

Abstract
BACKGROUND
The evaluation of procedures for drug susceptibility prediction of Mycobacterium tuberculosis based on genomic data against the conventional reference method test based on culture is realistic considering the scenario of growing number of tools proposals based on whole-genome sequences (WGS).
OBJECTIVES
This study aimed to evaluate drug susceptibility testing (DST) outcome based on WGS tools and the phenotypic methods performed on isolates of M. tuberculosis Lineage 1 from the state of Pará, Brazil, generally associated with low levels of drug resistance.
METHODOLOGY
Culture based DST was performed using the Proportion Method in Löwenstein-Jensen medium on 71 isolates that had been submitted to WGS. We analysed the seven main genome sequence-based tools for resistance and lineage prediction applied to M. tuberculosis and for comparison evaluation we have used the Kappa concordance test.
FINDINGS
When comparing the WGS-based tools against the DST, we observed the highest level of agreement using TB-profiler. Among the tools, TB-profiler, KvarQ and Mykrobe were those which identified the largest number of TB-MDR cases. Comparing the four most sensitive tools regarding resistance prediction, agreement was observed for 43 genomes.
MAIN CONCLUSIONS
Drug resistance profiling using next-generation sequencing offers rapid assessment of resistance-associated mutations, therefore facilitating rapid access to effective treatment.
Key words: tuberculosis, genomic, drug resistance, Mycobacterium tuberculosis, Lineage 1, Brazil
Tuberculosis (TB) is a millenary infectious disease caused by bacteria belonging to the classical taxonomy group Mycobacterium tuberculosis complex (MTBC). Within this group, M. tuberculosis and M. africanum are the main causative agents of the disease in humans which has been classified into eight phylogenetic lineages (L1-L8), presenting different patterns of geographical distribution and associated drug resistance profile. Among these, L1 is mostly restricted to Eastern Africa and the South of India. 1
Lineage 1 is infrequently reported in South America except for one study that described 98 such strains in Northern Brazil, representing 10% of the study population. 2 Through single nucleotide polymorphism (SNP) typing, we hypothesised to have been introduced during the trans-Atlantic slave trade. 3
The L1 has not been associated to the drug resistance (DR), 4 different to L2 and L4 that are strongly associated with DR. 4 We therefore determined the DR profile using the conventional phenotypical antimicrobial drug susceptibility tests (DST) and compared that with in silico DR prediction on whole-genome sequencing (WGS) of 71 isolates of M. tuberculosis L1 from Pará generated presently, together with three isolates with recently published genomes. 5 In silico procedures were: TB-profiler, 6 KvarQ, 7 PhyresSe, 8 Mykrobe, 9 MTBSEQ, 10 CASTB 11 and Resistance Sniffer (RSniffer). 12
MATERIALS AND METHODS
Sampling - Out of 980 M. tuberculosis isolates from the state of Pará, Brazil, 97 were classified as East-African-Indian (EAI) by Spoligotyping, 2 among which 71 were recovered and classified as Lineage 1 3 were used in this present study for DR evaluation.
Drug susceptibility testing - DST for isoniazid (INH), rifampicin (RIF), ethambutol (EMB) and streptomycin (SM) was performed using the Proportions Method in Löwenstein-Jensen (LJ) medium using the recommended critical concentrations and using the H37Rv strain as a control. The DST was performed according to the national manual for laboratory surveillance of TB and other mycobacteria 13 without modifications. This test consisted of detecting the proportion of resistant bacilli present in a sample of M. tuberculosis, given the concentration of the drug, capable of inhibiting the development of sensitive cells, but not that of resistant cells - “critical concentration”.
Whole-genome sequencing - After DNA extraction by Phenol-chloroform protocol 5 and library preparation using the Nextera XT DNA Library Prep Kit (Illumina, San Diego, USA), the isolates were sequenced using the Hiseq 2500 platform (Illumina, San Diego, USA) with a coverage of 250x. The raw reads were deposited at NCBI under the accession number PRJNA494931 5 and PRJNA630228.
Following the genome quality control by FastQC v0.11.9, reads were trimmed using Trimmomatic v0.35. 14 To compare the drug susceptibility profile based on SNPs obtained from WGS, we have used the following tools on the trimmed files: TB-profiler v2.8.6, 6 KvarQ v0.12.2, 7 PhyresSe, 8 Mykrobe v0.8.1, 9 MTBSEQ v1.0.4, 10 CASTB 11 and RSniffer. 12 All results are described in the Supplementary data (54.9KB, xlsx) (Table I).
Statistics - To compare the sensitivity, specificity and accuracy of DST as determined phenotypically or in silico, the Kappa Concordance Analysis was applied using the BioEstat 5.5 software. 15 This test is a measure of interobserver agreement and measures the degree of agreement beyond what would be expected only by chance. This measure of agreement has a maximum value of 1, where this value 1 represents total agreement and values close to and even below 0 indicate no agreement, or the agreement was exactly what was expected by chance. An eventual Kappa value less than zero or negative, suggests that the agreement found was lower than that expected by chance. Therefore, it indicates disagreement, but its value has no interpretation as a degree of disagreement. The p-value is considered significant when it is less than or equal to 5% (p ≤ 0.05).
Ethics - This study was approved by the Ethics Committee/IEC, Ananindeua, Pará, Brazil, under the number 018/2011 (CAAE: 0002.0.071.000-11).
RESULTS
Based on the DST, among the 71 isolates, 38 (53.5%) were drug susceptible, 17 (23.9%) were resistant to at least one of the drugs and 16 (22.5%) were TB-MDR. The summary of the results obtained with each of the WGS based tools for TB resistance prediction is described in Table I.
TABLE I. Drug resistance profile according to whole-genome sequencing tools for first-line anti-tuberculosis.
| Genomic tools | Multidrug resistant | Other resistance | Susceptible |
| TB-profiler | 18 (25.4%) | 14 (19.7%) | 39 (54.9%) |
| PhyResSea | 1 (1.4%) | 25 (36.2%) | 43 (62.4%) |
| KvarQ | 18 (25.4%) | 13 (18.3%) | 40 (56.3%) |
| CASTBb | 15 (25%) | 34 (56.7%) | 11 (18.3%) |
| RSniffer | 0 | 0 | 71 (100%) |
| Mykrobe | 18 (25.4%) | 17 (23.9%) | 36 (50.7%) |
| MTBSEQ | 17 (23.9%) | 23 (32.4%) | 31 (43.7%) |
a: n = 69; b: n = 60.
Among the tools TB-profiler, KvarQ and Mykrobe identified the largest number of TB-MDR cases, while PhyResSe presented a low capacity to find mutations related to the rpoB gene (k = 0,08). Due to technical issues by not generating data with the PhyResSe and CASTB softwares, we were unable to obtain results for all submitted genomes, reducing the total number of samples to 69 and 60, respectively. All samples submitted to RSniffer were determined as being drug susceptible.
When comparing the WGS based tools to DST (Table II), we observed the highest level of agreement on all drugs in the case of TB-profiler (Table III). The program that showed the least compatibility 16 with all antibiotics was RSniffer. The conclusion for each tool is described in Supplementary data (54.9KB, xlsx) (Table II).
TABLE II. Comparison among the seven whole-genome sequencing based tools against the drug susceptibility test.
| Drug-susceptibility test (Proportion Method) → | INH | RIF | PZA | EMB | SM | MDR: RIF+INH | ||||||||
| WGS tools ↓ | DST status | R | S | R | S | R | S | R | S | R | S | MDR-status | MDR | N-MDR |
| TB-profiler | R | 26 | 1 | 17 | 5 | 3 | 2 | 8 | 6 | 6 | 1 | MDR | 14 | 4 |
| S | 4 | 40 | 1 | 48 | 3 | 63 | 0 | 57 | 2 | 62 | N-MDR | 1 | 52 | |
| MTBSEQ | R | 28 | 10 | 15 | 5 | 2 | 5 | 7 | 5 | 6 | 2 | MDR | 13 | 4 |
| S | 2 | 31 | 3 | 48 | 4 | 60 | 1 | 58 | 2 | 61 | N-MDR | 3 | 51 | |
| Phyressea | R | 20 | 0 | 1 | 0 | 2 | 5 | 8 | 4 | 5 | 2 | MDR | 1 | 0 |
| S | 10 | 41 | 17 | 53 | 4 | 60 | 0 | 59 | 3 | 61 | N-MDR | 15 | 55 | |
| Kvarq | R | 26 | 1 | 17 | 5 | 1 | 4 | 7 | 2 | 6 | 1 | MDR | 14 | 4 |
| S | 4 | 40 | 1 | 48 | 5 | 61 | 1 | 61 | 2 | 62 | N-MDR | 2 | 51 | |
| CASTBb | R | 25 | 3 | 13 | 4 | 2 | 4 | 5 | 5 | 5 | 36 | MDR | 11 | 4 |
| S | 5 | 38 | 5 | 49 | 2 | 61 | 3 | 58 | 3 | 27 | N-MDR | 5 | 51 | |
| Resistance sniffer | R | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | MDR | 0 | 0 |
| S | 30 | 41 | 18 | 53 | 6 | 65 | 8 | 63 | 8 | 63 | N-MDR | 16 | 55 | |
| Mykrobe | R | 27 | 2 | 17 | 6 | 4 | 5 | 7 | 7 | 6 | 1 | MDR | 14 | 4 |
| S | 3 | 39 | 1 | 47 | 2 | 60 | 1 | 56 | 2 | 62 | N-MDR | 2 | 51 | |
INH: isoniazid; RIF: rifampicin; PZA: pyrazinamide; EMB: ethambutol; SM: streptomycin; MDR: multidrug-resistant; WGS: whole-genome sequencing; DST: drug susceptibility test. a: n = 69; b: n = 60.
TABLE III. TB-profiler versus MTBSEQ whole-genome sequencing tools according to Kappa Coefficient.
| Kappa (Interpretation) | Observed concordance | Replicability | 95% CI | p-value | ||||||
| Drugs | TB-profiler | MTBSEQ | TB-profiler | MTBSEQ | TB-profiler | MTBSEQ | TB-profiler | MTBSEQ | TB-profiler | MTBSEQ |
| INH | 0.854 | 0.692 | 92.96% | 84.51% | Almost perfect | Substantial | 0.731 to 0.977 | 0.528 to 0.856 | <0.0001 | <0.0001 |
| RIF | 0.792 | 0.713 | 91.55% | 88.73% | Substantial | Substantial | 0.635 to 0.949 | 0.528 to 0.898 | <0.0001 | <0.0001 |
| PZA | 0.508 | 0.238 | 92.96% | 87.32% | Moderate | Fair | 0.133 to 0.882 | -0.108 to 0.585 | <0.0001 | <0,0219 |
| EMB | 0.645 | 0.653 | 90.41% | 91.55% | Substantial | Substantial | 0.411 to 0.879 | 0.400 to 0.906 | <0.0001 | <0.0001 |
| SM | 0.776 | 0.718 | 95.77% | 94.73% | Substantial | Substantial | 0.533 to 1.000 | 0.457 to 0.980 | <0.0001 | <0.0001 |
INH: isoniazid; RIF: rifampicin; PZA: pyrazinamide; EMB: ethambutol; SM: streptomycin; 95% CI: 95% confidence interval.
Considering the performance of the in silico DST against the antibiotics separately, Mykrobe was the one with highest accuracy in relation to INH (k = 0.855 and p < 0.0001). Owing to the design of algorithm or technical runtime issues, CASTB presented a greater number of positive results for SM, influencing the agreement (k = 0.04 and p < 0.2025) together with EMB (k = 0.166 and p < 0.095), these results do not indicate statistical significance. For pyrazinamide (PZA), in silico analysis demonstrated a low agreement rate, with Kappa coefficient results ranging from 0.114 to 0.508 [Supplementary data (54.9KB, xlsx) (Table III)].
Among the evaluated tools in general, TB-profiler performed favorably. For identification of MDR samples however, sensitivity (77%), specificity (96%) and accuracy (14.71) were the same for KvarQ and Mykrobe [Supplementary data (54.9KB, xlsx) (Table III)].
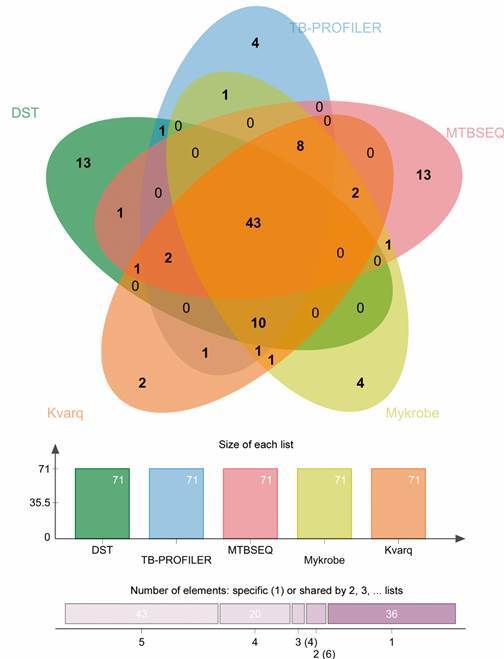
A Venn graphic illustrates the comparison among the four most sensible tools (TB-profiler; KvarQ; Mykrobe and MTBSEQ) and conventional DST, including 43 genomes as common elements: 12 MDR (G04875, G04876, G04877, G04878, G04882, G04893, G049162, G049222; G049392; G049442, G049512 and G049522), 26 susceptible (G04871, G04881, G04883, G04885, G04886, G04887, G04889, G04896, G049182, G049202, G049212, G049252, G049272, G049292, G049302, G049312, G049372, G049402, G049412, G049422, G049432, G049462, G049472, G049492, G049532 and G049542) and five INH monoresistant isolates (G04888, G049382, G049482, G049502 and G049582) (Figure).
Comparison among five drug resistance prediction tools based on whole-genome sequencing data against the drug resistance testing (DST) reference technique for 71 Mycobacterium tuberculosis Lineage 1 from Brazil.
DISCUSSION
One of the objectives of the Genomic Era is to replace the classic genotyping techniques for the detection and identification of MTBC species for diagnostic purposes and the phenotypic methods for DST, by in silico analysis of WGS data. 17 During the last decades, genotyping tools have been developed that identify both lineage and drug resistance and their validation is of major importance to evaluate their impact as a possible substitute for traditional methodologies.
This present study compared the widely used WGS based tools to predict antimicrobial resistance profile in 71 genomes from isolates of M. tuberculosis of the Lineage 1 from Pará, Brazil, using DST as the reference method. The DST based on the proportions method is mostly used in Brazil as an AST for mycobacteria, but it is a laborious and time-consuming method, requiring four to six weeks to obtain the results. 18 On the other hand, DR prediction from WGS data can be performed from early positive MGIT cultures after an average of 14 days, or even directly on sputum sample generating results within five days. 19
Lineage 1 (EAI) is not usually associated with DR and has also low correlation to transmissibility and virulence, presenting a restricted geographical distribution. 4 , 20 In this study however, the resistance profile by the DST demonstrated that 17 (23.9%) were resistant at least to one drug and 16 (22.5%) were MDR. This high frequency of MDR isolates might be related to the fact that the TB cases were from the reference hospital for MDR-TB Hospital Universitário João de Barros Barreto (HUJBB), including TB contacts (without a previous history of TB), and patients suspected of treatment failure or TB relapse. Compared the DR of Lineage 1 in the context of other lineages from the same region, the most predominant was Lineage 4, among of which T and X lineages, were associated to MDR-TB, while Lineage 1 the highest among ‘any resistance’ group. 2
Among all in silico based tools tested presently, we encountered difficulties to predict resistance to PZA, which can be partly due to alternative mechanisms of resistance to this drug (non pncA related) 21 and reports of low-frequency SNPs that may be associated with PZA resistance. 22 In the present study however, we did not include PZA in the conventional DST, a major limitation of the study.
The ability to correctly identify whether there is a mutation in the sample is called sensitivity and the ability to identify whether the sample does not actually have the mutation is specificity, when analysing these results it is important to generate the level of accuracy, thus it is easier to assess whether the results obtained were compared correctly.
TB-profiler showed that, in addition to good sensitivity and specificity, 23 it has a good statistical correlation with conventional DST, proving that it is a good resistance predictor tool. A recent study on isolates from patient from the state of São Paulo in Brazil and from province of Sofala in Mozambique compared DST performed in liquid medium MGIT-960 SIRE kit against TB-profiler prediction and the LPA tests Genotype-MTBDRplus 2.0 and MTBDRsl 2.0. The TB-profiler had the best performance among the genotypic DST as compared to the phenotypic test with a good concordance with phenotypic DST for RIF and SM (89.6%), INH (96.5%) and EMB (82.7%). WGS sensitivity and specificity for detection resistance were respectively 87.5 and 92.3% for RIF; 95.6 and 100% for INH; 85.7 and 93.3% for SM while 100 and 77.2% for EMB. 24
Moreover, our data is also in agreement with other studies 10 , 25 suggesting that the use of TB-profiler together with Mykrobre, MTBSeq and KvarQ may increase the chances to fully elucidate the mutations of the genomes under analysis.
Regarding RIF prediction by PhyResSe, it detected only one mutation (rpoB_His445Arg) in rpoB gene of a MDR strain by DST (G04893), therefore this tool presented a low sensitivity and specificity for this drug. In general, we observed a lower performance PhyResSe compared to other pipelines, much more pronounced that that described in other studies. 23 This might be a characteristic of the performance of this pipeline particular in genomes of Lineage 1 and needs further investigation.
Even though in some studies, CASTB has demonstrated a good performance in finding variants related to mutations, 11 , 21 we observed a high number of false positives for resistance to SM, in addition to not generating outputs for some samples resulting in inconclusive results.
Regarding RSniffer based genome analysis, all isolates were considered as susceptible to all drugs and this seems to be due to the fact that by default, this tool assumes Linage 1 as a drug pan-susceptible, 12 limiting its applicability in this strain population.
Since WGS is mainly done from a DNA pool of a culture, it is possible that there is a discrepancy between the phenotypic and genotypic tests for the same sample, as these strains can manifest themselves, thus tests can be influenced by mixed infections or mixtures of drug susceptible and resistant populations in phenomenon of heteroresistance. 26
The evaluated tools in this study were based on the technique of Direct Association (DA) which relies on the established correlation between the various resistance conferring mutations and their presence or absence in the MTB isolate under study. These pre-documented correlations are utilised by these tools to ascertain the drug resistance profile of the sample. 27 , 28 Studies which are valid as genomic analysis protocol for the detection of MTBC species and their genetic characterisation, especially for resistance analysis, are important for the progress of translational research in TB, with the goal to replace phenotypic tests by WGS.
Recently, WGS performed directly on clinical specimen has been proposed for an even more rapid TB surveillance, allowing researchers to do real-time genomic epidemiology and drug resistance surveillance in settings where culture and DST are not available. 29 , 30 However, this is still technically challenging and a under active study.
ACKNOWLEDGEMENTS
To the Centro de Inovações Tecnológicas at Instituto Evandro Chagas for technical assistance.
Footnotes
Financial support: CNPq (207,071/2014-4), CAPES (Finance Code 001), Swiss National Science Foundation (grants 310030_166687 and IZRJZ3_164171).
REFERENCES
- 1.Ngabonziza JCS, Loiseau C, Marceau M, Jouet A, Menardo F, Tzfadia O. A sister lineage of the Mycobacterium tuberculosis complex discovered in the African Great Lakes region. Nat Commun. 2020;11:1–11. doi: 10.1038/s41467-020-16626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Conceição EC, Rastogi N, Couvin D, Lopes ML, Furlaneto IP, Gomes HM. Genetic diversity of Mycobacterium tuberculosis from Pará, Brazil, reveals a higher frequency of ancestral strains than previously reported in South America. Infect Genet Evol. 2017;56:62–74. doi: 10.1016/j.meegid.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Conceição EC, Refregier G, Gomes HM, Olessa-Daragon X, Coll F, Ratovonirina NH. Mycobacterium tuberculosis lineage 1 genetic diversity in Pará, Brazil, suggests common ancestry with east-African isolates potentially linked to historical slave trade. Infect Genet Evol. 2019;73:337–341. doi: 10.1016/j.meegid.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis Vol. 26. Seminars in Immunology. 2014;26:431–444. doi: 10.1016/j.smim.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conceição EC, Guimarães AES, Lopes ML, Furlaneto IP, Rodrigues YC, da Conceição ML. Analysis of potential household transmission events of tuberculosis in the city of Belem, Brazil. Tuberculosis. 2018;113:125–129. doi: 10.1016/j.tube.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Phelan JE. O'Sullivan DM.Machado D.Ramos J.Oppong YEA.Campino S Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019;11:41–41. doi: 10.1186/s13073-019-0650-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steiner A, Stucki D, Coscolla M, Borrell S, Gagneux S. KvarQ targeted and direct variant calling from fastq reads of bacterial genomes. BMC Genomics. 2014;15(1):881–881. doi: 10.1186/1471-2164-15-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feuerriegel S, Schleusener V, Beckert P, Kohl TA, Miotto P, Cirillo DM. PhyResSE a web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J Clin Microbiol. 2015;53(6):1908–1914. doi: 10.1128/JCM.00025-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hunt M, Bradley P, Lapierre SG, Heys S, Thomsit M, Hall MB. Antibiotic resistance prediction for Mycobacterium tuberculosis from genome sequence data with Mykrobe. Wellcome Open Res. 2019;4:191–191. doi: 10.12688/wellcomeopenres.15603.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kohl TA, Utpatel C, Schleusener V, De Filippo MR, Beckert P, Cirillo DM. MTBseq a comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ. 2018;13(6):e5895. doi: 10.7717/peerj.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwai H, Kato-Miyazawa M, Kirikae T, Miyoshi-Akiyama T. CASTB (the comprehensive analysis server for the Mycobacterium tuberculosis complex) a publicly accessible web server for epidemiological analyses, drug-resistance prediction and phylogenetic comparison of clinical isolates. Tuberculosis. 2015;95(6):843–844. doi: 10.1016/j.tube.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 12.Muzondiwa D, Mutshembele A, Pierneef RE, Reva ON. Resistance sniffer an online tool for prediction of drug resistance patterns of Mycobacterium tuberculosis isolates using next generation sequencing data. Int J Med Microbiol. 2020;310:1513992–1513992. doi: 10.1016/j.ijmm.2020.151399. [DOI] [PubMed] [Google Scholar]
- 13.MS - Ministério da Saúde . Manual nacional de vigilância laboratorial da tuberculose e outras micobactérias (Internet) Brasília: MS; 2008. [Google Scholar]
- 14.Bolger AM, Lohse M, Usadel B. Trimmomatic a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayres M, Ayres MJ, Ayres DL. Aplicações estatísticas nas áreas das Ciências Biomédicas. BioEstat. 2007;Vol. 5 [Google Scholar]
- 16.Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33:159–174. [PubMed] [Google Scholar]
- 17.Nsofor CA, Jiang Q, Wu J, Gan M, Liu Q, Zuo T. Transmission is a noticeable cause of resistance among treated tuberculosis patients in Shanghai, China. Sci Rep. 2017;7:7691–7691. doi: 10.1038/s41598-017-08061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canetti G, Fox W, Khomenko A, Mahler HT, Menon NK, Mitchison DA. Advances in techniques of testing mycobacterial drug sensitivity, and the use of sensitivity tests in tuberculosis control programmes. Bull World Health Organ. 1969;41(1):21–43. [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen KA, Manson AL, Desjardins CA, Abeel T, Earl AM. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing progress, promise, and challenges. Genome Med. 2019;11:45–45. doi: 10.1186/s13073-019-0660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanveer M, Hasan Z, Siddiqui AR, Ali A, Kanji A, Ghebremicheal S. Genotyping and drug resistance patterns of M tuberculosis strains in Pakistan. BMC Infect Dis. 2008;8(1):171–171. doi: 10.1186/1471-2334-8-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwamoto T, Murase Y, Yoshida S, Aono A, Kuroda M, Sekizuka T, et al. Overcoming the pitfalls of automatic interpretation of whole genome sequencing data by online tools for the prediction of pyrazinamide resistance in Mycobacterium tuberculosis. PLoS One. 2019;14(2) doi: 10.1371/journal.pone.0212798. https://pubmed.ncbi.nlm.nih.gov/30817803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oppong YEA, Phelan J, Perdigão J, Machado D, Miranda A, Portugal I, et al. Genome-wide analysis of Mycobacterium tuberculosis polymorphisms reveals lineage-specific associations with drug resistance. BMC Genomics. 2019;20(1) doi: 10.1186/s12864-019-5615-3. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5615-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schleusener V, Köser CU, Beckert P, Niemann S, Feuerriegel S. Mycobacterium tuberculosis resistance prediction and lineage classification from genome sequencing comparison of automated analysis tools. Sci Rep. 2017;7:46327–46327. doi: 10.1038/srep46327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feliciano CS, Namburete EI, Plaça JR, Peronni K, Dippenaar A, Warren RM. Accuracy of whole genome sequencing versus phenotypic (MGIT) and commercial molecular tests for detection of drug-resistant Mycobacterium tuberculosis isolated from patients in Brazil and Mozambique. Tuberculosis. 2018;110:59–67. doi: 10.1016/j.tube.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Beek J, Haanperä M, Smit PW, Mentula S, Soini H. Evaluation of whole genome sequencing and software tools for drug susceptibility testing of Mycobacterium tuberculosis. Clin Microbiol Infect. 2019;25(1):82–86. doi: 10.1016/j.cmi.2018.03.041. [DOI] [PubMed] [Google Scholar]
- 26.Dheda K, Gumbo T, Maartens G, Dooley KE, McNerney R, Murray M, et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 2017;5(4) doi: 10.1016/S2213-2600(17)30079-6. http://linkinghub.elsevier.com/retrieve/pii/S2213260017300796 [DOI] [PubMed] [Google Scholar]
- 27.Seifert M, Catanzaro D, Catanzaro A, Rodwell TC. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis a systematic review. PLoS One. 2015;10(3):0119628–0119628. doi: 10.1371/journal.pone.0119628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goig GA, Cancino-Muñoz I, Torres-Puente M, Villamayor LM, Navarro D, Borrás R. Whole-genome sequencing of Mycobacterium tuberculosis directly from clinical samples for high-resolution genomic epidemiology and drug resistance surveillance an observational study. Lancet Microbe. 2020;1:e175–e183. doi: 10.1016/S2666-5247(20)30060-4. [DOI] [PubMed] [Google Scholar]
- 29.Walker TM, Kohl TA, Omar SV, Hedge J. Del Ojo Elias C.Bradley P Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance a retrospective cohort study. Lancet Infect Dis. 2015;15(10):1193–1202. doi: 10.1016/S1473-3099(15)00062-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown AC, Bryant JM, Einer-Jensen K, Holdstock J, Houniet DT, Chan JZM. Rapid whole-genome sequencing of Mycobacterium tuberculosis isolates directly from clinical samples. J Clin Microbiol. 2015;53(7):2230–2237. doi: 10.1128/JCM.00486-15. [DOI] [PMC free article] [PubMed] [Google Scholar]