

Abstract
Backgroud: Toll-like receptor 4 (TLR4), a key mediator of inflammatory responses, which is associated with vascular remodeling. The association between TLR4 and NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome in the regulation of vascular smooth muscle cell (VSMC) proliferation remains unclear. This study was to explore the role and underlying mechanisms of TLR4 in the proliferation of VSMC in hypertension. Methods: VSMC proliferation after TLR4 overexpression or downregulation was determined by CCK-8, EdU Incorporation and colony formation assays. Western blots were carried out to investigate the expression of TLR4 and NLRP3 inflammasome components in VSMCs. Next, blood pressure measurements and Hematoxylin and Eosin (HE) staining assays were performed in spontaneously hypertensive rats (SHR). Media thickness (M) and diameter lumen (L) were measured as indicators of vascular remodeling. The expression of TLR4, PCNA and NLRP3 inflammasome complex was analyzed by Western blots in the aorta of SHR. Results: We showed that TLR4 overexpression with cDNA enhanced, while knockdown of TLR4 with shRNA inhibited Ang II-induced VSMC proliferation. Besides, TLR4 overexpression upregulated the proteion expression of the NLRP3 inflammasome components including NLRP3, ASC and caspase-1, whereas their corresponding levels of expression were observed to decrease in TLR4 shRNA-transfected VSMCs. Knockdown of TLR4 attenuated vascular remodeling, blood pressure (BP) and the levels of NLRP3, ASC, caspase-1, IL-1β and IL-18 in SHR aortas. Conclusion: This study revealed that TLR4 regulated Ang II-induced VSMC proliferation through modulating the NLRP3 inflammasome. Knockdown of TLR4 attenuated the BP and vascular remodeling by inhibiting the expression of the NLRP3 inflammasome component in SHR. Our results support that TLR4 regulates VSMC proliferation in hypertension via triggering the NLRP3 inflammasome.
Keywords: TLR4, proliferation, hypertension, NLRP3 inflammasome
Introduction
Hypertension is a major risk factor in cardiovascular events such as atherosclerosis and myocardial infarction [1]. An excessive inflammatory response is critical to vascular smooth muscle cell (VSMC) proliferation, which is widely known for determining vascular remodeling and stiffening in the initiation and progression of hypertension [2,3]. Increasing evidence suggests that chronic low-grade vascular inflammation contributes to VSMC proliferation in hypertension [4,5].
The NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is a cytosolic complex thant is involved in inflammatory responses and is comprised of NLRP3, apoptosis-associated Speck-like Protein (ASC) and Pro-caspase-1 [6]. The integration featured by the inflammasome results in the eventual activation of pro-caspase-1 on active caspase-1 [7]. Furthermore, activated caspase-1 processes pro-interleukin (IL)-1β and pro-interleukin (IL)-18 into mature IL-1β and IL-18, respectively, triggering an inflammatory response [8]. The promotion of IL-1β and IL-18 occurs to hypertension, serving as potential mediators of vascular inflammation [9]. Angiotensin II (Ang II), another important proinflammatory cytokine involved in the pathogenesis of hypertension [10,11], is widely used to induce hypertension and vascular remodeling [12,13].
Toll-like receptor 4 (TLR4), a member of the toll-like receptor (TLR) family, attached to the interleukin receptor superfamily, which exerts an indispensable role in the activation of innate inflammatory responses [14]. Mounting evidence suggests that TLR4-mediated inflammatory reactions embrace relevance to cardiovascular diseases, including diabetic cardiomyopathy, atherosclerosis and hypertension [15,16]. Previous studies have also reported that activation of the NLRP3 inflammasome requires TLR4 stimulation and thus facilitates the secretion of the inflammatory cytokines [17,18]. Excessive proliferation of VSMCs is induced by various cytokines, such as angiotensin II and platelet-derived growth factors [19]. It has been established that Ang II is binds to Ang II type 1 receptor (AT1R) and consequently promotes VSMC proliferation [20]. Clinical and experiment studies report that inhibition of Ang II generation is an important strategy for the treatment of hypertension [21]. Therefore, Ang II is widely used as a model of VSMC proliferation and vascular remodeling.
However, whether TLR4 plays an important role in VSMC proliferation and vascular remodeling through the regulation of the NLRP3 inflammasome leading to hypertension remains unclear. To assess this mechanism, we first evaluated the role of TLR4 in Ang II-induced VSMC proliferation, and eamined the relationship between TLR4 and the NLRP3 inflammasome in VSMCs. Next, the effect of TLR4 inhibition on vascular remodeling was evaluated in SHR.
Materials and methods
Experimental animals
The spontaneous hypertensive rats (SHR) of 13 weeks, together with those normotensive Wistar-Kyoto (WKY) rats in age match as attained out of Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The animals were maintained on the condition of room temperature with a 12-hour light-dark cycle. With food and water available in random. The rats were given one week to acclimatize to environments. The approval of experimental procedures was from the Animal Care and Use Committee of China Medical University. The experiment was performed according to the Care and Use of Laboratory Animals guide under the publishment of the US National Institutes of Health (NIH Publication, 2011). At the end of the experiment, aortic tissues were collected for histopathology and Western blots analysis.
Cell culture
VSMCs were separated and cultured from the thoracic aorta of mice, as previously described in detail [22]. VSMCs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) based on 10% fetal bovine serum (FBS, Scientific, Noble Park, Australia), penicillin (100 units/ml) as well asstreptomycin (100 mg/ml) on the condition of 37°C in a 5% CO2 humidified incubator. Then, the medium was replaced with serum free medium (SFM) containing DMSO (1:1000) and MCC950 (50 nM, cat. PZ0276; Sigma-Aldric) for 24 h [23].
Overexpression of TLR4 in VSMCs
In order to perform the transient overexpression of TLR4, the corresponding cDNA from the pCMV6-AC-GFP-TLR4-cDNA plasmid was cloned using an iScript cDNA Synthesis Kit (TaKaRa, Kusatsu, Shiga, Japan); the plasmid sequence was verified via sequencing. VSMCs were seeded into the plates of 24 wells and then infected with the TLR4 overexpression plasmid in virtue of Lipofectamine 2000 (Invitrogen, Carlsbad, CA), in accordance with the directions from the manufacturer. In regard to the Ang II-induced inflammation model, VSMCs transfected with TLR4 cDNA were treated with Ang II (100 nM) for 48 h to establish an inflammation model, as previously reported [12].
Knockdown of TLR4 in VSMCs and rats
A small short hairpin RNA (shRNA) that specifically targets TLR4 was commercially constructed by Kangchen Biological Engineering & Technology Co., Ltd. (Shanghai, China). The VSMCs were infected with lentiviral particles that were packed with pGFP-V-RS-TLR-4-shRNA (5’-CCGGCCGCTGGTGTATCTTTGAATACTCGAGTATTCAAAGATACACCAGCGGTTTTTG-3’). For in vitro studies, VSMCs were transfected with TLR4 shRNA for 24 h in vitro and were then treated with Ang II (100 nM) or PBS for 48 hours. For in vivo studies, 13-week-old WKY and SHR were subjected to intravenous injection of lentivirus packaged TLR4 shRNA plasmid with a concentration of 2 × 109 TU/mL [24]. The scrambled lentivirus plasmid was prepared as the negative control. After 4 weeks of lentivirus induction, the experiments were carried out.
CCK-8 assay
Cell counting kit-8 assays (CCK-8, KGA317, KeyGen Biotech, Shanghai, China) were used to evaluate VSMC proliferation according to the manufacturer’s instructions. CCK-8 reagent (10 μL) was added to each well and was incubated for 2 hours at 37°C. Optical absorbance was monitored at a wavelength of 450 nm (BioTek Instruments, Winooski, VT, USA). The viability of VSMCs was calculated as the experimental OD value/control OD value.
Colony formation assay
VSMCs were seeded into 6-well plates (500 cells per well) after transfection. For colony formation assay, the cells were incubated for 10 days. Then, the colonies were fixed and stained with staining solution containing 0.05% crystal violet, 1% formaldehyde, and 10% methanol buffered with PBS. Images were taken by camera, and the process was performed in triplicate.
EdU incorporation assay
To quantify EdU incorporation, EdU reagent (Invitrogen) was added to the culture medium during the 18 hours of cell culturing. VSMCs were stained using a CLICK-iT assay according to the manufacturer’s protocol. EdU-positive cells were calculated and standardized with the total number of Hoechst 33342-stained cells.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of IL-1β and IL-18 were measured using the IL-1β ELISA kit (cat. CSB-E04563h, ExCell Biology, Shanghai, China) and IL-18 ELISA kit (cat. CSB-E04563h, R&D, Minneapolis, MN, USA) according to the manufacturer’s instructions. The data (pg protein) were normalized to mg of total protein.
Blood pressure measurements
Blood pressure (BP) was monitored using the noninvasive tail-cuff method (ADInstruments, New South Wales, Australia) as per recommendation of the American Heart Association. The measurement to systolic blood pressure (SBP) and the mean arterial pressure (MAP) were measured at 9 AM to avoid diurnal variation.
Hematoxylin and Eosin (HE) staining
Aorta tissues were fixed with 4% formalin and embeded with paraffin. 5 μm sections were prepared and stained with hematoxylin and eosin (HE) to discern their morphology. Images were acquired in virtue of a light microscope (Olympus, Tokyo, Japan). The media thickness, lumen diameter as well as ratio between the two were used as indexes for vascular remodeling.
Western blots analysis
Proteins were extracted using the RIPA lysis buffer (cat. KGP245, Beyotime Biotechnology, Shanghai, China). The protein concentration was determined by utilizing the BCA protein Assay Kit (Pierce, Appleton, WI, USA) according to the manufacturer’s protocol. Protein samples (40 μg) were separated by SDS-PAGE, and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were probed with 5% non-fat milk overnight at 4°C in TBST buffer, which were incubated with primary antibodies: TLR4 (cat. ab223588; 1:1200; Abcam, US), ASC (cat. ab223588; 1:500; Abcam, US), caspase-1 (cat. ab223588; 1:500; Abcam, US), NLRP3 (cat. MAB7578; 1:2000; Santa Cruz, CA, USA), GAPDH (cat. SC-25774; 1:5000; Santa Cruz, CA, USA), IL-1β (cat. SC-7864; 1:500; Santa Cruz, CA, USA) and IL-18 (cat. SC-656; 1:2000; Santa Cruz, CA, USA). The membranes were washed with TBST and incubated secondary antibodies for 2 hours. These results were normalized to GAPDH levels, and the bands were visualized using an enhanced chemiluminescence (ECL) kit (Future Biotech, China). The intensities of the protein bands were quantified using the Gel Doc 2000 image system.
Statistical analysis
Values were presented as mean ± standard error of the mean (SEM). Statistical analyses were performed in virtue of two-way analysis of variance (ANOVA) and followed the Multiple Comparison Test. P<0.05 was considered to be statistically significant.
Results
TLR4 shRNA attenuated Ang II-induced VSMC proliferation
To determine the role of TLR4 in VSMC proliferation, CCK-8 and EdU incorporation assay were carried out to assess the number of cells. According to the CCK-8 assay, VSMC proliferation was observed to be inhibited by TLR4 knockdown with shRNA (Figure 1A). Furthermore, an EdU incorporation assay was employed to detect DNA synthesis in VSMCs. Accordingly, TLR4 shRNA was found to reduce the number of EdU-positive cells during Ang II-induced VSMC proliferation (Figure 1B, 1C). The above findings demonstrated that TLR4 shRNA prevented the increase in proliferation capacity among Ang II-treated VSMCs.
Figure 1.

Effect of TLR4 cDNA or shRNA on Ang II-induced VSMC proliferation. A. Cell viability was detected by CCK-8 assay in VSMCs transfected with TLR4 cDNA or shRNA. B. Representative images showing EdU-positive cells measured with an EdU incorporation assay. Blue fluorescence indicateds cell nuclei and red fluorescence indicates cells undergoing DNA synthesis. C. Bar graph showing the percentage of Edu positive cells. D. Cell colony forming capabilities of transfected VSMCs with TLR4 cDNA were determined by colony forming assays. E. Bar graph showing the number of cell clones. Values are the mean ± SEM (n=6; *P<0.05 vs PBS; #P<0.05 vs control; +P<0.05 vs TLR4 cDNA).
TLR4 shRNA decreased the expression of the NLRP3 inflammasome in VSMCs
The NLRP3 inflammasome is a multiprotein complex consisting of NLRP3, ASC and caspase-1 [25]. In order to understand how TLR4 mediates NLRP3 component expression in Ang II-induced VSMCs, the protein levels of TLR4 and NLRP3 components were evaluated in VSMCs transfected with shRNA. TLR4 shRNA was found to downregulate the protein level of TLR4 in Ang II-induced VSMCs (Figure 2). In addition, the levels of NLRP3, ASC and caspase-1 were downregulated following TLR4 shRNA transfection (Figure 2). These results implied that TLR4 is association with the modulation of NLRP3 components in Ang II-induced VSMCs.
Figure 2.
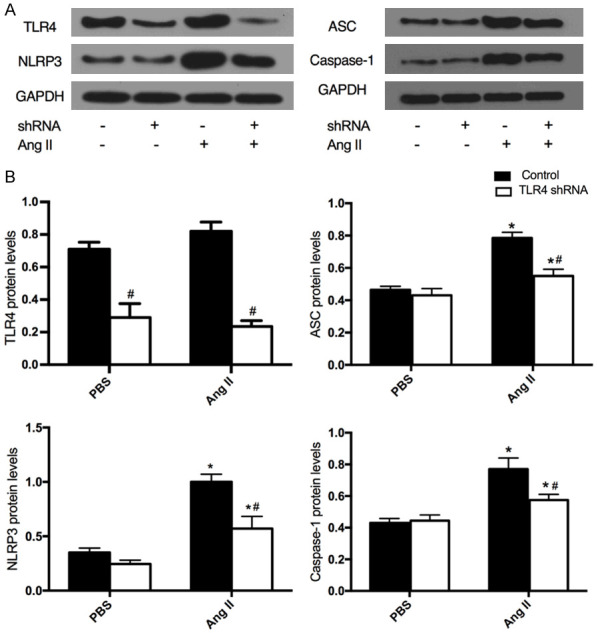
Protein expression of the NLRP3 inflammasome in Ang II-induced VSMCs transfected with TLR4 shRNA. A. Western blots analysis was performed to measure the protein expression of TLR4, NLRP3, ASC and caspase-1. B. Bar graphs showing the expression of TLR4, NLRP3, ASC and caspase-1. Values are mean ± SEM (n=6; *P<0.05 vs PBS; #P<0.05 vs control).
TLR4 cDNA promoted Ang II-induced VSMC proliferation
In view of the data obtained from the CCK-8 assay, TLR4 cDNA transfection was observed to aggravate Ang II-induced VSMC proliferation (Figure 1A), which was further confirmed by colony formation assay (Figure 1D, 1E). The small molecule MCC950 is known to be a potent and selective inhibitor of NLRP3 [26]. To further determine whether VSMC proliferation is dependent on NLRP3 activation, the formation assay was carried out to detect cell growth in VSMCs with MCC950 treatment after TLR4 cDNA transfection. The acquired data demonstrated that MCC950 inhibited the proliferation of VSMCs with TLR4 cDNA transfection, consistent with previous results (Figure 1D, 1E) [27]. Hence, this study puts forward that the activation of the NLRP3 inflammasome was involved in TLR4 cDNA-induced VSMC proliferation. Additionaly, treatment with MCC950 may effectively against the cell growth of VSMCs by inhibiting inflammation.
TLR4 cDNA increased the expression of the NLRP3 inflammasome in VSMCs
The protein expression of TLR4 and NLRP3 inflammasome components in PBS-treated or Ang II-induced VSMCs transfected with TLR4 cDNA were also investigated. TLR4 cDNA transfection was found to increase the protein expression of TLR4 in both PBS-treated and Ang II-induced VSMCs (Figure 3), confirming the efficiency of VSMC transfection with TLR4 cDNA. Meanwhile, protein levels of NLRP3, ASC and caspase-1 were upregulated in Ang II-induced VSMCs transfected with TLR4 cDNA (Figure 3). All corresponding data demonstrated that TLR4 cDNA transfection upregulated protein levels of the NLRP3 inflammasome in Ang II-induced VSMCs.
Figure 3.

Protein expression of the NLRP3 inflammasome in Ang II-induced VSMCs transfected with TLR4 cDNA. A. Western blots analysis was performed to measure the protein expression of TLR4, NLRP3, ASC and caspase-1. B. Bar graphs showing the expression of TLR4 NLRP3, ASC and caspase-1. Values are mean ± SEM (n=6; *P<0.05 vs PBS; #P<0.05 vs control).
TLR4 shRNA attenuated blood pressure in SHR
VSMC proliferation plays a key role in hypertension development [28]. Here, TLR4 shRNA was shown to inhibit Ang II-induced VSMC proliferation, which is implicated in the pathogenesis of hypertension. The SHR model was originally derived from the normal WKY strain. Hence, WKY served as a normal control group for SHR. Furthermore, the downregulation of TLR4 conferred no significant effects on systolic blood pressure (SBP) and mean arterial pressure (MAP) in WKY. However, TLR4 shRNA caused a constant decline on blood pressure (BP) in SHR (Figure 4A).
Figure 4.
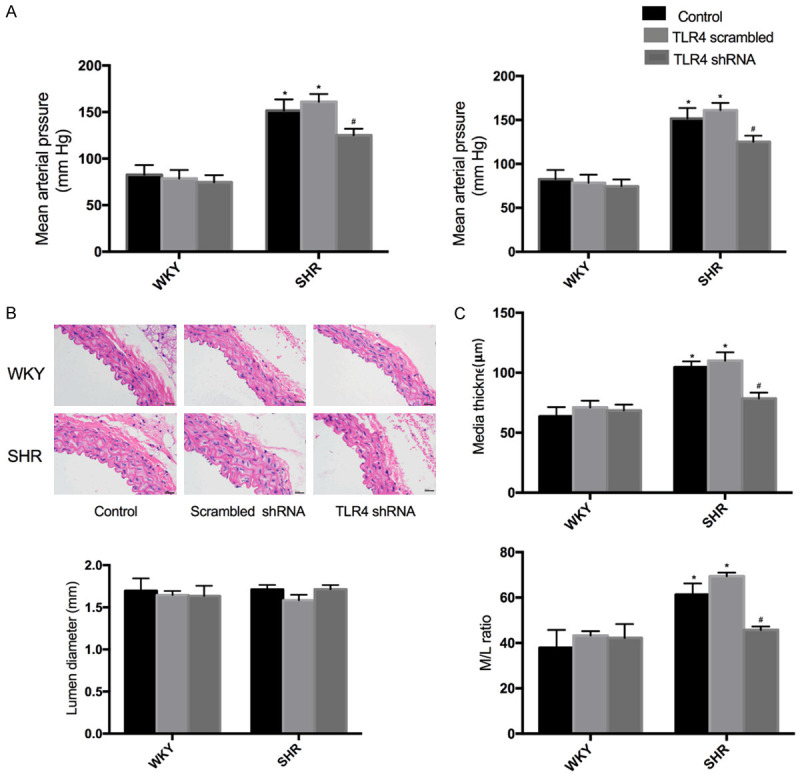
Effects of shTLR4 on blood pressure and vascular remodeling of the aortas of WKY and SHR. A. Systolic blood pressure (SBP) and mean arterial pressure (MAP). B. Representative transverse section images of aorta with hematoxylin eosin staining. C. Media thickness (M), Lumen diameter (L) and the ratio of M to L of aorta. Values are mean ± SEM (n=6; *P<0.05 vs control; #P<0.05 vs scrambled shRNA).
TLR4 shRNA attenuated vascular remodeling in SHR
HE staining and Western blots were performed to evalutate the effects of TLR4 shRNA on vascular remodeling in SHR. Knockdown of TLR4 had no significant effect on media thickness and the protein expression of PCNA in WKY. VSMC proliferation was inhibited by TLR4 knockdown, as the evidence of the decreased PCNA protein level (Figure 5A). In addition, the media thickness (M), lumen diameter (L) and their ratio to artery were used as the indexes of vascular remodeling in rats. Specifically, intervention with TLR4 shRNA reduced M and M/L in the aorta of SHR (Figure 4B).
Figure 5.
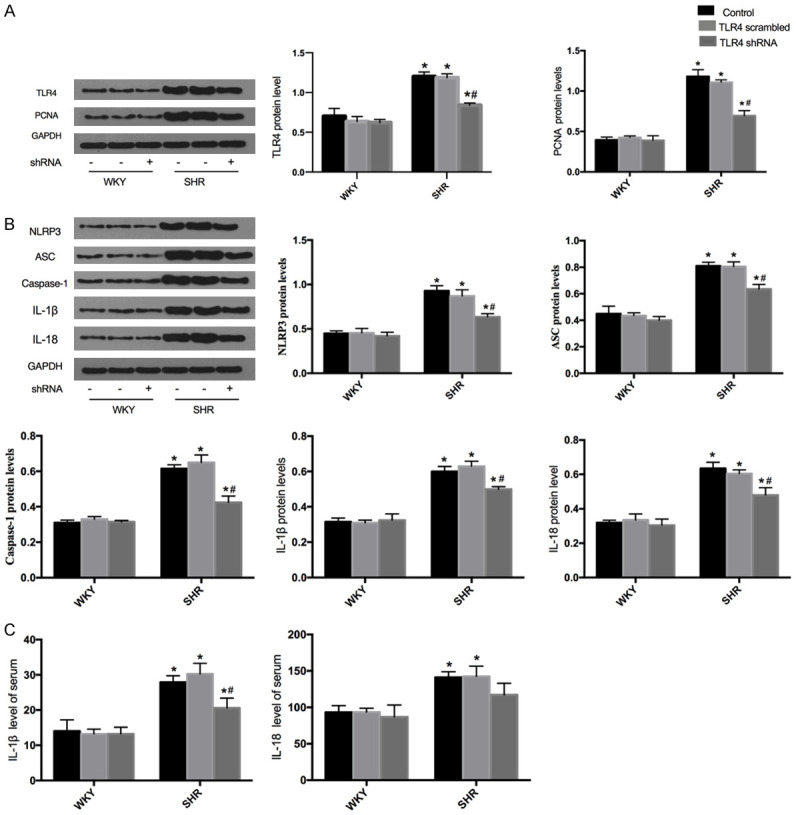
Effects of shTLR4 on the NLRP3 inflammasome and proliferation in the aortas of WKY and SHR. A. Western blots were performed to measure the protein expression of TLR4 and PCNA. B. The protein expression of NLRP3, ASC, caspase-1, IL-1β and IL-18. C. IL-1β and IL-18 levels of serum. Values are mean ± SEM (n=6; *P<0.05 vs PBS; #P<0.05 vs control or scrambled shRNA).
TLR4 shRNA inhibited the NLRP3 inflammasome in SHR
To confirm the above findings, TLR4 was genetically silenced with recombinant lentiviral shRNA in WKY and SHR. The levels of the NLRP3 inflammasome and subsequent inflammatory cytokine secretions were assessed using Western blots and ELISA analysis. After TLR4 silencing, no significant changes in the expression of the NLRP3 inflammasome components in WKY (Figure 5A). When SHR were treated with TLR4 shRNA, the protein levels of NLRP3, ASC, caspase-1, IL-1β and IL-18 were dramatically decreased compared with that of the control and scrambled shRNA-treated SHR (Figure 5B). Furthermore, ELISA assay revealed that TLR4 shRNA treatment significantly inhibited serum IL-1β levels (Figure 5C).
Discussion
As far as we know, inflammatory cell intrusion into destroyed tissues is followed by the increased production of inflammatory cytokines. triggering vascular remodeling disorders such as transplantation arteriopathy, diabetic vascular complications, hypertension, atherosclerosis and vascular restenosis [29,30]. Toll-like receptor family, especially TLR4, dissociates from its inhibitory protein, IκBα, and is transmitted into the nucleus, where it promotes the secretion out of pro-inflammatory cytokines [31]. TLR4 motivates pro-inflammatory signaling cascades, leading to the activation of protein kinase B (Akt or PKB) [32,33]. The positive feedback regulation involved in the proliferation of VSMCs has been shown to be mediated by the TLR4/Akt signaling pathway [34]. The mechanistic target of rapamycin (mTOR) is critical in TLR4 signaling, playing roles in cell proliferation and immune regulation. Studies have shown that vascular levels of NLRP3 inflammasome components is elevated in hypertension [35,36]. Moreover, previous studies have inllustrated that inhibition of TLR4 significantly downregulates the activation of the NLRP3 as well as inhibiting the secretion of mature IL-1β and IL-18 [37]. This study preliminarily probed that TLR4 regulates VSMC proliferation through modulation of the NLRP3 inflammasome both in vitro and in vivo.
Ang II binds to Ang II type I receptor (AT1R), promoting VSMC proliferation [38]. Earlier studies have show that Ang II induces VSMC proliferation and vascular remodeling in a hypertension model [39]. Hernanz et al demonstrated that up-regulation of TLR4 by Ang II participates in the pathogenesis of hypertension by affecting the structure and the mechanical properties of the vasculature by mechanisms likely to involve oxidative stress [40]. Besides, Han et al found that Ang II and TLR4 co-receptor binding mediates cardiovascular inflammation [41]. Previous researches demonstrated that knockdown of TLR4 attenuated Ang II-induced cardiac remodeling in vivo [42]. Here, the treatment of VSMCs with PBS had no detectable impact on proliferation, however, Ang II-induced the proliferation of VSMCs. This study primarily shows that knockdown of TLR4 inhibits, while TLR4 overexpression exacerbates Ang II-induced VSMC proliferation, which suggests that TLR4 is an important factor in regulating VSMC proliferation. The NLRP3 inflammasome is a critical positive regulator of VSMC proliferation in hypertension. The NLRP3 inflammasome may be activated by various signals as well as metabolic dysregulation including reactive oxygen species (ROS), mitochondrial impairment and Ang II [43]. The activation of NLRP3 inflammasome require transcription, which enables the assembly of the NLRP3 inflammasome components [44]. Increasing evidence has demonstrated that inflammatory responses are both caused and maintained by the NLRP3 inflammasome [45]. Some human VSMCs expressed the NLRP3 inflammasome components at the site of atherosclerosis [46]. These findings suggested that TLR4 might have an association with NLRP3 inflammasome in VSMCs. Accordingly, Ang II-induced VSMC proliferation was observed to be inhibited by MCC950 (an inhibitor of NLRP3 inflammasome) in this investigation. TLR4 is known to initiate the activation of the NLRP3 inflammasome, which has been identified within vascular tissues [47]. In the present study, Western blots analysis showed that TLR4 overexpression increased the release of the inflammatory cytokines NLRP3, ASC and caspase-1. However, this inflammatory response was suppressed by TLR4 knockdown in VSMCs. The vitro study provides evidence that TLR4 regulates VSMC proliferation through the regulation of the NLRP3 inflammasome as well as its associated inflammatory cytokines.
VSMC proliferation is widely known to contribute to vascular remodeling in hypertension [48]. The next step in the experiment was to determine whether the downregulation of TLR4 decreases the inflammatory reaction and counteracts hypertension in vivo. Spontaneously hypertensive rats (SHR) are an inbred strain of rats that develop hypertension, which are widely used as models for the study of hypertension in humans. Previous studies have confirmed the enhanced proliferation of VSMCs in the SHR model [49,50]. It has been well established that proliferating cell nuclear antigen (PCNA), a nuclear factor involved in DNA replication as well as the repair of proliferating cells, exert functions within only proliferating cells [51]. Here, treatment with TLR4 shRNA was found to decrease the PCNA protein level in SHR. However, TLR4 shRNA did not alter PCNA expression in WKY. Furthermore, treating SHR with TLR4 shRNA triggered a drop in BP. Treatment with TLR4 shRNA did not significantly decrease BP in WKY, indicating that SHR may be more sensitive to TLR4 than WKY. The obtained results clearly suggest that the hypotensive effect was more pronounced in SHR compared to WKY. In keeping with our findings, a previous report demonstrated that knockdown of TLR4 prevented the increase of BP in SHR [49]. Vascular remodeling is characterized by changes in vascular structure and function, contributing to both the development and complication of hypertension [52]. TLRs promote the flow-mediated inward remodeling of ductal artery through the adaptor protein MyD88. Intensive research have revealed that TLR4 is involved in the outward arterial remodeling after atherosclerotic plaque formation [53,54]. VSMC proliferation is closely related to vascular remodeling and hypertension. Thus, the effect of TLR4 shRNA on vascular remodeling was examined among SHR. In this study, SHR treated with TLR4 shRNA showed attenuation of vascular remodeling in the aortic media, but treatment had no detectable impact on that of WKY. These findings align with the notion that TLR4 shRNA inhibits Ang II-induced VSMC proliferation in vitro.
TLR4 is activated by lipopolysaccharide as well as by non-infectious host endogenous compounds, which is present in hypertension as a consequence of chronic cell damage and death. Hypertension is a chronic inflammatory condition. Numerous studies suggest that increased pro-inflammatory cytokines play an important role in the development of hypertension [55,56]. Moreover, relevant studies have reported that the NLRP3 inflammasome is activated within the aortic adventitia of SHR [57]. Considering that the activation of the NLRP3 inflammasome is closely associated with TLR4. it is reasonable to speculate that TLR4 shRNA exerts its effects via the NLRP3 inflammasome in order to suppress vascular remodeling in SHRs. In this study, the protein levels of TLR4 were downregulation in those aortic media of SHR were transfected with TLR4 shRNA. Then, TLR4 shRNA significantly reduced the activation of the NLRP3 inflammasome components in SHR, inhibiting the expression of NLRP3, ASC, caspase-1, IL-1β and IL-18. Similarly, previous studies revealed that a particular TLR4 signaling inhibitor almost completely abrogated the upregualation of NLRP3, caspase-1 and IL-1β [58]. However, the expression of these proteins in WKY was not affected. Futhermore, TLR4 shRNA also decreased serum IL-1β levels. Therefore, it is plausible to suggest that TLR4 shRNA attenuates hypertension in SHR, at least partly, antiproliferative effects on VSMCs and vascular remodeling by downregulation of the NLRP3 inflammasome.
Conclusions
We revealed that TLR4 overexpression aggravates, while TLR4 knockdown attenuates Ang II-induced VSMC proliferation through the modulation of the NLRP3 inflammasome. The knockdown of TLR4 attenuates proliferation and vascular remodeling in SHR by inhibiting the NLRP3 inflammasome. Overall, the findings of this study provide novel evidence that TLR4 triggers the NLRP3 inflammasome, which sheds new light on how TLR4 mediates VSMC proliferation in the pathogenesis of hypertension.
Acknowledgements
This work was supported by his project was supported by Chinese National Natural Science Fundation for Young Scientists [No. 81900642 (Q.L.)], Scientific Research Fundation for “Seeding” Project of Young Scientific and Technological Talents of Liaoning Provincial Education Department [No. QN2019006 (Q.L.)], Youth Backbone Foundation of China Medical University [No. 1210519033 (Q.L)].
Disclosure of conflict of interest
None.
References
- 1.Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. 2016;68:476–532. doi: 10.1124/pr.115.010652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi N, Chen SY. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J Biomed Res. 2014;28:40–46. doi: 10.7555/JBR.28.20130130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li M, Fukagawa NK. Age-related changes in redox signaling and VSMC function. Antioxid Redox Signal. 2010;12:641–655. doi: 10.1089/ars.2009.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown IAM, Diederich L, Good ME, DeLalio LJ, Murphy SA, Cortese-Krott MM, Hall JL, Le TH, Isakson BE. Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler Thromb Vasc Biol. 2018;38:1969–1985. doi: 10.1161/ATVBAHA.118.311229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, Montezano AC. Vascular smooth muscle contraction in hypertension. Cardiovasc Res. 2018;114:529–539. doi: 10.1093/cvr/cvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:262. doi: 10.3389/fphar.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Negash AA, Ramos HJ, Crochet N, Lau DT, Doehle B, Papic N, Delker DA, Jo J, Bertoletti A, Hagedorn CH, Gale M Jr. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013;9:e1003330. doi: 10.1371/journal.ppat.1003330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. 2015;8:15–27. doi: 10.2147/JIR.S51250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leng B, Zhang Y, Liu X, Zhang Z, Liu Y, Wang H, Lu M. Astragaloside IV suppresses high glucose-induced NLRP3 inflammasome activation by inhibiting TLR4/NF-κB and CaSR. Mediators Inflamm. 2019;2019:1082497. doi: 10.1155/2019/1082497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE, Kalergis AM. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int J Mol Sci. 2016;17:797. doi: 10.3390/ijms17070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malha L, Sison CP, Helseth G, Sealey JE, August P. Renin-angiotensin-aldosterone profiles in pregnant women with chronic hypertension. Hypertension. 2018;72:417–424. doi: 10.1161/HYPERTENSIONAHA.118.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC Jr, Llorens-Cortes C, Ehlers MR, Sturrock ED. Novel therapeutic approaches targeting the renin-angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev. 2019;71:539–570. doi: 10.1124/pr.118.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF, Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2017;312:F245–F253. doi: 10.1152/ajprenal.00178.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dange RB, Agarwal D, Teruyama R, Francis J. Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation. 2015;12:31. doi: 10.1186/s12974-015-0242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez H, Medina-Ortiz WE, Luan T, Clark AF, McDowell CM. Crosstalk between transforming growth factor beta-2 and toll-like receptor 4 in the trabecular meshwork. Invest Ophthalmol Vis Sci. 2017;58:1811–1823. doi: 10.1167/iovs.16-21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roshan MH, Tambo A, Pace NP. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int J Inflam. 2016;2016:1532832. doi: 10.1155/2016/1532832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okla M, Zaher W, Alfayez M, Chung S. Inhibitory effects of toll-like receptor 4, NLRP3 inflammasome, and interleukin-1β on white adipocyte browning. Inflammation. 2018;41:626–642. doi: 10.1007/s10753-017-0718-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koch KN, Müller A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes. 2015;6:382–387. doi: 10.1080/19490976.2015.1105427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moraes JA, Frony AC, Dias AM, Renovato-Martins M, Rodrigues G, Marcinkiewicz C, Assreuy J, Barja-Fidalgo C. Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis. 2015;243:477–485. doi: 10.1016/j.atherosclerosis.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 20.Touyz RM. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp Physiol. 2005;90:449–455. doi: 10.1113/expphysiol.2005.030080. [DOI] [PubMed] [Google Scholar]
- 21.Kurdi M, Booz GW. New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension. 2011;57:1034–1038. doi: 10.1161/HYPERTENSIONAHA.111.172700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren XS, Ling L, Zhou B, Han Y, Zhou YB, Chen Q, Li YH, Kang YM, Zhu GQ. Silencing salusin-β attenuates cardiovascular remodeling and hypertension in spontaneously hypertensive rats. Sci Rep. 2017;7:43259. doi: 10.1038/srep43259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pellegrini C, Fornai M, Antonioli L, Blandizzi C, Calderone V. Phytochemicals as novel therapeutic strategies for NLRP3 inflammasome-related neurological, metabolic, and inflammatory diseases. Int J Mol Sci. 2019;20:2876. doi: 10.3390/ijms20122876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Zang YH, Qiu Y, Zhang F, Chen AD, Wang JJ, Chen Q, Li YH, Kang YM, Zhu GQ. BCL6 attenuates proliferation and oxidative stress of vascular smooth muscle cells in hypertension. Oxid Med Cell Longev. 2019;2019:5018410. doi: 10.1155/2019/5018410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, Sohal SS, Robertson AAB, Schroder K, Kunde D, Eri R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8:8618. doi: 10.1038/s41598-018-26775-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Qian F, Liu H, Zhang Z. Elevated uric acid levels promote vascular smooth muscle cells (VSMC) proliferation via an nod-like receptor protein 3 (NLRP3)-inflammasome-dependent mechanism. Med Sci Monit. 2019;25:8457–8464. doi: 10.12659/MSM.916667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Bundy JD, Hamm LL, Hsu CY, Lash J, Miller ER 3rd, Thomas G, Cohen DL, Weir MR, Raj DS, Chen HY, Xie D, Rao P, Wright JT Jr, Rahman M, He J. Inflammation and apparent treatment-resistant hypertension in patients with chronic kidney disease. Hypertension. 2019;73:785–793. doi: 10.1161/HYPERTENSIONAHA.118.12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Ren J, Zhou B, Ding C, Chen J, Wang G, Gu G, Wu X, Liu S, Hu D, Li J. Gene silencing of non-obese diabetic receptor family (NLRP3) protects against the sepsis-induced hyper-bile acidaemia in a rat model. Clin Exp Immunol. 2015;179:277–293. doi: 10.1111/cei.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goulopoulou S, McCarthy CG, Webb RC. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol Rev. 2016;68:142–167. doi: 10.1124/pr.114.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain M, Dhanesha N, Doddapattar P, Chorawala MR, Nayak MK, Cornelissen A, Guo L, Finn AV, Lentz SR, Chauhan AK. Smooth muscle cell-specific fibronectin-EDA mediates phenotypic switching and neointimal hyperplasia. J Clin Invest. 2020;130:295–314. doi: 10.1172/JCI124708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malara A, Gruppi C, Abbonante V, Cattaneo D, De Marco L, Massa M, Iurlo A, Gianelli U, Balduini CL, Tira ME, Muro AF, Chauhan AK, Rosti V, Barosi G, Balduini A. EDA fibronectin-TLR4 axis sustains megakaryocyte expansion and inflammation in bone marrow fibrosis. J Exp Med. 2019;216:587–604. doi: 10.1084/jem.20181074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang D, Li D, Cao L, Wang L, Zhu S, Xu T, Wang C, Pan D. Positive feedback regulation of proliferation in vascular smooth muscle cells stimulated by lipopolysaccharide is mediated through the TLR 4/Rac1/Akt pathway. PLoS One. 2014;9:e92398. doi: 10.1371/journal.pone.0092398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol. 2016;13:148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun HJ, Ren XS, Xiong XQ, Chen YZ, Zhao MX, Wang JJ, Zhou YB, Han Y, Chen Q, Li YH, Kang YM, Zhu GQ. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8:e3074. doi: 10.1038/cddis.2017.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chi W, Li F, Chen H, Wang Y, Zhu Y, Yang X, Zhu J, Wu F, Ouyang H, Ge J, Weinreb RN, Zhang K, Zhuo Y. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1β production in acute glaucoma. Proc Natl Acad Sci U S A. 2014;111:11181–11186. doi: 10.1073/pnas.1402819111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jojima T, Uchida K, Akimoto K, Tomotsune T, Yanagi K, Iijima T, Suzuki K, Kasai K, Aso Y. Liraglutide, a GLP-1 receptor agonist, inhibits vascular smooth muscle cell proliferation by enhancing AMP-activated protein kinase and cell cycle regulation, and delays atherosclerosis in ApoE deficient mice. Atherosclerosis. 2017;261:44–51. doi: 10.1016/j.atherosclerosis.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Mishra JS, More AS, Gopalakrishnan K, Kumar S. Testosterone plays a permissive role in angiotensin II-induced hypertension and cardiac hypertrophy in male rats. Biol Reprod. 2019;100:139–148. doi: 10.1093/biolre/ioy179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernanz R, Martínez-Revelles S, Palacios R, Martín A, Cachofeiro V, Aguado A, García-Redondo L, Barrús MT, de Batista PR, Briones AM, Salaices M, Alonso MJ. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol. 2015;172:3159–3176. doi: 10.1111/bph.13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han J, Ye S, Zou C, Chen T, Wang J, Li J, Jiang L, Xu J, Huang W, Wang Y, Liang G. Angiotensin II causes biphasic STAT3 activation through TLR4 to initiate cardiac remodeling. Hypertension. 2018;72:1301–1311. doi: 10.1161/HYPERTENSIONAHA.118.11860. [DOI] [PubMed] [Google Scholar]
- 42.Singh MV, Cicha MZ, Meyerholz DK, Chapleau MW, Abboud FM. Dual activation of TRIF and MyD88 adaptor proteins by angiotensin II evokes opposing effects on pressure, cardiac hypertrophy, and inflammatory gene expression. Hypertension. 2015;66:647–656. doi: 10.1161/HYPERTENSIONAHA.115.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb. 2017;24:443–451. doi: 10.5551/jat.RV17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark SE, Schmidt RL, McDermott DS, Lenz LL. A Batf3/Nlrp3/IL-18 axis promotes natural killer cell IL-10 production during listeria monocytogenes infection. Cell Rep. 2018;23:2582–2594. doi: 10.1016/j.celrep.2018.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang ML, Kang YM, Li XG, Su Q, Li HB, Liu KL, Fu LY, Saahene RO, Li Y, Tan H, Yu XJ. Central blockade of NLRP3 reduces blood pressure via regulating inflammation microenvironment and neurohormonal excitation in salt-induced prehypertensive rats. J Neuroinflammation. 2018;15:95. doi: 10.1186/s12974-018-1131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono H, Ohta R, Kawasaki Y, Niwa A, Takada H, Nakahata T, Ohga S, Saito MK. Lysosomal membrane permeabilization causes secretion of IL-1β in human vascular smooth muscle cells. Inflamm Res. 2018;67:879–889. doi: 10.1007/s00011-018-1178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Li L, Deng S, Liu F, He Z. Ursolic acid ameliorates inflammation in cerebral ischemia and reperfusion injury possibly via high mobility group box 1/toll-like receptor 4/NFκB pathway. Front Neurol. 2018;9:253. doi: 10.3389/fneur.2018.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camargo LL, Harvey AP, Rios FJ, Tsiropoulou S, Da Silva RNO, Cao Z, Graham D, McMaster C, Burchmore RJ, Hartley RC, Bulleid N, Montezano AC, Touyz RM. Vascular nox (NADPH Oxidase) compartmentalization, protein hyperoxidation, and endoplasmic reticulum stress response in hypertension. Hypertension. 2018;72:235–246. doi: 10.1161/HYPERTENSIONAHA.118.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bou Daou G, Li Y, Anand-Srivastava MB. Enhanced expression of Giα proteins contributes to the hyperproliferation of vascular smooth muscle cells from spontaneously hypertensive rats via MAP kinase- and PI3 kinase-independent pathways. Can J Physiol Pharmacol. 2016;94:49–58. doi: 10.1139/cjpp-2015-0146. [DOI] [PubMed] [Google Scholar]
- 50.Madiraju P, Hossain E, Anand-Srivastava MB. Natriuretic peptide receptor-C activation attenuates angiotensin II-induced enhanced oxidative stress and hyperproliferation of aortic vascular smooth muscle cells. Mol Cell Biochem. 2018;448:77–89. doi: 10.1007/s11010-018-3316-x. [DOI] [PubMed] [Google Scholar]
- 51.Bouayad D, Pederzoli-Ribeil M, Mocek J, Candalh C, Arlet JB, Hermine O, Reuter N, Davezac N, Witko-Sarsat V. Nuclear-to-cytoplasmic relocalization of the proliferating cell nuclear antigen (PCNA) during differentiation involves a chromosome region maintenance 1 (CRM1)-dependent export and is a prerequisite for PCNA antiapoptotic activity in mature neutrophils. J Biol Chem. 2012;287:33812–33825. doi: 10.1074/jbc.M112.367839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. doi: 10.1136/bmj.j5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shinohara M, Hirata K, Yamashita T, Takaya T, Sasaki N, Shiraki R, Ueyama T, Emoto N, Inoue N, Yokoyama M, Kawashima S. Local overexpression of toll-like receptors at the vessel wall induces atherosclerotic lesion formation: synergism of TLR2 and TLR4. Arterioscler Thromb Vasc Biol. 2007;27:2384–2391. doi: 10.1161/ATVBAHA.106.139253. [DOI] [PubMed] [Google Scholar]
- 54.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–454. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 55.Agita A, Alsagaff MT. Inflammation, immunity, and hypertension. Acta Med Indones. 2017;49:158–165. [PubMed] [Google Scholar]
- 56.Sharma RK, Yang T, Oliveira AC, Lobaton GO, Aquino V, Kim S, Richards EM, Pepine CJ, Sumners C, Raizada MK. Microglial cells impact gut microbiota and gut pathology in angiotensin II-induced hypertension. Circ Res. 2019;124:727–736. doi: 10.1161/CIRCRESAHA.118.313882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. 2017;97:1127–1164. doi: 10.1152/physrev.00031.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang W, Lv H, Wang H, Wang D, Sun S, Jia Q, Wang P, Song B, Ni L. Activation of the NLRP3/caspase-1 inflammasome in human dental pulp tissue and human dental pulp fibroblasts. Cell Tissue Res. 2015;361:541–555. doi: 10.1007/s00441-015-2118-7. [DOI] [PMC free article] [PubMed] [Google Scholar]