

Summary
It has been suggested that aberrant activation of glycogen synthase kinase-3-beta (GSK-3β) can trigger abnormal tau hyperphosphorylation and aggregation, which ultimately leads to neuronal/synaptic damage and impaired cognition in Alzheimer disease (AD). We examined if isoform-selective partial reduction of GSK-3β can decrease pathological tau changes, including hyperphosphorylation, aggregation, and spreading, in mice with localized human wild-type tau (hTau) expression in the brain. We used adeno-associated viruses (AAVs) to express hTau locally in the entorhinal cortex of wild-type and GSK-3β hemi-knockout (GSK-3β-HK) mice. GSK-3β-HK mice had significantly less accumulation of hyperphosphorylated tau in synapses and showed a significant decrease of tau protein spread between neurons. In primary neuronal cultures from GSK-3β-HK mice, the aggregation of exogenous FTD-mutant tau was also significantly reduced. These results show that a partial decrease of GSK-3β significantly represses tau-initiated neurodegenerative changes in the brain, and therefore is a promising therapeutic target for AD and other tauopathies.
Subject areas: Biological Sciences, Neuroscience, Cellular Neuroscience
Graphical abstract
Highlights
-
•
Genetic reduction of GSK-3β decreases synaptic accrual of GSK-3β and p-Tau in mice
-
•
Reduction of GSK-3β lowers the trans-cellular spread of tau in vivo and in vitro
-
•
Reduction of GSK-3β diminishes the formation of tau aggregates in vitro
Biological Sciences; Neuroscience; Cellular Neuroscience
Introduction
Glycogen synthase kinase 3 (GSK-3) is an ubiquitously expressed, highly conserved serine/threonine kinase that regulates multiple major biological processes such as cell differentiation, metabolism, immunity, and cell survival (Maurer et al., 2014). GSK-3 is expressed as two highly homologous isoforms, α and β, that have partially overlapping functions. GSK-3β is the most abundant isoform in neurons (Leroy and Brion, 1999; Takahashi et al., 1994; Woodgett, 1990) and is involved in the pathogenesis of multiple neurological disorders including schizophrenia, bipolar disorder, fragile X syndrome, brain tumors, stroke, Parkinson disease, and Alzheimer disease (AD) (Chuang et al., 2011; Jope and Roh, 2006; Kozikowski et al., 2006; Mills et al., 2011; Mines and Jope, 2011; Takashima, 2006).
One of the hallmark pathological changes in the AD brain is the hyperphosphorylation and intraneuronal aggregation of the microtubule-associated protein tau in form of neurofibrillary tangles (NFTs) (reviewed in Gomez-Isla et al., 2008). Tau NFT pathology spreads through the brain with disease progression (Braak and Braak, 1991) and correlates with neuronal death and cognitive decline (Gomez-Isla et al., 1997). Furthermore, soluble phosphorylated forms of tau appear to be synaptotoxic (Tai et al., 2012) and can propagate between neurons across neuronal networks (de Calignon et al., 2012; Dujardin et al., 2014; Wegmann et al., 2019).
In AD, GSK-3β is a pivotal kinase responsible for tau phosphorylation, and multiple GSK-3β phosphorylated tau sites are present in NFTs (Hanger et al., 1992; Lucas et al., 2001; Mandelkow et al., 1992). It has been demonstrated that GSK-3β is aberrantly activated in human AD brains (DaRocha-Souto et al., 2012; Leroy et al., 2007) and is a key player in the pathogenesis of AD (Takashima, 2006). Overexpression of GSK-3β leads to tau hyperphosphorylation, microtubule dissociation, and cognitive impairment (Engel, 2006; Hanger et al., 1992; Hernández et al., 2002; Lovestone et al., 1994; Lucas et al., 2001; Spittaels et al., 2000), whereas treatment with GSK-3 inhibitors can significantly reduce tau hyperphosphorylation, halt neuronal and synaptic loss, and rescue memory deficits in AD mouse models (Caccamo et al., 2007; Engel et al., 2006; Liu et al., 2003; Nakashima et al., 2005; Noble et al., 2005; Peng et al., 2013; Pérez et al., 2003; Serenó et al., 2009). However, some studies alerted about potential adverse effects of pharmacological GSK-3 inhibitors, which include anatomical disruption of dendritic spines in neurons in culture (DaRocha-Souto et al., 2012), inflammation, and behavioral deficits in mice, likely resulting from an excessive inhibition of GSK-3 constitutive activity and/or non-specific inhibition of other kinases (Hu et al., 2009). Isoform-selective GSK-3β inhibition, however, is difficult to achieve and controversy on how much inhibition of GSK-3β can be safely achieved needs to be settled.
In the present work, we investigated whether partial isoform-selective reduction of GSK-3β (haplo-insufficiency) is safe and can alleviate AD-related tau pathological changes in the brain. Although GSK-3β full-knockout mice die during embryonic life due to severe liver degeneration, GSK-3β-HK mice, with a decrease of about 45% of GSK-3β expression, are viable and healthy (Hoeflich et al., 2000). We expressed human full-length wild-type tau (2N4R isoform) in the entorhinal cortex (EC) in GSK-3β-HK mice and wild-type (WT) littermate controls using AAVs and evaluated tau hyperphosphorylation, synaptic accumulation, aggregation, and trans-cellular spread in the brain. Detailed histopathological and biochemical analyses showed a decrease of all evaluated tau changes in GSK-3β-HK compared with WT littermate controls. Furthermore, human tauP301L aggregation was reduced in GSK-3β-HK primary neurons. Our data show that isoform-selective reduction of GSK-3β can be safely achieved in vivo and ameliorates pathological tau changes relevant for neurodegeneration in AD.
Results
Isoform-selective reduction of GSK-3β in GSK-3β-HK mice
In AD, hyperphosphorylation of tau by GSK-3β and other kinases is suggested to promote its aggregation into insoluble aggregates (Figure 1A) (Hanger et al., 1992) and is a strong correlate of synapse loss and neurodegeneration (Gomez-Isla et al., 1996; Gong and Iqbal, 2008). GSK-3β phosphorylates more than 30 sites in the human tau protein and is considered one of the main tau kinases (Figure 1B). Using immunohistochemistry (IHC), we confirmed in mice as well as in primary neurons that the constitutively active form of GSK-3β (pGSK-3β-Tyr216) co-localizes with tau in the neuronal cytosol and in some neurites (Figure 1C).
Figure 1.

Tau phosphorylation by GSK-3β
(A) Tau binds and stabilizes axonal microtubules in the brain, a process regulated by phosphorylation. Hyperphosphorylation of tau by different kinases, including GSK-3β, is associated with tau aggregation in AD.
(B) GSK-3β phosphorylates tau at several epitopes (pink stars) across the tau sequence. Two phospho-sites targeted by GSK-3β, S396 and S404 (highlighted), are commonly hyperphosphorylated in AD and are found in neurofibrillary tangles. R1 to R4 refer to the four sequence repeats in the microtubule binding domain of tau.
(C) Immunostaining of primary neurons from WT embryos after treatment with hTau (AAV added to culture media) and brain sections from an AAV-injected WT mouse showed that active GSK-3β (GSK-3β phosphorylated at Thr216) co-localizes with hTau (Y9) in the cytosol of neurons and in some neurites. Scale bars: 20 μm (C).
To study the effect of GSK-3β reduction on tau, we used GSK-3β-HK mice (Hoeflich et al., 2000). First, we confirmed that in these mice total GSK-3β levels were reduced by 45% (p < 0.0001), and the levels of pGSK-3β-Tyr216 (enzyme active form) and pGSK-3β-Ser9 (enzyme inhibited form) are reduced by about 40% compared with WT mice (Tyr216: p = 0.0004 and Ser9: p = 0.0045; Figures 2A and 2B). Importantly, no alteration in the protein levels of other relevant tau kinases—GSK-3α, pGSK-3α-Tyr279 (active form of GSK-3α), FYN, and CDK5—were detected (GSK-3α: p = 0.1035; pGSK-3α-Tyr279: p = 0.7100; FYN: p = 0.3213; CDK5: p = 0.9874; Figures 2A and 2B). These data confirmed that the reduction of GSK-3 levels in the brain of GSK-3β-HK mice is selective for the β-isoform of the enzyme, does not lead to compensatory upregulation of remaining GSK-3β activity, and does not induce significant upregulation of other kinases implicated in the pathologic phosphorylation of tau protein in AD.
Figure 2.

GSK-3β-HK mice model used in this study
(A) Representative images of western blots using EC lysates from WT and GSK-3β-HK mice probed for kinases known to phosphorylate tau.
(B) Quantification of western blots. Results showed a significant overall reduction of 45% of GSK-3β and 40% of pGSK-3β (active and inhibited forms, -Tyr216 and -Ser9, respectively) in GSK-3β-HK compared with WT mice. No differences were found in GSK-3α, pGSK-3α-Tyr279 (active GSK-3α), FYN, and CDK5 between WT and GSK-3β-HK mice. Data are presented as mean ± SEM, N = 14 mice: 7WT, 7HK. Two-tailed Student's t test, ∗∗p < 0.01, ∗∗∗p < 0.001.
(C) Representative images of western blots probed for hTau and Total Tau (mouse + human tau (ms + h)) in AAV-injected mice.
(D) Quantification of western blots showed no difference in Total Tau (ms + h) and hTau in the EC of WT and GSK-3β-HK mice (AAV-injected). GAPDH was used as loading control for all proteins. pGSK-3 and hTau were also normalized by total GSK-3β and Total Tau, respectively (as indicated in graphs). Data are presented as mean ± SEM, N = 14 mice: 7WT, 7HK. Two-tailed Student's t test, p > 0.05.
We also tested if WT and GSK-3β-HK mice had comparable total tau levels at baseline (non-injected: p = 0.9905; Figures 3K and 3N) as well as after AAV injections (AAV-injected: p = 0.9052; Figures 2C, 2D, 3K, and 3N) to ensure that genetic manipulation of GSK-3β did not affect tau protein expression.
Figure 3.

AAV-mediated human tau expression in the entorhinal cortex of GSK-3β-HK and WT mice
(A) AAV construct designed to express eGFP and hTau as individual proteins, separated by the self-cleaving 2a peptide under the CBA promoter (AAV CBA.eGFP-2a-hTau).
(B) Schematic representation of proteins expected to be identified in the EC of mice injected with AAV. AAV-transduced neurons express eGFP (green) and hTau (red) (“donor neurons”), whereas some neurons received hTau protein from other cells and do not have GFP ("recipient neurons" (red only)).
(C) Stereotactic coordinates for unilateral intracranial AAV injection into the left EC of adult WT and GSK-3β-HK mice.
(D) Representative image of a Nissl-stained horizontal section of a mouse brain with the approximate area corresponding to the EC shaded in green.
(E) Horizontal brain sections of a WT and a GSK-3β-HK-injected mouse with AAV expression in the EC and interconnected neighboring regions. Twelve weeks after AAV injection, brain sections were immunostained for hTau (antibodies HT7 or TauY9) and GFP.
(F) Primary neurons and brain sections immunostained for GFP and neuronal marker NeuN show that AAV is expressed in neurons (“AAV-transduced neurons”; white arrowheads).
(G) Stereologically based counts of neurons (Nissl stained) in the non-injected (right hemisphere) and (H) injected (left hemisphere) EC of WT and GSK-3β-HK mice showed no significant differences, indicating that there are no baseline differences in neuronal numbers between the two genotypes or neuronal cell death due to the AAV injections. Data are presented as mean ± SEM, N = 14 mice: 7WT, 7HK, 5 brain sections per mouse. Two-tailed Student's t test, p > 0.05.
(I) The volume of the transduced brain area (GFP+) and (J) the number of transduced (GFP+) neurons was similar in GSK-3β-HK and WT mice. Data are presented as mean ± SEM, N = 14 mice: 7WT, 7HK, 3–5 sections per mouse, two-tailed Student's t test, p > 0.05.
(K) Representative image of western blot from EC lysates from non-injected and AAV-injected WT and GSK-3β-HK mice probed for postsynaptic (PSD95) and presynaptic (synaptophysin, SYP) markers and Total Tau (ms + h). GAPDH was used as loading control.
(L) Quantification of western blots for Postsynaptic density protein 95 (PSD95), (M) Synaptophysin (SYP), and (N) Total Tau (ms + h). Levels of synaptic markers (PSD95 and SYP) and total tau were not significantly different between WT and GSK-3β-HK mice at baseline and 12 weeks after AAV injection. Data are presented as mean ± SEM, N = 6 non-injected mice: 3WT, 3HK, two-tailed Student's t test; N = 14 AAV-injected mice: 7WT, 7HK, two-tailed Student's t test, p > 0.05. Scale bars: 1,000 μm (E), 20 μm (F, in vitro and in vivo).
AAV-mediated expression of hTau in the EC of WT and GSK-3β-HK mice
To generate a model that allows us to study the effect of GSK-3β reduction on tau accumulation and spreading, we utilized targeted intracranial AAV injections to express hTau in the EC of WT and GSK-3β-HK mice. The AAV construct used was designed to express eGFP and hTau as individual proteins separated by a self-cleaving 2a-peptide under the CBA promoter (AAV CBA-eGFP-2a-hTau; Figure 3A) (Wegmann et al., 2015, 2017, 2019). AAV-transduced “donor neurons” express both eGFP and hTau, whereas “recipient neurons,” which acquired hTau protein from transduced donor neurons, contain only hTau (no eGFP) that is detectable by immunostaining (Figure 3B). AAV-injections into the superficial layers (II–III) of the EC (Figures 3C and 3D) successfully led to the expression of hTau around the injection site (Figure 3E). Twelve weeks after AAV injection, brain sections of WT and GSK-3β-HK mice were immunolabeled for hTau (HT7 or TauY9 antibody) and GFP to identify and distinguish tau-transduced donor from tau recipient neurons. Most neurons around the injection site were AAV-transduced donor neurons expressing both GFP (green) and hTau (red) (Figure 3E). Brain sections (and primary neurons) were immunolabeled for GFP and a neuronal marker (NeuN), to confirm neuronal expression of the AAV (Figure 3F).
Comparable AAV transduction and hTau expression in GSK-3β-HK and WT mice
Unbiased stereological counts of neurons (Nissl stained) in the medial and lateral EC showed no differences in the total number of neurons between GSK-3β-HK versus WT non-injected or injected mice (p = 0.9682 and p = 0.6685, respectively; Figures 3G and 3H). This excluded both the possibility of a baseline difference in neuronal numbers between the two genotypes, as well as a neurotoxic effect of the AAV injections themselves, which could have artificially impacted the rate of trans-cellular tau spreading in the brain.
Differences in AAV transduction and hTau expression could potentially result in differences in detected tau spreading. To rule out such artifacts, we compared the volume of the injection sites (area with GFP-positive neurons in consecutive brain sections; p = 0.6702. Figure 3I) and the total number of transfected neurons (number of GFP-positive neurons; p = 0.6414. Figure 3J) between WT and GSK-3β-HK mice. We found comparable AAV transduction and hTau expression in both groups of mice. This was further confirmed by western blot analysis of EC brain lysates, in which the amount of total tau (mouse and human tau (ms + h); indicator for general neuronal abundance) and hTau expression was comparable between the WT and GSK-3β-HK groups of mice (Figures 2C and 2D). Furthermore, the overall levels of post- and pre-synaptic markers, PSD95 and synaptophysin, respectively, were similar in WT and GSK-3β-HK mice both at baseline (PSD95: p = 0.7960; synaptophysin: p = 0.6304) and after AAV injections (PSD95: p = 0.9242; synaptophysin: p = 0.7402; Figures 3L and 3M), indicating no overt gain or loss of synapses upon GSK-3β reduction or as a result of AAV injections and hTau expression.
GSK-3β reduction diminishes tau spreading in vitro and in vivo
Next, we assessed if the reduction in GSK-3β changes the potential of tau to propagate between neurons in the brain. hTau propagation was quantified both in vitro in primary neuronal cultures from WT and GSK-3β-HK embryos and in vivo in adult WT and GSK-3β-HK mice unilaterally injected with AAV-CBA-eGFP-2a-hTau into the EC. We counted all neurons (in culture and in the EC) that were simultaneously positive for GFP and hTau (= transduced tau donor neurons) and neurons that were positive only for hTau (= tau recipient neurons). We observed in vitro that primary neurons derived from GSK-3β-HK embryos showed significant lower number of hTau recipient neurons compared with WT (0.68% ± 0.19 vs. 3.19% ± 0.72, respectively; p = 0.0356; Figure 4A). Furthermore, and as expected from previous in vivo studies (Wegmann et al., 2015, 2019), transduced donor neurons near the injection site expressed both GFP and hTau (Figure 4B: green and red neuron “b” in lower panel) and, after 12 weeks, a small number of hTau recipient neurons (expressing hTau only but not GFP) were also present both in WT and GSK-3β-HK mice (Figure 4B: red neuron “a” in lower panel). Even though tau propagation was a rare event in WT mice and less than 1% of all hTau-containing neurons in the EC were identified as “tau recipient” neurons (7.9 ± 2.1 of 829 hTau positive neurons (0.94%)), this percentage was found to be significantly reduced in GSK-3β-HK mice, with the number of tau recipient neurons being zero in some GSK-3β-HK mice (1.7 ± 0.8 of 826 hTau positive neurons (0.21%); p = 0.0063; Figure 4B). These results indicate that a partial decrease in GSK-3β levels can significantly diminish tau trans-cellular propagation in the brain.
Figure 4.

Reduction of GSK-3β diminishes tau propagation in vitro and in vivo
(A) Representative images of WT and GSK-3β-HK primary neurons immunostained for GFP and hTau (Y13) after addition of AAV to culture media. Arrowhead shows a tau-recipient neuron (red) amid AAV-transduced neurons (red and green). The percentage of tau recipient neurons was significantly reduced in primary neurons from GSK-3β-HK embryos compared with WT. Data are presented as mean ± SEM, N = 7: 4WT, 3HK, 6 wells of plated neurons per embryo. Two-tailed Student's t test, ∗p < 0.05.
(B) Immunofluorescence images of the injected EC of WT and GSK-3β-HK mice showed neurons that had hTau (red) but no GFP (green) (“tau recipient neurons”; neurons in insert “a”) and neurons that had both GFP (green) and hTau (red) (“AAV-transduced neurons”; neurons in insert “b”). The percentage of tau recipient neurons (normalized to GFP + neurons) was significantly reduced in the EC of GSK-3β-HK compared with WT mice. Data are presented as mean ± SEM, n = 15: 7WT, 7HK, two-tailed Student's t test, ∗∗p < 0.01. Scale bars: 20 μm (A) and 10 μm (B).
GSK-3β reduction decreases synaptic accrual of p-Tau in vivo
Levels of total tau and p-Tau (as reported by PHF-1 antibody) in total cell extracts from primary neuronal cultures did not show significant differences between GSK-3β-HK and WT neurons (total tau: p = 0.2437 and PHF-1: p = 0.2757. Figures 5A and 5B). It has been suggested that tau phosphorylation site-specific antibodies against pS202 and pS396/404 and against misfolded tau label progressive stages of disease-associated tau changes (Augustinack et al., 2002). In agreement with these observations, immunofluorescence labeling of GSK-3β-targeted tau phosphorylation sites in brain sections from WT and GSK-3β-HK mice showed that of all the neuronal cell bodies expressing hTau in the EC, approximately 30% were positive for pS202 (CP13 antibody), 18% were positive for pS396/pS404 (PHF-1 antibody), and 8% were positive for misfolded tau (Alz50 antibody), with no significant differences between WT and GSK-3β-HK AAV-injected mice (CP13: p = 0.3626; PHF-1: p = 0.8224; Alz50: p = 0.6490. Figures 5C and 5D). The spread of NFT pathology through the AD brain is thought to rely, at least in part, on synaptic transmission of pathological tau proteins between neurons. Because a reduction in GSK-3β levels diminished the trans-cellular transmission of tau in the EC of GSK-3β-HK mice compared with WT, we investigated the effect of the overall decrease in GSK-3β expression on the kinase levels, specifically within the synaptic compartment. We measured the levels of GSK-3β, pGSK-3β-Tyr216 (active form of the enzyme) and -Ser9 (inhibited form), total tau, and p-Tau in both the cytosol (CYT) and in synaptoneurosome (SNS) preparations from the EC of WT and GSK-3β-HK mice after AAV CBA-eGFP-2a-hTau injection (Figure 5E).
Figure 5.

Reduction of GSK-3β leads to lower levels of p-Tau tau in synapses
(A) Representative western blot images using homogenates from WT and GSK-3β-HK primary neurons after treatment with hTau (AAV added to culture media). Immunoblots were probed with antibodies for GSK-3β, total tau, and PHF-1 (pS396, pS404). GAPDH was used as loading control.
(B) Protein quantification showed that despite significantly lower levels of GSK-3β in total cell homogenates, GSK-3β-HK neurons displayed similar overall levels of total tau and p-Tau (PHF-1) compared with WT neurons. Data are presented as mean ± SEM, n = 10: 5WT, 5HK. Two-tailed Student's t test, ∗∗p < 0.001.
(C) Horizontal brain section showing an illustrative outline of the EC where AAV-transduced neurons can be easily identified by the presence of GFP labeling.
Brain sections of WT and GSK-3β-HK-injected mice were co-labeled for GFP and specific tau-phosphorylation sites targeted by GSK-3β: CP13: pS202; PHF-1: pS396/pS404, and Alz50: misfolded tau.
(D) Quantification of GFP + neurons that were also positive for CP13, PHF-1, and Alz50. About 27% of the transduced neurons were positive for CP13, 15% were positive for PHF-1, and only 6% were positive for Alz50. There were no significant differences between the number of neurons positive for any of the three markers between GSK-3β-HK and WT mice. Data are presented as mean ± SEM, n = 14: 7WT, 7HK, 3 sections per mouse. Two-tailed Student's t test, p > 0.05.
(E) Representative images of cytosolic (C) and synaptoneurosome (S) fractions on western blots probed for PSD95 (confirming appropriate separation of cytosolic and synaptic compartments), GSK-3β, total tau (ms + h), p- GSK-3β-Tyr216 (active form), p-GSK-3β-Ser9 (inhibited form), and p-Tau (PHF-1 antibody).
(F) There was a lower accumulation of GSK-3β and p-Tau in synapses in the EC of GSK-3β-HK compared with WT mice in the presence of equal amounts of tau. Lower levels of total GSK-3β in GSK-3β-HK mice led to redistribution of active and inhibited GSK-3β forms (with lower synaptic levels of active GSK-3β (Tyr216) and equivalent levels of inhibited GSK-3β (Ser9) compared with WT mice. Significantly lower levels of p-Tau (as reported by PHF-1 antibody) were detected in the synapses of GSK-3β-HK mice compared with WT. GAPDH on the same membrane was used for normalization. Data are presented as mean ± SEM, n = 14: 7WT, 7HK, one-way ANOVA, Holm-Sidak post-hoc multiple comparisons, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Scale bars: 500 μm (C, EC image) and 20 μm (C, phospho-tau).
Interestingly, GSK-3β-HK mice, compared with WT mice, not only had significantly lower levels of GSK-3β in both compartments (CYT and SNS) (CYT: p < 0.05 and SNS: p < 0.0001) but also exhibited a different distribution of GSK-3β between CYT and SNS. WT mice had higher levels of GSK-3β in SNS compared with CYT (p < 0.05), whereas there was no difference in GSK-3β levels between the two compartments in GSK-3β-HK mice (p > 0.05; Figure 5F). This suggests that neurons, in response to genetic reduction of total GSK-3β levels, may favor a decrease of synaptic over cytosolic GSK-3β, perhaps to preserve minimal required levels of this kinase in the cytosol for critical cellular functions. Total protein levels of pGSK-3β (-Tyr216 and -Ser9) were also significantly lower in GSK-3β-HK compared with WT mice (Figure 2B). Notably, the levels of active GSK-3β (-Tyr216) were significantly lower in both compartments (CYT and SNS) in GSK-3β-HK compared with WT mice (CYT and SNS: p < 0.0001; Figure 5F), however, without significant changes in the compartmental distribution between the two groups (p > 0.05). Interestingly, the levels of inactive GSK-3β (-Ser9) were significantly lower in the cytosol but not in synapses of GSK-3β-HK compared with WT mice (CYT: p < 0.05 and SNS: p > 0.05 for SNS; Figure 5F). These results further support the idea of a preferential reduction of total and active GSK-3β in synapses of GSK-3β-HK mice.
Overall levels of total tau were not significantly different between WT and GSK-3β-HK mice (Figures 2C and 2D), yet both genotypes had significantly more tau in the SNS fraction (42% and 36% higher levels in synapses, respectively; p < 0.01; Figure 5F). Importantly, p-Tau (PHF-1 antibody-detecting epitopes pS396/pS404 in mouse and human tau) levels were overall significantly lower in GSK-3β-HK compared with WT mice (p = 0.0233), whereby p-Tau levels were only slightly lower in CYT but significantly lower in SNS of GSK-3β-HK compared with WT mice (CYT: p > 0.05 and SNS: p < 0.05; Figure 5F).
Together, these results show that even though WT and GSK-3β-HK mice had similar amounts of total tau in the cytosolic and synaptic compartments, the 45% reduction of GSK-3β in the GSK-3β-HK mice resulted in the selective decrease of GSK-3β—followed by reduction of tau phosphorylation—within the synaptic compartment. Notably, this seems to be a specific effect of the isoform-selective reduction of GSK-3β because no compensatory up- or downregulation of other relevant tau kinases (GSK-3α, pGSK-3α-Tyr216 (active form), CDK5, and FYN) could be detected in the brain of GSK-3β-HK mice (Figures 2A and 2B).
Partial GSK-3β reduction decreases tau aggregation in vitro
GSK-3β has been found to be associated with tangles in AD brains (Imahori and Uchida, 1997; Pei et al., 1997, 1999; Yamaguchi et al., 1996) as well as accumulated in pre-tangle neurons (Pei et al., 1999).
In our model, in vivo expression of hTau upon AAV CBA-eGFP-2a-hTau injections did not trigger the formation of brain β-sheet-containing tau aggregates, as reported by Thioflavin S, either in WT or in GSK-3β-HK mice, even after long-term expression (6 months post-injection, Figure 6A). This is in agreement with previous findings that tau aggregation into β-sheet-containing aggregates is not a prerequisite for propagation (Dujardin et al., 2014; Wegmann et al., 2019).
Figure 6.

Reduced levels of GSK-3β decrease tau aggregation in vitro
(A) Representative images of brain sections from WT and GSK-3β-HK mice 6 months after AAV-injection, stained with Thioflavin S (marker of β-sheet-rich amyloid-like aggregated proteins) and immunolabeled for NeuN (neuronal marker). No tau aggregates were observed.
(B) Schematic representation of tau aggregation assay in neurons: primary neurons from WT and GSK-3β-HK embryos were co-transduced with lentiviruses encoding tauRDP301L-CFP and tauRDP301LCFP, and tau aggregation is initiated by subsequent treatment of the neurons with seeding-competent tau in brain extracts from tau transgenic mice (rTg4510). Tau aggregation in the neurons can be detected by fluorescent imaging as FRET signal.
(C) Timeline for tau aggregation assay: primary neurons from WT and GSK-3β-HK embryos are plated in 96-well plate (DIV0) and transduced with lentivirus (lenti-tauRD(P301L)-YFP and -CFP) (DIV1). Transduction is followed with treatment with rTg4510 brain lysate (DIV6) and imaging of FRET-positive tau aggregates (DIV8).
(D) Representative images and quantification of tau aggregates in primary neuronal cultures at DIV8. GSK-3β-HK neurons showed significantly fewer intracellular tau aggregates compared with WT neurons. Inserts show tau aggregates formed intracellularly in WT and HK neurons in culture. Data are presented as mean ± SEM, N = 22 embryos: 10WT, 12HK, 3–5 wells plated neurons per embryo. Two-tailed Student's t test, ∗∗∗p < 0.0001. Scale bars: 100 μm (A) and 20 μm (D).
We therefore tested if a reduction of GSK-3β could potentially decrease tau aggregation using a more aggressive in vitro model based on the expression of aggregation-prone FTD-mutant tau in primary neuronal cultures derived from WT and GSK-3β-HK embryos. In this in vitro model, primary neurons were transduced with lentiviral viruses (Lenti CBA.tauRDP301L-YFP/CFP) encoding the repeat domain of tau (tauRD) carrying a P301L FTD mutation fused to either CFP or YFP fluorescent protein, in order to monitor tauRD aggregation through fluorescence resonance energy transfer (FRET; Figure 6B) (Holmes et al., 2014; Nobuhara et al., 2017). Aggregation of tauRD was induced by the addition of brain lysates from tauP301L transgenic mice (rTg4510 line) containing pre-aggregated, seeding-competent tau (Figures 6B and 6C). Six days after aggregation seeding (at DIV8), we found that the number of intracellular tau aggregates was significantly reduced in GSK-3β-HK compared with WT neurons (p < 0.001; Figure 6D). Importantly, viral particle number (number of neurons transduced) and neuronal cell density were comparable in all experiments and treatment groups. These results show that the reduction of GSK-3β not only significantly lowers synaptic tau phosphorylation and propagation in the brain but it also significantly inhibits tau aggregation in neurons in culture.
Discussion
In the present work we show that AD-related tau pathological changes (hyperphosphorylation, aggregation, and propagation) can be diminished by the reduction of GSK-3β. Expressing unmodified human wild-type tau in WT and GSK-3β-HK mice using AAVs, we found that partial isoform-selective genetic decrease of GSK-3β could reduce synaptic tau hyperphosphorylation and propagation in the mouse brain. Furthermore, seeded tau aggregation was diminished in primary neurons from GSK-3β-HK embryos expressing aggregation prone FTD-mutant tau. Here, we show that the genetic decrease of GSK-3β reduces not only tau phosphorylation and aggregation but also neuronal propagation. Tau propagation across neural circuits is increasingly recognized to contribute to pathology in AD and other tauopathies (Gibbons et al., 2019; Mudher et al., 2017; Takeda, 2019a, 2019b; Walker et al., 2013). We conclude that a partial reduction of GSK-3β is sufficient to reduce pathological tau changes in the brain and therefore could be utilized to halt the tau-associated neurodegeneration.
We and others have previously shown aberrant activation of GSK-3β in human AD brains and AD transgenic mice (Blalock et al., 2009; DaRocha-Souto et al., 2012; Leroy et al., 2007; Serenó et al., 2009). Because there is a general consensus that pharmacological modulation of abnormal tau hyperphosphorylation is a promising therapeutic approach for AD (Engel, 2006; Engel et al., 2006; Le Corre et al., 2006; Nakashima et al., 2005; Noble et al., 2005; Pérez et al., 2003), the inhibition of GSK-3β seems an obvious target to achieve the reduction of pathological tau hyperphosphorylation (Cohen and Goedert, 2004). Pharmacological reduction of GSK-3 has been used by several groups, including our own, to reduce AD-like phenotypic changes in transgenic rodent and in vitro models (de Barreda et al., 2010; DaRocha-Souto et al., 2012; Engel, 2006; Houck et al., 2016; Muñoz-Montaño et al., 1997; Serenó et al., 2009; Wilson et al., 2020). However, the GKS-3 inhibitors used in those studies (e.g. thiadiazolidine derivatives and lithium, among others) are not selective for the GSK-3β isoform, and thus inhibition of other kinases could have potentially contributed to the observed results. The few thiadiazolidine derivative and lithium trials conducted in AD patients have yielded mixed results on potential benefits/side effects (Lovestone et al., 2015; Matsunaga et al., 2015; Mazanetz and Fischer, 2007; Muñoz-Montaño et al., 1997; Wilson et al., 2020). Thus, the development of a specific pharmacological inhibitor of GSK-3β is still an awaited feat (Gong and Iqbal, 2008). Efforts to develop selective, safe, tissue-specific, and dose-efficient GSK-3β inhibitors have been unsuccessful to date (Bhat et al., 2018; Matsunaga et al., 2019). However, the recent development of novel drug delivery strategies aimed at achieving adequate therapeutic concentrations to target organ or cells (i.e. nanoparticles or GSK-3β-cell knockouts), whereas avoiding potential off-target effects provides a new window of opportunity (reviewed in Bhat et al., 2018).
In the current study, we overcame the limitations of non-selective inhibition by using a mouse model with an isoform-selective reduction of GSK-3β activity through haploinsufficiency (GSK-3β-HK line).
Interestingly, GSK-3β-HK mice did not only show reduced tau pathological changes but also revealed new insights about the cellular distribution of GSK-3β in neurons. We found that GSK-3β, commonly referred to as mostly cytosolic (Bijur and Jope, 2001, 2003; Watcharasit et al., 2002), is present at even higher levels in synapses, pointing to its (yet insufficiently acknowledged) role in this cellular compartment. Even though a role of GSK-3β as a regulator of synaptic plasticity has been suggested (Bradley et al., 2012), particularly in long-term depression via NMDA receptors (Peineau et al., 2007, 2008, 2009), the physiological function of this enzyme in synapses remains largely unknown. GSK-3β-HK mice had a stronger reduction of GSK-3β levels in synapses compared with the cytosol, potentially explaining the partial protection against tau pathological changes in these mice. In fact, the function of GSK-3β may depend, at least in part, on its subcellular localization. For example, whereas nuclear and mitochondrial GSK-3β were reported to participate in pro-apoptotic signaling, cytosolic GSK-3β seems to mediate survival signals (Jacobs et al., 2012). In combination with our data, we postulate that synaptic GSK-3β levels may be reduced in GSK-3β-HK neurons in order to ensure essential amounts of GSK-3β in the cytosol.
In healthy neurons, tau is mostly restricted to axons where it binds to microtubules and promotes axonal transport (Avila et al., 2004; Buee et al., 2000; Papasozomenos and Binder, 1987). However, synaptic accumulation of p-Tau in AD (Brandt et al., 2005; Gendron and Petrucelli, 2009; Perez-Nievas et al., 2013; Tai et al., 2012) can lead to altered synaptic function (Hoover et al., 2010), correlates with preclinical stages of the disease, and seems to precede neuronal degeneration (Arvanitakis et al., 2007). Thus, the reduction of soluble hyperphosphorylated tau accumulation in synapses could protect against the tau-initiated neurodegenerative cascade in AD. Synaptic p-Tau could result either from local tau phosphorylation by synaptic GSK-3β activity or from cytosolic GSK-3β-mediated tau phosphorylation inducing p-Tau transport to the synapse.
In this study we were able to show that both soluble p-Tau accrual in synapses and tau protein spread between neurons in the brain can be successfully achieved through partial reduction of GSK3β. These findings support the idea that the presence of p-Tau in synapses may be directly linked to its ability to propagate trans-cellularly. We propose that GSK-3β activation lies upstream of neurodegenerative tau phenotypes in AD, not only by promoting tau phosphorylation and aggregation (Engel, 2006; reviewed in Mines et al., 2011) but also by promoting abnormal accrual of p-Tau into the synaptic compartment and trans-cellular spread in the brain, ultimately compromising the function and anatomical integrity of neurons and synapses in AD (see Figure 7 for proposed model).
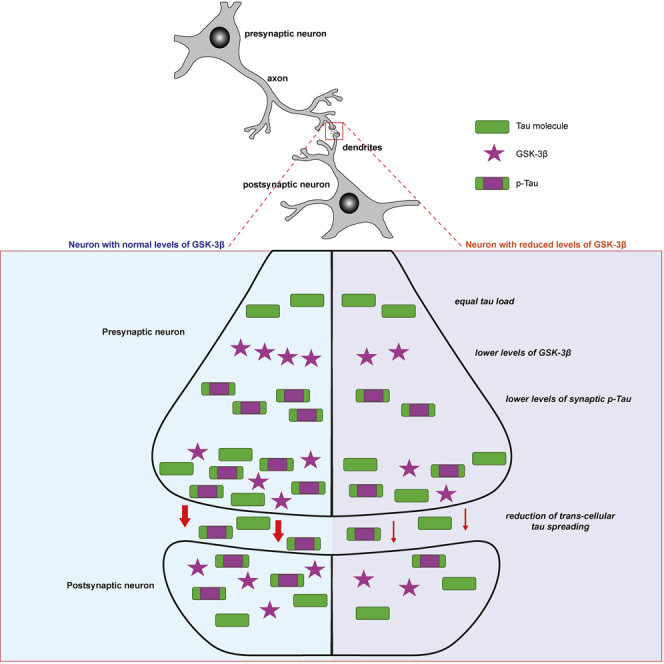
Figure 7.

Schematic of GSK-3β and p-Tau in the synapses
GSK-3β and tau co-localize in the soma and neurites of neurons. Genetic reduction of GSK-3β in mice leads to overall lower levels of GSK-3β in all cellular compartments and also changes the relative abundance of the kinase in the cytosolic and synaptic compartments. In WT mice, GSK-3β accumulates in higher levels in the synapse compared with the cytosol, whereas in GSK-3β-HK mice both compartments have similar amounts of GSK-3β. GSK-3β-HK mice have the same amount of total tau in the synapses as WT mice but significantly lower amounts of active GSK-3β and p-Tau.
Previous studies based on selective in vivo silencing of either GSK-3α or GSK-3β in transgenic mice suggested that the GSK-3α isoform contributes to both amyloid and tau pathology, whereas the GSK-3β isoform only modulates tau phosphorylation and neurofibrillary tangle formation (Hurtado et al., 2012). Our results show that GSK-3β-HK neurons (in vitro and in vivo) still render overall levels of tau phosphorylation that are similar to WT neurons and yet exhibit a targeted reduction of p-Tau levels in synapses that may confer protection against tau pathological changes.
After 12 weeks of human wild-type tau expression in vivo, we did not observe β-sheet-containing tau aggregates (Thioflavin-S staining) in WT or GSK-3β-HK mice, even in the presence of robust tau hyperphosphorylation (PHF1+) and misfolding (Alz50+). This observation, in agreement with our previous work and others (Dujardin et al., 2014; Mudher et al., 2017; Wegmann et al., 2019), provides further evidence that not necessarily aggregated but also soluble tau species can travel from a “donor” to a “recipient” cell. Our current results support the idea that a decrease in soluble synaptic tau phosphorylated at Ser396/404 may be linked to reduced tau spreading observed in GSK-3β-HK mice and primary neurons. Whether GSK-3β-mediated tau phosphorylation at this epitope directly promotes tau release and uptake (leading to tau spreading) remains to be further elucidated.
In conclusion, the current study shows that a partial isoform-selective decrease of GSK-3β is sufficient to diminish pathological phosphorylation and propagation of human tau in the mouse brain and is also effective against the aggregation of FTD-mutant tau in cultured neurons. We propose a mechanism, in which the reduction of GSK-3β in synapses lowers synaptic p-Tau, leading to (1) the reduction of tau trans-cellular propagation and (2) a decrease in seeded tau aggregate formation. Targeted manipulation of GSK-3β levels and/or its activity in the synapse may thus be a promising approach against tau-related pathological changes in the brain that are relevant in AD and other tauopathies.
Limitations of the study
We used AAV-mediated local expression of wild-type human tau expression to detect tau protein propagation and the effects of local tau overexpression. Our model has limitations for studying tau aggregation in vivo: AAV-injected WT and GSK-3β-HK mice did not develop β-sheet-containing tau aggregates upon expression of hTau in the EC, even at 6 months post-injection. To study the effect of GKS-3β reduction on tau aggregation, we therefore employed a previously established approach to measure tau aggregation propensity in cells through expression of the aggregation-prone repeat domain of FTLD-mutant tauP301S (Nobuhara et al., 2017). Of note, this tau construct does not occur in the etiology of AD and may be mechanistically different from wild-type tau aggregation in AD. However, this in vitro assay allowed us to demonstrate that the selective reduction of GKS-3β was protective against tau-induced pathological formation of neuronal aggregates.
Resource availability
Lead contact
Requests for additional information can be directed to the Lead Contact, Ana Claudia Amaral (aamaral@mgh.harvard.edu).
Materials availability
This study did not generate unique reagents.
Data and code availability
This study did not generate code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Dr. James R. Woodgett for the donation of the GSK-3β-HK mouse line and Dr. Peter Davies for the donation of the p-Tau antibodies (CP13, PHF-1, and Alz50) used in this study.
Author contributions
Conceptualization: A.C.A., B.P.N., S.W., and T.G.I.; Design of the Experiments: A.C.A and T.G.I.; Performance of the Majority of the Experiments: A.C.A. and M.S.T.C.; Methodology: A.C.A, E.H., Z.F., S.T., S.W., and T.G.I.; Additional Experimental Work: A.G.M., H.A.E., S.R.G., C.C., S.M., B.E., and P.R.; Construct Design and Production: E.H., Z.F., and S.W.; Funding Acquisition: T.G.I; Supervision: S.W. and T.G.I.
Declaration of interests
Teresa Gómez-Isla participated as speaker in an Eli Lilly and Company-sponsored educational symposium and serves in an Eli Lilly Data Monitoring Committee. All other authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102058.
Supplemental information
References
- Arvanitakis Z., Witte R.J., Dickson D.W., Tsuboi Y., Uitti R.J., Slowinski J., Hutton M.L., Lin S.-C., Boeve B.F., Cheshire W.P. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation) Parkinsonism Relat. Disord. 2007;13:230–239. doi: 10.1016/j.parkreldis.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Augustinack J.C., Schneider A., Mandelkow E.-M., Hyman B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- Avila J., Lucas J.J., Pérez M., Hernández F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004;84:361–384. doi: 10.1152/physrev.00024.2003. [DOI] [PubMed] [Google Scholar]
- de Barreda E.G., Pérez M., Ramos P.G., de Cristobal J., Martín-Maestro P., Morán A., Dawson H.N., Vitek M.P., Lucas J.J., Hernández F. Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 2010;37:622–629. doi: 10.1016/j.nbd.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Bhat R.V., Andersson U., Andersson S., Knerr L., Bauer U., Sundgren-Andersson A.K. The conundrum of GSK3 inhibitors: is it the dawn of a new beginning? J. Alzheimers Dis. 2018;64:S547–S554. doi: 10.3233/JAD-179934. [DOI] [PubMed] [Google Scholar]
- Bijur G.N., Jope R.S. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J. Biol. Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur G.N., Jope R.S.C. Glycogen synthase kinase-3[beta] is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Blalock W.L., Grimaldi C., Fala F., Follo M., Horn S., Basecke J., Martinelli G., Cocco L., Martelli A.M. PKR activity is required for acute leukemic cell maintenance and growth: a role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J. Cell. Physiol. 2009;221:232–241. doi: 10.1002/jcp.21848. [DOI] [PubMed] [Google Scholar]
- Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Bradley C.A., Peineau S., Taghibiglou C., Nicolas C.S., Whitcomb D.J., Bortolotto Z.A., Kaang B.-K., Cho K., Wang Y.T., Collingridge G.L. A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 2012;5:13. doi: 10.3389/fnmol.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R., Hundelt M., Shahani N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim. Biophys. Acta. 2005;1739:331–354. doi: 10.1016/j.bbadis.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Buee L., Bussiere T., Buee-Scherrer V., Delacourte A., Hof P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- Caccamo A., Oddo S., Tran L.X., LaFerla F.M. Lithium reduces tau phosphorylation but not Aβ or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol. 2007;170:1669–1678. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A., Polydoro M., Suárez-Calvet M., William C., Adamowicz D.H., Kopeikina K.J., Pitstick R., Sahara N., Ashe K.H., Carlson G.A. Propagation of tau pathology in a model of early alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang D.-M., Wang Z., Chiu C.-T. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front. Mol. Neurosci. 2011;4:15. doi: 10.3389/fnmol.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P., Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat. Rev. Drug Discov. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- Le Corre S., Klafki H.W., Plesnila N., Hubinger G., Obermeier A., Sahagun H., Monse B., Seneci P., Lewis J., Eriksen J. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc. Natl. Acad. Sci. U S A. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRocha-Souto B., Coma M., Pérez-Nievas B.G., Scotton T.C., Siao M., Sánchez-Ferrer P., Hashimoto T., Fan Z., Hudry E., Barroeta I. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol. Dis. 2012;45:425–437. doi: 10.1016/j.nbd.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S., Lécolle K., Caillierez R., Bégard S., Zommer N., Lachaud C., Carrier S., Dufour N., Aurégan G., Winderickx J. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014;2:14. doi: 10.1186/2051-5960-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T. Full reversal of alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006;26:5083–5090. doi: 10.1523/JNEUROSCI.0604-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T., Goñi-Oliver P., Lucas J.J., Avila J., Hernández F. Chronic lithium administration to FTDP-17 tau and GSK-3β overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 2006;99:1445–1455. doi: 10.1111/j.1471-4159.2006.04139.x. [DOI] [PubMed] [Google Scholar]
- Gendron T.F., Petrucelli L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons G.S., Lee V.M.Y., Trojanowski J.Q. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 2019;76:101–108. doi: 10.1001/jamaneurol.2018.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T., Price J.L., McKeel D.W., Jr., Morris J.C., Growdon J.H., Hyman B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild alzheimer’s disease. J. Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T., Hollister R., West H., Mui S., Growdon J.H., Petersen R.C., Parisi J.E., Hyman B.T. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T., Spires T., De Calignon A., Hyman B.T. Neuropathology of Alzheimer’s disease. Handb. Clin. Neurol. 2008;89:233–243. doi: 10.1016/S0072-9752(07)01222-5. [DOI] [PubMed] [Google Scholar]
- Gong C.-X., Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for alzheimer disease. Curr. Med. Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanger D.P., Hughes K., Woodgett J.R., Brion J.-P., Anderton B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Hernández F., Borrell J., Guaza C., Avila J., Lucas J.J. Spatial learning deficit in transgenic mice that conditionally over-express GSK-3β in the brain but do not form tau filaments. J. Neurochem. 2002;83:1529–1533. doi: 10.1046/j.1471-4159.2002.01269.x. [DOI] [PubMed] [Google Scholar]
- Hoeflich K.P., Jin O., Woodgett J.R. Requirement for glycogen synthase kinase-3b in cell survival and NF-kB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Holmes B.B., Furman J.L., Mahan T.E., Yamasaki T.R., Mirbaha H., Eades W.C., Belaygorod L., Cairns N.J., Holtzman D.M., Diamond M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U S A. 2014;111:E4376–E4385. doi: 10.1073/pnas.1411649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover B.R., Reed M.N., Su J., Penrod R.D., Kotilinek L.A., Grant M.K., Pitstick R., Carlson G.A., Lanier L.M., Yuan L.-L. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houck A.L., Hernández F., Ávila J. A simple model to study tau pathology. J. Exp. Neurosci. 2016;10 doi: 10.4137/JEN.S25100. JEN.S25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S., Begum A.N., Jones M.R., Oh M.S., Beech W.K., Beech B.H., Yang F., Chen P., Ubeda O.J., Kim P.C. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol. Dis. 2009;33:193–206. doi: 10.1016/j.nbd.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado D.E., Molina-Porcel L., Carroll J.C., MacDonald C., Aboagye A.K., Trojanowski J.Q., Lee V.M.-Y. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of alzheimer’s disease. J. Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imahori K., Uchida T. Physiology and pathology of tau protein kinases in relation to Alzheimer’s disease. J. Biochem. 1997;121:179–188. [PubMed] [Google Scholar]
- Jacobs K.M., Bhave S.R., Ferraro D.J., Jaboin J.J., Hallahan D.E., Thotala D. GSK-3: a bifunctional role in cell death pathways. Int. J. Cell Biol. 2012;2012:1–11. doi: 10.1155/2012/930710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope R.S., Roh M.-S. Glycogen synthase kinase-3 (GSK3) in psychiatric diseases and therapeutic interventions. Curr. Drug Targets. 2006;7:1421–1434. doi: 10.2174/1389450110607011421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozikowski A.P., Gaisina I.N., Petukhov P.A., Sridhar J., King L.T., Blond S.Y., Duka T., Rusnak M., Sidhu A. Highly potent and specific GSK-3β inhibitors that block tau phosphorylation and decrease α-synuclein protein expression in a cellular model of Parkinson’s disease. ChemMedChem. 2006;1:256–266. doi: 10.1002/cmdc.200500039. [DOI] [PubMed] [Google Scholar]
- Leroy K., Brion J.-P. Developmental expression and localization of glycogen synthase kinase-3β in rat brain. J. Chem. Neuroanat. 1999;16:279–293. doi: 10.1016/s0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- Leroy K., Yilmaz Z., Brion J.-P. Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007;33:43–55. doi: 10.1111/j.1365-2990.2006.00795.x. [DOI] [PubMed] [Google Scholar]
- Liu S.J., Zhang A.H., Li H.L., Wang Q., Deng H.M., Netzer W.J., Xu H., Wang J.Z. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J. Neurochem. 2003;87:1333–1344. doi: 10.1046/j.1471-4159.2003.02070.x. [DOI] [PubMed] [Google Scholar]
- Lovestone S., Reynolds C.H., Latimer D., Davis D.R., Anderton B.H., Gallo J.-M., Hanger D., Mulot S., Marquardt B., Stabel S. Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 1994;4:1077–1086. doi: 10.1016/s0960-9822(00)00246-3. [DOI] [PubMed] [Google Scholar]
- Lovestone S., Boada M., Dubois B., Hüll M., Rinne J.O., Huppertz H.-J., Calero M., Andrés M.V., Gómez-Carrillo B., León T. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015;45:75–88. doi: 10.3233/JAD-141959. [DOI] [PubMed] [Google Scholar]
- Lucas J.J., Hernández F., Gómez-Ramos P., Morán M.A., Hen R., Avila J. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E.-M., Drewes G., Biernat J., Gustke N., Van Lint J., Vandenheede J.R., Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- Matsunaga S., Kishi T., Annas P., Basun H., Hampel H., Iwata N. Lithium as a treatment for alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 2015;48:403–410. doi: 10.3233/JAD-150437. [DOI] [PubMed] [Google Scholar]
- Matsunaga S., Fujishiro H., Takechi H. Efficacy and safety of glycogen synthase kinase 3 inhibitors for alzheimer’s disease: a systematic review and meta-analysis. J. Alzheimers Dis. 2019;69:1031–1039. doi: 10.3233/JAD-190256. [DOI] [PubMed] [Google Scholar]
- Maurer U., Preiss F., Brauns-Schubert P., Schlicher L., Charvet C. GSK-3-at the crossroads of cell death and survival. J. Cell Sci. 2014;127:1369–1378. doi: 10.1242/jcs.138057. [DOI] [PubMed] [Google Scholar]
- Mazanetz M.P., Fischer P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- Mills C.N., Nowsheen S., Bonner J.A., Yang E.S. Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front. Mol. Neurosci. 2011;4:47. doi: 10.3389/fnmol.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines M.A., Jope R.S. Glycogen synthase kinase-3: a promising therapeutic target for fragile X syndrome. Front. Mol. Neurosci. 2011;4:35. doi: 10.3389/fnmol.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines M.A., Beurel E., Jope R.S. Regulation of cell survival mechanisms in alzheimer’s disease by glycogen synthase kinase-3. Int. J. Alzheimers Dis. 2011;2011:861072. doi: 10.4061/2011/861072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudher A., Colin M., Dujardin S., Medina M., Dewachter I., Alavi Naini S.M., Mandelkow E.-M., Mandelkow E., Buée L., Goedert M. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017;5:99. doi: 10.1186/s40478-017-0488-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Montaño J.R., Moreno F.J., Avila J., Díaz-Nido J. Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett. 1997;411:183–188. doi: 10.1016/s0014-5793(97)00688-1. [DOI] [PubMed] [Google Scholar]
- Nakashima H., Ishihara T., Suguimoto P., Yokota O., Oshima E., Kugo A., Terada S., Hamamura T., Trojanowski J.Q., Lee V.M.-Y. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005;110:547–556. doi: 10.1007/s00401-005-1087-4. [DOI] [PubMed] [Google Scholar]
- Noble W., Planel E., Zehr C., Olm V., Meyerson J., Suleman F., Gaynor K., Wang L., LaFrancois J., Feinstein B. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U S A. 2005;102:6990–6995. doi: 10.1073/pnas.0500466102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobuhara C.K., DeVos S.L., Commins C., Wegmann S., Moore B.D., Roe A.D., Costantino I., Frosch M.P., Pitstick R., Carlson G.A. Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of tau in vitro. Am. J. Pathol. 2017;187:1399–1412. doi: 10.1016/j.ajpath.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasozomenos S.C., Binder L.I. Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- Pei J.J., Tanaka T., Tung Y.C., Braak E., Iqbal K., Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- Pei J.J., Braak E., Braak H., Grundke-Iqbal I., Iqbal K., Winblad B., Cowburn R.F. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Peineau S., Taghibiglou C., Bradley C., Wong T.P., Liu L., Lu J., Lo E., Wu D., Saule E., Bouschet T. LTP inhibits LTD in the Hippocampus via regulation of GSK3β. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Peineau S., Bradley C., Taghibiglou C., Doherty A., Bortolotto Z.A., Wang Y.T., Collingridge G.L. The role of GSK-3 in synaptic plasticity: GSK-3 and synaptic plasticity. Br. J. Pharmacol. 2008;153:S428–S437. doi: 10.1038/bjp.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peineau S., Nicolas C.S., Bortolotto Z.A., Bhat R.V., Ryves W.J., Harwood A.J., Dournaud P., Fitzjohn S.M., Collingridge G.L. A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain. 2009;2:22. doi: 10.1186/1756-6606-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C.-X., Hu J., Liu D., Hong X.-P., Wu Y.-Y., Zhu L.-Q., Wang J.-Z. Disease-modified glycogen synthase kinase-3β intervention by melatonin arrests the pathology and memory deficits in an Alzheimer’s animal model. Neurobiol. Aging. 2013;34:1555–1563. doi: 10.1016/j.neurobiolaging.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Pérez M., Hernández F., Lim F., Díaz-Nido J., Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimers Dis. 2003;5:301–308. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- Perez-Nievas B.G., Stein T.D., Tai H.-C., Dols-Icardo O., Scotton T.C., Barroeta-Espar I., Fernandez-Carballo L., de Munain E.L., Perez J., Marquie M. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain J. Neurol. 2013;136:2510–2526. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serenó L., Coma M., Rodríguez M., Sánchez-Ferrer P., Sánchez M.B., Gich I., Agulló J.M., Pérez M., Avila J., Guardia-Laguarta C. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009;35:359–367. doi: 10.1016/j.nbd.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Spittaels K., Van den Haute C., Van Dorpe J., Geerts H., Mercken M., Bruynseels K., Lasrado R., Vandezande K., Laenen I., Boon T. Glycogen synthase kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J. Biol. Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- Tai H.-C., Serrano-Pozo A., Hashimoto T., Frosch M.P., Spires-Jones T.L., Hyman B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Tomizawa K., Kato R., Sato K., Uchida T., Fujita S.C., Imahori K. Localization and developmental changes of τ protein kinase I/glycogen synthase kinase-3β in rat brain. J. Neurochem. 1994;63:245–255. doi: 10.1046/j.1471-4159.1994.63010245.x. [DOI] [PubMed] [Google Scholar]
- Takashima A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers Dis. 2006;9:309–317. doi: 10.3233/jad-2006-9s335. [DOI] [PubMed] [Google Scholar]
- Takeda S. Progression of Alzheimer’s disease, tau propagation, and its modifiable risk factors. Neurosci. Res. 2019;141:36–42. doi: 10.1016/j.neures.2018.08.005. [DOI] [PubMed] [Google Scholar]
- Takeda S. Tau propagation as a diagnostic and therapeutic target for dementia: potentials and unanswered questions. Front. Neurosci. 2019;13:1274. doi: 10.3389/fnins.2019.01274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L.C., Diamond M.I., Duff K.E., Hyman B.T. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P., Bijur G.N., Zmijewski J.W., Song L., Zmijewska A., Chen X., Johnson G.V.W., Jope R.S. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc. Natl. Acad. Sci. U S A. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S., Maury E.A., Kirk M.J., Saqran L., Roe A., DeVos S.L., Nicholls S., Fan Z., Takeda S., Cagsal-Getkin O. Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 2015;34:3028–3041. doi: 10.15252/embj.201592748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S., Bennett R.E., Amaral A.C., Hyman B.T. Academic Press; 2017. Studying tau protein propagation in the mouse brain using adeno-associated viruses. In Methods in Tau Cell Biology; pp. 307–322. [DOI] [PubMed] [Google Scholar]
- Wegmann S., Bennett R.E., Delorme L., Robbins A.B., Hu M., McKenzie D., Kirk M.J., Schiantarelli J., Tunio N., Amaral A.C. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019;5:eaaw6404. doi: 10.1126/sciadv.aaw6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson E.N., Carmo S.D., Welikovitch L.A., Hall H., Aguilar L.F., Foret M.K., Iulita M.F., Jia D.T., Marks A.R., Allard S. NP03, a microdose lithium formulation, blunts early amyloid post-plaque neuropathology in McGill-R-Thy1-APP alzheimer-like transgenic rats. J. Alzheimers Dis. 2020;73:723–739. doi: 10.3233/JAD-190862. [DOI] [PubMed] [Google Scholar]
- Woodgett J.R. Molecular cloning and expression of glycogen synthase kinase-3/Factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H., Ishiguro K., Uchida T., Takashima A., Lemere C.A., Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3β and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate code.