

Abstract
A novel and efficient route for the preparation of (2S)-2-chloro-2-fluorolactone 29 is described. This approach takes advantage of a highly efficient diastereoselective electrophilic fluorination reaction (94% yield; >50:1 dr)
Graphical Abstract
INTRODUCTION
Hepatitis C virus (HCV) a flavivirus that causes significant morbidity and mortality, affects at least 71 million people worldwide.1 Globally, this major public health concern is responsible for approximately 27% of liver cirrhosis and around 25% of hepatocellular carcinoma.2 Since the discovery of HCV in 1989, several antiviral drug regimens have been developed.3 Until 2011, subcutaneous pegylated interferon alfa and oral ribavirin were used as the main HCV treatment. Since then, the addition of protease inhibitors (PIs) and the development of interferon-free therapies considerably increased the rates of treatment tolerability and sustained virological response. Further improvement was recently achieved with Epclusa, the first all-oral, pan-genotypic, single tablet regimen for chronic HCV infection (combination of NS5B inhibitor sofosbuvir and NS5A inhibitor velpatasvir).4 Despite these advances, new potent pan-genotypic nucleosides analogues that can reduce treatment duration (~12 weeks) are needed. Recently, our group and other laboratories paid special attention to the (S)-2′-chloro-2′-fluoro-deoxyribouridine prodrug 1 (Figure 1)5 which displays high pan-genotypic potency against HCV in subgenomic 1a replicon assays (EC50 = 0.1 μM).5d This prodrug was rapidly metabolized in human hepatocytes to its active metabolite, 2′-chloro-2′-fluoro-deoxyribonucleoside 5′-triphosphate, which inhibits the NS5B polymerase by competing with natural uridine triphosphate. With a sofosbuvir-like C2′-exopuckered sugar, this prodrug also exhibited a favorable resistance and safety profile which makes it a potential candidate for clinical development.
Figure 1.
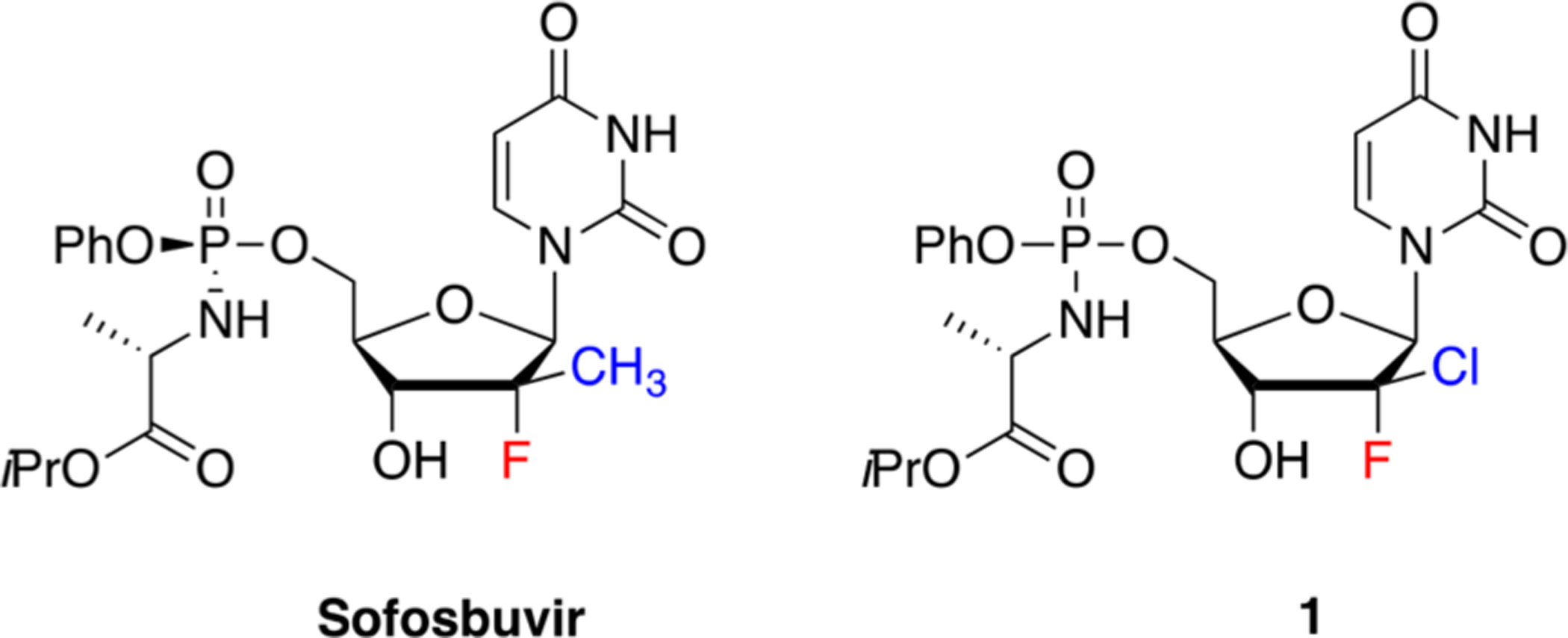
Structure of sofosbuvir and novel 2′-Cl,2′-F nucleoside analogue 1.
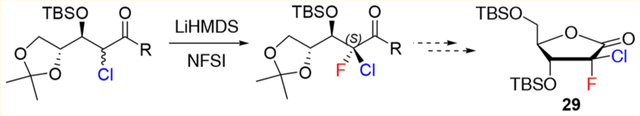
The synthesis of this nucleoside analogue is particularly challenging due to the unusual (2′S)-Cl,F substitution on the sugar moiety. One of the key intermediates required for the synthesis of 1 is lactone 4, which can be prepared in four steps from 2-deoxy-d-ribose (Scheme 1a).5d,6 However, the reported synthesis is hampered by the lack of stereoselectivity of the fluorination/chlorination sequence. These halogenation reactions also suffer from undesired side reactions such as β-elimination6 or overfluorination, resulting in low yields and difficult chromatographic separations.
Scheme 1.
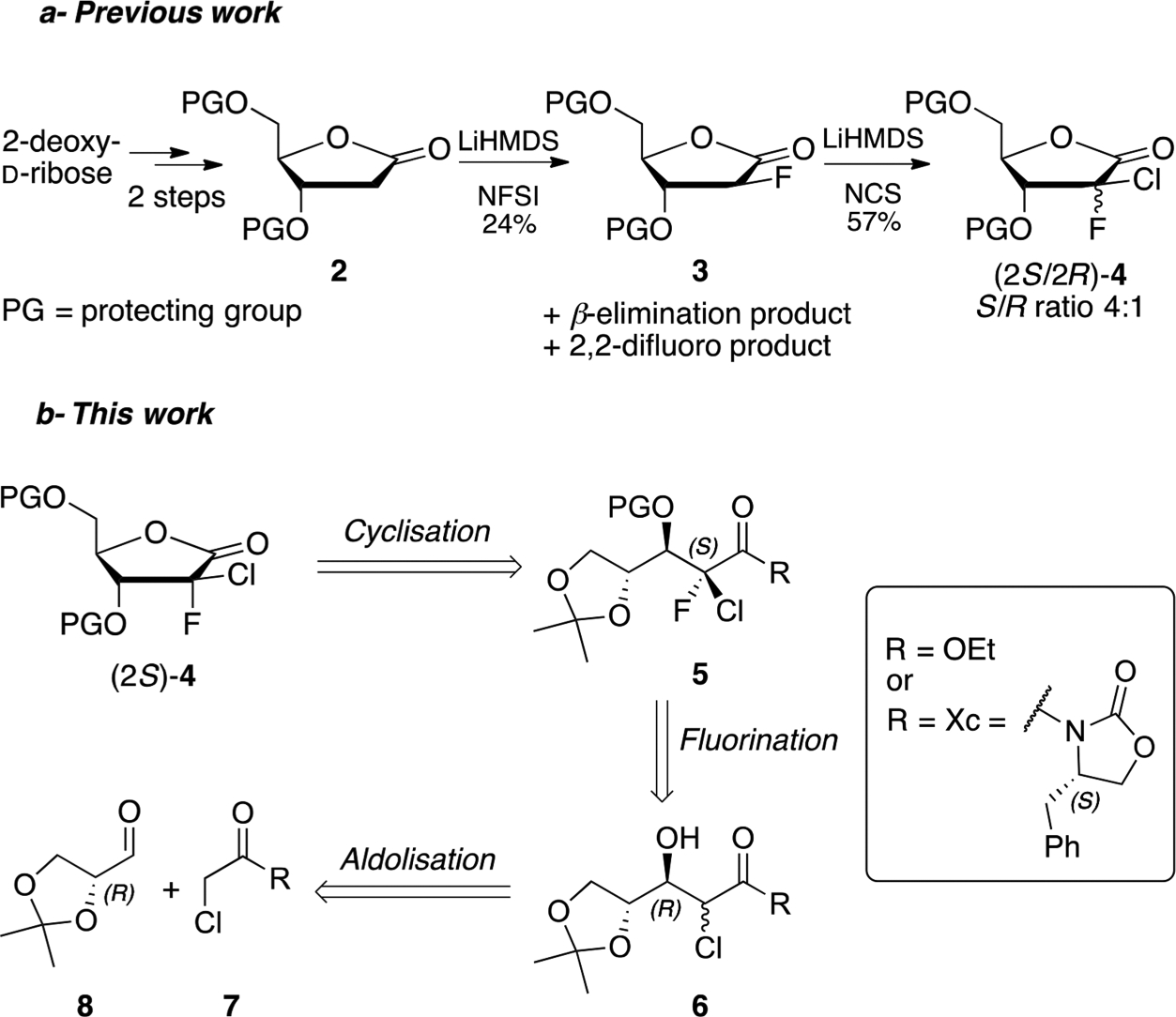
Reported Synthesis and Retrosynthetic Approach for the Preparation of (2S)-2-Chloro-2-fluororibolactone 4
Modified riboside moieties are generally prepared from natural sugars such as ribose or 2′-deoxyribose but also from d-glyceraldehyde via Reformatsky, Wittig, or aldol reactions. The latter approach was successfully used for the synthesis of several 2′-modified nucleosides such as sofosbuvir7 or gemcitabine.8 Regarding the formation of gem-Cl-F-carbons, there are only a few examples in the literature and most of them are synthesized via electrophilic fluorination of an α-chloro carbonyl compound.9 Partial stereoselectivity is usually achieved by using chiral transition metal complexes (Ni, Pd, Ti)10 with or without organocatalysts such as spiro oxazoline ligands,11 cinchona alkaloid esters,12 N-acyloxazolidinones,13 and the Jørgensen catalyst.14 It is worth mentioning that there are only a few examples reporting the stereoselective electrophilic chlorination of α-fluoro carbonyl compounds.14a
Herein we report an alternative route for the preparation of the (S)-2-chloro-2-fluorolactone intermediate 4 using stereoselective aldolization and fluorination reactions (Scheme 1b). This strategy allows us to introduce directly the chlorine atom during the aldol step and therefore avoid an additional chlorination step. Using this approach, we expect the stereochemistry of the C3 hydroxyl group to be controlled by the C4 chiral center, mostly favoring a 3,4-trans configuration. Also, according to literature precedents, the use of (S)-4-benzyloxazolidinone (Xc) as chiral auxiliary could potentially improve the chiral induction toward the desired (3R) configuration.15
RESULTS AND DISCUSSION
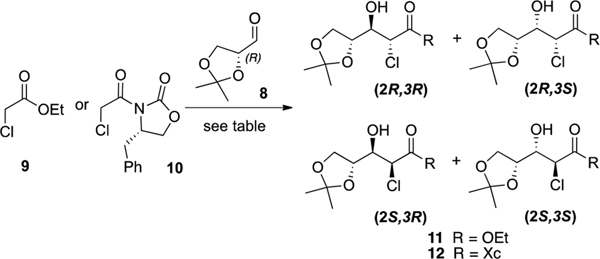
Initially, we investigated the aldolization between ethyl chloroacetate 9 and glyceraldehyde 8 (Table 1). The reaction of a lithium enolate of 9, generated by using either LiHMDS or LDA, gave the aldol product 11 in moderate yields as a mixture of the four possible diastereomers (entries 1 and 2).16 The yield increased to 62% when we used a boron enolate but, as mentioned earlier, no apparent diastereoselectivity was observed (entry 4).17 On the other hand, the use of a titanium enolate was found to be incompatible with our reactants as only degradation was observed (entry 3).18 The lack of selectivity of this reaction along with the possible side reactions (such as self-Claisen7b or Darzens16 condensations) that could occur during the process led us to investigate an aldol reaction between N-(α-chloroacetyl)-oxazolidinone 10 and glyceraldehyde 8. Unfortunately, standard conditions with either TiCl4 or Bu2BOTf only gave traces of the desired compound 12 with complete recovery of the starting materials (entries 5 and 6).19 However, when the reaction was performed with a lithiated base such as LiHMDS or LDA, only one diastereomer was obtained in 54% and 44% yield, respectively (entries 7 and 8). Despite our efforts, we were not able to improve this yield, as compound 12 seems to undergo partial retroaldolization during workup.20
Table 1.
Aldol Reaction Optimization
 | |||
|---|---|---|---|
| entry | Rx | conditions | product (yield, ratio)a,b |
| 1 | 9 | LiHMDS, THF, −78 °C | 11 (39%, 2:2:5:1) |
| 2 | 9 | LDA, THF, −78 °C | 11 (47%, 6:1:8:1) |
| 3 | 9 | TiCl4, DIPEA, CH2Cl2, −78 °C | 11 (0%) |
| 4 | 9 | Bu2BOTf, DIPEA, CH2Cl2, −78 to 0 °C | 11 (62%, 3:1:3:3) |
| 5 | 10 | Bu2BOTf, DIPEA, CH2Cl2, −78 to 0 °C | 12 (traces) |
| 6 | 10 | TiCl4, DIPEA, CH2Cl2, −78 °C | 12 (traces) |
| 7 | 10 | LiHMDS, THF, −78 °C | (2R,3R)-12 (54%, >50:1) |
| 8 | 10 | LDA, THF, −78 °C | (2R,3R)-12 (44%, >50:1) |
Isolated yield.
Ratio determined by 1H NMR analysis.
Although our ultimate goal was to introduce a fluorine atom at C2, it was important to fully define the newly set stereocenters in compound 12. Unfortunately, we were unable to assign the absolute configuration at this stage, and therefore product 12 was further converted to the lactone 15 using the chemistry depicted in Scheme 2. Thus, compound 12 was first silylated with TBSOTf in the presence of 2,6-lutidine to give compound 13 in 94% yield. Our initial attempt to directly convert 13 to 15 using a standard LiOH-H2O2 hydrolysis resulted in epimerization at C2.21 Therefore, the oxazolidinone was first cleaved by careful treatment with 0.5 equiv of sodium ethoxide at −78 °C to afford ester 14 in 91% yield as a single diastereomer.22 Compound 14 was finally cyclized in acidic conditions followed by 5-TBS-protection to give the 2-chlorolactone 15 which was identified as the (2R,3R) isomer by X-ray crystallography (Figure 2).
Scheme 2.
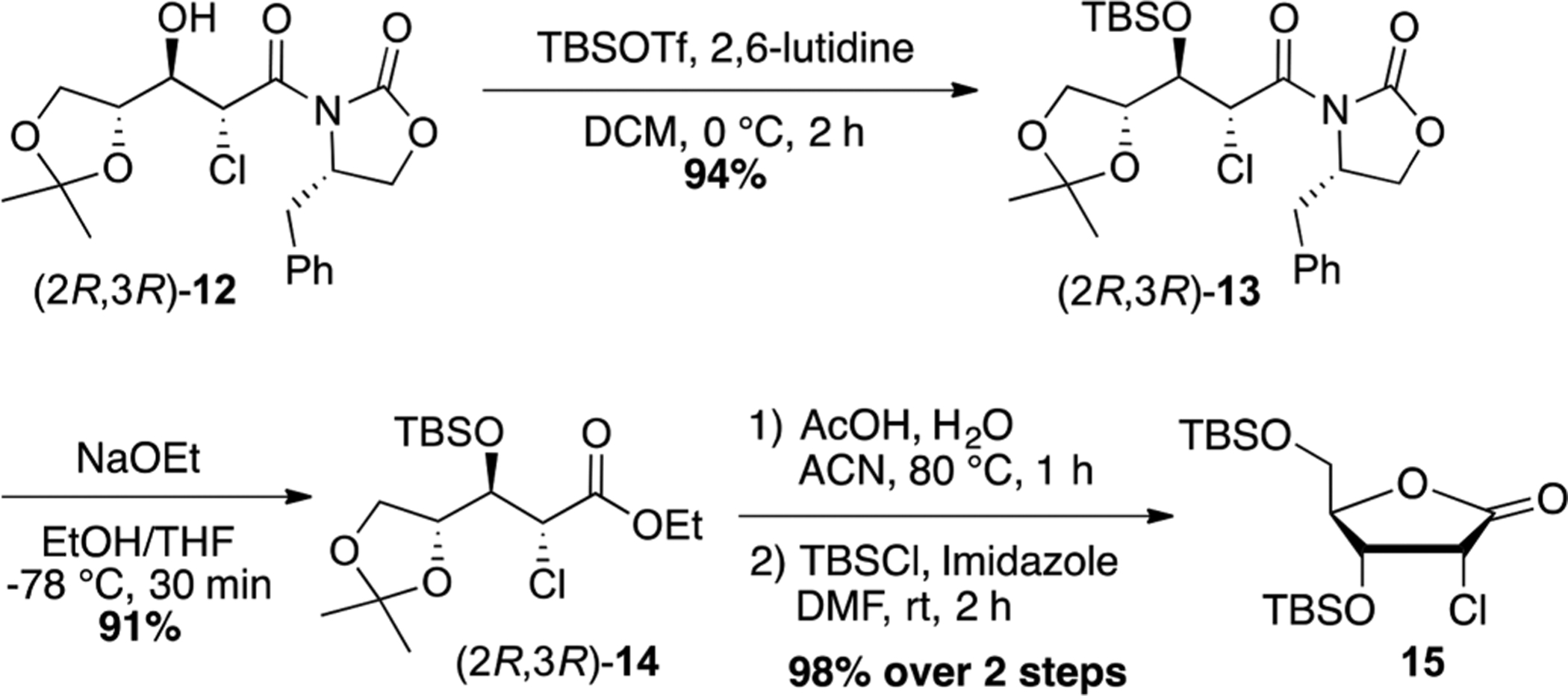
Synthesis of 2-Cl Lactone 15
Figure 2.
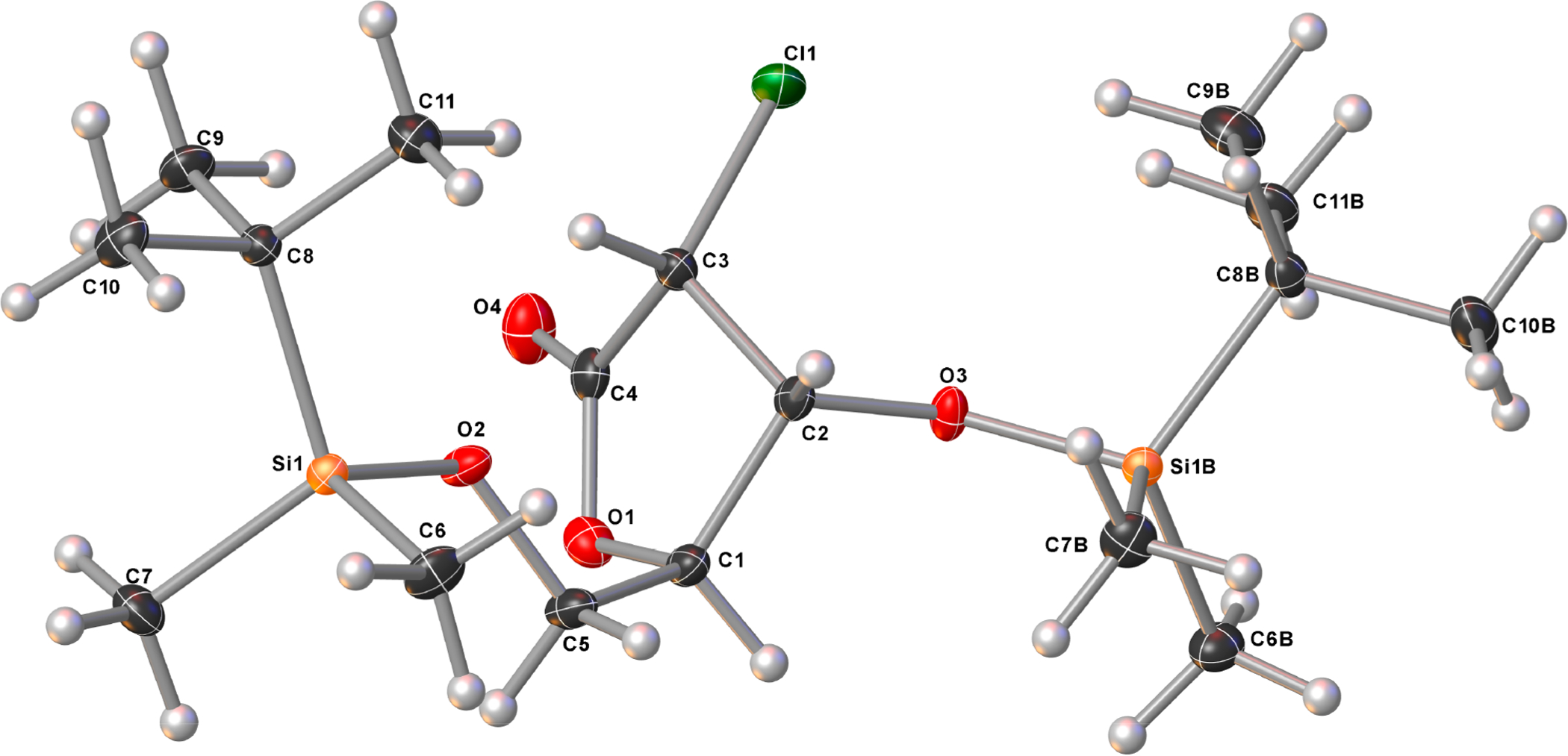
Crystal structure of (2R,3R)-2-chlorolactone 15 showing thermal ellipsoids at the 50% probability level.
Fluorination of oxazolidinone derivative 13 was attempted using 1.3 equiv of LiHMDS and 1.5 equiv of NFSI (Scheme 3), but unfortunately no fluorinated product was observed under these standard conditions.23 We hypothesized that the approach of the fluorinating reagent toward the C2 center might be restricted by the presence of the nearby bulky oxazolidinone and therefore prevent the fluorination.
Scheme 3.
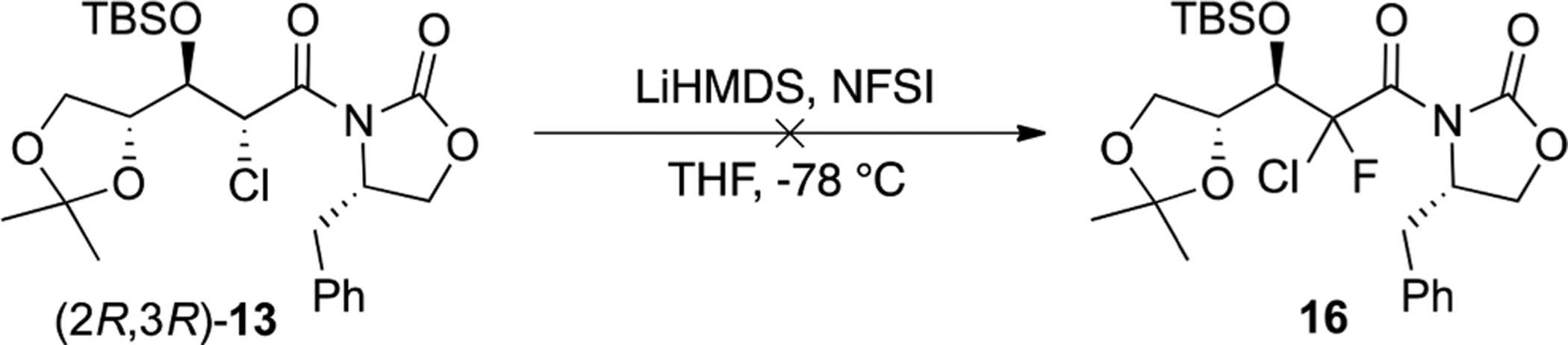
Fluorination Attempt on Compound 13
To confirm this hypothesis, the less hindered ethyl ester 11 was prepared from the Evans oxazolidinone 10 through a one-pot aldolization/auxiliary cleavage sequence (Scheme 4). Thus, the aldol reaction of 10 with glyceraldehyde 8 was performed in the presence of LiHMDS and quenched by addition of ethanol at −78 °C to provide ester 11 in 67% with an excellent stereoselectivity at C3. However, despite an improvement in yield and simpler one-pot reaction versus Table 1 entry 7, ethanol quenching led to significant epimerization at C2 (ratio 7:3, determined by 1H NMR). As the subsequent fluorination step in our approach goes through an enolate intermediate, racemization had limited consequences and 11 was directly reacted with TBSOTf and 2,6-lutidine to afford protected compound 14 almost quantitatively.
Scheme 4.
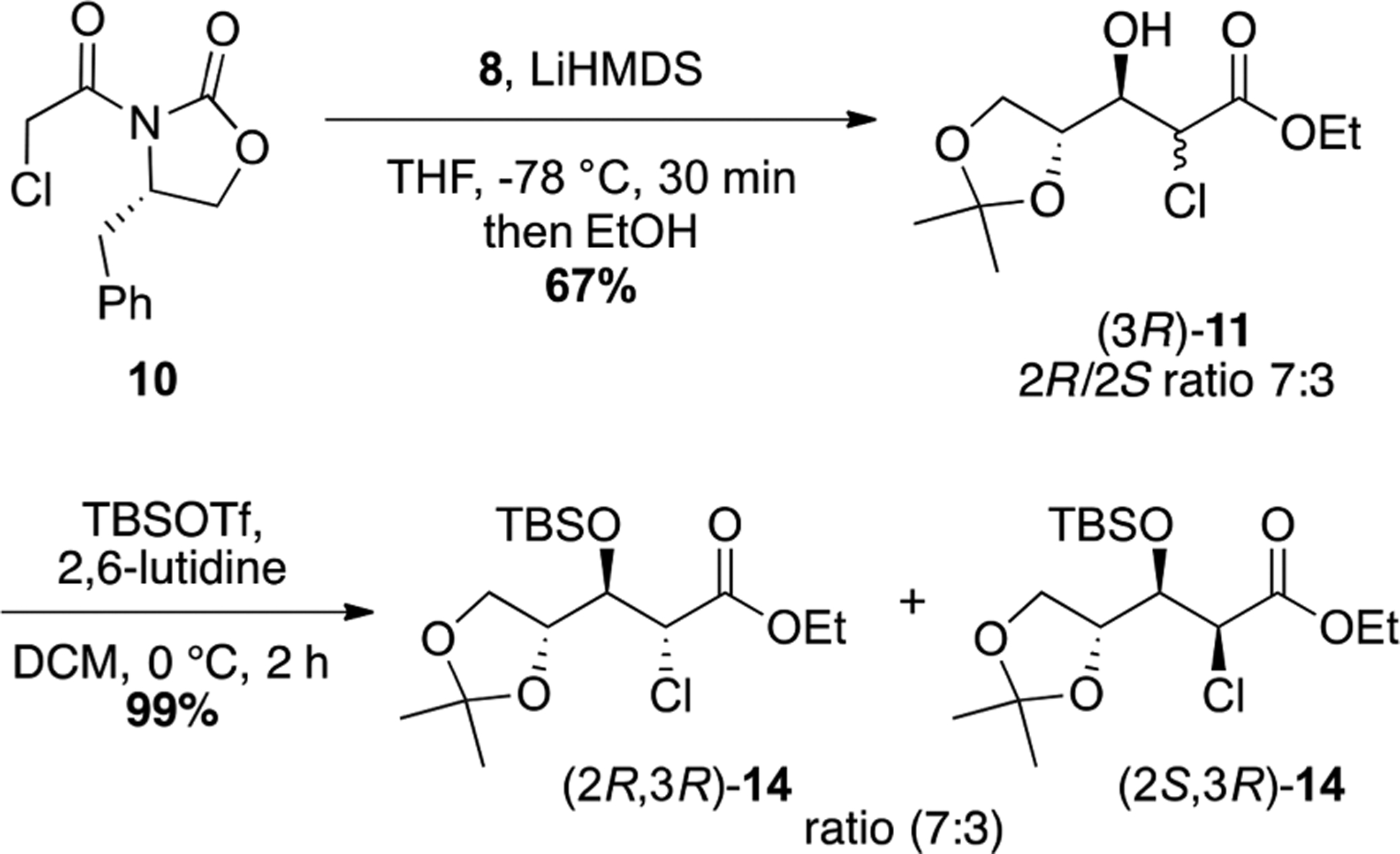
Synthesis of Compound 14
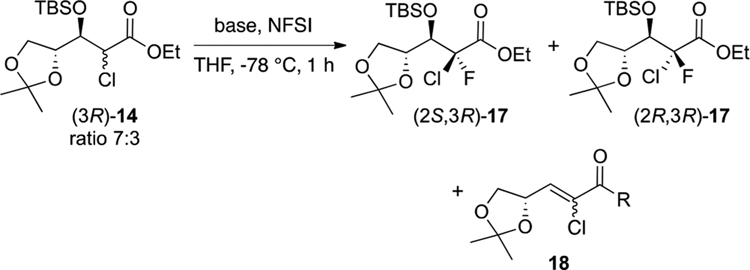
Fluorination of the epimeric mixture 14 was attempted following the conditions described above (Table 2, entry 1). Surprisingly, the reaction afforded only a single diastereomer, which was later confirmed to be the desired (2S)-isomer. Unfortunately, the reaction was not complete after 1 h at −78 °C and gave 15% of the elimination product 18.24 Interestingly, we were able to remove the undesired olefin 18 during workup by treatment of the crude mixture with solid potassium permanganate.7a It is noteworthy that warming the reaction to room temperature or increasing the reaction time affected neither the conversion rate nor the diastereoselectivity.
Table 2.
Fluorination Conditions Screening
 | ||||
|---|---|---|---|---|
| entry | base (equiv) | NFSI (equiv) | conversion (%)a | S/R-17b |
| 1 | LiHMDS, 1.3 | 1.5 | 70 | >50:1 |
| 2 | LiHMDS, 2.5 | 2.5 | 100 (94) | >50:1 |
| 3 | NaHMDS, 2.5 | 2.5 | 100 (79) | 16:1 |
| 4 | KHMDS, 2.5 | 2.5 | 96 (92) | 10:1 |
| 5 | LDA, 1.3 | 1.5 | 100 (33) | >50:1 |
Determined by 1H NMR, isolated yield in parentheses.
(2S:2R) ratio determined by 1H NMR or 19F NMR.
Nevertheless, complete conversion was finally achieved using 2.5 equiv of both LiHMDS and NFSI (Table 2, entry 2) which afforded (2S)-17 in 94% yield along with approximately 5% of elimination product 18 (ratio Z/E, 33:1). To investigate the role of the chelating effect of the counterion in the diastereoselectivity of this particular reaction, the fluorination of compound 14 was attempted using NaHMDS and KHMDS (entries 3 and 4). Although these reactions yielded the fluorinated product in good yields, only partial diastereoselectivity was observed (16:1 and 10:1 with NaHMDS and KHMDS, respectively). Interestingly, fluorination in the presence of a stronger base such as LDA25 also gave compound 17 as a single diastereomer but with a low yield due to rapid elimination of the β-OTBS group and formation of compound 18 (entry 5).
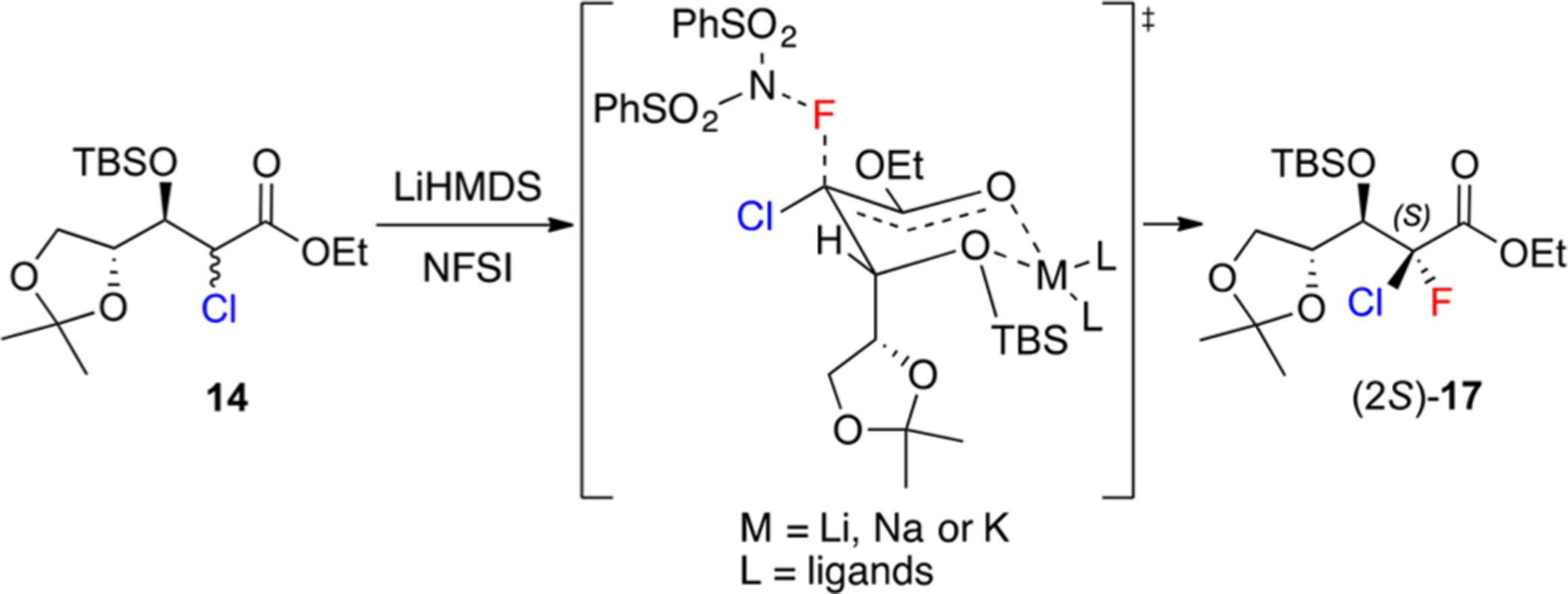
The mechanism illustrated in Scheme 5 was postulated in an attempt to rationalize the stereoselectivity of this fluorination step. Thus, the reaction performed with LiHMDS might go through a transition state in which the lithium ion simultaneously chelates the enolate and the oxygen atom at the β-position, forming a Zimmerman–Traxler-like six-membered ring. The loss of diastereoselectivity observed with sodium and potassium cations could be explained by their weaker interaction with oxygen relative to lithium,26 which might destabilize the aforementioned six-membered ring transition state. This hypothesis is supported by the fact that LDA, another lithiated base, led to complete diastereoselectivity as well. Additionally, this transition state allows the axial attack of the electrophilic fluorinating agent without steric hindrance from the ethoxy moiety, leading only to the formation of the 2-chloro-2-fluoro isomer (2S)-17.
Scheme 5.
Proposed Mechanism for the Fluorination of Compound 14
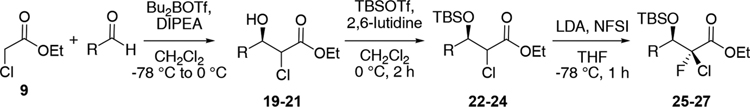
As a side note, the γ-isopropylidene group and its C4 chiral center seems to have no effect on the diastereoselectivity because similar selectivities were observed on substrates lacking a chiral center at this position (Table 3). α-Chloro-β-hydroxyester derivatives 22–24 were prepared by aldol reaction between ethyl chloroacetate 9 and achiral alkyl or aromatic aldehydes followed by protection with TBSOTf.27,28 Interestingly, unlike for compound 14, treatment of substrates 22–24 with LiHMDS resulted only in partial deprotonation and prompted us to use the stronger base, LDA, instead. Obtaining the desired 2-chloro-2-fluoro compounds 25–27 as single S diastereomers at the 2 position (ratio >50:1, 24–39%) indicates that the isopropylidene moiety and its chirality at the C4-center are not influencing the diastereoselectivity of the fluorination of compounds such as 14.
Table 3.
Preparation of the γ-Modified Chloroacetate Derivatives 25–27
 | ||||
|---|---|---|---|---|
| yielda (ratio anti/syn) | ||||
| entry | R | aldolization | protection | yielda (ratio) of fluorinationb |
| 1 | i-Pr | 19, 88% (2.3:1) | 22, 92% (2.4:1) | 25, 33% (>50:1) |
| 2 | cyclopentyl | 20, 76% (2.3:1) | 23, 96% (2:1) | 26, 24% (>50:1) |
| 3 | Ph | 21, 88% (1.8:1) | 24, 95% (1.6:1) | 27, 39% (>50:1) |
Isolated yield; ratio determined by 1H NMR.
LDA (2.5 equiv), NFSI (2.5 equiv).
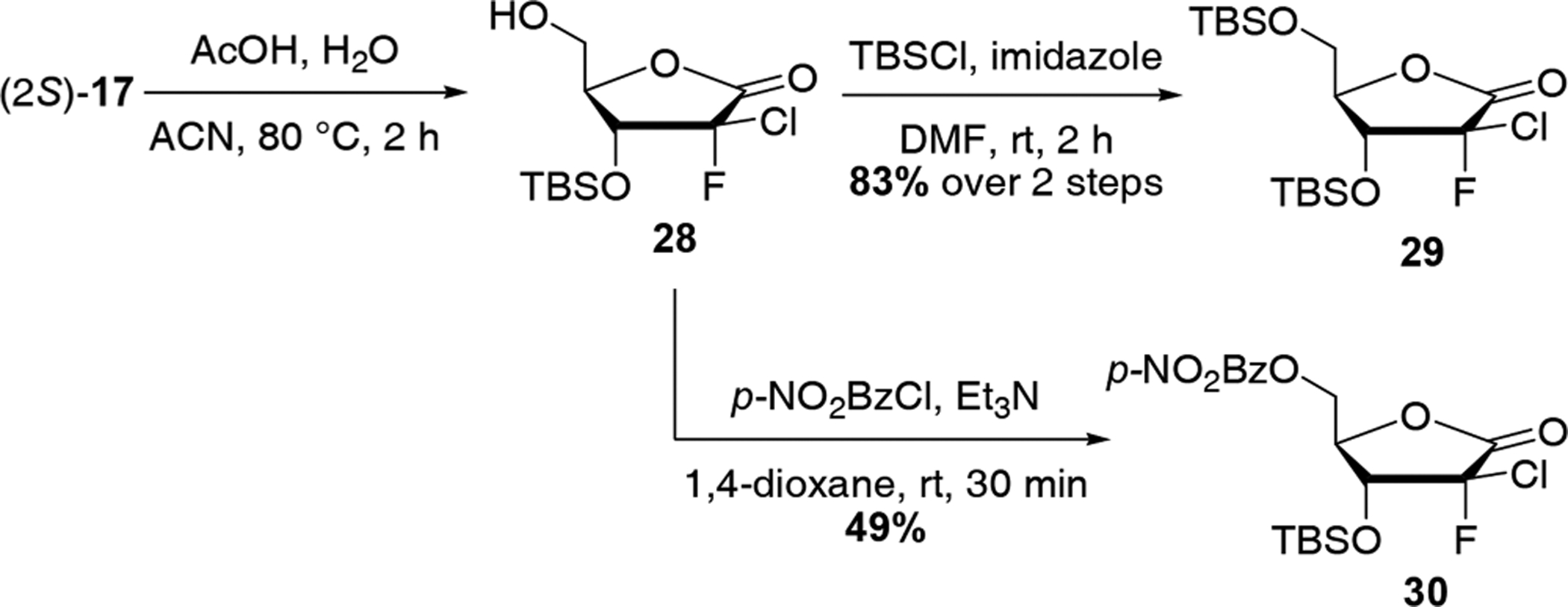
Finally, with compound 17 in hand, the desired 2-chloro-2-fluorolactone 29 was readily obtained by cyclization and 5-TBS-protection (Scheme 6). As our attempts to crystallize the TBS-lactone 29 to determine the absolute configuration at C2 were unsuccessful, introduction of different protecting groups at position 5 was investigated. X-ray crystallography of the 5-p-nitrobenzoyl-protected lactone 30, prepared from intermediate 28 by reaction with p-nitrobenzoyl chloride and triethylamine in 1,4-dioxane (Figure 3), revealed the S configuration of the C2 position.
Scheme 6.
Synthesis of Ribolactones 29 and 30
Figure 3.
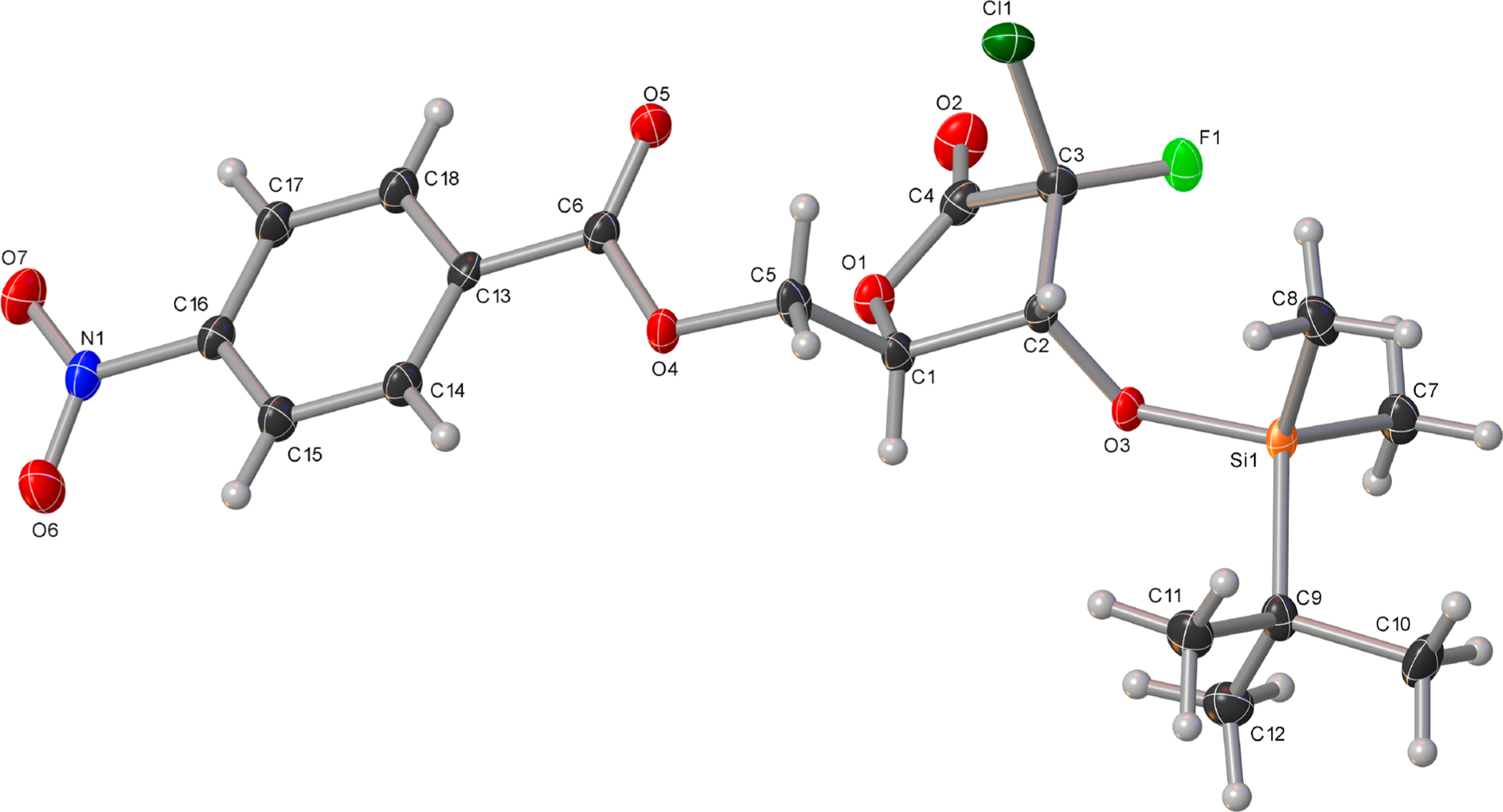
Crystal structure of (2S,3R)-2-Cl,2-F-lactone 30 showing thermal ellipsoids at the 50% probability level.
CONCLUSION
We developed an alternative pathway to prepare a key intermediate required for the synthesis of the prodrug 1. The 2-chloro-2-fluorolactone 29 was obtained in five steps with an improved 40% overall yield compared to the 10% overall yield of the original four-step synthesis. This new route starts with an efficient one-pot synthesis of 2-chloroacetate 11 from 10 and takes advantage of the diastereoselective electrophilic fluorination of this derivative to form the (2S)-Cl,F-stereocenter in high yield. Further investigations are underway to determine the scope and limitations of this ligand-free diastereoselective fluorination.
EXPERIMENTAL SECTION
General Information.
NMR spectra were recorded on a Bruker Ascend 400 MHz Fourier transform spectrometer at rt in CDCl3 or DMSO-d6 as indicated. Chemical shifts are reported in parts per million (ppm) with tetramethylsilane (TMS) as an internal standard. NMR treatment was performed with MestReNova version 10.0.2–15465 using the following abbreviations for signal characterization: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), bs (broad signal), dd (doublet of doublets), or heptd (heptuplet of doublet). High-resolution mass spectra (HRMS) were recorded on a Thermo-Fisher Q Exactive Plus orbitrap high-resolution mass spectrometer with electrospray ionization positive (ESI+) or negative (ESI−). Reactions were monitored by thin-layer chromatography (TLC) and visualized by ultraviolet irradiation and/or KMnO4 dip. Purifications were performed on silica gel column chromatography (60 Å, 63–200 μm, or 40–75 μm) with CombiFlash Rf 200. Anhydrous solvents were purchased from Aldrich Chemical Co., Inc. (Milwaukee). Reagents were purchased from commercial sources and used without further purification.
(S)-4-Benzyl-3-(2-chloroacetyl)oxazolidin-2-one (10).
To a solution of (S)-4-benzyl-2-oxazolidinone (5 g, 28.2 mmol) in dry THF (150 mL) at −78 °C under N2 was added n-BuLi (2.5 M in hexanes, 13.5 mL, 33.9 mmol). The solution was stirred at −78 °C for 15 min, and chloroacetyl chloride (2.47 mL, 31.0 mmol) was added in one portion. After full conversion (typically 30 min), the reaction was quenched by addition of a saturated aqueous solution of NH4Cl and allowed to reach rt. The mixture was diluted with EtOAc and washed with water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude residue was purified via column chromatography on silica gel to afford a colorless syrup (5.1 g, 71%), which crystallized while standing. Alternatively, precipitation could be initiated by dilution of the syrup in cold diethyl ether to afford a white solid after filtration. 1H and 13C NMR data matched those reported in the literature.29
Ethyl 2-Chloro-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-3-hydroxypropanoate (11).
Procedure A: Ethyl chloroacetate 9 (5.0 g, 40.8 mmol) was added to a solution of LDA (1 M in THF/hexanes, 53 mL, 53.0 mmol) in dry THF (300 mL) at −78 °C under N2. After 15 min, (R)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde 8 (6.1 mL, 49.0 mmol)30 was introduced to the mixture and the reaction was stirred for an additional 1 h at −78 °C. The reaction was quenched with 1 M HCl at −78 °C. The mixture was extracted with ethyl acetate, washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography (hexanes/EtOAc 9:1 to 8:2) to afford compound 11 (colorless oil, 4.8 g, 47%) as a diastereomeric mixture of four isomers. NMR data matched those reported in the literature.31 Procedure B: To a solution of LiHMDS (1 M in THF, 5.9 mL, 5.90 mmol) in dry THF (20 mL) at −78 °C under N2 was added dropwise via cannula a solution of 10 (1 g, 3.94 mmol) in THF (2 mL). The mixture was stirred 30 min at this temperature, and a solution of (R)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde 8 (736 μL, 5.90 mmol) in THF (2 mL) was added dropwise via cannula. After 30 min, anhydrous ethanol (5 mL) was added at −78 °C and the reaction mixture was further stirred for 15 min before addition of silica (10 g). The mixture was filtered on a silica gel pad prior to concentration under reduced pressure. The crude residue was purified via column chromatography on silica gel (hexanes/EtOAc 95:5 to 70:30) to afford the desired aldol product 11 as a mixture of two diastereomers in 67% yield (670 mg, 2R/2S ratio 7:3, colorless oil). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H18ClO5 253.0843, found 253.0838.
(S)-4-Benzyl-3-((2R,3R)-2-chloro-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-3-hydroxypropanoyl)oxazolidin-2-one (12).
To a solution of LiHMDS (1 M in THF, 1.48 mL, 1.48 mmol) in dry THF (5 mL) at −78 °C under N2 was added dropwise via cannula a solution of 10 (250 mg, 0.99 mmol) in THF (2 mL). The mixture was stirred 30 min at this temperature, and a solution of (R)-2,2-dimethyl-1,3-dioxolane-4-carbaldehyde 8 (115 μL, 1.28 mmol) in THF (2 mL) was added dropwise via cannula. After 30 min, the reaction was quenched at −78 °C by addition of 1 M HCl. The mixture was rapidly diluted with EtOAc and then washed with water and brine. The organic layer was dried over MgSO4 and filtered on a silica gel pad prior to concentration under reduced pressure. The crude residue was purified via column chromatography on silica gel (hexanes/EtOAc 95:5 to 70:30) to afford the desired aldol product as a single diastereoisomer in 54% yield as a white solid (205 mg). 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 3H), 7.25 (d, J = 6.9 Hz, 2H), 5.77 (d, J = 6.1 Hz, 1H), 4.79–4.72 (m, 1H), 4.38–4.08 (m, 6H), 3.31 (dd, J = 13.6, 3.3 Hz, 1H), 3.27 (bs, 1H), 2.85 (dd, J = 13.5, 9.2 Hz, 1H), 1.47 (s, 3H), 1.39 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 168.5, 152.4, 134.7, 129.5, 129.1, 127.6, 109.9, 75.9, 73.7, 66.4, 65.9, 55.5, 52.7, 37.6, 26.6, 25.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H23ClNO6 384.1214, found 384.1201.
(S)-4-Benzyl-3-((2R,3R)-3-((tert-butyldimethylsilyl)oxy)-2-chloro-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)propanoyl)-oxazolidin-2-one (13).
To a solution of compound 12 (230 mg, 0.60 mmol) in dry CH2Cl2 (6 mL) at 0 °C under N2 were sequentially added 2,6-lutidine (280 μL, 2.4 mmol) and TBSOTf (275 μL, 1.2 mmol). The mixture was stirred 1 h at 0 °C and quenched by addition of 1 M HCl (5 mL). The mixture was extracted with CH2Cl2 (2 × 10 mL). Combined organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude residue was purified via column chromatography on silica gel (hexanes/EtOAc, 95:5) to afford the desired silylated derivative 13 in 94% yield as a white solid (281 mg). 1H NMR (400 MHz, CDCl3) δ 7.47–7.23 (m, 5H), 5.71 (d, J = 4.0 Hz, 1H), 4.78–4.70 (m, 1H), 4.54 (dd, J = 6.1, 4.0 Hz, 1H), 4.43 (dd, J = 13.4, 6.3 Hz, 1H), 4.32–4.22 (m, 1H), 4.10 (dd, J = 8.2, 6.4 Hz, 1H), 3.92 (t, J = 7.8 Hz, 1H), 3.52 (dd, J = 13.3, 2.8 Hz, 1H), 2.65 (dd, J = 13.2, 10.6 Hz, 1H), 1.40 (s, 3H), 1.32 (s, 3H), 0.93 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 165.9, 152.9, 135.2, 129.3, 129.1, 127.5, 109.3, 74.6, 72.4, 67.1, 66.3, 62.0, 56.1, 38.0, 26.6, 25.7, 25.4, 18.1, −4.5, −4.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H37ClNO6Si 498.2079, found 498.2075.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)propanoate (14).
Procedure A: To a solution of compound 13 (52 mg, 0.10 mmol) in a mixture of anhydrous EtOH (1.5 mL) and THF (1.5 mL) at −78 °C was added NaOEt (21 wt % in EtOH, 0.02 mL, 0.5 mmol). The mixture was stirred 30 min at −78 °C and quenched by addition of 1 M HCl (5 mL). After dilution with EtOAc, the organic layer was washed with a saturated aqueous solution of NaHCO3 and brine. The organic layer was dried over MgSO4 and concentrated in vacuo. The crude residue was purified via column chromatography on silica gel (hexanes/EtOAc, 95:5) to afford the desired ethyl ester 14 as a colorless oil (35 mg, 91%). 1H NMR (400 MHz, CDCl3) δ 4.60 (d, J = 2.7 Hz, 1H), 4.34–4.28 (m, 1H), 4.25 (qd, J = 7.2, 1.1 Hz, 2H), 4.20 (dd, J = 7.2, 2.7 Hz, 1H), 4.07 (dd, J = 8.4, 6.3 Hz, 1H), 3.89 (dd, J = 8.4, 5.4 Hz, 1H), 1.40 (s, 3H), 1.36–1.30 (m, 6H), 0.90 (s, 9H), 0.19 (s, 3H), 0.15 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 166.8, 109.4, 75.8, 75.6, 66.6, 62.2, 61.3, 26.6, 25.7, 25.1, 18.0, 14.1, −4.3, −4.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H32ClO5Si 367.1708, found 367.1701. Procedure B: To a solution of compound (3R)-11 (2.32 g, 9.18 mmol) in dichloromethane (10 mL) at 0 °C was added 2,6-lutidine (4.28 mL, 36.72 mmol) followed by TBSOTf (4.22 mL, 18.36 mmol). The reaction mixture was stirred for 2 h and then quenched with a saturated aqueous solution of NaHCO3. The crude mixture was extracted with ethyl acetate, washed with 1 M HCl and brine, dried over MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes/EtOAc, 95/5) afforded the two major diastereoisomers (2R,3R)-14 and (2S,3R)-14 in 99% yield (colorless oil, 3.33 g, ratio 7:3).
(3R,4R,5R)-4-((tert-Butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-chlorodihydrofuran-2(3H)-one (15).
To a solution of compound 14 (29 mg, 0.08 mmol) in acetonitrile (0.2 mL) were added water (0.01 mL) and acetic acid (0.05 mL). The reaction mixture was refluxed for 1 h and coevaporated with toluene under reduced pressure. The crude 2-chlorolactone was dissolved in DMF (0.2 mL), and TBSCl (24 mg, 0.16 mmol) and imidazole (14 mg, 0.20 mmol) were added. The reaction was stirred at rt for 2 h and quenched with water (5 mL). The mixture was extracted with EtOAc/Et2O (1:1). The organic layer was washed with water (3 × 10 mL), a saturated aqueous solution of NH4Cl (10 mL), and brine (10 mL), dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (hexanes/EtOAc, 95:5) to afford the protected lactone 15 as a white solid (30.7 mg, 98%). 1H NMR (400 MHz, CDCl3) δ 4.62 (d, J = 5.4 Hz, 1H), 4.51 (dd, J = 5.6, 2.7 Hz, 1H), 4.41 (d, J = 2.5 Hz, 1H), 3.94 (dd, J = 12.0, 2.7 Hz, 1H), 3.82 (dd, J = 12.0, 2.0 Hz, 1H), 0.93 (s, 9H), 0.90 (s, 9H), 0.17 (s, 3H), 0.14 (s, 3H), 0.10 (s, 3H), 0.09 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 171.1, 85.8, 70.4, 61.4, 55.7, 25.7, 25.6, 18.3, 18.2, −4.9, −5.1, −5.5, −5.6; mp 85–86 °C; HRMS (ESI-TOF) m/z: [M − H]− calcd for C17H36ClO4Si2 395.1841, found 395.1830.
Ethyl (2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-2-chloro-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-2-fluoropropanoate (17).
A solution of LiHMDS (1 M in THF, 0.34 mL, 0.34 mmol) was added dropwise to a mixture of compound (3R)-14 (50 mg, 0.14 mmol) and NFSI (107 mg, 0.34 mmol) in anhydrous THF (0.5 mL) at −78 °C. After 1 h, the reaction was quenched with a saturated aqueous solution of NH4Cl and extracted with EtOAc. Solid potassium permanganate (300 mg) and water (0.5 mL) were added to the organic layer (consumption of the elimination compound was monitored by TLC, hexanes/EtOAc 9:1, Rf = 0.57). A saturated aqueous solution of Na2S2O3 (20 mL) was added to the heterogeneous mixture, and formation of a brown precipitate was observed after vigorous shaking. The mixture was treated with 1 M HCl (10 mL). The organic layer was separated and washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (hexanes/EtOAc, 95/5) to give compound 17 as a colorless oil (49 mg, 94%, ratio 2S/2R > 50:1). 1H NMR (400 MHz, CDCl3) δ 4.42–4.21 (m, 4H), 4.10–4.00 (m, 1H), 3.93 (dd, J = 8.2, 6.4 Hz, 1H), 1.41 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H), 1.33 (s, 3H), 0.93 (s, 9H), 0.20 (s, 3H), 0.17 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −127.12 (d, J = 18.2 Hz); 13C NMR (101 MHz, CDCl3) δ 164.8 (d, J = 27.6 Hz), 108.9, 104.8 (d, J = 261.4 Hz), 77.2 (d, J = 21.4 Hz), 75.3 (d, J = 4.0 Hz), 65.6 (d, J = 1.5 Hz), 63.1, 25.9, 25.8, 24.6, 18.3, 13.9, −4.0, −4.2, −4.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H31ClFO5Si 385.1613, found 385.1607.
Ethyl 2-Chloro-3-hydroxy-4-methylpentanoate (19).
To a solution of 9 (0.22 mL, 2.04 mmol) in CH2Cl2 (20 mL) at −78 °C were added dibutylboryl trifluoromethanesulfonate (1.0 M in CH2Cl2, 4.1 mL, 4.08 mmol) and DIPEA (1 mL, 6.12 mmol). The mixture was stirred 2 h at −78 °C, and isobutyraldehyde (0.28 mL, 3.06 mmol) was added. The reaction was stirred at this temperature for 1 h and for another 1 h at 0 °C. The mixture was quenched with a (1:1) mixture of methanol/phosphate buffer (20 mL, pH 7.2) followed by H2O2 (10 mL, 30% w/w in water), and the solution was stirred 16 h at rt. After removal of the solvent under reduced pressure, the solution was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude compound was purified by flash column chromatography (hexanes/EtOAc, 9:1) to give compound 19 as an anti/syn mixture (2.3:1) (colorless oil, 350 mg, 88%). 1H and 13C NMR data matched those reported in the literature.32 Major isomer: 1H NMR (400 MHz, CDCl3) δ 4.30–4.22 (m, 2H), 4.20 (d, J = 7.9 Hz, 1H), 3.81 (dd, J = 8.0, 4.1 Hz, 1H), 2.72 (bs, 1H), 2.12– 2.04 (m, 1H), 1.31 (t, J = 7.2 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C8H16ClO3 195.0788, found 195.0785.
Ethyl 2-Chloro-3-cyclopentyl-3-hydroxypropanoate (20).
Compound 20 was prepared following the procedure described for 19 using cyclopentanecarboxaldehyde to give 20 as an anti/syn mixture (2.3:1) (colorless oil, 76%). Major isomer: 1H NMR (400 MHz, CDCl3) δ 4.29–3.85 (m, 4H), 2.75 (bs, 1H), 2.26–2.14 (m, 1H), 1.94–1.35 (m, 8H), 1.32 (t, J = 7.2 Hz, 3H); mixture: 13C NMR (101 MHz, CDCl3) δ 168.8, 168.7, 76.1, 76.0, 62.4, 62.2, 61.9, 58.7, 43.1, 41.8, 29.0, 28.9, 28.9, 27.1, 25.6, 25.4, 25.4, 25.4, 14.0, 13.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H18ClO3 221.0944, found 221.0939.
Ethyl 2-Chloro-3-hydroxy-3-phenylpropanoate (21).
Compound 21 was prepared following the procedure described for 19 using benzaldehyde to give 21 as an anti/syn mixture (1.8:1) (colorless oil, 88%). 1H and 13C NMR data matched those reported in the literature.32 Major isomer: 1H NMR (400 MHz, CDCl3) δ 7.45–7.33 (m, 5H), 5.07 (dd, J = 7.7, 5.5 Hz, 1H), 4.41 (d, J = 7.5 Hz, 1H), 4.28 (q, J = 7.2 Hz, 2H), 3.07 (d, J = 5.0 Hz, 1H), 1.30 (t, J = 7.1 Hz, 3H).
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-4-methylpentanoate (22).
Compound 22 was prepared following conditions described for 14 (procedure B) with 19 (250 mg, 1.28 mmol), TBSOTf (0.6 mL, 2.56 mmol), and 2,6-lutidine (0.6 mL, 5.12 mmol) in CH2Cl2 (6 mL) to afford 22 (colorless oil, 366 mg, 92%) as an anti/syn mixture (2.4:1). Major isomer: 1H NMR (400 MHz, CDCl3) δ 4.27–4.19 (m, 2H), 4.17 (d, J = 7.7 Hz, 1H), 4.00 (dd, J = 7.7, 3.3 Hz, 1H), 2.17 (heptd, J = 6.9, 3.2 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H), 0.99–088 (m, 15H), 0.10–0.03 (m, 6H); mixture: 13C NMR (101 MHz, CDCl3) δ 169.0, 168.8, 61.9, 61.8, 61.2, 58.2, 31.9, 30.3, 26.1, 26.0, 20.2, 19.5, 18.5, 18.4, 16.5, 16.0, 13.9, 13.9, −3.8, −4.0, −4.2, −4.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H30ClO3Si 309.1653, found 309.1647.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-3-cyclopentylpropanoate (23).
Compound 23 was prepared following conditions described for 14 (procedure B) with 20 (250 mg, 1.13 mmol), TBSOTf (0.5 mL, 2.26 mmol) and 2,6-lutidine (0.5 mL, 4.52 mmol) in CH2Cl2 (5 mL) to afford 23 (colorless oil, 364 mg, 96%) as an anti/syn mixture (2:1). Major isomer: 1H NMR (400 MHz, CDCl3) δ 4.31–4.29 (m, 1H), 4.27–4.20 (m, 2H), 4.10–4.04 (m, 1H), 2.38–2.24 (m, 1H), 1.80–1.36 (m, 7H), 1.32 (t, J = 7.2 Hz, 3H), 1.28–1.16 (m, 1H), 0.89 (s, 9H), 0.12–0.08 (m, 6H); mixture: 13C NMR (101 MHz, CDCl3) δ 168.7, 168.2, 62.5, 62.0, 61.9, 61.2, 43.9, 43.2, 29.7, 28.9, 28.0, 27.8, 26.0, 25.9, 25.5, 25.9, 25.2, 25.1, 18.4, 18.3, 14.0, −3.9, −4.0, −4.2, −4.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H32ClO3Si 335.1809, found 335.1801.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-3-phenylpropanoate (24).
Compound 24 was prepared following conditions described for 14 (procedure B) with 21 (410 mg, 1.79 mmol), TBSOTf (0.82 mL, 3.58 mmol) and 2,6-lutidine (0.83 mL, 7.16 mmol) in CH2Cl2 (20 mL) to afford 24 (colorless oil, 582 mg, 95%) as an anti/syn mixture (1.6:1). Major isomer: 1H NMR (400 MHz, CDCl3) δ 7.44–7.29 (m, 5H), 4.95 (d, J = 9.1 Hz, 1H), 4.33–4.25 (m, 2H), 4.23 (d, J = 9.1 Hz, 1H), 1.36 (t, J = 7.1 Hz, 3H), 0.83 (s, 9H), 0.12–−0.24 (m, 6H); mixture: 13C NMR (101 MHz, CDCl3) δ 168.9, 167.9, 139.9, 139.7, 128.5, 128.2, 128.1, 127.5, 127.1, 63.7, 61.9, 61.7, 60.3, 25.6, 25.5, 18.1, 17.9, 14.0, 13.7, −4.7, −5.0, −5.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H28ClO3Si 343.1496, found 343.1491.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-2-fluoro-4-methylpentanoate (25).
To a solution of diisopropylamine (0.09 mL, 0.65 mmol) in dry THF (1 mL) at −78 °C under N2 was added dropwise n-BuLi (2.5 M in hexanes, 0.26 mL, 0.65 mmol). After 10 min at −78 °C, compound 22 (100 mg, 0.32 mmol) in dry THF (0.3 mL) was introduced dropwise to the reaction mixture. A solution of NFSI (224 mg, 0.70 mmol) in anhydrous THF (0.5 mL) was immediately added. After 1 h at −78 °C, the reaction was quenched with a saturated aqueous solution of NH4Cl and extracted with EtOAc. Solid potassium permanganate (300 mg) and water (0.5 mL) were added to the organic layer (consumption of the elimination product was monitored by TLC, hexanes/EtOAc 95:5, Rf = 0.54). A saturated aqueous solution of Na2S2O3 (20 mL) was added to the heterogeneous mixture, and formation of a brown precipitate was observed after vigorous shaking. The mixture was treated with 1 M HCl (10 mL). The organic layer was separated, washed with brine, dried over MgSO4, and concentrated under reduced pressure. The crude residue was purified by flash column chromatography (hexanes/EtOAc 95:5 as eluent) to give compound 25 (colorless oil, 17.3 mg, 33%). 1H NMR (400 MHz, CDCl3) δ 4.35 (q, J = 7.1 Hz, 2H), 4.04 (dd, J = 20.0, 2.2 Hz, 1H), 2.00 (heptd, J = 6.8, 2.3 Hz, 1H), 1.38 (t, J = 7.1 Hz, 3H), 1.04 (d, J = 7.0 Hz, 3H), 0.97 (s, 9H), 0.94 (dd, J = 6.8, 1.8 Hz, 3H), 0.21 (s, 3H), 0.15 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −126.02 (d, J = 20.2 Hz); 13C NMR (101 MHz, CDCl3) δ 165.5 (d, J = 27.9 Hz), 106.9 (d, J = 263.9 Hz), 80.3 (d, J = 19.2 Hz), 63.0, 30.9, 26.0, 22.1, 18.6, 16.2 (d, J = 3.6 Hz), 13.9, −4.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H29ClFO3Si 327.1559, found 327.1553.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-3-cyclopentyl-2-fluoropropanoate (26).
Compound 26 was prepared following the procedure described for 25 with 23 (50 mg, 0.15 mmol), LDA (2 M in THF/heptane/ethylbenzene, 0.1 mL, 0.20 mmol), and NFSI (118 mg, 0.37 mmol) in THF (1.5 mL) to afford 26 (colorless oil, 12.6 mg, 24%). 1H NMR (400 MHz, CDCl3) δ 4.34 (qd, J = 7.1, 1.1 Hz, 2H), 4.19 (dd, J = 17.8, 3.6 Hz, 1H), 2.25–2.08 (m, 1H), 1.85–1.73 (m, 1H), 1.72–1.40 (m, 7H), 1.37 (t, J = 7.1 Hz, 3H), 0.94 (s, 9H), 0.19 (d, J = 1.0 Hz, 3H), 0.14 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −123.05 (d, J = 18.0 Hz); 13C NMR (101 MHz, CDCl3) δ 165.3 (d, J = 28.1 Hz), 106.8 (d, J = 261.3 Hz), 78.9 (d, J = 20.4 Hz), 63.0, 42.3 (d, J = 1.8 Hz), 31.1, 26.5 (d, J = 2.3 Hz), 26.0, 25.3 (d, J = 16.1 Hz), 18.6, 13.9, −4.0 (d, J = 2.7 Hz), −4.1; HRMS (ESI-TOF) m/z: [M +H]+ calcd for C16H31ClFO3Si 353.1715, found 353.1703.
Ethyl 3-((tert-Butyldimethylsilyl)oxy)-2-chloro-2-fluoro-3-phenylpropanoate (27).
Compound 27 was prepared following the procedure described for 25 with 24 (50 mg, 0.15 mmol), LDA (2 M in THF/heptane/ethylbenzene, 0.15 mL, 0.30 mmol), and NFSI (118 mg, 0.37 mmol) in THF (1.5 mL) to afford 30 (colorless oil, 20.4 mg, 39%). 1H NMR (400 MHz, CDCl3) δ 7.45–7.39 (m, 2H), 7.37–7.31 (m, 3H), 5.17 (d, J = 17.0 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H), 0.91 (s, 9H), 0.14 (s, 3H), −0.10 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −130.11 (d, J = 17.2 Hz); 13C NMR (101 MHz, CDCl3) δ 164.8 (d, J = 26.9 Hz), 136.6, 128.9, 128.5, 128.5, 127.9, 106.3 (d, J = 263.0 Hz), 78.2 (d, J = 21.3 Hz), 63.0, 25.6, 18.2, 13.8, −4.9, −5.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H27ClFO3Si 361.1402, found 361.1396.
(2S)-2-Deoxy-2-chloro-2-fluoro-3-O-(tert-butyldimethylsilyl)-5-hydroxy-d-ribonolactone (28).
To a solution of compound 9 (150 mg, 0.39 mmol) in acetonitrile (0.5 mL) were added water (0.05 mL) and acetic acid (0.3 mL). The reaction mixture was stirred at 80 °C for 2 h and coevaporated with toluene under reduced pressure. Compound 28 was obtained in 88% yield and used in the next step without further purification. A sample was purified for analysis by flash column chromatography (hexanes/EtOAc, 8:2) to afford chlorofluorolactone 28 as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 4.56 (dd, J = 13.2, 6.5 Hz, 1H), 4.41 (dt, J = 6.4, 3.1 Hz, 1H), 4.09 (dd, J = 13.0, 2.3 Hz, 1H), 3.83 (d, J = 12.9 Hz, 1H), 2.02 (bs, 1H), 0.95 (s, 9H), 0.25 (s, 3H), 0.20 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −134.91 (d, J = 13.3 Hz); 13C NMR (101 MHz, CDCl3) δ 164.9 (d, J = 25.7 Hz), 102.4 (d, J = 258.3 Hz), 83.4, 74.2 (d, J = 16.3 Hz), 59.2, 25.5, 18.0, −4.3, −5.2; HRMS (ESI-TOF) m/z: [M − H]− calcd for C11H19ClFO4Si 297.0725, found 297.0732.
(2S)-2-Deoxy-2-chloro-2-fluoro-3,5-di-O-(tert-butyldimethylsilyl)-d-ribonolactone (29).
To a solution of compound 28 (3.89 g, 13 mmol) in DMF (30 mL) were added imidazole (2.3 g, 52 mmol) and TBSCl (3.9 g, 26 mmol). The mixture was stirred 2 h at 25 °C and quenched with water. The solution was extracted with ethyl acetate/diethyl ether (1:1), and the organic layer was washed with water (3 × 30 mL), a saturated aqueous solution of NH4Cl (30 mL), and brine (30 mL), dried over MgSO4, and concentrated under reduced pressure. The crude was purified by flash column chromatography (hexanes/EtOAc, 95:5) to afford protected lactone 29 as a colorless oil (5.0 g, 94%). 1H NMR (400 MHz, CDCl3) δ 4.58 (dd, J = 12.0, 5.7 Hz, 1H), 4.37–4.32 (m, 1H), 3.98 (dd, J = 12.1, 3.7 Hz, 1H), 3.84 (dd, J = 12.1, 2.9 Hz, 1H), 0.92 (s, 9H), 0.89 (s, 9H), 0.21 (s, 3H), 0.16 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −134.67 (d, J = 11.9 Hz); 13C NMR (101 MHz, CDCl3) δ 165.2 (d, J = 26.0 Hz), 102.1 (d, J = 259.6 Hz), 83.6, 74.1 (d, J = 16.0 Hz), 59.5, 25.7, 25.5, 18.2, 18.0, −4.4, −5.2, −5.4, −5.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H35ClFO4Si2 413.1746, found 413.1739.
((2R,3R,4S)-3-((tert-Butyldimethylsilyl)oxy)-4-chloro-4-fluoro-5-oxotetrahydrofuran-2-yl)methyl 4-Nitrobenzoate (30).
To a solution of lactone 28 (140 mg, 0.47 mmol) in 1,4-dioxane (2 mL) were added p-nitrobenzoyl chloride (93 mg, 0.50 mmol) and triethylamine (0.09 mL, 0.65 mmol). The reaction mixture was stirred at rt for 45 min and quenched with 1 M HCl (10 mL). The organic layer was washed with a saturated aqueous solution of NaHCO3 and brine, dried over MgSO4, and concentrated under reduced pressure. Purification by flash column chromatography (hexanes/EtOAc, 95:5) afforded compound 30 (102 mg, 49%) as a white solid. A small amount was recrystallized by slow evaporation from pentane/Et2O for X-ray analysis. 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 8.9 Hz, 2H), 8.24 (d, J = 8.9 Hz, 2H), 4.79 (dd, J = 12.3, 3.5 Hz, 1H), 4.75–4.69 (m, 1H), 4.62 (dd, J = 12.3, 5.9 Hz, 1H), 4.45 (dd, J = 8.4, 4.9 Hz, 1H), 0.95 (s, 9H), 0.24 (s, 3H), 0.19 (s, 3H); 19F NMR (377 MHz, CDCl3) δ −136.48 (d, J = 8.5 Hz); 13C NMR (101 MHz, CDCl3) δ 164.4 (d, J = 26.6 Hz), 164.0, 151.0, 134.2, 130.9, 123.8, 100.6 (d, J = 263.7 Hz), 81.4, 75.6 (d, J = 16.9 Hz), 62.6, 25.5, 18.1, −4.4, −5.2; mp 99–100 °C; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H24ClFNO7Si 448.0995, found 448.0984.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Bryan Cox for fruitful discussions. This work was supported in part by NIH grant 5P30-AI-50409 (CFAR).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b02245.
1H, 13C, and 19F NMR spectra for newly synthesized compounds and crystallography data for compound 15 and 30 (PDF)
CIF data for 15 (CIF)
CIF data for 30 (CIF)
The authors declare the following competing financial interest(s): Dr. Schinazi is the Chairman and a major shareholder of Cocrystal Pharma, Inc. Emory received no funding from Cocrystal Pharma, Inc. to perform this work and vice versa.
REFERENCES
- 1.http://www.who.int/mediacentre/factsheets/fs164/en/, accessed July 3, 2017.
- 2.Perz JF; Armstrong GL; Farrington LA; Hutin YJ; Bell BP J. Hepatol 2006, 45, 529–538. [DOI] [PubMed] [Google Scholar]
- 3.Webster DP; Klenerman P; Dusheiko GM Lancet 2015, 385, 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.http://www.gilead.com/news/press-releases/2016/6/us-food-and-drug-administration-approves-gileads-epclusa-sofosbuvirvelpatasvir-for-the-treatment-of-all-genotypes-of-chronic-hepatitis-c, accessed May 5, 2017.
- 5.(a) Kalayanov G; Torssell S; Wälhing H WO 2015/034420, March 12, 2015.; (b) Paparin J-L; Badaroux E; Pierra C; Dousson CB WO 2015/081297, June 4, 2015.; (c) Coats SJ; Zhou S; Amblard F; Schinazi RF; Khalil A WO 2015/164812, October 29, 2015.; (d) Zhou S; Mahmoud S; Liu P; Zhou L; Ehteshami M; Bassit L; Tao S; Domaoal R; Sari O; De Schutter C; Amiralaei S; Khalil A; Ollinger Russell O; McBrayer T; Whitaker T; Abotaleb N; Amblard F; Coats SJ; Schinazi RF J. Med. Chem 2017, 60, 5424–5437. [DOI] [PubMed] [Google Scholar]
- 6.Cen Y; Sauve AA J. Org. Chem 2009, 74, 5779–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Wang P; Chun B-K; Rachakonda S; Du J; Khan N; Shi J; Stec W; Cleary D; Ross BS; Sofia MJ J. Org. Chem 2009, 74, 6819–6824. [DOI] [PubMed] [Google Scholar]; (b) Zhang P; Iding H; Cedilote M; Brunner S; Williamson T; Cleary TP Tetrahedron: Asymmetry 2009, 20, 305–312. [Google Scholar]; (c) Chen R; Li Y; Zhao J; Zheng J; Zhu G WO 2014/108525.; (d) Peifer M; Berger R; Shurtleff VW; Conrad JC; MacMillan DWC J. Am. Chem. Soc 2014, 136, 5900–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown K; Dixey M; Weymouth-Wilson A; Linclau B Carbohydr. Res 2014, 387, 59–73. [DOI] [PubMed] [Google Scholar]
- 9.(a) Baudoux J; Cahard D Organic Reactions 2008, 69, 1–326. [Google Scholar]; (b) Lectard S; Hamashima Y; Sodeoka M Adv. Synth. Catal 2010, 352, 2708–2732. [Google Scholar]; (c) Kwiatkowski P; Beeson TD; Conrad JC; MacMillan DWC J. Am. Chem. Soc 2011, 133, 1738–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Kang SH; Kim DY Adv. Synth. Catal 2010, 352, 2783–2786. [Google Scholar]; (b) Woo SB; Suh CW; Koh KO; Kim DY Tetrahedron Lett 2013, 54, 3359–3362. [Google Scholar]; (c) Frantz R; Hintermann L; Perseghini M; Broggini D; Togni A Org. Lett 2003, 5, 1709–1712. [DOI] [PubMed] [Google Scholar]
- 11.Shibatomi K; Narayama A; Soga Y; Muto T; Iwasa S Org. Lett 2011, 13, 2944–2947. [DOI] [PubMed] [Google Scholar]
- 12.Yi W-B; Huang X; Zhang Z; Zhu D-R; Cai C; Zhang W Beilstein J. Org. Chem 2012, 8, 1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarado J; Herrmann AT; Zakarian AJ Org. Chem 2014, 79, 6206–6220. [DOI] [PubMed] [Google Scholar]
- 14.(a) Shibatomi K; Yamamoto H Angew. Chem., Int. Ed 2008, 47, 5796–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shibatomi K; Okimi T; Abe Y; Narayama A; Nakamura N; Iwasa S Beilstein J. Org. Chem 2014, 10, 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hayes MD; Rodríguez-Alvarado M; Brenner-Moyer SE Tetrahedron Lett 2015, 56, 4718–4720. [Google Scholar]
- 15.Kumaraswamy G; Satish Kumar R Helv. Chim. Acta 2013, 96, 1366–1375. [Google Scholar]
- 16.Krafft ME; Twiddle SJR; Cran JW Tetrahedron Lett 2011, 52, 1277–1280. [Google Scholar]
- 17.Inoue T; Liu J-F; Buske DC; Abiko A J. Org. Chem 2002, 67, 5250–5256. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK; Kim J-H Tetrahedron Lett 2001, 42, 1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdel-Magid A; Pridgen LN; Eggleston DS; Lantos I J. Am. Chem. Soc 1986, 108, 4595–4602. [Google Scholar]
- 20.Cheeseman M; Davies IR; Axe P; Johnson AL; Bull SD Org. Biomol. Chem 2009, 7, 3537–3548. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmeister L; Persich P; Fürstner A Chem. - Eur. J 2014, 20, 4396–4402. [DOI] [PubMed] [Google Scholar]
- 22.Albler C; Schmid W Eur. J. Org. Chem 2014, 2014, 2451–2459. [Google Scholar]
- 23.Ma J-A; Cahard D Chem. Rev 2008, 108, PR1–PR43. [DOI] [PubMed] [Google Scholar]
- 24.Flores R; Rustullet A; Alibés R; Álvarez-Larena A; de March P; Figueredo M; Font J J. Org. Chem 2011, 76, 5369–5383. [DOI] [PubMed] [Google Scholar]
- 25.Fraser RR; Mansour TS; Savard S J. Org. Chem 1985, 50, 3232–3234. [Google Scholar]
- 26.Clayden J; Greeves N; Warren S; Wothers P Organic Chemistry, 1st ed.; Oxford University Press: Oxford, U.K., 2001; pp 663, 769. [Google Scholar]
- 27.Inoue T; Liu J-F; Buske DC; Abiko A J. Org. Chem 2002, 67, 5250–5256. [DOI] [PubMed] [Google Scholar]
- 28.Ramachandran PV; Parthasarathy G; Gagare PD Org. Lett 2010, 12, 4474–4477. [DOI] [PubMed] [Google Scholar]
- 29.Abdel-Magid A; Pridgen LN; Eggleston DS; Lantos I J. Am. Chem. Soc 1986, 108, 4595–4602. [Google Scholar]
- 30.Zhong Y-L; Shing TKM J. Org. Chem 1997, 62, 2622–2624. [DOI] [PubMed] [Google Scholar]
- 31.Murakami M; Mukaiyama T Chem. Lett 1982, 11, 1271–1274. [Google Scholar]
- 32.Cabon O; Buisson D; Larcheveque M; Azerad R Tetrahedron: Asymmetry 1995, 6, 2199–2210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.