

Highlights
-
•
In this review, we detail the cellular function of tumor suppressors essential in DNA damage repair pathways, which explain their tumor suppressor function.
-
•
We present the mechanisms of action of inhibitors used to create synthetic lethality in BRCA carriers, and review the major molecular sources of drug resistance that can happen in response to treatment.
-
•
Finally, we present examples of the many strategies being developed to circumvent drug resistance.
Keywords: DNA damage responses, BRCA1, BRCA2, PARP-1, Synthetic lethality, Drug response, Familial breast cancer, Inherited cancer
Abstract
Cells are continuously subjected to DNA damaging agents. DNA damages are repaired by one of the many pathways guarding genomic integrity. When one or several DNA damage pathways are rendered inefficient, cells can accumulate mutations, which modify normal cellular pathways, favoring abnormal cell growth. This supports malignant transformation, which can occur when cells acquire resistance to cell cycle checkpoints, apoptosis, or growth inhibition signals. Mutations in genes involved in the repair of DNA double strand breaks (DSBs), such as BRCA1, BRCA2, or PALB2, significantly increase the risk of developing cancer of the breast, ovaries, pancreas, or prostate. Fortunately, the inability of these tumors to repair DNA breaks makes them sensitive to genotoxic chemotherapies, allowing for the development of therapies precisely tailored to individuals’ genetic backgrounds. Unfortunately, as with many anti-cancer agents, drugs used to treat patients carrying a BRCA1 or BRCA2 mutation create a selective pressure, and over time tumors can become drug resistant. Here, we detail the cellular function of tumor suppressors essential in DNA damage repair pathways, present the mechanisms of action of inhibitors used to create synthetic lethality in BRCA carriers, and review the major molecular sources of drug resistance. Finally, we present examples of the many strategies being developed to circumvent drug resistance.
Graphical abstract
Introduction
Mammalian cells are exposed daily to a variety of endogenous and exogenous genotoxic agents. Exposure to radiation and chemicals of natural or human origin introduces breaks or modifications in the DNA. These need to be repaired in a timely fashion to avoid mutations from being passed on to daughter cells following DNA replication and cell division. Several distinct mechanisms recognize DNA damage, process DNA ends or modified bases, and ensure restoration of the correct genetic information (Fig. 1).
Fig. 1.

DNA damages and their major repair mechanisms. Several different types of DNA damage can be discerned. Depending on the type of damage and the cellular and molecular context, they can be repaired via various different DNA repair mechanisms.
Out of all types of DNA damage, two events can be considered most deleterious for human cells: the double strand break (DSB), and replication fork collapse (RFC). If not properly repaired, both can lead to extensive loss of genetic material, genome rearrangement, and ultimately cancer.
Homologous recombination (HR) and Non-Homologous End Joining (NHEJ) are the two major pathways to repair DSBs. HR is considered more faithful than NHEJ, as it involves extensive homology search prior to repair. Conversely, NHEJ requires little homology, and ensures the ligation of DNA ends at the break. While NHEJ is operational throughout most of the cell cycle, homologous recombination is only effective during the S and G2 phases of the cell cycle, limiting its utility [1,2]. Since eukaryotes rely heavily on NHEJ to repair DSBs, efficient mechanisms are in place to limit errors and increase fidelity of the NHEJ [3], [4], [5]. However, NHEJ is still considered error-prone, as it results in frequent deletions/insertions (indels), and for this reason can be used with high efficiency to create targeted mutations using the CRISPR-Cas9 system.
Interestingly, while HR is considered faithful, mutations in HR proteins and inefficient HR are a major cause of genomic instability and are responsible for many familial cancer predispositions. Efficient DNA damage repair relies on a complex and well-orchestrated sequence of events. Upon DSB, the damage is sensed and signaled, cell cycle checkpoints are activated, and DNA damage repair (DDR) pathways are activated [6]. HR is initiated with resection of the DSBs to create 3′ single-stranded DNA (ssDNA) strands that are essential for HR and inhibit NHEJ. The recombinase RAD51 is a DNA strand-exchange protein that provides the defining step of HR, by binding to the ssDNA overhang, forming a structure named the presynaptic filament [7]. RAD51 bound to DNA is responsible for homology search, duplex capture, invasion and strand exchange [8,9]. Utilizing the sister chromatid within the homologous duplex, restoration of the original DNA sequence prior to the damage is possible [10]. RAD51 cannot carry this complex sequence of events without help from mediator proteins [11], [12], [13], [14], which facilitate all steps of HR. Sequencing and mutation analysis in vivo and in vitro have demonstrated that the complex formed by three tumor suppressors BRCA1, PALB2, and BRCA2 plays an essential role in HR.
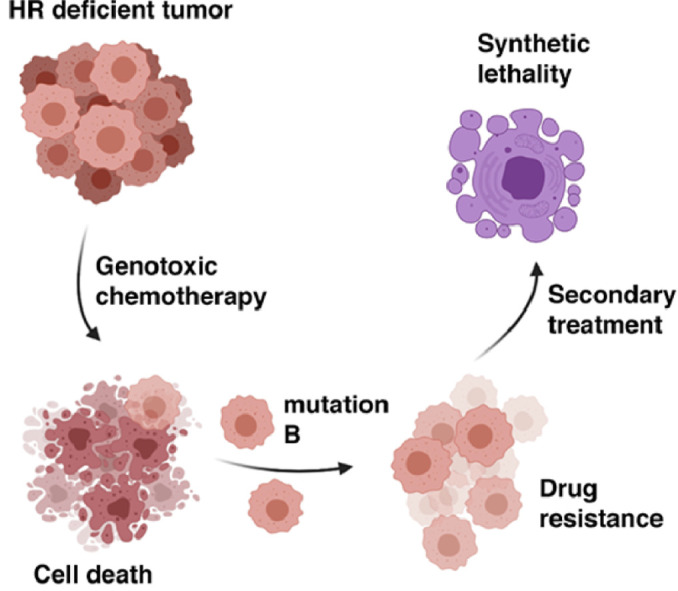
BRCA1 functions in the process of HR by promoting resection of DSBs to create 3′ ssDNA overhangs. The BRCA1-PALB2-BRCA2 complex is critical for the strand exchange reaction and loading of the RAD51 recombinase protein onto the ssDNA to create the presynaptic filament [15]. Genetically engineered mutations in RAD51 phenocopy BRCA2 mutations. While cells with defective BRCA1, BRCA2, or other HR genes accumulate mutations at a high rate, they also exhibit exquisite sensitivity to radiation, platinum-derived drugs that crosslink the DNA, or to any other form of DNA damage that induces DSBs directly or indirectly. In recent years, molecules that target DNA repair pathways other than HR have been developed to specifically kill HR-deficient cells. All adducts, base modifications and single stranded DNA breaks (SSBs) can eventually become DSBs upon passage of the replisome, in the DNA synthesis phase of the cell cycle. For this reason, increasing the lesion burden, blocking their repair, or causing the stalling and collapse of replication forks all increase DSBs, which cannot be repaired in a BRCA1/2 deficient background. Apoptosis then occurs. Of note, the use of principle, referred to as 'synthetic lethality', is extremely attractive for patients carrying a BRCA-deficiency, as they often inherit one mutated allele from a parent, and are heterozygous for the BRCA gene in all cells but the tumor. This potentially allows usage of low doses of chemotherapy and limits side effects for healthy cells. While HR deficiency can be exploited to design efficient therapeutic courses tailored to BRCA1/2 mutation carriers, such cancers are extremely unstable, commonly recur, and prone to drug resistance.
In this review, we present an overview of the links between DNA damage repair machineries and cancer; we detail the mode of action of PARP inhibitors and their potential role in non-BRCA cancers. We then review the complex mechanisms that make a cell resistant to therapy, and finally present recent advances being developed to counter drug resistance.
DNA damage repair and familial cancer
Frequently mutated DNA damage repair pathways
Homologous recombination
Mutations in the BRCA1 and BRCA2 genes are associated with an increased lifetime risk for breast and ovarian cancers. The lifetime risk for breast cancers in women with mutations in either BRCA1 or BRCA2 is 45–80%, while the lifetime risk for ovarian cancer in BRCA1-mutation carriers is 45–60% and 11–35% for BRCA2-mutation carriers [16]. While mono-allelic inactivation of these tumor suppressors leads to cancer predisposition, biallelic inactivation of BRCA1, BRCA2, or their partner PALB2, causes the rare Fanconi Anemia (FA) syndrome [17], [18], [19]. Often found in 'triple-negative' breast cancer tumors, so called due to their lack of expression of the estrogen receptor (ER), progesterone receptor (PR), and HER2 receptor, BRCA1 mutations are predominantly associated with basal-like subtypes of breast cancers. As 70% of BRCA1 mutant breast cancers lack ER expression [20], this makes them difficult to treat. BRCA2-associated breast cancers, however, are usually ER-positive and p53-negative [21].
The BRCA1 gene is located on chromosome 17q21 and encodes a multi-functional protein that is involved in DNA damage repair, cell-cycle arrest, transcriptional activation, tumorigenesis and genetic instability [22], [23], [24]. Mutations in BRCA1 are mostly observed in three particular domains, the N-terminal RING domain, exons 11–13 and the C-terminus. The RING domain interacts with the protein BARD1 and plays a key role in the ubiquitination pathway during S phase of the cell cycle [25], [26], [27]. Exons 11–13 are critical for the tumor suppressor function of BRCA1 [28,29]. The C-terminal domain plays a key role by interacting with various substrates of the DNA damage-activated kinases ATM and ATR, as well as transcription regulators, such as p53 and damage repair proteins, such as CCDC98/CtIP [30,31].
The BRCA2 gene is also highly involved in much of the same processes as BRCA1 and consists of 27 exons with eight internally repeated sequences called BRC motifs that are essential for RAD51 interaction, and thus for HR [32,33].
While BRCA1 and BRCA2 are probably the most frequently mutated HR genes in familial cancer and increase risk for cancer in the breast, ovaries, pancreas, lung, prostate, and other organs, these are not the only HR genes conferring predisposition to cancer. ATM, PALB2, CHK2, BARD1, RAD51C to cite a few, are also essential players of HR and have been found mutated in cancer (Table 1). Cells deficient for repair by HR and exhibiting typical BRCA-deficient phenotypes, such as sensitivity to DSB-creating drugs, are often referred to as exhibiting “BRCA-ness”. While the exact criteria for BRCA-ness have not been defined, gene signatures and major genome rearrangement events have been identified in whole-genome sequencing studies and this can help profile patient samples and predict their response to genotoxic chemotherapy [34], [35], [36], [37].
Table 1.
DNA repair genes deregulated in familial cancer, and their penetrance in breast cancer.
| Genea | Function/Pathway | Penetranceb |
|---|---|---|
| AKT1 | AKT signaling | Low |
| ATM | Double Strand Break (DSB) repair | Intermediate |
| BARD1 | BRCA1-associated protein complex | Intermediate |
| BRCA1 | BRCA1-associated protein complex | High |
| BRCA2 | Fanconi/BRCA | High |
| BRIP1 | Fanconi/BRCA | Intermediate |
| CDH1 | Cell adhesion | High |
| CHEK2 | DSB repair | Intermediate |
| EXO1 | DSB repair | Unknown |
| FAM175A/Abraxas | DSB repair | Intermediate |
| GEN1 | DSB repair | Unknown |
| MRE11/RAD50/NBS1 | DSB repair | Intermediate |
| PALB2 | Fanconi/BRCA | Intermediate |
| PIK3CA | AKT signaling | Unknown |
| PTEN | PI3K/MAPK Signaling | High |
| RAD51 | DSB Repair | High |
| RAD51C | Fanconi/BRCA | Intermediate |
| RAD51D | Fanconi/BRCA | Intermediate |
| STK11 | Cell Cycle/p53 regulation | High |
| TP53 | Cell growth | High |
| XRCC2 | DSB repair | Intermediate |
Some of the major DNA repair genes found mutated, silenced, or over-expressed in familial breast cancer.
Penetrance is indicated as low (<5%) intermediate (5–20%) or high (>20%) increased lifetime risk of developing breast cancer.
Other DNA repair pathways
For the purpose of this review, we will focus on HR-deficient cancers and their treatment with PARPi and platinum-derived drugs. However, one cannot ignore the essential role other DNA repair mechanisms play to prevent cancer development, by limiting mutations and maintaining genomic stability. For example, DNA mismatch repair is linked to colon cancer suppression [38,39]. Breast, gastric, and colorectal cancers are all suppressed by Base Excision Repair mechanisms (BER), while Nucleotide Excision Repair is essential for the removal of UV-damaged nucleotides and cross-link repair, thus limiting the risks of skin melanoma [40], [41], [42], [43].
In addition, it has recently become evident that another, more discreet attack on the DNA can be as deleterious and oncogenic as external genotoxic substances, if not more. Endogenous increase of replication stress, as defined by a difference in the replication fork processivity, excessive firing of origins, or stalled forks that cannot be rescued, is a major source of genomic instability and carcinogenesis [44]. Accumulation of SSB or adducts in the DNA, a deficient replisome, which processivity and/or accuracy is not fully controlled, and improper firing of origins of replication, are some of the mechanisms that can increase the risk of fork stalling. Inactivation of any of the many proteins protecting the fork against extensive resection, or promoting its restart over collapse, all contribute to increased replicative stress. Early stages of carcinogenesis often exhibit high levels of replicative stress and lack of cell cycle checkpoints, and for years genotoxic chemotherapy has been exploiting the fast growth of cancer by enhancing replicative stress.
Following major advances in our understanding of human replication mechanisms and their coupling with DNA repair machineries, therapies are now being specifically designed to target endogenous replication stress, especially those caused by identified mutations in tumor suppressors and oncogenes [45,46].
PARP1, PARP2, and PARG
Poly (ADP-ribose) polymerases (PARPs) are enzymes that are important for the cellular response to single-stranded DNA damage [47]. Specifically, PARP1 and PARP2 are recruited to sites of DNA damage via signals from unligated Okazaki fragments [48] and become activated when binding to the regions of DNA damage. Upon activation, PARP uses DNA nicking to recruit other repair proteins and subsequently uses energy from NAD+ to add polymers of PAR to itself and the other repair proteins. This process, referred to as PARylation [49,50], leads to the generation of a repair complexes that mend the damaged DNA and allows the cell to survive. The degradation of PAR, also known as dePARylation, is an important step in allowing the repair process to proceed after the repair complex has been formed. This process is mediated by a molecule known as poly (ADP-ribose) glycol-hydrolase (PARG) [48,51]. Accumulation of unrepaired DNA insults has dire consequences for the human genome and its stability, and eukaryotic cells have evolved redundant mechanisms to survive the loss of critical functions. Interestingly, while the loss of PARG in mouse cells increases sensitivity to cellular stress [52], it may lead to PARP-inhibitor resistance via the permission of the accumulation of PAR in BRCA2-deficient cells, highlighting the role of PARG molecules in facilitating DNA repair processes [48]. The many mechanisms of drug resistance will be detailed in a later section.
Exploiting DNA repair deficiencies for medicine
Synthetic lethality and PARP inhibition: a success story for HR deficient cancers
HR-deficient cells are extremely prone to accumulation of mutations, loss of genetic information, and genome rearrangement. As a result, these cells are at a high risk of losing tumor suppressors and growth inhibitions, and undergo tumorigenesis. Such cancers can be difficult to treat for many reasons. First, inherited mutations predispose one to early onset cancers. Early onset cancers are generally more advanced cancers at diagnosis [53], which are harder to treat than early-stage cancers. Second, the genome of HR deficient cells, such as cells from BRCA1/2 mutations carriers, are unstable, and this can give rise to a variety of individual clones of heterogenous genetic background. Last, the penetrance of inherited mutations is such that primary tumors can arise rarely concomitantly or more often sequentially in various tissues within the same patient [54], [55], [56], dramatically complicating diagnosis and treatment. Interestingly, these tumors, which can be aggressive in a patient, are often difficult to culture in vitro. This stems from their heightened sensitivity to exogenous stressors and their heavy reliance on biological molecules, such as specific growth factors and hormones. Moreover, their incapacity to repair DSBs makes them very sensitive to radiotherapy in vitro and in animal models, and genotoxic chemotherapy in vitro and in clinical settings. Inactivating a second DNA repair pathway ensures active and efficient killing of cells homozygous for BRCA1/2 mutations. The best example of this strategy has been the development of PARP inhibitors and their approval in the clinics. Most clinically relevant PARP-inhibitors work by binding to activated PARP1 molecules and trapping them onto damaged DNA [57] leading to stalling of the replication fork and the accumulation of single-stranded DNA breaks (SSBs). PARP inhibition also deregulates fork speed [58] and stability. Last, PARP inhibition can favor NHEJ above HR, which causes increased mutation rates. These unrepaired SSBs progress into double-stranded breaks (DSBs) upon passage of the replication fork. While cells that have intact HR are capable of repairing these DSBs that are created by PARPi in S-phase, cells with mutations in BRCA1, BRCA2 or PALB2 are unable to effectively perform HR, promoting the persistence of these DSBs [59], [60], [61]. As a result, BRCA1- or BRCA2-deficient cells have traditionally demonstrated synthetic lethality with the use of PARP-inhibitor treatment, a concept defined as causing cell death with the combination of two defects, here simultaneous HR deficiency and PARP inhibition, but cell survival when each occurs alone [57] (Fig. 2).
Fig. 2.

PARPi exploits HR deficiency to induce synthetic lethality. Concomitant HR deficiency and inhibition of PARP using PARP inhibitors (PARPi) causes cell death, while on their own, neither leads to cell death.
Ultimately, cancer cell death is the goal of PARPi treatment, and thus BRCA1/2 deficient patients have been the primary target population for treatment with this class of drugs. PARPi also shows promises in treating carriers of a PALB2 mutation, and has been tested with some success on triple-negative breast cancers, serous carcinomas of the ovaries, tumors with high replicative stress levels, and in combination with immune checkpoint blockade [62], [63], [64], [65], [66]. This cautiously offers hope for the development of novel combination therapies. Currently, three third generation PARPi molecules have been approved for the treatment of BRCA1/2 deficient breast and ovarian cancers: olaparib, niraparib, and talazoparib. A fourth, veliparib, is under investigation for the treatment of breast, lung, and ovarian cancer. In the clinic, PARPi have been mostly successful, but also exhibited a range of efficacy based on the molecule used and the cancer treated. This is likely due to slightly different mechanisms of action of the various inhibitors, and the way they impact the PARPi-DNA complex, specifically, whether they trap or exclude PARP1 to make it non-functional. The full extent of these mechanism has only started to be understood, thanks to elegant and recent functional [67] and structural studies [68].
PARPi combination therapies for HR proficient cancers
Pin1 inhibition via ATRA
Pin1 is a molecule that has been found to stabilize BRCA1 and inhibition of Pin1 increases sensitivity to PARP-inhibitor treatment, causing a near-complete block in cell proliferation [69,70]. All-trans retinoic acid (ATRA), a drug that is well-known for its treatment in acute promyelocytic leukemia, has been found to have Pin1 inhibitory effects. Combining ATRA with olaparib has been shown to decrease tumor growth and induce apoptosis in BRCA1/2 proficient triple-negative breast cancer cells. ATRA has the potential to be used to disrupt homologous recombination and sensitize cancer cells to treatment with PARP-inhibitors. Studies to assess the toxicity of the combination therapy are warranted.
Alantolactone
Another agent that can be used to induce PARP-inhibitor sensitivity in HR-proficient cells is alantolactone, via its mechanism of inhibition of thioredoxin reductase leading to selective accumulation of oxidative DNA damage in cancer cells [71]. Of note, this particular drug sensitizes cancer cells to PARPi that induce PARP trapping. Veliparib is a PARP-inhibitor that causes limited DNA damage signaling and lacks potent ability to cause PARP trapping, and thus its combination with alantolactone does not produce synergistic effects. However, combination of alantolactone with olaparib, a strong PARP-trapping drug, has been shown to induce significant synergistic effects. This finding has the potential to broaden the cancer population that can be treated with PARP-inhibitors [72]. The combination of the two drugs requires further assessment in clinical trials.
CDK12 inhibitors
CDK12 is thought to be involved in promoting the expression of DNA-damage response genes such as BRCA1, ATR, FAN1, and FANCD2 [73]. Inhibition of CDK12 with the drug dinaciclib has the potential to sensitize cells to PARP-inhibitors in both BRCA-deficient and BRCA wild type triple-negative breast cancer cells. It probably does so by promoting genomic instability and increase sensitivity to DNA damage [74]. A phase I clinical trial is currently underway studying dinaciclib and veliparib combination therapy (trial number NCT01434316, clinicaltrials.gov). Importantly, CDK12 deficiency was noted to be a significant biomarker for PARPi sensitivity in ovarian cancer cells, inducing synthetic lethality similar to BRCA1/2 mutations [75].
PI3K-Akt-mTOR pathway inhibitors
The PI3K-Akt pathway is a cellular pathway that promotes survival and proliferation [76]. PI3K inhibitors in combination with PARPi treatment have been shown to have synergistic therapeutic effects in BRCA1 deficient breast cancer and BRCA1-proficient triple negative breast cancer [77]. Another study found that PI3KCA-deficient ovarian cancer cells were more responsive to combination therapy with the PI3K inhibitor BKM120 and olaparib than monotherapy with either drug [78]. Akt can potentially be inhibited for cancer treatment, but no drugs have been FDA-approved at this time [79]. Inhibition of mTOR using everolimus is also an effective treatment option for several different cancer types, and has been approved for treatment in advanced breast cancer. Akt activates a signaling cascade that promotes the survival of the cell, and it could interfere with the ability of PARP-inhibitor to induce cellular death [80]. However, the role of Akt-inhibitors and mTOR-inhibitors in relationship with PARP-inhibitor resistance is not fully understood yet.
Molecular basis of drug resistance
Causes and mechanisms of drug resistance
Restoration of the BRCA1-PALB2-BRCA2 axis
Defective HR that occurs as a result of BRCA1, BRCA2, or PALB2 mutations is a key component for the overall efficacy of PARP inhibitors. The combination of PARP inhibition and loss of HR leads to cell death, and without one or the other, the cell will survive. Despite excellent clinical response to PARP inhibition, HR-deficient cells almost always become resistant by developing reversion mutations that either restore or bypass HR [81,82]. The mutations that cause HR-deficiency in BRCA1/2 or PALB2 genes are often frameshift mutations, leading to non-functioning proteins. With reversion mutations, the reading frame is restored and thus, so is some of the protein function. Since new mutations rarely occur at the original mutation sites, newly synthetized proteins are chimeric versions and contain deletions (Fig. 3). However, as critical functional domains are restored in the C-terminus, HR is no longer defective and cells become resistant to PARPi (Fig. 3). Acquisition of secondary mutations is a prominent mechanism of PARPi resistance, which has been observed in breast, ovarian, and pancreatic cancers. Such acquisition also confers resistance to platinum-derived agents, like cisplatin and carboplatin [83], [84], [85].
Fig. 3.

Major mechanisms of resistance to PARPi. Top panel: Restoration of HR by acquisition of secondary mutations in HR genes, or in NHEJ genes. Bottom left panel: drug efflux pumps can decrease intracellular concentration of drugs such as PARPi. Bottom right: Stabilization of forks allow rescue even in the absence of other cellular components, and reverse the synthetic lethality, thus rendering cells resistant to PARPinhibition.
Demethylation of the promoter of the BRCA1 gene, which is often hypermethylated in cancer and turned off, is another means to restore the expression of a fully functional BRCA1 [86] and thus reactivate HR. When HR is intact, RAD51 is recruited to sites of DNA damage to assist in the initiation of HR. This can be visualized by indirect immunofluorescence in cells [87,88] or immunohistochemistry (IHC) on tissue samples [89]. The restoration of HR in BRCA1/2 or PALB2 mutants is associated with detectable increases in RAD51 foci [90], [91], [92], suggesting that these foci may be used as screening markers for potential resistance to PARP inhibition or platinum therapies.
Reactivation of HR through expression of RAD51
In addition to the reversion mutations mentioned above, secondary somatic mutations in RAD51 paralogs have been identified and associated with PARPi resistance in BRCA1/2 mutated serous ovarian cancer cells [93]. These mutations, as well as mutations in RAD51 itself [94], are thought to restore RAD51’s ability to mediate homologous sequence invasion in the homologous recombination process. This increase in HR cancels the synthetic lethal effects of PARPi treatment, and cells become resistant to the drug.
Similarly, downregulation of Early Mitotic Inhibitor 1 (EMI1) is associated with RAD51 stabilization and accumulation, subsequent restoration of HR and thus PARPi resistance. Mutations that cause downregulation of EMI1 are selected with a high frequency in triple-negative breast cancer order enabling them to evade Olaparib-induced cell death [95].
Interestingly, RAD51 is often found overexpressed in cancer and this is associated with poor prognosis [96], [97], [98], suggesting that high levels confer a selective advantage to tumor cells, possibly through drug resistance.
Mutations in NHEJ can compensate for lack of HR
Mutations in the components of the NHEJ pathway can lead to reactivation of HR in BRCA1/2 mutant cells. In normal cells, BRCA1 favors HR directly by interacting with PALB2 and BRCA2 to promote strand exchange. However, it also prevents NHEJ by antagonizing 53BP1 [99]. 53BP1 is a key component of the NHEJ machinery, and plays an essential role in the DSB repair pathway choice [100], [101], [102]. It promotes NHEJ by limiting DNA end-resection, which is required for HR to occur. 53BP1 interacts with RIF1, REV7, and the Shieldin complex (SHLD1, SHLD2, SHLD3) and the resulting complex, referred to as the 53BP1-RIF1-REV7-Shieldin axis, inhibits resection [103], [104], [105]. Loss of any of the factors in this complex has been associated with PARPi resistance in BRCA1-deficient cells, but interestingly not in BRCA2-deficient cells [106]. It is thought that the inhibition of the 53BP1-RIF1-REV7-Shieldin axis allows for end resection then HR to occur in a BRCA1-independent and RNF168-dependent fashion. RNF168 is an E3 ubiquitin ligase that can bypass BRCA1 requirement through its direct interaction with PALB2 to initiate HR mechanism.
Another molecule that has been linked to PARPi resistance is Dynein Light Chain 1 (DYNLL1), which, like 53BP1, functions to inhibit DNA end resection in normal cells and thus favors NHEJ. While it has been suggested that DYNLL1 interacts with 53BP1 to inhibit end resection, or that DYNLL1 works by inhibiting components of the resection machinery such as MRE11 [107], [108], [109], the exact relationship between DYNLL1 and the 53BP1 axis is still unclear. Similar to the inactivation of 53BP1, loss of DYNLL1 is associated with PARPi-resistance [109].
Stabilization of the replication fork
Stalling of the replication fork is extremely dangerous for genome integrity, as unprotected forks that do not restart eventually collapse, leading to DSBs and loss of genetic information. In normal cells, BRCA2 and other DNA repair proteins contribute to fork protection [110,111]. In BRCA1/2 mutants or cells exhibiting BRCA phenotypes in the absence of BRCA mutations (so-called “BRCA-like” cells), PARPi functionality leads to the accumulation of single stranded breaks that stall replication forks. PTIP and EZH2 then recruit the nucleases MRE11 and MUS81 to stalled forks, where they resect nascent strands and induce replication fork collapse (RFC) [110,112], resulting in double stranded breaks [57].
Stabilization of the replication fork through a variety of mechanisms has been seen as a potential mechanism of resistance to PARP-inhibitors. A frequent culprit is FANCD2, which is often expressed in BRCA1/2 mutant ovarian, breast, and uterine cancer cells. It also functions to protect the fork by suppressing MRE11-mediated fork collapse, and by stabilizing the fork, it leads to PARPi resistance [113]. Other proteins that promote resection of the fork in the absence of BRCA2, such as SMARCAL1, or replication forks remodelers such as HTLF and ZRANB3, also induce PARPi resistance when deleted [114,115]. SLFN11 is another important protein that has been linked to PARP-inhibitor resistance. SLFN11 functions to prolong S-phase of the replication cycle upon replication fork stress by inhibiting replication. This ultimately causes prolonged and irreversible stalling of the replication fork that is irreversible and can render a cell more susceptible to the effects of PARP-inhibitors [116]. Loss of SLFN11 can lead to resistance to PARPi-Temozolomide combination therapy [117].
Drug efflux pumps
A readily acquired mechanism of resistance to any class of drugs is the use of efflux pumps that transport drugs out of the cell, thus limiting their intracellular availability. ABCB1 genes, also referred to as multidrug resistance (MDR1) genes, encode for p-glycoprotein efflux pumps that ultimately reduce the available amount of PARPi drug in the cell, leading to reduced efficacy of the drug and PARPi resistance, especially in BRCA1-deficient breast cancer and ovarian cancer cell lines [118,119]. Patients treated with chemotherapy prior to initiating PARPi therapy may be at increased risk for this resistance mechanism, as it has been demonstrated that the ABCB1 transporters are upregulated in non-naïve (e.g., previously exposed to therapies) tumors as a result of chromosomal translocations that occur upon paclitaxel treatment [120]. MDR1 inhibitors, such as verapamil and elacridar, can reverse ABCB1-mediated drug-resistance in both olaparib- and paclitaxel-treated ovarian cancer cells [118]. Interestingly, paclitaxel-resistant ovarian cancer cell lines were also cross-resistant to olaparib, rucaparib, and doxorubicin but were not resistant to veliparib, which is not a substrate of p-glycoprotein efflux pumps. The above evidence highlights the difference between PARPi molecules, and the need for caution when prescribing olaparib and paclitaxel, with the upregulation of MDR1 being a significant risk factor in the resistance of cancer cells to treatment. Clinical trials involving MDR1 inhibitors have yielded poor outcomes, and recent studies point toward MDR1 being essential for an efficient immune response [121]. While further studies are needed to delineate the role of MDR1 gene screening in designing treatment course, it remains very uncertain how effective regimens that combine low-dose MDR1 inhibitors with PARP inhibitors – to limit or reverse drug resistance – will be.
PARP-1 and PARG mutations
Mutations in the PARP1 molecule itself have been linked with the development of PARPi resistance, particularly by decreasing PARP trapping on damage DNA segments, the main mechanism by which PARPi work [122]. Interestingly, the specific types of BRCA1 mutations can tolerate mutations in PARP better than others. Mutations in exon-11 of BRCA1 leave the mutant BRCA1 protein partially functional, while frameshift mutations leading the deletion of C-terminal BRCT domains cause complete loss of BRCA1 function. Experimental evidence showed that the partially functional BRCA1 mutants were able to tolerate PARP mutations and survive, while cells with the frameshift mutated BRCA1 genes were not able to survive with PARP mutations. Because of this, the partially functional mutant BRCA1 genes with concomitant PARP1 mutations pose the risk of developing PARPi resistance through decreased PARP trapping. Frameshift BRCA1 mutations theoretically will not be resistant to PARPi through the mechanism of PARP mutation, because these cells would not be viable in the first place [122].
As briefly mentioned previously, PARG plays a role in removing PAR groups from multiple proteins in a process referred to as dePARylation. This process is necessary for the DNA repair process to occur. Loss of PARG in BRCA2-deficient cells that are being treated with a PARPi is thought to lead to partial restoration of PARylation so that there is decreased PARP trapping and also some return of DNA-damage repair signaling, ultimately leading to PARPi resistance [123]. Studies conducted to assess mechanisms of resistance to PARPi in ovarian cancer cells detected reduced PARG levels in both HR-proficient and HR-deficient cell lines [124], supporting the notion that loss of PARG can contribute to the development of PARPi drug resistance. Interestingly, in cells that have not been previously treated with PARPi, inhibition of PARG appears to be synthetically lethal with HR deficiency [125], restoring PARPi sensitivity. It is postulated that PARG inhibition leads to dysregulation of PARylation and the accumulation of unrepaired DNA with subsequent replication fork stalling [126]. Further studies are warranted to define the relationship between PARG and PARP and what role PARG inhibition has in the treatment of HR-deficient or HR-proficient malignancies. In addition, TARG1 is a molecule that plays a similar role to PARG with respect to dePARylation and may be another potential target to prevent PARPi resistance, but further studies regarding this are warranted [127].
Therapeutic options for aquired drug resistance
Drug resistance is extremely common in advanced cancers. Overcoming this problem is therefore of paramount importance for improving treatment efficacy. With the success using PARP inhibition, yet the many potential mechanisms for PARPi resistance, there is an abundant body of research that looks to address interventions to restore PARPi sensitivity. Fortunately, a variety of pathways can be targeted and drugs under study show great potential for the reduction and even the reversal of PARP-inhibitor resistance.
ATR inhibitors
Three major kinases control the majority of the DNA damage responses in vertebrates, by acting both as sensors of the break and effectors of the repair signaling [128]. The kinases: ATM, ATR, and DNA-PK, are responsible for phosphorylating most DNA repair proteins, and by doing so activate proteins, allow protein-protein or protein-DNA interactions, and eventually DNA damage repair to occur. While mutations in their respective genes cause human disease, molecules have been developed against each of these kinases offer hope as combination therapy with genotoxic chemo-regimen. ATM deficiency has been shown to increase sensitivity to PARPi in lung adenocarcinoma, metastatic gastric cancer, metastatic prostate cancer, and lymphoid tumor cells, likely due to genomic instability and increased DNA damage in the absence of ATM's counteraction of toxic end-joining to allow the repair of DSBs [129], [130], [131], [132], [133]. This highlights the importance of screening for ATM-deficiency as a potential marker for PARPi-sensitivity. Additionally, combination of ATR-inhibitors with PARP-inhibitors in the treatment of ATM-deficient BRCA1/2 mutated tumors achieved therapeutic response at lower concentrations than monotherapy with either drug [134]. There may be a role for the combination of ATR-inhibitors and PARP-inhibitors in ATM-deficient cancer cells to reduce drug toxicities and resistance that is associated with prolonged use of high-dose drugs.
In addition to their treatment of ATM-deficient BRCA1/2 mutant cancer cells, ATR inhibitors in combination with PARP-inhibitors have been shown effective in the treatment of cancers with acquired BRCA1/2 mutations and those with wild-type BRCA1/2. While ATM is the DNA repair upstream kinase, ATR's role is replication centric. Following DNA damages, ATR activates DNA replication checkpoints and rescues operations of stalled replication forks by signaling DNA damage and activating DNA repair proteins for their recruitment to damaged fork. ATR recruits PALB2 to sites of DNA-damage, both in the BRCA-dependent and BRCA-independent pathways [135]. Increased PALB2 recruitment to DNA damage sites restores HR and is linked to PARPi resistance [136,137]. Inhibition of ATR has the potential to reverse PARPi caused by secondary mutations in BRCA1/2, as it limits HR through PALB2 [138].
Combination treatment with ATRi and PARPi is a promising therapeutic avenue for drug resistant cancers. Currently, several clinical trials are underway to assess the role of AZD6738 (ceralasertib) in future cancer drug regimens (see [139] for a list of clinical interventions using this ATRi).
Cell cycle kinases inhibitors
WEE1 kinase functions to prolong the G2 phase of the cell cycle by phosphorylating CDK1, thus inhibiting it and preventing it from initiating progression of the cell cycle while cells repair DNA damages. One study showed that both HR-deficient and HR-proficient high-grade serous ovarian cancer cells with resistance to olaparib were sensitive to treatment with WEE1 kinase inhibitor, AZD1775 [124]. It is thought that WEE1 inhibitors promote more DNA damage and thus cell death in the absence of inhibitor CDK1 phosphorylation. Administration of a WEE1 inhibitor concurrently with a PARP inhibitor in ovarian cells is highly effective but toxic. Sequential therapy where PARPi are followed by administration of WEE1i is to be preferred as it is less toxic than the combination, and targets specifically cells with high replication stress thus killing preferentially tumor cells [140]. Clinical trials are currently underway to further assess the clinical role of AZD1775 in cancer treatment.
Another cell cycle kinase, CDK12 is thought to be involved in promoting the expression of DNA-damage response genes, such as BRCA1, ATR, FAN1, and FANCD2. Inhibition of CDK12 by dinaciclib has the potential to reverse primary and acquired PARP-inhibitor resistance in both BRCA-deficient and BRCA wild type triple-negative breast cancer cells, likely through increasing genomic instability [74]. A phase I clinical trial is currently underway studying dinaciclib and veliparib combination therapy. Moreover, CDK12 deficiency was noted to be a significant biomarker for PARPi sensitivity in ovarian cancer cells, inducing synthetic lethality similar to BRCA1/2 mutated cells when treated with a PARPi [75]. This highlights the importance of the use of genomic sequencing to better predict a specific cancer's likelihood of response to PARPi treatment. Although sequencing is not systematic in patient's management, the identification of specific signatures and the recent development of specific antibodies for immuno-histochemistry [141,142] have made it easier to use specific markers of drug response or drug resistance. Interestingly, deletion of CDK12 has been shown to reverse PARPi resistance regardless of the BRCA mutation status and is one of the most promising therapeutic options to counter PARPi resistance [[73], [74], [75],143].
PARG inhibitors
BRCA2 mutation carrier cells that have defective HR demonstrate sensitivity to inhibition of PARG, a potential treatment avenue for HR-deficient cells. This effect from PARG-inhibition is not seen in cells with intact homologous recombination [144]. It is thought that PARG-inhibitors can induce accumulation of DNA damage and stalling of the replication fork by blocking the essential dePARYlation step that is required for efficient DNA repair [126]. To assess the synthetic lethality of PARG-inhibitors with homologous recombination deficiency, one study assessed the effects of PARG-inhibitors and PARP-inhibitors on PTEN-mutated glioblastoma multiforme cells. The study found that neither drug induced synthetic lethality in PTEN-mutated or PTEN-wildtype cells [126]. While PARG knock down is synthetic lethal with many HR genes, such as BRCA1, BRCA2, PALB2, FAM175A (ABRAXAS) and BARD1, studies have suggested that PARG inhibition in vivo might not be efficient to kill BRCA1-deficient tumors [145], and could even promote PARPi resistance. In the light of these conflicting data, and as new PARGi are being developed [146], further studies are required to uncover the potential role for PARG-inhibitors in cancer therapy and more specifically their role, if any, in PARPi resistance or reversion of it.
Direct inhibition of oncogene transcription
BRD4 promotes the transcription of several oncogenes by the recruitment of transcription factors or by binding to their enhancers. Inhibition of BRD4 causes deficiency of homologous recombination, resulting in synthetic lethality with PARP-inhibitor treatment. Interestingly, BRD4-inhibitors were found to reverse PARP-inhibitor resistance related to reversion of HR function, specifically through secondary BRCA1, RAD51C, and RAD51D mutations, loss of 53BP1, decreased PARP1 levels, and KRAS mutations. [147,148] Clinical trials assessing the safety of BRD4-inhibitors in combination with PARP-inhibitors are required for further evaluation of this treatment regimen in the role of clinical PARP-inhibitor resistance treatment.
Similar to BRD4-inhibitors, BET-inhibitors function by binding to and inhibiting bromodomains that are responsible for oncogenesis. BET-inhibitors have been found to inhibit BRCA1 and RAD51 expression, and have demonstrated synthetic lethality with PARP-inhibitors in the treatment of HR-proficient tumor xenografts [149,150]. Clinical trials are currently underway to study these drugs.
Conclusion
Whole genome sequencing and data mining using large datasets has contributed a better understanding of cancer etiology, the sub-classification of patients into risk groups, and the establishment of genomic signatures. All this new information, when available, can help predict patient survival, response to drug treatment, and possibly even short- and long- term drug resistance.
While these advances in understanding specific predisposition to certain types of cancers for any given individual is powerful and can help design a therapeutic course, it has shown limitations. Not all mutations within the same gene yield similar cellular consequences, and researchers and clinicians alike are now trying to classify patients based on the clinicopathological features of their tumors rather than solely based on their inactivated genes. For example, while BRCA1 and BRCA2 genes are well-known tumor suppressors through their role in HR, their mutations can be mimicked by inactivation of other genes, and differentially impact on BRCA1/2 protein function. The shared clinical features and especially the sensitivity of such tumors for genotoxic agents (of physical and chemical) is defined as ‘BRCAness’. The definition of BRCAness is a defect in HR repair, and high genetic instability that ensues, phenocopying deficiencies in BRCA1 or BRCA2. As our understanding of BRCA1 and BRCA2 function grows, other roles, such as contribution to ubiquitination and replication fork protection respectively, can be included in BRCAness. Defining a “behavioral” pattern and predicting drug response for all tumors is the ultimate goal, and a major barrier in attaining patient- tailored, or personalized medicine for all.
Two major reasons that impair perfect prediction of long-term cancer behavior and drug response are (i) the complex biology of carcinogenesis (ii) the multiple mechanisms that can lead to drug resistance, and the difficulty to provide toxic prophylactic treatment in anticipation of resistances and recurrence. The development of combination therapies will hopefully identify additional candidates for tailored therapy by PARPi, ATRi, and other promising molecules.
Declaration of Competing Interest
All authors declared that there are no conflicts of interest.
Acknowledgments
Authors’ contribution
All authors wrote the manuscript, E.D and P.D edited it, D.J prepared figures.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by a grant from the San Antonio Area Foundation. The Mays Cancer Center is supported by NCI cancer center support core grant P30 CA054174.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
References
- 1.Rothkamm K., Kruger I., Thompson L.H., Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23(16):5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biehs R., Steinlage M., Barton O., Juhasz S., Kunzel J., Spies J., Shibata A., Jeggo P.A., Lobrich M. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell. 2017;65(4):671–684. doi: 10.1016/j.molcel.2016.12.016. e675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betermier M., Bertrand P., Lopez B.S. Is non-homologous end-joining really an inherently error-prone process. PLos Genet. 2014;10(1) doi: 10.1371/journal.pgen.1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emerson C.H., Lopez C.R., Ribes-Zamora A., Polleys E.J., Williams C.L., Yeo L., Zaneveld J.E., Chen R., Bertuch A.A. Ku DNA end-binding activity promotes repair fidelity and influences end-processing during nonhomologous end-joining in saccharomyces cerevisiae. Genetics. 2018;209(1):115–128. doi: 10.1534/genetics.117.300672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y., Wang J., Zhou G., Lajeunesse M., Le N., Stawicki B.N., Corcino Y.L., Berkner K.L., Runge K.W. Nonhomologous end-joining with minimal sequence loss is promoted by the Mre11-Rad50-Nbs1-Ctp1 complex in schizosaccharomyces pombe. Genetics. 2017;206(1):481–496. doi: 10.1534/genetics.117.200972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sancar A., Lindsey-Boltz L.A., Unsal-Kacmaz K., Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 7.Short J.M., Liu Y., Chen S., Soni N., Madhusudhan M.S., Shivji M.K., Venkitaraman A.R. High-resolution structure of the presynaptic RAD51 filament on single-stranded DNA by electron cryo-microscopy. Nucleic. Acids. Res. 2016;44(19):9017–9030. doi: 10.1093/nar/gkw783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J., Ehmsen K.T., Heyer W.D., Morrical S.W. Presynaptic filament dynamics in homologous recombination and DNA repair. Crit. Rev. Biochem. Mol. Biol. 2011;46(3):240–270. doi: 10.3109/10409238.2011.576007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan M.R., Bernstein K.A. Rad-ical new insights into RAD51 regulation. Genes (Basel) 2018;(12):9. doi: 10.3390/genes9120629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morrical S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015;7(2) doi: 10.1101/cshperspect.a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godin S., Wier A., Kabbinavar F., Bratton-Palmer D.S., Ghodke H., Van Houten B., VanDemark A.P., Bernstein K.A. The Shu complex interacts with Rad51 through the Rad51 paralogues Rad55-Rad57 to mediate error-free recombination. Nucleic. Acids. Res. 2013;41(8):4525–4534. doi: 10.1093/nar/gkt138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tavares E.M., Wright W.D., Heyer W.D., Le Cam E., Dupaigne P. In vitro role of Rad54 in Rad51-ssDNA filament-dependent homology search and synaptic complexes formation. Nat. Commun. 2019;10(1):4058. doi: 10.1038/s41467-019-12082-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor M.R.G., Spirek M., Chaurasiya K.R., Ward J.D., Carzaniga R., Yu X., Egelman E.H., Collinson L.M., Rueda D., Krejci L. Rad51 paralogs remodel pre-synaptic RAD51 filaments to stimulate homologous recombination. Cell. 2015;162(2):271–286. doi: 10.1016/j.cell.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zelensky A., Kanaar R., Wyman C. Mediators of homologous DNA pairing. Cold Spring Harb. Perspect. Biol. 2014;6(12) doi: 10.1101/cshperspect.a016451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prakash R., Zhang Y., Feng W., Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015;7(4) doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King M.C., Marks J.H., Mandell J.B. New York breast cancer study G: breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 17.Sawyer S.L., Tian L., Kahkonen M., Schwartzentruber J., Kircher M., University of Washington Centre for Mendelian G., Consortium F.C., Majewski J., Dyment D.A., Innes A.M. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–142. doi: 10.1158/2159-8290.CD-14-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Andrea A.D. Susceptibility pathways in Fanconi's anemia and breast cancer. N. Engl. J. Med. 2010;362(20):1909–1919. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tischkowitz M., Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70(19):7353–7359. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lakhani S.R., Reis-Filho J.S., Fulford L., Penault-Llorca F., van der Vijver M., Parry S., Bishop T., Benitez J., Rivas C., Bignon Y.J. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005;11(14):5175–5180. doi: 10.1158/1078-0432.CCR-04-2424. [DOI] [PubMed] [Google Scholar]
- 21.Veronesi A., de Giacomi C., Magri M.D., Lombardi D., Zanetti M., Scuderi C., Dolcetti R., Viel A., Crivellari D., Bidoli E. Familial breast cancer: characteristics and outcome of BRCA 1-2 positive and negative cases. BMC Cancer. 2005;5:70. doi: 10.1186/1471-2407-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narod S.A., Foulkes W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer. 2004;4(9):665–676. doi: 10.1038/nrc1431. [DOI] [PubMed] [Google Scholar]
- 23.Tarsounas M., Sung P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020;21(5):284–299. doi: 10.1038/s41580-020-0218-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida K., Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 2004;95(11):866–871. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meza J.E., Brzovic P.S., King M.C., Klevit R.E. Mapping the functional domains of BRCA1. Interaction of the ring finger domains of BRCA1 and BARD1. J. Biol. Chem. 1999;274(9):5659–5665. doi: 10.1074/jbc.274.9.5659. [DOI] [PubMed] [Google Scholar]
- 26.Baer R., Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002;12(1):86–91. doi: 10.1016/s0959-437x(01)00269-6. [DOI] [PubMed] [Google Scholar]
- 27.Stewart M.D., Duncan E.D., Coronado E., DaRosa P.A., Pruneda J.N., Brzovic P.S., Klevit R.E. Tuning BRCA1 and BARD1 activity to investigate RING ubiquitin ligase mechanisms. Protein Sci. 2017;26(3):475–483. doi: 10.1002/pro.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorodetska I., Kozeretska I., Dubrovska A. BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J. Cancer. 2019;10(9):2109–2127. doi: 10.7150/jca.30410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seong M.W., Cho S.I., Kim K.H., Chung I.Y., Kang E., Lee J.W., Park S.K., Lee M.H., Choi D.H., Yom C.K. A multi-institutional study of the prevalence of BRCA1 and BRCA2 large genomic rearrangements in familial breast cancer patients. BMC Cancer. 2014;14:645. doi: 10.1186/1471-2407-14-645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohammad D.H., Yaffe M.B. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair (Amst.) 2009;8(9):1009–1017. doi: 10.1016/j.dnarep.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aparicio T., Gautier J. BRCA1-CtIP interaction in the repair of DNA double-strand breaks. Mol Cell Oncol. 2016;3(4) doi: 10.1080/23723556.2016.1169343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen P.L., Chen C.F., Chen Y., Xiao J., Sharp Z.D., Lee W.H. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc. Natl. Acad. Sci. U. S. A. 1998;95(9):5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galkin V.E., Esashi F., Yu X., Yang S., West S.C., Egelman E.H. BRCA2 BRC motifs bind RAD51-DNA filaments. Proc. Natl. Acad. Sci. U. S. A. 2005;102(24):8537–8542. doi: 10.1073/pnas.0407266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birkbak N.J., Wang Z.C., Kim J.Y., Eklund A.C., Li Q., Tian R., Bowman-Colin C., Li Y., Greene-Colozzi A., Iglehart J.D. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012;2(4):366–375. doi: 10.1158/2159-8290.CD-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popova T., Manie E., Rieunier G., Caux-Moncoutier V., Tirapo C., Dubois T., Delattre O., Sigal-Zafrani B., Bollet M., Longy M. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012;72(21):5454–5462. doi: 10.1158/0008-5472.CAN-12-1470. [DOI] [PubMed] [Google Scholar]
- 36.Abkevich V., Timms K.M., Hennessy B.T., Potter J., Carey M.S., Meyer L.A., Smith-McCune K., Broaddus R., Lu K.H., Chen J. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer. 2012;107(10):1776–1782. doi: 10.1038/bjc.2012.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquard A.M., Eklund A.C., Joshi T., Krzystanek M., Favero F., Wang Z.C., Richardson A.L., Silver D.P., Szallasi Z., Birkbak N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015;3:9. doi: 10.1186/s40364-015-0033-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S.K.H., Martin A. Mismatch repair and colon cancer: mechanisms and therapies explored. Trends Mol. Med. 2016;22(4):274–289. doi: 10.1016/j.molmed.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Kheirelseid E.A., Miller N., Chang K.H., Curran C., Hennessey E., Sheehan M., Kerin M.J. Mismatch repair protein expression in colorectal cancer. J. Gastrointest. Oncol. 2013;4(4):397–408. doi: 10.3978/j.issn.2078-6891.2013.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Limpose K.L., Corbett A.H., Doetsch P.W. BERing the burden of damage: pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair (Amst.) 2017;56:51–64. doi: 10.1016/j.dnarep.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szalat R., Samur M.K., Fulciniti M., Lopez M., Nanjappa P., Cleynen A., Wen K., Kumar S., Perini T., Calkins A.S. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia. 2018;32(1):111–119. doi: 10.1038/leu.2017.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeulen W., Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013;5(8) doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mulderrig L., Garaycoechea J.I. XPF-ERCC1 protects liver, kidney and blood homeostasis outside the canonical excision repair pathways. PLos Genet. 2020;16(4) doi: 10.1371/journal.pgen.1008555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanjiv K., Hagenkort A., Calderon-Montano J.M., Koolmeister T., Reaper P.M., Mortusewicz O., Jacques S.A., Kuiper R.V., Schultz N., Scobie M. Cancer-specific synthetic lethality between ATR and CHK1 kinase activities. Cell Rep. 2016;14(2):298–309. doi: 10.1016/j.celrep.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilad O., Nabet B.Y., Ragland R.L., Schoppy D.W., Smith K.D., Durham A.C., Brown E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010;70(23):9693–9702. doi: 10.1158/0008-5472.CAN-10-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heale J.T., Ball A.R., Jr., Schmiesing J.A., Kim J.S., Kong X., Zhou S., Hudson D.F., Earnshaw W.C., Yokomori K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell. 2006;21(6):837–848. doi: 10.1016/j.molcel.2006.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Noordermeer S.M., van Attikum H. PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019;29(10):820–834. doi: 10.1016/j.tcb.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Dziadkowiec K.N., Gasiorowska E., Nowak-Markwitz E., Jankowska A. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny. 2016;15(4):215–219. doi: 10.5114/pm.2016.65667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Plummer R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer? Breast Cancer Res. 2011;13(4):218. doi: 10.1186/bcr2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koh D.W., Lawler A.M., Poitras M.F., Sasaki M., Wattler S., Nehls M.C., Stoger T., Poirier G.G., Dawson V.L., Dawson T.M. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. U. S. A. 2004;101(51):17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortes U., Tong W.M., Coyle D.L., Meyer-Ficca M.L., Meyer R.G., Petrilli V., Herceg Z., Jacobson E.L., Jacobson M.K., Wang Z.Q. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 2004;24(16):7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abualkhair W.H., Zhou M., Ahnen D., Yu Q., Wu X.C., Karlitz J.J. Trends in incidence of early-onset colorectal cancer in the united states among those approaching screening age. JAMA Netw. Open. 2020;3(1) doi: 10.1001/jamanetworkopen.2019.20407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Markowska A., Lubin J., Markowska J., Kasprzak B., Chajewska-Czekanska M., Madry R., Stawicka M. Multiple primary cancers in BRCA 1/2 carriers - a review of literature and our observations. Eur. J. Gynaecol. Oncol. 2017;38(3):361–363. [PubMed] [Google Scholar]
- 55.Gangi A., Cass I., Paik D., Barmparas G., Karlan B., Dang C., Li A., Walsh C., Rimel B.J., Amersi F.F. Breast cancer following ovarian cancer in BRCA mutation carriers. JAMA Surg. 2014;149(12):1306–1313. doi: 10.1001/jamasurg.2014.1081. [DOI] [PubMed] [Google Scholar]
- 56.Molina-Montes E., Perez-Nevot B., Pollan M., Sanchez-Cantalejo E., Espin J., Sanchez M.J. Cumulative risk of second primary contralateral breast cancer in BRCA1/BRCA2 mutation carriers with a first breast cancer: a systematic review and meta-analysis. Breast. 2014;23(6):721–742. doi: 10.1016/j.breast.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 57.Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 2011;5(4):387–393. doi: 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maya-Mendoza A., Moudry P., Merchut-Maya J.M., Lee M., Strauss R., Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559(7713):279–284. doi: 10.1038/s41586-018-0261-5. [DOI] [PubMed] [Google Scholar]
- 59.Scully R. Role of BRCA gene dysfunction in breast and ovarian cancer predisposition. Breast Cancer Res. 2000;2(5):324–330. doi: 10.1186/bcr76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buisson R., Dion-Cote A.M., Coulombe Y., Launay H., Cai H., Stasiak A.Z., Stasiak A., Xia B., Masson J.Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010;17(10):1247–1254. doi: 10.1038/nsmb.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dray E., Etchin J., Wiese C., Saro D., Williams G.J., Hammel M., Yu X., Galkin V.E., Liu D., Tsai M.S. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat. Struct. Mol. Biol. 2010;17(10):1255–1259. doi: 10.1038/nsmb.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee C.K., Scott C., Lindeman G.J., Hamilton A., Lieschke E., Gibbs E., Asher R., Badger H., Paterson R., Macnab L. Phase 1 trial of olaparib and oral cyclophosphamide in BRCA breast cancer, recurrent BRCA ovarian cancer, non-BRCA triple-negative breast cancer, and non-BRCA ovarian cancer. Br. J. Cancer. 2019;120(3):279–285. doi: 10.1038/s41416-018-0349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pierce A., McGowan P.M., Cotter M., Mullooly M., O'Donovan N., Rani S., O'Driscoll L., Crown J., Duffy M.J. Comparative antiproliferative effects of iniparib and olaparib on a panel of triple-negative and non-triple-negative breast cancer cell lines. Cancer Biol. Ther. 2013;14(6):537–545. doi: 10.4161/cbt.24349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu H., Wei M., Xu J., Hua J., Liang C., Meng Q., Zhang Y., Liu J., Zhang B., Yu X. PARP inhibitors in pancreatic cancer: molecular mechanisms and clinical applications. Mol. Cancer. 2020;19(1):49. doi: 10.1186/s12943-020-01167-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiao S., Xia W., Yamaguchi H., Wei Y., Chen M.K., Hsu J.M., Hsu J.L., Yu W.H., Du Y., Lee H.H. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 2017;23(14):3711–3720. doi: 10.1158/1078-0432.CCR-16-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cesaire M., Thariat J., Candeias S.M., Stefan D., Saintigny Y., Chevalier F. Combining PARP inhibition, radiation, and immunotherapy: a possible strategy to improve the treatment of cancer? Int. J. Mol. Sci. 2018;(12):19. doi: 10.3390/ijms19123793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michelena J., Lezaja A., Teloni F., Schmid T., Imhof R., Altmeyer M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018;9(1):2678. doi: 10.1038/s41467-018-05031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zandarashvili L., Langelier M.F., Velagapudi U.K., Hancock M.A., Steffen J.D., Billur R., Hannan Z.M., Wicks A.J., Krastev D.B., Pettitt S.J. Structural basis for allosteric PARP-1 retention on DNA breaks. Science. 2020;(6486):368. doi: 10.1126/science.aax6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cai C. A novel mechanism to induce BRCAness in cancer cells. Cancer Res. 2020;80(14):2977–2978. doi: 10.1158/0008-5472.CAN-20-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo M.L., Zheng F., Chen W., Liang Z.M., Chandramouly G., Tan J., Willis N.A., Chen C.H., Taveira M.O., Zhou X.Z. Inactivation of the prolyl isomerase Pin1 sensitizes BRCA1-proficient breast cancer to PARP inhibition. Cancer Res. 2020;80(14):3033–3045. doi: 10.1158/0008-5472.CAN-19-2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rasul A., Khan M., Ali M., Li J., Li X. Targeting apoptosis pathways in cancer with alantolactone and isoalantolactone. Sci. World J. 2013;2013 doi: 10.1155/2013/248532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang H., Zhang S., Song L., Qu M., Zou Z. Synergistic lethality between PARP-trapping and alantolactone-induced oxidative DNA damage in homologous recombination-proficient cancer cells. Oncogene. 2020;39(14):2905–2920. doi: 10.1038/s41388-020-1191-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25(20):2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson S.F., Cruz C., Greifenberg A.K., Dust S., Stover D.G., Chi D., Primack B., Cao S., Bernhardy A.J., Coulson R. CDK12 inhibition reverses De Novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 2016;17(9):2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bajrami I., Frankum J.R., Konde A., Miller R.E., Rehman F.L., Brough R., Campbell J., Sims D., Rafiq R., Hooper S. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74(1):287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gallyas F., Jr., Sumegi B., Szabo C. Role of Akt Activation in PARP Inhibitor Resistance in Cancer. Cancers (Basel) 2020;12(3) doi: 10.3390/cancers12030532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Juvekar A., Burga L.N., Hu H., Lunsford E.P., Ibrahim Y.H., Balmana J., Rajendran A., Papa A., Spencer K., Lyssiotis C.A. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–1063. doi: 10.1158/2159-8290.CD-11-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang D., Wang M., Jiang N., Zhang Y., Bian X., Wang X., Roberts T.M., Zhao J.J., Liu P., Cheng H. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget. 2016;7(11):13153–13166. doi: 10.18632/oncotarget.7549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alzahrani A.S. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin. Cancer Biol. 2019;59:125–132. doi: 10.1016/j.semcancer.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 80.Datta S.R., Brunet A., Greenberg M.E. Cellular survival: a play in three Akts. Genes Dev. 1999;13(22):2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 81.Barber L.J., Sandhu S., Chen L., Campbell J., Kozarewa I., Fenwick K., Assiotis I., Rodrigues D.N., Reis Filho J.S., Moreno V. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013;229(3):422–429. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 82.Edwards S.L., Brough R., Lord C.J., Natrajan R., Vatcheva R., Levine D.A., Boyd J., Reis-Filho J.S., Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 83.Dhillon K.K., Swisher E.M., Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011;102(4):663–669. doi: 10.1111/j.1349-7006.2010.01840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pishvaian M.J., Biankin A.V., Bailey P., Chang D.K., Laheru D., Wolfgang C.L., Brody J.R. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br. J. Cancer. 2017;116(8):1021–1026. doi: 10.1038/bjc.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sakai W., Swisher E.M., Karlan B.Y., Agarwal M.K., Higgins J., Friedman C., Villegas E., Jacquemont C., Farrugia D.J., Couch F.J. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ter Brugge P., Kristel P., van der Burg E., Boon U., de Maaker M., Lips E., Mulder L., de Ruiter J., Moutinho C., Gevensleben H. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J. Natl. Cancer Inst. 2016;(11):108. doi: 10.1093/jnci/djw148. [DOI] [PubMed] [Google Scholar]
- 87.Castroviejo-Bermejo M., Cruz C., Llop-Guevara A., Gutierrez-Enriquez S., Ducy M., Ibrahim Y.H., Gris-Oliver A., Pellegrino B., Bruna A., Guzman M. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018;(12):10. doi: 10.15252/emmm.201809172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de la Pena Avalos B., Dray E. Visualization of DNA repair proteins interaction by immunofluorescence. J. Vis. Exp. 2020;(160) doi: 10.3791/61447. [DOI] [PubMed] [Google Scholar]
- 89.Leonardi S., Buttarelli M., De Stefano I., Ferrandina G., Petrillo M., Babini G., Scambia G., Marino C., Mancuso M., Gallo D. The relevance of prelamin A and RAD51 as molecular biomarkers in cervical cancer. Oncotarget. 2017;8(55):94247–94258. doi: 10.18632/oncotarget.21686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chopra N., Tovey H., Pearson A., Cutts R., Toms C., Proszek P., Hubank M., Dowsett M., Dodson A., Daley F. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020;11(1):2662. doi: 10.1038/s41467-020-16142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waks A.G., Cohen O., Kochupurakkal B., Kim D., Dunn C.E., Buendia Buendia J., Wander S., Helvie K., Lloyd M.R., Marini L. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020;31(5):590–598. doi: 10.1016/j.annonc.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yazinski S.A., Comaills V., Buisson R., Genois M.M., Nguyen H.D., Ho C.K., Todorova Kwan T., Morris R., Lauffer S., Nussenzweig A. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–332. doi: 10.1101/gad.290957.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kondrashova O., Nguyen M., Shield-Artin K., Tinker A.V., Teng N.N.H., Harrell M.I., Kuiper M.J., Ho G.Y., Barker H., Jasin M. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–998. doi: 10.1158/2159-8290.CD-17-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu Y., Burness M.L., Martin-Trevino R., Guy J., Bai S., Harouaka R., Brooks M.D., Shang L., Fox A., Luther T.K. RAD51 mediates resistance of cancer stem cells to PARP inhibition in triple-negative breast cancer. Clin. Cancer Res. 2017;23(2):514–522. doi: 10.1158/1078-0432.CCR-15-1348. [DOI] [PubMed] [Google Scholar]
- 95.Marzio A., Puccini J., Kwon Y., Maverakis N.K., Arbini A., Sung P., Bar-Sagi D., Pagano M. The F-box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol. Cell. 2019;73(2):224–237. doi: 10.1016/j.molcel.2018.11.003. e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rugo H.S., Rumble R.B., Macrae E., Barton D.L., Connolly H.K., Dickler M.N., Fallowfield L., Fowble B., Ingle J.N., Jahanzeb M. Endocrine therapy for hormone receptor-positive metastatic breast cancer: American society of clinical oncology guideline. J. Clin. Oncol. 2016;34(25):3069–3103. doi: 10.1200/JCO.2016.67.1487. [DOI] [PubMed] [Google Scholar]
- 97.Tennstedt P., Fresow R., Simon R., Marx A., Terracciano L., Petersen C., Sauter G., Dikomey E., Borgmann K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Cancer. 2013;132(9):2118–2126. doi: 10.1002/ijc.27907. [DOI] [PubMed] [Google Scholar]
- 98.Zhang X., Ma N., Yao W., Li S., Ren Z. RAD51 is a potential marker for prognosis and regulates cell proliferation in pancreatic cancer. Cancer Cell Int. 2019;19:356. doi: 10.1186/s12935-019-1077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Isono M., Niimi A., Oike T., Hagiwara Y., Sato H., Sekine R., Yoshida Y., Isobe S.Y., Obuse C., Nishi R. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 dephosphorylation. Cell Rep. 2017;18(2):520–532. doi: 10.1016/j.celrep.2016.12.042. [DOI] [PubMed] [Google Scholar]
- 100.Bunting S.F., Callen E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mirman Z., de Lange T. 53BP1: a DSB escort. Genes Dev. 2020;34(1–2):7–23. doi: 10.1101/gad.333237.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zimmermann M., de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24(2):108–117. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu G., Chapman J.R., Brandsma I., Yuan J., Mistrik M., Bouwman P., Bartkova J., Gogola E., Warmerdam D., Barazas M. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521(7553):541–544. doi: 10.1038/nature14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339(6120):700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Noordermeer S.M., Adam S., Setiaputra D., Barazas M., Pettitt S.J., Ling A.K., Olivieri M., Alvarez-Quilon A., Moatti N., Zimmermann M. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560(7716):117–121. doi: 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bouwman P., Aly A., Escandell J.M., Pieterse M., Bartkova J., van der Gulden H., Hiddingh S., Thanasoula M., Kulkarni A., Yang Q. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010;17(6):688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lo K.W., Kan H.M., Chan L.N., Xu W.G., Wang K.P., Wu Z., Sheng M., Zhang M. The 8-kDa dynein light chain binds to p53-binding protein 1 and mediates DNA damage-induced p53 nuclear accumulation. J. Biol. Chem. 2005;280(9):8172–8179. doi: 10.1074/jbc.M411408200. [DOI] [PubMed] [Google Scholar]
- 108.Becker J.R., Cuella-Martin R., Barazas M., Liu R., Oliveira C., Oliver A.W., Bilham K., Holt A.B., Blackford A.N., Heierhorst J. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018;9(1):5406. doi: 10.1038/s41467-018-07855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.He Y.J., Meghani K., Caron M.C., Yang C., Ronato D.A., Bian J., Sharma A., Moore J., Niraj J., Detappe A. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563(7732):522–526. doi: 10.1038/s41586-018-0670-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145(4):529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feng W., Jasin M. Homologous recombination and replication fork protection: BRCA2 and more! Cold Spring Harb. Symp. Quant. Biol. 2017;82:329–338. doi: 10.1101/sqb.2017.82.035006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rondinelli B., Gogola E., Yucel H., Duarte A.A., van de Ven M., van der Sluijs R., Konstantinopoulos P.A., Jonkers J., Ceccaldi R., Rottenberg S. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017;19(11):1371–1378. doi: 10.1038/ncb3626. [DOI] [PubMed] [Google Scholar]
- 113.Kais Z., Rondinelli B., Holmes A., O'Leary C., Kozono D., D'Andrea A.D., Ceccaldi R. FANCD2 maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA repair. Cell Rep. 2016;15(11):2488–2499. doi: 10.1016/j.celrep.2016.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.W., Madubata C., Anand R., Levy B., Rabadan R. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell. 2017;68(2):414–430. doi: 10.1016/j.molcel.2017.09.036. e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Betous R., Couch F.B., Mason A.C., Eichman B.F., Manosas M., Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3(6):1958–1969. doi: 10.1016/j.celrep.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Murai J., Tang S.W., Leo E., Baechler S.A., Redon C.E., Zhang H., Al Abo M., Rajapakse V.N., Nakamura E., Jenkins L.M.M. SLFN11 blocks stressed replication forks independently of ATR. Mol. Cell. 2018;69(3):371–384. doi: 10.1016/j.molcel.2018.01.012. e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murai J., Feng Y., Yu G.K., Ru Y., Tang S.W., Shen Y., Pommier Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–76550. doi: 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vaidyanathan A., Sawers L., Gannon A.L., Chakravarty P., Scott A.L., Bray S.E., Ferguson M.J., Smith G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer. 2016;115(4):431–441. doi: 10.1038/bjc.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rottenberg S., Jaspers J.E., Kersbergen A., van der Burg E., Nygren A.O., Zander S.A., Derksen P.W., de Bruin M., Zevenhoven J., Lau A. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. U. S. A. 2008;105(44):17079–17084. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marzolini C., Paus E., Buclin T., Kim R.B. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin. Pharmacol. Ther. 2004;75(1):13–33. doi: 10.1016/j.clpt.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 121.Chen M.L., Sun A., Cao W., Eliason A., Mendez K.M., Getzler A.J., Tsuda S., Diao H., Mukori C., Bruno N.E. Physiological expression and function of the MDR1 transporter in cytotoxic T lymphocytes. J. Exp. Med. 2020;(5):217. doi: 10.1084/jem.20191388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pettitt S.J., Krastev D.B., Brandsma I., Drean A., Song F., Aleksandrov R., Harrell M.I., Menon M., Brough R., Campbell J. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018;9(1):1849. doi: 10.1038/s41467-018-03917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gogola E., Duarte A.A., de Ruiter J.R., Wiegant W.W., Schmid J.A., de Bruijn R., James D.I., Guerrero Llobet S., Vis D.J., Annunziato S. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33(6):1078–1093. doi: 10.1016/j.ccell.2018.05.008. e1012. [DOI] [PubMed] [Google Scholar]
- 124.Gomez M.K., Illuzzi G., Colomer C., Churchman M., Hollis R.L., O'Connor M.J., Gourley C., Leo E., Melton D.W. Identifying and overcoming mechanisms of parp inhibitor resistance in homologous recombination repair-deficient and repair-proficient high grade serous ovarian cancer cells. Cancers (Basel) 2020;12(6) doi: 10.3390/cancers12061503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fisher A.E., Hochegger H., Takeda S., Caldecott K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell. Biol. 2007;27(15):5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hermanowski H., Huebert B., Aldrighetti C., Hurley J.K., Quénet D. Role of PARylation and PTEN mutation on PARP and PARG inhibitor efficacy on glioblastoma. bioRxiv. 2020 [Google Scholar]
- 127.Sharifi R., Morra R., Appel C.D., Tallis M., Chioza B., Jankevicius G., Simpson M.A., Matic I., Ozkan E., Golia B. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013;32(9):1225–1237. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Blackford A.N., Jackson S.P. ATM,ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017;66(6):801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 129.Schmitt A., Knittel G., Welcker D., Yang T.P., George J., Nowak M., Leeser U., Buttner R., Perner S., Peifer M. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017;77(11):3040–3056. doi: 10.1158/0008-5472.CAN-16-3398. [DOI] [PubMed] [Google Scholar]
- 130.Bang Y.J., Im S.A., Lee K.W., Cho J.Y., Song E.K., Lee K.H., Kim Y.H., Park J.O., Chun H.G., Zang D.Y. Randomized, double-blind phase II Trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of olaparib plus paclitaxel in patients with recurrent or metastatic gastric cancer. J. Clin. Oncol. 2015;33(33):3858–3865. doi: 10.1200/JCO.2014.60.0320. [DOI] [PubMed] [Google Scholar]
- 131.Mateo J., Carreira S., Sandhu S., Miranda S., Mossop H., Perez-Lopez R., Nava Rodrigues D., Robinson D., Omlin A., Tunariu N. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015;373(18):1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weston V.J., Oldreive C.E., Skowronska A., Oscier D.G., Pratt G., Dyer M.J., Smith G., Powell J.E., Rudzki Z., Kearns P. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116(22):4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 133.Balmus G., Pilger D., Coates J., Demir M., Sczaniecka-Clift M., Barros A.C., Woods M., Fu B., Yang F., Chen E. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019;10(1):87. doi: 10.1038/s41467-018-07729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lloyd R.L., Wijnhoven P.W.G., Ramos-Montoya A., Wilson Z., Illuzzi G., Falenta K., Jones G.N., James N., Chabbert C.D., Stott J. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene. 2020;39(25):4869–4883. doi: 10.1038/s41388-020-1328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Buisson R., Niraj J., Rodrigue A., Ho C.K., Kreuzer J., Foo T.K., Hardy E.J., Dellaire G., Haas W., Xia B. Coupling of homologous recombination and the checkpoint by ATR. Mol. Cell. 2017;65(2):336–346. doi: 10.1016/j.molcel.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luijsterburg M.S., Typas D., Caron M.C., Wiegant W.W., van den Heuvel D., Boonen R.A., Couturier A.M., Mullenders L.H., Masson J.Y., van Attikum H. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. Elife. 2017;6 doi: 10.7554/eLife.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Belotserkovskaya R., Raga Gil E., Lawrence N., Butler R., Clifford G., Wilson M.D., Jackson S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020;11(1):819. doi: 10.1038/s41467-020-14563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim H., Xu H., George E., Hallberg D., Kumar S., Jagannathan V., Medvedev S., Kinose Y., Devins K., Verma P. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020;11(1):3726. doi: 10.1038/s41467-020-17127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.sources M. 2020. Clinical Trials Intervention: ATR Kinase Inhibitor AZD6738. [Google Scholar]
- 140.Fang Y., McGrail D.J., Sun C., Labrie M., Chen X., Zhang D., Ju Z., Vellano C.P., Lu Y., Li Y. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35(6):851–867. doi: 10.1016/j.ccell.2019.05.001. e857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Naidoo K., Wai P.T., Maguire S.L., Daley F., Haider S., Kriplani D., Campbell J., Mirza H., Grigoriadis A., Tutt A. Evaluation of CDK12 protein expression as a potential novel biomarker for DNA damage response-targeted therapies in breast cancer. Mol. Cancer Ther. 2018;17(1):306–315. doi: 10.1158/1535-7163.MCT-17-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wu Y.M., Cieslik M., Lonigro R.J., Vats P., Reimers M.A., Cao X., Ning Y., Wang L., Kunju L.P., de Sarkar N. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell. 2018;173(7):1770–1782. doi: 10.1016/j.cell.2018.04.034. e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Alagpulinsa D.A., Ayyadevara S., Yaccoby S., Shmookler Reis R.J. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol. Cancer Ther. 2016;15(2):241–250. doi: 10.1158/1535-7163.MCT-15-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fathers C., Drayton R.M., Solovieva S., Bryant H.E. Inhibition of poly(ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell Cycle. 2012;11(5):990–997. doi: 10.4161/cc.11.5.19482. [DOI] [PubMed] [Google Scholar]
- 145.Noll A., Illuzzi G., Ame J.C., Dantzer F., Schreiber V. PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer Cell Int. 2016;16:53. doi: 10.1186/s12935-016-0333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Houl J.H., Ye Z., Brosey C.A., Balapiti-Modarage L.P.F., Namjoshi S., Bacolla A., Laverty D., Walker B.L., Pourfarjam Y., Warden L.S. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 2019;10(1):5654. doi: 10.1038/s41467-019-13508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun C., Yin J., Fang Y., Chen J., Jeong K.J., Chen X., Vellano C.P., Ju Z., Zhao W., Zhang D. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401–416. doi: 10.1016/j.ccell.2018.01.019. e408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005;19(4):535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 149.Karakashev S., Zhu H., Yokoyama Y., Zhao B., Fatkhutdinov N., Kossenkov A.V., Wilson A.J., Simpkins F., Speicher D., Khabele D. BET bromodomain inhibition synergizes with PARP inhibitor in epithelial ovarian cancer. Cell Rep. 2017;21(12):3398–3405. doi: 10.1016/j.celrep.2017.11.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wilson A.J., Stubbs M., Liu P., Ruggeri B., Khabele D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018;149(3):575–584. doi: 10.1016/j.ygyno.2018.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.