

In this study, Morimoto et al. sought to investigate how reactive oxygen species (ROS) are regulated in spermatogonial stem cells (SSCs). Using in vivo and genomic approaches, the authors report that hypoxic niche sets the ROS levels in SSCs, and spermatogenesis depends on an intricate balance between NOX1 and mitochondria-derived ROS.
Keywords: Hif1, reactive oxygen species, spermatogonia
Abstract
Reactive oxygen species (ROS) produced by NADPH1 oxidase 1 (NOX1) are thought to drive spermatogonial stem cell (SSC) self-renewal through feed-forward production of ROS by the ROS-BCL6B-NOX1 pathway. Here we report the critical role of oxygen on ROS-induced self-renewal. Cultured SSCs proliferated poorly and lacked BCL6B expression under hypoxia despite increase in mitochondria-derived ROS. Due to lack of ROS amplification under hypoxia, NOX1-derived ROS were significantly reduced, and Nox1-deficient SSCs proliferated poorly under hypoxia but normally under normoxia. NOX1-derived ROS also influenced hypoxic response in vivo because Nox1-deficient undifferentiated spermatogonia showed significantly reduced expression of HIF1A, a master transcription factor for hypoxic response. Hypoxia-induced poor proliferation occurred despite activation of MYC and suppression of CDKN1A by HIF1A, whose deficiency exacerbated self-renewal efficiency. Impaired proliferation of Nox1- or Hif1a-deficient SSCs under hypoxia was rescued by Cdkn1a depletion. Consistent with these observations, Cdkn1a-deficient SSCs proliferated actively only under hypoxia but not under normoxia. On the other hand, chemical suppression of mitochondria-derived ROS or Top1mt mitochondria-specific topoisomerase deficiency did not influence SSC fate, suggesting that NOX1-derived ROS play a more important role in SSCs than mitochondria-derived ROS. These results underscore the importance of ROS origin and oxygen tension on SSC self-renewal.
Stem cells are characterized by a unique ability to undergo self-renewal division. Compared with more actively dividing progenitor cells, division of stem cells is generally slow and requires a special microenvironment with the necessary self-renewal factors (Scadden 2006). The analysis of stem cells is hampered by their small number and the lack of specific markers. The interactions of stem cells with their environment also complicate analysis because the niche is composed of several somatic cell types. Although stem cells are biologically unique and show promise for clinical application, little progress has been made in our understanding of the role of niche and its regulation of stem cells.
Stem cells are typically sensitive to environmental changes and lose their stem cell potential after in vitro culture. However, spermatogonial stem cells (SSCs) can proliferate in vitro for more than 5 yr (Kanatsu-Shinohara et al. 2019). In the presence of GDNF and FGF2, germ cells from postnatal testes form grapelike clusters of spermatogonia, which, upon microinjection into the seminiferous tubules, restart spermatogenesis (Kanatsu-Shinohara et al. 2003). These cells, designated germline stem (GS) cells, can be genetically manipulated to produce knockout (KO) animals and have a stable karyotype and DNA methylation pattern (Kanatsu-Shinohara and Shinohara 2013). Although much remains unknown about SSC self-renewal mechanism (Meistrich and van Beek 1993; de Rooij 2017), we showed that reactive oxygen species (ROS) play an important role in driving self-renewal (Lee et al. 2009; Morimoto et al. 2019). Compared with Asingle (As) spermatogonia that divide only once per 10 d, doubling time of GS cells is 2.5 d (Kanatsu-Shinohara et al. 2003). This suggests that in vitro culture significantly enhances proliferation. Adding GDNF and FGF2 increased the ROS levels, while ROS depletion compromised SSC self-renewal, suggesting that the ROS levels have a close relationship with self-renewal division.
The major sources of ROS are NADPH oxidases (NOXs) and mitochondria. The involvement of Nox1 in SSC self-renewal was demonstrated using Nox1 KO mice (Morimoto et al. 2013). NOXs catalyze the generation of O2 by single-electron transfer from NADPH to O2. Nox1 KO mice have smaller testes and a significantly reduced number of GFRA1+ undifferentiated spermatogonia. Transplantation experiments showed that self-renewal division of Nox1-deficient SSCs is less efficient than that of wild-type (WT) SSCs. Subsequent studies revealed that “ROS amplification” drives SSC self-renewal in vitro (Morimoto et al. 2019). Upon stimulation with GDNF and FGF2, MAPK14 and MAPK7 are phosphorylated, promoting the nuclear translocation of BCL6B and subsequent induction of Nox1 for ROS generation. Because addition of ROS not only induced phosphorylation of MAPK14 and MAPK7 but also promoted BCL6B nuclear translocation and Nox1 induction, we proposed that the ROS-BCL6B-NOX1 pathway creates a positive feedback loop to sustain self-renewal division. Because ROS are considered harmful to spermatogenesis, enhancement of SSC self-renewal by ROS was unexpected.
Our recent study implicated a role for mitochondria-derived ROS in GS cells. GS cells cultured for 5 yr showed enhanced proliferation (Kanatsu-Shinohara et al. 2019). Although aged GS cells expressed Nox1 more abundantly than young GS cells, the ROS levels were significantly lower in aged GS cells. We found that excessive activation of self-renewal division in aged GS cells significantly reduced mitochondrial activity by increasing WNT7B expression. These results raised questions on whether mitochondria-derived ROS influences SSCs and their relationship with NOX1-derived ROS. In fact, much remains unknown about the functional role of mitochondria-derived ROS in tissue-specific stem cells (Bigarella et al. 2014).
Here, we evaluated the effect of Nox1 in GS cells by generating Nox1 KO GS cells. Contrary to our expectation, proliferation of Nox1 KO GS cells was comparable with that of WT GS cells when cultured under normoxic conditions. This observation led us to study the role of oxygen and HIF1A in SSC self-renewal. HIF1A is a key transcription factor that mediates hypoxic response. Although previous in vitro studies suggested that hypoxia is beneficial for SSCs (Kubota et al. 2009; Helsel et al. 2017), HIF1A expression is controversial in both mouse and human spermatogonia (Marti et al. 2002; Depping et al. 2004; Takahashi et al. 2016), and the role of oxygen in SSCs has remained unknown. Our study revealed that the hypoxia modulates NOX1-derived ROS levels and increases the expression of CDKN1A via MYC down-regulation.
Results
Defective proliferation of Nox1 GS cells under hypoxia
We previously reported that Nox1 was important for SSC self-renewal in vivo (Morimoto et al. 2013). To confirm this, we generated Nox1 KO GS cells. Nox1 KO testes were dissociated into single cells, and the SSC self-renewal factors GDNF and FGF2 were added to enhance self-renewal division and establish GS cells. However, GS cell colonies appeared normal and their proliferation was comparable with that of WT GS cells (Fig. 1A). To explain this discrepancy between in vivo and in vitro, we analyzed factors potentially involved in the phenotypic change. The factors analyzed included temperature, self-renewal factor or serum concentrations, culture substrate, etc. We eventually found out that oxygen concentration (1%) induces a marked difference in proliferation between Nox1 KO and WT GS cells (Fig. 1B). Although low-oxygen culture suppressed the proliferation of both Nox1 KO and WT cells, the difference between cell types becoming evident when oxygen concentration was lowered to 1% O2 (Fig. 1B). However, expression of several markers of undifferentiated spermatogonia and differentiating spermatogonia showed only modest changes by real-time polymerase chain reaction (PCR) (Supplemental Fig. S1). Nevertheless, the results of the hypoxic cultures suggested that oxygen tension plays a role in the changes observed in Nox1 KO GS cells.
Figure 1.
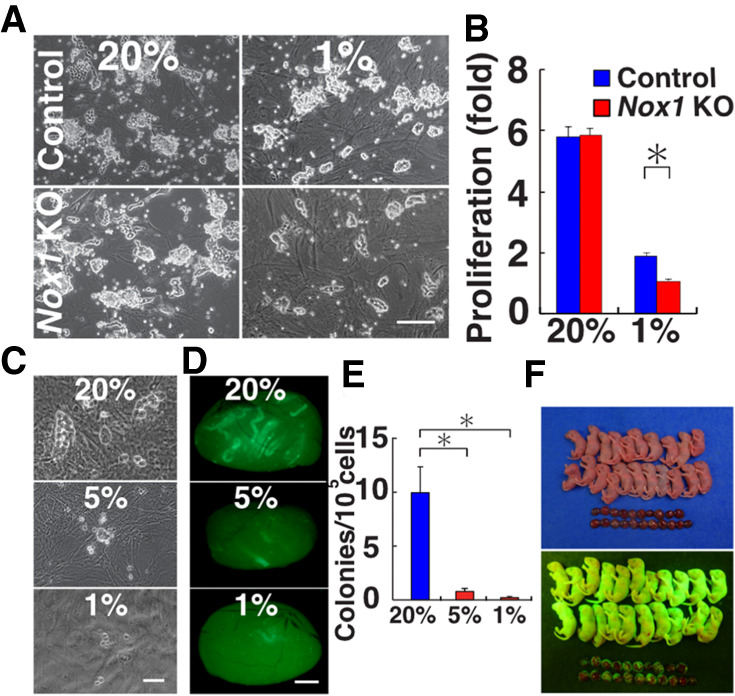
Proliferation of Nox1 KO GS cells. (A) Appearance of Nox1 KO GS cells. (B) Proliferation of Nox1 KO GS cells (n = 3). Cells were cultured under 1% or 20% O2 for 8 d. Relative increase of cell recovery after culture initiation is shown. (C) Culture of green pup testis cells under 1%, 5%, and 20% O2 for 7 d on MEFs. (D) Appearance of recipient testes after transplantation of pup testis cells under hypoxia. (E) Colony counts (n = 12). (F) Offspring born after microinsemination using sperm derived from GS cells cultured under hypoxia. Scale bars: A,C, 200 µm; D, 1 mm. Asterisk indicates statistical significance (P < 0.05).
To examine the impact of hypoxia on SSCs, we performed spermatogonial transplantation using fresh testis cells (Brinster and Zimmermann 1994), because abnormalities in SSCs are difficult to evaluate by morphological analysis. Testis cells from immature C57BL6/Tg14(act-EGFP-OsbY01) (designated green) pups, which express Egfp ubiquitously, were cultured under 1%, 5%, and 20% O2. After 1 wk of culture, germ cell colonies were found in 20% O2 cultures (Fig. 1C). In contrast, such colonies were rare when the cells were cultured under lower oxygen concentrations. We microinjected cultured cells into the seminiferous tubules of busulfan-treated infertile mice. Analysis of recipient mice showed increased colony formation in cultures maintained under 20% O2 (Fig. 1D). The number of colonies generated from 1%, 5%, and 20% O2 cultures was 0.2, 0.8, and 10.0 per 105 transplanted cells, respectively (Fig. 1E). Although the number of colonies did not differ significantly between 1% and 5% O2, the differences between 20% O2 and 1% or 5% O2 were significant.
Because the results suggested a growth-suppressive effect of hypoxia, we analyzed the long-term impact of hypoxia on the proliferation of WT GS cells. GS cells were maintained under 1% O2 and transplanted into the seminiferous tubules of infertile mouse testes at different time points. GS cells cultured under 20% O2 expanded by 1.8 × 105-fold over 43 d, whereas GS cells cultured under 1% O2 expanded by 2.9 × 103-fold during the same period, indicating a 62.1-fold enhancement of proliferation under normoxia (Supplemental Fig. S2A). The doubling times under 20% and 1% O2 were 2.5 and 3.7 d, respectively. Analysis of recipient testes showed that the SSC concentration under 20% O2 was significantly higher than that under 1% O2 (Supplemental Fig. S2B,C).
Because these results suggested the poor quality of GS cells under hypoxia and hypoxia can cause genomic instability in some cell types (Rodríguez-Jiménez et al. 2008), we examined the full developmental potential of GS cells cultured under hypoxia. We cultured GS cells for 77 d in vitro and transplanted them into the seminiferous tubules of congenitally infertile WBB6F1-W/Wv (W) mice. Within 3 mo, the testes became larger (Supplemental Fig. S2D), and immunostaining of recipient testes showed production of peanut agglutinin (PNA)+ haploid cells (Supplemental Fig. S2E). Elongated spermatids or spermatozoa were retrieved and microinjected into oocytes. A total of 92 embryos were created, and 69 two-cell embryos were transferred into pseudopregnant mothers. Twenty-one offspring were born by cesarean section (Fig. 1F). These results confirmed that GS cells cultured under hypoxia are fertile.
Increased mitochondria-derived ROS under hypoxia
To evaluate the mechanism of hypoxia-induced growth suppression, we examined the ROS levels in WT and Nox1 KO GS cells by flow cytometry. Despite the lack of Nox1, Nox1 KO GS cells showed increased ROS levels under normoxia. When cultured under hypoxic conditions (1% O2), their ROS levels were comparable with those of WT cells (Fig. 2A). Although Nox1 deficiency induced significant changes in other Nox gene expression levels under normoxia, these changes were not observed under hypoxia (Supplemental Fig. S3A). We next compared the ROS levels in WT GS cells cultured under hypoxic and normoxic conditions. Culture under hypoxic conditions significantly increased the ROS levels in WT GS cells (Fig. 2B). However, Nox1 expression in WT GS cells was down-regulated upon transfer to hypoxic conditions (Supplemental Fig. S3B). Because addition of H2O2 also increased ROS levels in Nox1 KO GS cells (Fig. 2C), these results suggested that ROS from sources other than NOX1 contributed to the increased ROS production under hypoxia.
Figure 2.
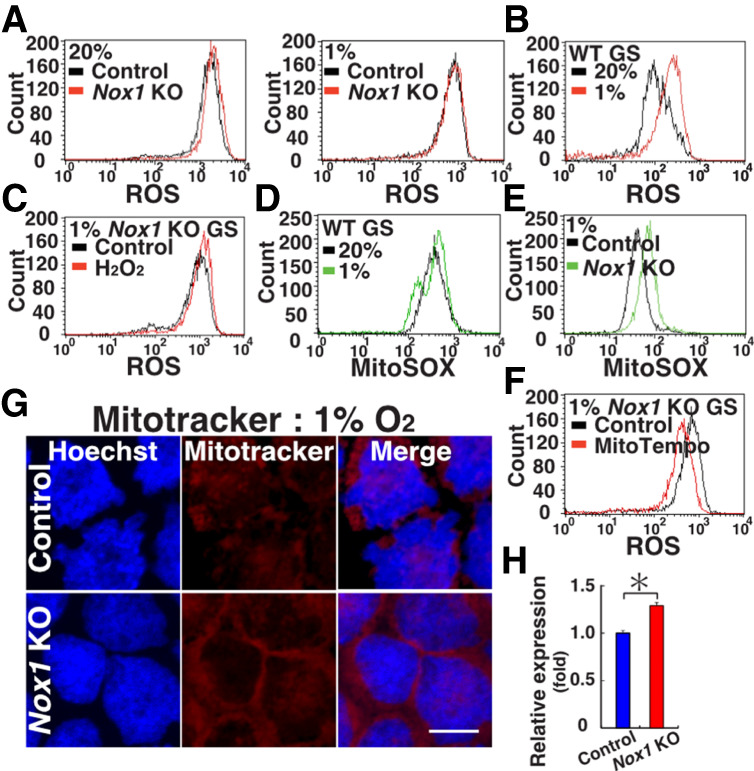
Oxygen-dependent changes in ROS levels. (A) Flow cytometric analysis of ROS levels in Nox1 KO GS cells under hypoxic and normoxic conditions by CellROX Deep Red. (B) Flow cytometric analysis of ROS levels in WT GS cells under hypoxic and normoxic conditions by CellROX Deep Red. (C) Flow cytometric analysis of ROS levels in Nox1 KO GS cells by CellROX Deep Red 2 d after H2O2 supplementation under hypoxia. (D,E) Flow cytometric analysis of mitochondria-derived ROS levels in WT (D) or Nox1 KO (E) GS cells using MitoSox. (F) Flow cytometric analysis of ROS levels in Nox1 KO GS cells by CellROX Deep Red 2 d after H2O2 treatment. (G,H) Mitotracker staining (G) and quantification (H) of its staining intensity in GS cells (n = 10). Scale bar in G, 20 µm. Stain in G is Hoechst 33342. Asterisk indicates statistical significance (P < 0.05).
Because mitochondria are a major source of ROS (Zorov et al. 2014), we reasoned that the increase in ROS levels was a result of increased ROS production by mitochondria. Therefore, we used MitoSox to assay mitochondria-derived ROS. MitoSox staining showed a significant increase in the mitochondria-derived ROS levels in WT cells under hypoxia (Fig. 2D; Supplemental Table S1). Although mitochondria-derived ROS activate NOX proteins and create a feed-forward cycle in somatic cells (Dikalov 2011), Nox1 deficiency further enhanced mitochondria-derived ROS levels under hypoxia (Fig. 2E), suggesting that NOX1-derived ROS suppress mitochondria-derived ROS in GS cells. To directly examine the effect of mitochondrial ROS on GS cells, we treated GS cells with MitoTempo, a mitochondrion-targeted antioxidant (Trnka et al. 2008). As expected, MitoTempo treatment down-regulated ROS levels in Nox1 KO GS cells under hypoxia (Fig. 2F). Moreover, the Mitotracker staining intensity was stronger in Nox1 KO GS cells under hypoxia (Fig. 2G,H). These results suggested that hypoxia increases ROS production by mitochondria and that Nox1 suppresses mitochondria-derived ROS.
Because an increase in ROS levels induces Nox1 and promotes GS cell proliferation by ROS amplification (Morimoto et al. 2019), impaired proliferation under hypoxia was likely caused by decreased ROS production by Nox1. Therefore, we examined the effect of exogenous ROS on GS cells under hypoxic conditions. As expected, the addition of hydrogen peroxide significantly increased the number of GS cells under hypoxia (Supplemental Fig. S3C). However, mitochondria-derived ROS also play a role in normoxic cultures because treatment with MitoTempo significantly reduced the proliferation of WT GS cells cultured under normoxia (Supplemental Fig. S3D). Down-regulation of ROS levels was also confirmed by MitoTempo treatment (Supplemental Fig. S3E). Because these results suggested that mitochondria-derived ROS also contribute to GS cell proliferation, we carried out spermatogonial transplantation to assess the effect of mitochondria-derived ROS on normoxic GS cell cultures. The numbers of colonies generated by MitoTempo-treated and control GS cells did not change significantly (Supplemental Fig. S3F,G). Therefore, mitochondria-derived ROS affect GS cell proliferation but do not influence the SSC concentration.
SSCs in Top1mt KO mice
The results in the preceding section suggested that mitochondria-derived ROS have relatively little impact on SSC fate determination. This observation agrees with our recent study that showed robust SSC activity of GS cells that have significantly fewer mitochondria after 5 yr of normoxic culture (Kanatsu-Shinohara et al. 2019). To study the impact of defective mitochondria on SSCs directly in vivo, we next examined Top1mt KO mice (Khiati et al. 2015). Top1mt is the only mitochondria-specific topoisomerase and plays a role in mitochondrial DNA maintenance and topology. Top1mt KO mouse embryonic fibroblasts (MEFs) exhibit activation of the DNA damage repair pathway and hyperpolarization of mitochondrial membrane (Douarre et al. 2012).
Top1mt KO mice were initially fertile and showed complete spermatogenesis (data not shown). However, they gradually lost spermatogenesis, and their testes were significantly smaller at >1 yr old (Fig. 3A,B). Histological analysis showed almost complete lack of spermatogenesis at this stage (Fig. 3C,D). When immunostaining was carried out using relatively young adult testes (<3 mo) with complete spermatogenesis, the number of KIT+ differentiating spermatogonia was significantly reduced (Supplemental Fig. S4A), suggesting impaired spermatogonial differentiation. In contrast, the number of GFRA1+ (As, Apaired, and some Aaligned) and CDH1+ (total undifferentiated) spermatogonia slightly increased in Top1mt KO mice (Supplemental Fig. S4B).
Figure 3.
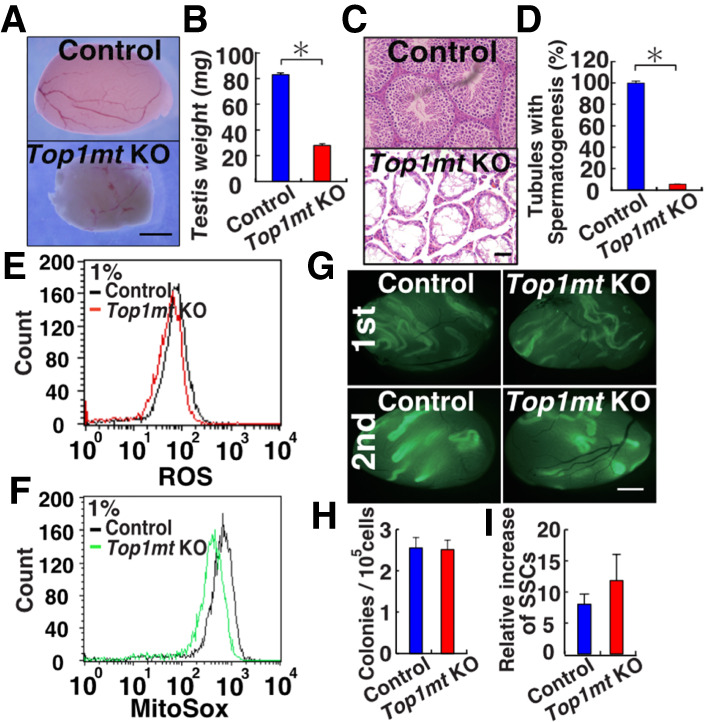
Functional analysis of SSCs in Top1mt KO mice. (A) Appearance of Top1mt KO mouse testis. (B) Testis weight (n = 6). (C) Histological appearance of Top1mt KO mouse testis. (D) Number of seminiferous tubules with spermatogenesis. At least 504 tubules were counted. (E,F) Flow cytometric analysis of ROS levels in Top1mt KO GS cells by CellROX Deep Red (E) and MitoSox (F). (G) Appearance of recipient testes. (H) Colony counts in primary recipients (n = 10). (I) Total increase in colony number (total regenerated colony number × 10)/(primary colony number used for serial transplantation) (n = 6). Scale bars: A,G, 1 mm; C, 50 µm. Stain in C is hematoxylin and eosin. Asterisk indicates statistical significance (P < 0.05).
We derived GS cells from Top1mt KO mice to examine the impact of this gene. Top1mt KO GS cells proliferated normally under normoxia. However, hypoxic culture showed a modest but significant proliferative defect (Supplemental Fig. S4C). Flow cytometric analysis showed that these cells have reduced ROS levels under hypoxia (Fig. 3E). MitoSox staining revealed that this reduction in ROS levels was caused by a decrease in mitochondria-derived ROS (Fig. 3F). To understand the mechanism of poor GS cell proliferation, we carried out RNA sequencing (RNA-seq) and found that 59 genes were differentially expressed in Top1mt KO GS cells under hypoxia (Supplemental Fig. S5A; Supplemental Table S2). One of the down-regulated genes was Gfra1, which is a critical component of the GDNF receptor and influences GS cell proliferation (Supplemental Fig. S5A). Because real-time PCR confirmed this result (Supplemental Fig. S1), we also checked expression of GFRA1 by flow cytometry and found significant down-regulation of GFRA1 expression (Supplemental Fig. S4D). Therefore, GFRA1 down-regulation might have caused slow proliferation under hypoxia.
Because SSCs comprise only a small population in GFRA1+ undifferentiated spermatogonia and in GS cells, we carried out spermatogonial transplantation to examine the effect of Top1mt deficiency on SSCs. The number of colonies generated by Top1mt KO and control cells did not show statistical difference (Fig. 3G,H). To confirm the impact on SSC self-renewal in the niche, we carried out serial transplantation. Although the exact location of the niche is unclear in vivo, transplantation analysis of SSC activity in the primary recipient allows evaluation on the impact of the microenvironment on self-renewal activity because self-renewal division is thought to occur in the niche. We used these primary recipient testes for serial transplantation. Analysis of the secondary recipients also failed to show significant changes in the number of germ cell colonies (Fig. 3I). Because serial transplantation of Nox1 KO testis showed impaired colonization (Morimoto et al. 2013), these results suggested that Nox1-derived ROS are more important than mitochondria-derived ROS in SSC fate determination.
Hif1a deficiency compromises self-renewal division
To identify the mechanism of slow proliferation under hypoxia, we next analyzed Hif genes because they are essential mediators of hypoxic response. Although the Hif1a and Epas1 (Hif2a) mRNA levels were not influenced by the oxygen level (Supplemental Fig. S6A), Western blot analysis showed that only HIF1A was up-regulated under hypoxia (Fig. 4A). Because Epas1 KO mice did not show a phenotype in spermatogonia (Gruber et al. 2010), we reasoned that Hif1a plays an important role in the hypoxic response. To examine the degree of hypoxia in spermatogonia, we first used pimonidazole, which forms covalent bonds with cellular macromolecules at oxygen levels <1.3% (Varia et al. 1998). Immunostaining of WT testis with spermatogonia markers showed that a larger number of CDH1+ undifferentiated spermatogonia exhibit pimonidazole staining compared with EPCAM+ (whole spermatogonia) or KIT+ (differentiating spermatogonia) cell populations (Fig. 4B,C).
Figure 4.
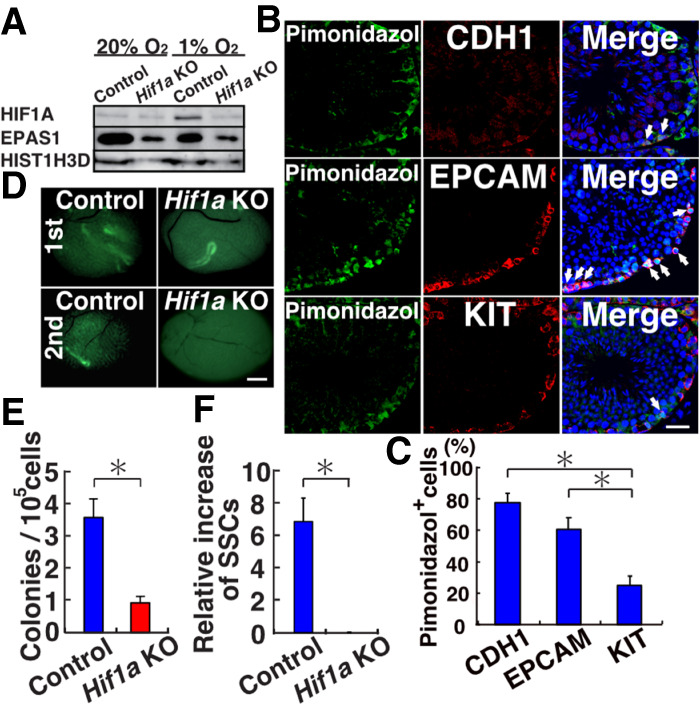
Functional analysis of Hif1a in SSCs. (A) Western blot analysis of HIF1A and EPAS1 in WT GS cells. (B,C) Immunostaining (B) and quantification (C) of WT testis with pimonidazole and spermatogonia markers. At least 10 tubules were counted. (D) Appearance of recipient testes. (E) Colony counts in primary recipients (n = 16). (F) Total increase in colony numbers (total regenerated colony number × 10)/(primary colony number used for serial transplantation) (n = 7 for Hif1a KO; n = 11 for control). Scale bars: B, 20 µm; D, 1 mm. Stain in B is Hoechst 33342. Asterisk indicates statistical significance (P < 0.05).
Because these results suggested that hypoxia plays an important role in SSC maintenance, we analyzed the involvement of ROS and mitochondria by examining HIF1A expression in Nox1 and Top1mt KO mouse testes. Compared with control testes, Nox1 KO testes showed reduction in spermatogonia numbers (Supplemental Fig. S6B,C). Also, the expression of HIF1A in GFRA1+ and CDH1+ spermatogonia was low in Nox1 KO testes (Supplemental Fig. S6D). In contrast to undifferentiated spermatogonia, very few KIT+ spermatogonia expressed HIF1A, and there was no significant difference in the number of cells expressing the two antigens between WT and Nox1 KO mice. These results suggested that ROS produced by NOX1 enhance HIF1A expression in undifferentiated spermatogonia. On the other hand, the numbers of HIF1A-expressing spermatogonia in Top1mt KO mouse testes were significantly reduced in both undifferentiated spermatogonia and differentiating spermatogonia stages (Supplemental Fig. S4E,F). Therefore, mitochondrial activity also influences HIF1A expression.
To examine the impact of Hif1a in vivo, Hif1af/f mice were crossed with green mice to introduce a donor cell marker (Ryan et al. 2000). Testes were collected from immature Hif1af/f green mice and cells were exposed to a Cre-expressing adenovirus (AxCANCre) before transplantation (Takehashi et al. 2007). Deletion efficiency, as estimated by southern blot analysis, was 75.7% at the time of transplantation (Supplemental Fig. S6E). Analysis of recipient mice showed a significant reduction in the number of colonies from Hif1a KO donor cells (Fig. 4D,E). Histological analyses of recipient testes showed a significantly reduced number of seminiferous tubules with Hif1a KO testes (Supplemental Fig. S6F). These results suggested that loss of Hif1a impairs SSC activity.
To examine the role of Hif1a in self-renewal division, we carried out serial transplantation. Analysis of the secondary recipients showed a complete lack of secondary colonies from Hif1a KO testes (Fig. 4F). However, control testis cells generated normal appearing secondary colonies by serial transplantation. These results suggested that loss of Hif1a significantly impairs self-renewal division in vivo.
Analysis of GS cell phenotype under hypoxia
To understand the molecular impact of hypoxia in GS cells, we carried out RNA-seq analysis of WT GS cells cultured under hypoxia. The RNA-seq analysis identified 228 genes whose expression levels were up-regulated or down-regulated under hypoxic conditions (Supplemental Fig. S5B; Supplemental Table S3). Next, we performed ingenuity pathway analysis (IPA) to identify transcriptional regulators linked to the hypoxia-induced changes in gene expression profiles. This analysis revealed several transcription regulators that were significantly enriched upstream of the hypoxia-regulated genes (Supplemental Table S4). They included HIF1A, EPAS1, VHL1, and CITED2, all of which are involved in the oxygen response in somatic cells. Of these candidate genes, we focused on MYC/MYCN because they enhance spermatogonial proliferation (Kanatsu-Shinohara et al. 2016). Both real-time PCR analysis confirmed down-regulation of Mycn/Myc under hypoxia (Supplemental Fig. S7A). Western blot analysis showed that both MYC and MYCN proteins are down-regulated under hypoxia (Fig. 5A; Supplemental Fig. S7B).
Figure 5.
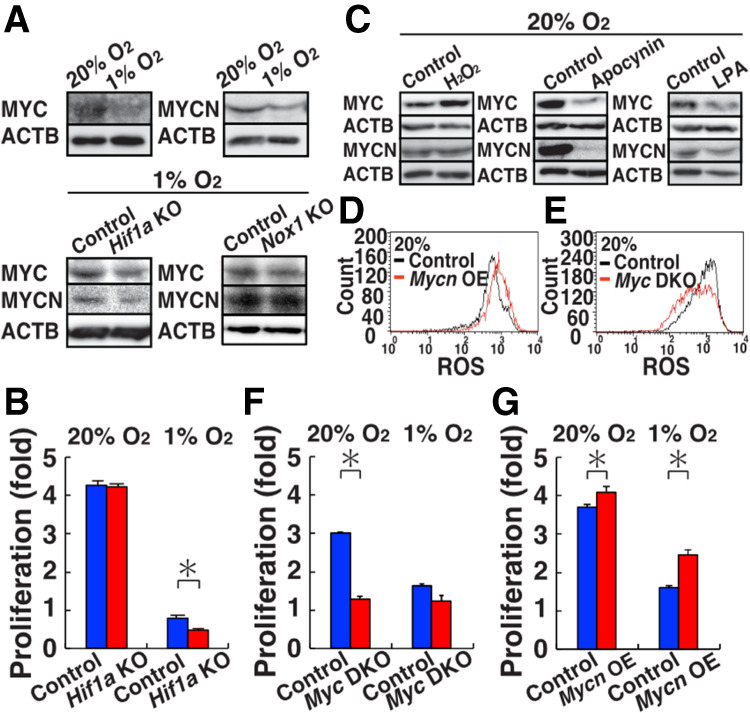
Induction of MYC/MYCN by ROS. (A) Western blot analysis of MYC/MYCN in WT (20% and 1% O2), Hif1a KO (1% O2), and Nox1 KO (1% O2) GS cells (n = 3). (B) Proliferation of Hif1a KO GS cells (n = 3). Cell recovery was determined after culturing under hypoxia for 3 d. (C) Western blot analysis of MYC/MYCN in WT GS cells after exposure to H2O2, apocynin or LPA (n = 3). (D) Flow cytometric analysis of ROS levels by CellROX Deep Red after Mycn overexpression in WT GS cells 11 d after transfection (n = 3). (E) Flow cytometric analysis of ROS levels in Myc DKO GS cells by CellROX Deep Red. (F) Proliferation of Myc DKO GS cells (n = 3). Cells were recovered after 4 d. (G) Proliferation of Myc DKO GS cells transfected with Mycn (n = 3). Cells were recovered 8 d after transfection. Asterisk indicates statistical significance (P < 0.05).
We also derived GS cells from Hif1af/f mice and deleted Hif1a by AxCANCre. Consistent with the transplantation experiments, these cells proliferated poorly under hypoxia (Fig. 5B). However, real-time PCR analysis of spermatogonia markers did not show apparent changes that might explain this phenotype (Supplemental Fig. S1). Therefore, we carried out RNA-seq analysis to understand the mechanism of poor proliferation by Hif1a deficiency (Supplemental Fig. S5C; Supplemental Tables S5, S6). Under normoxic culture, we found only three genes that are expressed differentially between WT and Hif1a KO GS cells, which is consistent with the fact that HIF1A was absent in normoxic culture (Fig. 4A). On the other hand, we noted 534 genes that are expressed differentially under hypoxia. As expected, Myc was one of the top candidate transcription factors by IPA analysis (Supplemental Table S7). Western blot analysis showed significant down-regulation of MYC/MYCN in Hif1a KO GS cells (Fig. 5A; Supplemental Fig. S7C). We also confirmed that MYC is down-regulated in Nox1 KO GS cells, although MYCN did not show statistical significance (Fig. 5A; Supplemental Fig. S7D). These results suggested that down-regulation of MYC is responsible for poor proliferation of Hif1a and Nox1 KO GS cells.
To identify the mechanism of the increased MYC/MYCN expression under normoxia, we examined the impact of ROS. Although ROS suppression by chemical agents, such as lipoic acid (LPA) and apocynin, decreased MYC/MYCN expression, hydrogen peroxide significantly increased MYC expression (Fig. 5C; Supplemental Fig. S7E). In contrast, Mycn overexpression under normoxia increased the ROS levels (Fig. 5D). Because these results suggested a close relationship between ROS and MYC/MYCN, we analyzed Myc/Mycn double KO (Myc DKO) GS cells and analyzed their ROS levels by flow cytometry (Kanatsu-Shinohara et al. 2016). As expected, Myc DKO GS cells exhibited significant down-regulation of ROS (Fig. 5E). Moreover, the rate of proliferation of Myc DKO GS cells did not change under hypoxia (Fig. 5F). However, transfection of Mycn rescued the defective proliferation of Myc DKO GS cells (Fig. 5G). Taken together, these results suggested that HIF1A enhances the proliferation of GS cells under hypoxia via MYC/MYCN.
Lack of ROS amplification and increased CDK inhibitor expression under hypoxia
Although these results suggested that ROS promote MYC/MYCN expression under normoxia, we focused our analysis under hypoxia to understand the mechanism of self-renewal in vivo. Because our results suggested that down-regulation of MYC/MYCN is responsible for slow proliferation under hypoxia, it was likely that GS cells lack expression of genes involved in ROS amplification under hypoxia. We found by our RNA-seq analysis that Bcl6b and Etv5, both of which act downstream from MAPK7 and MAPK14 (Morimoto et al. 2019), were down-regulated (0.4-fold and 0.66-fold, respectively) under hypoxia. We confirmed down-regulation of Bcl6b and Etv5 by real-time PCR (Fig. 6A). Western blot analysis also showed a significant reduction in the phosphorylation level of MAPK7 and MAPK14 (Fig. 6B; Supplemental Fig. S8A). These results suggested that ROS amplification does not occur under hypoxia.
Figure 6.
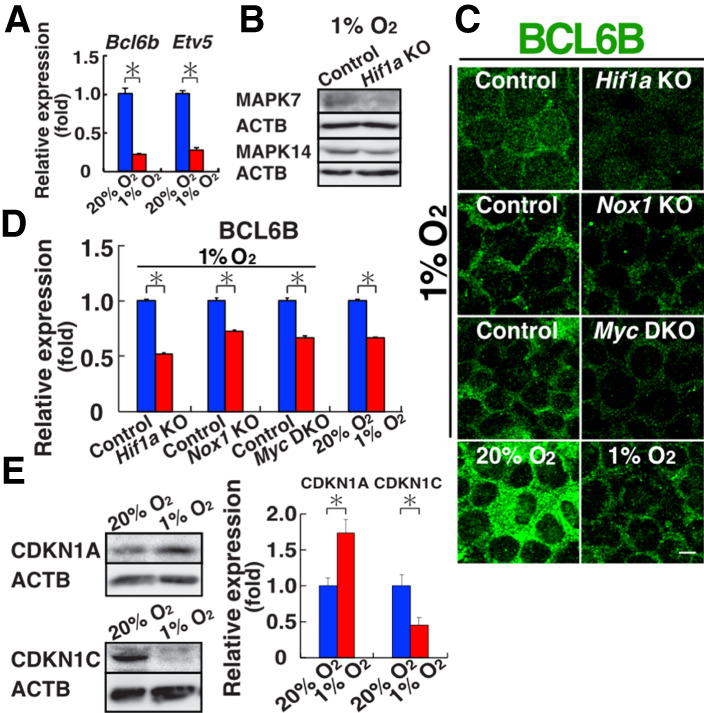
Lack of ROS amplification and increased CDKN1A expression under hypoxia. (A) Real-time PCR analysis of Etv5 and Bcl6b expression (n = 3). (B) Western blot analysis of MAPK7 and MAPK14 in WT GS cells (n = 3). (C,D) Immunostaining (C) and quantification (D) of BCL6B in WT, Hif1a, Nox1, and Myc DKO KO GS cells. Relative staining intensity in the nucleus was quantified (n = 10). (E) Western blot analysis of CDKN1A and CDKN1C in WT GS cells (n = 3). Scale bar in C, 10 µm. Stain in C is Hoechst 33342. Asterisk indicates statistical significance (P < 0.05).
Because nuclear translocation of BCL6B was critical for Nox1 expression and ROS amplification (Morimoto et al. 2019), we carried out immunostaining of BCL6B. Although BCL6B was localized predominantly in the cytoplasm in GS cells, the amount of nuclear BCL6B was significantly decreased in Hif1a KO GS cells (Fig. 6C,D). We also checked the distribution of BCL6B in WT GS cells (1% vs. 20% O2), Nox1 KO GS cells (1% O2), and Myc DKO GS cells (1% O2). As expected, WT GS cells down-regulated BCL6B expression when the cells were cultured under hypoxia. The amount of nuclear BCL6B under hypoxia was further reduced in both Nox1 KO and Myc DKO cells. Because Bcl6b is indispensable for SSCs (Oatley et al. 2006), these results suggested that reduced nuclear BCL6B expression is responsible for the poor proliferation of GS cells under hypoxia.
To understand how a lack of nuclear BCL6B leads to poor proliferation under hypoxia, we analyzed the expression of Ccnd2, which plays a critical role in GS cell proliferation (Lee et al. 2009). Real-time PCR analysis of WT GS cells showed down-regulation of Ccnd2 (Supplemental Fig. S8B). However, Myc DKO and Hif1a KO GS cells expressed Ccnd2 strongly, although Nox1 KO GS cells did not show significant changes. Therefore, the expression level of Ccnd2 was not necessarily correlated with GS cell proliferation under hypoxia. We next focused on CDK inhibitors and reanalyzed RNA-seq data of WT and Hif1a KO GS cells. Analysis revealed significant increase in Cdkn1c and Cdkn1a by hypoxia treatment. Western blot analysis showed that CDKN1C was down-regulated in WT cells, whereas CDKN1A was significantly up-regulated (Fig. 6E). These results suggested that CDKN1A is responsible for the slow proliferation of GS cells.
Cdkn1a deficiency increases GS cell proliferation under hypoxia
Based on these observations, we analyzed the expression of Cdkn1a in Myc DKO GS cells. Irrespective of the oxygen concentration, Myc DKO GS cells showed increased expression of Cdkn1a (Supplemental Fig. S9A). Because proliferation of Myc DKO GS cells was not influenced by oxygen levels (Fig. 5F), we reasoned that up-regulation of Cdkn1a might be responsible for the hypoxia-induced proliferative defect. As expected, Western blot analysis showed that Myc DKO, Hif1a KO, and Nox1 KO GS cells expressed CDKN1A strongly (Fig. 7A; Supplemental Fig. S9B). Based on these results, we carried out knockdown (KD) experiments using short hairpin RNA (shRNA). As expected, Cdkn1a depletion rescued Hif1a KO and Nox1 KO GS cells. However, Myc DKO GS cells still showed poor proliferation, which suggested that additional target genes are involved in the poor proliferation of Myc DKO GS cells (Fig. 7B). Real-time PCR analysis confirmed down-regulation of Cdkn1a by shRNA (Supplemental Fig. S9C). These results suggested that up-regulation of Cdkn1a is responsible for the defective proliferation of WT GS cells under hypoxia.
Figure 7.
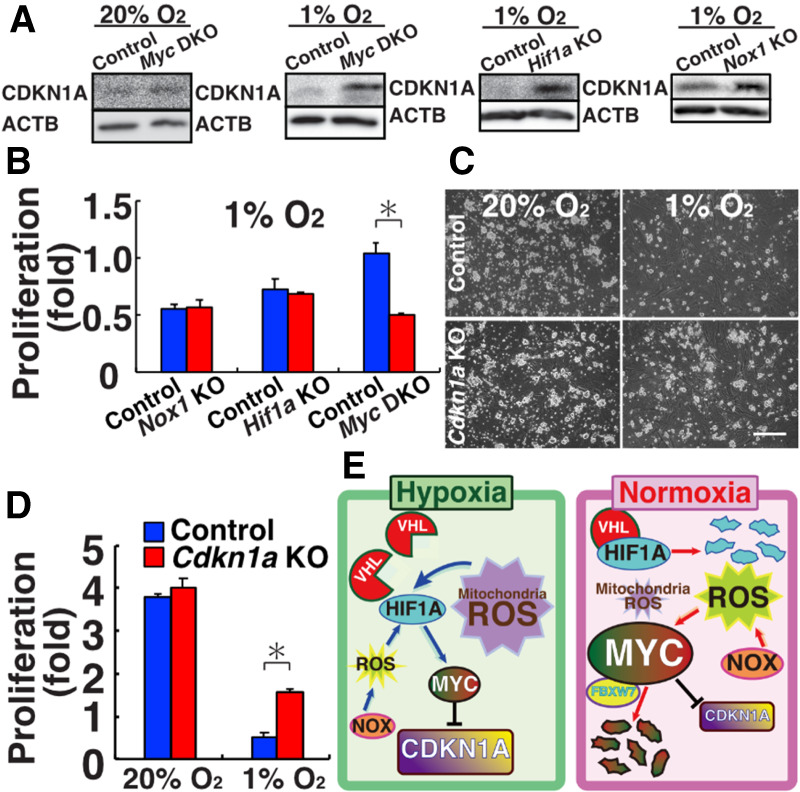
Improvement of GS cell proliferation by Cdkn1a deficiency under hypoxia. (A) Western blot analysis of CDKN1A in Myc DKO, Hif1a KO, and Nox1 KO GS cells (n = 3). (B) Rescue of proliferative defect of KO GS cells after Cdkn1a KD. Cells were counted 6 d (Nox1 KO and Myc DKO) or 8 d (Hif1a KO) after transfection. (C) Appearance of Cdkn1a KO GS cells. (D) Proliferation of Cdkn1a KO GS cells (n = 6). Cells were cultured for 6 d. (E) Summary of experiments. Under hypoxic conditions, NOX1-derived ROS and mitochondria-derived ROS enhance expression of HIF1A, which induces MYC to inhibit CDKN1A expression, thereby promoting self-renewal. Under normoxia, however, VHL degrades HIF1A, and the contribution of mitochondria-derived ROS is decreased under normoxia. Because ROS induce MYC expression (Fig. 5C), GS cells can proliferate actively. How ROS stabilize MYC is still unknown, but FBXW7 may be involved. Scale bar in C, 200 µm. Asterisk indicates statistical significance (P < 0.05).
To confirm the above, we derived Cdkn1a KO GS cells. Cdkn1a KO mice have significantly larger testes than do WT mice, which has been attributed to an increased number of Sertoli cells (Holsberger et al. 2005). Indeed, Cdkn1a KO GS cells appeared very similar to WT cells in terms of morphology or proliferation (Fig. 7C). However, because hypoxia induced a phenotype in Nox1 KO GS cells, we cultured Cdkn1a KO GS cells under hypoxia. As expected, Cdkn1a KO GS cells proliferated more efficiently than WT cells under hypoxia (Fig. 7D). These results suggested that CDKN1A inhibits GS cell proliferation under hypoxia.
Discussion
Several previous studies highlight the beneficial effects of hypoxic culture, including spermatogonia. For example, Kit-deficient spermatogonia proliferated only when cultured under hypoxia (Kubota et al. 2009). Proliferation of bull spermatogonia was also promoted under hypoxia (Oatley et al. 2016). Although these results suggested that hypoxia is beneficial for SSC cultures, these experiments were carried out under 10% O2. However, the oxygen level in the seminiferous tubules is estimated to be <1.5% (Gruber et al. 2010). Therefore, our analysis of cells under 1% O2 probably more closely reflects the in vivo environment. Because doubling time of As spermatogonia in vivo is significantly longer than that of GS cells (10 d vs. 2.5 d) (de Rooij 2017), it was likely that the current GS cell culture conditions are significantly different from the physiological environment. We initiated this study by analyzing the apparently normal proliferation of Nox1 KO GS cells.
Our analysis revealed significant changes in mitochondria-derived ROS under hypoxia. Using MitoTempo and Top1mt KO mice, however, we showed that mitochondria-derived ROS do not have a significant impact on the SSC fate. The involvement of mitochondria in differentiating spermatogonia was suggested in a recent study using mice deficient in Mfn1/2, which mediate mitochondrial fusion (Varuzhanyan et al. 2019). These mice lacked differentiating spermatogonia and exhibited a meiotic defect. However, because impairment of mitochondrial function was analyzed only in Stra8-expressing germ cells, the impact on SSCs was unclear. These results suggest that mitochondria-derived ROS are likely more important for proliferation of committed progenitor cells. Although one might think that mitochondria are dispensable for SSCs, they are still required for SSCs because adding MitoTempo or rotenone inhibited GS cell proliferation (Kanatsu-Shinohara et al. 2019). It also should be noted that the impact of Top1mt deficiency was caused by the lack of mitochondria-derived ROS alone because it is difficult to distinguish the impact of mitochondria-derived ROS from the basic function of mitochondria that would have been impaired by Top1mt deficiency. However, because Top1mt KO SSCs are still capable of undergoing self-renewal with reduced mitochondria-derived ROS generation, NOX1-derived ROS play more important roles in SSC self-renewal. The two ROS-producing systems are not totally independent because NOX1 inhibited mitochondria-derived ROS production (Fig. 2E). Thus, contribution of ROS from the two different sources change dynamically during spermatogenesis, and disturbance of this balance results in infertility.
The seminiferous tubules are poorly vascularized and are under low oxygen tension, and previous studies have suggested the importance of oxygen on spermatogenesis. For example, Epas1 (Hif2a) deficiency compromises spermatogenesis (Gruber et al. 2010). In these testes, defects were found in spermatids and spermatozoa, but not in spermatogonia. Impaired spermatogenesis is considered to be caused by a defective blood-testis barrier formed between Sertoli cells. Likewise, loss of Vhl, which degrades HIF1A and EPAS1 proteins, also inhibits spermatogenesis (Ma et al. 2003). In these testes, very few spermatozoa were found in the lumen of seminiferous tubules with severely reduced numbers of germinal cells. Because Vhl was mutated in both germ cells and somatic cells and the number of Sertoli cells was reduced in these experiments, it is not clear whether the defects are caused by germ cells.
Our analysis of Hif1a KO mice revealed that Hif1a plays a critical role in self-renewal division. Compared with Nox1 KO SSCs, defects in Hif1a KO SSCs were more severe because no secondary colonies were generated upon serial transplantation. Because no defect was observed in spermatogonia of Vhl KO mice, increased HIF1A expression may not hinder self-renewal, but its deficiency has a strong impact on SSCs. Importantly, Nox1 KO and Top1mt KO mice showed decreased expression of HIF1A in GFRA1+ spermatogonia. This suggests that the amount of HIF1A is not simply determined by the oxygen level but is also influenced by the ROS levels. HIF1A regulation by oxidative stress has been suggested (Movafagh et al. 2015), but the mechanism is unclear. Therefore, while oxygen tension changes the ROS levels in SSCs, ROS are also influencing hypoxic response. Thus, our results suggest that the amount of HIF1A is regulated in a sophisticated manner involving ROS and oxygen levels.
Our analysis of the hypoxic response in GS cells revealed significant changes in gene expression. Of the several candidate genes, we focused on Myc/Mycn because MYC binds to the promoter of many genes that showed significant changes in expression under hypoxia. In addition, Myc/Mycn promoted SSC self-renewal in our previous studies (Kanatsu-Shinohara et al. 2016). Our analysis revealed ROS-dependent expression of MYC/MYCN; addition of H2O2 increased the MYC/MYCN level, whereas MYCN overexpression up-regulated ROS levels, suggesting that Myc/Mycn are critical for increases in ROS under normoxia. Moreover, Etv5 and Bcl6b, both of which are important in SSC self-renewal (Simon et al. 2007; Lee et al. 2009), were down-regulated under hypoxia. Therefore, ROS amplification is severely compromised under hypoxic conditions, which may explain the poor proliferation of Myc DKO GS cells even under normoxia.
In our analysis of poor proliferation under hypoxia, we found that increased CDKN1A expression is responsible for poor proliferation under hypoxia. Cdkn1a has been implicated in spermatogonial proliferation. Although Cdkn1a KO mice do not show fertility defects (Holsberger et al. 2005), the impact of Cdkn1a in germ cells became evident when the same number of WT and Cdkn1a KO testis cells were cotransplanted simultaneously. This competitive spermatogonial transplantation revealed that offspring were derived exclusively from Cdkn1a KO SSCs (Kanatsu-Shinohara et al. 2010). Although the mechanism of the increased germline transmission efficiency of Cdkn1a KO SSCs has long remained unexplained, the enhancement of Cdkn1a KO GS cell proliferation under hypoxia suggests that Cdkn1a KO spermatogonia outcompeted WT spermatogonia in the hypoxic seminiferous tubules and produced more progenitors, thus contributing to fertilization. However, depletion of Cdkn1a alone may not be sufficient for promoting Myc DKO GS cells. While it is possible that Cdkn1a depletion efficiency might not have been sufficient, it is likely that Myc DKO GS cells failed to stimulate expression of positive drivers of cell cycles, such as Cdk4 or Cdc25a. Alternatively, they might show increased expression of other CDK inhibitors.
Our analysis of ROS-mediated self-renewal uncovered an unexpected role of hypoxia in modulating ROS levels in SSCs. To our knowledge, this is the first report of a direct link between ROS regulation and oxygen tension in self-renewal division (Fig. 7E). Modulation of ROS amplification under hypoxia is reasonable because ROS can damage DNA (Lu et al. 2001), which needs to be minimized to prevent transmission of de novo mutations to the next generation. Given the lack of a phenotype of Nox1 and Cdkn1a KO GS cells in vitro and considering that HIF1A is degraded under normoxia, the regulation of MYC/MYCN under normoxia is apparently different. Although much remains unknown about how ROS stabilize MYC/MYCN under normoxia, Fbxw7 may be involved because Fbxw7 deficiency significantly increased MYC, but not MYCN, under normoxia (Kanatsu-Shinohara et al. 2014a). Considering the significant difference of phenotypes of KO GS cells under hypoxia, caution is necessary when SSCs are analyzed under normoxic conditions. Because increased HIF1A expression inhibits spermatogenesis and ROS induce de novo mutations (Lu et al. 2001; Ma et al. 2003), further analysis of oxygen and ROS metabolism in the germline will increase our knowledge of the molecular basis of male infertility. Thus, the current study provides new insight into the mechanism of SSC self-renewal and has important implications for treating male infertility.
Materials and methods
Cell culture
Green GS cells were derived from C57BL6/Tg14(act-EGFP-OsbY01) transgenic mice on a DBA/2 background (Kanatsu-Shinohara et al. 2003). We also derived GS cells from 7- to 10-d-old Nox1 KO, B6.129-Hif1atm3Rsjo/J, or Cdkn1a KO mice, as described previously (Kanatsu-Shinohara et al. 2014b). Myc DKO GS cells have been described (Kanatsu-Shinohara et al. 2016). For hypoxic culture, GS cells were placed into plastic bags and gassed with 1% O2/5% CO2/94% N2. When cells were cultured under 5% or 10% O2, the CO2 level was maintained at 5%, but the N2 level was reduced to 90% or 85%, respectively. Where indicated, GS cells were cultured with 30 µM hydrogen peroxide (Wako), 100 µM MitoTempo, 2 mM LPA (both from Sigma), and 1 mM apocynin (Tokyo Chemical Industry).
For Cdkn1a and KD, p21-shRNA-1, p21-shRNA-2, and empty control vectors were used (Dr. Sally Temple, Albany Medical College, NY) (Fasano et al. 2007). A mixture of lentiviral particles was used for transfection. For cDNA overexpression, mouse Mycn cDNA was inserted into the CSII-Ef1a-IRES-Venus vector. Virus particles were prepared as described previously (Shinohara and Kanatsu-Shinohara 2020). The multiplicity of infection was adjusted to 10.0. AxCANCre was used for CRE-mediated deletion of target genes in GS cells (MOI = 2.0).
Statistical analysis
Results are presented as the means ± SEM. Data were analyzed by Student's t-test. Multiple comparison analyses were performed by ANOVA followed by Tukey's HSD test.
Accession numbers
All the sequencing data in this study have been deposited in the GEO (GSE148706).
Supplementary Material
Acknowledgments
We thank Dr. R.N. Eisenman for providing Mycn KO mice, and Ms. S. Ikeda for technical assistance. Financial support for this research was provided by the Uehara Memorial Foundation; the Agency for Medical Research and Development (17933225 and JP20gm1110008); and the Ministry of Education, Culture, Sports, Science, and Technology (19K22512, 19H05750, 19H04906, 18H04882, 18H05281, and 18H02935). The Institute for Advanced Study of Human Biology is supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan.
Author contributions: H.M. carried out most of the experiments. T.Y. analyzed sequence data. T.M. carried out real-time PCR and flow cytometry of KO GS cells. N.O. and A.O. performed microinsemination. T.T. carried out analysis of mutant mice. C.Y.-N., H.Z., Y.P., and A.T. provided new reagents. T.S. carried out spermatogonial transplantation. M.K.-S. and T.S. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.339903.120.
References
- Bigarella CL, Liang R, Ghaffari S. 2014. Stem cells and the impact of ROS signaling. Development 141: 4206–4218. 10.1242/dev.107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster RL, Zimmermann JW. 1994. Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci 91: 11298–11302. 10.1073/pnas.91.24.11298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depping R, Hägele S, Wagner KF, Wiesner RJ, Camenisch G, Wnger RH, Katschinski DM. 2004. A dominant-negative isoform of hypoxia-inducible factor-1α specifically expressed in human testis. Biol Reprod 71: 331–339. 10.1095/biolreprod.104.027797 [DOI] [PubMed] [Google Scholar]
- de Rooij DG. 2017. The nature and dynamics of spermatogonial stem cells. Development 144: 3022–3030. 10.1242/dev.146571 [DOI] [PubMed] [Google Scholar]
- Dikalov S. 2011. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301. 10.1016/j.freeradbiomed.2011.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douarre C, Sourbier C, Dalla Rosa I, Brata Das B, Redon CE, Zhang H, Neckers L, Pommier Y. 2012. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS One 7: e41094 10.1371/journal.pone.0041094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. 2007. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 1: 87–99. 10.1016/j.stem.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Gruber M, Mathew LK, Runge AC, Garcia JA, Simon MC. 2010. EPAS1 is required for spermatogenesis in the postnatal mouse testis. Biol Reprod 82: 1227–1236. 10.1095/biolreprod.109.079202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsel AR, Oatley MJ, Oatley JM. 2017. Glycolysis-optimized conditions enhance maintenance of regenerative integrity in mouse spermatogonial stem cells during long-term culture. Stem Cell Reports 8: 1430–1441. 10.1016/j.stemcr.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsberger DR, Buchold GM, Leal MC, Kieseweter SE, O'Brien DA, Hess RA, França LR, Kiyokawa H, Cooke PS. 2005. Cell-cycle inhibitors p27Kip1 and p21Cip1 regulate murine Sertoli cell proliferation. Biol Reprod 72: 1429–1436. 10.1095/biolreprod.105.040386 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Shinohara T. 2013. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol 29: 163–187. 10.1146/annurev-cellbio-101512-122353 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ogonuki N, Inoue K, Miki H, Ogura A, Toyokuni S, Shinohara T. 2003. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod 69: 612–616. 10.1095/biolreprod.103.017012 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Takashima S, Shinohara T. 2010. Transmission distortion by loss of p21 or p27 cyclin-dependent kinase inhibitors following competitive spermatogonial transplantation. Proc Natl Acad Sci 107: 6210–6215. 10.1073/pnas.0914448107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Onoyama I, Nakayama KI, Shinohara T. 2014a. Skp1-Cullin-F-box (SCF)-type ubiquitin ligase FBXW7 negatively regulates spermatogonial stem cell self-renewal. Proc Natl Acad Sci 111: 8826–8831. 10.1073/pnas.1401837111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Ogonuki N, Matoba S, Morimoto H, Ogura A, Shinohara T. 2014b. Improved serum- and feeder-free culture of mouse germline stem cells. Biol Reprod 91: 88 10.1095/biolreprod.114.122317 [DOI] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Tanaka T, Ogonuki N, Ogura A, Morimoto H, Cheng PF, Eisenman RN, Trumpp A, Shinohara T. 2016. Myc/mycn-mediated glycolysis enhances mouse spermatogonial stem cell self-renewal. Genes Dev 30: 2637–2648. 10.1101/gad.287045.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu-Shinohara M, Yamamoto T, Toh H, Kazuki Y, Kazuki K, Imoto J, Ikeo K, Oshima M, Shirahige K, Iwama A, et al. 2019. Aging of spermatogonial stem cells by Jnk-mediated glycolysis activation. Proc Natl Acad Sci 116: 16404–16409. 10.1073/pnas.1904980116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khiati S, Baechler SA, Factor VM, Zhang H, Huang SN, Rosa ID, Sourbier C, Neckers L, Thorgeirsson SS, Pommier Y. 2015. Lack of mitochondrial topoisomerase I (TOP1mt) impairs liver regeneration. Proc Natl Acad Sci 112: 11282–11287. 10.1073/pnas.1511016112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H, Avarbock MR, Schmidt JA, Brinster RL. 2009. Spermatogonial stem cells derived from infertile Wv/Wv mice self-renew in vitro and generate progeny following transplantation. Biol Reprod 81: 293–301. 10.1095/biolreprod.109.075960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kanatsu-Shinohara M, Morimoto H, Kazuki Y, Takashima S, Oshimura M, Toyokuni S, Shinohara T. 2009. Genetic reconstruction of mouse spermatogonial stem cell self-renewal in vitro by Ras-cyclin D2 activation. Cell Stem Cell 5: 76–86. 10.1016/j.stem.2009.04.020 [DOI] [PubMed] [Google Scholar]
- Lu AL, Li X, Gu Y, Wright PM, Chang DY. 2001. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys 35: 141–170. 10.1385/CBB:35:2:141 [DOI] [PubMed] [Google Scholar]
- Ma W, Tessarollo L, Hong SB, Baba M, Southon E, Back TC, Spence S, Lobe CG, Sharma N, Maher G, et al. 2003. Hepatic vascular tumors, angiectasis in multiple organs, and impaired spermatogenesis in mice with conditional inactivation of the VHL gene. Cancer Res 63: 5320–5328. [PubMed] [Google Scholar]
- Marti HH, Katschinski DM, Wagner KF, Schäffer L, Stier B, Wenger RH. 2002. Isoform-specific expression of hypoxia-inducible factor-1α during the late stages of mouse spermiogenesis. Mol Endocrinol 16: 234–243. [DOI] [PubMed] [Google Scholar]
- Meistrich ML, van Beek MEAB. 1993. Spermatogonial stem cells. In Cell and molecular biology of the testis (ed. Desjardins C, Ewing LL), pp. 266–297. Oxford University Press, New York, NY. [Google Scholar]
- Morimoto H, Iwata K, Ogonuki N, Inoue K, Atsuo O, Kanatsu-Shinohara M, Morimoto H, Yabe-Nishimura C, Shinohara T. 2013. ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell 12: 774–786. 10.1016/j.stem.2013.04.001 [DOI] [PubMed] [Google Scholar]
- Morimoto H, Kanatsu-Shinohara M, Ogonuki N, Kamimura S, Ogura A, Yabe-Nishimura C, Mori Y, Morimoto T, Watanabe S, Otsu K, et al. 2019. ROS amplification drives mouse spermatogonial stem cell self-renewal. Life Sci Alliance 2: e201900374 10.26508/lsa.201900374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movafagh S, Crook S, Vo K. 2015. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem 116: 696–703. 10.1002/jcb.25074 [DOI] [PubMed] [Google Scholar]
- Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL. 2006. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc Natl Acad Sci 103: 9524–9529. 10.1073/pnas.0603332103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley MJ, Kaucher AV, Yang QE, Waqas MS, Oatley JM. 2016. Conditions for long-term culture of cattle undifferentiated spermatogonia. Biol Reprod 95: 14 10.1095/biolreprod.116.139832 [DOI] [PubMed] [Google Scholar]
- Rodríguez-Jiménez FJ, Moreno-Manzano V, Lucas-Dominguez R, Sánchez-Puelles J. 2008. Hypoxia causes downregulation of mismatch repair system and genomic instability in stem cells. Stem Cells 26: 2052–2062. 10.1634/stemcells.2007-1016 [DOI] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty S, Elson D, Gassmann M, Arbeit JM, Johnson RS. 2000. Hypoxia-inducible factor-1α is a positive factor in solid tumor growth. Cancer Res 60: 4010–4015. [PubMed] [Google Scholar]
- Scadden DT. 2006. The stem-cell niche as an entity of action. Nature 441: 1075–1079. 10.1038/nature04957 [DOI] [PubMed] [Google Scholar]
- Shinohara T, Kanatsu-Shinohara M. 2020. Transgenesis and genome editing of mouse spermatogonial stem cells by lentivirus pseudotyped with Sendai virus F protein. Stem Cell Reports 14: 447–461. 10.1016/j.stemcr.2020.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon L, Ekman GC, Tyagi G, Hess RA, Murphy KM, Cooke PS. 2007. Common and distinct factors regulate expression of mRNA for ETV5 and GDNF, Sertoli cell proteins essential for spermatogonial stem cell maintenance. Exp Cell Res 313: 3090–3099. 10.1016/j.yexcr.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Takahashi N, Davy PM, Gardner LH, Mathews J, Yamazaki Y, Allsopp RC. 2016. Hypoxia inducible factor 1α is expressed in germ cells throughout the murine life cycle. PLoS One 11: e0154309 10.1371/journal.pone.0154309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takehashi M, Kanatsu-Shinohara M, Inoue K, Ogonuki N, Miki H, Toyokuni S, Ogura A, Shinohara T. 2007. Adenovirus-mediated gene delivery into mouse spermatogonial stem cells. Proc Natl Acad Sci 104: 2596–2601. 10.1073/pnas.0609282104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka J, Blaikie FH, Smith RA, Murphy MP. 2008. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radc Biol Med 44: 1406–1419. 10.1016/j.freeradbiomed.2007.12.036 [DOI] [PubMed] [Google Scholar]
- Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC Jr, Raleigh JA. 1998. Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol 71: 270–277. 10.1006/gyno.1998.5163 [DOI] [PubMed] [Google Scholar]
- Varuzhanyan G, Rojansky R, Sweredoski MJ, Graham RLJ, Hess S, Ladinsky MS, Chan DC. 2019. Mitochondrial fusion is required for spermatogonial differentiation and meiosis. Elife 8: e51601 10.7554/eLife.51601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Juhaszova M, Sollott SJ. 2014. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950. 10.1152/physrev.00026.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.