

Abstract
Glioma is the most commonly observed primary intracranial tumour and is associated with massive angiogenesis. Glioma neovascularization provides nutrients for the growth and metabolism of tumour tissues, promotes tumour cell division and proliferation, and provides conditions ideal for the infiltration and migration of tumour cells to distant places. Growing evidence suggests that there is a correlation between the activation of nuclear factor (NF)‐κB and the angiogenesis of glioma. In this review article, we highlighted the functions of NF‐κB in the angiogenesis of glioma, showing that NF‐κB activation plays a pivotal role in the growth and progression of glioma angiogenesis and is a rational therapeutic target for antiangiogenic strategies aimed at glioma.
Keywords: angiogenesis, Glioma, NF‐κB
Growing evidence suggests that there is a correlation between the activation of nuclear factor (NF)‐κB and the angiogenesis of glioma. Herein, we sought to discuss the current understanding of the molecular mechanisms of NF‐κB in diverse glioma microenvironments such as hypoxia, inflammation, and oxidative stress, and its function as a therapeutic target for antiangiogenic strategies aimed at glioma.
1. INTRODUCTION
Glioma, which originates from the glial cells surrounding the neurons, is the most commonly observed intracranial tumour with the greatest degree of malignancy and accounts for approximately 80% of all brain malignancies. 1 The median survival of malignant glioma patients is only about 1 year, even after common treatments including surgical resection, radiotherapy and chemotherapy are performed. 2 Angiogenesis is among the factors vital to tumour development. 3 As is the case with most solid tumours, the survival and growth of fast‐growing gliomas with an avascular area volume exceeding 2 mm 3 require newly generated blood vessels for the provision of the necessary oxygen, growth factors and nutrients. 4 Several studies have shown that angiogenesis has the strongest prognostic significance among all the clinical and pathological features of glioma, and that widespread angiogenesis tends to be associated with worse prognoses. 5 Based on the clinical significance and potentialities of the therapeutic interventions for glioma, it is necessary to identify the targets and underlying molecular mechanisms that regulate glioma angiogenesis.
Tumour necrosis factor (TNF, also referred to as TNF‐α) exerts its function using two receptors—TNF receptor I (TNFR1, p55 receptor) and TNF receptor II (TNFR2, p75 receptor)—which are members of the TNF superfamily. 6 TNF plays a role in the promotion of tumour cell apoptosis through TNFR1 binding; however, it also promotes tumour cell growth through TNFR2. 7 , 8 TNFR2 activation leads to the recruitment of TNF receptor‐associated factor 2 and motivates the pro‐survival nuclear factor (NF)‐κB pathway. 9 , 10 NF‐κB regulates the genes involved in tumour microenvironment development and proangiogenic and pro‐inflammatory cytokine synthesis. 11 Abnormal or constitutive NF‐κB activity in glioma 12 and a remarkable correlation between NF‐κB activation level and glioma grade have been previously demonstrated. 13 Furthermore, accumulating evidence shows that constitutive NF‐κB activity could regulate the proangiogenic context of glioma. Notably, NF‐κB restraint even led to the blocking of the angiogenesis of glioma in nude mice. 14
Herein, we sought to discuss the current understanding of the molecular mechanisms of NF‐κB in diverse glioma microenvironments such as hypoxia, inflammation and oxidative stress, and its function as a therapeutic target for antiangiogenic strategies aimed at glioma.
2. MOLECULAR MECHANISMS AND TARGETED THERAPIES
2.1. NF‐κB in hypoxia‐induced glioma angiogenesis
During the entire process of angiogenesis, new capillaries sprout from the current capillaries, and endothelial cells (ECs) are released from their stroma and migrate and transfer to areas without capillaries, thereby allowing them to differentiate into tubular structures. The newly generated capillaries provide a large amount of necessary oxygen and nutrients for fast‐growing malignant tumours that have an avascular area volume greater than 2 mm.315
Due to abnormalities in the structure of malignant tumours, local or temporary hypoxia and a lack of nutritional components may lead to EC apoptosis and tumour angiogenesis inhibition. 16 , 17 Nevertheless, migrating ECs often overcome these adverse conditions to boost tumour angiogenesis. ECs are stimulated by vascular endothelial growth factor (VEGF) or adhere to extracellular matrix (ECM) molecules, leading to the augmentation of anti‐apoptotic genes via the phosphatidylinositol 3‐kinase (PI3K)/Akt or NF‐κB signalling pathways. 18 , 19 Akt induces the transcription function of NF‐κB by stimulating the RelA/p65 transactivation subunit via IκB kinase and activation of the protein kinase p38. 20 Studies have reported that the induction of cell survival signals by PI3K/Akt partially mediates the activation of NF‐κB transcription factors. 20 The VEGF released by glioma cells stimulates EC proliferation, resulting in angiogenesis. 21 Interestingly, TNF, which could induce the apoptosis of ECs, was detected in glioma but did not inhibit the associated angiogenesis. 22 , 23 It was reported that human umbilical vein ECs must activate NF‐κB in order to avoid undergoing TNF‐induced apoptosis. 24 Using a human brain microvascular endothelial cell (HBMVEC) and U251 glioma cell co‐culture system, investigators found that EC apoptosis was induced by serum starvation and reversed by recombinant VEGF protein and a culture medium of hypoxic U251 glioma cells. In addition, hypoxia treatment activated TNF‐induced VEGF and NF‐κB to upregulate the antiapoptotic gene expressions, such as those of Bcl‐2, Bcl‐XL, survivin and X‐chromosome‐linked inhibitor of apoptosis protein (XIAP) in ECs 25 (Figure 1). Therefore, it is clear that the hypoxic environment of glioma, in addition to not killing ECs, promotes NF‐κB‐dependent angiogenesis.
Figure 1.
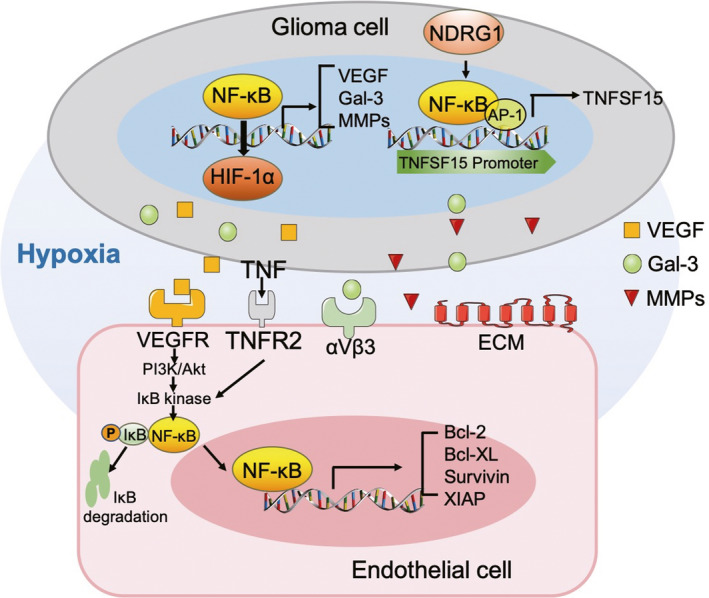
Role of NF‐κB in glioma angiogenesis under a hypoxic microenvironment. Hypoxia‐induced NF‐κB is conducive to the upregulation of the HIF‐1α, VEGF, gal‐3 and MMP genes, which are released by tumour cells to induce EC chemotaxis and motility and stimulate angiogenesis. Hypoxia activated TNFR2 and VEGFR induced NF‐κB to upregulate the expression of antiapoptotic genes, such as Bcl‐2, Bcl‐XL, survivin and XIAP in ECs. NDRG1 induced the upregulation of anti‐angiogenesis gene TNFSF15 by the activation of NF‐κB and AP‐1 in the TNFSF15 promoter region. TNFSF: tumour necrosis factor super family; TNFR: tumour necrosis factor receptor; NF‐κB: nuclear factor‐κB; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; PI3K: phosphatidylinositol 3‐kinase; Gal‐3: galectin‐3; XIAP: X‐chromosome‐linked inhibitor of apoptosis protein; HIF: hypoxia‐inducible factor; MMP: matrix metalloproteinase; AP: activator protein; ECM: extracellular matrix; NDRG1: N‐myc downstream‐regulated gene‐1
Galectin‐3 (gal‐3) is a b‐galactoside binding protein that is involved in several types of pathological tumour progression, such as angiogenesis, cell proliferation and anti‐apoptosis. 26 , 27 , 28 Evidence shows that gal‐3 is visibly upregulated in a hypoxia‐inducible factor (HIF)‐1α‐dependent manner in mouse fibroblasts and nucleus pulposus cells under hypoxic conditions. 29 , 30 In addition, HIF‐1α is a pivotal transcriptional regulator of the hypoxic response, which upregulates its target genes including vascular endothelial growth factor (VEGF) and matrix metalloproteinase (MMP) to boost tumour angiogenesis and invasion. 31 Hypoxia is a commonly observed feature of solid tumours such as gliomas, in which a high proportion of gal‐3 accumulates. 32 , 33 Gal‐3 is released by tumour cells for the induction of EC chemotaxis and motility and the stimulation of angiogenesis (Figure 1). Gal‐3 knockout U87 glioma cells implanted subcutaneously in nude mice blocked tumour growth in vivo. 34 In vitro, Ikemori et al found that gal‐3 protected T98G glioma cells from apoptosis in the absence of oxygen and nutrition, and the knockdown of gal‐3 induced double apoptosis. It is worth noting that the upregulation of gal‐3 was dependent not only on HIF‐1α but also on NF‐κB. 35 Hypoxia‐induced NF‐κB was conducive to the regulation of the HIF‐1α and gal‐3 genes and prevention of cell death caused by hypoxia. 36 , 37 Based on the above‐stated literature, it can be concluded that hypoxia in glioma protects both ECs and tumour cells against death, which facilitates angiogenesis and leads to tumour aggravation, directly or indirectly.
N‐myc downstream‐regulated gene‐1 (NDRG1) is considered a regulatory gene in the hypoxic microenvironment of glioma. 38 In untreated glioma patients, high NDRG1 expression was associated with increased survival, and the gene also reduced the rate of angiogenesis. 39 Thomas et al indicated that the expression of NDRG1 was markedly upregulated during hypoxia in glioma, and that an NDRG1‐overexpressing glioma implantation model with reduced angiogenic activity reduced the rate of glioma growth and resistance to antiangiogenic treatment. The anti‐angiogenesis gene TNFSF15 showed a 30‐fold increase in glioma development, with an increasing expression of NDRG1, and demonstrated that anti‐angiogenesis was positively correlated with TNFSF15. Interestingly, mutated NF‐κB and activator protein (AP‐1) in the TNFSF15 promoter region reversed the anti‐angiogenesis of NDRG1 40 (Figure 1). The research demonstrated that NF‐κB and AP‐1 are positively correlated with TNFSF15 expression in glioma.
Based on the aforementioned evidence, hypoxia promotes NF‐κB‐dependent angiogenesis in glioma. However, the hypoxic regulatory gene NDRG1 induces the upregulation of the anti‐angiogenesis gene TNFSF15 also depends on the transcriptional activity of NF‐κB. Therefore, NF‐κB exerts multiple effects on the angiogenic system in glioma under a hypoxic microenvironment.
2.2. NF‐κB in inflammation‐induced glioma angiogenesis
Some interactions exist between inflammation and angiogenesis in the course of glioma progression. 41 The angiogenesis of glioma depends on the interaction between tumour cells, ECs and surrounding inflammatory cells. In the tumour environment, newly formed blood vessels are allowed to recruit inflammatory cells continuously, leading to the release of proangiogenic cytokines, including VEGF‐A, MMP, chemokines and pro‐inflammatory factors. In this manner, a larger number of blood vessels are formed, ending in a vicious cycle. 42 , 43 The process requires the induction of a large number of pro‐inflammatory factors, such as TNF, interleukin (IL)‐8 and IL‐6, leading to capillary budding. NF‐κB, activated by TNF through the phosphorylation of IκB, 44 binds to the promoters of IL‐8 and IL‐6 to activate them 45 , 46 , 47 (Figure 2).
Figure 2.
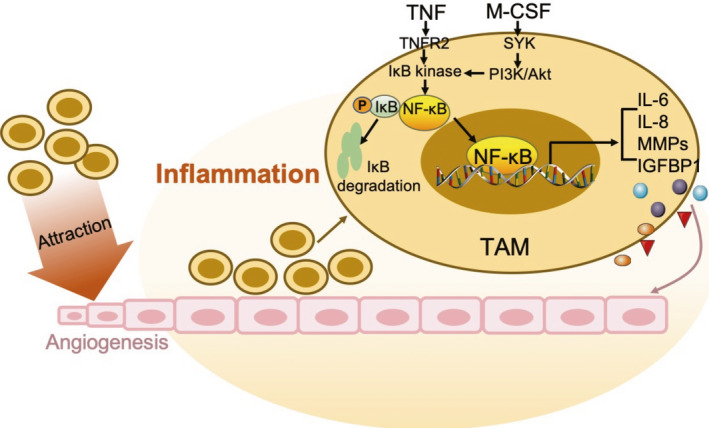
Role of NF‐κB in glioma angiogenesis under an inflammatory microenvironment. TNF activates the transcriptional activity of NF‐κB for IL‐6, IL‐8, MMPs and IGFBP1, by TNFR2, leading to angiogenesis. The newly formed blood vessels are allowed to recruit a larger number of inflammatory cells such as macrophages, leading to the further release of proangiogenic cytokines. TAM: tumour‐associated macrophages; M‐CSF: macrophage colony‐stimulating factor; SYK: spleen tyrosine kinase; IGFBP: insulin‐like growth factor binding protein; IL: interleukin; TNF: tumour necrosis factor; TNFR: tumour necrosis factor receptor; PI3K: phosphatidylinositol 3‐kinase; NF‐κB: nuclear factor‐κB; MMP: matrix metalloproteinase
IL‐8 is a chemical attractant cytokine that attracts and activates neutrophils at the site of inflammation and has an angiogenic effect. 47 It has been reported that the binding of IL‐8 to its receptors in ECs in vitro, CXCR1 and CXCR2, can activate ECs and facilitate tumour angiogenesis. 48 , 49 The expression level of IL‐8 is directly related to the degree of glioma angiogenesis. Furthermore, Xie et al used NF‐κB inhibitors to suppress IL‐8 secretion in vitro and in vivo. 14 Studies focusing on IL‐6 in vitro indicated that it affects the human inflammatory response and is related to tumour angiogenesis through the transcriptional activity of angiogenic growth factors such as VEGF and MMP. 50 , 51 Meanwhile, the IL‐4 and IL‐10 released by microglia have anti‐inflammatory effects in the glioma environment. 52 The IL‐4 released by macrophages can be used as anti‐inflammatory agents of TNF. 53 Importantly, IL‐4 inhibits the migration of ECs to inflammatory regions and inhibits their differentiation into organized vascular structures. 54
As mentioned above, angiogenesis is the formation of new capillaries based on existing blood vessels and is the result of inflammation. Macrophages are among the most important inflammatory immune cells in the tumour matrix. Tumour‐associated macrophages (TAMs) are recruited to the periphery of tumour cells, have immune function, and release a wide array of inflammatory mediators and cytokines. 55 Therefore, TAMs are considered as bridges that link inflammation and tumours. 56
It has been confirmed that glioma‐derived macrophage colony‐stimulating factor (M‐CSF) induces microglia and macrophages towards the M2 phenotype, thereby increasing the rate of tumour growth. 57 Another study suggested that the level of M‐CSF was upregulated in both glioma tissue and its serum, and that it induced angiogenesis in vivo and in vitro through the macrophage/microglia‐secreted insulin‐like growth factor binding protein 1 (IGFBP1). Notably, investigators found that spleen tyrosine kinase (SYK) activated the PI3K/Akt pathway, further leading to the NF‐κB‐dependent upregulation of M‐CSF in glioma. 58 Thus, the upregulation of M‐CSF induces angiogenesis through a SYK‐PI3K‐NF‐κB‐dependent mechanism. Additionally, TAMs increase the expression of IL‐8 through the NF‐κB pathway to promote angiogenesis (Figure 2). Interestingly, anti‐inflammatory drugs, including pentoxifylline, pyrrolidine dithiocarbamate and dexamethasone block, the expression of the IL‐8 induced by macrophages at least partially through the NF‐κB pathway. 59
Human cytomegalovirus (HCMV) has been shown to be associated with glioblastoma, with more than 90% of glioblastomas (GBMs) showing HCMV infection. 60 , 61 Previous reports have shown that HCMV pp71 is a viral protein that boosts the progression of the cell cycle and promotes the angiogenic glioma microenvironment through the induction of the stem cell factor (SCF). 62 , 63 Lisa et al found that pp71, by the activation of the NF‐κB pathway, led to the upregulation of SCF and induction of pro‐inflammatory responses with the upregulation of some pro‐inflammatory cytokines (IL‐8, IL‐1B, IL‐6, LIF, PTGS2 and IL‐1A), MMPs (MMP‐3, 12, 1 and 7) and angiopoietins. 64 The poor prognoses associated with human GBM may be attributed to the selective enhancement of pp71 levels and NF‐κB activation in pro‐inflammatory environments.
Manoj et al demonstrated that T11‐target structure (T11TS), sheep red blood cell membrane protein with immune‐enhancing and cell cycle‐regulating effects, 65 , 66 exerted antiangiogenic and anti‐tumour functions in an animal model and clinical glioma samples. 67 Their latest results indicate that the pro‐inflammatory cytokine expression of TNF, IL‐8, IL‐6 and NF‐κB is enhanced in glioma‐associated ECs. T11TS treatment repressed the NF‐κB signalling pathway in glioma‐induced animal models and thus induced the downregulation of pro‐inflammatory cytokines and upregulation of anti‐inflammatory cytokines, IL‐4 and IL‐10, for glioma angiogenesis elimination 68 (Table 1). Therefore, the expression of IL‐8 and IL‐6 in the glioma‐associated ECs induced by TNF is blocked by the NF‐κB‐mediated pathway, which has important implications for anti‐angiogenesis therapy.
Table 1.
List of NF‐κB‐dependent therapies against glioma angiogenesis
| Therapeutic targets | Targeted therapies/agents | Clinical development | Antiangiogenic pathways | Related transcription factors | References |
|---|---|---|---|---|---|
| TNF, IL‐8, IL‐6 and NF‐κB | T11‐target structure | Pre‐clinical study | Repressing the NF‐κB signalling pathway to induce downregulation of pro‐inflammatory cytokines and upregulation of anti‐inflammatory cytokines | 67, 68 | |
| TNF, VEGF | Thalidomide | Pre‐clinical study | Reducing inflammatory stimulation and interfering with the transcriptional regulation of NF‐κB | 71 | |
| ROS, IL‐6, IL‐8, GROα and MCP‐1 | NG and nGO | Pre‐clinical study | Suppressing proangiogenic cytokines and inhibiting the levels of ROS depending on the p53 mutation status and NF‐κB | p53 | 83, 84 |
| VEGF, HIF‐1α, MMP‐2, MMP‐9 | Tumour treating field | VEGF/VEGR inhibitor: Avastin (bevacizumab), Genentech | Downregulating NF‐κB, MAPK, and PI3K/Akt signalling pathways | 110 | |
| MMP‐3 and MMP‐9 | Glycitein | Pre‐clinical study | Suppressing the transcriptional activity of NF‐κB and AP‐1 on transcription of MMP‐3 or MMP‐9 | AP‐1 | 111 |
| MMP‐9 | Mangiferin | Pre‐clinical study | Inhibiting the binding of NF‐κB and AP‐1 to MMP‐9 promoter | AP‐1 | 112 |
| VEGF‐C | Bmi‐1 inhibitor | Pre‐clinical study | Blocking the transcriptional activity of NF‐κB/VEGF‐C | 119 | |
| Resistance to anti‐VEGF | PDGF inhibitor | Pre‐clinical study | Targeting NF‐κB/snail‐dependent PDGF | Snail | 122, 123 |
| EGFR | EGFRvIII inhibitor | Pre‐clinical study | Suppressing the transcriptional activity of NF‐κB, AP‐1, and C/EBP | AP‐1, C/EBP | 125, 126 |
| VEGF, MMP‐9 | Parthenolide derivative | Australian Clinical Trials: ACTRN12616000228482 | Attenuating the NF‐κB, Akt phosphorylation and VEGF and MMP‐9 expression; activating mitochondrial signalling | 128 | |
| MMP‐9, VEGF, urokinase‐type plasminogen activator receptor and urokinase‐type plasminogen activator | Anti‐p65 intrabody construct (pFv/nu) | Pre‐clinical study | Downregulating expression of p65, and NF‐κB‐dependent genes | 131 | |
| CA IX, HIF‐1α, hypoxia, ROS and VEGFR2 | Ketogenic diet | Pre‐clinical study | Reducing the activation of NF‐κB and NF‐κB‐mediated regulators; inhibiting the levels of ROS | HIF‐1α | 132, 133, 134 |
Abbreviations: AP, activator protein; CA IX, carbonic anhydrase IX; EBP, enhancer‐binding protein; EGFR, endothelial growth factor receptor; GROα, growth‐regulated oncogene α; HIF, hypoxia‐inducible factor; IL, interleukin; MAPK, mitogen‐activated protein kinase; MCP, monocyte chemotactic protein; MMP, matrix metalloproteinase; NF‐κB, nuclear factor‐κB; NG, graphite nanoparticles; nGO, graphene oxide nanoplatelets; PDGF, platelet‐derived growth factor; PI3K, phosphatidylinositol 3‐kinase; ROS, reactive oxygen species; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
As early as 1991, thalidomide was shown to be a potent TNF inhibitor that inhibited NF‐κB activation with anti‐inflammatory effects; in 1994, it was demonstrated to inhibit VEGF with antiangiogenic effects. 69 , 70 Investigators found that thalidomide inhibited the proliferation of ECs in vitro without affecting their viability, but did not suppress the proliferation of U251 glioma cells. 71 NF‐κB also controlled the genes related to vascular endothelial growth factor receptor (VEGFR) expression, 72 and the anti‐inflammatory and antiangiogenic effects of thalidomide were regulated to a certain extent by NF‐κB. Thalidomide reduced inflammatory stimulation, including the production of TNF indirectly, and interfered with the transcriptional regulation of NF‐κB in ECs directly for the simultaneous inhibition of glioma angiogenesis (Table 1). The inflammatory suppression and antiangiogenic function offered by thalidomide may be beneficial for glioma patients with severe inflammatory factors infiltration‐dependent angiogenesis, which further highlights the crucial role of inflammation in angiogenesis and regulatory role of NF‐κB.
In numerous types of tumours including gliomas, the interaction between inflammation and tumours has been recognized, and inflammation is considered the ‘seventh sign of cancer’. 73 , 74 Growing evidence shows that TNF is a key mediator of inflammation and tumour growth. Furthermore, the NF‐κB activated by TNF further releases pro‐inflammatory and proangiogenic factors to promote tumour vessel formation and tumour cell survival. Thus, understanding the mechanisms of NF‐κB in the interaction and mutual promotion between inflammation and angiogenesis will provide new ideas for glioma treatment.
2.3. NF‐κB in oxidative stress‐induced glioma angiogenesis
Interactions between inflammation and angiogenesis have been observed in the course of pathological progression. 41 One of the characteristics of the cellular inflammatory process is a respiratory burst, which generates and accumulates a large amount of extracellular reactive oxygen species (ROS), thereby preventing the invasion of pathogens. 75 , 76 However, the excessive accumulation of extracellular ROS leads to an imbalance of aerobic cells and tissues, called ‘oxidative stress’, which is related to ageing and several heart and vascular diseases. 77 , 78 Intracellular and extracellular ROS are involved in the angiogenesis process in many pathophysiological processes. 79 , 80 Intracellular ROS plays a crucial role in VEGF signalling in ECs. 81 In the tumour microenvironment, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase family, plasma membrane‐bound enzymes that generate superoxide, is a major source of ROS. 82
ROS affect the angiogenesis of tumours in numerous ways, the most important of which is the regulation of NF‐κB transcriptional activation. Since NF‐κB facilitates the synthesis of proangiogenic factors including IL‐6 and IL‐8, ROS could promote glioma angiogenesis. 46 , 47 The regulation of the NF‐κB signalling pathway by ROS is very complex and depends on multiple processes. However, the main regulatory mechanism is the phosphorylation and direct oxidation of the NF‐κB subunit. Since antioxidants have been shown to decrease Ser‐276 phosphorylation to inhibit p65 transcriptional activity and oxidation of p50 by ROS suppress its DNA‐binding ability, increased intracellular ROS reduce the p50 subunit activation and increase p65 subunit activation. 11 , 83 , 84 The mutation status of the tumour suppressor gene p53 results in further NF‐κB activation commands. 11 , 85 P53‐mutated tumours tend to show a greater degree of malignancy, with enhanced invasion and reduced sensitivity to apoptotic signals. 86 Mateusz et al stated that graphite nanoparticles and graphene oxide nanoplatelets could reduce intracellular ROS‐induced angiogenesis via the downregulation of NF‐κB‐dependent proangiogenic cytokines including IL‐6, IL‐8, growth‐regulated oncogene α (GROα) and monocyte chemotactic protein 1 (MCP‐1) in a p53wt glioma cell line (U87); however, they had no effect in a p53mut cell line (U118) 84 (Figure 3 and Table 1).
Figure 3.
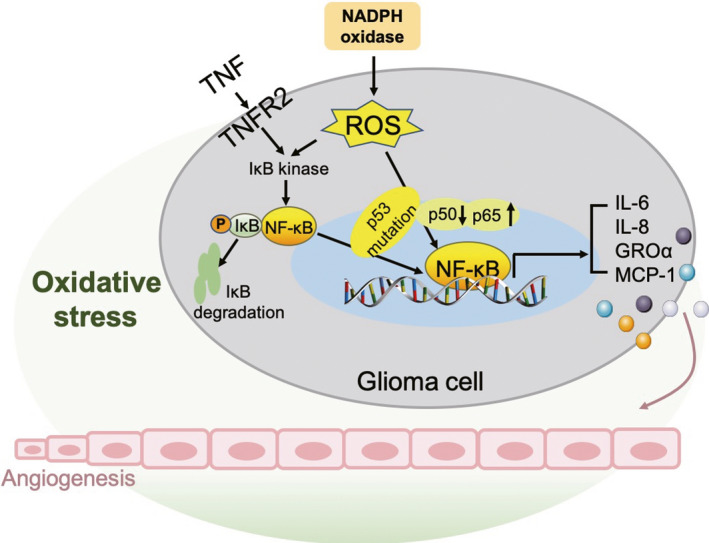
Role of NF‐κB in glioma angiogenesis under a microenvironment of oxidative stress. The ROS produced by NADPH oxidase and TNFR2 mediates the activation of NF‐κB and the main regulatory mechanism of ROS is the phosphorylation and direct oxidation of the NF‐κB subunit (reduce p50 activation and increase p65 activation), and the activation of NF‐κB further depends on the p53 mutation status. The transcriptional activity of NF‐κB for IL‐6, IL‐8, GROα and MCP‐1 promotes glioma angiogenesis. NF‐κB: nuclear factor‐κB; ROS: reactive oxygen species; IL: interleukin; TNFR: tumour necrosis factor receptor; NADPH: nicotinamide adenine dinucleotide phosphate; GRO: growth‐regulated oncogene; MCP: monocyte chemotactic protein
In this regard, oxidative stress‐induced angiogenesis depends on p53 mutation status and NF‐κB regulation, which provides novel strategies in the field of nanoparticle treatment for glioma angiogenesis.
2.4. Caspase in NF‐κB‐dependent glioma angiogenesis
Caspase‐8 was initially identified as participating in death receptor‐induced apoptosis. 87 Apoptosis signalling is usually absent in cancer, and caspase‐8 expression is also suppressed. 88 , 89 However, caspase‐8 shows high expression in glioma and may be associated with poorer prognoses. In glioma models, caspase‐8 could facilitate the expression of NF‐κB‐dependent proangiogenic cytokines and tumour promoters. 90 Further, it has been confirmed that it exerts growth‐promoting effects in several conditions, such as fibrosis, 91 wound healing, tissue regeneration 92 and tumour reunion. 93 Feng et al found that dying glioma cells, following radiation, built a proangiogenic microenvironment by the caspase 3‐dependent NF‐κB/COX‐2/PGE axis. 94 These results demonstrate that certain cancers such as glioma may reverse caspase‐8 or the caspase‐3 pro‐apoptotic function that is dependent on NF‐κB, leading to the promotion of blood vessel formation (Figure 4).
Figure 4.
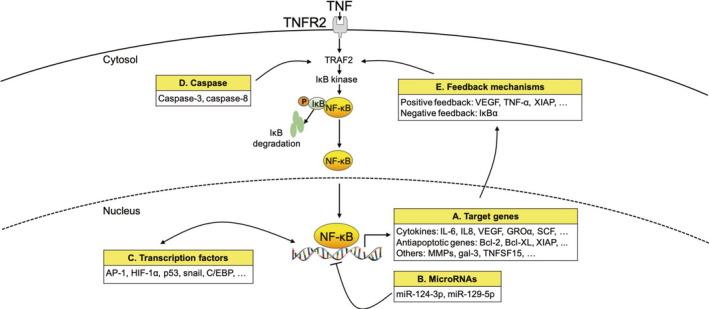
Crosstalk of the NF‐κB pathway involved in glioma angiogenesis with other signalling processes. (A) Target genes of NF‐κB associated with glioma angiogenesis include cytokines such as IL‐6, IL8, VEGF, GROα and SCF; antiapoptotic genes such as Bcl‐2, Bcl‐XL and XIAP; and other genes such as MMP, gal‐3 and TNFSF15. (B) MiR‐124‐3p and miR‐129‐5p block the NF‐κB activation pathways. (C) Numerus transcription factors such as AP‐1, HIF‐1α, p53, snail and C/EBP affect the NF‐κB activation pathways or directly activate the target genes of NF‐κB. (D) Caspase‐3 and caspase‐8 enhance the transcriptional activity of NF‐κB. (E) Positive feedback target genes of NF‐κB such as VEGF, TNF‐α and XIAP further activate the NF‐κB pathway. A significant negative feedback molecule is IκBα. GRO: growth‐regulated oncogene; SCF: stem cell factor; IL: interleukin; TNF: tumour necrosis factor; NF‐κB: nuclear factor‐κB; VEGF: vascular endothelial growth factor; XIAP: X‐chromosome‐linked inhibitor of apoptosis protein; HIF: hypoxia‐inducible factor, MMP: matrix metalloproteinase; TNFSF: tumour necrosis factor super family; Gal‐3: galectin‐3; AP: activator protein; EBP: enhancer‐binding protein
2.5. MicroRNAs in NF‐κB‐dependent glioma angiogenesis
MicroRNAs (miRNAs) are a type of small, non‐coding RNA molecules that participate in cell differentiation, maturation and signal transduction by imperfectly base‐pairing with complementary sites of their target genes, leading to target mRNA degradation or translation inhibition. 95 Zhang et al found that miR‐124‐3p was reduced in human glioma, which led to the negative regulation of neuropilin‐1 (NRP‐1). NRP‐1 is expressed as a multifunctional receptor in various human tumours, including gliomas, and the degree of expression is related to the clinicopathological characteristics of the host tumours. 96 The expression of p‐PI3K, p‐Akt and p‐p65 (NF‐κB) was markedly reduced when miR‐124‐3p was overexpressed in glioma cells compared to the control group, and the total protein levels of PI3K, Akt and p65 were unchanged. These authors also found that the overexpression of miR‐124‐3p led to the inhibition of glioma development and blood vessel formation in vivo, in a glioma‐bearing patient‐derived xenograft (PDX) model. Therefore, the overexpression of miR‐124‐3p significantly inhibited glioma cell growth and angiogenesis by targeting the PI3K/Akt/NF‐κB pathway in both in vitro and in vivo PDX models. 97
Similarly, other investigators suggested that the overexpression of miR‐129‐5p in glioma cells medium culturing HBMVEC showed fewer capillaries, branches and shorter tube lengths, and that the overexpression of miR‐129‐5p in glioma cells was associated with lower VEGF expression than that in the control group. 98 MiR‐129‐5p overexpression obviously reduced the MMP‐2 and MMP‐9 protein levels and the luciferase activities of NF‐κB, indicating that miR‐129‐5p blocked the NF‐κB pathways to suppress glioma angiogenesis and growth (Figure 4).
There is an urgent need to investigate the effects of dysregulated miRNAs on NF‐κB‐induced angiogenesis in glioma, to gain a better understanding of the biological basis of the occurrence and development of glioma angiogenesis.
2.6. Target NF‐κB‐dependent angiogenesis for glioma therapy
2.6.1. Matrix metalloproteinase
Angiogenesis is related to invasion and is used for glioma grading. 99 A recent study showed similar molecular mechanisms for angiogenesis and invasion. 100 The new formation of blood vessels can be considered to an invasive course in which activated ECs proliferate, adhere to the ECM molecules and migrate. 101 MMPs are involved in angiogenesis, invasion and ECM degradation for the promotion of tumour development. 102 , 103 , 104 Among MMPs, MMP‐2 and MMP‐9 have been indicated as having an upregulated expression in glioma. The upregulation and activation of MMP‐2 in association with HIF‐1α expression enhance tumour cell infiltration and blood‐brain barrier permeability. 105 MMP‐1 and MMP‐3 levels also increase as the tumour grade increases. 106 , 107 It is also to be noted that the NF‐κB binding sites in the MMPs promoter regions are closely related to tumour cell invasion and angiogenesis. 108 Therefore, effective MMP inhibitors may show promise for use in therapeutic strategies for glioma angiogenesis (Table 1).
The use of tumour treating field (TTF) therapy, entailing an alternating electric field with an intermediate‐frequency (100‐300 kHz) for tumour treatment, led to glioma suppression. 109 It has been found that TTF suppresses the metastatic ability of glioma by the downregulation of the NF‐κB, MAPK and PI3K/Akt signalling pathways. Second, TTF application decreases the levels of VEGF, HIF1α, MMP‐2 and MMP‐9 via the suppression of NF‐κB, thus suppressing glioma angiogenesis. These results suggest that TTF is an effective MMP and NF‐κB‐related treatment for glioma invasion and angiogenesis. 110
Glycitein, a bacterial metabolite of the isoflavone glycitein, inhibits the expression of MMP‐3 and MMP‐9 in phorbol myristate acetate (PMA)‐stimulated U87MG glioma cells. Furthermore, glycitein suppresses the transcriptional activity of NF‐κB and AP‐1 for MMP‐3 and MMP‐9. 111 A previous study focused on mangiferin, a natural polyphenol compound isolated from Anemarrhena asphodeloides, which could be widely found in several higher plants including Mangifera indica L. 112 Mangiferin specifically suppresses MMP‐9 mRNA and protein expression in PMA‐stimulated U87MG, U373MG and CRT‐MG glioma cells. Further mechanistic studies indicated that mangiferin inhibits MMP‐9 by the inhibition of the binding of NF‐κB and AP‐1 to MMP‐9 promoters to block glioma invasion and angiogenesis. 113
MMPs have a vital role in the invasion and angiogenesis of malignant glioma. The inhibition of NF‐κB DNA‐binding activity and the interference of NF‐κB‐activated signal cascade leading to the inhibition of MMPs gene expression may be a promising therapeutic strategy for the blocking of glioma angiogenesis.
2.6.2. Vascular endothelial growth factor
ECs are stimulated by VEGF or adhere to ECM molecules, leading to the augmentation of anti‐apoptotic genes via the PI3K/Akt or NF‐κB signalling pathways. 18 , 19 Therefore, anti‐angiogenesis therapy, that is, the inhibition of tumour‐associated ECs, has become a major strategy for tumour treatment. 88 VEGF blocking and VEGF receptor inhibition have been used as anti‐angiogenesis therapies for glioma; however, their effects are weak. 114 , 115 B cell–specific moloney murine leukemia virus integration site 1 (Bmi‐1) is expressed in numerous cancers types, such as breast, lung and ovarian cancers, and could serve as an oncogene. 116 , 117 , 118 Jiang et al found that the upregulation of Bmi‐1 induced the expression of NF‐κB target genes by the activation of NF‐κB‐induced VEGF‐C, which plays a major role in angiogenesis, thus promoting glioma angiogenesis in vitro and in vivo. It is worth noting that the angiogenesis and VEGF‐C stimulated by the upregulation of Bmi‐1 were significantly blocked after the inhibition of NF‐κB activity. 119 VEGF‐C plays an important role in angiogenesis and EC growth and survival. It is upregulated in glioma and involved in tumour progression and prognoses 120 (Table 1). These findings indicate that Bmi‐1 could promote angiogenesis in glioma via NF‐κB/VEGF‐C, further suggesting that NF‐κB/VEGF‐C‐dependent Bmi‐1 may represent a novel therapeutic target for antiangiogenic strategies aimed at glioma.
2.6.3. Platelet‐derived growth factor (PDGF)
PDGF, a proangiogenic factor, is the major mitogen for many mesenchymal‐derived cell types, such as fibroblasts and pericytes. 121 PDGF‐mediated endothelial‐mesenchymal transformation (EMT) reduced the expression of VEGFR‐2 in ECs. With the loss of VEGFR‐2 expression, ECs convert to a VEGF‐independent state for the maintenance of their growth and survival in glioma, leading to the resistance of ECs to anti‐VEGF treatment. 122 The expression of snail, a pivotal downstream regulator of EMT in the glioma environment, is regulated by NF‐κB. 123 In addition, PDGF induces NF‐κB‐dependent snail expression, resulting in resistance to anti‐VEGF treatment with the downregulation of VEGFR‐2. The inhibition of PDGF receptor sensitized VEGF/VEGFR‐2 targeted therapy in glioma‐bearing mice model (Table 1). Collectively, targeting NF‐κB/snail‐dependent PDGFs may serve as a promising strategy for cases with resistance to anti‐VEGF in glioma.
2.6.4. Epidermal growth factor receptor (EGFR)
The amplification of the EGFR gene occurs in almost half of all glioblastoma cases and is related to gene rearrangement. 124 The rearrangement is often related to activating mutations such as the loss of exons 2‐7 (EGFRvIII or EGFRde2‐7). EGFRvIII overexpression in human glioma cells or primary mouse astrocytes can lead to the significantly faster formation of tumours in animal models by intracranial or subcutaneous injection than in the control group, demonstrating that EGFRvIII enhances the carcinogenic capacity. 125 Bonavia et al suggested that EGFRvIII facilitated high levels of IL‐8 expression in glioma clinical samples and cell lines mediated by NF‐κB, AP‐1 and C/EBP. Additionally, the knocking down of NF‐κB suppressed the EGFRvIII overexpressing glioma cell bearing‐tumour growth in vivo with the inhibition of angiogenesis, indicating the crucial role of NF‐κB in EGFRvIII enhancing the carcinogenic capacity 126 (Table 1). In conclusion, EGFRvIII facilitates glioma angiogenesis and growth by the NF‐κB pathway. Thus, the inhibition of EGFR gene amplification and kinase activating mutants or the targeting of a unique EGFR epitope such as EGFRvIII with monoclonal antibodies for the inhibition of NF‐κB as well as tumour angiogenesis and growth may be rational strategies for the development of glioma therapy.
2.6.5. Other NF‐κB ‐targeted therapeutic strategies for glioma angiogenesis
Parthenolide reportedly has the potential to cross the blood‐brain barrier and alleviate brain inflammation. 127 Nakabayashi et al found that parthenolide inhibited U87MG glioma cell proliferation and invasion and induced angiogenesis in a dose‐dependent manner in vitro. Mechanically, parthenolide attenuates NF‐κB transcriptional activity and the expression of NF‐κB targets, VEGF and MMP‐9, in glioma cells. Moreover, parthenolide suppresses Akt phosphorylation and activated mitochondrial signalling, demonstrating that it inhibits angiogenesis by the inhibition of NF‐κB, and further suppresses glioma growth by the inhibition of the Akt signal and activation of the apoptosis signal. 128
The transcriptional activity of NF‐κB can be induced by IκB cytoplasmic segregation and RelA/p65 phosphorylation. 129 , 130 Using the phage display technique to frame a single‐chain fragment of anti‐p65 antibody variable region (scFv), investigators cloned the scFv‐encoding sequence into the mammalian nuclear‐targeting vector, pCMV/myc/nuc, to fabricate an anti‐p65 intrabody construct (pFv/nu). U251 and U87 glioma cells transfected with pFv/nu dramatically suppressed the expression of p65, and NF‐κB‐dependent genes such as MMP‐9, VEGF, urokinase‐type plasminogen activator receptor and urokinase‐type plasminogen activator. Additionally, U251 and U87 glioma cells transfected with pFv/nu‐bearing intracranial tumours were almost restrained. 131 Thus, inhibition of the transcriptional activity of NF‐κB by nuclear‐targeting intrabody could serve as a promising antiangiogenic strategy for glioma.
The ketogenic diet (KD) is a novel high‐fat, low‐carbohydrate, protein‐rich diet that targets tumour metabolism and has been used in non‐drug therapy for intractable epilepsy. Of note, first, mouse glioma models fed a KD showed higher survival values than those on a normal diet. 132 Glioma models fed a KD at will demonstrated observable reductions in NF‐κB activation and reductions in the levels of NF‐κB‐mediated regulators in the hypoxic context, such as carbonic anhydrase IX (CA IX) and HIF‐1α. 36 Second, the KD inhibits the levels of ROS, which boosts angiogenesis by the activation of NF‐κB transcription in tumours. 25 Third, the KD blocks the expression of VEGFR2, the major receptor involved in tumour angiogenesis regulation 133 , 134 (Table 1). KD therapy that targets tumour metabolism and represses the NF‐κB‐mediated hypoxic response may provide a low‐toxic, easy‐to‐implement method for glioma aimed at angiogenesis inhibition.
3. CLINICAL RELEVANCE AND FUTURE PERSPECTIVES
Bevacizumab, a recombinant humanized anti‐VEGF monoclonal antibody, is the only FDA‐approved anti‐glioblastoma angiogenesis drug. 135 Although it has been used in clinical treatment, it usually causes serious adverse reactions, and its clinical efficacy remains controversial. 136 Bevacizumab increases the hypoxic area and boosts the rate of MMP‐2 activation, resulting in a more invasive, treatment‐resistant glioma state. 137 Whereas the inhibition of NF‐κB transcriptional activation reduces hypoxia‐induced angiogenesis and the levels of NF‐κB‐mediated regulators in hypoxic context. In this regard, if the beneficial effects of bevacizumab can be mimicked by the inhibition of the transcriptional activity of NF‐κB in vivo, it could provide a low‐toxic method for glioma to block angiogenesis even with the inhibition of the invasive potential.
Structurally and functionally altered glioma blood vessels may impede the delivery of therapeutic agents, promote the outward leakage of tumour cells, and aid in the rapid infiltration of numerous inflammatory cells. 138 Chronic inflammation leads to oedema development around the tumour, which aggravates the pathological progression of glioma. Although dexamethasone is used to treat glioma peripheral inflammation and oedema, its severe side effects, such as electrolyte disorders, osteoporosis, mental excitement, elevated blood pressure, menstrual disorders and weight gain, distinctly lower the quality of life and even interfere with the effectiveness of adjuvant chemoradiotherapy in glioma settings. 139 , 140 Based on the current literature, it is clear that the key role of NF‐κB is the interaction and mutual promotion between inflammation and angiogenesis that it offers. Thus, the inhibition of the transcriptional activity of NF‐κB may restrain the continuous recruitment and permeation of inflammatory cells, which may inhibit angiogenesis as well as peripheral inflammation and oedema development. NF‐κB is associated with angiogenetic signal transduction pathways that regulate hypoxia, oxidative stress and the production of pro‐inflammatory cytokines, proangiogenic cytokines and MMPs. Therefore, targeting NF‐κB could be a potential method for the simultaneous targeting of multiple glioma features.
Considering the wide range of the regulatory responses covered by NF‐κB, which also mediates tumour cells infiltration, proliferation and migration besides angiogenesis, the more reasonable disposal of specific subsets of NF‐κB responses could show greater efficacy in glioma treatment, in terms of angiogenesis inhibition. In addition, accumulating evidence indicates that NF‐κB participates in numerous targeted therapies, including those with MMPs, VEGF and PDGF. Further clinical study of the molecular mechanisms between angiogenesis and NF‐κB in glioma is warranted to broaden the options of targeted therapies for the prevention of NF‐κB‐dependent angiogenesis.
4. CONCLUSIONS
Angiogenesis in glioma accelerates tumour growth and increases the degree of malignancy. NF‐κB plays a pivotal role in the growth and progression of glioma angiogenesis. Interference with the transcriptional activity of NF‐κB that leads to alterations in the proangiogenic context and the inhibition of proangiogenic gene expression may be promising therapeutic strategies aimed at glioma angiogenesis blocking.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest.
AUTHOR CONTRIBUTION
JJT and YLF drafted the manuscript. DFH, XWT, HFJ, XG, XMW, WMH and WW revised the manuscript.
Tu J, Fang Y, Han D, et al. Activation of nuclear factor‐κB in the angiogenesis of glioma: Insights into the associated molecular mechanisms and targeted therapies. Cell Prolif.2021;54:e12929 10.1111/cpr.12929
Jiajie Tu and Yilong Fang authors equally contribute to this paper.
Funding informationThis study was supported by the National Natural Science Foundation of China (31900616, 81673444) and Natural Science Foundation of Anhui Province for young scholars (1908085QH379).
Contributor Information
Wenming Hong, Email: 524594636@qq.com.
Wei Wei, Email: wwei@ahmu.edu.cn.
DATA AVAILABILITY STATEMENT
No new data generated.
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803‐820. [DOI] [PubMed] [Google Scholar]
- 2. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492‐507. [DOI] [PubMed] [Google Scholar]
- 3. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Ann Surg. 1966;164(3):491‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wong ML, Prawira A, Kaye AH, Hovens CM. Tumour angiogenesis: its mechanism and therapeutic implications in malignant gliomas. J Clin Neurosci. 2009;16(9):1119‐1130. [DOI] [PubMed] [Google Scholar]
- 6. Aggarwal BB. Signalling pathways of the TNF superfamily: a double‐edged sword. Nat Rev Immunol. 2003;3(9):745‐756. [DOI] [PubMed] [Google Scholar]
- 7. Ham B, Fernandez MC, D'Costa Z, Brodt P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016;11(1):1‐27. [PMC free article] [PubMed] [Google Scholar]
- 8. Shivakumar P, Mizuochi T, Mourya R, et al. Preferential TNFalpha signaling via TNFR2 regulates epithelial injury and duct obstruction in experimental biliary atresia. JCI Insight. 2017;2(5):e88747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2‐mediated activation of NF‐kappa B by TNF receptor 2 and CD40. Science. 1995;269(5229):1424‐1427. [DOI] [PubMed] [Google Scholar]
- 10. Fotin‐Mleczek M, Henkler F, Hausser A, et al. Tumor necrosis factor receptor‐associated factor (TRAF) 1 regulates CD40‐induced TRAF2‐mediated NF‐kappaB activation. J Biol Chem. 2004;279(1):677‐685. [DOI] [PubMed] [Google Scholar]
- 11. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF‐kappaB signaling. Cell Res. 2011;21(1):103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Madrid LV, Baldwin AS Jr. Regulation of NF‐kappaB by oncoproteins and tumor suppressor proteins. Methods Mol Biol. 2003;223:523‐532. [DOI] [PubMed] [Google Scholar]
- 13. Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84(8):941‐951. [DOI] [PubMed] [Google Scholar]
- 14. Xie TX, Xia Z, Zhang N, Gong W, Huang S. Constitutive NF‐kappaB activity regulates the expression of VEGF and IL‐8 and tumor angiogenesis of human glioblastoma. Oncol Rep. 2010;23(3):725‐732. [PubMed] [Google Scholar]
- 15. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen consumption and tissue oxygenation of human tumors. Adv Exp Med Biol. 1990;277:895‐905. [DOI] [PubMed] [Google Scholar]
- 16. Soini Y, Paakko P, Lehto VP. Histopathological evaluation of apoptosis in cancer. Am J Pathol. 1998;153(4):1041‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brat DJ, Van Meir EG. Vaso‐occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest. 2004;84(4):397‐405. [DOI] [PubMed] [Google Scholar]
- 18. Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3'‐kinase/Akt signal transduction pathway. Requirement for Flk‐1/KDR activation. J Biol Chem. 1998;273(46):30336‐30343. [DOI] [PubMed] [Google Scholar]
- 19. Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF‐kappaB mediates alphavbeta3 integrin‐induced endothelial cell survival. J Cell Biol. 1998;141(4):1083‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Madrid LV, Mayo MW, Reuther JY, Baldwin AS Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF‐kappa B through utilization of the Ikappa B kinase and activation of the mitogen‐activated protein kinase p38. J Biol Chem. 2001;276(22):18934‐18940. [DOI] [PubMed] [Google Scholar]
- 21. Plate KH, Breier G, Millauer B, Ullrich A, Risau W. Up‐regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. 1993;53(23):5822‐5827. [PubMed] [Google Scholar]
- 22. Robaye B, Mosselmans R, Fiers W, Dumont JE, Galand P. Tumor necrosis factor induces apoptosis (programmed cell death) in normal endothelial cells in vitro. Am J Pathol. 1991;138(2):447‐453. [PMC free article] [PubMed] [Google Scholar]
- 23. Roessler K, Suchanek G, Breitschopf H, et al. Detection of tumor necrosis factor‐alpha protein and messenger RNA in human glial brain tumors: comparison of immunohistochemistry with in situ hybridization using molecular probes. J Neurosurg. 1995;83(2):291‐297. [DOI] [PubMed] [Google Scholar]
- 24. Zen K, Karsan A, Stempien‐Otero A, et al. NF‐kappaB activation is required for human endothelial survival during exposure to tumor necrosis factor‐alpha but not to interleukin‐1beta or lipopolysaccharide. J Biol Chem. 1999;274(40):28808‐28815. [DOI] [PubMed] [Google Scholar]
- 25. Ueda Y, Nakagawa T, Kubota T, Ido K, Sato K. Glioma cells under hypoxic conditions block the brain microvascular endothelial cell death induced by serum starvation. J Neurochem. 2005;95(1):99‐110. [DOI] [PubMed] [Google Scholar]
- 26. Debray C, Vereecken P, Belot N, et al. Multifaceted role of galectin‐3 on human glioblastoma cell motility. Biochem Biophys Res Commun. 2004;325(4):1393‐1398. [DOI] [PubMed] [Google Scholar]
- 27. Nangia‐Makker P, Honjo Y, Sarvis R, et al. Galectin‐3 induces endothelial cell morphogenesis and angiogenesis. Am J Pathol. 2000;156(3):899‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshii T, Fukumori T, Honjo Y, Inohara H, Kim HR, Raz A. Galectin‐3 phosphorylation is required for its anti‐apoptotic function and cell cycle arrest. J Biol Chem. 2002;277(9):6852‐6857. [DOI] [PubMed] [Google Scholar]
- 29. Greijer AE, van der Groep P, Kemming D, et al. Up‐regulation of gene expression by hypoxia is mediated predominantly by hypoxia‐inducible factor 1 (HIF‐1). J Pathol. 2005;206(3):291‐304. [DOI] [PubMed] [Google Scholar]
- 30. Zeng Y, Danielson KG, Albert TJ, Shapiro IM, Risbud MV. HIF‐1 alpha is a regulator of galectin‐3 expression in the intervertebral disc. J Bone Miner Res. 2007;22(12):1851‐1861. [DOI] [PubMed] [Google Scholar]
- 31. Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia‐inducible‐factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7(2):134‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olbryt M, Jarzab M, Jazowiecka‐Rakus J, Simek K, Szala S, Sochanik A. Gene expression profile of B 16(F10) murine melanoma cells exposed to hypoxic conditions in vitro. Gene Expr. 2006;13(3):191‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu R, Jin H, Zhou S, Yang P, Li X. Proteomic analysis of hypoxia‐induced responses in the syncytialization of human placental cell line BeWo. Placenta. 2007;28(5–6):399‐407. [DOI] [PubMed] [Google Scholar]
- 34. Machado CM, Andrade LN, Teixeira VR, et al. Galectin‐3 disruption impaired tumoral angiogenesis by reducing VEGF secretion from TGFbeta1‐induced macrophages. Cancer Med. 2014;3(2):201‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ikemori RY, Machado CM, Furuzawa KM, et al. Galectin‐3 up‐regulation in hypoxic and nutrient deprived microenvironments promotes cell survival. PLoS One. 2014;9(11):e111592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nam SY, Ko YS, Jung J, et al. A hypoxia‐dependent upregulation of hypoxia‐inducible factor‐1 by nuclear factor‐kappaB promotes gastric tumour growth and angiogenesis. Br J Cancer. 2011;104(1):166‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Melvin A, Mudie S, Rocha S. Further insights into the mechanism of hypoxia‐induced NFkappaB. [corrected]. Cell Cycle. 2011;10(6):879‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weiler M, Blaes J, Pusch S, et al. mTOR target NDRG1 confers MGMT‐dependent resistance to alkylating chemotherapy. Proc Natl Acad Sci U S A. 2014;111(1):409‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson TA, Karajannis MA, Harter DH. Glioblastoma multiforme: State of the art and future therapeutics. Surg Neurol Int. 2014;5:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Broggini T, Wustner M, Harms C, et al. NDRG1 overexpressing gliomas are characterized by reduced tumor vascularization and resistance to antiangiogenic treatment. Cancer Lett. 2016;380(2):568‐576. [DOI] [PubMed] [Google Scholar]
- 41. Kim YW, West XZ, Byzova TV. Inflammation and oxidative stress in angiogenesis and vascular disease. J Mol Med (Berl). 2013;91(3):323‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor‐associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549‐555. [DOI] [PubMed] [Google Scholar]
- 43. Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8(4):1008‐1013. [PubMed] [Google Scholar]
- 44. Cheng N, Chen J. Tumor necrosis factor‐alpha induction of endothelial ephrin A1 expression is mediated by a p38 MAPK‐ and SAPK/JNK‐dependent but nuclear factor‐kappa B‐independent mechanism. J Biol Chem. 2001;276(17):13771‐13777. [DOI] [PubMed] [Google Scholar]
- 45. Yamaguchi S, Tanabe K, Takai S, et al. Involvement of Rho‐kinase in tumor necrosis factor‐alpha‐induced interleukin‐6 release from C6 glioma cells. Neurochem Int. 2009;55(6):438‐445. [DOI] [PubMed] [Google Scholar]
- 46. Libermann TA, Baltimore D. Activation of interleukin‐6 gene expression through the NF‐kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327‐2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roebuck KA. Regulation of interleukin‐8 gene expression. J Interferon Cytokine Res. 1999;19(5):429‐438. [DOI] [PubMed] [Google Scholar]
- 48. Silwedel C, Speer CP, Haarmann A, et al. Ureaplasma species modulate cytokine and chemokine responses in human brain microvascular endothelial cells. Int J Mol Sci. 2019;20(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Salcedo R, Ponce ML, Young HA, et al. Human endothelial cells express CCR2 and respond to MCP‐1: direct role of MCP‐1 in angiogenesis and tumor progression. Blood. 2000;96(1):34‐40. [PubMed] [Google Scholar]
- 50. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL‐6 in the tumor microenvironment. Semin Immunol. 2014;26(1):38‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kishimoto T. Interleukin‐6: from basic science to medicine–40 years in immunology. Annu Rev Immunol. 2005;23:1‐21. [DOI] [PubMed] [Google Scholar]
- 52. Hart PH, Vitti GF, Burgess DR, Whitty GA, Piccoli DS, Hamilton JA. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proc Natl Acad Sci U S A. 1989;86(10):3803‐3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee IY, Kim J, Ko EM, Jeoung EJ, Kwon YG, Choe J. Interleukin‐4 inhibits the vascular endothelial growth factor‐ and basic fibroblast growth factor‐induced angiogenesis in vitro. Mol Cells. 2002;14(1):115‐121. [PubMed] [Google Scholar]
- 54. Wong HL, Lotze MT, Wahl LM, Wahl SM. Administration of recombinant IL‐4 to humans regulates gene expression, phenotype, and function in circulating monocytes. J Immunol. 1992;148(7):2118‐2125. [PubMed] [Google Scholar]
- 55. Kimura YN, Watari K, Fotovati A, et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007;98(12):2009‐2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro‐environment in tumor progression: the role of tumor‐associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 57. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nijaguna MB, Patil V, Urbach S, et al. Glioblastoma‐derived Macrophage Colony‐stimulating Factor (MCSF) Induces Microglial Release of Insulin‐like Growth Factor‐binding Protein 1 (IGFBP1) to Promote Angiogenesis. J Biol Chem. 2015;290(38):23401‐23415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hong TM, Teng LJ, Shun CT, Peng MC, Tsai JC. Induced interleukin‐8 expression in gliomas by tumor‐associated macrophages. J Neurooncol. 2009;93(3):289‐301. [DOI] [PubMed] [Google Scholar]
- 60. Bhattacharjee B, Renzette N, Kowalik TF. Genetic analysis of cytomegalovirus in malignant gliomas. J Virol. 2012;86(12):6815‐6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. Significant association of multiple human cytomegalovirus genomic Loci with glioblastoma multiforme samples. J Virol. 2012;86(2):854‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hwang J, Kalejta RF. Proteasome‐dependent, ubiquitin‐independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus‐infected cells. Virology. 2007;367(2):334‐338. [DOI] [PubMed] [Google Scholar]
- 63. Sun L, Hui AM, Su Q, et al. Neuronal and glioma‐derived stem cell factor induces angiogenesis within the brain. Cancer Cell. 2006;9(4):287‐300. [DOI] [PubMed] [Google Scholar]
- 64. Matlaf LA, Harkins LE, Bezrookove V, Cobbs CS, Soroceanu L. Cytomegalovirus pp71 protein is expressed in human glioblastoma and promotes pro‐angiogenic signaling by activation of stem cell factor. PLoS One. 2013;8(7):e68176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Begum Z, Ghosh A, Sarkar S, et al. Documentation of immune profile of microglia through cell surface marker study in glioma model primed by a novel cell surface glycopeptide T11TS/SLFA‐3. Glycoconj J. 2004;20(9):515‐523. [DOI] [PubMed] [Google Scholar]
- 66. Acharya S, Chatterjee S, Kumar P, Bhattacharjee M, Chaudhuri S, Chaudhuri S. Induction of G1 arrest in glioma cells by T11TS is associated with upregulation of Cip1/Kip1 and concurrent downregulation of cyclin D (1 and 3). Anticancer Drugs. 2010;21(1):53‐64. [DOI] [PubMed] [Google Scholar]
- 67. Bhattacharya D, Singh MK, Chaudhuri S, Acharya S, Basu AK, Chaudhuri S. T11TS impedes glioma angiogenesis by inhibiting VEGF signaling and pro‐survival PI3K/Akt/eNOS pathway with concomitant upregulation of PTEN in brain endothelial cells. J Neurooncol. 2013;113(1):13‐25. [DOI] [PubMed] [Google Scholar]
- 68. Singh MK, Chaudhuri S, Bhattacharya D, Kumar P, Datta A, Chaudhuri S. T11 target structure induced modulations of the pro‐inflammatory and anti‐infammatorycytokine expressions in experimental animals for glioma abrogation. Int Immunopharmacol. 2015;24(2):198‐207. [DOI] [PubMed] [Google Scholar]
- 69. Ridings JE. The thalidomide disaster, lessons from the past. Methods Mol Biol. 2013;947:575‐586. [DOI] [PubMed] [Google Scholar]
- 70. D'Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91(9):4082‐4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moreira AL, Friedlander DR, Shif B, Kaplan G, Zagzag D. Thalidomide and a thalidomide analogue inhibit endothelial cell proliferation in vitro. J Neurooncol. 1999;43(2):109‐114. [DOI] [PubMed] [Google Scholar]
- 72. Shukla K, Sonowal H, Saxena A, Ramana KV. Didymin by suppressing NF‐kappaB activation prevents VEGF‐induced angiogenesis in vitro and in vivo. Vascul Pharmacol. 2019;115:18‐25. [DOI] [PubMed] [Google Scholar]
- 73. Mantovani A. Cancer: Inflaming metastasis. Nature. 2009;457(7225):36‐37. [DOI] [PubMed] [Google Scholar]
- 74. Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer‐related inflammation and treatment effectiveness. Lancet Oncol. 2014;15(11):e493‐503. [DOI] [PubMed] [Google Scholar]
- 75. Filippin LI, Vercelino R, Marroni NP, Xavier RM. Redox signalling and the inflammatory response in rheumatoid arthritis. Clin Exp Immunol. 2008;152(3):415‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bulua AC, Simon A, Maddipati R, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med. 2011;208(3):519‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49(11):1603‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Amanso A, Lyle AN, Griendling KK. NADPH oxidases and measurement of reactive oxygen species. Methods Mol Biol. 2017;1527:219‐232. [DOI] [PubMed] [Google Scholar]
- 79. Prieto‐Bermejo R, Hernandez‐Hernandez A. The importance of NADPH oxidases and redox signaling in angiogenesis. Antioxidants (Basel). 2017;6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ushio‐Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266(1):37‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Colavitti R, Pani G, Bedogni B, et al. Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor‐2/KDR. J Biol Chem. 2002;277(5):3101‐3108. [DOI] [PubMed] [Google Scholar]
- 82. Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47(9):1239‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha‐induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120(5):649‐661. [DOI] [PubMed] [Google Scholar]
- 84. Wierzbicki M, Sawosz E, Strojny B, Jaworski S, Grodzik M, Chwalibog A. NF‐kappaB‐related decrease of glioma angiogenic potential by graphite nanoparticles and graphene oxide nanoplatelets. Sci Rep. 2018;8(1):14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Weisz L, Damalas A, Liontos M, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67(6):2396‐2401. [DOI] [PubMed] [Google Scholar]
- 86. Oren M, Rotter V. Mutant p53 gain‐of‐function in cancer. Cold Spring Harb Perspect Biol. 2010;2(2):a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Muzio M, Chinnaiyan AM, Kischkel FC, et al. FLICE, a novel FADD‐homologous ICE/CED‐3‐like protease, is recruited to the CD95 (Fas/APO‐1) death–inducing signaling complex. Cell. 1996;85(6):817‐827. [DOI] [PubMed] [Google Scholar]
- 88. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 89. Stupack DG, Teitz T, Potter MD, et al. Potentiation of neuroblastoma metastasis by loss of caspase‐8. Nature. 2006;439(7072):95‐99. [DOI] [PubMed] [Google Scholar]
- 90. Fianco G, Mongiardi MP, Levi A, et al. Caspase‐8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife. 2017;6:e22593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Laplante P, Sirois I, Raymond MA, et al. Caspase‐3‐mediated secretion of connective tissue growth factor by apoptotic endothelial cells promotes fibrosis. Cell Death Differ. 2010;17(2):291‐303. [DOI] [PubMed] [Google Scholar]
- 92. Li F, Huang Q, Chen J, et al. Apoptotic cells activate the "lphoenix rising" pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3(110):ra13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Huang Q, Li F, Liu X, et al. Caspase 3‐mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17(7):860‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Feng X, Yu Y, He S, et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017;385:12‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281‐297. [DOI] [PubMed] [Google Scholar]
- 96. Osada H, Tokunaga T, Nishi M, et al. Overexpression of the neuropilin 1 (NRP1) gene correlated with poor prognosis in human glioma. Anticancer Res. 2004;24(2B):547‐552. [PubMed] [Google Scholar]
- 97. Zhang G, Chen L, Khan AA, et al. miRNA‐124‐3p/neuropilin‐1(NRP‐1) axis plays an important role in mediating glioblastoma growth and angiogenesis. Int J Cancer. 2018;143(3):635‐644. [DOI] [PubMed] [Google Scholar]
- 98. Zeng A, Yin J, Li Y, et al. miR‐129‐5p targets Wnt5a to block PKC/ERK/NF‐kappaB and JNK pathways in glioblastoma. Cell Death Dis. 2018;9(3):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995;15(3):211‐221. [DOI] [PubMed] [Google Scholar]
- 100. Skobe M, Rockwell P, Goldstein N, Vosseler S, Fusenig NE. Halting angiogenesis suppresses carcinoma cell invasion. Nat Med. 1997;3(11):1222‐1227. [DOI] [PubMed] [Google Scholar]
- 101. Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267(16):10931‐10934. [PubMed] [Google Scholar]
- 102. Haas TL, Milkiewicz M, Davis SJ, et al. Matrix metalloproteinase activity is required for activity‐induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2000;279(4):H1540‐1547. [DOI] [PubMed] [Google Scholar]
- 103. Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. J Natl Cancer Inst. 1997;89(17):1260‐1270. [DOI] [PubMed] [Google Scholar]
- 104. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161‐174. [DOI] [PubMed] [Google Scholar]
- 105. Ishihara H, Kubota H, Lindberg RL, et al. Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol. 2008;67(5):435‐448. [DOI] [PubMed] [Google Scholar]
- 106. Rao JS, Steck PA, Tofilon P, et al. Role of plasminogen activator and of 92‐KDa type IV collagenase in glioblastoma invasion using an in vitro matrigel model. J Neurooncol. 1994;18(2):129‐138. [DOI] [PubMed] [Google Scholar]
- 107. McCready J, Broaddus WC, Sykes V, Fillmore HL. Association of a single nucleotide polymorphism in the matrix metalloproteinase‐1 promoter with glioblastoma. Int J Cancer. 2005;117(5):781‐785. [DOI] [PubMed] [Google Scholar]
- 108. Eberhardt W, Huwiler A, Beck KF, Walpen S, Pfeilschifter J. Amplification of IL‐1 beta‐induced matrix metalloproteinase‐9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF‐kappa B and activating protein‐1 and involves activation of the mitogen‐activated protein kinase pathways. J Immunol. 2000;165(10):5788‐5797. [DOI] [PubMed] [Google Scholar]
- 109. Davies AM, Weinberg U, Palti Y. Tumor treating fields: a new frontier in cancer therapy. Ann N Y Acad Sci. 2013;1291:86‐95. [DOI] [PubMed] [Google Scholar]
- 110. Kim EH, Song HS, Yoo SH, Yoon M. Tumor treating fields inhibit glioblastoma cell migration, invasion and angiogenesis. Oncotarget. 2016;7(40):65125‐65136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lee EJ, Kim SY, Hyun JW, Min SW, Kim DH, Kim HS. Glycitein inhibits glioma cell invasion through down‐regulation of MMP‐3 and MMP‐9 gene expression. Chem Biol Interact. 2010;185(1):18‐24. [DOI] [PubMed] [Google Scholar]
- 112. Jung K, Lee B, Han SJ, Ryu JH, Kim DH. Mangiferin ameliorates scopolamine‐induced learning deficits in mice. Biol Pharm Bull. 2009;32(2):242‐246. [DOI] [PubMed] [Google Scholar]
- 113. Jung JS, Jung K, Kim DH, Kim HS. Selective inhibition of MMP‐9 gene expression by mangiferin in PMA‐stimulated human astroglioma cells: involvement of PI3K/Akt and MAPK signaling pathways. Pharmacol Res. 2012;66(1):95‐103. [DOI] [PubMed] [Google Scholar]
- 114. Batchelor TT, Sorensen AG, di Tomaso E, et al. AZD2171, a pan‐VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell. 2007;11(1):83‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733‐4740. [DOI] [PubMed] [Google Scholar]
- 116. Guo BH, Feng Y, Zhang R, et al. Bmi‐1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011;10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vonlanthen S, Heighway J, Altermatt HJ, et al. The bmi‐1 oncoprotein is differentially expressed in non‐small cell lung cancer and correlates with INK4A‐ARF locus expression. Br J Cancer. 2001;84(10):1372‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zhang FB, Sui LH, Xin T. Correlation of Bmi‐1 expression and telomerase activity in human ovarian cancer. Br J Biomed Sci. 2008;65(4):172‐177. [DOI] [PubMed] [Google Scholar]
- 119. Jiang L, Song L, Wu J, et al. Bmi‐1 promotes glioma angiogenesis by activating NF‐kappaB signaling. PLoS One. 2013;8(1):e55527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Pang XH, Tian H, Liu ZY, Li SM, Liu ST, Tian GP. Significance and expression of vascular endothelial growth factor‐C (VEGF‐C) in esophageal squamous carcinoma and glioma. Ai Zheng. 2003;22(11):1166‐1169. [PubMed] [Google Scholar]
- 121. Andrae J, Gallini R, Betsholtz C. Role of platelet‐derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Liu T, Ma W, Xu H, et al. PDGF‐mediated mesenchymal transformation renders endothelial resistance to anti‐VEGF treatment in glioblastoma. Nat Commun. 2018;9(1):3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Julien S, Puig I, Caretti E, et al. Activation of NF‐kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445‐7456. [DOI] [PubMed] [Google Scholar]
- 124. Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60(5):1383‐1387. [PubMed] [Google Scholar]
- 125. Bachoo RM, Maher EA, Ligon KL, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269‐277. [DOI] [PubMed] [Google Scholar]
- 126. Bonavia R, Inda MM, Vandenberg S, et al. EGFRvIII promotes glioma angiogenesis and growth through the NF‐kappaB, interleukin‐8 pathway. Oncogene. 2012;31(36):4054‐4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Rummel C, Gerstberger R, Roth J, Hubschle T. Parthenolide attenuates LPS‐induced fever, circulating cytokines and markers of brain inflammation in rats. Cytokine. 2011;56(3):739‐748. [DOI] [PubMed] [Google Scholar]
- 128. Nakabayashi H, Shimizu K. Involvement of Akt/NF‐kappaB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer. 2012;12:453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr. Tumor necrosis factor alpha‐induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem. 2000;275(42):32592‐32597. [DOI] [PubMed] [Google Scholar]
- 130. Schmitz ML, dos Santos Silva MA, Baeuerle PA. Transactivation domain 2 (TA2) of p65 NF‐kappa B. Similarity to TA1 and phorbol ester‐stimulated activity and phosphorylation in intact cells. J Biol Chem. 1995;270(26):15576‐15584. [DOI] [PubMed] [Google Scholar]
- 131. Li L, Gondi CS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Transfection with anti‐p65 intrabody suppresses invasion and angiogenesis in glioma cells by blocking nuclear factor‐kappaB transcriptional activity. Clin Cancer Res. 2007;13(7):2178‐2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Woolf EC, Scheck AC. The ketogenic diet for the treatment of malignant glioma. J Lipid Res. 2015;56(1):5‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Navis AC, Bourgonje A, Wesseling P, et al. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c‐MET and VEGFR2. PLoS One. 2013;8(3):e58262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Woolf EC, Curley KL, Liu Q, et al. The ketogenic diet alters the hypoxic response and affects expression of proteins associated with angiogenesis, invasive potential and vascular permeability in a mouse glioma model. PLoS One. 2015;10(6):e0130357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Reardon DA, Herndon JE 2nd, Peters K, et al. Outcome after bevacizumab clinical trial therapy among recurrent grade III malignant glioma patients. J Neurooncol. 2012;107(1):213‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy‐temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):709‐722. [DOI] [PubMed] [Google Scholar]
- 137. Hu YL, DeLay M, Jahangiri A, et al. Hypoxia‐induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773‐1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Seynhaeve ALB, Amin M, Haemmerich D, van Rhoon GC, Ten Hagen TLM. Hyperthermia and smart drug delivery systems for solid tumor therapy. Adv Drug Deliv Rev. 2020;S0169‐409X(20)30010‐7. [DOI] [PubMed] [Google Scholar]
- 139. Kumar AA, Abraham Koshy A. Regression of recurrent high‐grade glioma with temozolomide, dexamethasone, and levetiracetam: case report and review of the literature. World Neurosurg. 2017;108(990):990.e11‐990.e16. [DOI] [PubMed] [Google Scholar]
- 140. Mayer A, Vaupel P, Struss HG, Giese A, Stockinger M, Schmidberger H. Strong adverse prognostic impact of hyperglycemic episodes during adjuvant chemoradiotherapy of glioblastoma multiforme. Strahlenther Onkol. 2014;190(10):933‐938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data generated.