

Flavonoid glycosides constitute a class of secondary metabolites widely distributed in nature. These compounds are involved in bitter taste or clouding in plant-based foods or beverages, respectively.
KEYWORDS: crystal structure, rutinosidase, Aspergillus oryzae, substrate specificity, N-glycosylation
ABSTRACT
The rutinosidase (Rut)-encoding gene Aorut has been expressed in Pichia pastoris with its native signal sequence from Aspergillus oryzae. Biochemical and structural investigation of the purified recombinant mature A. oryzae Rut (AoRut), designated rAoRutM, was performed in this study. A 1.7-Å resolution crystal structure of rAoRutM was determined, which is an essential step forward in the utilization of AoRut as a potential catalyst. The crystal structure of rAoRutM was represented by a (β/α)8 TIM barrel fold with structural similarity to that of rutinosidase from Aspergillus niger (AnRut) and an exo-β-(1,3)-glucanase from Candida albicans. The crystal structure revealed that the catalytic site was located in a deep cleft, similarly to AnRut, and that internal cavities and water molecules were also present. Purified rAoRutM hydrolyzed not only 7-O-linked and 3-O-linked flavonoid rutinosides but also 7-O-linked and 3-O-linked flavonoid glucosides. rAoRutM displayed high catalytic activity toward quercetin 3-O-linked substrates such as rutin and isoquercitrin, rather than to the 7-O-linked substrate, quercetin-7-O-glucoside. Unexpectedly, purified rAoRutM exhibited increased thermostability after treatment with endo-β-N-acetylglucosaminidase H. Circular dichroism (CD) spectra of purified intact rAoRutM and of the enzyme after N-deglycosylation showed a typical α-helical CD profile; however, the molar ellipticity values of the peaks at 208 nm and 212 nm differed. The Km and kcat values for the substrates modified by rutinose were higher than those for the substrates modified by β-d-glucose.
IMPORTANCE Flavonoid glycosides constitute a class of secondary metabolites widely distributed in nature. These compounds are involved in bitter taste or clouding in plant-based foods or beverages, respectively. Flavonoid glycoside degradation can proceed through two alternative enzymatic pathways: one that is mediated by monoglycosidases and another that is catalyzed by a diglycosidase. The present report on the biochemical and structural investigation of A. oryzae rutinosidase provides a potential biocatalyst for industrial applications of flavonoids.
INTRODUCTION
Flavonoids have been gaining attention as valuable nutritional factors due to their antioxidant, anticancerous, antibacterial, and antifungal properties (1, 2). Flavonoids are a widely distributed class of secondary metabolites and exist almost exclusively as β-glycosides in nature. The major flavonoid glycosides found in plants or fruits are diglycosides such as quercetin 3-O-rutinoside (rutin), hesperetin 7-O-rutinoside (hesperidin), kaempferol-3-O-rutinoside, and naringenin 7-O-neohesperidoside (naringin) (3). In the citrus processing industry, the deglycosylation of flavonoids plays an important role in enhancing product quality, such as in reducing bitterness and in clarification of juices (4, 5). Flavonoid glycosides exhibit reduced nutrient absorption in humans, and the gut microbiota often metabolizes the heterocyclic pyran ring of free flavonoid aglycones to yield phenolic acids (6). However, there are no endogenous glycosidases in the small intestine of humans to hydrolyze the glycosidic bond of the flavonoid glycosides.
The 6-O-α-l-rhamnopyranosyl-β-d-glucosidase (rutinosidase; EC 3.2.1.168) enzyme is interesting from a practical perspective because its substrates such as rutin and hesperidin are inexpensive by-products of the food industry. The rutin hydrolysis reaction scheme catalyzed by rutinosidase is shown in Fig. S1 in the supplemental material. Based on amino acid sequence similarity, the carbohydrate active enzymes database (CAZy) (7) classifies the fungal diglycosidases into the glycoside hydrolase (GH) families GH3 and GH5 subfamily 23 (8–10). Rutinosidases, belonging to GH 5 subfamily 23, from Acremonium sp. DSM 24697, Aspergillus niger K2, and Aspergillus oryzae RIB40 have previously been reported (10–12). Recently, the crystal structure of rutinosidase from A. niger (AnRut) was deciphered, and its substrate specificity was higher for quercetin 3-O-glucoside (isoquercitrin) than for rutin (13).
A. oryzae is a filamentous fungus widely used in the fermentation industry for manufacturing sake, soy sauce, and miso in Japan. In a previous study, we identified AoRut, the α-l-rhamnosyl-β-d-glucosidase-encoding gene in A. oryzae, and performed biochemical characterization of the purified recombinant enzyme expressed in Pichia pastoris using the α-factor secretion signal sequence of Saccharomyces cerevisiae (12). For this report, we present the crystal structure of recombinant AoRut protein newly expressed in P. pastoris using the native A. oryzae signal sequence and identified the catalytic sites of the enzyme by mutational analysis. We also investigated its substrate specificity with various flavonoid β-glucosides and the role of their N-linked oligosaccharides in specificity. Our study revealed A. oryzae rutinosidase to be a novel potential biocatalyst for industrial applications.
RESULTS
Preparation of rAoRutM and mutational variants.
rAoRutM was secreted by P. pastoris transformants using the native A. oryzae signal sequence and accumulated as an active enzyme in the P. pastoris culture supernatant. Additionally, the rAoRutM E210Q and E317Q mutant variants were also secreted using the native signal sequence and accumulated as inactive enzymes in the culture supernatant. The protein expression level of rAoRutM in P. pastoris was similar to that of rAoRut fusing with the Saccharomyces cerevisiae α-factor secretion signal sequence instead of the native signal sequence. rAoRutM and its variants produced by P. pastoris were purified in a two-step procedure using anion-exchange chromatography and gel filtration chromatography as described in Materials and Methods.
General properties of purified rAoRut.
The NetNglyc 1.0 server (http://www.cbs.dtu.dk/services/NetNGlyc/) predicted 5 putative N-glycosylation sites in the AoRut protein (12). Our previous study showed that purified rAoRut appears as 2 protein bands with molecular masses of 40 kDa and 42 kDa, respectively, after treatment with endo-β-N-acetylglucosaminidase H (endo-H) and N-terminal amino acid sequences VQSPE and EFAPH. The sequence of VQSPE corresponds to the internal AoRut protein sequence that follows the N-terminal signal peptidase cleavage site. The sequence of EFAPH corresponds to the AoRut N-terminal amino acid sequence, including the sequence derived from the EcoRI recognition site (GAATTC) engineered in PCR primers, before the N terminus of the signal peptidase cleaving site. In the present study, purified rAoRutM exhibited one protein band with a molecular mass of 40 kDa after treatment with endo-H (see Fig. S2 in the supplemental material) and VQSPE N-terminal amino acid sequences, indicating the homogeneity of the protein and suitability for crystallization trials.
The optimal pH, temperature, and thermal stability of purified rAoRutM were similar to that of rAoRut. The optimal pH and temperature for rAoRutM were observed to be 5.0 and 45 to 50°C, respectively. The enzyme retained more than 90% of its initial activity after incubation at 50°C for 2 h. Interestingly, after treatment with endo-H, rAoRutM retained 82% and 32% of its activity after incubation at 50°C for 4 h and 8 h, respectively, whereas the levels of residual activity of the intact enzyme were 41% and 16% under identical conditions (Fig. 1), indicating higher thermal stability of endo-H-treated rAoRutM than of the intact enzyme.
FIG 1.
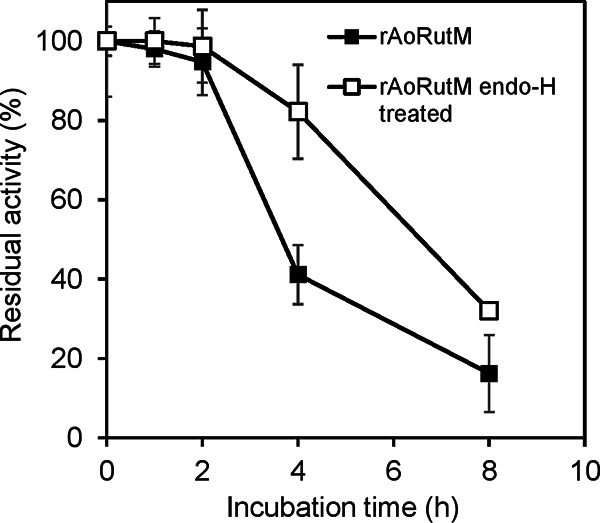
Effects of N-glycosylation on the thermal stability of purified rAoRutM (closed symbols for native enzyme and open symbols for the endo-H-treated enzyme). Aliquots of purified enzymes were incubated at 50°C for different times. After cooling on ice, the residual activity was measured at pH 5.0 and 45°C.
CD spectra.
To evaluate the secondary structure of AoRut, we measured circular dichroism (CD) spectra at pH 5.0. The CD spectra of intact rAoRutM and rAoRutM after endo-H treatment are shown in Fig. 2. Though both spectra show typical α-helical CD profiles with peaks at 208 nm and 212 nm, the molar ellipticity values at the peaks differ.
FIG 2.
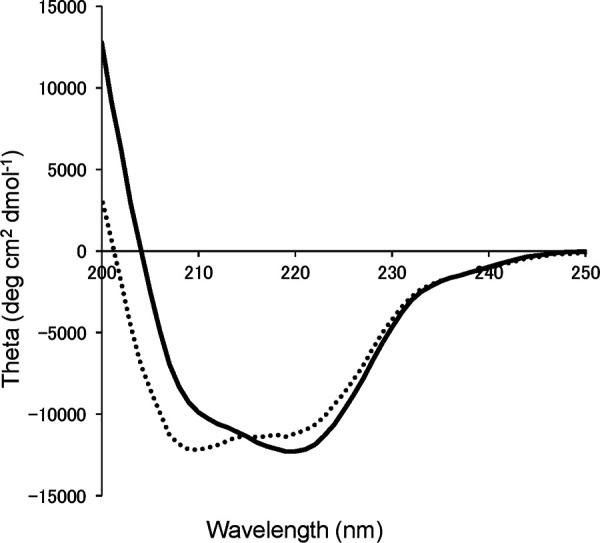
CD spectra of intact rAoRutM (solid line) and endo-H-treated rAoRutM (dotted line).
Crystal structure of rAoRutM.
The crystal structure of N-deglycosylated rAoRutM was determined to a resolution of 1.7 Å. The asymmetric unit contained two monomers (molecules A and B) which have almost identical structures with a root mean square deviation (RMSD) value of 0.16 Å by superposition of Cα atoms. Here, we focus on molecule A for detailed structure analysis. The monomer A had five N-linked N-acetylglucosamine molecules linked to Nδ2 atoms of Asn32, Asn128, Asn176, Asn288, and Asn359 (Fig. 3a). Of 373 entire residues, 360 residues of AoRut were resolved in the structure whereas nine N-terminal and four C-terminal residues were disordered. The overall structure of rAoRutM consisted of a classical (β/α)8 TIM barrel fold, typical for GH-5 family enzymes (8). The N-terminal α helix-2 and -3 were stabilized by a disulfide bond between Cys72 and Cys79. The characteristic feature of the structure was a deep cleft on the C-terminal side of the β-barrel (Fig. 3a). The amino acid residues at the bottom of the cleft were well conserved, with negative electrostatic potential, indicating their important role in enzymatic activity (Fig. 3b and c).
FIG 3.

Structure of rAoRutM. (a) Ribbon (left) and surface (right) representations with side views (top) and top views (bottom) are shown. Five N-acetyl-d-glucosamine molecules are shown as red CPK (Corey-Pauling-Koltun) spheres in the ribbon representation. A catalytic cleft is also indicated. (b) Sequence conservation among homologous proteins is depicted using the ConSurf server with the top view of the surface representation. Conserved residues are shown with purple, while nonconserved variable residues are shown with green. (c) Electrostatic potential of enzyme surface (red, negative; blue, positive) calculated using the PBEQ-Solver server on a solvent-accessible surface with monosaccharide molecules.
Comparison of structure of rAoRutM with that of rutinosidase from Aspergillus niger (AnRut).
AoRut has 71% amino acid sequence identity with rutinosidase from A. niger (AnRut; GenBank accession number MN393234), and both AnRut and AoRut belong to GH-5 subfamily 23. Recently, a crystal structure of AnRut was reported (PDB identifier [ID] 6I1A [13]), and the superimposition of the rAoRutM structure onto that of AnRut resulted in a 0.34-Å RMSD value of Cα atoms. This result indicated a high degree of structural similarity between rutinosidase from A. oryzae and that from A. niger (Fig. 4a). Thus far, structures have been determined in GH-5 subfamily 23 for only these two enzymes. As shown in Fig. 4b, the catalytic residues were found at invariant positions in the structures of rAoRutM and AnRut: E210 as the putative acid/base catalyst and E317 as the putative catalytic nucleophile in AoRut (8, 13). Analyses of E210Q and E317Q variants were performed to find further evidence for essential interactions in catalysis. E210Q and E317Q variants were purified from each culture supernatant. The levels of specific activities of E210Q and E317Q variants toward rutin were 0.15 ± 0.02 μmol/min/mg and 0.044 ± 0.006 μmol/min/mg, respectively, indicating that the activities of two variants drastically decreased (2.9% and 0.8%, respectively) relative to the intact enzyme (Table 1).
FIG 4.

Superposition of the rAoRutM structure (shown in green) onto that of AnRut (shown in gray). (a) A side view of the superposed structures with the ribbon representation. Catalytic E210 and E317 are shown as red CPK spheres. (b) Close-up view of the superposition of active site residues of rAoRutM and AnRut. (c) Ribbon representation of rAoRutM with B-factor data represented by color (red represents the highest level and blue the lowest level) and thickness (thicker lines represent the highest level and thinner lines the lowest). E210 and E317 are shown in blue spheres. (d) Internal cavities and water molecules in rAoRutM near the catalytic site. Cavities and the substrate binding cleft are shown as green surfaces. Water molecules are shown as blue spheres.
TABLE 1.
Specific activities of wild-type rAoRutM and its E210Q and E317Q variants with flavonoid glucoside or rutinosidea
| Substrate | Specific activity (μmol/min/mg) |
||
|---|---|---|---|
| Wild type | E210Q | E317Q | |
| Quercetin-3-O-rutinoside (rutin) | 5.2 ± 0.2 | 0.15 ± 0.02 | 0.044 ± 0.006 |
| Hespertin-7-O-rutinoside (hesperidin) | 1.8 ± 0.3 | ND | ND |
| Naringenin-7-O-rutinoside (narirutin) | 4.5 ± 0.3 | ND | ND |
| Quercetin-3-O-glucoside (isoquercitrin) | 4.5 ± 0.7 | ND | ND |
| Quercetin-7-O-glucoside | 2.3 ± 0.5 | ND | ND |
| Hespertin-7-O-glucoside | 1.6 ± 0.1 | ND | ND |
| Naringenin-7-O-glucoside (prunin) | 3.3 ± 0.2 | ND | ND |
ND, not determined.
Substrate specificity of purified rAoRutM.
Specific activities of endo-H-treated rAoRutM and intact rAoRutM measured 6.5 ± 0.6 μmol/min/mg and 5.2 ± 0.2 μmol/min/mg, respectively, toward rutin and 1.4 ± 0.01 μmol/min/mg and 1.8 ± 0.3 μmol/min/mg, respectively, toward hesperidin (Table 1). No significant change in activity was found between deglycosylated rAoRutM and intact enzyme for other tested substrates. Substrate specificity was tested toward not only 7-O-linked and 3-O-linked flavonoid rutinosides but also 7-O-linked and 3-O-linked flavonoid β-glucosides, such as naringenin-7-O-glucoside (prunin), hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside (isoquercitrin) (Fig. 5). Of these, rAoRutM hydrolyzed both 7-O-linked and 3-O-linked flavonoid rutinosides and their β-glucosides (Table 1). Interestingly, rAoRutM did not hydrolyze naringin [naringenin-7-O-linked α-l-rhamnosyl-(1,2)-β-d-glucose], whereas it hydrolyzed prunin (naringenin-7-O-linked β-d-glucose). However, purified rAoRutM did not hydrolyze p-nitrophenyl-β-d-glucoside, galactoside, or cellobioside.
FIG 5.
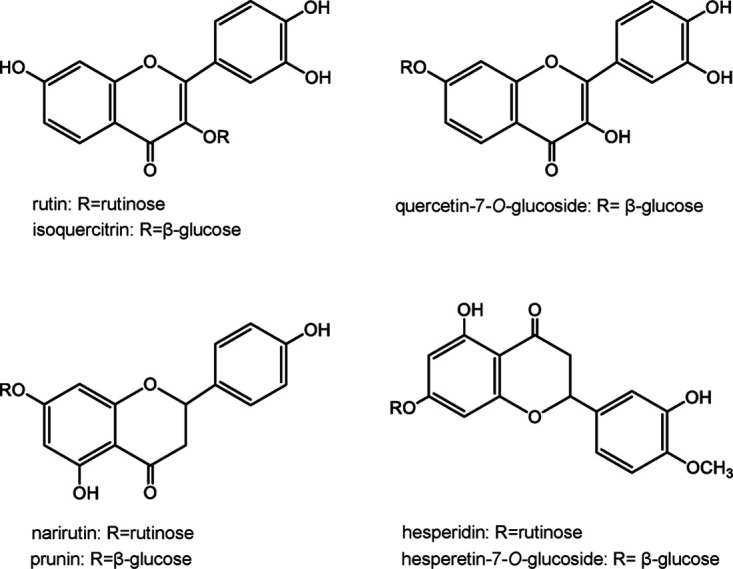
Chemical structure of the substrates used for the analysis of substrate specificity in purified rAoRutM.
The rAoRutM Km and kcat values were determined for 7-O-linked and 3-O-linked flavonoid rutinosides and their β-d-glucoside derivatives (Table 2). The enzyme exhibited typical Michaelis-Menten kinetics; however, the kinetic parameters varied depending on the substrates. The Km values for naringenin-7-O-glucoside (prunin), hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside (isoquercitrin) were 0.67 ± 0.10 mM, 0.29 ± 0.04 mM, 0.85 ± 0.35 mM, and 0.76 ± 0.01 mM, respectively, indicating higher affinity (or lower Km) for hesperetin-7-O-glucoside than for prunin, quercetin-7-O-glucoside, and isoquercitrin. However, the kcat values for prunin, hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and isoquercitrin were 0.95 × 103 ± 0.08 × 103 s−1, 0.22 × 103 ± 0.01 × 103 s−1, 0.98 × 103 ± 0.17 × 103 s−1, and 1.11 × 103 ± 0.02 × 103 s−1, respectively, indicating the lowest substrate turnover for hesperetin-7-O-glucoside among the above four substrates. Meanwhile, the Km values for rutin, hesperidin, and narirutin were 1.50 ± 0.03 mM, 1.38 ± 0.03 mM, and 1.17 ± 0.21 mM, indicating higher affinity than for isoquercitrin, hesperetin-7-O-glucoside, and prunin. However, the kcat values for rutin, hesperidin, and narirutin (2.52 × 103 ± 0.02 × 103 s−1, 1.78 × 103 ± 0.16 × 103 s−1, and 2.91 × 103 ± 0.34 × 103 s−1) were considerably higher than those for isoquercitrin, hesperetin-7-O-glucoside, and prunin, respectively.
TABLE 2.
Kinetic constants of rAoRutM toward flavonoid glucoside or rutinoside
| Substrate | Km (mM) | Kcat (×103 s−1) | Kcat/Km (×103 s−1 · mM−1) |
|---|---|---|---|
| Quercetin-3-O-rutinoside (rutin) | 1.50 ± 0.03 | 2.52 ± 0.04 | 1.68 |
| Hesperetin-7-O-rutinoside (hesperidin) | 1.38 ± 0.03 | 1.78 ± 0.16 | 1.29 |
| Naringenin-7-O-rutinoside (narirutin) | 1.17 ± 0.21 | 2.91 ± 0.34 | 2.50 |
| Quercetin-3-O-glucoside (isoquercitrin) | 0.76 ± 0.01 | 1.11 ± 0.02 | 1.46 |
| Quercetin-7-O-glucoside | 0.85 ± 0.35 | 0.98 ± 0.17 | 1.21 |
| Hesperetin-7-O-glucoside | 0.29 ± 0.04 | 0.22 ± 0.01 | 0.76 |
| Naringenin-7-O-glucodeside (prunin) | 0.67 ± 0.10 | 0.95 ± 0.08 | 1.42 |
DISCUSSION
The use of heterologous expression is an effective approach for the production of industrially useful enzymes. P. pastoris, a methylotrophic yeast, is the most extensively exploited yeast for recombinant protein production (14–16). However, the level of expression depends on the secretion signal peptide used in the yeast expression system. Sequencing analysis revealed incorrect processing of the α-mating factor prepropeptide in recombinant proteins expressed in P. pastoris, such as feruloyl esterase from Talaromyces stipitatus (17), xylanase from Thermomyces lanuginosus (18), and lipase from Candida antarctica (19); however, their catalytic properties were not affected. Additionally, it has been reported that the modification at the N-terminal residue of the expressed protein, adjacent to the signal peptide sequence, influences the activity, stability, and folding of recombinant proteins (20–22). The signal sequence of AoRut derived from A. oryzae was functional in the heterologous host P. pastoris as AoRut was successfully expressed with its native signal sequence in P. pastoris as an active extracellular protein via normal processing. rAoRutM was purified to homogeneity and was found to be suitable for crystallization trials.
It has been shown that the levels of thermostability of some N-glycosylated enzymes in fungi are higher than those of N-deglycosylated enzymes (23–26); in contrast, it has been reported that the long N-glycan chains weaken the enzyme activity and reduce thermostability (27). In accordance with those findings, in the present study, a rutinosidase from A. oryzae was heterologously expressed in P. pastoris, wherein five asparagine residues were all glycosylated. N-Deglycosylated rAoRutM exhibited a greater than 15% increase in thermal stability. The molar ellipticity values at peaks of 208 nm and 212 nm in the CD spectra differed between intact rAoRutM and endo-H-treated rAoRutM, suggesting that the conformation of α-helices is affected by hyperglycosylation in P. pastoris. Some glycosylation sites were located near the α-helices, which were speculated to affect the structure of the α-helices (Fig. 3a). Therefore, it is possible that N-linked hyperglycosylation is involved in the stability and folding of rAoRutM. AnRut is modified by glycans attached to five asparagine residues, i.e., N128, N176, N181, N214, and N361 (13), and AoRut also has five N-linked N-acetylglucosamine molecules linked to Nδ2 atoms of N32, N128, N176, N288, and N359, indicating that the two positions at N32 and N288 are significantly different from those of AnRut.
The crystal structure of rAoRutM revealed that the catalytic amino acids, Glu210 and Glu317, are located at the bottom of the substrate binding cleft (Fig. 4a and b). The amino acid residues around the cleft are well conserved (Fig. 3b), indicating similar catalytic mechanisms among the members of GH-5 subfamily 23. Loop atoms located at the substrate entrance showed high B-factors (Fig. 4c). B-factor is an indicator of protein dynamics and structural fluctuation of atoms about their average positions. The main-chain trace colored by the B-factor value is shown in Fig. 4c. According to the model, a loop located near the entrance of the catalytic cleft (residues 214 to 222) showed high crystallographic B-factor levels. This result indicated that this loop in rAoRutM is very flexible. Phe220 in this loop is believed to be involved in the recognition of aglycone through π-π staking (28). The other interesting feature is the presence of internal cavities and water molecules in rAoRutM. Figure 4d shows an internal view of rAoRutM beneath the substrate-binding cleft, where several internal cavities were observed near the cleft with hydrated water molecules (green lumps in Fig. 4d). Several water molecules were also positioned without forming a cavity near the bottom of the cleft. A W33 water molecule was positioned in the middle of the substrate-binding cleft, cavity1, and cavity2 (Fig. 4d). Polar side chains surround these cavities and water molecules; therefore, we speculate that these areas are accessible to substrates via thermal fluctuation of rAoRutM. Thus, structural determination of the substrate bond state within rAoRutM will be necessary to clarify the role of these cavities and water molecules in future studies, and, on the basis of these speculations, we have initiated the crystallization of the E317Q variant with rutin (in progress).
Quercetin-3-O-glucoside (isoquercitrin) has been reported to be a better substrate than rutin in AnRut (13). We studied the substrate specificity of AoRut protein by biochemical assays performed with various substrates. Specific activity and kinetic constants were determined for rAoRutM with respect to not only 7-O-linked and 3-O-linked flavonoid rutinosides using rutin, hesperidin, and narirutin but also 7-O-linked and 3-O-linked flavonoid β-glucosides using naringenin-7-O-glucoside (prunin), hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside (isoquercitrin). As shown in Table 1, rAoRutM hydrolyzed the substrates containing 6-O-α-l-rhamnosyl-β-d-glucoside or β-d-glucoside as the glycone moiety. rAoRutM was found to be catalytically more active for quercetin 3-O-linked substrates such as rutin and isoquercitrin than for its 7-O-linked substrates such as quercetin-7-O-glucoside. rAoRutM also exhibited higher catalytic activity toward narirutin than toward prunin. However, no significant differences in rAoRut activity were observed between rutin and isoquercitrin or between hesperidin and hesperetin-7-O-glucoside. The former substrates are modified by rutinose, whereas the latter substrates are modified by β-d-glucose. Additionally, no activity was determined with p-nitrophenyl-β-d-glucoside as a substrate. These observations suggest that rAoRutM recognizes rutinose and β-d-glucose equally as the glycone moieties of flavonoid glycosides and that the aglycone moiety also determines enzyme specificity. The Km value was higher for the substrates modified by rutinose than for the substrates modified by β-d-glucose. Furthermore, the kcat value was considerably higher for the substrates modified by rutinose than for the substrates modified by β-d-glucose. Hence, its higher catalytic efficiency (kcat/Km) for rutin, hesperidin, and narirutin than for isoquercitrin, hesperetin-7-O-glucoside, and prunin, respectively, was attributed to the higher kcat value.
Rutinosidase, which breaks down the plant monoglycoconjugated and diglycoconjugated flavonoids, is an important enzyme with many biotechnological applications, including pharmaceutical and medicinal uses. Moreover, it is used as an effective catalyst in food industries for aroma modulation. The biochemical characterization of AoRut elucidated in this study could help in the utilization of AoRut as a potential catalyst in the food fermentation industry.
Conclusion.
In the present study, Aorut, along with the native signal sequence derived from A. oryzae, was successfully expressed in P. pastoris. The recombinant mature form of AoRut, designated rAoRutM, accumulated as an active extracellular protein in the P. pastoris culture supernatant. The 1.7-Å-resolution crystal structure of AoRut was represented by a (β/α)8 TIM barrel fold with structural similarity to that of AnRut and of exo-β-(1,3)-glucanase from C. albicans (CaExg). The catalytic site was located in a deep cleft, displaying a striking structural similarity to AnRut. An interesting feature is the presence of internal cavities and water molecules in AoRut. The catalytic residues were found at invariant positions in the structures of mutant variants of AoRut (E210 and E317) and AnRut (E210 and E319). Purified rAoRutM hydrolyzed not only 7-O-linked and 3-O-linked flavonoid rutinosides but also 7-O-linked and 3-O-linked flavonoid glucosides, such as naringenin-7-O-glucoside, hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside. rAoRutM displayed high catalytic activity for quercetin 3-O-linked substrates such as rutin and isoquercitrin rather than for its 7-O-linked substrates such as quercetin-7-O-glucoside.
MATERIALS AND METHODS
Chemicals.
Quercetin-3-O-rutinoside (rutin) and hesperetin-7-O-rutinoside (hesperidin) were purchased from Fujifilm Wako Pure Chemical Industries (Osaka, Japan). Naringenin-7-O-rutinoside (narirutin) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Naringenin-7-O-glucoside (prunin), hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside (isoquercitrin) were obtained from Funakoshi Co., Ltd. (Tokyo, Japan). A High Pure plasmid isolation kit (Roche, Mannheim, Germany) was used for plasmid miniprep isolation.
Cloning and expression of the Aorut gene.
The Aorut candidate gene was PCR amplified using the genomic DNA of A. oryzae RIB40 as a template with the following primers: forward primer, 5′-GCGAATTCATGCGGGCGTCACTACCG-3′ (the EcoRI site is underlined); reverse primer, 5′-GTTCTAGATCACTTCGTATCATGGAA-3′ (the XbaI site is underlined). The purified PCR product was subjected to restriction digestion with EcoRI and XbaI and cloned into Pichia expression vector pPICZB (Invitrogen, Waltham, MA, USA). The resulting construct, pPICZB-Rut, was used to transform Escherichia coli DH5α, and the positive clone was sequenced to confirm the correct gene sequence. The recombinant plasmid (pPICZB-Rut) was linearized with SacI and transformed into P. pastoris KM71H per the protocols from a Pichia EasyComp transformation kit (Invitrogen). This procedure yielded an expression plasmid vector containing Aorut with the native signal sequence under the control of the AOX1 promoter and terminator. The P. pastoris transformant thus obtained was cultivated at 30°C in BMGY medium (1% yeast extract, 2% peptone, 1% glycerol, 1.34% yeast nitrogen base without amino acids, and 0.1 M potassium phosphate [pH 6.0]). After the optical density of the cell suspensions reached 2 to 6 at a wavelength of 600 nm, the cells were harvested and cultivated at 30°C in BMMY medium (same composition as BMGY medium except for the presence of 0.5% methanol instead of 1% glycerol to induce AoRut expression).
Purification of recombinant rutinosidase (rAoRutM).
The 7-day cell culture was harvested by centrifugation (5,000 × g, 15 min). The culture supernatant was used for the purification of rAoRutM. Enzyme purification was performed as described previously (12). The enzyme purity and molecular mass were evaluated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie brilliant blue staining. The protein concentration of rAoRutM was measured using a bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific Inc., USA). After boiling, the purified rAoRutM was treated overnight with 0.5 mU of endo-β-N-acetylglucosaminidase H (endo-H; Fujifilm Wako Pure Chemical Industries)–50 mM sodium acetate buffer (pH 5.0) at 37°C for 18 h. For N-terminal sequencing, the protein bands from 12% SDS-PAGE gel were transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). N-terminal amino acid sequences of peptides were analyzed at Hokkaido System Science Co., Ltd. (Sapporo, Japan).
The nondenatured protein was treated with 0.5 mU of endo-H–50 mM sodium acetate buffer (pH 5.0) at 30°C overnight for crystallization purposes and to figure out the effect of N-glycosylation on enzyme activity and thermostability of rAoRutM. After endo-H treatment, rAoRutM was further purified using a gel filtration chromatography.
Crystallization and structure determination.
N-Deglycosylated rAoRutM was concentrated to 6.5 mg/ml in 10 mM Tris-HCl (pH 8.0). Screening for crystallization conditions using a JCSG Plus screen (Molecular Dimensions, United Kingdom) was performed. Single crystals formed under conditions that included the use of 12% polyethylene glycol 4000 (PEG 4000) and 0.1 M sodium acetate (pH 4.6) with a hanging-drop-crystallization setup at 20°C. The crystals were flash frozen in liquid nitrogen with 50% PEG 3350 cryoprotectant. The samples were initially evaluated by the use of an automatic data collection system at Spring-8 beamline BL32XU (Harima, Japan), and the results confirmed that the crystals diffracted beyond 2.0-Å resolution (29, 30). We then measured the diffraction using Photon Factory BL-5A (Tsukuba, Japan). The data were processed with XDS (31) and AIMLESS in CCP4i (32). An initial structure was deciphered by the use of Molrep and CCP4i, a molecular replacement program, using homology modeling based on an exo‐β‐(1,3)‐glucanase structure (sequence identity, 26%; PDB ID 2PF0 [28]), as the crystal structure of the most homologous AnRut protein had not been reported at that time. Refinement was performed using Phenix.Refine (33), and the model was built using Coot (34). Molecular structures were depicted with PyMol software (35). The amino acid conservation shown in the structure was depicted using ConSurf (36). The inner cavity was depicted by PyMol. The statistics for data collection and refinement are provided in Table 3. The final structure was deposited in the Protein Data Bank (PDB ID 6LA0).
TABLE 3.
| Parameter | Result |
|---|---|
| Data collection statistics | |
| Space group | P21 |
| Cell parameters | a=47.91 b=169.99 c=50.49 β=115.62 |
| Beamline | PF-BL5A |
| Wavelength (Å) | 1.0000 |
| Resolution (Å)b | 20.0–1.75 (1.85–1.75) |
| Completeness (%) | 98.9 (95.4) |
| I/σ (I) | 13.28 (2.06) |
| Rmerge | 0.097 (0.747) |
| Rpim | 0.041 (0.379) |
| CC1/2 | 0.998 (0.803) |
| Average redundancy | 5.8 (5.5) |
| Refinement statistics | |
| Resolution range (Å) | 19.6 to 1.75 |
| No. of reflections used (free) | 72,673 (3,563) |
| R factor | 0.196 |
| Rfreec | 0.229 |
| RMSD | |
| Bonds (Å) | 0.007 |
| Angles (°) | 0.815 |
| Average B factor (Å2) | 23.1 |
| Ramachandran plot statistics | |
| Most favored (%) | 98 |
| Additionally allowed (%) | 2 |
| Generally allowed (%) | 0 |
Values in parentheses represent the highest resolution shell unless otherwise indicated. CC1/2, 50% correlation coefficient.
The highest-resolution shell is shown in parentheses.
Rfree is R with 5% of reflections sequestered before refinement.
Enzyme assays.
The enzyme activity was assayed as follows. A 440-μl volume of 50 mM sodium citrate buffer (pH 4.0) was mixed with 50 μl of the substrate (final concentration of 0.2 mg/ml) dissolved in methanol. Next, 10 μl of the purified enzyme (5.2 μg) was added to this mixture. The reaction mixture was incubated at 45°C for 10 min. The release of hydrolysis products was detected at 280 nm by high-performance liquid chromatography (HPLC) with an UV detector. Reaction products were separated on a TSKgel ODS-120T HPLC column (Tosoh Co., Tokyo, Japan) (4.6 by 250 mm) using an isocratic elution gradient of 50 mM sodium acetate buffer (pH 4.0)–acetonitrile (7:3) at a flow rate 0.5 ml/min at 45°C. One unit of enzyme activity was defined as the amount of enzyme required to release saccharides at the rate of 1 μmol/min at a reaction temperature of 45°C and pH of 4.0.
Effect of pH and temperature on enzyme activity.
The effect of pH and temperature on rutinosidase activity was analyzed using rutin as a substrate. The optimum pH for enzyme activity was identified in the following buffers (50 mM): sodium citrate (pH 3.0 to 6.0) and sodium phosphate (pH 6.0 to 7.0). The effect of temperature was investigated at temperatures in the range of 40 to 55°C (50 mM sodium citrate buffer; pH 4.0). The thermal stabilities of the enzymes, with or without endo-H treatments, were determined after incubation at 30 to 60°C for 1 h or at 50°C for 1 to 8 h. The enzyme solutions were immediately cooled on ice, and their residual activities were measured at 45°C (pH 5.0), using rutin as a substrate according to a previously described method (12). The percentage of residual activity was reported relative to the highest observed rAoRutM activity in our assays, defined as 100%.
Substrate specificity and kinetic constants.
The substrate specificity of purified rAoRutM for 7-O-linked and 3-O-linked flavonoid rutinosides, such as rutin, hesperidin, and narirutin, and 7-O-linked and 3-O-linked flavonoid β-glucosides, such as naringenin-7-O-glucoside (prunin), hesperetin-7-O-glucoside, quercetin-7-O-glucoside, and quercetin-3-O-glucoside (isoquercitrin), was investigated. Kinetic constants were also determined using the substrates specified above. The enzyme assay was monitored for each substrate using the HPLC-based method described above, and kinetic constants were fitted using GraFit 5.0 software (Erithacus Software, Surrey, United Kingdom).
Site-directed mutagenesis.
Site-directed mutagenesis was performed using a QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). For these experiments, base substitutions were introduced into the Aorut gene docked on the pPICZB-Rut plasmid using the following primers: primers E210Q-F (5′-GAGCCGATGAACCAACCTACCGATA-3′) and E210Q-R (5′-TATCGGTAGGTTGGTTCATCGGCTC-3′) and primers E317Q-F (5′-GTGTTCACGGGACAATGGGCTATTC-3′) and E317Q-R (5′-GAATAGCCCATTGTCCCGTGAACAC-3′) (that mutations that were introduced are underlined). The desired sequences were verified by DNA sequencing analysis.
Circular dichroism (CD) spectroscopy analysis.
CD spectra were observed using a Jasco J-820 instrument (Tokyo, Japan) with 50 mM sodium acetate (pH 5.0)–150 mM NaCl buffer at 25°C. A 1-mm-path-length quartz cuvette was used, and the protein concentration was set to approximately 7 μM. Far-UV 200-to-250-nm-wavelength spectra of the protein samples were obtained, and the buffer background was subtracted.
Data availability.
Newly determined protein data were deposited in the Protein Data Bank under accession number 6LA0.
Supplementary Material
ACKNOWLEDGMENTS
We thank the beamline staff of KEK Photon Factory, Tsukuba, Japan, for technical assistance. We thank Kunio Hirata for the automated measurements at Spring-8, Harima, Japan.
We declare no competing financial interest.
K.M., Y.S., and T.K. conceived and designed the work. K.M., R.H., and Y.T. performed experiments and analyzed data. K.M. and T.K. wrote the manuscript. All of us reviewed and approved the manuscript.
This research was (partially) supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research [BINDS]) from AMED under grant number JP19am0101070 (support number 2139).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Nielsen ILF, Chee WSS, Poulsen L, Offord-Cavin E, Rasmussen SE, Frederiksen H, Enslen M, Barron D, Horcajada M, Williamson G. 2006. Bioavailability is improved by enzymatic modification of the citrus flavonoid hesperidin in humans: a randomized, double-blind, crossover trial. J Nutr 136:404–408. doi: 10.1093/jn/136.2.404. [DOI] [PubMed] [Google Scholar]
- 2.Williamson G. 2017. The role of polyphenols in modern nutrition. Nutr Bull 42:226–235. doi: 10.1111/nbu.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mazzaferro L, Breccia JD. 2011. Functional and biotechnological insights into diglycosidases. Biocat Biotrans 29:103–112. doi: 10.3109/10242422.2011.594882. [DOI] [Google Scholar]
- 4.Wang D, Kurasawa E, Yamaguchi Y, Kubota K, Kobayashi A. 2001. Analysis of glycosidically bound aroma precursors in tea leaves. 2. Changes in glycosidic contents and glycosidase activities in tea leaves during the black tea manufacturing process. J Agric Food Chem 49:1900–1903. doi: 10.1021/jf001077+. [DOI] [PubMed] [Google Scholar]
- 5.Genovés S, Gil JV, Vallés S, Casas JA, Manzanares P. 2005. Assessment of the aromatic potential of palomino fino grape must using glycosidases. Am J Enol Vitic 56:188–191. [Google Scholar]
- 6.Hollman PCH. 2004. Absorption, bioavailability, and metabolism of flavonoids. Pharm Biol 42:74–83. doi: 10.3109/13880200490893492. [DOI] [Google Scholar]
- 7.Lombard V, Golaconda RH, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aspeborg H, Coutinho PM, Wang Y, Brumer H, Henrissat B. 2012. Evolution, substrate specificity and subfamily classification of glycoside hydrolase family 5 (GH5). BMC Evol Biol 12:186. doi: 10.1186/1471-2148-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuzawa T, Mitsuishi Y, Kameyama A, Yaoi K. 2016. Identification of the gene encoding isoprimeverose-producing oligoxyloglucan hydrolase in Aspergillus oryzae. J Biol Chem 291:5080–5087. doi: 10.1074/jbc.M115.701474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiz G, Mazzaferro LS, Kotik M, Neher BD, Halada P, Křen V, Breccia JD. 2019. The flavonoid degrading fungus Acremonium sp. DSM 24697 produces two diglycosidases with different specificities. Appl Microbiol Biotechnol 103:9493–9504. doi: 10.1007/s00253-019-10180-y. [DOI] [PubMed] [Google Scholar]
- 11.Šimčíková D, Kotik M, Weignerová L, Halada P, Pelantová H, Adamcová K, Křen V. 2015. α-L-Rhamnosyl-β-D-glucosidase (rutinosidase) from Aspergillus niger: characterization and synthetic potential of a novel diglycosidase. Adv Synth Catal 357:107–117. doi: 10.1002/adsc.201400566. [DOI] [Google Scholar]
- 12.Ishikawa M, Kawasaki M, Shiono Y, Koseki T. 2018. A novel Aspergillus oryzae diglycosidase that hydrolyzes 6-O-α-L-rhamnosyl-β-D-glucoside from flavonoids. Appl Microbiol Biotechnol 102:3193–3201. doi: 10.1007/s00253-018-8840-9. [DOI] [PubMed] [Google Scholar]
- 13.Pachl P, Kapešová J, Brynda J, Biedermannová L, Pelantová H, Bojarová P, Křen V, Řezáčová P, Kotik M. 2020. Rutinosidase from Aspergillus niger: crystal structure and insight into the enzymatic activity. FEBS J 287:3315–3327. doi: 10.1111/febs.15208. [DOI] [PubMed] [Google Scholar]
- 14.Cereghino JL, Cregg JM. 2000. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev 24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- 15.Macauley-Patrick K, Fazenda ML, McNeil B, Harvey LM. 2005. Heterologous protein production using the Pichia pastoris expression system. Yeast 22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad M, Hirz M, Pichler H, Schwab H. 2014. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol 98:5301–5317. doi: 10.1007/s00253-014-5732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crepin VF, Faulds CB, Connerton IF. 2003. Production and characterization of the Talaromyces stipitatus feruloyl esterase FAEC in Pichia pastoris: identification of the nucleophilic serine. Protein Expr Purif 29:176–184. doi: 10.1016/S1046-5928(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 18.Damaso MCT, Almeida MS, Kurtenbach E, Martins OB, Pereira N Jr, Andrade CMMC, Albano RM. 2003. Optimized expression of a thermostable xylanase from Thermomyces lanuginosus in Pichia pastoris. Appl Environ Microbiol 69:6064–7072. doi: 10.1128/aem.69.10.6064-6072.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen MW, Bornscheuer UT, Hult K. 2008. Expression of Candida antarctica lipase B in Pichia pastoris and various Escherichia coli systems. Protein Expr Purif 62:90–97. doi: 10.1016/j.pep.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Steinlein LM, Graf TN, Ikeda RA. 1995. Production and purification of N-terminal half-treaferrin in Pichia pastoris. Protein Expr Purif 6:619–624. doi: 10.1006/prep.1995.1081. [DOI] [PubMed] [Google Scholar]
- 21.Shuichiro G, Kazufumi T, Yuriko Y, Yoshio K, Katsuhide Y. 2000. Effect of extra N-terminal residues on the stability and folding of human lysozyme expressed in Pichia pastoris. Protein Eng 13:299–307. doi: 10.1093/protein/13.4.299. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa K, Shiono Y, Shintani T, Watanabe A, Kanzaki H, Gomi K, Koseki T. 2020. Efficient production of recombinant tannase in Aspergillus oryzae using an improved glucoamylase gene promoter. J Biosci Bioeng 129:150–154. doi: 10.1016/j.jbiosc.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Hanson SR, Culyba EK, Hsu T-L, Wong C-H, Kelly JW, Powers ET. 2009. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhancers stability. Proc Natl Acad Sci U S A 106:3131–3136. doi: 10.1073/pnas.0810318105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benoit I, Asther M, Sulzenbacher G, Record E, Marmuse L, Parsiegla G, Gimbert I, Asther M, Bignon C. 2006. Respective importance of protein folding and glycosylation in the thermal stability of recombinant feruloyl esterase A. FEBS Lett 580:5815–5821. doi: 10.1016/j.febslet.2006.09.039. [DOI] [PubMed] [Google Scholar]
- 25.Jafari-Aghdam J, Khajeh K, Ranjbar B, Nemat-Gorgani M. 2005. Deglycosylation of glucoamylase from Aspergillus niger: effects on structure, activity and stability. Biochim Biophys Acta 1750:61–68. doi: 10.1016/j.bbapap.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 26.Mizuno T, Shiono Y, Koseki T. 2014. Biochemical characterization of Aspergillus oryzae native tannase and the recombinant enzyme expressed in Pichia pastoris. J Biosci Bioeng 118:392–395. doi: 10.1016/j.jbiosc.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Wei W, Chen L, Zou G, Wang Q, Yan X, Zhang J, Wang C, Zhou Z. 2013. N-glycosylation affects the proper folding, enzymatic characteristics and production of a fungal β-glucosidase. Biotechnol Bioeng 110:3075–3084. doi: 10.1002/bit.24990. [DOI] [PubMed] [Google Scholar]
- 28.Patrick WM, Nakatani Y, Cutfield SM, Sharpe ML, Ramsay RJ, Cutfield JF. 2010. Carbohydrate binding sites in Candida albicans exo-β-1,3-glucanase and the role of the Phe-Phe ‘clamp’ at the active site entrance. FEBS J 277:4549–4561. doi: 10.1111/j.1742-4658.2010.07869.x. [DOI] [PubMed] [Google Scholar]
- 29.Hirata K, Yamashita K, Ueno G, Kawano Y, Hasegawa K, Kumasaka T, Yamamoto M. 2019. ZOO: an automatic data-collection system for high-throughput structure analysis in protein microcrystallography. Acta Crystallogr D Struct Biol 75:138–150. doi: 10.1107/S2059798318017795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita K, Hirata K, Yamamoto M. 2018. KAMO: towards automated data processing for microcrystals. Acta Crystallogr D Struct Biol 74:441–449. doi: 10.1107/S2059798318004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D Struct Biol 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.DeLano WL. 2009. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 36.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. 2016. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Newly determined protein data were deposited in the Protein Data Bank under accession number 6LA0.