

Abstract
In B lymphocytes, the uracil N-glycosylase (UNG) excises genomic uracils made by activation-induced deaminase (AID), thus underpinning antibody gene diversification and oncogenic chromosomal translocations, but also initiating faithful DNA repair. Ung−/− mice develop B-cell lymphoma (BCL). However, since UNG has anti- and pro-oncogenic activities, its tumor suppressor relevance is unclear. Moreover, how the constant DNA damage and repair caused by the AID and UNG interplay affects B-cell fitness and thereby the dynamics of cell populations in vivo is unknown. Here, we show that UNG specifically protects the fitness of germinal center B cells, which express AID, and not of any other B-cell subset, coincident with AID-induced telomere damage activating p53-dependent checkpoints. Consistent with AID expression being detrimental in UNG-deficient B cells, Ung−/− mice develop BCL originating from activated B cells but lose AID expression in the established tumor. Accordingly, we find that UNG is rarely lost in human BCL. The fitness preservation activity of UNG contingent to AID expression was confirmed in a B-cell leukemia model. Hence, UNG, typically considered a tumor suppressor, acquires tumor-enabling activity in cancer cell populations that express AID by protecting cell fitness.
Graphical Abstract
Graphical Abstract.
Activation-induced deaminase (AID) and uracil N-glycosylase (UNG) have several different functions and effects in normal and cancer B cells, with UNG protecting cell fitness from AID in both contexts and the net outcome being context dependent.
INTRODUCTION
Activation-induced deaminase (AID, encoded by Aicda) transforms cytosine DNA bases into uracil at the immunoglobulin (Ig) genes, thereby initiating somatic hypermutation (SHM) to modify the antibody affinity in germinal center (GC) B cells (1). Mutagenic processing of these uracils by DNA repair enzymes expands the spectrum of SHM (1), while at the Ig heavy chain gene (Igh) it triggers antibody class switch recombination (CSR) by producing DNA breaks (2). The same AID-initiated mechanisms can produce collateral DNA damage at other loci, causing mutations and chromosomal translocations (3). Accordingly, AID is oncogenic in GC-derived and other murine models of B-cell neoplasm (4–12). Human B-cell lymphomas (BCLs) are mostly GC experienced (13) and many express AID, especially diffuse large BCL (DLBCL) and Burkitt’s lymphoma (14–18). The oncogenic potential of AID is mitigated by several layers of regulation. First, AID expression and activity are regulated by multiple mechanisms (2,3). Second, DNA repair pathways can counter AID-induced off-target DNA lesions (19,20). A third line of defense is provided by tumor suppressors that allow time for DNA repair or eliminate B cells displaying genomic instability, with p53 playing a prominent role against AID-induced transformation (8,11,21).
AID-born uracils are recognized by only two pathways (22). The major pathway starts with the uracil-DNA glycosylase UNG, which excises uracil to produce an abasic site. Abasic sites expand the SHM spectrum by forcing mutations when replicated over, or are converted to DNA double-strand breaks by abasic site-specific nucleases at the Igh (2). Alternatively, the MSH2/6 heterodimer of the mismatch repair (MMR) pathway recognizes U:G mispairs and initiates mutagenic noncanonical MMR, which expands SHM to A:T pairs and contributes to Igh DNA breaks (23). AID is most highly expressed in activated and GC B cells, which proliferate rapidly (24). Off-target DNA breaks caused by the consecutive action of AID and UNG or MSH2/6 are repaired by homologous recombination to prevent cell cycle arrest in GC B cells (20). Despite their mutagenic roles in SHM and CSR, the canonical function of UNG and MSH2/6 is to initiate error-free base excision repair (BER) and MMR, respectively (2,23), whereby they also prevent a proportion of AID-induced lesions at the Ig and genome-wide (19,22,25,26). Since the constant repair of AID-induced lesions would exact an energy cost and can generate toxic intermediates, AID activity reduces cell fitness, understood as the potential to thrive in a given condition. Fitness is most important in vivo, where cell populations are subjected to a stringent or competitive environment. Accordingly, Aicda−/− mice show GC hyperplasia and Aicda−/− GC B cells display a fitness advantage over wild type (WT) in mixed bone marrow (BM) chimera experiments, partly due to reduced apoptosis (27,28). One would expect that the uracil sensors UNG and MSH2/6 would also impact fitness in AID+ cells. Ung−/− Msh2−/− GC B cells are at a disadvantage in BM chimeras and display more apoptosis than WT (28), but this could be attributed to the MSH2 deficiency. Indeed, Msh2−/− mice show reduced GC expansion and increased GC B-cell apoptosis (29), and ablating MMR in a B-cell line causes severe proliferation defects independently of AID (30). The effect of UNG on B-cell fitness in vivo has not been addressed. A similar logic would apply to AID+ BCL. However, while MMR deficiency is associated with cancer predisposition (26,31–33) and BCL (34), the links between UNG and B-cell transformation or population dynamics of cells expressing AID are unclear.
The main function of UNG in vertebrate cells is to repair uracil that DNA polymerases can misincorporate opposite adenine during DNA replication, which is not directly mutagenic (35,36). This specialization, and partial redundancy with the uracil-DNA glycosylase SMUG1, explains why UNG-deficient tissues show only a modest mutation increase in vertebrates (35,36). Nonetheless, UNG could be considered a tumor suppressor by contributing to canonical uracil BER. Accordingly, Ung−/− mice spontaneously develop BCL (37,38). A causal role for AID in BCL in Ung−/− mice has been suggested but never tested. On the other hand, UNG can be oncogenic. UNG mediates the chromosomal translocations initiated by AID (21,39). Moreover, UNG deficiency impaired the development of DLBCL in the IμHABcl6 transgenic mice, and of plasmacytoma in Bcl-xL transgenic mice (39,40). In both mouse models, neoplasia is also prevented by AID deficiency (4,10), suggesting that UNG is required for AID-mediated cancer initiation. In addition, UNG can protect the integrity of the telomeres from uracil misincorporation in hematopoietic cells (41) and from AID- and MMR-mediated truncation in cultured B cells and BCL cell lines (42), which after the generation of oncogenic mutations could impinge on tumor expansion. The latter has not been tested in vivo, as it requires a model in which AID and UNG are expressed but not required for oncogenesis.
Here, we have tested the relevance and net outcome of the multiple AID-triggered molecular transactions by which UNG can either protect or compromise genome stability, on the dynamics of normal and neoplastic B-cell populations in vivo. We find that, in the physiological GC setting, UNG promotes B-cell fitness. In pathological settings, we confirm that UNG is a BCL suppressor, but show that this activity is relatively weak and not necessarily due to opposing AID activity, as shown by BCL predisposition in Aicda−/− Ung−/− mice. Nonetheless, many Ung−/− BCLs do originate from B cells with previous AID activity, but the established tumor loses AID expression. Finally, by ablating Ung in a mouse B-cell leukemia model, we provide direct evidence of a tumor-enabling activity of UNG contingent on AID expression. Thus, while UNG is a tumor suppressor in B cells, it also protects the fitness of AID+ B cells, whereby it favors cancer promotion after cancer initiation.
MATERIALS AND METHODS
Mice
Ung−/− mice (35), a gift from Dr H. Krokan (NUST, Trondheim, Norway), Aicda−/− mice (1), a gift from Dr T. Honjo (Kyoto University, Japan), Smug1−/− (43) and Ung−/− Msh2−/− (22) were in C57BL6/J background. C57BL6/J WT, CD45.1, Tp53−/− (44) and NOD Rag1−/− Il2rgtm1Wjl/SzJ (NRG) mice were from the Jackson Laboratory (Bar Harbor, MN). Mice used for experiments and the prospective cancer cohorts were housed at the IRCM, Montréal, Canada. We had previously analyzed the AID status of some of the Ung−/− BCLs in this cohort (42), but they were reanalyzed for this work in parallel with additional mice and BCLs from the WT controls. Retrospective survival data were compiled from historical records of mice housed at the MRC-LMB animal facility, Cambridge, UK. Animal work was reviewed and approved by the IRCM animal protection committee (protocols 2009-15 and 2013-15) in accordance to the guidelines from the Canadian Council of Animal Care or covered by UK home office project licenses 70/7571 and 80/2226 in accordance with guidelines in effect at the LMB-MRC. Mice were housed in specific pathogen-free facilities. For GC B-cell analysis, age- and sex-matched mice, 40–120 days old, were immunized with 109 sheep red blood cells (SRBCs; Innovative Research, IC100-0210) in 200 μl phosphate-buffered saline (PBS) by the tail vein. For cancer follow-up, mice were observed regularly until they spontaneously developed signs of disease (paralysis, rapid loss of weight, ataxia, lack of cleaning, general posture, visible masses), or reached an age of 30 months, and then sacrificed in a CO2 chamber and analyzed. Bone marrow (BM) chimeras: BM cells were isolated from WT, Ung −/− and Aicda−/− Ung−/− mice as described (45) and mixed with identical numbers of BM cells from CD45.1+ WT mice. Lethally irradiated (9 Gy) C57BL6/J (CD45.2) recipient mice were injected by the tail vein with 100 μl of PBS containing 2 × 106 mixed BM cells. All donor and recipient mice were females. Hematological reconstitution was confirmed in blood by flow cytometry 4 weeks post-injection. Mice were immunized intraperitoneally with 50 μg of nitrophenyl-chicken gamma globulin in alum 40–50 days after reconstitution and analyzed 11 days later. The proportion of CD45.1 and CD45.2 cells for each B-cell subset was determined by flow cytometry. The CD45.2/CD45.1 ratios of all subsets were normalized to the ratio of the BM mix used to reconstitute the corresponding group of recipient mice. The individual GC B-cell ratios were further normalized to the ratio of splenic total B220 B cells in the same mouse. Finally, for each cell subset, the ratios in individual mice were normalized to the average ratio of the wt:wt control chimeras, to account for biases known to occur in these type of experiments (46).
Flow cytometry
Mononuclear cells were extracted by mashing tumors or organs through a cell strainer with a syringe plunger. BM cells were flushed out of decapped long bones using a syringe. Single-cell suspensions (1–2 × 106 for spleen, 5 × 106 for BM) or whole blood (50 μl) were stained with antibody combinations as per Supplementary Table S1. To assess proliferation, 3 × 106 splenocytes purified and stained for GC markers were treated with fixation/permeabilization solution (eBioscience, cat. #00-5523) for 1 h at 4°C in the dark, followed by 1 h incubation with anti-Ki67-PECY7 (eBioscience) at 4°C. To assess apoptosis, 106 splenocytes were incubated with FITC-conjugated CaspGLOW pan-caspase substrate (BioVision) for 60 min at 37°C in warm culture media followed by surface marker staining and flow cytometry analysis. Data were acquired using FACS Caliber 2, BD LSR I or BD LSR Fortessa (BD Biosciences) and analyzed using FlowJo.
FISH and telomere loss scoring
Resting splenic B cells were purified using anti-CD43 magnetic beads following the manufacturer’s instructions (Miltenyi Biotec), activated with 5 μg/ml lipopolysaccharide (LPS, Sigma-Aldrich) and 20 ng/ml IL-4 (PeproTech), and processed for metaphase spreads 24 h later. Cells were incubated with 50 ng/ml colcemide in cell culture media for 3 h, harvested, incubated for 10 min at room temperature in 75 mM KCl and fixed in methanol/glacial acetic acid (3:1 vol/vol). Cells were dropped onto wet slides and air dried for metaphase spreading. Metaphases were processed for FISH and hybridized to a Fluor 488-conjugated PNA probe (Alexa Fluor 488-OO-[CCCTAA]3) in hybridization solution (70% deionized formamide, 2.5% 50× Denhardt solution, 10 mM Tris–HCl, pH 7.5, and 1.5 mM MgCl2), followed by DAPI staining, exactly as previously described (42). Hybridized samples were mounted in Slow-Fade Gold mounting reagent (Thermo Fisher Scientific). Telomere truncations were scored as unequal telomere signals from at least 50 metaphases per sample per experiment from pictures taken in a fluorescence microscope.
Histopathology
Tumors or enlarged organs were removed during necropsy, fixed in 10% formalin at room temperature and embedded in paraffin. Sections were stained with hematoxylin–eosin and analyzed by a pathologist. Sections of paraffin-embedded tissues (5 μm) were deparaffinized in xylene (2 × 5 min) and then rehydrated to distilled water using graded alcohols. Antigen was retrieved by steaming the slides for 20 min and then cooling for 20 min in either 1 mM EDTA, 0.05% Tween 20, pH 8 (for AID and BCL6), or 10 mM acid citric, 0.05% Tween 20, pH 6.0, for all other antigens. Blockings were as follows: 0.3% H2O2 for 10 min for endogenous peroxidase, avidin/biotin blocking buffer (#SP2001, Vector Laboratories, Burlingame, CA) for 15 min and/or 3% normal goat serum and 1% bovine serum albumin for 60 min at room temperature. Carbo-free blocking buffer (#SP5040, Vector Laboratories) was used prior to PNA staining for 60 min at room temperature. Sections were incubated with PNA (Vector Laboratories) or primary antibodies (Supplementary Table S1) for 60 min at room temperature or overnight at 4°C, followed by biotin-conjugated secondary antibodies and detection with Vectastain ABC Kit (PK-6100, Vector Laboratories). Peroxidase activity was developed using ImmPACT NovaRED HRP substrate (Vector Laboratories).
Spectral karyotyping
Lymphoma cells (5 × 106) recovered from enlarged organs of moribund mice were injected via tail vein into NRG mice. Transplanted mice were sacrificed when showing signs of disease and lymphoma cells recovered from spleen and mesenteric lymph nodes. Lymphoma cells were resuspended in culture media after red blood cell lysis, incubated with colchicine and processed for spectral karyotyping on representative metaphases. Slide pretreatment, hybridization with mouse chromosomes probes (SkyPaint™ DNA Kit) and detection with the Concentrated Antibodies Detection Kit were performed using the protocol provided by Applied Spectral Imaging with minor modifications. Spectral images were acquired with a GenASIs HiSKY™ Hyperspectral Karyotyping system mounted on an Olympus BX53 microscope and analyzed using the ASI software version 7.2.6.
IgV and Sμ sequencing
Genomic DNA from fresh or frozen lymphoma cells purified from NRG mice transplanted with BCL samples #4681 and #19-087-2016 was extracted with DirectPCR lysis buffer (Viagen) or Gentra Puregene DNA isolation (QIAGEN), respectively. For all other BCL samples, genomic DNA was prepared from FFPE laser capture microdissection samples using the Cobas® DNA Sample Preparation Kit (Roche, cat. #05985536190). The rearranged VHDJ junction was determined by PCR amplification with a mixture of degenerate forward primers for most VH gene families (V1:V5:V3:V7:V9, ratio 6:3:1:1:1) located in FR2/3 (47) and a reverse primer in JH4. The complete IgVH was amplified using V-specific forward primers located in FR1 and reverse primers located in or downstream of the closest JH region, according to the VDJ rearrangement. The Sμ was amplified with forward primer OJ521 and reverse OJ299 or OJ354. See Supplementary Table S2 for all primer sequences. PCR amplification was done using KOD Hot Start DNA Polymerase (SIGMA Millipore, cat. #71086) in 25 μl reactions using 0.02 U/μl KOD polymerase, 2 mM MgSO4, 5% DMSO, 0.3 μM each primer and 50 ng DNA. Cycling conditions were as follows: 95°C for 2 min + 40 cycles of (95°C for 20 s, 56°C for 10 s and 70°C for 10 s). DNA fragments of the right size were purified from agarose gel electrophoresis with Monarch® DNA Gel Extraction Kit (New England Biolabs, cat. #T1020), A-tailed using home-made Taq polymerase, cloned in pGEM-T-Easy (Promega, cat. #A1360) and transformed in DH5-α Escherichia coli. Multiple individual clones were Sanger sequenced. Sequences were visually inspected, and primers trimmed off using SnapGene, and then aligned for mutation analyses and phylogenetic tree building in MEGA v7.0.14. Trees were made by the minimum evolution statistical method with 1000 bootstrap replicates. Evolutionary distances were computed using the number of differences method. Rate variation among site was modeled with a gamma distribution (shape parameter = 1). All sequences are available in Supplementary Data Set 1.
Whole genome sequencing and mutation analysis
Library preparation was performed as described (48). Sequencing was performed at the Kinghorn Centre of Clinical Genomics at the Garvan Institute of Medical Research, Sydney, Australia, to a minimum of 30× coverage using Illumina HiSeq technology with 150-bp paired end reads. Each tumor sample (Ung−/− BCL #16836 from mouse #4689 and Ung−/− Aicda−/− BCL #16840 from mouse #19-087-2016) was compared to a common control non-tumor (tail) sample to allow unequivocal assignment of mutations. Each FASTQ file obtained was quality checked using FastQC (S. Andrews, FastQC version 0.11.2, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Further analysis was based on the Broad Institute best practices. Reads were aligned to the mouse GRCm38 (mm10) reference genome sequence using BWA-MEM 0.7.10 (arXiv:1303.3997) (49). Mates were then fixed and results converted to bam format and sorted using samtools 1.1 (50). Duplicates were marked using biobambam markduplicates2 (51) and indexed using samtools. Realignment around indels including known indels in dbSNP 138 (obtained from EnsEMBL release 74) and subsequent base recalibration was performed using GATK 3.2.2 (52,53). Mutations were scored as variants that appeared in the tumor but were absent in non-tumor tissue. Single-nucleotide variants were called using MuTect 1.1.7 (54). Mutations detected in regions of the reference genome containing simple repeats or low-complexity DNA identified by VSEARCH (55) using the Dust method were ignored. Single-nucleotide variants were classified into the six possible base pair substitutions based on the pyrimidine of the mutated base pair (C>A, C>G, C>T, T>A, T>C and T>G) and counted. The UCSC Genome Browser and IGV 2.3.26 were used for visual inspection of mapped reads and mutated sites. The Ensembl Variant Effect Predictor (56) was used to assess possible functional impact of mutations. Mutational signatures were detected by de novo extraction based on somatic substitutions and their immediate sequence context. The classification of single-base mutations into six types was further elaborated by including both the 5′ and 3′ base immediately next to the mutation, resulting in 96 possible mutation types. The mutations extracted from the mouse data were also renormalized to the trinucleotide frequency of the mouse genome. Plots of normalized mutations frequencies in different trinucleotide sequence contexts were plotted using Galaxy and the Genomic Hyperbrowser (57). To identify mutations at C within WRCY sequence context, frequencies were counted by overlapping genomic locations for the three possible mutations from C (C>T, C>A, C>G) with occurrences of the 4-mer pattern WRCY. Subsequently, their normalized counts were plotted separately for all mutation types. The log-transformed distribution of distances between mutation following pattern WRCY and the rest of mutations from the datasets was calculated. Rainfall plots (58) were used to visualize the distribution of mutations and mutation hotspot (kategis) regions along the reference genome. A list of AID off-targets was compiled from (19,59) and intersected with the list of genes with mutations in coding regions in each BCL sample.
Acute B-cell leukemia model
A retroviral vector encoding BCR-ABL1p210-ires-GFP was transfected into the ecotropic packaging cell line PLAT-E (60) and cultured 24 h in DMEM containing 10% FBS, and then supplemented with 0.5 M sodium butyrate (Sigma) for additional 8 h. The supernatant was collected 48 h later, concentrated using Vivaspin 20 (GE Healthcare) and used to infect purified total BM cells, and then cultured in StemSpan medium (Stemcell Technologies) for 48 h with 2 mM glutamine, 50 ng/ml Flt3L and 10 ng/ml rIL-7 (PeproTech). Pelleted BM cells were resuspended in 0.2 ml of concentrated viral supernatant containing 8 μg/ml polybrene (Sigma) and incubated for 2 h in 37°C before adding 1 ml of complete StemSpan, spinning for 30 min at 600 × g and incubating overnight in six-well plates. Sublethally irradiated (5 Gy) NRG mice were injected with 106 cells in 0.1 ml PBS by tail vein. Mice were sacrificed when reaching one of the endpoints (paralysis, rapid loss of weight, ataxia, lack of cleaning, general posture, visible masses). Leukemia cells were isolated and analyzed by flow cytometry. RNA was isolated from freshly isolated tumor cells using TRIzol (Life Technologies) and cDNA synthesized using the ProtoScript™ M-MuLV Taq RT-PCR kit (New England Biolabs). Endpoint PCR was performed on the cDNA using Taq DNA polymerase with Aicda primers AIDfwd and AIDrev for mouse AID (389-bp fragment), 35 cycles of (94°C for 30 s, 55°C for 30 s and 68°C for 1 min), and OJ651 and OJ652 for Gapdh (121 bp fragment), 30 cycles of (94°C for 30 s, 51°C for 20 s and 68°C 1 min). Oligo sequences are provided in Supplementary Table S2.
Statistical analyses
Data were analyzed in Prism (GraphPad Soft). Tests used are indicated in the text, using parametric or non-parametric methods depending on the result of normality test. P-values <0.05 were considered significant.
RESULTS
UNG protects GC B-cell fitness
To test the effect of AID–UNG interplay on GC B cells, we first analyzed GC formation in 2–4-month-old Ung−/−, Aicda−/− and Ung−/− Aicda−/− mice immunized with SRBCs. None of the mice showed B-cell hyperplasia at this age (Supplementary Figure S1A). There were no significant differences in the GC B-cell proportion or number, or in the GC B-cell kinetics between Ung−/− and WT mice after immunization (Figure 1A and B). Compared to WT, Aicda−/− mice displayed up to >4-fold more GC B cells, as previously shown (1,27), and the GC reaction persisted for much longer after immunization. Neither of these phenotypes was affected by compounded UNG deficiency in Ung−/− Aicda−/− mice (Figure 1A and B).
Figure 1.

Reduced fitness of UNG-deficient GC B cells. (A) Representative flow cytometry of GC B-cell (CD3− B220+ GL7+ CD95+) proportions in the spleen of mice immunized with SRBCs. Means + SEM GC B-cell proportion for two to nine mice per group per time point compiled from multiple experiments are plotted. (B) Absolute number of GC B cells in individual mice (symbols) and mean values (bars) from two pooled experiments done as in (A) at day 14 post-SRBC immunization. (C) Representative flow cytometry gating for dark (DZ) and light zone (LZ) GC B cells (B220+ GL7+ CD95+) and compiled DZ to LZ ratio for the individual mice in (B) (symbols) and mean values (bars). (D) Representative flow cytometry histogram of Ki67 (color) and isotype control (gray) staining of GC B cells. Means ± SEM proportion of Ki67low cells for ≥2 mice per group per time point are plotted from two to three pooled independent experiments. (E) Representative flow cytometry histogram for activated caspase staining in GC B cells. Means + SD values in follicular (FO) and GC B cells for ≥3 mice per group from two pooled independent experiments are plotted. (B–E) One-way ANOVA test, P-values for significant differences are shown. (F) Analysis of FO and GC B cells in the spleen of radiation chimeras reconstituted with a 1:1 ratio of BM cells from WT, Ung−/− or Ung−/− Aicda−/− CD45.2 and WT CD45.1 mice, immunized 6 weeks after reconstitution and analyzed 11 days post-immunization. The CD45.2 to CD45.1 ratio for 8–10 mice (symbols) per group and mean values (bars) from two pooled experiments are plotted. P-values for significant differences by two-way ANOVA with Sidak’s multiple comparisons. Representative CD45.1 versus CD45.2 flow cytometry staining gated on GC B cells (CD3− B220+ GL7+ CD95+; see gating in Supplementary Figure S1C).
GC B cells have two major populations, centroblasts in the DZ, where most proliferation takes place, and centrocytes in the LZ, which undergo antibody affinity-based selection and differentiation to effector cells (24,61). Ung−/− showed a small reduction in DZ/LZ ratio (Figure 1C), but there were no differences in Ki67 staining between Ung−/− and WT mice (Figure 1D). In contrast, the DZ/LZ in Ung−/− Aicda−/− mice was much lower than WT, reflecting the predominance of centrocytes typical of Aicda−/− GC (1,27) (Figure 1C). Accordingly, Aicda−/− GC consistently showed a larger fraction of Ki67low B cells than WT, especially at late time points, which was less noticeable in Ung−/− Aicda−/− GC B cells (Figure 1D). Reduced B-cell apoptosis contributes to GC hyperplasia in Aicda−/− mice (27), which we also verified in Ung−/− Aicda−/− GC B cells (Figure 1E). Interestingly, Ung−/− GC B cells had similarly reduced apoptosis (Figure 1E), despite the lack of GC hyperplasia.
As a more sensitive assay for B-cell fitness, we set up competitive BM chimeras by reconstituting irradiated WT host mice with a 1:1 mix of WT and Ung−/− or Ung−/- Aicda−/− BM cells distinguishable by their CD45 allele, which were compared to WT:WT control chimeras. For the most part, Ung−/− and Ung−/- Aicda−/− BM contributed to T- and B-cell subsets similarly to WT, or even showed a slight advantage in pre-B cells and blood B cells (Supplementary Figure S1B and C). In marked contrast, Ung−/− GC B cells were underrepresented by 3-fold relative to WT, revealing a disadvantage (Figure 1F). This GC-specific B-cell defect disappeared in the Ung−/− Aicda−/− GC B cells, which outcompeted their WT counterparts (Figure 1F), similar to the GC phenotype of Aicda−/−:WT BM chimeras (27,28).
We conclude that, despite both UNG and AID deficiencies reducing apoptosis of GC B cells, their effects are not additive but have different consequences on GC B cells, as Ung−/− mice do not develop the GC hyperplasia typical of Aicda−/−. Moreover, in contrast to AID-deficient GC B cells (27), Ung−/− B cells have in fact a fitness disadvantage in the GC, which is eliminated when AID is simultaneously ablated.
Activated Ung−/− B cells undergo extensive telomere damage
The lack of GC B-cell hyperplasia in Ung−/− mice compared to Aicda−/− and Ung−/− Aicda−/−, despite equally reduced GC B-cell apoptosis in all three, suggested that another process offsets the benefit of less apoptosis in activated Ung−/− B cells. We have shown that Ung−/− B cells activated ex vivo undergo telomere truncations that are AID dependent and can reduce cell proliferation (42). We hypothesized that this DNA damage, which would affect Ung−/− but not Ung−/− Aicda−/− B cells, might contribute to the GC phenotype in Ung−/− mice. To assess the real extent of ongoing telomere damage, we activated Ung−/− Trp53−/− and control primary B cells ex vivo with LPS and IL-4, which induces AID and proliferation. While AID-dependent telomere losses were found in ∼20% Ung−/− B cells, these aberrations were visible in ∼60% of Ung−/− Trp53−/− B cells (Figure 2). The difference between the proportion of Ung−/− and Ung−/− Trp53−/− B cells with telomere loss represented cells that do not reach metaphase, and hence cannot be scored by telomere FISH, due to p53-dependent checkpoint activation (62). Thus, ongoing AID-dependent telomere DNA damage in Ung−/− B cells could offset reduced apoptosis and explain the lack of GC hyperplasia in the mice, as well as the defect in GC B-cell fitness in vivo.
Figure 2.

Telomere loss in Ung-deficient B cells activates p53-dependent checkpoints. Pictures of FISH with a telomeric probe on metaphases of primary B cells from the indicated mouse lines, 24 h post-stimulation with LPS and IL-4. FISH signal in green (FITC) is overlaid on DNA stain in blue (DAPI). Numbered arrows indicate telomere loss events, identified as unequal hybridization on sister chromatids. Pictures showing telomere loss events for most genotypes were selected and individual chromosomes with either intact telomeres or representative telomere loss events (arrows) are magnified below. The proportions of mitoses showing at least one telomere loss, scored from two independent experiments and 50 metaphases per experiment per genotype, are plotted.
AID ablation does not prevent BCL in Ung−/− mice
Besides GC B cells, the other in vivo setting in which AID is well expressed is in the context of BCL (14–16). To analyze the interplay between AID and UNG in BCL, we used Ung−/− mice, which develop spontaneous BCL (37). To test whether the accrued incidence of BCL in Ung−/− mice was caused by AID, we followed prospective cohorts of C57BL6/J WT, Ung−/−, Aicda−/− and Ung−/− Aicda−/− mice.
The median lifespan of Ung−/− mice was 8 weeks shorter than WT (Figures 2B and 3A), in line with the 15-week difference reported for a larger cohort (>100 mice) (37). Most mice reached endpoint due to age-related morbidities but, as expected, there was a significant increase in lymphoma incidence in Ung−/− versus WT mice (Figure 3C). Ung−/− mice with lymphoma succumbed at a median age of 782 days (Figure 3D), indicating that the Ung−/− lymphomas were not aggressive and/or appeared late. Accordingly, four mice that were euthanized between 13 and 30 months of age due to ulcerative dermatitis or final endpoint showed lymphoid tissue histopathology consistent with either preneoplastic expansion or early lymphoma (Figure 3C and Supplementary Table S3).
Figure 3.
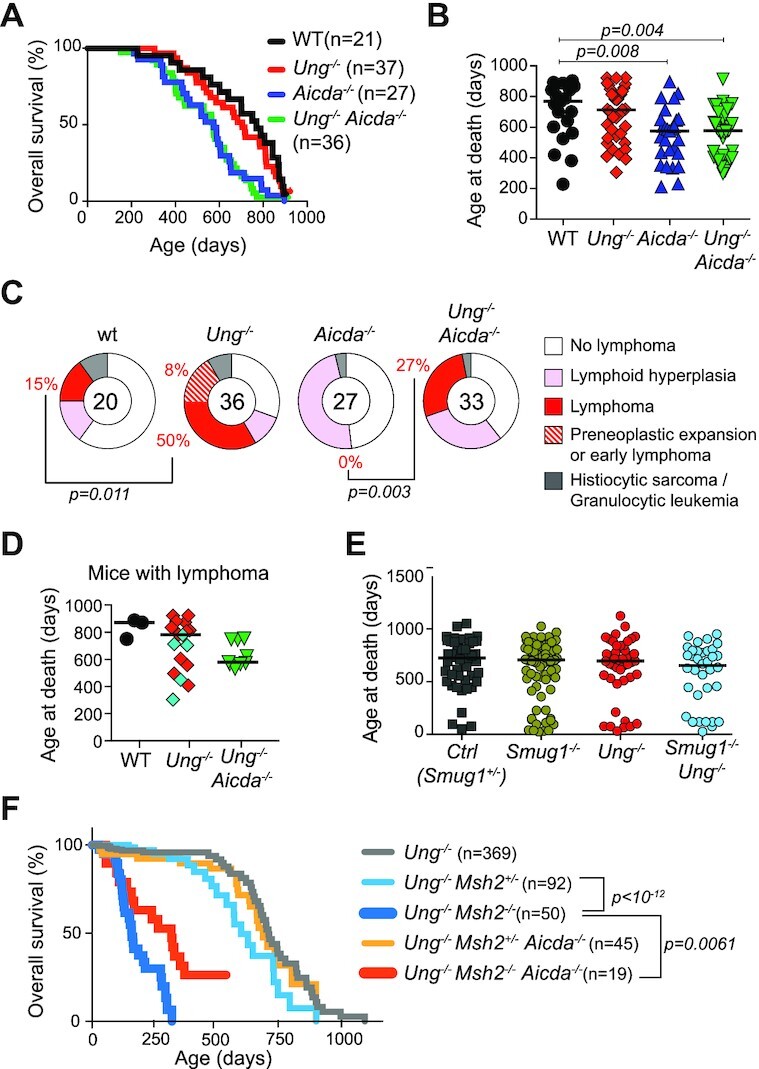
Survival and lymphoma incidence in Ung−/− and Ung−/− Aicda−/− mice. (A) Kaplan–Meier survival plot of prospective mouse cohorts of the indicated genotypes. Significant differences by the Mantel–Cox log-rank test (wt versus Aicda−/−, P = 0.0043; WT versus Ung−/− Aicda−/−, P = 0.0011; Ung−/− versus Aicda−/−, P = 0.0031; Ung−/− versus Ung−/− Aicda−/−, P = 0.0011). (B) Age at death of individual mice (symbols) and median values (bars) for the mouse groups in (A). (C) Incidence of hematological neoplasms in the mouse groups from (A). Significant P-values by Fisher’s exact test. (D) Age at death of individual mice (symbols) and median values for the mice diagnosed with lymphoma. Non-B-cell neoplasms in Ung−/− are shown in cyan. (E) Age at death of individual mice (symbols) and median values (bars) from the indicated mouse genotypes retrospectively compiled. (B, D, E) P-values for significant differences by one-way ANOVA are indicated. (F) Kaplan–Meier plot compiling retrospective survival data for the indicated mouse cohorts [Ung−/− Msh2−/−: n = 27/41 (endpoint observed/subjects at risk); Aicda−/− Ung−/− Msh2−/−: n = 14/19; Ung−/− Msh2+/−: n = 20/91; Aicda−/− Ung−/− Msh2+/−: n = 20/39; Ung−/−: n = 45/249]. Comparisons by the Mantel–Cox test: Ung−/− versus Ung−/− Msh2+/−, P = 0.0376; Aicda−/− Ung−/− Msh2+/− versus Ung−/− Msh2+/−, P = 0.2247; Ung−/− Msh2−/− versus Ung−/− Msh2+/−, P< 10−12; Aicda−/− Ung−/− Msh2−/− versus Ung−/− Msh2−/−, P = 0.0061.
Surprisingly, the median lifespan of Aicda−/− and Ung−/− Aicda−/− mice was significantly shorter (∼575 days) than WT (Figure 3A and B). These mice did not show any difference in hematological parameters up to 1 year of age (Supplementary Figure S2A) and the cause of death was not obvious during necropsy. The exceptions were a significant proportion of Ung−/- Aicda−/− mice (9/36) displaying lymphoid tissue hyperplasia secondary to the presence of lymphoma (Figure 3C, Supplementary Figure S2B and Supplementary Table S3). Ung−/− Aicda−/− mice presenting lymphoma were younger than the corresponding Ung−/− mice (Figure 3D) but still around the median life expectancy of the line, again indicating late incidence. In contrast, none of the 27 Aicda−/− mice analyzed developed any neoplasm.
SMUG1 can contribute some CSR in mice, but it mostly initiates error-free uracil repair in B cells (48,63–65). To rule out a putative protective role of SMUG1 in the relatively modest effect of UNG deficiency on mouse survival, we retrospectively analyzed the lifespan of Ung−/−, Smug1−/− and Ung−/− Smug1−/− mice cohorts housed in the same facility. Overall survival was identical for all groups (Figure 3E).
Since the deletion of tumor suppressors like MSH6 in p53-deficient mice is sufficient to change the tumor spectrum from T-cell lymphoma (TCL) to BCL, and accelerate lymphoma onset (66,67), we asked whether the lymphoma suppressor activity of UNG might have a similar effect. However, irrespective of UNG status, all Trp53−/− groups developed exclusively TCL, with no obvious difference in type, incidence or overall survival to the Trp53−/− control mice (Supplementary Figure S3A–D). UNG and/or AID deficiencies also failed to alter the kinetic or spectrum of cancer in p53-haploinsufficient mice, which develop a broader spectrum of tumors with longer latency than Trp53−/− mice (44) (Supplementary Figure S3E–G).
MSH2 deficiency drastically reduces mouse life expectancy, and Ung−/− Msh2−/− show the same survival as Msh2−/− mice (43). Interestingly, a retrospective analysis showed that ablating AID significantly increased the survival of Ung−/− Msh2−/− mice (Figure 3F), suggesting that MMR may limit the oncogenic activity of AID.
We conclude that UNG deficiency specifically causes spontaneous lymphoma predisposition in mice, but this lymphoma suppressor activity does not synergize with p53 deficiency, and produces lymphomas that develop slowly, are incompletely penetrant and can be AID independent.
Ung−/− and Ung−/− Aicda−/− mice develop mature BCL
Since the reduced lifespan of AID-deficient mice prevented a conclusion about the etiological role of AID in Ung−/− mice BCL, we sought to gain insight by detailed BCL phenotyping in Ung−/− and Ung−/− Aicda−/− mice.
Aged WT C57BL6/J mice rarely developed lymphoma but the three that did showed isotype switched (μ− κ/λ+) mature BCL (Figure 4A, Supplementary Figure S4 and Supplementary Table S3). Aged Ung−/− mice developed lymphoma but hardly any other neoplasm. Most mice displayed BCL, as shown by B220 staining (13/18), but there were also TCL (3/18) and B220− CD3− non-BCL/TCL (2/18) (Figure 4A and B, and Supplementary Figure S4). Ung−/− BCLs were of large B-cell type, with diffuse, nodular or mixed histopathology (Figure 4B and Supplementary Table S3), consistent with previous reports (37,38). Those Ung−/− BCLs that could be unambiguously typed by flow cytometry showed a mature B-cell phenotype (IgM+ IgD+) (Figure 4B and Supplementary Figure S4). Ung−/− Aicda−/− mice exclusively developed BCL, all but one being mature with diffuse or nodular histopathology (Figure 4C, Supplementary Figure S4 and Supplementary Table S3). Ung−/− BCLs were mostly Ki67−, but a majority of Ung−/− Aicda−/− BCLs were Ki67+ (Supplementary Figure S5A), suggesting they were relatively more aggressive, which is consistent with their earlier appearance compared to Ung−/− BCL (Figure 3D).
Figure 4.

Lymphoma spectrum in Ung−/− and Ung−/− Aicda−/− mice. (A) Proportion of lymphoma types found in each mouse line from the prospective cohort. (B) Examples of BCL and TCL in Ung−/− mice. Representative microscopy images of tissue sections stained by IHC with anti-B220 or anti-CD3 (bars = 50 μm) and flow cytometry plots. (C) Examples of BCL in Ung−/− Aicda−/− mice. Representative microscopy images of tissue sections stained with hematoxylin and eosin or by IHC with anti-B220 (bars = 50 μm), and flow cytometry plots. (D) Summary of the expression of the indicated markers monitored in BCL by IHC (Bcl6, PNA; see Supplementary Figure S5) or flow cytometry (CD21, CD23; see Supplementary Figure S4). Features typically found in activated (PNA+ Bcl6low CD21− CD23+), GC (Bcl6+ PNA+ CD21low CD23low/−) or post-GC (PNA− Bcl6− CD21− CD23−) B cells are shaded.
We focused on the mature BCLs to define whether they were derived from GC or activated B cells, which express AID. BCL6 expression was rare in BCLs from this cohort, whether from WT (1/3), Ung−/− (0/10) or Ung−/− Aicda−/− (2/8) mice, yet all WT BCLs were PNAHIGH and switched (Figure 4D and Supplementary Figure S5B), consistent with an activated or GC B-cell origin. The BCLs from Ung−/− and Ung−/− Aicda−/− were more heterogeneous, with 30% and 50%, respectively, expressing the PNAHIGH activation marker (Figure 4D and Supplementary Figure S5C). We also scored CD21 and CD23 expression, as within mature B cells these markers are downregulated in activated and GC versus FO B cells (Supplementary Figure S5D). BCLs from all three genotypes often showed low or absent CD21 and CD23 expression (Figure 4D and Supplementary Figure S4).
We conclude that full-body UNG deficiency specifically predisposes to lymphoma, mostly but not only mature BCL. The lymphoma-specific tumor suppressor activity of UNG is not prevented by AID deficiency, but rather seems to synergize with it to produce only BCL in Ung−/− Aicda−/− mice and earlier than in Ung−/−. Mature BCLs in both mice are heterogeneous but express markers of B-cell activation (Figure 4D).
Ung−/− BCLs show transient AID activity
To obtain more conclusive indications of an activated or GC origin for Ung−/− BCL, we analyzed AID expression and activity. We first stained by IHC all the cohort’s mature BCLs in parallel, together with normal GCs to compare their AID protein expression. The three WT BCLs expressed AID at levels comparable to that of GC B cells (Figure 5A). In contrast, only 1 out of 10 Ung−/− BCLs analyzed was positive for AID (Figure 5A). We analyzed more in detail Ung−/− mouse #4681, diagnosed with a relatively aggressive (496 days old at endpoint) mature PNAHIGH BCL, which we were able to amplify by transplantation into recipient mice that developed disease (Supplementary Figure S6A). Western blot of Ung−/− #4681 BCL cells confirmed the lack of AID expression, even after LPS stimulation (Figure 5A). Despite not expressing AID, the monoclonal VDJ rearrangement of this BCL showed SHM, as confirmed by the typical G:C to A:T pattern and the lack of mutations in a similarly analyzed Ung−/−Aicda−/− BCL (Supplementary Figure S6B and Figure 5B). Isotype switching was not expected in the absence of UNG, but this does not prevent AID from mutating the Igh switch (S)μ region (22), which was indeed the case in this tumor (Figure 5C). We analyzed mutations at the Sμ of additional Ung−/− BCL cells obtained from paraffin-embedded samples by laser capture microdissection. We were able to amplify the Sμ from three BCL samples, two of which showed mutations above the background determined from three samples of Ung−/−Aicda−/− BCL Sμ regions similarly obtained (Figure 5C). The two Ung−/− BCL Sμ regions with high mutation frequency also showed signs of intraclonal diversification (Figure 5D), suggesting AID was active for some time in the tumors before being downregulated.
Figure 5.

AID expression and activity in Ung-deficient BCL. (A) Representative IHC for AID in BCL tissue sections from WT or Ung−/− mice. A splenic GC from an immunized WT mouse serves as a positive control. Bars = 20 μm. The pie charts compile the proportion of AID+ BCL out of the mice analyzed (total n at the center). All samples were stained simultaneously, imaged and processed identically for comparison. AID western blot for one Ung−/− BCL sample is shown next to WT B cells activated with LPS and IL-4. (B) SHM frequency per nucleotide position of the represented IgVH region of the indicated BCL. (C) Mutation frequency in the Sμ region of the indicated Ung−/− BCL. The pie chart shows the proportion of sequences with the indicated number of mutations. The dot plot (top right) shows the mutation frequency calculated for each Ung−/− BCL and three Ung−/− Aicda−/− BCLs used as controls for mutation background. (D) Dynastic relationship between the Sμ sequences obtained from each BCL. Circles indicate the number of sequences for each unique variant bearing the indicated number of mutations. (E) Frequency of mutations at C within a WRCY sequence context, normalized to the frequency of all mutations identified at C by whole genome sequencing of the indicated BCL. (F) Log-transformed distribution of distances between WRCY mutations from (F). (G) Pie charts with the proportion of AID off-targets containing coding region mutations in each BCL. P-value by Fisher’s exact test.
We analyzed the presence of genomic instability in the Ung−/− #4681 BCL. Spectral karyotyping revealed the clonal aberrations Chr15 trisomy and T(1;6) translocation, but a Ung−/− Aicda−/− BCL (#19-087-2016) showed similar levels of clonal aberrations with a Der(4)T(X;4) translocation, large Chr10 deletion and tetraploid ChrX (Supplementary Figure S6C). The mutation spectra of these Ung−/− and Ung−/− Aicda−/− BCL, determined by whole genome sequencing, were dominated by C to T transitions (Supplementary Figure S6D). While TCL from Ung−/− Smug1−/− mice show a predominance of mutations at CpG (48), this was not the case in either UNG-deficient tumor, as shown by trinucleotide frequency plots (Supplementary Figure S6E). The mutations in Ung−/− and Ung−/− Aicda−/− BCLs differed in three aspects. First, kataegis events were apparent only in the Ung−/− BCL (Supplementary Figure S6F). Second, the fraction of mutations at C within the preferred AID context (WRCY) in the Ung−/− BCL was twice as high, and these mutations were more closely spaced than in the Ung−/− Aicda−/− BCL (Figure 5E and F). Third, the two tumors showed a non-overlapping set of coding gene mutations with distinct predicted functional effects (Supplementary Table S4), with mutations in coding regions in the Ung−/− BCL overlapping significantly more with AID off-target genes identified in mouse B cells (19,59) (Figure 5G).
We conclude that spontaneous BCL in either Ung−/− and Ung−/− Aicda−/− can accumulate complex genomic alterations and abundant mutations. In addition, and even acknowledging the caveat of a limited number of tumors analyzed, the presence of SHM in three out of four randomly chosen Ung−/− BCLs, one of them additionally showing a WRCY mutation signature, allows us to conclude that a good proportion of mature BCLs in Ung−/− mice originates from activated B cells that expressed AID.
Intact UNG expression in human BCL
Given the lymphoma suppressor activity for UNG in mice and that it could protect from AID-induced mutagenesis, we asked whether UNG loss might be prevalent in BCL patients. However, UNG mutations were very rare in human cancer. Among 36 157 cancer samples, encompassing human cancers of any tissue origin in the COSMIC database (GRCh38 v87), there were only 61 coding mutations, none of them in the 4226 hematopoietic neoplasms available (Supplementary Table S5). Similarly, there was a single UNG mutation (R269Q) among 3225 lymphoid neoplasms (1170 DLBCL) in the cBioPortal database and none in 38 cases of resistant/relapsed DLBCL we had sequenced (68) (Figure 6A). In contrast, mutations in MMR genes (MSH2, MSH6, PMS2, MLH1) were more common in DLBCL (0.3–3%), consistent with a report that found MMR genes mutated in 9/27 BCL samples but a single UNG mutation (34). UNG was not downregulated either in BCL. UNG transcript levels largely correlated with the proliferative characteristics of the neoplasm, being the highest in DLBCL, Burkitt’s and follicular lymphomas (Figure 6B). Burkitt’s lymphoma and DLBCL most frequently express AID (16). However, RNA-seq data of 48 DLBCL samples from TCGA showed consistent UNG expression and variable AICDA expression (Figure 6C). Nonetheless, UNG expression was significantly higher in activated B-cell-like DLBCL type, which express higher AID levels than the GC B-cell-like type (Figure 6D). We conclude that UNG is seldom mutated and typically well expressed in human BCL.
Figure 6.

UNG is expressed and unmutated in human BCL. (A) Summary of alterations in the indicated genes found by exome sequencing of B-cell neoplasms. Data from cBioPortal and from (68) for rrDLBCL (resistant/relapsed DLBCL). (B) UNG transcript levels by microarray in normal human B-cell subpopulations and B-cell neoplasm (GSE2350). (C) AICDA and UNG transcript levels by RNA-seq for a TCGA dataset of 48 cases (GSE2350). HPRT1, a well-expressed gene, and CDA, a low to not expressed gene, were included as references. (D) Comparative expression of UNG and AICDA by microarray in activated B-cell (ABC) versus germinal center B-cell (GCB) DLBCL (GSE22470). P-values by an unpaired two-tailed t-test.
Ung enables AID+ B-cell neoplasm
The paucity of UNG mutations in human BCL patients and the protective effect of UNG in normal GC B cells suggested that a potential tumor-enabling role of UNG might supersede its tumor suppressor role in AID+ cells. We therefore tested the effect of UNG in vivo in a mouse model of B-cell neoplasm cells that express AID. To dissociate AID from cancer initiation, we used an acute B-cell leukemia model that is driven by the expression of the BCR-ABL oncogene (69). In this model, AID is expressed at 5–10% levels compared to activated B cells but causes off-target mutations and acts as a disease progression factor (69,70). Indeed, Aicda−/− BM cells transduced with BCR-ABL caused a less aggressive leukemia than BCR-ABL WT BM in recipient mice (Figure 7A). In this setting, if the role of UNG in counteracting AID activity prevailed, we would have expected a more aggressive leukemia when using Ung−/− BM cells. Instead, mice transplanted with BCR-ABL Ung−/− BM cells showed significantly less aggressive leukemia than the WT (Figure 7A). AID transcript was detectable in 13/15 (86.7%) of the WT and 11/17 (64%) of the Ung−/− leukemia obtained from moribund animals that were tested by RT-PCR (Figure 7B). Importantly, ablation of Ung did not improve the survival of AID-deficient leukemia (Figure 7A). These results provide good evidence of a tumor-enabling effect of Ung for B-cell leukemia in vivo, which is specific to AID sufficiency.
Figure 7.
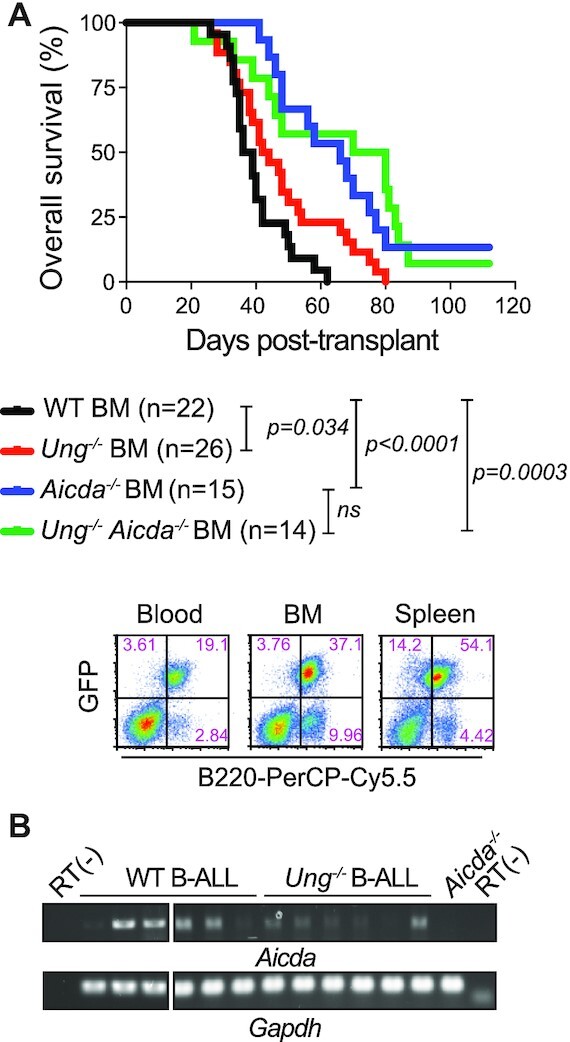
Tumor-enabling effect of UNG on AID+ B-cell leukemia. (A) Overall survival of NRG mice transplanted with BM from the indicated donor genotypes previously transduced with the BCR-ABL oncogene. Significant differences by the Mantel–Cox log-rank test. Representative flow cytometry plots of the typical leukemia phenotype at endpoint are shown below. (B) Aicda and Gapdh transcripts detected by endpoint RT-PCR in total splenocytes from moribund mice (43–80% GFP+ cells).
DISCUSSION
The role of UNG has been especially scrutinized in B cells because of its function in CSR and SHM. UNG also mediates some of AID oncogenic effects by enabling off-target DNA breaks that lead to chromosomal translocations (2,21,22,39). On the other hand, UNG funnels a proportion of the AID-catalyzed uracils into error-free BER, which prevents mutations, genomic rearrangements and telomere truncations (19,25,39,42,71). After cancer initiation, the ongoing DNA damage and repair transactions arising from AID and UNG could still affect cell fitness and thereby cancer progression. We show that UNG protects B-cell fitness from AID, which has an impact on the population dynamics of GC and cancer B cells. UNG could protect B cells from transformation, as evidenced by the predominance of AID-experienced BCL in Ung−/− mice, but after cancer initiation UNG becomes an enabler of tumor growth by protecting the cells from the detrimental effect of AID activity, as suggested by the loss of AID in Ung−/− BCL and shown by the slower growth of Ung−/− B-ALL.
Our conclusion that UNG promotes B-cell fitness in vivo, and that this effect is linked to the expression of AID, is supported by three lines of evidence. First, UNG has a positive effect on fitness specifically in GC but not any other B-cell subset, as shown in competitive BM chimeras. Others had shown that AID has the opposite effect on GC B-cell fitness, with Aicda−/− GC B cells outcompeting WT in BM chimeras (27,28). Since Ung−/− Aicda−/− GC B cells also outcompeted the WT, AID most likely causes the fitness defect of Ung−/− GC B cells. Ung−/− mice show normal GC B-cell numbers (further discussed below). In contrast, Ung−/− mice that overexpress AID by ∼3-fold generated 2–3-fold less GC B cells than the UNG-proficient controls after immunization (42). We posit that B-cell fitness is sensitive to the cellular AID:UNG ratio, to minimize the risk of genomic instability in GC B cells with high AID:UNG ratio (42). Second, and consistent with the latter, the spontaneous BCLs arising in Ung−/− mice lack AID expression despite showing signs of AID activity (SHM and genome-wide WRCY signature). Even acknowledging that we analyzed a modest number of samples, these were randomly chosen (based on success to amplify the IgVH or Sμ), yet most showed SHM. In contrast, all three BCLs found in WT mice showed high AID expression. These data allow us to conclude that AID was lost in the Ung−/− BCL, rather than never expressed, further suggesting that, in the absence of UNG, AID expression becomes a liability and is counterselected. In one BCL sample we could analyze further, Aicda could not be induced with LPS and IL-4, which induce AID expression in normal B cells. This suggests that Aicda might be silenced epigenetically, which remains to be confirmed. Third, ablating UNG in B-ALL cells results in a population disadvantage in vivo, as judged by the slower disease kinetics, specifically when the leukemia cells were AID proficient. This is the first experimental demonstration of a tumor-enabling activity of UNG. Two reports have shown that UNG is essential for AID-initiated oncogenesis in mouse models. Crossing Ung−/− mice with the IμHABcl6 transgenic model of DLBCL, or the Bcl-xL transgenic model of plasmacytoma, greatly reduces the incidence of the neoplasms (39,40). Neither model allows to test the effect of UNG on cancer progression as a function of AID expression because either enzyme is required for oncogenesis (4,10). Collectively, our results provide good evidence that UNG protects normal and cancer B-cell fitness from AID activity, thereby revealing a tumor-enabling activity of UNG that is at play during cancer progression. This activity is distinct from the roles AID and UNG might have during cancer initiation.
The tumor-enabling function of UNG does not rule out that UNG acts as a tumor suppressor by reducing the mutation load caused by AID off-target activity, as shown in normal B cells (37,38,72). The reduced lifespan of AID-deficient mice (see below) hampered our effort to directly test this. Interestingly, AID deficiency significantly reduced the mortality of Ung−/− Msh2−/− mice in retrospective analyses. Together with the report that MSH2 deficiency increases lymphoma incidence in the IμHABcl6 transgenic mouse model (40), our observation suggests that MMR might more robustly mitigate the oncogenic effect of AID, which remains to be investigated. Surprisingly, we also find evidence of an AID-independent lymphoma suppressor activity for UNG. First, we find some lymphomas originating from cells that are not expected to express AID in our Ung−/− cohort. Second, Ung−/− Aicda−/− mice developed mature BCL, often displaying activation markers, albeit not Bcl6. Combined with the fact that AID deficiency should affect the tissue where the enzyme is normally expressed, BCL in Ung−/− Aicda−/− is likely derived from activated B cells. The lack of lymphoma in Aicda−/− mice and earlier incidence of BCL in Ung−/− Aicda−/− than in Ung−/− mice suggest the two deficiencies synergize. The underlying mechanism is unknown but must be independent of the function of UNG in antibody diversification, in which both enzymes are epistatic. Perhaps the mild mutagenic effect of compromised uracil BER caused by UNG deficiency (35,36) compounds with reduced apoptosis and protracted GC caused by AID deficiency to increase the probability of B-cell transformation in Ung−/− Aicda mice. We did not analyze this further because this double deficiency has no correlate in human patients. On the other hand, our findings could have implications for AID- or UNG-deficient patients. First, we find that AID deficiency can shorten lifespan. The reason for this was not apparent but we ruled out the previously reported mutation in Lag3 acquired during backcrossing Aicda−/− to Balb/c (73,74), which is absent in our C57BL6/J mice (not shown). Immunodeficiency is also an unlikely cause, as the mice were kept in clean facilities and showed no signs of infection. However, AID-deficient mice fail to control the gut flora (75), and show autoimmune traits due to defects in B-cell tolerance (76,77), both of which could contribute to reduce lifespan. In any case, our observation calls for closer follow-up of AID-deficient patients. Second, our data emphasize that UNG is a weak tumor suppressor in mice and suggest it is not at all in humans. Thus, Ung−/− BCLs are not aggressive (i.e. incomplete penetrance, long latency, Ki67-negative, no synergy with p53 deficiency), consistent with the modest effect on overall mouse survival (37). Human BCLs invariably show intact UNG expression, in contrast to MMR genes that are frequently mutated, suggesting that cancer cells prefer to retain UNG expression. None of the five reported patients with congenital UNG deficiency, some of which are >40 years old, has developed cancer yet (78). These patients should be monitored, but it is possible that the oncogenic activity UNG (39,40), and/or the tumor-enabling activity described here, predominates over its tumor suppressor activity in humans.
We show that UNG protects the fitness of normal GC B cells, yet Ung−/− mice show normal GC B-cell numbers. Two AID-related functions of UNG likely underlie this apparent discrepancy. First, AID-dependent DNA damage induces apoptosis, which explains the GC hyperplasia in Aicda−/− mice (27,28). UNG mediates most or the apoptogenic intrachromosomal DNA breaks downstream from AID, for both Igh CSR and off-target (21,22,79). The similar decrease in GC B-cell apoptosis that we find in Ung−/−, Aicda−/− and Ung−/−Aicda−/− mice is consistent with AID and UNG being epistatic for intrachromosomal DNA damage (21,22,79). The lack of GC hyperplasia in Ung−/− mice compared to Aicda−/− and Ung−/− Aicda−/− is likely explained by a second mechanism, in which AID and UNG are not epistatic. We posit that the fitness defect in Ung−/− B cells is caused by AID-induced telomere damage. AID can target the telomere C-rich strand, but UNG prevents any DNA damage (42). Instead, in UNG-deficient B cells and BCL cell lines, MMR transforms the U:G mismatches into single-stranded DNA gaps, leading to telomere truncation upon replication (42). We now show that AID-induced telomere loss is very frequent and activates p53-dependent checkpoints, which likely compromises cell fitness. We cannot measure telomere loss in GC B cells because they do not replicate in vitro, but it is fair to assume it happens based on the ex vivo evidence. Importantly, while intrachromosomal DNA breaks typically induce apoptosis, DNA breaks at the telomeres induce autophagy (80). Thus, the normal GC B-cell numbers in Ung−/− mice likely reflect the net result of reduced apoptosis from less intrachromosomal breaks, offset by increased telomere damage that hampers population expansion. The latter becomes apparent in competitive BM chimera system and in the B-ALL model.
In summary, we have identified and suggest the mechanism of a new UNG function, whereby it protects cell fitness to favor the expansion of normal and neoplastic B-cell populations expressing AID. This activity reveals a novel cancer-promoting aspect of UNG, which so far has been considered a tumor suppressor. AID is commonly expressed in certain human BCL types, and is linked to clonal evolution, accelerated disease and poor outcome (12,17,18,81). Our findings suggest that inhibiting UNG could eliminate AID+ cells and thereby aggressive lymphoma clones.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the assistance of Simone Terouz with histology; Eric Massicotte and Julie Lord with flow cytometry; Manon Laprise, Marie-Claude Lavallée and Caroline Dubé with animal experimentation; Mylène Cawthorn and Evelyn Thivièrge for animal care; Lene Alsøe for coordinating WGS; and Geir Kjetil Sandve for informatics support. We thank Ludivine Litzler, P. Ganesh Subramani, Damien Montamat-Sicotte and Stephen Methot for discussions and/or technical advice at various stages of the project.
Author contributions: S.S., A.L., A.Z., A.-M.P., F.D., K.D., J.H., Y.L. and R.E.V. performed experiments; S.S., A.L., D.D., S.-K.K., T.R., J.H., R.E.V., C.A.R., F.V., N.J., H.N. and J.M.D.N. analyzed data; S.S., A.L. and J.M.D.N. analyzed results and made figures; H.N. and J.M.D.N. designed research; J.M.D.N. wrote the manuscript; all authors commented on and approved the manuscript.
Contributor Information
Shiva Safavi, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada; Division of Experimental Medicine, Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada.
Ariane Larouche, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada.
Astrid Zahn, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada.
Anne-Marie Patenaude, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada.
Diana Domanska, Department of Informatics, University of Oslo, PO Box 1080, Blindern, 0316 Oslo, Norway; Section for Clinical Molecular Biology, Akershus University Hospital, PO 1000, 1478 Lørenskog, Norway.
Kiersten Dionne, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada.
Torbjørn Rognes, Department of Informatics, University of Oslo, PO Box 1080, Blindern, 0316 Oslo, Norway; Department of Microbiology, Oslo University Hospital, Rikshospitalet, Oslo NO-0027, Norway.
Felix Dingler, MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, UK.
Seong-Kwi Kang, ITR Laboratories Canada, Inc., 19601 Clark Graham Ave, Baie-D’Urfe, QC H9X 3T1, Canada.
Yan Liu, Section for Clinical Molecular Biology, Akershus University Hospital, PO 1000, 1478 Lørenskog, Norway.
Nathalie Johnson, Division of Experimental Medicine, Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada; Jewish General Hospital, Montreal, QC H3T 1E2, Canada.
Josée Hébert, Department of Medicine, Université de Montréal, C.P. 6128, Montreal, QC H3C 3J7, Canada; The Quebec Leukemia Cell Bank, Research Centre and Division of Hematology–Oncology, Maisonneuve-Rosemont Hospital, 5415 l’Assomption blvd, Montreal, QC H1T 2M4, Canada.
Ramiro E Verdun, Division of Hematology, Department of Medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL 33136, USA; Geriatric Research Education and Clinical Center, Miami VA Healthcare System, Miami, FL 33136, USA.
Cristina A Rada, MRC Laboratory of Molecular Biology, Cambridge CB2 0QH, UK.
Francisco Vega, Division of Hematology, Department of Medicine, Sylvester Comprehensive Cancer Center, University of Miami, Miami, FL 33136, USA; Division of Hematopathology, Department of Pathology and Laboratory Medicine, University of Miami and Sylvester Comprehensive Cancer Center, Miami, FL 33136, USA.
Hilde Nilsen, Section for Clinical Molecular Biology, Akershus University Hospital, PO 1000, 1478 Lørenskog, Norway; Institute of Clinical Medicine, University of Oslo,P.O box 1171,Oslo, Norway.
Javier M Di Noia, Institut de Recherches Cliniques de Montréal, 110 Av des Pins Ouest, Montréal, QC H2W 1R7, Canada; Division of Experimental Medicine, Department of Medicine, McGill University, Montréal, QC H4A 3J1, Canada; Department of Medicine, Université de Montréal, C.P. 6128, Montreal, QC H3C 3J7, Canada.
DATA AVAILABILITY
Whole genome sequence data were deposited to the EMBL-EBI European Nucleotide Archive (primary accession: PRJEB31113; samples’ primary accession: ERS3128659; title: ‘Mouse C57BL6/J Ung−/− BCL lymphoma #16836’; primary accession: ERS3128660; title: ‘Mouse C57BL6/J Ung−/− Aicda−/− BCL lymphoma #16840’; primary accession: ERS3128661; title: ‘Mouse C57BL6/J Ung−/− Aicda−/− tail’). All other data are available in the supplementary data file or as supplementary tables. Igh V and Sμ sequences analyzed in Figure 5B–D are provided in the supplementary data file. Flow cytometry data were deposited at FlowRepository.org under accession numbers FR-FCM-Z2RR, FR-FCM-Z2TZ, FR-FCM-Z2RH, FR-FCM-Z2R7 and FR-FCM-Z2S6.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Canadian Cancer Society Research Institute [700348 to J.M.D.N.]; Leukemia and Lymphoma Society of Canada [514661 to J.M.D.N.]; Norwegian Cancer Society [182793-2016 to H.N.]; Southern and Eastern Norway Regional Health Authority [276940 to Y.L.]; Fonds de Recherche du Québec—Santé "Bourse de Merite" [to J.M.D.N.]; NIH/NIGMS [R01GM121595 to R.E.V.]; NHI/NCI [P30CA240139 to R.E.V.].
Conflict of interest statement. J.M.D.N. and R.E.V. are named as inventors in US Patent Appl. 15/569,916, ‘Modulating uracil-DNA glycosylase and uses thereof’, 2018.
REFERENCES
- 1. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000; 102:553–563. [DOI] [PubMed] [Google Scholar]
- 2. Methot S.P., Di Noia J.M. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv. Immunol. 2017; 133:37–87. [DOI] [PubMed] [Google Scholar]
- 3. Casellas R., Basu U., Yewdell W.T., Chaudhuri J., Robbiani D.F., Di Noia J.M. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat. Rev. Immunol. 2016; 16:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ramiro A.R., Jankovic M., Eisenreich T.R., Difilippantonio S., Chen-Kiang S., Muramatsu M., Honjo T., Nussenzweig A., Nussenzweig M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004; 118:431–438. [DOI] [PubMed] [Google Scholar]
- 5. Chesi M., Robbiani D.F., Sebag M., Chng W.J., Affer M., Tiedemann R., Valdez R., Palmer S.E., Haas S.S., Stewart A.K. et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008; 13:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiarle R., Zhang Y., Frock R.L., Lewis S.M., Molinie B., Ho Y.-J., Myers D.R., Choi V.W., Compagno M., Malkin D.J. et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011; 147:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klein I.A., Resch W., Jankovic M., Oliveira T., Yamane A., Nakahashi H., Di Virgilio M., Bothmer A., Nussenzweig A., Robbiani D.F. et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011; 147:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Robbiani D.F., Deroubaix S., Feldhahn N., Oliveira T.Y., Callen E., Wang Q., Jankovic M., Silva I.T., Rommel P.C., Bosque D. et al. Plasmodium infection promotes genomic instability and AID-dependent B cell lymphoma. Cell. 2015; 162:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Komeno Y., Kitaura J., Watanabe-Okochi N., Kato N., Oki T., Nakahara F., Harada Y., Harada H., Shinkura R., Nagaoka H. et al. AID-induced T-lymphoma or B-leukemia/lymphoma in a mouse BMT model. Leukemia. 2010; 24:1018–1024. [DOI] [PubMed] [Google Scholar]
- 10. Pasqualucci L., Bhagat G., Jankovic M., Compagno M., Smith P., Muramatsu M., Honjo T., Morse H.C., Nussenzweig M.C., Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008; 40:108–112. [DOI] [PubMed] [Google Scholar]
- 11. Robbiani D.F., Bunting S., Feldhahn N., Bothmer A., Camps J., Deroubaix S., McBride K.M., Klein I.A., Stone G., Eisenreich T.R. et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol. Cell. 2009; 36:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Swaminathan S., Klemm L., Park E., Papaemmanuil E., Ford A., Kweon S.-M., Trageser D., Hasselfeld B., Henke N., Mooster J. et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat. Immunol. 2015; 16:766–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basso K., Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015; 15:172–184. [DOI] [PubMed] [Google Scholar]
- 14. Greeve J., Philipsen A., Krause K., Klapper W., Heidorn K., Castle B.E., Janda J., Marcu K.B., Parwaresch R. Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood. 2003; 101:3574–3580. [DOI] [PubMed] [Google Scholar]
- 15. Pasqualucci L., Guglielmino R., Houldsworth J., Mohr J., Aoufouchi S., Polakiewicz R., Chaganti R.S.K., Dalla-Favera R. Expression of the AID protein in normal and neoplastic B cells. Blood. 2004; 104:3318–3325. [DOI] [PubMed] [Google Scholar]
- 16. Montamat-Sicotte D., Palacios F., Di Noia J.M., Oppezzo P. Origins and consequences of AID expression in lymphoid neoplasms. Curr. Immunol. Rev. 2013; 9:72–85. [Google Scholar]
- 17. Klemm L., Duy C., Iacobucci I., Kuchen S., von Levetzow G., Feldhahn N., Henke N., Li Z., Hoffmann T.K., Kim Y. et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell. 2009; 16:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palacios F., Moreno P., Morande P., Abreu C., Correa A., Porro V., Landoni A.I., Gabus R., Giordano M., Dighiero G. et al. High expression of AID and active class switch recombination might account for a more aggressive disease in unmutated CLL patients: link with an activated microenvironment in CLL disease. Blood. 2010; 115:4488–4496. [DOI] [PubMed] [Google Scholar]
- 19. Álvarez-Prado Á.F., Pérez-Durán P., Pérez-García A., Benguria A., Torroja C., de Yébenes V.G., Ramiro A.R. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J. Exp. Med. 2018; 215:761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hasham M.G., Donghia N.M., Coffey E., Maynard J., Snow K.J., Ames J., Wilpan R.Y., He Y., King B.L., Mills K.D. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat. Immunol. 2010; 11:820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramiro A.R., Jankovic M., Callen E., Difilippantonio S., Chen H.T., McBride K.M., Eisenreich T.R., Chen J., Dickins R.A., Lowe S.W. et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006; 440:105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rada C., Di Noia J.M., Neuberger M.S. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 2004; 16:163–171. [DOI] [PubMed] [Google Scholar]
- 23. Zanotti K.J., Gearhart P.J. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair. 2016; 38:110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cyster J.G., Allen C.D.C. B cell responses: cell interaction dynamics and decisions. Cell. 2019; 177:524–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu M., Duke J.L., Richter D.J., Vinuesa C.G., Goodnow C.C., Kleinstein S.H., Schatz D.G. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008; 451:841–845. [DOI] [PubMed] [Google Scholar]
- 26. Roa S., Li Z., Peled J.U., Zhao C., Edelmann W., Scharff M.D. MSH2/MSH6 complex promotes error-free repair of AID-induced dU:G mispairs as well as error-prone hypermutation of A:T sites. PLoS One. 2010; 5:e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zaheen A., Boulianne B., Parsa J.-Y., Ramachandran S., Gommerman J.L., Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood. 2009; 114:547–554. [DOI] [PubMed] [Google Scholar]
- 28. Boulianne B., Rojas O.L., Haddad D., Zaheen A., Kapelnikov A., Nguyen T., Li C., Hakem R., Gommerman J.L., Martin A. AID and caspase 8 shape the germinal center response through apoptosis. J. Immunol. 2013; 191:5840–5847. [DOI] [PubMed] [Google Scholar]
- 29. Vora K.A., Tumas-Brundage K.M., Lentz V.M., Cranston A., Fishel R., Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J. Exp. Med. 1999; 189:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Campo V.A., Patenaude A.M., Kaden S., Horb L., Firka D., Jiricny J., Di Noia J.M. MSH6- or PMS2-deficiency causes re-replication in DT40 B cells, but it has little effect on immunoglobulin gene conversion or on repair of AID-generated uracils. Nucleic Acids Res. 2013; 41:3032–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Wind N., Dekker M., Berns A., Radman M., te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995; 82:321–330. [DOI] [PubMed] [Google Scholar]
- 32. Edelmann W., Yang K., Umar A., Heyer J., Lau K., Fan K., Liedtke W., Cohen P.E., Kane M.F., Lipford J.R. et al. Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell. 1997; 91:467–477. [DOI] [PubMed] [Google Scholar]
- 33. Whiteside D., McLeod R., Graham G., Steckley J.L., Booth K., Somerville M.J., Andrew S.E. A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple café-au-lait spots. Cancer Res. 2002; 62:359–362. [PubMed] [Google Scholar]
- 34. de Miranda N.F., Peng R., Georgiou K., Wu C., Sörqvist E.F., Berglund M., Chen L., Gao Z., Lagerstedt K., Lisboa S. et al. DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J. Exp. Med. 2013; 210:1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nilsen H., Rosewell I., Robins P., Skjelbred C.F., Andersen S., Slupphaug G., Daly G., Krokan H.E., Lindahl T., Barnes D.E. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol. Cell. 2000; 5:1059–1065. [DOI] [PubMed] [Google Scholar]
- 36. An Q., Robins P., Lindahl T., Barnes D.E. C → T mutagenesis and γ-radiation sensitivity due to deficiency in the Smug1 and Ung DNA glycosylases. EMBO J. 2005; 24:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nilsen H., Stamp G., Andersen S., Hrivnak G., Krokan H.E., Lindahl T., Barnes D.E. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. 2003; 22:5381–5386. [DOI] [PubMed] [Google Scholar]
- 38. Andersen S., Ericsson M., Dai H.Y., Peña-Diaz J., Slupphaug G., Nilsen H., Aarset H., Krokan H.E. Monoclonal B-cell hyperplasia and leukocyte imbalance precede development of B-cell malignancies in uracil-DNA glycosylase deficient mice. DNA Repair. 2005; 4:1432–1441. [DOI] [PubMed] [Google Scholar]
- 39. Kovalchuk A.L., Ansarah-Sobrinho C., Hakim O., Resch W., Tolarová H., Dubois W., Yamane A., Takizawa M., Klein I., Hager G.L. et al. Mouse model of endemic Burkitt translocations reveals the long-range boundaries of Ig-mediated oncogene deregulation. Proc. Natl Acad. Sci. U.S.A. 2012; 109:10972–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gu X., Booth C.J., Liu Z., Strout M.P. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood. 2016; 127:102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vallabhaneni H., Zhou F., Maul R.W., Sarkar J., Yin J., Lei M., Harrington L., Gearhart P.J., Liu Y. Defective repair of uracil causes telomere defects in mouse hematopoietic cells. J. Biol. Chem. 2015; 290:5502–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cortizas E.M., Zahn A., Safavi S., Reed J.A., Vega F., Di Noia J.M., Verdun R.E. UNG protects B cells from AID-induced telomere loss. J. Exp. Med. 2016; 213:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kemmerich K., Dingler F.A., Rada C., Neuberger M.S. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/− mice. Nucleic Acids Res. 2012; 40:6016–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jacks T., Remington L., Williams B.O., Schmitt E.M., Halachmi S., Bronson R.T., Weinberg R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994; 4:1–7. [DOI] [PubMed] [Google Scholar]
- 45. Litzler L.C., Zahn A., Meli A.P., Hébert S., Patenaude A.-M., Methot S.P., Sprumont A., Bois T., Kitamura D., Costantino S. et al. PRMT5 is essential for B cell development and germinal center dynamics. Nat. Commun. 2019; 10:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jafri S., Moore S.D., Morrell N.W., Ormiston M.L. A sex-specific reconstitution bias in the competitive CD45.1/CD45.2 congenic bone marrow transplant model. Sci. Rep. 2017; 7:6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Delbos F., De Smet A., Faili A., Aoufouchi S., Weill J.C., Reynaud C.A. Contribution of DNA polymerase η to immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2005; 201:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alsøe L., Sarno A., Carracedo S., Domanska D., Dingler F., Lirussi L., SenGupta T., Tekin N.B., Jobert L., Alexandrov L.B. et al. Uracil accumulation and mutagenesis dominated by cytosine deamination in CpG dinucleotides in mice lacking UNG and SMUG1. Sci. Rep. 2017; 7:7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H., Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tischler G., Leonard S. biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 2014; 9:13. [Google Scholar]
- 52. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. DePristo M.A., Banks E., Poplin R., Garimella K. V, Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013; 31:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rognes T., Flouri T., Nichols B., Quince C., Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016; 4:e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simovski B., Vodák D., Gundersen S., Domanska D., Azab A., Holden L., Holden M., Grytten I., Rand K., Drabløs F. et al. GSuite HyperBrowser: integrative analysis of dataset collections across the genome and epigenome. Gigascience. 2017; 6:gix032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Domanska D., Vodák D., Lund-Andersen C., Salvatore S., Hovig E., Sandve G.K. The rainfall plot: its motivation, characteristics and pitfalls. BMC Bioinformatics. 2017; 18:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Qian J., Wang Q., Dose M., Pruett N., Kieffer-Kwon K.-R., Resch W., Liang G., Tang Z., Mathe E., Benner C. et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014; 159:1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Morita S., Kojima T., Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000; 7:1063–1066. [DOI] [PubMed] [Google Scholar]
- 61. Victora G.D., Schwickert T.A., Fooksman D.R., Kamphorst A.O., Meyer-Hermann M., Dustin M.L., Nussenzweig M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010; 143:592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Palm W., de Lange T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008; 42:301–334. [DOI] [PubMed] [Google Scholar]
- 63. Nilsen H., Haushalter K.A., Robins P., Barnes D.E., Verdine G.L., Lindahl T. Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J. 2001; 20:4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Di Noia J.M., Rada C., Neuberger M.S., M D.N.J., Rada C., Neuberger M.S. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. EMBO J. 2006; 25:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dingler F.A., Kemmerich K., Neuberger M.S., Rada C. Uracil excision by endogenous SMUG1 glycosylase promotes efficient Ig class switching and impacts on A:T substitutions during somatic mutation. Eur. J. Immunol. 2014; 44:1925–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Young L.C., Keuling A.M., Lai R., Nation P.N., Tron V.A., Andrew S.E. The associated contributions of p53 and the DNA mismatch repair protein Msh6 to spontaneous tumorigenesis. Carcinogenesis. 2007; 28:2131–2138. [DOI] [PubMed] [Google Scholar]
- 67. Cranston A., Fishel R. Female embryonic lethality in Msh2-Trp53 nullizygous mice is strain dependent. Mamm. Genome. 1999; 10:1020–1022. [DOI] [PubMed] [Google Scholar]
- 68. Morin R.D., Assouline S., Alcaide M., Mohajeri A., Johnston R.L., Chong L., Grewal J., Yu S., Fornika D., Bushell K. et al. Genetic landscapes of relapsed and refractory diffuse large B-cell lymphomas. Clin. Cancer Res. 2016; 22:2290–2300. [DOI] [PubMed] [Google Scholar]
- 69. Gruber T.A., Chang M.S., Sposto R., Müschen M. Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia. Cancer Res. 2010; 70:7411–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Montamat-Sicotte D., Litzler L.C., Abreu C., Safavi S., Zahn A., Orthwein A., Müschen M., Oppezzo P., Muñoz D.P., Di Noia J.M. HSP90 inhibitors decrease AID levels and activity in mice and in human cells. Eur. J. Immunol. 2015; 45:2365–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zahn A., Daugan M., Safavi S., Godin D., Cheong C., Lamarre A., Di Noia J.M. Separation of function between isotype switching and affinity maturation in vivo during acute immune responses and circulating autoantibodies in UNG-deficient mice. J. Immunol. 2013; 190:5949–5960. [DOI] [PubMed] [Google Scholar]
- 72. Gearhart P.J. B cells pay a price. Oncogene. 2003; 22:5379–5380. [DOI] [PubMed] [Google Scholar]
- 73. Hase K., Takahashi D., Ebisawa M., Kawano S., Itoh K., Ohno H. Activation-induced cytidine deaminase deficiency causes organ-specific autoimmune disease. PLoS One. 2008; 3:e3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Okazaki T., Okazaki I., Wang J., Sugiura D., Nakaki F., Yoshida T., Kato Y., Fagarasan S., Muramatsu M., Eto T. et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011; 208:395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fagarasan S., Muramatsu M., Suzuki K., Nagaoka H., Hiai H., Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002; 298:1424–1427. [DOI] [PubMed] [Google Scholar]
- 76. Cantaert T., Schickel J.-N., Bannock J.M., Ng Y.-S., Massad C., Oe T., Wu R., Lavoie A., Walter J.E., Notarangelo L.D. et al. Activation-induced cytidine deaminase expression in human B cell precursors is essential for central B cell tolerance. Immunity. 2015; 43:884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kuraoka M., Holl T.M., Liao D., Womble M., Cain D.W., Reynolds A.E., Kelsoe G. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc. Natl Acad. Sci. U.S.A. 2011; 108:11560–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cantaert T., Schickel J.-N., Bannock J.M., Ng Y.-S., Massad C., Delmotte F.R., Yamakawa N., Glauzy S., Chamberlain N., Kinnunen T. et al. Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J. Clin. Invest. 2016; 126:4289–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yang D., Sun Y., Chen J., Zhang Y., Fan S., Huang M., Xie X., Cai Y., Shang Y., Gui T. et al. REV7 is required for processing AID initiated DNA lesions in activated B cells. Nat. Commun. 2020; 11:2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nassour J., Radford R., Correia A., Fuste J.M., Schoell B., Jauch A., Shaw R.J., Karlseder J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019; 565:659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kawamura K., Wada A., Wang J.-Y., Li Q., Ishii A., Tsujimura H., Takagi T., Itami M., Tada Y., Tatsumi K. et al. Expression of activation-induced cytidine deaminase is associated with a poor prognosis of diffuse large B cell lymphoma patients treated with CHOP-based chemotherapy. J. Cancer Res. Clin. Oncol. 2016; 142:27–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequence data were deposited to the EMBL-EBI European Nucleotide Archive (primary accession: PRJEB31113; samples’ primary accession: ERS3128659; title: ‘Mouse C57BL6/J Ung−/− BCL lymphoma #16836’; primary accession: ERS3128660; title: ‘Mouse C57BL6/J Ung−/− Aicda−/− BCL lymphoma #16840’; primary accession: ERS3128661; title: ‘Mouse C57BL6/J Ung−/− Aicda−/− tail’). All other data are available in the supplementary data file or as supplementary tables. Igh V and Sμ sequences analyzed in Figure 5B–D are provided in the supplementary data file. Flow cytometry data were deposited at FlowRepository.org under accession numbers FR-FCM-Z2RR, FR-FCM-Z2TZ, FR-FCM-Z2RH, FR-FCM-Z2R7 and FR-FCM-Z2S6.