

Regulation of cell wall assembly is essential for bacterial survival and contributes to pathogenesis and antibiotic tolerance in mycobacteria, including pathogens such as Mycobacterium tuberculosis. However, little is known about how the cell wall is regulated in stress.
KEYWORDS: PstP, serine/threonine phosphatase, peptidoglycan metabolism, mycolic acid metabolism, antibiotic tolerance, Mycobacteria, dephosphorylation, starvation, cell wall metabolism, CwlM
ABSTRACT
Mycobacterium tuberculosis and its relatives, like many bacteria, have dynamic cell walls that respond to environmental stresses. Modulation of cell wall metabolism in stress is thought to be responsible for decreased permeability and increased tolerance to antibiotics. The signaling systems that control cell wall metabolism under stress, however, are poorly understood. Here, we examine the cell wall regulatory function of a key cell wall regulator, the serine/threonine phosphatase PstP, in the model organism Mycobacterium smegmatis. We show that the peptidoglycan regulator CwlM is a substrate of PstP. We find that a phosphomimetic mutation, pstP T171E, slows growth, misregulates both mycolic acid and peptidoglycan metabolism in different conditions, and interferes with antibiotic tolerance. These data suggest that phosphorylation on PstP affects its activity against various substrates and is important in the transition between growth and stasis.
IMPORTANCE Regulation of cell wall assembly is essential for bacterial survival and contributes to pathogenesis and antibiotic tolerance in mycobacteria, including pathogens such as Mycobacterium tuberculosis. However, little is known about how the cell wall is regulated in stress. We describe a pathway of cell wall modulation in Mycobacterium smegmatis through the only essential Ser/Thr phosphatase, PstP. We showed that phosphorylation on PstP is important in regulating peptidoglycan metabolism in the transition to stasis and mycolic acid metabolism in growth. This regulation also affects antibiotic tolerance in growth and stasis. This work helps us to better understand the phosphorylation-mediated cell wall regulation circuitry in Mycobacteria.
INTRODUCTION
Tuberculosis (TB), an infectious disease caused by the bacterium Mycobacterium tuberculosis, is one of the leading causes of death from infectious diseases (1). The fact that TB treatment requires at least a six-month regimen with four antibiotics is partly due to the intrinsic antibiotic tolerance of M. tuberculosis (2, 3). Stressed M. tuberculosis cells can achieve a dormant or slow-growing state (4, 5) that exhibits antibiotic tolerance (6), cell wall thickening (7), and altered cell wall staining (4).
The currently accepted cell wall structure of M. tuberculosis (8) is composed of three covalently linked layers (9) as follows: surrounding the plasma membrane, a (i) peptidoglycan (PG) layer is covalently bound to an (ii) arabinogalactan layer; a (iii) lipid layer composed of mycolic acids surrounds the arabinogalactan layer, and the inner leaflet of this layer is covalently linked to the arabinogalactan (10); and the outer leaflet of the mycolic acid layer contains free mycolic acids, trehalose mycolates and other lipids, glycolipids, glycans and proteins (11). The mycolic acid layer, or mycomembrane, is the outer membrane of mycobacteria and is the major contributor to impermeability of the cell wall (12–14).
In addition to serving as a permeability barrier, the regulation of the cell wall likely contributes to antibiotic tolerance either through further changes in permeability (15) or by changing the activity of antibiotic targets (16). Several studies have observed changes in the cell wall under stress (7, 15, 17, 18). These cell wall changes have been shown to correlate with increased antibiotic tolerance (19–21). This has led the prevalent model that stress-induced regulation of the cell wall contributes to antibiotic tolerance (22). While most of the extant data to support this model is correlative, we recently identified a mutant in M. smegmatis that specifically upregulates peptidoglycan metabolism in starvation and also causes decreased antibiotic tolerance in that condition (23). This shows there is a causal relationship between cell wall regulation and antibiotic tolerance, at least in limited conditions in M. smegmatis.
Reversible protein phosphorylation is a key regulatory tool used by bacteria for environmental signal transduction to regulate cell growth (24–27). In M. tuberculosis, serine/threonine phosphorylation is important in cell wall regulation (28). M. tuberculosis has 11 serine/threonine protein kinases (STPKs) (PknA, PknB, and PknD to PknL) and only one serine/threonine protein phosphatase (PstP) (29, 30).
Among the STPKs, PknA and PknB are essential for growth and phosphorylate many substrates involved in cell growth and division (23, 31–34). Some of these substrates are enzymes whose activity is directly altered by phosphorylation. For example, all the enzymes in the fatty acid synthase II (FAS-II) system of mycolic acid biosynthesis are inhibited by threonine phosphorylation (35–40). There are also cell wall regulators that are not enzymes, but whose phosphorylation by STPKs affects cell shape and growth. For example, the regulator CwlM, once it is phosphorylated by PknB, activates MurA (23), the first enzyme in PG precursor biosynthesis (41). In the transition to starvation, CwlM is rapidly dephosphorylated in M. smegmatis (23). Misregulation of MurA activity increases sensitivity to antibiotics in early starvation (23), implying that phosphoregulation of CwlM promotes antibiotic tolerance. CwlM may also regulate other steps of PG synthesis (42). A recent phosphoproteomic study showed that transcriptional repression of the operon that contains both pstP and pknB leads to increased phosphorylation of CwlM (43). While the effects of the individual genes were not separated (43), this suggests that PstP could dephosphorylate CwlM.
PstP is essential in M. tuberculosis and M. smegmatis (44, 45). It is a member of the protein phosphatase 2C (PP2C) subfamily of metal-dependent protein serine/threonine phosphatases (46) that strictly require divalent metal ions for activity (47, 48). PP2C phosphatases are involved in responding to environmental signals, regulating metabolic processes, sporulation, cell growth, division, and stress response in a diverse range of prokaryotes and eukaryotes (49–54). PstPMtb shares structural folds and conserved residues with the human PP2Cα (55), which serves as the representative of the PP2C family. PstPMtb has an N-terminal cytoplasmic enzymatic domain, a transmembrane pass, and a C-terminal extracellular domain (46).
Many of the proteins known to be dephosphorylated by PstP (35, 46, 56–59) are involved in cell wall metabolism; however, the effects of this activity seem to differ. For example, dephosphorylation of CwlM should decrease PG metabolism in stasis (23), but dephosphorylation of the FAS-II enzymes (35–40) should upregulate lipid metabolism in growth. However, PG and lipid metabolism are expected to be coordinated (22). Therefore, PstP must be able to alter substrate specificity in growth and in stasis.
PstPMtb is itself phosphorylated on threonine residues 137, 141, 174, and 290 (56). We hypothesized that phosphorylation of the threonine residues of PstP might help coordinate activity against different substrates through changes in access to substrates, or through toggling catalytic activity against substrates.
We report here that phosphoablative and phosphomimetic mutations at the phosphosite T171 of PstPMsmeg (T174 in PstPMtb) alter growth rate, cell length, cell wall metabolism, and antibiotic tolerance in M. smegmatis. Strains of M. smegmatis with pstP T171E alleles grow slowly, are unable to properly downregulate PG metabolism, and upregulate antibiotic tolerance in the transition to starvation. We observed that the same mutation has nearly opposite effects on mycolic acid layer metabolism. We also report that PstPMtb dephosphorylates CwlMMtb.
RESULTS
Phosphosite T171 of PstPMsmeg impacts growth rate.
PstP is necessary for cell growth in M. smegmatis (43, 44) and phosphorylation increases the activity of PstPMtb against small molecule substrates in vitro (43, 44, 56). To see if the phosphorylations on PstP regulate cell growth, we made M. smegmatis strains with either phosphoablative (T to A) or phosphomimetic (T to E) alleles (60) at each of the three conserved phosphorylation sites of PstPMtb (55, 56) (Fig. 1A) and performed growth curves. We found that biological replicates of the T134A, T134E, T138A, and T138E mutant strains had bimodal distributions of doubling times (Fig. 1B). Phosphosites T134 and T138 in PstPMtb map to the flap subdomain (55) (Fig. 1A). This subdomain varies greatly in sequence and structure across different PP2C family members and has been shown to be important in regulating substrate binding, specificity, and catalytic activity (55, 61–63). Particularly, T138A and T138E variants of the serine/threonine phosphatase tPphA from Thermosynechococcus elongatus showed differences in substrate reactivity (61). This suggests that phosphorylation at T134 and T138 could be very important in regulating the normal activity of PstPMsmeg in the cell. We think the inconsistent doubling times of those strains may result from the formation of suppressor mutants, which we will study in future work.
FIG 1.
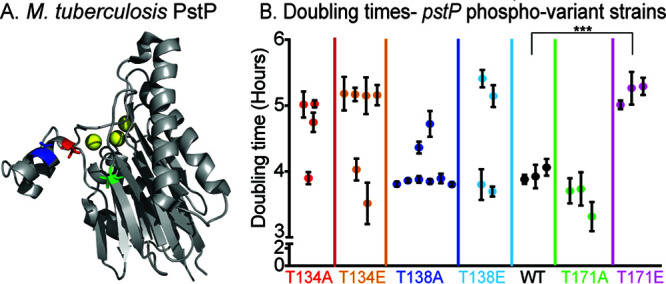
Phosphosite T171 on PstP affects growth. (A) Schematic of the crystal structure of PstP from M. tuberculosis (PstPMtb) (55) (PDB code 1TXO). The threonine (T) sites on PstPMtb phosphorylated by the kinases PknA and PknB (56) are highlighted on the structure as follows: red, PstPMtb T137 (T134 in PstPMsmeg); blue, PstPMtb T141 (T138 in PstPMsmeg); and green, PstPMtb T174 (T171 in PstPMsmeg). (B) Doubling times of strains containing pstPMsmeg WT, phosphoablative mutant alleles pstPMsmeg T134A, T138A, and T171A, and phosphomimetic mutant alleles pstPMsmeg T134E, T138E, and T171E. Each dot is the mean of doubling times from two or three different experiments on different dates of a single isolated clone. The error bars represent the standard deviation. ***, P = 0.0003.
The M. smegmatis strains with pstP T171A and T171E mutations showed consistent and reproducible growth rates (Fig. 1B). The T171A mutants showed no significant difference in doubling time compared to the wild type (WT), but the T171E strain grew more slowly than the wild type. Since T171E mimics constitutive phosphorylation, this result suggests that the continuous presence of a phosphate on T171 downregulates or interferes with cell growth.
PstPMtb WT and PstPMtb T174E dephosphorylate CwlMMtb in vitro.
Only a few substrates of PstP have been biochemically verified, including STPKs PknA and PknB (46, 56, 58, 59), KasA and KasB (35), and EmbR (57). The STPK PknB phosphorylates CwlM, which is an activator of PG biosynthesis (23). CwlM is rapidly dephosphorylated in the transition to starvation in M. smegmatis (23) and becomes hyperphosphorylated when PstP is depleted in M. smegmatis (43), which suggests PstP dephosphorylates CwlM.
To test whether PstP and its T174 (T171 in M. smegmatis) phosphomimetic variant directly dephosphorylate CwlM, we performed an in vitro biochemical assay with purified M. tuberculosis proteins. We purified His-MBP-PknBMtb, His-SUMO-CwlMMtb, and the cytoplasmic region of PstPMtb that has the catalytic domain (His-PstPcWTMtb or PstPcT174EMtb). PstP dephosphorylates itself rapidly (56), so the purified form is unphosphorylated. We phosphorylated His-SUMO-CwlMMtb by His-MBP-PknBMtb, stopped the phosphorylation reaction with calf intestinal phosphatase, and then added His-PstPcWTMtb or PstPcT174EMtb to His-SUMO-CwlMMtb∼P. Our control assay with His-SUMO-CwlM∼P without PstPcWTMtb or PstPcT174EMtb showed that the phosphorylation on the substrate is stable (Fig. 2A, bottom panel). The phosphorylation signal on His-SUMO-CwlMMtb started decreasing within 5 min after addition of His-PstPcWTMtb and kept decreasing over a period of 90 min (Fig. 2A, top panel). This is direct biochemical evidence that the PG-regulator CwlMMtb is a substrate of PstPMtb. We observed that the WT and T174 phosphomimetic forms of PstPMtb have no significant differences in activity against His-SUMO-CwlMMtb∼P in vitro (Fig. 2B).
FIG 2.
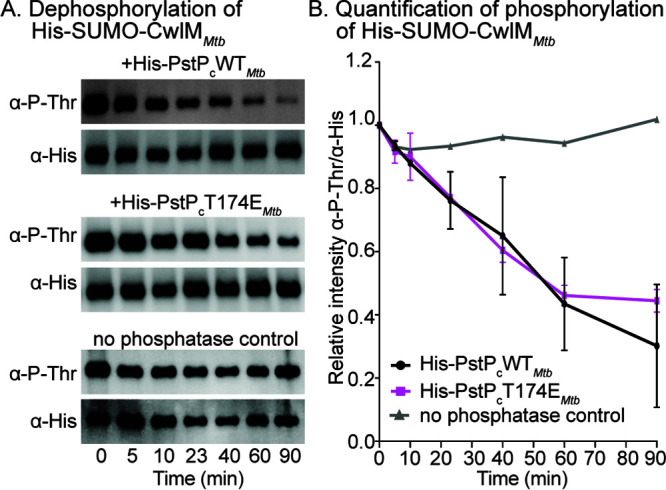
PstPMtb dephosphorylates CwlMMtb. (A) Antiphosphothreonine (anti-P-Thr) and anti-His Western blots of in vitro phosphatase reactions with His-PstPcWTMtb (top panel), His-PstPcT174EMtb (middle panel), and no-phosphatase control (bottom panel) against phosphorylated His-SUMO-CwlMMtb. The assay was performed at least twice with two individually purified batches of each phosphatase; one set of images is shown here. (B) Quantification of relative intensities of anti-P-Thr over anti-His on Western blots. P values were calculated using two-tailed, unpaired t test. All the P values of WT versus T171E at any given time were nonsignificant. P values of WT versus T171E at 5 min = 0.683; 10 min = 0.809; 23 min = 0.934; 40 min = 0.831; 60 min = 0.876; and 90 min = 0.545. The error bars represent standard error of means.
These data show that, in vitro, the activity of the catalytic domain of PstP against a single substrate is not affected by a negative charge on T171Msmeg/T174Mtb. The phenotypes of the full-length pstP T171 phosphoalleles (Fig. 1B, 3, 4 and 5) indicate that these in vitro data do not reflect the full complexity of the regulation of PstP in vivo.
FIG 3.
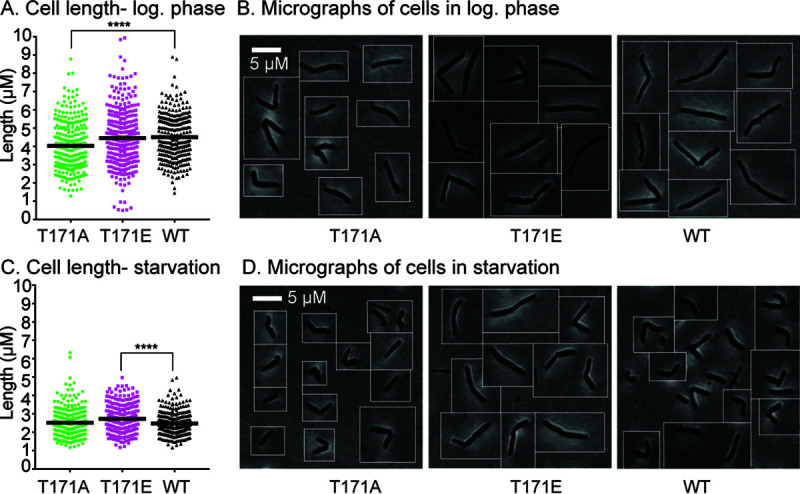
Phosphosite T171 on PstPMsmeg is important in regulating cell length. (A) Quantification of cell lengths of isogenic pstP allele strains (T17A, T171E, and WT) grown in 7H9 medium in log phase. One hundred cells from each of three biological replicates of each pstP allelic variant were measured. P values were calculated by unpaired t test; ****, P = 0.000005. (B) Representative phase images of cells from panel A. (C) Quantification of cell lengths of isogenic pstP allele strains (T17A, T171E, and WT) after starvation in HdB with no glycerol for 5.5 h. One hundred cells from each of three biological replicates of each pstP allelic genotype were measured. P values were calculated by unpaired t test; ****, P = 0.000003. (D) Representative phase images of cells from panel C.
FIG 4.

Phosphosite T171 of PstP alters cell wall staining. (A and B) Quantification of mean intensities of HADA (A) and DMN-Tre (B) signals of pstP allele strains (WT, T17A, and T171E) in log-phase cells, with representative cells below. In panel B, P values of both WT versus T171A and WT versus T171E were 0.000001. (C and D) Quantification of mean intensities of HADA (C) and DMN-Tre (D) signals of starved pstP allele strains (WT, T17A, and T171E) after 5.5 h in HdB with no glycerol, with representative cells below. In panel C, P values of WT versus T171A = 0.0002 and WT versus T171E = 0.000001. (E and F) Intensity profiles of HADA (E) and DMN-Tre (F) signals in cells from pstP allele strains (WT, T17A, and T171E) after starving for 5.5 h in HdB with no glycerol. The shaded regions denote standard deviations and the solid lines represent the mean intensity values. Signal intensities from at least 100 cells from each of three biological replicates of every pstP allelic variant genotype were analyzed in MicrobeJ. In panels A to D, the values of the intensities are represented in percentages of the maximum value of all intensities for all strains either in log phase or during starvation. P values were calculated by a two-tailed, unpaired t test on all 300 values of each pstP allelic variant genotype (WT, T17A, and T171E) using GraphPad Prism (v7.0d).
FIG 5.
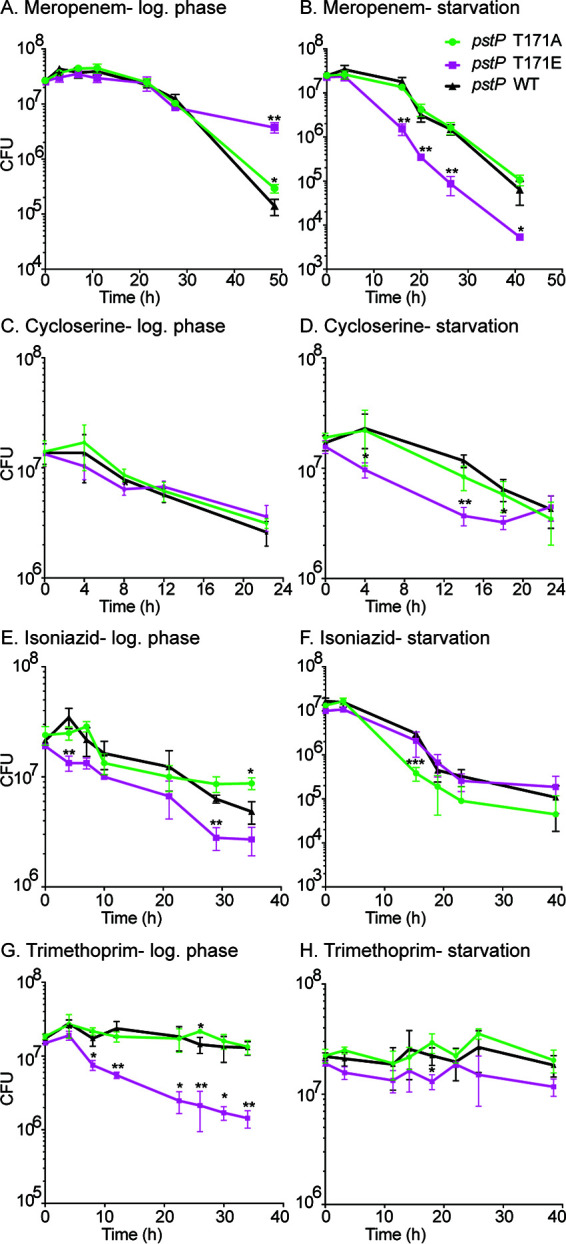
Phosphosite T171 of PstP plays a role in antibiotic sensitivity. Survival of pstP allele strains (WT, T17A, and T171E) in different media and antibiotics. (A) Log-phase culture in 7H9 medium treated with 8 μg/ml of meropenem. P values of WT versus T171E at 3 h = 0.043; WT versus T171A at 48.5 h = 0.001; and WT versus T171E at 48.5 h = 0.018. (B) Starvation culture in HdB medium (no glycerol, 0.05% Tween) for 5.5 h and then treated with 45 μg/ml of meropenem. P values of WT versus T171E at 16 h = 0.003; 23 h = 0.007; 26.3 h = 0.004; and 41 h = 0.046. (C) Log-phase culture in 7H9 medium treated with 100 μg/ml of d-cycloserine. P values of WT versus T171E at 8 h = 0.032. (D) Starvation culture in HdB medium (no glycerol, 0.05% Tween) for 5.5 h and then treated with 900 μg/ml of d-cycloserine. P values of WT versus T171E at 3.5 h = 0.046; 14 h = 0.001; and 18 h = 0.022. (E) Log-phase culture in 7H9 medium treated with 10 μg/ml of isoniazid. P values of WT versus T171E at 4 h = 0.008; WT versus T171E at 29 h = 0.002; and WT versus T171A at 35 h = 0.052. (F) Starvation culture in HdB medium (no glycerol, 0.05% Tween) for 5.5 h and then treated with 90 μg/ml of isoniazid. P values of WT versus T171A at 15.3 h = 0.009. (G) Log-phase culture in 7H9 medium treated with 50 μg/ml of trimethoprim. P values of WT versus T171E at 4 h = 0.023; 8 h = 0.013; 12 h = 0.005; 22.5 h = 0.015; 26 h = 0.005; 30 h = 0.017; and 34 h = 0.002. (H) Starvation culture in HdB medium (no glycerol, 0.05% Tween) for 5.5 h and then treated with 360 μg/ml of trimethoprim. P values of WT versus T171A at 18 h = 0.023. All experiments were done with three biological replicate strains of each pstP allelic variant (T171A, T171E, and WT) at least twice. One representative trial (performed with three biological replicate strains of each pstP allelic variant genotype) is shown. All P values were calculated using two-tailed, unpaired t test. All error bars represent standard deviation (SD).
Phosphosite T171 of PstPMsmeg regulates cell length.
To assess how the phosphosite T171 affects growth, we examined the cell morphology of pstP T171 mutants and wild type (Fig. 3A and B). The quantification of cell length revealed that the pstP T171A cells were shorter (mean = 4.026 ± 0.076) in log phase than the wild-type cells (mean = 4.503 ± 0.07) (Fig. 3A). The pstP T171E strain had cell lengths similar to the wild type (difference between means = 0.053 ± 0.116) (Fig. 3A and B) despite the slower growth (difference between means = −1.037 ± 0.116) (Fig. 1B) in log phase.
PstP could promote the transition from growth to stasis by downregulating the activity of some growth regulatory substrates, such as CwlM, PknA, or PknB, all of which promote growth when phosphorylated (59). PstP likely has dozens of other substrates that may be regulated similarly (23, 43, 46, 56, 59). To test if phosphosite T171 of PstPMsmeg affects the transition to stasis, we transferred the strains from log phase to minimal Hartmans-de Bont (HdB) medium with Tween 80 as the only source of carbon, which leads Msmeg cells to reductively divide (64). We aerated the cultures for 5.5 h before imaging (Fig. 3C and D). The effects of phosphomutations of PstPMsmeg on starved cells were the inverse of what we saw in the log phase. pstPMsmeg T171E cells in starvation were longer than the wild-type and T171A cells, and looked like log phase cells. These data imply that phosphorylation on T171 of PstPMsmeg is involved in cell size regulation upon carbon starvation.
Phosphosite T171 of PstPMsmeg regulates cell wall metabolism.
Since pstPMsmeg T171 seems to play a role in regulating cell length in growth and stasis, we hypothesized that it affects cell wall metabolism in different phases. To test this, we used fluorescent dyes that preferentially stain metabolically active cell wall material (65, 66). We stained T171 allele variant cells from log phase and after 5.5 h of carbon starvation with both the fluorescent d-amino acid HADA, which is incorporated into the PG (65, 67, 68) (Fig. 4A and C; see Fig. S1A and B in the supplemental material) and the fluorescent trehalose DMN-Tre, which stains the mycomembrane (66) (Fig. 4B and D; Fig. S1A and B).
The PG-staining intensity between the strains was the same in log phase (Fig. 4A; Fig. S2A and S3A). In starvation, the pstPMsmeg T171E mutant stained much more brightly with HADA than the other strains (Fig. 4C and E; Fig. S3C). This suggests that phosphorylation on PstPMsmeg T171 may inhibit the downregulation of PG layer biosynthesis in the transition to stasis but that this phosphosite is not important in modulating PG metabolism during rapid growth. The HADA staining is higher at the poles in starved T171E cells compared to T171A and wild-type cells (Fig. 4E; Fig. S3C).
Staining with DMN-Tre showed the inverse pattern to HADA staining. DMN-Tre stains the trehalose mycolates leaflet of the mycomembrane (66). The mean intensities of the pstPMsmeg allelic variants were similar in starvation (Fig. 4D). The intensity profiles of DMN-Tre signal (Fig. 4F; Fig. S3D) showed that the T171E cells had higher DMN-Tre signal at the poles and lower signal at the septal region compared to T171A and wild type. Our HADA and DMN-Tre signal intensity profile analysis in starvation (Fig. 4E and F; Fig. S3C and D) suggests that the T171E cells prioritize elongation, which may account for their greater length in starvation (Fig. 3C and D).
In log phase, however, both mutants showed a significant decrease in DMN-Tre signal compared to the wild type (Fig. 4B; Fig. S2B and S3B), although the pstPMsmeg T171E mutant had the weakest staining. DMN-Tre is incorporated via Ag85-mediated trehalose mycolate metabolism of the mycomembrane (66). Inhibition of cytoplasmic mycolic acid synthesis through isoniazid treatment decreases DMN-Tre staining, and DMN-Tre fluorescence is sensitive to the hydrophobicity of the membrane (66).
Our data (Fig. 4A, C, and F; Fig. S2A and S3A and C) suggest that phosphorylation on PstPMsmeg T171 impacts PG layer metabolism in starvation but not during growth. But the same phosphorylation appears to regulate the trehalose mycolate metabolism in growth but not during starvation (Fig. 4B, D, and F; Fig. S2B and S3B and D).
Phosphosite T171 of PstPMsmeg affects antibiotic tolerance.
Stresses that arrest cell growth in mycobacteria are associated with increased antibiotic tolerance (15, 18, 69–71). We hypothesized that if M. smegmatis fails to downregulate PG synthesis in starvation (Fig. 4C and E; Fig. S3C), then it might be more susceptible to a PG-targeting drug. We treated pstPMsmeg WT, T171A, and T171E strains in log phase and starvation with meropenem, which targets the cross-linking in the PG cell wall (72), and quantified survival by CFU. We saw that the pstPMsmeg T171E strain was more susceptible in starvation, compared to pstPMsmeg T171A and wild-type strains (Fig. 5B), but survived similarly in log phase (Fig. 5A) except at very late time points, when it was more tolerant.
We also treated pstPMsmeg wild-type, T171A, and T171E strains in log phase and starvation with d-cycloserine, which inhibits incorporation of d-alanine into PG pentapeptides in the cytoplasm (73, 74). The results were similar to those in meropenem, where the strains survived similarly in log phase (Fig. 5C) but the pstPMsmeg T171E strain was more sensitive in starvation (Fig. 5D). The apparent failure of the pstPMsmeg T171E strain to downregulate PG synthesis (Fig. 4C and E; Fig. S3C) likely makes it more sensitive to both PG inhibitors in starvation (Fig. 5B and D).
Next, we treated our wild-type and pstPMsmeg T171 mutant strains with isoniazid, which targets InhA in the FAS-II pathway of mycolic acid synthesis (75). We did not see significant differences in isoniazid sensitivity between the strains in starvation (Fig. 5F). In log phase, though, we saw that the pstPMsmeg T171E strain was more susceptible to isoniazid than the pstPMsmeg T171A and the wild-type strains (Fig. 5E). Our data (Fig. 5E) suggest that phosphorylation on PstPMsmeg T171 misregulates the mycolic acid biosynthesis pathway of mycomembrane metabolism (Fig. 4B; Fig. S2B and S3B), thus increasing isoniazid susceptibility.
To see if the PstP T171 phosphosite affects susceptibility to a drug that does not target the cell wall, we treated the strains with trimethoprim, which targets thymidine biosynthesis in the cytoplasm (76). We saw that, in log phase, pstP T171E was very susceptible to this drug (Fig. 5G), while the wild-type and T171A strains were tolerant. All strains were tolerant to trimethoprim in starvation (Fig. 5H). This shows that misregulation of PstP may impact processes beyond cell wall metabolism.
Trimethoprim is a hydrophobic drug which is taken up via passive diffusion (77). Therefore, permeability to trimethoprim is expected to be affected by changes in mycomembrane metabolism in log phase (Fig. 4B and 5E; Fig. S2B and S3B). It is notable that the pstP phosphoallele strains in starvation did not exhibit differences in either DMN-Tre staining (Fig. 4D and F; Fig. S3D) or isoniazid (Fig. 5F) or trimethoprim sensitivity (Fig. 5H), which suggests that susceptibility to trimethoprim could be determined largely by the permeability of the mycomembrane layer. d-Cycloserine, on the other hand, is hydrophilic and therefore its uptake is likely dependent on porins (73, 78), and therefore less sensitive to changes in the mycomembrane. So, sensitivity to d-cycloserine (Fig. 5C and D) appears to be largely dependent on regulation of PG metabolism (Fig. 4A, C, and F; Fig. S2A, S3A and B).
Our data thus show that phosphosite T171 of PstP regulates mycolic acid layer biosynthesis in growth, and PG layer metabolism in starvation. Misregulation of PstP can increase sensitivity to cell wall-targeting drugs in both growth and stasis.
DISCUSSION
Previous studies on mycobacterial phosphoregulation suggest that PstP could play a critical role in modulating cell wall metabolism in the transition between growth and stasis (18, 22, 23, 35, 43, 56, 59, 79). In this work, we explored how the phosphorylation of PstP contributes to this regulation. We report here that the phosphosite T171 of PstPMsmeg impacts growth, cell wall metabolism, and antibiotic tolerance. We found that the PG master regulator CwlMMtb is a substrate of PstPMtb. Our findings indicate that the phosphorylation on PstP affects PG metabolism in stasis and mycolic acid metabolism during growth.
PG is regulated by phosphorylation factors at several points along the biosynthesis pathway (23, 42, 80, 81), mostly by PknB. PknB’s kinase activity is responsive to lipid II that it detects in the periplasm (82). PstP is a global negative regulator of STPK phosphorylation (43) and has been proposed to be the cognate phosphatase of PknB in regulating cell growth (22, 43, 59, 83). Our data suggest that mutations at T171 of PstP do not affect PG metabolism in growth (Fig. 4A; see Fig. S2A and S3A in the supplemental material) but that the PstPMsmeg T171E strain fails to downregulate PG in starvation (Fig. 4C and E; Fig. S3C). We expect that the activity of PstP against the PG regulator CwlM (Fig. 2A, top panel) should be critical for this downregulation because it should deactivate MurA, the first enzyme in PG precursor synthesis (23, 41).
The in vitro biochemistry (Fig. 2A and B) predicts the log-phase staining data (Fig. 4A; Fig. S2A and S3A), where the pstP T171E variant shows no difference in apparent PG activity. The proximity of a phosphosite to the substrate-binding site of an enzyme may affect the catalytic activity directly (84), but T174 maps to the β-sheet core (β8) in PstPMtb, which is distant from the active site (Fig. 1A) (55). The PG staining in starvation (Fig. 4C and E; Fig. S3C) suggests that the PstPMsmeg T171E phosphomimetic variant might dephosphorylate CwlM more slowly in vivo (Fig. 5B and D), but this is not what we see in vitro (Fig. 2A and B). Therefore, it is possible that, in starvation, phosphorylation at this site affects interaction (85) with other regulatory proteins (86–88) that could modulate PstP’s activity against PG substrates, or it could affect access to substrates via localization changes.
Synthesis of the various mycobacterial cell wall layers are likely synchronized (22, 68). PknB almost surely plays a crucial role in connecting PG and mycolic acid metabolism during growth. If PG metabolism is slowed, PknB could sense the accumulation of periplasmic lipid II (82) and signal to halt mycolic acid biosynthesis by inactivating the FAS-II enzymes and the trehalose monomycolate transporter MmpL3 (89) via phosphorylation (35–40, 83). Our data imply that PstP helps balance the effects of these inhibitory phosphorylations to allow coordinated synthesis of mycolic acids in log phase (Fig. 4B and 5E; Fig. S2B and S3B). Misphosphorylation of PstP likely disrupts this coordination and seems to decrease mycolic acid layer metabolism. This may partly explain the slow growth of the pstP T171E mutants (Fig. 1B).
DMN-Tre incorporation into the mycomembrane is directly catalyzed by secreted (90) Ag85 enzymes (66). DMN-Tre fluorescence depends on mycomembrane hydrophobicity and is affected by inhibition of cytoplasmic mycolic acid synthesis (66). Hydrophobicity can be affected by the glycolipid composition of the mycomembrane (91). The differences in DMN-Tre fluorescence that we see (Fig. 4B; Fig. S2B and S3B) could be due to changes in mycolic acid synthesis or due to changes in other glycolipids that affect hydrophobicity (91). Our DMN-Tre data clearly indicate that the trehalose mycolate leaflet of the mycomembrane is affected due to phosphomisregulation of PstP, although it does not yield detailed information about how it is affected.
We propose that PstP’s regulation of mycolic acid layer biosynthesis occurs in the cytoplasm. PstP and all the STPKs work in the cytoplasm, and there are currently no known systems whereby secreted proteins like Ag85 can be regulated by phosphorylation. All the enzymes of the FAS-II complex, which elongates fatty acids into the long lipids used in mycolic acids (75), are downregulated by phosphorylation (35–40), and two are biochemically verified substrates of PstP (35). MmpL3, the mycolic acid flippase (89), is also inhibited by phosphorylation (83). It is likely that PstP could affect the activity of the entire FAS-II complex, including the target of isoniazid, InhA, which is inactivated by threonine phosphorylation (36, 38). Isoniazid is a small hydrophilic drug and undergoes active diffusion via the porins (78, 92); therefore, alterations in mycomembrane permeability are not likely to contribute substantially to differences in isoniazid sensitivity. Although our data (Fig. 5E) do not reveal the exact misregulated spot in the mycolic acid synthesis and transport pathway, the higher susceptibility of the phosphomimetic strain (Fig. 5E) to isoniazid suggests that this metabolic pathway is affected. Our DMN-Tre staining also suggests there should be a balance of the non-phospho- and phosphoforms of PstPMsmeg T171 (Fig. 4B) during growth to regulate mycomembrane biosynthesis.
PstP may dephosphorylate the cell wall substrates directly and/or by deactivating their kinases (93) in both the PG and mycolic acid biosynthesis pathways. All these data combined suggest a complex cross-talk of the STPKs and PstP to regulate diverse cell wall substrates.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All Mycobacterium smegmatis mc2155 ATCC 700084 cultures were started in 7H9 (Becton, Dickinson, Franklin Lakes, NJ) medium containing 5 g/liter bovine serum albumin (BSA), 2 g/liter dextrose, 0.003 g/liter catalase, 0.85 g/liter NaCl, 0.2% glycerol, and 0.05% Tween 80 and incubated at 37°C until log phase. Hartmans-de Bont (HdB) minimal medium made as described previously (94) without glycerol was used for starvation assays. Serial dilutions of all CFU counts were plated on LB Lennox agar (Fisher BP1427-2).
E. coli Top10, XL1-Blue, and Dh5α strains were used for cloning and E. coli strain BL21 Codon Plus was used for protein expression. Antibiotic concentrations for M. smegmatis were 25 μg/ml kanamycin, 50 μg/ml hygromycin, and 20 μg/ml zeocin. Antibiotic concentrations for E. coli were 50 μg/ml kanamycin, 25 μg/ml zeocin, 20 μg/ml chloramphenicol, and 140 μg/ml ampicillin.
Strain construction.
The PstPMsmeg knockdown strain was made first by creating a merodiploid strain and then by deleting the native pstPMsmeg gene from its chromosomal location. The merodiploid strain was generated by introducing a constitutively expressing pstPMtb gene cloned on an StrR plasmid at the L5 attB integration site. The pstPMsmeg gene (MSMEG_0033) at the native locus was then deleted by RecET-mediated double-stranded recombineering approach using a 1.53-kb loxP-hyg-loxP fragment carrying a 125-bp region flanking the pstPMsmeg gene, as described previously (95). The recombineering substrate was generated by two sequential overlapping PCRs of the loxP-hyg-loxP substrate present in the plasmid pKM342. The downstream flanking primer used in the first PCR also carried an optimized mycobacterial ribosome binding site in front of the start codon of MSMEG_0032 to facilitate the expression of the genes present downstream of pstPMsmeg in the M. smegmatis pstP-pknB operon.
Deletion of the pstPMsmeg gene was confirmed by PCR amplification and sequencing of the 5′ and 3′ recombinant junctions, and the absence of an internal wild-type pstPMsmeg PCR product. The pstPMtb allele present at the L5 site was then swapped, as described previously (96), with a Tet-regulatable pstPMtb allele (RevTetR-P750-pstPMtb-DAS tag-L5-Zeo plasmid). The loxP-flanked hyg marker present in the chromosomal locus was then removed by expressing Cre from pCre-sacB-Kan, and the Cre plasmid was subsequently cured from this strain by plating on sucrose. We named this strain CB1175.
Different alleles of pstP were attained by swapping the wild-type (WT) allele at the L5 site of CB1175 as described previously (97). In order to do so, WT and the phosphoablative alleles of pstPMsmeg were first cloned individually into a kanamycin resistance-marked L5 vector pCT94 under the control of a TetO promoter to generate vectors pCB1206-1208 and pCB1210, which would swap out the zeocin resistance-marked vector at the L5 site in CB1175. The strong TetO promoter in the vectors pCB1206-1208 and pCB1210 was swapped with an intermediate-strength promoter p766TetON6 (cloned from the vector pCB1030 [pGMCgS-TetON-6 sspB]) to generate the L5 vectors pCB1282 to 85. pCB1285 was used as the parent vector later on to clone in the phosphomimetic pstPMsmeg alleles under the control of p766TetON6.
These kanamycin resistance-marked vector constructs were then used to swap out the zeocin resistance-marked vector at the L5 site of CB1175 to attain different allelic strains of pstPMsmeg as described previously (97).
Growth curve assay.
At least three biological replicates of different pstPMsmeg allele variants (T171A, T171E, and WT) were grown in 7H9 medium up to log phase. The growth curves were performed in nontreated 96-well plates using a plate reader (BioTek Synergy neo2 multi mode reader) in 200 μl of 7H9 medium starting at an optical density at 600 nm (OD600) of 0.1. An exponential growth equation was used to calculate the doubling times of each strain using the least squared ordinary fit method in GraphPad Prism (version 7.0d). P values were calculated using two-tailed, unpaired t tests.
Cell staining.
Three biological replicate strains of each pstP allelic variant (T171A, T171E, and WT) were used for this assay. For staining cells in log phase, 100 μl of culture in 7H9 medium was incubated at 37°C with 1 μl of 10 mM DMN-Tre for 30 min and 1 μl of 10 mM HADA for 15 min. Cells were then pelleted and resuspended in 1× phosphate-buffered saline (PBS) supplemented with 0.05% Tween 80 and fixed with 10 μl of 16% paraformaldehyde (PFA) for 10 min at room temperature. Cells were then washed and resuspended in PBS plus Tween 80.
For starvation microscopy, cultures were shaken for 4 h in HdB medium without glycerol at 37°C. Aliquots (500 μl) of each culture were pelleted and concentrated to 100 μl, then incubated at 37°C with 1 μl of 10 mM DMN-Tre for 1 h and 3 μl of 10 mM HADA for 30 min. Cells were then washed and fixed as above. The total time of starvation before fixation was 5.5 h.
Microscopy and image analysis.
Cells were imaged with a Nikon Ti-2 widefield epifluorescence microscope with a Photometrics Prime 95B camera and a Plan Apo 100×, 1.45 -numerical-aperture (NA) lens objective. The green fluorescence images for DMN-Tre staining were taken with a 470/40 nm excitation filter and a 525/50 nm emission filter. Blue fluorescence images for HADA staining were taken using a 350/50 nm excitation filter and a 460/50 nm emission filter. All images were captured using NIS Elements software and analyzed using FIJI and MicrobeJ (98). For cell detection in MicrobeJ, appropriate parameters for length, width, and area were set. The V-snapping cells were split at the septum so that each daughter cell could be considered a single cell. Any overlapping cells were excluded from analysis.
Length and mean intensities of HADA and DMN-Tre signals of 300 cells from each of pstPMsmeg T171A, pstPMsmeg T171E, and pstPMsmeg WT (100 cells from each of three biological replicate strains of each genotype) were quantified using MicrobeJ. The values of the mean intensities of 300 cells of each pstP allelic mutant and WT are represented in the graph as percentages of the highest mean intensity from all the cells in that experiment. GraphPad Prism (v7.0d) was used to generate the graphs and perform t tests. To compare two groups (pstPMsmeg T171A versus pstPMsmeg WT and pstPMsmeg T171E versus pstPMsmeg WT), the lengths and the percentage-intensity values of 300 cells per pstP allelic variant genotype (100 per biological replicate strain per genotype) were used as data array inputs to perform unpaired, two-tailed t tests in GraphPad Prism.
Medial intensity profiles of DMN-Tre and NADA signals in cells from different pstP allelic strains in log phase and starvation analyzed with MicrobeJ were plotted on the y axis over relative positions of cells using the “XStatProfile” plotting feature in MicrobeJ to show subcellular localization of fluorescent intensities.
Demographs of DMN-Tre and NADA signal intensity across cell lengths in log phase and starvation were built using the “Demograph” feature of MicrobeJ by plotting the medial intensity profiles of DMN-Tre and NADA signals.
Western blotting.
Cultures were grown in 7H9 medium to OD600 = 0.8 in 10 ml of 7H9 medium, pelleted and resuspended in 500 μl PBS with 1 mM phenylmethylsulfonyl fluoride (PMSF), and lysed (MiniBeadBeater-16, model 607, Biospec). Supernatants from the cell lysates were run on 12% resolving Tris-glycine gels and then transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare). Rabbit anti-Strep-tag II antibody (1:1,000, Abcam, ab76949) in Tris-buffered saline with Tween 20 (TBST) buffer with 0.5% milk and goat anti-rabbit IgG (H+L) horseradish peroxidase (HRP)-conjugated secondary antibody (1:10,000, Thermo Fisher Scientific 31460) in TBST were used to detect PstP–Strep-tag II. For starvation experiments, cultures were first grown to log phase, then starved in HdB no-glycerol medium starting at OD600 = 0.5 for 1.5 h.
For Western blotting of in vitro assays, samples were run on 12% SDS gel (Mini-Protean TGX, Bio-Rad, 4561046) and then transferred onto a PVDF membrane (GE Healthcare). Mouse anti-His antibody (1:1,000, Genscript A00186) in TBST buffer with 0.5% BSA and goat anti-mouse IgG (H+L) HRP-conjugated secondary antibody (1:10,000, Invitrogen A28177) were used to detect His-tagged proteins on the blot. The blots were stripped (Thermo Fisher Scientific, 21059) and reprobed with rabbit antiphosphothreonine antibody (1:1,000, Cell Signaling number 9381) and goat anti-rabbit IgG (H+L) HRP-conjugated secondary antibody (1:10,000, Thermo Fisher Scientific 31460) to detect phosphorylation on the blots.
Antibiotic assays.
Biological triplicates of each pstP allelic variant were used for all antibiotic assays. For antibiotic assays in log phase, log phase cultures were diluted in 7H9 medium to OD600 = 0.05 before treatment. For starvation assays, cells were grown to OD600 = 0.5, pelleted, washed, and resuspended in HdB starvation medium (with no glycerol and 0.05% Tween) at OD600 = 0.3 and incubated at 37°C for a total of 5.5 h. The cultures were then diluted to OD600 = 0.05 before antibiotic treatment. Meropenem (8 μg/ml and 45 μg/ml), isoniazid (10 μg/ml and 90 μg/ml), d-cycloserine (100 μg/ml and 900 μg/ml), and trimethoprim (50 μg/ml and 360 μg/ml) were used separately to treat log-phase and starved cultures, respectively. Samples from the cultures were serially diluted and plated on LB agar before and after treatment, and CFU were calculated.
Protein purification.
All the proteins were expressed using E. coli BL21 Codon Plus cells.
N-terminally His-MBP-tagged PknBMtb was expressed and purified as described previously (80). His-PstPcWTMtb (1 to 300 amino acids of the cytosolic domain [99]) and His-SUMO-CwlMMtb were both expressed overnight by IPTG (isopropyl-β-d-thiogalactopyranoside) induction (1 mM and 1.3 mM, respectively), purified on Ni-nitrilotriacetic acid (Ni-NTA) resin (G-Biosciences, number 786-940 in 5 ml Bio-Scale Mini Cartridges, Bio-Rad number 7324661), then dialyzed, concentrated, and run over size exclusion resin (GE Biosciences Sephacryl S-200 in HiPrep 26/70 column) to obtain soluble proteins. The buffer for His-SUMO-CwlMMtb was 50 mM Tris pH 8, 350 mM NaCl, 1 mM dithiothreitol (DTT), and 10% glycerol. The buffer for His-PstPcWTMtb was 50 mM Tris pH 7.5, 350 mM NaCl, 1 mM DTT, and 10% glycerol. Imidazole (20 mM) was added to each buffer for lysis and application to the Ni-NTA column, and 250 mM imidazole was added for elution. His-PstPcT174EMtb was expressed and purified using the same conditions and buffers used for His-PstPcWTMtb.
In vitro dephosphorylation assay.
Purified His-SUMO-CwlMMtb was phosphorylated with the purified kinase His-MBP-PknBMtb for 1 h at room temperature in the presence of 0.5 mM ATP, 1 mM MnCl2, and buffer (50 mM Tris [pH 7.5], 250 mM NaCl, and 1 mM DTT). The amount of kinase was one-tenth of the amount of substrate in the phosphorylation reaction. To stop the kinase reaction by depleting ATP, 0.434 unit of calf intestinal alkaline phosphatase (Quick CIP; New England BioLabs, MO525S) per μg of His-SUMO-CwlMMtb was added to the reaction mixture and incubated for 1 h at 37°C. The reaction mixture was then divided into five parts for the different phosphatase samples and a control with buffer.
Two individually expressed and purified batches of both His-PstPcWTMtb and His-PstPcT1714EMtb were used as biological replicates to perform the dephosphorylation assay. The reaction was carried out at room temperature for up to 90 min in the presence of phosphatase buffer (50 mM Tris [pH 7.5], 10 mM MnCl2, and 1 mM DTT). The amount of phosphatase used was half the amount of His-SUMO-CwlMMtb.
The intensities of the anti-His and the antiphosphothreonine signals on the blots were quantified with FIJI. The intensities of the anti-His and the antiphosphothreonine signals at each time point were normalized against the respective antibodysignal intensity at 0 min. These relative intensities were used to calculate antiphosphothreonine/anti-His for each time point and the values were plotted over time using GraphPad Prism (version 7.0d).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants 1R15GM131317-01 and R01AI148917-01A1 to C.C.B. from the National Institutes of Health and by startup funds from the University of Texas at Arlington.
We thank Kenan Murphy for the plasmids pDE54MCZD and pKM55 and Dirk Schnappinger for the pDE43-MCS and RevTetR promoter plasmids used in this study.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.World Health Organization. 2019. Global tuberculosis report 2019. https://www.who.int/publications/i/item/global-tuberculosis-report-2019.
- 2.Nguyen L. 2016. Antibiotic resistance mechanisms in M. tuberculosis: an update. Arch Toxicol 90:1585–1604. doi: 10.1007/s00204-016-1727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarlier V, Nikaido H. 1994. Mycobacterial cell wall: structure and role in natural resistance to antibiotics. FEMS Microbiol Lett 123:11–18. doi: 10.1111/j.1574-6968.1994.tb07194.x. [DOI] [PubMed] [Google Scholar]
- 4.Seiler P, Ulrichs T, Bandermann S, Pradl L, Jörg S, Krenn V, Morawietz L, Kaufmann SHE, Aichele P. 2003. Cell-wall alterations as an attribute of Mycobacterium tuberculosis in latent infection. J Infect Dis 188:1326–1331. doi: 10.1086/378563. [DOI] [PubMed] [Google Scholar]
- 5.Muñoz-Elías EJ, Timm J, Botha T, Chan WT, Gomez JE, McKinney JD. 2005. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice. Infect Immun 73:546–551. doi: 10.1128/IAI.73.1.546-551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallis RS, Patil S, Cheon SH, Edmonds K, Phillips M, Perkins MD, Joloba M, Namale A, Johnson JL, Teixeira L, Dietze R, Siddiqi S, Mugerwa RD, Eisenach K, Ellner JJ. 1999. Drug tolerance in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:2600–2606. doi: 10.1128/AAC.43.11.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham AF, Spreadbury CL. 1998. Mycobacterial stationary phase induced by low oxygen tension: cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J Bacteriol 180:801–808. doi: 10.1128/JB.180.4.801-808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 83:91–97. doi: 10.1016/s1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 9.Minnikin DE. 1991. Chemical principles in the organization of lipid components in the mycobacterial cell envelope. Res in Microbiologoy 142:423–427. doi: 10.1016/0923-2508(91)90114-P. [DOI] [PubMed] [Google Scholar]
- 10.Kieser KJ, Rubin EJ. 2014. How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol 12:550–562. doi: 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marrakchi H, Lanéelle M-A, Daffé M. 2014. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol 21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Hett EC, Rubin EJ. 2008. Bacterial growth and cell division: a mycobacterial perspective. Microbiol Mol Biol Rev 72:126–156. doi: 10.1128/MMBR.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc Natl Acad Sci U S A 105:3963–3967. doi: 10.1073/pnas.0709530105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarlier V, Nikaido H. 1990. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J Bacteriol 172:1418–1423. doi: 10.1128/jb.172.3.1418-1423.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarathy J, Dartois V, Dick T, Gengenbacher M. 2013. Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 57:1648–1653. doi: 10.1128/AAC.02202-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batt SM, Burke CE, Moorey AR, Besra GS. 2020. Antibiotics and resistance: the two-sided coin of the mycobacterial cell wall. Cell Surf 6:100044. doi: 10.1016/j.tcsw.2020.100044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhamidi S, Shi L, Chatterjee D, Belisle JT, Crick DC, McNeil MR. 2012. A bioanalytical method to determine the cell wall composition of Mycobacterium tuberculosis grown in vivo. Anal Biochem 421:240–249. doi: 10.1016/j.ab.2011.10.046. [DOI] [PubMed] [Google Scholar]
- 18.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 19.Sarathy JP, Via LE, Weiner D, Blanc L, Boshoff H, Eugenin EA, Clifton BE III, Dartois VA. 2017. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob Agents Chemother 62:e02266-17. doi: 10.1128/AAC.02266-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Z, Siddiqi N, Rubin EJ. 2005. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother 49:4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Tan S, Huang L, Abramovitch RB, Rohde KH, Zimmerman MD, Chen C, Dartois V, VanderVen BC, Russell DG. 2016. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med 213:809–825. doi: 10.1084/jem.20151248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dulberger CL, Rubin EJ, Boutte CC. 2020. The mycobacterial cell envelope—a moving target. Nat Rev Microbiol 18:47–59. doi: 10.1038/s41579-019-0273-7. [DOI] [PubMed] [Google Scholar]
- 23.Boutte CC, Baer CE, Papavinasasundaram K, Liu W, Chase MR, Meniche X, Fortune SM, Sassetti CM, Ioerger TR, Rubin EJ, Laub M. 2016. A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. Elife 5:e14590. doi: 10.7554/eLife.14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Echenique J, Kadioglu A, Romao S, Andrew PW, Trombe MC. 2004. Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect Immun 72:2434–2437. doi: 10.1128/iai.72.4.2434-2437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juris SJ, Rudolph AE, Huddler D, Orth K, Dixon JE. 2000. A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc Natl Acad Sci U S A 97:9431–9436. doi: 10.1073/pnas.170281997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galyov EE, Håkansson S, Forsberg Å, Wolf-Watz H. 1993. A secreted protein kinase of Yersinia pseudotuberculosis is an indispensable virulence determinant. Nature 361:730–732. doi: 10.1038/361730a0. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Li C, Yang H, Mushegian A, Jin S. 1998. A novel serine/threonine protein kinase homologue of Pseudomonas aeruginosa is specifically inducible within the host infection site and is required for full virulence in neutropenic mice. J Bacteriol 180:6764–6768. doi: 10.1128/JB.180.24.6764-6768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gee CL, Papavinasasundaram KG, Blair SR, Baer CE, Falick AM, King DS, Griffin JE, Venghatakrishnan H, Zukauskas A, Wei JR, Dhiman RK, Crick DC, Rubin EJ, Sassetti CM, Alber T. 2012. A phosphorylated pseudokinase complex controls cell wall synthesis in Mycobacteria. Sci Signal 5:ra7. doi: 10.1126/scisignal.2002525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Av-Gay Y, Everett M. 2000. The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol 8:238–244. doi: 10.1016/s0966-842x(00)01734-0. [DOI] [PubMed] [Google Scholar]
- 30.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 31.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 32.Kang C-M, Abbott DW, Park ST, Dascher CC, Cantley LC, Husson RN. 2005. The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev 19:1692–1704. doi: 10.1101/gad.1311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez P, Saint-Joanis B, Barilone N, Jackson M, Gicquel B, Cole ST, Alzari PM. 2006. The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol 188:7778–7784. doi: 10.1128/JB.00963-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kusebauch U, Ortega C, Ollodart A, Rogers RS, Sherman DR, Moritz RL, Grundner C. 2014. Mycobacterium tuberculosis supports protein tyrosine phosphorylation. Proc Natl Acad Sci U S A 111:9265–9270. doi: 10.1073/pnas.1323894111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Molle V, Brown AK, Besra GS, Cozzone AJ, Kremer L. 2006. The Condensing Activities of the Mycobacterium tuberculosis type II fatty acid synthase are differentially regulated by phosphorylation. J Biol Chem 281:30094–30103. doi: 10.1074/jbc.M601691200. [DOI] [PubMed] [Google Scholar]
- 36.Molle V, Gulten G, Vilchèze C, Veyron-Churlet R, Zanella-Cléon I, Sacchettini JC, Jacobs WR Jr., Kremer L. 2010. Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol Microbiol 78:1591–1605. doi: 10.1111/j.1365-2958.2010.07446.x. [DOI] [PubMed] [Google Scholar]
- 37.Slama N, Leiba J, Eynard N, Daffé M, Kremer L, Quémard A, Molle V. 2011. Negative regulation by Ser/Thr phosphorylation of HadAB and HadBC dehydratases from Mycobacterium tuberculosis type II fatty acid synthase system. Biochem Biophys Res Commun 412:401–406. doi: 10.1016/j.bbrc.2011.07.051. [DOI] [PubMed] [Google Scholar]
- 38.Khan S, Nagarajan SN, Parikh A, Samantaray S, Singh A, Kumar D, Roy RP, Bhatt A, Nandicoori VK. 2010. Phosphorylation of enoyl-acyl carrier protein reductase InhA impacts mycobacterial growth and survival. J Biol Chem 285:37860–37871. doi: 10.1074/jbc.M110.143131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilchèze C, Molle V, Carrère-Kremer S, Leiba J, Mourey L, Shenai S, Baronian G, Tufariello J, Hartman T, Veyron-Churlet R, Trivelli X, Tiwari S, Weinrick B, Alland D, Guérardel Y, Jacobs WR, Kremer L. 2014. Phosphorylation of KasB regulates virulence and acid-fastness in Mycobacterium tuberculosis. PLoS Pathog 10:e1004115. doi: 10.1371/journal.ppat.1004115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veyron-Churlet R, Zanella-Cléon I, Cohen-Gonsaud M, Molle V, Kremer L. 2010. Phosphorylation of the Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein reductase MabA regulates mycolic acid biosynthesis. J Biol Chem 285:12714–12725. doi: 10.1074/jbc.M110.105189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marquardt JL, Siegele DA, Kolter R, Walsh CT. 1992. Cloning and sequencing of Escherichia coli murZ and purification of its product, a UDP-N-acetylglucosamine enolpyruvyl transferase. J Bacteriol 174:5748–5752. doi: 10.1128/jb.174.17.5748-5752.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turapov O, Forti F, Kadhim B, Ghisotti D, Sassine J, Straatman-Iwanowska A, Bottrill AR, Moynihan PJ, Wallis R, Barthe P, Cohen-Gonsaud M, Ajuh P, Vollmer W, Mukamolova GV. 2018. Two faces of CwlM, an essential PknB substrate, in Mycobacterium tuberculosis. Cell Rep 25:57–67. doi: 10.1016/j.celrep.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iswahyudi Mukamolova GV, Straatman-Iwanowska AA, Allcock N, Ajuh P, Turapov O, O'Hare HM. 2019. Mycobacterial phosphatase PstP regulates global serine threonine phosphorylation and cell division. Sci Rep 9:8337. doi: 10.1038/s41598-019-44841-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma AK, Arora D, Singh LK, Gangwal A, Sajid A, Molle V, Singh Y, Nandicoori VK. 2016. Serine/threonine protein phosphatase PstP of Mycobacterium tuberculosis is necessary for accurate cell division and survival of pathogen. J Biol Chem 291:24215–24230. doi: 10.1074/jbc.M116.754531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8:e02133-16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chopra P, Singh B, Singh R, Vohra R, Koul A, Meena LS, Koduri H, Ghildiyal M, Deol P, Das TK, Tyagi AK, Singh Y. 2003. Phosphoprotein phosphatase of Mycobacterium tuberculosis dephosphorylates serine-threonine kinases PknA and PknB. Biochem Biophys Res Commun 311:112–120. doi: 10.1016/j.bbrc.2003.09.173. [DOI] [PubMed] [Google Scholar]
- 47.Barford D. 1996. Molecular mechanisms of the protein serine/threonine phosphatases. Trends Biochem Sci 21:407–412. doi: 10.1016/s0968-0004(96)10060-8. [DOI] [PubMed] [Google Scholar]
- 48.Cohen P. 1989. The structure and regulation of protein phosphatases. Annu Rev Biochem 58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- 49.Mougous JD, Gifford CA, Ramsdell TL, Mekalanos JJ. 2007. Threonine phosphorylation post-translationally regulates protein secretion in Pseudomonas aeruginosa. Nat Cell Biol 9:797–803. doi: 10.1038/ncb1605. [DOI] [PubMed] [Google Scholar]
- 50.Irmler A, Forchhammer K. 2001. A PP2C-type phosphatase dephosphorylates the PII signaling protein in the cyanobacterium Synechocystis PCC 6803. Proc Natl Acad Sci U S A 98:12978–12983. doi: 10.1073/pnas.231254998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bradshaw N, Losick R. 2015. Asymmetric division triggers cell-specific gene expression through coupled capture and stabilization of a phosphatase. Elife 4:e08145. doi: 10.7554/eLife.08145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bradshaw N, Levdikov VM, Zimanyi CM, Gaudet R, Wilkinson AJ, Losick R. 2017. A widespread family of serine/threonine protein phosphatases shares a common regulatory switch with proteasomal proteases. Elife 6:e26111. doi: 10.7554/eLife.26111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu G, Wang Y. 2008. Functional diversity of mammalian type 2C protein phosphatase isoforms: new tales from an old family. Clin Exp Pharmacol Physiol 35:107–112. doi: 10.1111/j.1440-1681.2007.04843.x. [DOI] [PubMed] [Google Scholar]
- 54.Vijay K, Brody MS, Fredlund E, Price CW. 2000. A PP2C phosphatase containing a PAS domain is required to convey signals of energy stress to the sigmaB transcription factor of Bacillus subtilis. Mol Microbiol 35:180–188. doi: 10.1046/j.1365-2958.2000.01697.x. [DOI] [PubMed] [Google Scholar]
- 55.Pullen KE, Ng H-L, Sung P-Y, Good MC, Smith SM, Alber T. 2004. An alternate conformation and a third metal in PstP/Ppp, the M. tuberculosis PP2C-family Ser/Thr protein phosphatase. Structure 12:1947–1954. doi: 10.1016/j.str.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Sajid A, Arora G, Gupta M, Upadhyay S, Nandicoori VK, Singh Y. 2011. Phosphorylation of Mycobacterium tuberculosis Ser/Thr phosphatase by PknA and PknB. PLoS One 6:e0017871. doi: 10.1371/journal.pone.0017871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma K, Gupta M, Krupa A, Srinivasan N, Singh Y. 2006. EmbR, a regulatory protein with ATPase activity, is a substrate of multiple serine/threonine kinases and phosphatase in Mycobacterium tuberculosis. FEBS J 273:2711–2721. doi: 10.1111/j.1742-4658.2006.05289.x. [DOI] [PubMed] [Google Scholar]
- 58.Durán R, Villarino A, Bellinzoni M, Wehenkel A, Fernandez P, Boitel B, Cole ST, Alzari PM, Cerveñansky C. 2005. Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem Biophys Res Commun 333:858–867. doi: 10.1016/j.bbrc.2005.05.173. [DOI] [PubMed] [Google Scholar]
- 59.Boitel B, Ortiz-Lombardía M, Durán R, Pompeo F, Cole ST, Cerveñansky C, Alzari PM. 2003. PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol Microbiol 49:1493–1508. doi: 10.1046/j.1365-2958.2003.03657.x. [DOI] [PubMed] [Google Scholar]
- 60.Cottin V, Van Linden A, Riches DW. 1999. Phosphorylation of tumor necrosis factor receptor CD120a (p55) by p42(mapk/erk2) induces changes in its subcellular localization. J Biol Chem 274:32975–32987. doi: 10.1074/jbc.274.46.32975. [DOI] [PubMed] [Google Scholar]
- 61.Su J, Forchhammer K. 2013. Determinants for substrate specificity of the bacterial PP2C protein phosphatase tPphA from Thermosynechococcus elongatus. FEBS J 280:694–707. doi: 10.1111/j.1742-4658.2011.08466.x. [DOI] [PubMed] [Google Scholar]
- 62.Greenstein AE, Grundner C, Echols N, Gay LM, Lombana TN, Miecskowski CA, Pullen KE, Sung P-Y, Alber T. 2005. Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J Mol Microbiol Biotechnol 9:167–181. doi: 10.1159/000089645. [DOI] [PubMed] [Google Scholar]
- 63.Schlicker C, Fokina O, Kloft N, Grüne T, Becker S, Sheldrick GM, Forchhammer K. 2008. Structural analysis of the PP2C phosphatase tPphA from Thermosynechococcus elongatus: a flexible flap subdomain controls access to the catalytic site. J Mol Biol 376:570–581. doi: 10.1016/j.jmb.2007.11.097. [DOI] [PubMed] [Google Scholar]
- 64.Wu M-L, Gengenbacher M, Dick T. 2016. Mild nutrient starvation triggers the development of a small-cell survival morphotype in Mycobacteria. Front Microbiol 7:947. doi: 10.3389/fmicb.2016.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, VanNieuwenhze MS. 2012. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl 51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamariza M, Shieh P, Ealand CS, Peters JS, Chu B, Rodriguez-Rivera FP, Babu Sait MR, Treuren WV, Martinson N, Kalscheuer R, Kana BD, Bertozzi CR. 2018. Rapid detection of Mycobacterium tuberculosis in sputum with a solvatochromic trehalose probe. Sci Transl Med 10:eaam6310. doi: 10.1126/scitranslmed.aam6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baranowski C, Welsh MA, Sham L-T, Eskandarian HA, Lim HC, Kieser KJ, Wagner JC, McKinney JD, Fantner GE, Ioerger TR, Walker S, Bernhardt TG, Rubin EJ, Rego EH. 2018. Maturing Mycobacterium smegmatis peptidoglycan requires non-canonical crosslinks to maintain shape. Elife 7:e37516. doi: 10.7554/eLife.37516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.García-Heredia A, Pohane AA, Melzer ES, Carr CR, Fiolek TJ, Rundell SR, Lim HC, Wagner JC, Morita YS, Swarts BM, Siegrist MS. 2018. Peptidoglycan precursor synthesis along the sidewall of pole-growing mycobacteria. Elife 7:e37243. doi: 10.7554/eLife.37243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deb C, Lee C-M, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, Pawar S, Rogers L, Kolattukudy PE. 2009. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y. 2004. Persistent and dormant tubercle bacilli and latent tuberculosis. Front Biosci 9:1136–1156. doi: 10.2741/1291. [DOI] [PubMed] [Google Scholar]
- 71.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64:2062–2069. doi: 10.1128/IAI.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cordillot M, Dubee V, Triboulet S, Dubost L, Marie A, Hugonnet JE, Arthur M, Mainardi JL. 2013. In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L,D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob Agents Chemother 57:5940–5945. doi: 10.1128/AAC.01663-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neuhaus FC, Hammes WP. 1981. Inhibition of cell wall biosynthesis by analogues and alanine. Pharmacol Ther 14:265–319. doi: 10.1016/0163-7258(81)90030-9. [DOI] [PubMed] [Google Scholar]
- 74.Feng Z, Barletta RG. 2003. Roles of Mycobacterium smegmatis D-alanine:D-alanine ligase and D-alanine racemase in the mechanisms of action of and resistance to the peptidoglycan inhibitor D-cycloserine. Antimicrob Agents Chemother 47:283–291. doi: 10.1128/aac.47.1.283-291.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marrakchi H, Lanéelle G, Quémard AK. 2000. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology (Reading) 146:289–296. doi: 10.1099/00221287-146-2-289. [DOI] [PubMed] [Google Scholar]
- 76.Brogden RN, Carmine AA, Heel RC, Speight TM, Avery GS. 1982. Trimethoprim: a review of its antibacterial activity, pharmacokinetics and therapeutic use in urinary tract infections. Drugs 23:405–430. doi: 10.2165/00003495-198223060-00001. [DOI] [PubMed] [Google Scholar]
- 77.Hancock RE, Bell A. 1988. Antibiotic uptake into gram-negative bacteria. Eur J Clin Microbiol Infect Dis 7:713–720. doi: 10.1007/BF01975036. [DOI] [PubMed] [Google Scholar]
- 78.Stephan J, Mailaender C, Etienne G, Daffé M, Niederweis M. 2004. Multidrug resistance of a porin deletion mutant of Mycobacterium smegmatis. Antimicrob Agents Chemother 48:4163–4170. doi: 10.1128/AAC.48.11.4163-4170.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ortega C, Liao R, Anderson LN, Rustad T, Ollodart AR, Wright AT, Sherman DR, Grundner C. 2014. Mycobacterium tuberculosis Ser/Thr protein kinase B mediates an oxygen-dependent replication switch. PLoS Biol 12:e1001746. doi: 10.1371/journal.pbio.1001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kieser KJ, Boutte CC, Kester JC, Baer CE, Barczak AK, Meniche X, Chao MC, Rego EH, Sassetti CM, Fortune SM, Rubin EJ. 2015. Phosphorylation of the peptidoglycan synthase PonA1 governs the rate of polar elongation in Mycobacteria. PLoS Pathog 11:e1005010. doi: 10.1371/journal.ppat.1005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arora D, Chawla Y, Malakar B, Singh A, Nandicoori VK. 2018. The transpeptidase PbpA and noncanonical transglycosylase RodA of Mycobacterium tuberculosis play important roles in regulating bacterial cell lengths. J Biol Chem 293:6497–6516. doi: 10.1074/jbc.M117.811190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaur P, Rausch M, Malakar B, Watson U, Damle NP, Chawla Y, Srinivasan S, Sharma K, Schneider T, Jhingan GD, Saini D, Mohanty D, Grein F, Nandicoori VK. 2019. LipidII interaction with specific residues of Mycobacterium tuberculosis PknB extracytoplasmic domain governs its optimal activation. Nat Commun 10:1231. doi: 10.1038/s41467-019-09223-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Le N-H, Locard-Paulet M, Stella A, Tomas N, Molle V, Burlet-Schiltz O, Daffé M, Marrakchi H. 2020. The protein kinase PknB negatively regulates biosynthesis and trafficking of mycolic acids in mycobacteria. J Lipid Res 61:1180–1191. doi: 10.1194/jlr.RA120000747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veyron-Churlet R, Molle V, Taylor RC, Brown AK, Besra GS, Zanella-Cléon I, Fütterer K, Kremer L. 2009. The Mycobacterium tuberculosis beta-ketoacyl-acyl carrier protein synthase III activity is inhibited by phosphorylation on a single threonine residue. J Biol Chem 284:6414–6424. doi: 10.1074/jbc.M806537200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li K-K, Qu D-H, Zhang H-N, Chen F-Y, Xu L, Wang M-Y, Su H-Y, Tao S-C, Wu F-L. 2020. Global discovery the PstP interactions using Mtb proteome microarray and revealing novel connections with EthR. J Proteomics 215:103650. doi: 10.1016/j.jprot.2020.103650. [DOI] [PubMed] [Google Scholar]
- 86.Bollen M, Peti W, Ragusa MJ, Beullens M. 2010. The extended PP1 toolkit: designed to create specificity. Trends Biochem Sci 35:450–458. doi: 10.1016/j.tibs.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roy J, Cyert MS. 2009. Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal 2:re9. doi: 10.1126/scisignal.2100re9. [DOI] [PubMed] [Google Scholar]
- 88.Lin K, Hwang PK, Fletterick RJ. 1997. Distinct phosphorylation signals converge at the catalytic center in glycogen phosphorylases. Structure 5:1511–1523. doi: 10.1016/s0969-2126(97)00300-6. [DOI] [PubMed] [Google Scholar]
- 89.Xu Z, Meshcheryakov VA, Poce G, Chng S-S. 2017. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc Natl Acad Sci U S A 114:7993–7998. doi: 10.1073/pnas.1700062114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harth G, Lee BY, Wang J, Clemens DL, Horwitz MA. 1996. Novel insights into the genetics, biochemistry, and immunocytochemistry of the 30-kilodalton major extracellular protein of Mycobacterium tuberculosis. Infect Immun 64:3038–3047. doi: 10.1128/IAI.64.8.3038-3047.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Viljoen A, Viela F, Kremer L, Dufrêne YF. 2020. Fast chemical force microscopy demonstrates that glycopeptidolipids define nanodomains of varying hydrophobicity on mycobacteria. Nanoscale Horiz 5:944–953. doi: 10.1039/c9nh00736a. [DOI] [PubMed] [Google Scholar]
- 92.Mailaender C, Reiling N, Engelhardt H, Bossmann S, Ehlers S, Niederweis M. 2004. The MspA porin promotes growth and increases antibiotic susceptibility of both Mycobacterium bovis BCG and Mycobacterium tuberculosis. Microbiology (Reading) 150:853–864. doi: 10.1099/mic.0.26902-0. [DOI] [PubMed] [Google Scholar]
- 93.Bhaskara GB, Wong MM, Verslues PE. 2019. The flip side of phospho-signalling: regulation of protein dephosphorylation and the protein phosphatase 2Cs. Plant Cell Environ 42:2913–2930. doi: 10.1111/pce.13616. [DOI] [PubMed] [Google Scholar]
- 94.Hartmans S, De Bont J. 1992. The genus Mycobacterium—nonmedical In Balows A. (ed), The prokaryotes, 2nd edition, Springer-Verlag, New York, NY. [Google Scholar]
- 95.Murphy KC, Papavinasasundaram K, Sassetti CM. 2015. Mycobacterial recombineering. Methods Mol Biol 1285:177–199. doi: 10.1007/978-1-4939-2450-9_10. [DOI] [PubMed] [Google Scholar]
- 96.Schnappinger D, O'Brien KM, Ehrt S. 2015. Construction of conditional knockdown mutants in mycobacteria. Methods Mol Biol 1285:151–175. doi: 10.1007/978-1-4939-2450-9_9. [DOI] [PubMed] [Google Scholar]
- 97.Pashley CA, Parish T. 2003. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol Lett 229:211–215. doi: 10.1016/S0378-1097(03)00823-1. [DOI] [PubMed] [Google Scholar]
- 98.Ducret A, Quardokus EM, Brun YV. 2016. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat Microbiol 1:671–677. doi: 10.1038/nmicrobiol.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gupta M, Sajid A, Arora G, Tandon V, Singh Y. 2009. Forkhead-associated domain-containing protein Rv0019c and polyketide-associated protein PapA5, from substrates of serine/threonine protein kinase PknB to interacting proteins of Mycobacterium tuberculosis. J Biol Chem 284:34723–34734. doi: 10.1074/jbc.M109.058834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.