.1080/17425255.2017.1252749. [DOI] [PubMed] [Google Scholar]
- 128.U.S. Food & Drug Administration (FDA). Baraclude (entecavir) film coated tablets & oral solution. 2005. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/21797_21798_BaracludeTOC.cfm.
- 129.Chang TT, Liaw YF, Wu SS, Schiff E, Han KH, Lai CL, et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology. 2010;52(3):886–893. doi: 10.1002/hep.23785. [DOI] [PubMed] [Google Scholar]
- 130.McKenzie R, Fried MW, Sallie R, Conjeevaram H, Di Bisceglie AM, Park Y, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med. 1995;333(17):1099–1105. doi: 10.1056/NEJM199510263331702. [DOI] [PubMed] [Google Scholar]
- 131.Honkoop P, Scholte HR, de Man RA, Schalm SW. Mitochondrial injury. Lessons from the fialuridine trial. Drug Saf. 1997;17(1):1–7. doi: 10.2165/00002018-199717010-00001. [DOI] [PubMed] [Google Scholar]
- 132.Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378(9785):73–85. doi: 10.1016/S0140-6736(10)61931-9. [DOI] [PubMed] [Google Scholar]
- 133.Tassopoulos NC, Volpes R, Pastore G, Heathcote J, Buti M, Goldin RD, et al. Efficacy of lamivudine in patients with hepatitis B e antigen-negative/hepatitis B virus DNA-positive (precore mutant) chronic hepatitis B. Lamivudine precore mutant study group. Hepatology. 1999;29(3):889–896. doi: 10.1002/hep.510290321. [DOI] [PubMed] [Google Scholar]
- 134.Pan CQ, Duan Z, Dai E, Zhang S, Han G, Wang Y, et al. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. N Engl J Med. 2016;374(24):2324–2334. doi: 10.1056/NEJMoa1508660. [DOI] [PubMed] [Google Scholar]
- 135.Fontana RJ, Avigan MI, Regev A, Mishra P, Gaggar A, et al. Liver safety assessment in clinical trials of new agents for chronic hepatitis B. J Viral Hepat. 2020;27(2):96–109. doi: 10.1111/jvh.13223. [DOI] [PubMed] [Google Scholar]
- 136.Merli M, Rattotti S, Gotti M, Arcaini L. Antiviral therapies for managing viral hepatitis in lymphoma patients. Expert Opin Pharmacother. 2017;18(4):363–76. doi: 10.1080/14656566.2017.1288718. [DOI] [PubMed] [Google Scholar]
- 137.U.S. Food & Drug Administration (FDA). FDA drug safety communication: FDA warns about the risk of hepatitis B reactivating in some patients treated with direct-acting antivirals for hepatitis C. 2016. https://www.fda.gov/Drugs/DrugSafety/ucm522932.htm.
- 138.Yeo W, Zee B, Zhong S, Chan PKS, Wong WL, Ho WM, et al. Comprehensive analysis of risk factors associating with hepatitis B virus (HBV) reactivation in cancer patients undergoing cytotoxic chemotherapy. Br J Cancer. 2004;90(7):1306–1311. doi: 10.1038/sj.bjc.6601699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yuen MF, Wong DK, Fung J, Ip P, But D, Hung I, et al. HBsAg seroclearance in chronic hepatitis B in Asian patients: replicative level and risk of hepatocellular carcinoma. Gastroenterology. 2008;135(4):1192–1199. doi: 10.1053/j.gastro.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 140.Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381(9865):468–475. doi: 10.1016/S0140-6736(12)61425-1. [DOI] [PubMed] [Google Scholar]
- 141.Bachofner JA, Valli PV, Kroger A, Bergamin I, Kunzler P, Baserga A, et al. Direct antiviral agent treatment of chronic hepatitis C results in rapid regression of transient elastography and fibrosis markers fibrosis-4 score and aspartate aminotransferase-platelet ratio index. Liver Int. 2017;37(3):369–376. doi: 10.1111/liv.13256. [DOI] [PubMed] [Google Scholar]
- 142.Dufour J-F, Delellis R, Kaplan MM. Regression of hepatic fibrosis in hepatitis C with long-term interferon treatment. Dig Dis Sci. 1998;43(12):2573–2576. doi: 10.1023/a:1026601904609. [DOI] [PubMed] [Google Scholar]
- 143.Singh S, Facciorusso A, Loomba R, Falck-Ytter YT. Magnitude and kinetics of decrease in liver stiffness after antiviral therapy in patients with chronic hepatitis C: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2018;16(1):27–38.e4. doi: 10.1016/j.cgh.2017.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754–1767. doi: 10.1002/hep.29477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garcia-Tsao G, Fuchs M, Shiffman ML, Chan JL, Morris M, Yamashita M, et al. Emricasan (IDN-6556) administered orally for 28 days lowers portal pressure in patients with compensated cirrhosis and severe portal hypertension: LB-6. J Hepatol. 2015;62(6):1382A–A1383. doi: 10.1002/hep.30199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, et al. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther. 2016;44(11–12):1183–98. doi: 10.1111/apt.13816. [DOI] [PubMed] [Google Scholar]
- 147.Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018;67(2):549–559. doi: 10.1002/hep.29514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.National Institutes of Health (NIH). Clinical trial to evaluation the safety and efficacy of GR-MD-02 for the treatment of liver fibrosis and resultant portal hypertension in patients with nash cirrhosis (NASH-CX). 2015. https://clinicaltrials.gov/ct2/show/NCT02462967.
- 149.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cavallin M, Kamath PS, Merli M, Fasolato S, Toniutto P, Salerno F, et al. Terlipressin plus albumin versus midodrine and octreotide plus albumin in the treatment of hepatorenal syndrome: a randomized trial. Hepatology. 2015;62(2):567–574. doi: 10.1002/hep.27709. [DOI] [PubMed] [Google Scholar]
- 151.Duan XZ, Liu FF, Tong JJ, Yang HZ, Chen J, Liu XY, et al. Granulocyte-colony stimulating factor therapy improves survival in patients with hepatitis B virus-associated acute-on-chronic liver failure. World J Gastroenterol. 2013;19(7):1104–1110. doi: 10.3748/wjg.v19.i7.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gustot T, Fernandez J, Garcia E, Morando F, Caraceni P, Alessandria C, et al. Clinical course of acute-on-chronic liver failure syndrome and effects on prognosis. Hepatology. 2015;62(1):243–252. doi: 10.1002/hep.27849. [DOI] [PubMed] [Google Scholar]
- 153.National Institutes of Health (NIH). Emricasan, a caspase inhibitor, for treatment of subjects with decompensated NASH cirrhosis (ENCORE-LF). 2017. https://clinicaltrials.gov/ct2/show/NCT03205345?term=emricasan&draw=1&rank=1.
- 154.Rockey DC, Vierling JM, Mantry P, Ghabril M, Brown RS, Jr, Alexeeva O, et al. Randomized, double-blind, controlled study of glycerol phenylbutyrate in hepatic encephalopathy. Hepatology. 2014;59(3):1073–1083. doi: 10.1002/hep.26611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stravitz RT, Gottfried M, Durkalski V, Fontana RJ, Hanje AJ, Koch D, et al. Safety, tolerability, and pharmacokinetics of l-ornithine phenylacetate in patients with acute liver injury/failure and hyperammonemia. Hepatology. 2018;67(3):1003–1013. doi: 10.1002/hep.29621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64(12):1147–1161. doi: 10.1007/s00228-008-0553-z. [DOI] [PubMed] [Google Scholar]
- 157.U.S. Food & Drug Administration (FDA). Guidance for industry pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072123.pdf.
- 158.Desai A, Schmitt-Hoffmann A-H, Mujais S, Townsend R. Population pharmacokinetics of isavuconazole in subjects with mild or moderate hepatic impairment. Antimicrob Agents Chemother. 2016;60(5):3025–3031. doi: 10.1128/AAC.02942-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rhee SJ, Chung H, Yi S, Yu KS, Chung JY. Physiologically based pharmacokinetic modelling and prediction of metformin pharmacokinetics in renal/hepatic-impaired young adults and elderly populations. Eur J Drug Metab Pharmacokinet. 2017;42(6):973–980. doi: 10.1007/s13318-017-0418-x. [DOI] [PubMed] [Google Scholar]
- 160.Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology. 2010;51(4):1445–1449. doi: 10.1002/hep.23478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Muir AJ. Understanding the complexities of cirrhosis. Clin Ther. 2015;37(8):1822–1836. doi: 10.1016/j.clinthera.2015.05.507. [DOI] [PubMed] [Google Scholar]
- 162.U.S. Food & Drug Administration (FDA). Noncirrhotic nonalcoholic steatohepatitis with liver fibrosis: developing drugs for treatment guidance for industry. https://www.fda.gov/media/119044/download.
- 163.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sorrentino P, Tarantino G, Conca P, Perrella A, Terracciano ML, Vecchione R, et al. Silent non-alcoholic fatty liver disease-a clinical-histological study. J Hepatol. 2004;41(5):751–757. doi: 10.1016/j.jhep.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 165.Neuschwander-Tetri BA, Clark JM, Bass NM, Van Natta ML, Unalp-Arida A, Tonascia J, et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology. 2010;52(3):913–924. doi: 10.1002/hep.23784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Turco L, Garcia-Tsao G, Magnani I, Bianchini M, Costetti M, Caporali C, et al. Cardiopulmonary hemodynamics and C-reactive protein as prognostic indicators in compensated and decompensated cirrhosis. J Hepatol. 2018;68(5):949–958. doi: 10.1016/j.jhep.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 167.Sheth SG, Flamm SL, Gordon FD, Chopra S. AST/ALT ratio predicts cirrhosis in patients with chronic hepatitis C virus infection. Am J Gastroenterol. 1998;93(1):44–8. doi: 10.1111/j.1572-0241.1998.044_c.x. [DOI] [PubMed] [Google Scholar]
- 168.Nyblom H, Berggren U, Balldin J, Olsson R. High AST/ALT ratio may indicate advanced alcoholic liver disease rather than heavy drinking. Alcohol Alcohol. 2004;39(4):336–339. doi: 10.1093/alcalc/agh074. [DOI] [PubMed] [Google Scholar]
- 169.Giannini E, Botta F, Fasoli A, Ceppa P, Risso D, Lantieri PB, et al. Progressive liver functional impairment is associated with an increase in AST/ALT ratio. Dig Dis Sci. 1999;44(6):1249–1253. doi: 10.1023/a:1026609231094. [DOI] [PubMed] [Google Scholar]
- 170.Udell JA, Wang CS, Tinmouth J, FitzGerald JM, Ayas NT, Simel DL, et al. Does this patient with liver disease have cirrhosis? JAMA. 2012;307(8):832–842. doi: 10.1001/jama.2012.186. [DOI] [PubMed] [Google Scholar]
- 171.Foucher J, Chanteloup E, Vergniol J, Castera L, Le Bail B, Adhoute X, et al. Diagnosis of cirrhosis by transient elastography (FibroScan): a prospective study. Gut. 2006;55(3):403–408. doi: 10.1136/gut.2005.069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vallet-Pichard A, Mallet V, Nalpas B, Verkarre V, Nalpas A, Dhalluin-Venier V, et al. FIB-4: An inexpensive and accurate marker of fibrosis in HCV infection. Comparison with liver biopsy and fibrotest. Hepatology. 2007;46(1):32–6. doi: 10.1002/hep.21669. [DOI] [PubMed] [Google Scholar]
- 173.Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38(2):518–526. doi: 10.1053/jhep.2003.50346. [DOI] [PubMed] [Google Scholar]
- 174.U.S. National Institute of Health (NIH). Liver tox: clinical and research information on drug-induced liver injury. 2018. https://livertox.nlm.nih.gov/.
- 175.Bjornsson ES, Hoofnagle JH. Categorization of drugs implicated in causing liver injury: critical assessment based on published case reports. Hepatology. 2016;63(2):590–603. doi: 10.1002/hep.28323. [DOI] [PubMed] [Google Scholar]
- 176.Martinez MA, Vuppalanchi R, Fontana RJ, Stolz A, Kleiner DE, Hayashi PH, et al. Clinical and histologic features of azithromycin-induced liver injury. Clin Gastroenterol Hepatol. 2015;13(2):369–76.e3. doi: 10.1016/j.cgh.2014.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sarges P, Steinberg JM, Lewis JH. Drug-induced liver injury: highlights from a review of the 2015 literature. Drug Saf. 2016;39(9):801–821. doi: 10.1007/s40264-016-0427-8. [DOI] [PubMed] [Google Scholar]
- 178.Chalasani N. DILI in chronic liver disease—the next major hurdle. In: The AASLD liver meeting 2018; San Francisco; 2018.
- 179.Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132–1156. doi: 10.1111/apt.12324. [DOI] [PubMed] [Google Scholar]
- 180.Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): A randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075–1086. doi: 10.1016/S0140-6736(14)61795-5. [DOI] [PubMed] [Google Scholar]
- 181.Leroy V, Angus P, Bronowicki JP, Dore GJ, Hezode C, Pianko S, et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: a randomized phase III study (ALLY-3+) Hepatology. 2016;63(5):1430–1441. doi: 10.1002/hep.28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT-450/r–ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–1982. doi: 10.1056/NEJMoa1402869. [DOI] [PubMed] [Google Scholar]
- 183.Reddy KR, Bourliere M, Sulkowski M, Omata M, Zeuzem S, Feld JJ, et al. Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: an integrated safety and efficacy analysis. Hepatology. 2015;62(1):79–86. doi: 10.1002/hep.27826. [DOI] [PubMed] [Google Scholar]
- 184.Wilder JM, Muir AJ. Strategies for treating chronic HCV infection in patients with cirrhosis: latest evidence and clinical outcomes. Ther Adv Chronic Dis. 2015;6(6):314–327. doi: 10.1177/2040622315603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shen H, Ding F, Wang Z, Sun F, Yu Y, Zhou J, et al. Comparison of telbivudine and entecavir therapy on nephritic function and drug resistance in patients with hepatitis B virus-related compensated cirrhosis. Cell Physiol Biochem. 2016;40(1–2):370–378. doi: 10.1159/000452552. [DOI] [PubMed] [Google Scholar]
- 186.Tsai NC, Marcellin P, Buti M, Washington MK, Lee SS, Chan S, et al. Viral suppression and cirrhosis regression with tenofovir disoproxil fumarate in Asians with chronic hepatitis B. Dig Dis Sci. 2015;60(1):260–268. doi: 10.1007/s10620-014-3336-7. [DOI] [PubMed] [Google Scholar]
- 187.Okada M, Enomoto M, Kawada N, Nguyen MH. Effects of antiviral therapy in patients with chronic hepatitis B and cirrhosis. Expert Rev Gastroenterol Hepatol. 2017;11(12):1095–1104. doi: 10.1080/17474124.2017.1361822. [DOI] [PubMed] [Google Scholar]
- 188.Tsai MC, Yu HC, Hung CH, Lee CM, Chiu KW, Lin MT, et al. Comparing the efficacy and clinical outcome of telbivudine and entecavir naive patients with hepatitis B virus-related compensated cirrhosis. J Gastroenterol Hepatol. 2014;29(3):568–575. doi: 10.1111/jgh.12436. [DOI] [PubMed] [Google Scholar]
- 189.Wu X, Shi Y, Zhou J, Sun Y, Piao H, Jiang W, et al. Combination of entecavir with thymosin alpha-1 in HBV-related compensated cirrhosis: a prospective multicenter randomized open-label study. Expert Opin Biol Ther. 2018;18(sup1):61–9. doi: 10.1080/14712598.2018.1451511. [DOI] [PubMed] [Google Scholar]
- 190.Buster EH, Hansen BE, Buti M, Delwaide J, Niederau C, Michielsen PP, et al. Peginterferon alpha-2b is safe and effective in HBeAg-positive chronic hepatitis B patients with advanced fibrosis. Hepatology. 2007;46(2):388–394. doi: 10.1002/hep.21723. [DOI] [PubMed] [Google Scholar]
- 191.Liaw YF, Sung JJ, Chow WC, Farrell G, Lee CZ, Yuen H, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351(15):1521–1531. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 192.D'Amico G, Garcia-Pagan JC, Luca A, Bosch J. Hepatic vein pressure gradient reduction and prevention of variceal bleeding in cirrhosis: a systematic review. Gastroenterology. 2006;131(5):1611–1624. doi: 10.1053/j.gastro.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 193.Garcia-Tsao G, Abraldes JG, Berzigotti A, Bosch J. Portal hypertensive bleeding in cirrhosis: risk stratification, diagnosis, and management: 2016 practice guidance by the American association for the study of liver diseases. Hepatology. 2017;65(1):310–335. doi: 10.1002/hep.28906. [DOI] [PubMed] [Google Scholar]
- 194.Ripoll C, Groszmann R, Garcia-Tsao G, Grace N, Burroughs A, Planas R, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology. 2007;133(2):481–488. doi: 10.1053/j.gastro.2007.05.024. [DOI] [PubMed] [Google Scholar]
- 195.Dultz G, Seelhof M, Herrmann E, Welker M-W, Friedrich-Rust M, Teuber G, et al. Baseline MELD score predicts hepatic decompensation during antiviral therapy in patients with chronic hepatitis C and advanced cirrhosis. PLoS ONE. 2013;8(8):e71262. doi: 10.1371/journal.pone.0071262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Avigan M. Premarketing clinical evaluation of DILI. Guidances: Challenges in chronic liver disease. Presentation at the FDA/Critical Path annual Drug Induced Liver Injury conference XVI, Hyattsville, MD. 2016. https://www.aasld.org/events-professional-development/drug-induced-liver-injury-2016-program.
- 197.Konerman MA, Yapali S, Lok AS. Systematic review: Identifying patients with chronic hepatitis C in need of early treatment and intensive monitoring predictors and predictive models of disease progression. Aliment Pharmacol Ther. 2014;40(8):863–79. doi: 10.1111/apt.12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jalan R, Gines P, Olson JC, Mookerjee RP, Moreau R, Garcia-Tsao G, et al. Acute-on chronic liver failure. J Hepatol. 2012;57(6):1336–1348. doi: 10.1016/j.jhep.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 199.Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144(7):1426–37, 37.e1-9. [DOI] [PubMed]
- 200.Vierling JM. Can study protocols protect patients with liver disease from serious DILI? In: Presentation at the FDA/critical path annual drug induced liver injury conference XVI. Hyattsville. 2016.
- 201.Lewis JH. Diagnosing DILI in patients with active or advanced liver disease. In: Presentation at the FDA/critical path annual drug induced liver injury conference XVII, Hyattsville. 2017.
- 202.Muir AJ, Poordad F, Lalezari J, Everson G, Dore GJ, Herring R, et al. Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. JAMA. 2015;313(17):1736–1744. doi: 10.1001/jama.2015.3868. [DOI] [PubMed] [Google Scholar]
- 203.Mishra PK. Recognizing, assessing managing acute DILI in HCVLD—FDA challenges. 2016. https://aasld.org/events-professional-development/drug-induced-liver-injury-2016-program.
- 204.Brunt EM, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94(9):2467–74. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 205.D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231. doi: 10.1016/j.jhep.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 206.Curry MP, O’Leary JG, Bzowej N, Muir AJ, Korenblat KM, Fenkel JM, et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med. 2015;373(27):2618–2628. doi: 10.1056/NEJMoa1512614. [DOI] [PubMed] [Google Scholar]
- 207.Singal AK, Fontana RJ. Meta-analysis: oral anti-viral agents in adults with decompensated hepatitis B virus cirrhosis. Aliment Pharmacol Ther. 2012;35(6):674–689. doi: 10.1111/j.1365-2036.2011.04990.x. [DOI] [PubMed] [Google Scholar]
- 208.Lian J-S, Zeng L-Y, Chen J-Y, Jia H-Y, Zhang Y-M, Xiang D-R, et al. De novo combined lamivudine and adefovir dipivoxil therapy vs entecavir monotherapy for hepatitis B virus-related decompensated cirrhosis. World J Gastroenterol. 2013;19(37):6278–6283. doi: 10.3748/wjg.v19.i37.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Liaw YF, Raptopoulou-Gigi M, Cheinquer H, Sarin SK, Tanwandee T, Leung N, et al. Efficacy and safety of entecavir versus adefovir in chronic hepatitis B patients with hepatic decompensation: a randomized, open-label study. Hepatology. 2011;54(1):91–100. doi: 10.1002/hep.24361. [DOI] [PubMed] [Google Scholar]
- 210.Poordad F, Schiff ER, Vierling JM, Landis C, Fontana RJ, Yang R, et al. Daclatasvir with sofosbuvir and ribavirin for hepatitis C virus infection with advanced cirrhosis or post-liver transplantation r. Hepatology. 2016;63(5):1493–1505. doi: 10.1002/hep.28446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Shim JH, Lee HC, Kim KM, Lim YS, Chung YH, Lee YS, et al. Efficacy of entecavir in treatment-naive patients with hepatitis B virus-related decompensated cirrhosis. J Hepatol. 2010;52(2):176–182. doi: 10.1016/j.jhep.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 212.Peng H, Liu J, Yang M, Tong S, Yin W, Tang H, et al. Efficacy of lamivudine combined with adefovir dipivoxil versus entecavir monotherapy in patients with hepatitis B-associated decompensated cirrhosis: a meta-analysis. J Clin Pharmacol. 2014;54(2):189–200. doi: 10.1002/jcph.181. [DOI] [PubMed] [Google Scholar]
- 213.Mishra P, Murray J, Birnkrant D. Direct-acting antiviral drug approvals for treatment of chronic hepatitis C virus infection: Scientific and regulatory approaches to clinical trial designs. Hepatology. 2015;62(4):1298–1303. doi: 10.1002/hep.27880. [DOI] [PubMed] [Google Scholar]
- 214.Marathe D, Yang Y, Shang E, Li S, Zhao P, Lee SC, et al. Dosing considerations for obeticholic acid (OcA) in the treatment of primary biliary cholangitis (PBC). Clin Pharmacol Ther. 2017;101(S 1):S77-S.
- 215.U.S. Food & Drug Administration (FDA). FDA drug safety communication: FDA warns about serious liver injury with ocaliva (obeticholic acid) for rare chronic liver disease. https://www.fda.gov/Drugs/DrugSafety/ucm576656.htm.
- 216.Mogalian E, Brainard DM, Osinusi A, Moorehead L, Murray B, Ling KHJ, et al. Pharmacokinetics and Safety of Velpatasvir and Sofosbuvir/Velpatasvir in Subjects with Hepatic Impairment. Clin Pharmacokinet. 2018;57(11):1449–1457. doi: 10.1007/s40262-018-0645-6. [DOI] [PubMed] [Google Scholar]
- 217.Jeong R, Lee YS, Sohn C, Jeon J, Ahn S, Lim KS. Model for end-stage liver disease score as a predictor of short-term outcome in patients with drug-induced liver injury. Scand J Gastroenterol. 2015;50(4):439–446. doi: 10.3109/00365521.2014.958094. [DOI] [PubMed] [Google Scholar]
- 218.Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology. 2010;52(6):2065–76. doi: 10.1002/hep.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.