

Here, Purohit et al. investigated the mechanisms that drive innate therapeutic resistance in pancreatic cancer. They show that increased ataxia-telangiectasia group D-associated gene (ATDC), which is overexpressed in pancreatic cancer and promotes tumor growth and metastasis, protects cancer cells from reactive oxygen species (ROS) via stabilization of nuclear factor erythroid 2-related factor 2 (NRF2). Their findings define a novel role for ATDC in regulating redox balance and chemotherapeutic resistance by modulating NRF2 activity.
Keywords: ATDC (Trim29), chemotherapeutic resistance, pancreatic cancer, tumor growth and invasion
Abstract
Pancreatic ductal adenocarcinoma is a lethal disease characterized by late diagnosis, propensity for early metastasis and resistance to chemotherapy. Little is known about the mechanisms that drive innate therapeutic resistance in pancreatic cancer. The ataxia-telangiectasia group D-associated gene (ATDC) is overexpressed in pancreatic cancer and promotes tumor growth and metastasis. Our study reveals that increased ATDC levels protect cancer cells from reactive oxygen species (ROS) via stabilization of nuclear factor erythroid 2-related factor 2 (NRF2). Mechanistically, ATDC binds to Kelch-like ECH-associated protein 1 (KEAP1), the principal regulator of NRF2 degradation, and thereby prevents degradation of NRF2 resulting in activation of a NRF2-dependent transcriptional program, reduced intracellular ROS and enhanced chemoresistance. Our findings define a novel role of ATDC in regulating redox balance and chemotherapeutic resistance by modulating NRF2 activity.
Pancreatic ductal adenocarcinoma (PDA), because of its often late diagnosis, propensity to metastasize early and resistance to chemotherapy, has the worst prognosis of any major malignancy (9% 5-yr survival rate) (Kleeff et al. 2016). Recent advances in therapy have had a modest impact on the mortality rate of the disease, and more effective therapies are needed. The gemcitabine/abraxane (G/A) and FOLFIRINOX chemotherapy regimens are the cornerstones of PDA treatment, but the effects of these treatments are largely short-lived, and resistance typically develops within weeks to months of treatment (Conroy et al. 2011, 2018; Von Hoff et al. 2013; Amrutkar and Gladhaug 2017). Therefore, understanding the mechanisms of therapeutic resistance is paramount to develop novel and more effective strategies to treat PDA.
Regulation of oxidative stress is essential in both tumor development and response to anti-cancer therapies. Alterations in the levels of reactive oxygen species (ROS) can result in different biological outcomes (Martin and Barrett 2002; Gorrini et al. 2013b). At low levels, ROS act as signaling molecules to sustain proliferation and transformation (Mitsushita et al. 2004; Hole et al. 2010; Weinberg et al. 2010; Jeong et al. 2016; Liou et al. 2016), while excessive ROS production damages DNA, proteins, and lipids, inducing cell death (Gao et al. 2009; Holmström and Finkel 2014; Maya-Mendoza et al. 2015). Therefore, modulation of ROS levels is critical for cellular homeostasis. In cancer cells, aberrant activation of signaling and metabolic pathways often results in elevated ROS levels (Vafa et al. 2002; Nogueira et al. 2008). However, cancer cells adapt to increased oxidative stress by elevating multiple antioxidant defense mechanisms (Sablina et al. 2005; Bensaad et al. 2009; DeNicola et al. 2011). Conventional chemotherapy often elevates ROS to induce cell death in cancer cells (Kong et al. 2000; Conklin 2004; Pelicano et al. 2004; Mizutani et al. 2005; Wang et al. 2013; Li et al. 2016). Thus, cytoprotective antioxidation can be a barrier to effective cancer treatment. An improved understanding of the determinants that govern ROS balance may allow identification of new targets to overcome therapeutic resistance.
The transcription factor nuclear factor erythroid-2-related factor 2 (NRF2) is the master regulator of the cellular antioxidant stress response (Menegon et al. 2016; Rojo de la Vega et al. 2018). Under normal conditions, NRF2 protein is rapidly degraded by the ubiquitin proteasome system. This process is mediated by the Kelch-Like ECH associated protein 1 (KEAP1), a component of the cullin 3 (CUL3)-based E3 ubiquitin ligase complex that controls NRF2 levels. KEAP1 is a direct sensor of ROS levels. When ROS levels increase, cysteine residues on KEAP1 are oxidized. This leads to conformational changes that dissociate NRF2 from the KEAP1/CUL3 ubiquitination complex (Tebay et al. 2015). Once released, NRF2 translocates to the nucleus where it binds to genes whose promoters contain an antioxidant response element (ARE) consensus sequence and promotes their expression (Zhu and Fahl 2001). NRF2 target genes include antioxidants, xenobiotic detoxification enzymes, and drug efflux pumps. The collective activation of these transcriptional programs results in a coordinated, cytoprotective response (Tonelli et al. 2018).
Constitutive activation of NRF2 is frequent in human cancers and is related to therapeutic resistance. NRF2 activation in cancer cells results from events that impair KEAP1/NRF2 interaction, such as (1) somatic mutations in KEAP1, CUL3, or NRF2 genes, which occur in 1% of all cancers (Taguchi and Yamamoto 2017); (2) epigenetic silencing of the KEAP1 gene (Wang et al. 2008); (3) accumulation of KEAP1 or NRF2 interacting proteins, such as BRCA1 (Gorrini et al. 2013a), p62 (Xia et al. 2014), and PALB2 (Ma et al. 2012); or (4) cysteine modification of KEAP1 by metabolites, such as fumarate (Akino et al. 2019) and methylglyoxal (Bollong et al. 2018).
Despite the fact that NRF2 and KEAP1 are rarely mutated in PDA (DeNicola et al. 2011; The Cancer Genome Atlas Research Network 2017), NRF2 expression is detectable in ∼90% of tumors and is significantly up-regulated compared with matching normal pancreatic epithelium (Lister et al. 2011b; Soini et al. 2014). The mechanisms regulating NRF2 expression and stability in PDA are only partially known. Activation of oncogenic KRAS is observed in ∼90%–95% of human PDA (The Cancer Genome Atlas Research Network 2017) and is considered an early and driving event (di Magliano and Logsdon 2013). Oncogenic KRAS requires ROS to promote the development of PDA precursor lesions (pancreatic intraepithelial neoplasia [PanIN]) (Liou et al. 2016) and transcriptionally enhances the expression of NRF2 to lower intracellular ROS and prevent the accumulation of toxic levels of ROS (DeNicola et al. 2011). Consistent with this observation, the genetic ablation of NRF2 inhibits oncogenic KRAS-driven proliferation and tumorigenesis (DeNicola et al. 2011; Hamada et al. 2017), indicating that elevated ROS levels represent a major barrier to pancreatic cancer initiation and progression.
We have previously demonstrated that ataxia-telangiectasia group D complementing gene (ATDC, gene symbol TRIM29), is highly overexpressed in PDA (Logsdon et al. 2003; Wang et al. 2009). The ATDC gene was initially described as a candidate gene responsible for the genetic disorder ataxia-telangiectasia (AT) because its expression increased radiation resistance of AT5BI (AT-D) cells (Kapp et al. 1992). However, ATDC was later dismissed as the AT gene after the gene responsible for ataxia-telangiectasia mutated (ATM) was identified (Savitsky et al. 1995). We have observed that ATDC overexpression promotes KRAS-driven tumorigenesis and stimulates epithelial-to-mesenchymal transition (EMT) and invasion through activation of the β-catenin pathway (Wang et al. 2009, 2015). Loss of ATDC completely prevents PDA development in a PDA genetically engineered mouse model driven by the expression of oncogenic KRASG12D (Wang et al. 2019). ATDC also acts as a mediator of resistance to multiple forms of DNA damage, including ionizing radiation (Wang et al. 2014; Yang et al. 2015).
Since ATDC is a potential mediator of oncogenic KRAS signaling and imparts resistance to DNA damage, we sought to determine whether ATDC mediates innate resistance to chemotherapy in PDA and to define the associated mechanism by which this occurs. This study revealed a novel, previously unknown role of ATDC as a critical modulator of NRF2 protein stability and antioxidant activity in PDA and a predictive biomarker of chemoresistance in PDA.
Results
ATDC mediates resistance to chemotherapy and regulates endogenous ROS levels in vitro
To evaluate whether ATDC promotes resistance to chemotherapy, we screened a panel of PDA cell lines, including patient-derived primary PDA cell lines (referred to here as NYU lines), and selected three cell lines each with low (S2-013, MIA PaCa-2, and NYU32) and elevated (Capan-2, HPAC, and NYU28) endogenous levels of ATDC expression (Supplemental Fig. S1A). In each model, either shRNA-mediated knockdown or overexpression of ATDC was used to generate isogenic cell lines (Supplemental Fig. S1B,C), which differed only by the levels of ATDC expression. To evaluate whether ATDC could promote resistance to chemotherapy, cells were treated with increasing doses of gemcitabine (Gem) or 5-fluorouracil (5-FU), two agents commonly used for PDA treatment in the clinical setting. In all cases, cells with high ATDC levels showed increased survival after 72 h of Gem or 5-FU treatment (Fig. 1A,B; Supplemental Table S1), while ATDC knockdown decreased cell survival with these treatments. Similar results were obtained with hydrogen peroxide (H2O2) treatment (Supplemental Fig. S1D). Since all of these treatments induce ROS (Hwang et al. 2001; Donadelli et al. 2007; Arora et al. 2013; Palmbos et al. 2015), we tested whether ATDC expression might regulate ROS levels as a mechanism to protect cells against chemotherapy-induced cell death. We evaluated ROS levels using the fluorogenic probe CellROX and measured the ratio between reduced and oxidized glutathione (GSH/GSSG). ATDC levels inversely correlated with ROS levels, and positively correlated with the GSH/GSSG ratio (Fig. 1C,D). Similar findings were observed in patient-derived NYU cell lines (Supplemental Fig. S1E). Oxidizing cytosolic conditions may lead to ROS-mediated damage of macromolecules, including lipid peroxidation (Gaschler and Stockwell 2017). We therefore evaluated lipid oxidation in the presence and absence of ATDC and confirmed that overexpression of ATDC reduced levels of lipid peroxidation (Fig. 1E). Overall, these data indicated that elevation of ATDC levels imparts resistance to oxidative stress in PDA, both at steady state as well as under the oxidizing conditions of treatment with chemotherapy or H2O2.
Figure 1.
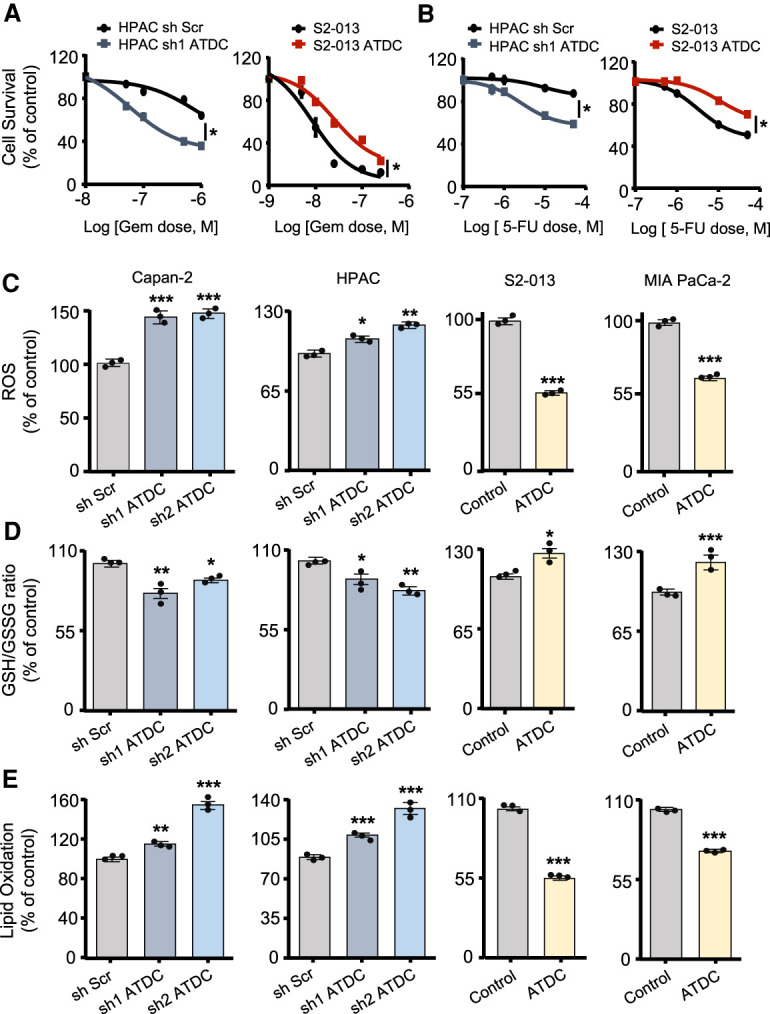
ATDC regulates oxidative stress and chemoresistance in pancreatic cancer cells. Dose response curves for gemcitabine (GEM) (A) and 5-fluorouracil (5-FU) (B) treatment in S2-013 or HPAC cells with either ATDC overexpression (S2-013) or ATDC shRNA expression (HPAC). (C) Steady state ROS levels in control (Scramble, Scr) and sh ATDC Capan-2 and HPAC cells, and control (vector [Vec]) and ATDC-overexpressing S2-013 and MIA Paca-2 cells. (D) Ratio of reduced (GSH) to oxidized (GSSG) glutathione in the PDA cell lines listed in C. (E) Lipid peroxidation levels in the same isogenic PDA cell lines listed in C. Data are representative of three independent experiments. (*) P < 0.05, (**) P < 0.01, (***) P < 0.005. Mean ± SEM.
ATDC-induced alterations in ROS levels are mediated by NRF2
Previous work has established that oncogenic KRAS regulates cellular antioxidant response in PDA cells through increased NRF2 gene (NFE2L2) mRNA expression (DeNicola et al. 2011). Since ATDC offers protection to redox stressors and both of these proteins are up-regulated in oncogenic KRAS driven tumors (Wang et al. 2009; DeNicola et al. 2011), we sought to determine whether there might be a functional relationship between ATDC and NRF2 levels. The silencing of ATDC reduced, while overexpressing ATDC increased NRF2 protein levels in four established PDA cell lines and the two low-passage NYU PDA cell lines (Fig. 2A; Supplemental Fig. S2A). There was, however, no change in the transcript levels of NFE2L2 (NRF2) upon ATDC modulation (Fig. 2B; Supplemental Fig. S2B), suggesting that ATDC regulates NRF2 expression post-transcriptionally. NRF2 knockdown did not affect ATDC levels, indicating that ATDC functions upstream of NRF2 (Supplemental Fig. S2C). As we have previously reported (Wang et al. 2015, 2019), we did not observe changes in KRAS expression levels or activity upon modulation of ATDC expression (Supplemental Fig. S2D), excluding the possibility that the observed changes in NRF2 levels were due to effects of ATDC on KRAS. We did observe that knockdown of KRAS significantly decreased ATDC and NRF2 protein levels (Supplemental Fig. S2E,G). Interestingly, while ATDC knockdown did not affect NRF2 mRNA levels, KRAS knockdown decreased NRF2 mRNA levels by ∼30%, and this knockdown was not rescued by overexpression of ATDC (Supplemental Fig. S2F), suggesting that KRAS may have an alternative contributing mechanism of increasing NRF2 transcription independent of ATDC (DeNicola et al. 2011).
Figure 2.

ATDC-induced alterations in ROS levels are mediated by NRF2. (A) Western blot assays of NRF2 levels in PDA cells with stable knockdown (Capan-2 and HPAC) or overexpression (S2-013 and MIA PaCa-2) of ATDC ([Scr] Scramble, [control] vector). (B) Expression of ATDC and NRF2 mRNA levels in response to modulation of ATDC expression. (C) After cell fractionation, nuclear and cytoplasmic levels of NRF2 in Capan2 or S2-013 cells with altered levels of ATDC were determined by Western blotting. (D) Quantification of nuclear and cytoplasmic levels of NRF2 in C. (E,F) Effect of ATDC knockdown (Capan-2 and HPAC cells) (E) or overexpression (S2-013 and MIA PaCa-2 cells) (F) on the mRNA expression of the NRF2-regulated genes. (NOQ1) NAD(P)H quinone dehydrogenase 1, (PRDX1) peroxiredoxin 1, (HMOX1) hemoxygenase 1, (GCLC) glutamate-cysteine ligase catalytic subunit, (TXN) thioredoxin, (GSTM1) glutathione S-transferase µ1, (GSTM3) glutathione S-transferase µ3, (FTL) ferritin light chain. (G) Expression of NRF2 target proteins HMOX1 and NQO1 in PDA cells with ATDC knockdown (Capan-2) and overexpression (S2-013). (H) Effect of NRF2 knockdown on intracellular ROS levels in control (vector) and ATDC-overexpressing S2-013 and MIA PaCa-2 cells. (#) P < 0.05 versus sh Scr, (ns) not significant. All experiments were repeated three times. (*) P < 0.05, (**) P < 0.01, (***) P < 0.005. Mean ± SEM
Consistent with an increase in total NRF2 levels, we observed that high ATDC levels correlated with an increase in the nuclear levels of NRF2 (Fig. 2C,D; Supplemental Fig. S2HI). Modulation of ATDC levels caused a corresponding alteration in expression of NRF2-regulated genes, such as NQO1, PRDX1, HMOX1, GCLC, TXN1, and FTL1 (Fig. 2E,F; Supplemental Fig. S2J) as well as a luciferase reporter gene driven by a promoter containing multiple antioxidant response elements (ARE) (Supplemental Fig. S2K). A concurrent decrease or increase in the protein levels of NRF2 targets NQO1 and HMOX1 was also observed in isogenic cells upon ATDC silencing or overexpression, respectively (Fig. 2G; Supplemental Fig. S2L). To evaluate whether ATDC-induced changes in ROS were mediated by NRF2, we silenced NRF2 in ATDC-overexpressing PDA cells and found NRF2 knockdown reversed ATDC-mediated reduction in ROS levels (Fig. 2H; Supplemental Fig. S2M). These data established that ATDC regulates ROS levels in cancer cells and suggest that this occurs by post-transcriptional regulation of NRF2.
ATDC stabilizes NRF2 through binding with KEAP1
To determine the mechanism by which ATDC regulates NRF2 protein levels, we first tested whether manipulation of ATDC levels altered KEAP1 or CUL3 protein levels in PDA cells, but no changes were observed (Supplemental Fig. S3A). Because we did not see changes in NRF2 mRNA levels in response to alteration of ATDC levels, we next hypothesized that ATDC might promote NRF2 activity by binding to either KEAP1 or NRF2 and disrupting the KEAP1/NRF2 interaction. Indeed, KEAP1 and ATDC proteins were reciprocally coimmunoprecipitated when tagged constructs were coexpressed in HEK293 cells lacking endogenous ATDC expression (Fig. 3A,B; Supplemental Fig. S3B) and endogenously in Capan-2 PDA cells (Fig. 3C). ATDC did not bind to NRF2 (Supplemental Fig. S3C). To delineate the ATDC domain that binds with KEAP1, we generated HEK293 cells expressing wild-type (WT) FLAG-tagged ATDC or a series of FLAG-tagged ATDC deletion mutants (Fig. 3D). In coimmunoprecipitation assays, we observed that the N-terminal 220 amino acids of ATDC were needed to bind to KEAP1 (Fig. 3E). Likewise, using HEK293 cells expressing FLAG-tagged WT or FLAG-tagged KEAP1 mutants, we confirmed that KEAP1 binds to ATDC through its C-terminal Kelch domain (Fig. 3F,G). Furthermore, using purified GST-ATDC and MYC-KEAP1 in GST pull-down assays, we established the direct interaction of these proteins in vitro (Kd = 2.865 nM) (Fig. 3H; Supplemental Fig. S3D), suggesting KEAP1/ATDC binding in a high-affinity mode. To determine whether an increase in ATDC levels might decrease the interaction of KEAP1 and NRF2, FLAG-ATDC, HA-KEAP1, and MYC-NRF2 were cotransfected into HEK293 cells. Coimmunoprecipitation assays confirmed that an increase in ATDC competitively inhibited the KEAP1 and NRF2 interaction and stabilized MYC-NRF2 levels (Fig. 3I). To support this finding, coimmunoprecipitation assays were also performed in HPAC cells expressing control or ATDC shRNA. Indeed, knockdown of endogenous ATDC significantly increased KEAP1/NRF2 binding and subsequently decreased NRF2 protein levels in HPAC cells (Supplemental Fig. S3E). The fraction of KEAP1 bound to endogenous ATDC was ∼ 65% of total KEAP1 in HPAC cells (Supplemental Fig. S3F). To further verify ATDC/KEAP1 binding specificity, FLAG-ATDC and HA-KEAP1 transfected HEK293 cells were pretreated with KI-696 (1 µM, 6 h), a small molecule inhibitor that specifically disrupts the KEAP1-NRF2 interaction (Davies et al. 2016; Sayin et al. 2017). ATDC-KEAP1 binding was significantly inhibited by KI-696 treatment (Supplemental Fig. S3G). Coimmunoprecipitation experiments with HEK293 cells transfected with FLAG-ATDC and MYC-CUL3 revealed no evidence of binding of ATDC to CUL3, confirming that ATDC regulation of NRF2 levels is through its interaction with KEAP1 and not CUL3 (Supplemental Fig. S3H). Furthermore, coimmunoprecipitation assays revealed that the KEAP1-CUL3 interaction was not altered by ATDC expression (Supplemental Fig. S3I), supporting the specificity of the ATDC/KEAP1 interaction in NRF2 regulation.
Figure 3.

ATDC stabilizes NRF2 through binding with KEAP1. (A–C) Cell lysates from HEK293 cells transfected with FLAG-ATDC and HA-KEAP1, or Capan2 cells transfected with KEAP1 antibody were subjected to immunoprecipitation (IP) with FLAG (A), HA (B), or ATDC (C) antibodies. Immunocomplexes were resolved by SDS-PAGE and subjected to Western analysis with HA (A), FLAG (B), or KEAP1 (C) antibodies. Whole-cell lysates (WCL) were probed with HA, FLAG (A,B), or ATDC (C) antibodies in Western blot assays. IP with IgG served as a negative control. (D) Schematic of FLAG-ATDC mutants used to determine KEAP1-binding region in ATDC. (E) Cell lysates from HEK293 cells transfected with FLAG-tagged ATDC, ATDC mutants or HA-KEAP1 were subjected to IP with FLAG antibody. Immunocomplexes and WCL were resolved by SDS-PAGE and subjected to Western analysis with HA and FLAG antibodies. IP with IgG served as a negative control. The ATDC N-terminal is required for binding with KEAP1. (F) Schematic of KEAP1 deletion mutants used to determine the ATDC-binding domain of KEAP1. (G) Cell lysates from HEK293 cells transfected with FLAG-tagged KEAP1 or FLAG-tagged KEAP1 mutants were subjected to IP with FLAG antibody. Immunocomplexes and WCL were resolved by SDS-PAGE and subjected to Western analysis with ATDC and FLAG antibodies. IP with IgG served as a negative control. Immunoblot results confirm KEAP1 interacts with ATDC through its KELCH domain. (H) In vitro binding assay for GST-ATDC with Myc-KEAP1. (I) Cell lysates from HEK293 cells transfected with FLAG-ATDC, HA-KEAP1, and Myc-NRF2 were subjected to IP with HA antibody. Immunocomplexes and WCL were resolved by SDS-PAGE and subjected to Western analysis with FLAG, HA, Myc, or β-actin antibodies. (J) Cell lysates from S2-013 cells transfected with ATDC and HA-ubiquitin (HA-Ub) were subjected to IP with HA antibody. Immunocomplexes and WCL were resolved by SDS-PAGE and subjected to Western analysis with NRF2, ATDC, or β-actin antibodies. IP with IgG served as a negative control. (K) Densitometric quantification of Ub-NRF2 from J. All experiments were repeated three times. (*) P < 0.05. Mean ± SEM.
KEAP1 degrades NRF2 by facilitating its ubiquitination (Ub) by Cul3 ubiquitin ligase (Sykiotis and Bohmann 2010). To test whether ATDC inhibits the ubiquitination of NRF2 by interrupting its interaction with KEAP1, we transfected HA-Ubiquitin (HA-Ub) into S2-013 cells and performed coimmunoprecipitation assays. Overexpression of ATDC significantly decreased the ubiquitination of NRF2 (Fig. 3J,K). We reasoned that if ATDC inhibited proteasomal degradation of NRF2 by regulating its interaction with KEAP1, treatment with the proteasome inhibitor MG132 would increase NRF2 protein to comparable levels independent of ATDC expression. Indeed, treatment with the proteasome inhibitor MG132 (10 µM, 6 h) increased NRF2 levels both in control and ATDC-overexpressing cells to comparable levels (Supplemental Fig. S3J). MG132 treatment also re-established NRF2 to similar levels in cells with and without stable knockdown of ATDC, indicating that loss of ATDC results in increased NRF2 proteasomal degradation (Supplemental Fig. S3K). To further prove that ATDC mediates NRF2 protein stabilization by inhibiting the KEAP1-NRF2 interaction, we treated S2-013 cells overexpressing ATDC with KI-696. ATDC's effects on NRF2 expression were abolished in the presence of KI-696 (1 µM, 6 h) (Supplemental Fig. S3L,M), indicating that ATDC's ability to regulate NRF2 levels requires an intact KEAP1-NRF2 interaction. We also observed a positive correlation between ATDC levels and NRF2 protein stability when cells were treated with the protein synthesis inhibitor cycloheximide (CHX), supporting our finding that ATDC regulates NRF2 expression levels post-transcriptionally (Supplemental Fig. S3N,O). Overall these findings demonstrate that binding of ATDC with KEAP1 stabilizes NRF2 in PDA cells by diminishing NRF2 proteasomal degradation.
ATDC-mediated increase in NRF2 regulates PDA cell growth and invasion
We have previously shown that ATDC regulates growth and invasion in PDA cells (Wang et al. 2009, 2015, 2019). To evaluate whether ATDC effects on cell growth and invasion are mediated by NRF2, we first silenced ATDC in the absence or presence of NRF2 rescue (Fig. 4A,B). In HPAC cells, ATDC knockdown decreased cell proliferation, invasion, and increased ROS levels, and these effects were recovered by NRF2 rescue (Fig. 4B–D). In S2-013 PDA cells, ATDC overexpression significantly increased cell proliferation and invasion, and decreased ROS levels (Fig. 4E–H; Supplemental Fig. S4C,D). These effects were blocked, at least in part, by NRF2 knockdown. Indeed, we observed similar effects of ATDC and NRF2 knockdown on cell proliferation and ROS levels in HPAC and Capan-2 cells (Supplemental Fig. S4A,B,G,H). Treatment with the antioxidants N-acetyl cysteine (NAC; 0–10 mM, 24 h) or trolox (0–200 µM, 24 h) reversed the increase in ROS due to ATDC or NRF2 knockdown and rescued ROS and cell proliferation in Capan-2 and HPAC cells when ATDC was silenced (Fig. 4I; Supplemental Fig. S4E–H). Conversely, NAC treatment did not further increase S2-013 cell proliferation when ATDC was overexpressed (Fig. 4I; Supplemental Fig. S4E). Overall, these data indicate that ATDC-mediated regulation of NRF2 and ROS is critical for PDA cell proliferation.
Figure 4.

ATDC-mediated increase in NRF2 regulates growth and invasion in PDA cells in vitro. (A,E) Immunoblotting showing levels of ATDC, NRF2, KEAP1, and NRF2 target NQO1 in HPAC cells with ATDC knockdown with or without subsequent NRF2 overexpression (A) and S2-013 cells overexpressing ATDC with or without subsequent knockdown of NRF2 (E). (B–D) Cell proliferation (B), invasion (C), and ROS formation (D) in HPAC cells with or without sh ATDC and NRF2 rescue. (F–H) Cell proliferation (F), invasion (G), and ROS formation (H) in S2-013 cells with or without ATDC overexpression and sh NRF2 rescue. (I) Cell proliferation in ATDC overexpressing S2-013 or Capan2 or HPAC cells with ATDC knockdown treated with increasing concentrations of NAC (0–10 mM). (J) Western blot analysis of ATDC and NRF2 expression in S2-013 cells expressing full-length ATDC or the ATDCΔ220N deletion. β-actin was used as a loading control. (K–M) Quantification of cell proliferation (K), invasion (L), and ROS levels (M) in S2-013 cells expressing ATDC or ATDCΔ220N. All experiments were repeated three times. (*) P < 0.05, (**) P < 0.01, (***) P < 0.005. Mean ± SEM.
To confirm that the ATDC protumorigenic function requires NRF2, we generated S2-013 cells expressing an ATDC-Δ220N mutant unable to bind KEAP1 and compared it with cells overexpressing WT ATDC (Fig. 4J). Notably, this mutation does not prevent ATDC from increasing the expression of a TCF4 reporter, indicating that this mutant is still able to activate the β-catenin pathway as previously reported (Supplemental Fig. S4I; Wang et al. 2009) and does not cause a complete loss of function. ATDC-overexpressing S2-013 cells had significantly increased NRF2 protein levels, cell proliferation, and invasion as compared with isogenic cells overexpressing ATDC-Δ220N (Fig. 4K,L). ATDC-mediated reduction in ROS was not observed when using the ATDC-Δ220N mutant (Fig. 4M).
An ATDC-KEAP1 interaction is required for ATDC-mediated PDA growth and invasion in vivo
We next evaluated the role of ATDC/NRF2 signaling on growth and metastasis in vivo by orthotopically implanting 0.5 × 105 S2-013 cells expressing vector control, ATDC alone or in combination with shNRF2, and ATDC-Δ220N in pancreata of NSG mice. As shown in Figure 5, A and B, overexpression of ATDC, but not ATDC-Δ220N, significantly promoted tumor growth (measured at 4 wk) compared with vector controls, and this effect was blocked when ATDC was coexpressed with shNRF2. Treatment of mice with NAC rescued the effect of NRF2 silencing in ATDC-overexpressing tumors, confirming that NRF2-dependent reduction of ROS, promoted by ATDC, is critical for ATDC-mediated PDA growth (Fig. 5A,B). ATDC overexpression in tumor cells increased in vivo expression of NRF2 protein and the target gene NQO1, an observation not seen with ATDC-Δ220N or coexpression of ATDC with NRF2 shRNA (Fig. 5C). ATDC also promoted lung and liver metastasis in mice in a NRF2- and ROS-dependent manner (Fig. 5E,F).
Figure 5.

ATDC-mediated increase in NRF2 regulates growth and invasion in vivo. NSG mouse pancreata (n=10/group) were implanted with S2-013 control (empty vector), ATDC, ATDCΔ220N, ATDC + shNRF2 cells. N-acetyl cysteine was provided in drinking water (1 g/L) to a cohort of mice implanted with ATDC + shNRF2 cells. (A) Representative images of tumors harvested at the end of the study. (B) Measurement of tumor volume in experimental groups depicted as scatter plots with error bars denoting SEM. (C) Hematoxylin and eosin (H&E) staining and immunohistochemical staining for NRF2, ATDC, and NQO1 in harvested tumors. (D) Hematoxylin and eosin (H&E) staining of liver metastases. Quantification of liver metastases (E) and lung metastases (F) in all groups. All experiments were performed three times. (**) P < 0.01, (***) P < 0.005. Mean ± SEM.
In a complementary set of experiments, we evaluated the effect of ATDC knockdown on tumor growth in vivo. HPAC cells (5 × 105) either expressing a control scramble shRNA (shScr) or a shRNA targeting ATDC with or without overexpression of NRF2 were orthotopically implanted in the pancreata of NSG mice. At the end of 6 wk, the mice were sacrificed, and tumor weight and volume were determined. We observed that sh Scr tumors had increased size, weight, and volume compared with sh-ATDC tumors, while NRF2 overexpression in the presence of sh-ATDC partially rescued tumor growth (Supplemental Fig. S5A,B). Consistent with our in vitro data, the silencing of ATDC significantly reduced NRF2 levels and NQO1 expression in vivo (Supplemental Fig. S5C). The silencing of ATDC also diminished lung and liver metastases in mice (Supplemental Fig. S5D,E), an effect that was mitigated by coexpression of NRF2.
To verify the impact of ATDC silencing in established tumors, HPAC cells expressing inducible shRNA (ish) ATDC (1 × 106) were orthotopically implanted in the pancreata of NSG mice. Mice were imaged by ultrasound weekly to monitor tumor size. When the ish-ATDC tumors reached the size of 200 mm3, doxycycline (625 mg/kg) was added into the diet in the experimental group. At the end of 4 wk, the mice were sacrificed and tumor volume, ATDC, and NRF2 protein levels were determined. Inducible knockdown of ATDC in established tumors resulted in a significant decrease in NRF2 protein levels and tumor volume compared with control tumors (Supplemental Fig. S5F,G).
We have previously demonstrated that ATDC promotes PDA growth and metastasis in ATDC; KrasG12D; p48-Cre (AKC) mice (Wang et al. 2015), and knockout of ATDC blocks PDA initiation and progression in KrasG12D; p53−/+; pdx1-Cre; ATDC−/− (KPCA) mice (Wang et al. 2019). To determine whether ATDC regulates NRF2 protein levels in genetically engineered mouse models of cancer, we performed Western blotting and coimmunofluorescent staining of pancreatic tissues from 3-mo-old KC and AKC mice and found that elevation in ATDC levels correlated with elevated NRF2 protein levels (Supplemental Fig. S5H–L). Conversely, knockout of ATDC in KPC mice (KPCA) markedly decreased NRF2 protein levels in pancreatic tissue (Supplemental Fig. S5M–O).
High ATDC expression correlates with poor survival in PDA patients
To determine the clinical relevance of our findings, we evaluated the correlation between ATDC expression, NRF2 expression, and overall survival in the TCGA PDA cohort. We found that ATDC expression positively correlated with NQO1 expression as well as expression of a previously described NRF2 core target gene signature (Supplemental Fig. S6A,B; Romero et al. 2017), chosen as proxies of NRF2 activity. Because the majority of PDA express ATDC, we examined the distribution of log-transformed values for ATDC and found them to be negatively skewed (skewness of −1.1). All of the outlier (median ± 1.5 median absolute deviation) samples, representing ∼15% of the total samples, had low expression levels (Supplemental Fig. S6C). Therefore, we grouped TCGA samples based on high and low (low = bottom 20 percentile) ATDC or NQO1 gene expression and found that elevated expression of either ATDC or NQO1 was associated with decreased survival (Fig. 6A,B). Similar results were obtained either when the ATDC-high and ATDC-low 20 percentile samples were directly compared (Supplemental Fig. S6D), or when samples were separated equally (Supplemental Fig. S6E).
Figure 6.

ATDC mediates chemoresistance in PDA by NRF2 signaling. Relative survival in patients with elevated ATDC (A) or NQO1 (B) expression based on the TCGA PAAD data set. (C) Cell proliferation in control S2-013 cells or S2-013 cells overexpressing ATDC in the absence or presence of shNRF2. Cells were left untreated or treated with gemcitabine (GEM; 5 nM) for 3 d. (D) Effect of NRF2 overexpression on chemoresistance in HPAC control (sh Scr) and sh ATDC cells as measured by cell proliferation. Cells were treated with GEM (5 nM) for 3 d. (E) Effect of the ATDCΔ220N mutant on chemoresistance in S2-013 cells. Cells were treated with GEM (5 nM) for 3 d. (F) Quantification of tumor volume in control (vector), ATDC, ATDC shNRF2, and Δ220N ATDC groups treated with vehicle or GEM. (G) Representative images of tumors indicating tumor sizes in the above study groups. All experiments were performed in triplicate. (*) P < 0.05, (**) P < 0.01, (***) P < 0.005. Mean + SEM. (H) Schematic representation showing the mechanism of ATDC-mediated regulation of NRF2 protein levels and antioxidant response resulting in increased growth, metastasis, and chemotherapy resistance.
NRF2 mediates chemoresistance in ATDC expressing PDA tumors
One of the defining features of PDA is an innate resistance to chemotherapy. Gemcitabine (Gem) treatment has been shown to generate ROS (Donadelli et al. 2011; Arora et al. 2013; Ju et al. 2015; Zhang et al. 2016). Gem-mediated ROS induction activates a feedback loop, which results in NRF2-dependent detoxification and resistance to treatment, and this is abolished by silencing of NRF2 (Palmbos et al. 2015). Because ATDC overexpression increases the resistance to Gem in vitro (Fig. 1A; Supplemental Table S1), we evaluated whether ATDC overexpression could reduce the amount of ROS generated by Gem treatment. Treatment of S2-013 cells with increasing doses of GEM (0.1–1 µM, 24 h) resulted in a significant increase in ROS, which was blocked by overexpression of ATDC and was dependent on NRF2 (Supplemental Fig. S6F). We hypothesized that the ability of ATDC to regulate ROS through NRF2 signaling would impact cell survival in response to Gem. We tested this by modulating NRF2 expression in S2-013 and HPAC isogenic cell lines expressing different levels of ATDC. Treatment of S2-013 cells with Gem caused a significant reduction in cell proliferation (Fig. 6C), while overexpression of ATDC promoted cell growth and increased resistance to Gem-induced cell death. Notably, the effects of ATDC were blocked by NRF2 silencing, indicating that ATDC-mediated chemoresistance was dependent on NRF2 (Fig. 6C). Conversely, the coexpression of NRF2 in the setting of ATDC silencing rescued the effects on cell proliferation in GEM-treated cells (Fig. 6D). Of note, knockdown of either ATDC or NRF2 in HPAC cells had similar effects on reducing cell proliferation and increasing sensitivity to Gem (Supplemental Fig. S6G). To test whether an ATDC-KEAP1 interaction was required for ATDC-mediated cell survival and chemoresistance in vitro, we compared the effects of overexpression of ATDC and ATDC-Δ220N and found that the deletion of the KEAP1 interaction domain abolished the ability of ATDC to promote cell proliferation and chemoresistance in S2-013 cells (Fig. 6E).
To test our hypothesis in vivo, we orthotopically implanted S2-013 cells expressing vector control, ATDC alone or in combination with shNRF2, and ATDC-Δ220N in the pancreata of NSG mice and evaluated tumor growth and response to Gem treatment. As shown in Figure 6, F and G, ATDC overexpression significantly promoted tumor growth compared with vector control, and this was partially reduced when ATDC was coexpressed with shNRF2 or by removal of the KEAP1 interaction domain (ATDC-Δ220N). Importantly, both ATDC + shNRF2 and ATDC-Δ220N tumors demonstrated growth inhibition in response to Gem similar to vector control tumors, while ATDC-overexpressing tumors were resistant to Gem treatment. Overall, these data support our hypothesis that ATDC drives enhanced PDA cell survival as well as chemoresistance through activation of a NRF2-dependent signaling pathway (Fig. 6H).
Discussion
Oncogenic mutations elevate the cellular antioxidant response to impart resistance to tumor cells against genotoxic stressors, including chemotherapy treatment (Weinberg et al. 2010; Collins et al. 2012). To orchestrate this response, cancer cells increase the expression of the NRF2-mediated transcriptional program, therefore increasing protection from oxidative stress (DeNicola et al. 2011). The major regulator of NRF2 protein stability is KEAP1, a dimeric protein that facilitates the proteasome degradation of NRF2 by Cullin-3 ubiquitin ligase (Sykiotis and Bohmann 2010). Our present study provides conclusive evidence that ATDC binds to and sequesters KEAP1, promoting NRF2 stabilization, reduction in intracellular ROS, and ultimately an increased survival of cancer cells.
NRF2 expression is elevated in human PDA (Lister et al. 2011a; Soini et al. 2014). However, unlike other tumor types, KEAP1 and NRF2 are rarely mutated in PDA (DeNicola et al. 2011), suggesting that other mechanisms regulate NRF2 expression levels. Oncogenic KRAS has been reported to increase NRF2 mRNA levels in mouse embryonic fibroblasts (DeNicola et al. 2011), and NRF2 is critical for KRAS-driven initiation and development in a PDA mouse model (DeNicola et al. 2011; Hayes et al. 2015; Chio et al. 2016; Hamada et al. 2017). It has also been shown that p62 accumulation promotes pancreatic malignant progression and reprogramming by controlling the p62-NRF2/Axis (Todoric et al. 2017). We provide evidence that ATDC represents an additional critical regulator of NRF2 expression levels in PDA. In agreement with our findings, mutations in KEAP1 or NRF2 are not mutually exclusive with KRAS mutations in lung adenocarcinoma (Singh et al. 2006; The Cancer Genome Atlas Research Network 2014; Berger et al. 2016), and >20% of KRAS mutant samples carry additional mutations in either KEAP1 or NRF2 gene, arguing that KRAS expression itself is not sufficient to elicit a full activation of the NRF2 transcriptional program in human tumors. Consistent with these observations, genetic inactivation of KEAP1 further enhanced KRAS driven tumorigenesis (Romero et al. 2017) and metastasis (Lignitto et al. 2019) in a lung adenocarcinoma mouse model. Notably, ATDC-mediated regulation of NRF2 is independent of KRAS, since neither changes in KRAS expression or activity, nor changes in NRF2 mRNA levels were observed upon modulation of ATDC expression. Here we highlight the importance of ATDC up-regulation as a major mechanism for modulation of NRF2 activity in PDA, in which ATDC overexpression is observed in the majority of PDA cases (Wang et al. 2009).
Our data demonstrate that ATDC positively regulates NRF2 through competing with KEAP1 binding. Mechanistically, the N-terminal domain of ATDC binds to the C-terminal Kelch domain of KEAP1, thereby competing with NRF2 and inhibiting its interaction with KEAP1. The NRF2-Neh2 domain contains two binding motifs, DLG and ETGE, which are responsible for KEAP1/NRF2 binding (Davies et al. 2016; Rojo de la Vega et al. 2018). In an analysis of previously published proteomic/mass spectrometry studies performed to identify KEAP1 protein interaction partners with or without DLG and ETGE motifs, ATDC was not identified as an interacting protein (Hast et al. 2013; Goldfarb et al. 2014; Tamir et al. 2016). However, these studies were performed in cell lines known to either not express ATDC, such as the HEK293 cell line, supporting the notion that experiments performed in cells with low ATDC or altered ATDC localization may not identify ATDC as a KEAP1 binidng partner. Of note, 38 of the 65 identified KEAP1 binding partners, like ATDC, do not possess a DLG or ETGE motif.
Our previous work suggests that ATDC promotes proliferation and invasiveness through activation of the β-Catenin pathway (Wang et al. 2009, 2015, 2019). However, ATDC domains involved in the interaction with KEAP1 or DVL2 (Wang et al. 2009), an activator of β-Catenin pathway, are distinct. Here we find that the ATDC-Δ220N mutant is unable to execute the tumorigenic activities of ATDC, despite being able to activate β-catenin signaling, suggesting that activation of NRF2 is required for key aspects of ATDC function. Additional experiments will be needed to investigate the relative contributions of these unique interactions to ATDC biology and the settings they are operative.
Previously, we and others revealed a role of ATDC in regulating the cellular response to radiation-induced DNA damage in PDA (Yuan et al. 2010; Wang et al. 2014; Yang et al. 2015). In this study, we provide evidence that ATDC also mediates resistance to chemotherapy by regulating NRF2 expression. Our data are consistent with previous studies reporting a role of NRF2 in contributing to chemoresistance in multiple cancers, including PDA (Jaramillo and Zhang 2013). While previous work using organoids derived from tumors of KrasP53R172H NRF2−/− mice showed no difference in Gem responsiveness as compared with their counterparts with intact NRF2 (Chio et al. 2016), a more recent analysis by Hamada et al. (2017) in cells derived from the same mouse model demonstrated increased sensitivity to Gem in the absence of NRF2. The reason for the difference in observed responses is not clear and may be due to differences in cell culture conditions or methods in which the models were generated. Nevertheless, other studies, including those from Hong et al. (2010) demonstrated that NRF2 knockdown increases sensitivity to cisplatin, as well as other chemotherapeutic agents in PDA cells. In our study, we show that ATDC overexpression promotes resistance to Gem and other chemotherapeutic agents, and that down-regulation of NRF2 alone prevents ATDC-mediated resistance to Gem both in vitro and in vivo.
In a recent study (Cheung et al. 2020) interrogating the role of TIGAR and ROS regulation in pancreatic tumorigenesis, the authors show at the early stages of pancreatic tumorigenesis, high TIGAR levels decrease ROS to promote cancer initiation, but at later stages, low TIGAR levels increase ROS and activate MAPK signaling to support metastasis. These behaviors are quite distinct from those we observe with ATDC modulation in both mouse and human systems, highlighting that TIGAR and ATDC appear to play distinct roles in pancreatic tumorigenesis, and the contributions of NRF2 signaling and ROS in this context are also likely distinct. Collectively, these data highlight the complexity of understanding the contribution of ROS to PDA biology, which is likely dependent on the model systems used, genetic and epigenetic changes in the tumor itself, and stage of tumor. All of the experiments performed in our study used human model systems, which may possess distinct differences from the KPC model system. The authors include data that NRF2 deficiency in the KPC model does not improve survival. This is countered by previously published data that deletion of NRF2 in the KPC model decreased the incidence of invasive and metastatic tumors and that NRF2 knockdown in human PDA organoids was essential for human PDA organoid maintenance (Chio et al. 2016). Also, others have shown that NRF2 promotes pancreatic carcinogenesis in the KPC model (Hamada et al. 2017). Our data clearly show that tumor growth and metastasis of human PDA models driven by ATDC is dependent on ROS and NRF2 (Supplemental Table S2). To support this contention, the examination of the TCGA data set shows that patients with an elevated ATDC and a NRF2 core target signature have worse survival. Thus, while there may be conflicting data in GEMM models, data in human systems collectively support an ATDC-driven protumorigenic NRF2 signal that is associated with worse outcomes. Our data also suggests that ATDC expression or an “activated” NRF2 signature represents a novel biomarker for resistance to Gem or FOLFIRINOX regimens. Further studies will be required to formally test this hypothesis in human samples.
In conclusion, our data extend the understanding of ATDC biology and demonstrate that ATDC is a critical upstream regulator of NRF2 activity as well as redox balance in PDA. Since most normal tissues have a low or absent expression of ATDC, targeting the ATDC-KEAP1 interaction may offer a new therapeutic opportunity to block PDA growth and metastasis and enhance responsiveness to chemotherapy regimens, not only in PDA but possibly other types of malignancies with high ATDC expression.
Materials and methods
Cell culture and reagents
The Capan-2, HPAC, MIA PaCa-2, H23, and SW900 cancer cell lines were obtained from the American Type Culture Collection (ATCC). NYU28 and NYU32 cell lines are primary patient-derived pancreatic adenocarcinoma cell lines developed in the Simeone laboratory. S2-013 cells were kindly provided by Dr. Michael Hollingsworth (University of Nebraska Medical Center, Omaha). All cell lines were maintained in Roswell Park Memorial Institute Medium from Gibco. The growth media for all cell lines was supplemented with 10% FBS. Trolox and N-acetyl cysteine were obtained from Sigma.
Creation of isogenic cell lines
ATDC knockdown cells were generated by shRNA-mediated knockdown using ATDC sh1RNA, ATDC sh2RNA, and control shRNA expression vectors as described previously (Wang et al. 2009). Similarly, ATDC-overexpressing S2-013, MIA PaCa-2, and NYU32 cells were generated by lentiviral-mediated overexpression of CMV-ATDC-PLX304 (Genocoepia) and selected for blasticidin resistance (1 µg/mL). NRF2 knockdowns were generated by using Sigma shRNAs SHCLNNV-NM_006164 (TRCN0000007555 as sh1 and TRCN0000007558 as sh2) and selected for puromycin resistance. NRF2 overexpression was generated via CMV-NRF2-PLX304 (Genocoepia) and selected for blasticidin resistance. The full-length ATDC or KEAP1 cDNAs, and the N-terminal or C-terminal mutants of ATDC (Δ220N-, Δ260N-, Δ348N-, and ΔC-) or KEAP1 (ΔIVR-, ΔCTR-, and CTR-) were generated using standard PCR and restriction enzyme procedures into N-terminal p3xFLAG-CMV expression vector as described (Yang et al. 2015). The MYC-NRF2, MYC-tagged CUL3, and HA-tagged KEAP1 cDNA were purchased from Addgene. Lentiviral ATDC (LPP-Z7667-Lv197) or NRF2 (LPP-T3128-Lv197) in pReceiver-Lv197 vectors were purchased from GeneCopeia, and the lentiviral ATDC-Δ220N mutant construct was subcloned into pReceiver-Lv197 vector (GeneCopeia). Full-length recombinant GST-ATDC (LS-G32941) and full-length recombinant MYC-KEAP1 (LS-G66144) proteins used in the GST pull-down binding assays were purchased from LifeSpan BioSciences, Inc. (LsBio).
Creation of HPAC cells with inducible ATDC shRNA
SMARTvector-inducible human ATDC shRNA (ish ATDC; V3SH11252-227098378) was purchased from Horizon. Following the manufacturer's instructions, HPAC cells were infected with ish ATDC. HPAC cells stably expressing ish ATDC were established by selection with medium containing puromycin. ATDC knockdown efficiency in HPACish ATDC cells was verified by Western blot assays 72 h after treatment with 1 µg/mL doxycycline (Doxy).
ROS assays
For total cellular ROS measurement, 100,000 cells per well were plated in 12-well plates. Each sample was plated in quadruplet. After 24 h, cells were trypsinized and three samples were stained with 5 μM CellROX for 15 min, followed by flow cytometric analysis. A fourth sample was used as an unstained background control. Raw values for stained samples were normalized to values for unstained cells and plotted as percent of sh Scr or control cells.
Glutathione assays
Total oxidized and reduced glutathione levels were determined by performing GSH/GSSG-Glo assays (Promega) as per the manufacturer's protocol. Ten-thousand cells/well were plated in 96-well plates. Raw luminescence counts were normalized to cell counts per well.
Peroxidation assays
Cellular oxidation levels were measured by performing BODIPY 493/503 assays as per the manufacturer's protocol (Thermo Fisher Scientific). Raw values for stained samples were normalized to values for unstained cells and plotted as percent of sh Scr or control cells.
Cell proliferation and cytotoxicity assays
Fifty-thousand cells were plated in 24-well plates and treated with indicated agents. At the end of 48 h, cell proliferation and cytotoxicity were determined by performing Cell-Titre Glo assays (Promega) as per the manufacturer's protocol.
Annexin V assays
Annexin V assays were performed by flow cytometry using Annexin V Pacific Blue conjugate antibody according to the manufacturer's protocol (Thermo Fisher Scientific).
Immunoblotting
Immunoblotting was performed as previously described (Zhang et al. 2004) using antibodies to detect ATDC, HMOX1, Fibrillin, GAPDH (SCBT), NRF2 (GeneTex and Abcam), NRF2, NQO1 (Abcam), or FLAG (Sigma Aldrich). For loading controls, blots were simultaneously probed for β-actin (Cell Signaling). All antibodies were used at a dilution of 1:1000. For MG132 and cycloheximide assays, cells were treated with 10 μM MG132 or 100 μg/mL cycloheximide for indicated times and immunoblotting was performed to assess NRF2 stability.
Nuclear and cytoplasmic protein extraction
Nuclear and cytoplasmic protein was extracted from cells by using a compartmental protein extraction kit as per the manufacturer's protocol (Millipore). Extracts were resolved by SDS-PAGE and immunoblotting was performed to determine nuclear and cytoplasmic levels of NRF2 (GeneTex) using GAPDH and fibrillrin (SCBT) as cytoplasmic and nuclear controls, respectively.
Real-time PCR
Total RNA was isolated from 2 × 106 cells using Qiagen RNAeasy kits as per the manufacturer's protocol. Pretreatment with DNase was done to ensure the purity of RNA. Three micrograms of RNA per sample was used to prepare complementary DNA by using a Thermo Scientific Verso cDNA synthesis kit. Real-time PCR was done using specific primers detailed in Supplemental Table S3. Values for each gene were normalized to the expression of β-Actin.
Antioxidant response element (ARE) activity assays
Isogenic cell lines were cotransfected with an ARE luciferase reporter vector and a Renilla luciferase internal control. The ARE-luc vector constitutes a firefly luciferase gene under the control of ARE responsive elements located upstream of a minimal promoter. ARE activity was measured by performing dual-luciferase activity assays as per the manufacturer's protocol (BPS Biosciences).
Invasion assays
Serum-starved cells (150,000) were plated in rehydrated Corning Biocoat Matrigel invasion chambers in serum-free DMEM/RPMI. Medium containing 0.1% FBS was added to the lower chamber as a chemoattractant. Twenty-four hours following plating, invasion was determined by staining with Shandon Kwik-Diff stain from Thermo Scientific. Invasion was calculated as % cells invaded through the matrix as compared with the control membranes as per the manufacturer's protocol (Fisher Scientific).
Protein complex immunoprecipitation (co-IP) assays
Coimmunoprecipitations were performed as described previously (Yang et al. 2015). Briefly, 293Tcells were transiently transfected with FLAG-tagged ATDC, HA-tagged KEAP1, Myc-tagged Nrf2, or deletion mutants of FLAG-ATDC or HA-KEAP1. Cells were lysed in a buffer containing 50 mM Tris, 150 mM NaCl, and 0.5% Igepal (pH 7.5), followed by sonication for 5 min. Lysates were used to perform immunoprecipitation with anti-FLAG, anti-HA or anti-ATDC antibodies. After overnight incubation at 4°C, the immunoprecipitates were resolved by SDS-PAGE, and detected by anti-FLAG (Sigma) anti-ATDC (Sigma) or anti-Myc (Cell Signaling) antibodies.
NRF2 ubiquitination assays
To detect ubiquitinated NRF2, S2-013 cells were transfected with expression vectors for ATDC and HA-ubiquitin (Ub). At 24 h following transfection, cells treated with10 µM MG132 (Sigma) for 6 h to inhibit the proteasome function. Cells were then lysed by boiling in a buffer containing 2% SDS, 150 mM NaCl, 10 mM Tris-HCl, and 1 mM DTT. This rapid lysis procedure inactivated cellular ubiquitin hydrolases and therefore preserved ubiquitin-NRF2 (Ub-NRF2) conjugates present in cells prior to lysis. Lysates were then immunoprecipitated (IP) with anti-HA antibody. The washed immunoprecipitates and 50 µg of whole cell lysates (WCL) were resolved by SDS-PAGE, followed by immunoblotting with NRF2, ATDC, and β-actin antibodies.
Fluorescence anisotropy binding assays
Fluorescence anisotropy binding assays were performed as described previously (Gijsbers et al. 2016). Briefly, purified human GST-Keap1 protein was fluorescein-labeled using a fluorescein-EX protein labeling kit according to the manufacturer's instructions (Thermofisher Scientific). For the binding experiments, 20 nM fluorescein-labeled GST-Keap1 was titrated with purified human ATDC protein. The binding assays were performed for 30 min at 25°C. Changes in the fluorescence anisotropy values (495-nm excitation and 520-nm emission) were measured using a stopped-flow model SF-300X (KinTek Corporation) set up with an automated titration module. The assays were repeated five times and the data were plotted using GraphPad Prism 8 (GraphPad Software).
Colony formation assays
S2-013 pancreatic cancer cells were plated in 12-well plates (500 cells/well). At the end of 2 wk, cells were fixed, stained with 0.2% crystal violet, and imaged to determine the number of colonies in S2-013 control, ATDC, or ATDC Δ220N overexpressing cells.
Animal studies
All animal experiments were approved by New York University Langone Health Institutional Animal Care and Use Committee (IACUC) under protocol number 170805-02. Isogenic pancreatic cancer cells (5 × 105; S2-013 or HPAC) were orthotopically implanted in the pancreas of 8-wk-old female immunocompromised NSG mice (NOD.Cg-Prkdcscid;Il2rgtm1Wjl/SzJ; The Jackson Laboratory 005557) (n = 10 animals/group). Mice were monitored for tumor growth weekly via ultrasound measurements. Mice treated with NAC were given NAC in drinking water at 1g/L. At the end of 27 d (S2-013) or 42 d (HPAC) animals were sacrificed and necropsies were performed. Relative tumor size and weight were measured. Primary tumor, lung, and liver were harvested and formalin-fixed or flash frozen for further analyses.
To evaluate whether NRF2 mediates chemoresistance in ATDC-expressing PDA tumors, 5 × 105 S2-013 cells expressing vector control, ATDC alone or in combination with shNRF2, and ATDC-Δ220N were orthotopically implanted in the pancreas of 8-wk-old female immunocompromised NSG mice (n = 10/group). Xenografted tumors were imaged by ultrasound to monitor and measure tumor sizes weekly. When the tumor reached the size of 200 mm3, 100 mg/kg gemcitabine (Gem) was administrated by intraperitoneal injection (IP) once per week for 2 wk. At the end of 2 wk, the mice were sacrificed, and tumor volume was determined. Metastasis analysis was done by 5-µm serial sectioning of lung and liver samples (n = 5/group) and analysis of metastasis by two independent researchers using a Zeiss Axio Observer microscope. Data were summarized as a scatter plot and representative sections were included in the figures.
To examine the effect of knocking down ATDC in established tumors, 5 × 105 HPACish ATDC cells (S2-013 or HPAC) were orthotopically implanted in the pancreas of 8-wk-old female immunocompromised NSG mice. Mice were distributed randomly into two groups (10 animals each) for the subsequent analysis of tumor development with and without doxycycline (Doxy)-treatment. Xenografted tumors were imaged by ultrasound to measure tumor sizes weekly. When the ish-ATDC-tumor reached the size of 200 mm3, control or doxy diets (625 mg/kg) were administered. At the end of 4 wk, the mice were sacrificed and tumor volume, ATDC, and NRF2 protein levels were determined.
Immunohistochemistry/coimmunofluorescence
Four-micron-thick formalin-fixed paraffin-embedded tumor and tissue sections were obtained and used for immunohistochemical and coimmunofluorescence analysis as described previously (Wang et al. 2009). IHC was performed to determine the tissue levels of NRF2 (antibody generously provided by J.R. Prigge and E.E. Schmidt, Montana State University), ATDC (Sigma HPA020053), NQO1 (Sigma HPA007308), Ki67 and cleaved caspase-3 (Cell Signaling). ATDC and NRF2 expression in PanIN lesions from KC or AKC mice were measured by co-IF staining using ATDC (Sigma HPA020053) and NRF2 (provided by Dr. Thales Papagiannakopoulos, New York University) antibodies.
TCGA patient data
TCGA pancreatic cancer batch effects-normalized mRNA data and curated clinical data were retrieved from the UCSC Xena Pan-Cancer Atlas hub. Survival curve estimates were calculated with the Kaplan–Meier method and the significance was evaluated with the log-rank test using the “survival” R package.
Statistical analysis
Data are presented as mean ± SEM. The experiments have repeated a minimum of three times to demonstrate reproducibility. Statistical significance was determined by the Student's t-test or one-way analysis of variance (ANOVA) using GraphPad Prism 8 (GraphPad Software). P-values < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank members of the Simeone and Lyssiotis laboratories for technical assistance and helpful discussions. C.A.L. was supported by a Pancreatic Cancer Action Network/AACR Pathway to Leadership award (13-70-25-LYSS), a 2017 AACR NextGen grant for Transformative Cancer Research (17-20-01-LYSS), and an ACS Research Scholar grant (RSG-18-186-01). V.P. was supported by a Peer Reviewed Cancer Research Program Horizon award by the Department of Defense (W81XWH-17-1-0497). The project was supported by National Cancer Institute grants 2R01CA131045 and 1R01CA174836 to D.M.S. We appreciate the help of the New York University Histopathology Core.
Author contributions: The study was conceptualized, conducted, and finalized by V.P., L.W., C.A.L., and D.M.S. In vitro experiments were perfomed by V.P., H.Y., L.W., E.R.G., A.Z., A.J.V., G.M.N., and A.W. Mechanistic studies were performed by L.W., H.Y., J.L., G.M.N., A.B., E.V.A., and V.P. Animal studies were performed by L.W., V.P., E.Z., and A.W. Bioinformatics analyses were perfomed by I.D. Resources were obtained by M.P.D. and H.C.C. V.P., D.D., L.W., and D.M.S. wrote the manuscript. T.Y.P., C.A.L., and D.M.S. edited the manuscript. C.A.L. and D.M.S. supervised the study.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.344184.120.
References
- Akino N, Wada-Hiraike O, Isono W, Terao H, Honjo H, Miyamoto Y, Tanikawa M, Sone K, Hirano M, Harada M, et al. 2019. Activation of Nrf2/Keap1 pathway by oral dimethylfumarate administration alleviates oxidative stress and age-associated infertility might be delayed in the mouse ovary. Reprod Biol Endocrinol 17: 23 10.1186/s12958-019-0466-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrutkar M, Gladhaug IP. 2017. Pancreatic cancer chemoresistance to gemcitabine. Cancers (Basel) 9: 157 10.3390/cancers9110157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora S, Bhardwaj A, Singh S, Srivastava SK, McClellan S, Nirodi CS, Piazza GA, Grizzle WE, Owen LB, Singh AP. 2013. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor κB- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. J Biol Chem 288: 21197–21207. 10.1074/jbc.M113.484576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH. 2009. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28: 3015–3026. 10.1038/emboj.2009.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M, Hogstrom L, et al. 2016. High-throughput phenotyping of lung cancer somatic mutations. Cancer Cell 30: 214–228. 10.1016/j.ccell.2016.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL, et al. 2018. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 562: 600–604. 10.1038/s41586-018-0622-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2014. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550. 10.1038/nature13385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2017. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32: 185–203.e13. 10.1016/j.ccell.2017.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, Vousden KH. 2020. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 37: 168–182.e4. 10.1016/j.ccell.2019.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, Wilson J, Sangar V, Hao Y, Öhlund D, et al. 2016. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 166: 963–976. 10.1016/j.cell.2016.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. 2012. Oncogenic kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 122: 639–653. 10.1172/JCI59227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin KA. 2004. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther 3: 294–300. 10.1177/1534735404270335 [DOI] [PubMed] [Google Scholar]
- Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul J-L, Gourgou-Bourgade S, de la Fouchardière C, et al. 2011. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364: 1817–1825. [DOI] [PubMed] [Google Scholar]
- Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J-L, Choné L, Francois E, Artru P, Biagi JJ, et al. 2018. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med 379: 2395–2406. [DOI] [PubMed] [Google Scholar]
- Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, et al. 2016. Monoacidic inhibitors of the kelch-like ECH-associated protein 1: nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein–protein interaction with high cell potency identified by fragment-based discovery. J Med Chem 59: 3991–4006. 10.1021/acs.jmedchem.6b00228 [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. 2011. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475: 106–109. 10.1038/nature10189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Magliano MP, Logsdon CD. 2013. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 144: 1220–1229. 10.1053/j.gastro.2013.01.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donadelli M, Costanzo C, Beghelli S, Scupoli MT, Dandrea M, Bonora A, Piacentini P, Budillon A, Caraglia M, Scarpa A, et al. 2007. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim Biophys Acta 1773: 1095–1106. 10.1016/j.bbamcr.2007.05.002 [DOI] [PubMed] [Google Scholar]
- Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, et al. 2011. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Cell Death Dis 2: e152 10.1038/cddis.2011.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. 2009. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458: 762–765. 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Stockwell BR. 2017. Lipid peroxidation in cell death. Biochem Biophys Res Commun 482: 419–425. 10.1016/j.bbrc.2016.10.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijsbers A, Nishigaki T, Sanchez-Puig N. 2016. Fluorescence anisotropy as a tool to study protein-protein interactions. J Vis Exp. 54640 10.3791/54640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb D, Hast BE, Wang W, Major MB. 2014. Spotlite: web application and augmented algorithms for predicting co-complexed proteins from affinity purification-mass spectrometry data. J Proteome Res 13: 5944–5955. 10.1021/pr5008416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B, et al. 2013a. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J Exp Med 210: 1529–1544. 10.1084/jem.20121337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, Mak TW. 2013b. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947. 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- Hamada S, Taguchi K, Masamune A, Yamamoto M, Shimosegawa T. 2017. Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 38: 661–670. 10.1093/carcin/bgx043 [DOI] [PubMed] [Google Scholar]
- Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, Hayes DN, Major MB. 2013. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res 73: 2199–2210. 10.1158/0008-5472.CAN-12-4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes AJ, Skouras C, Haugk B, Charnley RM. 2015. Keap1-Nrf2 signalling in pancreatic cancer. Int J Biochem Cell Biol 65: 288–299. 10.1016/j.biocel.2015.06.017 [DOI] [PubMed] [Google Scholar]
- Hole PS, Pearn L, Tonks AJ, James PE, Burnett AK, Darley RL, Tonks A. 2010. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34+ hematopoietic progenitor cells. Blood 115: 1238–1246. 10.1182/blood-2009-06-222869 [DOI] [PubMed] [Google Scholar]
- Holmström KM, Finkel T. 2014. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15: 411–421. 10.1038/nrm3801 [DOI] [PubMed] [Google Scholar]
- Hong YB, Kang HJ, Kwon SY, Kim HJ, Kwon KY, Cho CH, Lee JM, Kallakury BV, Bae I. 2010. Nuclear factor (erythroid-derived 2)-like 2 regulates drug resistance in pancreatic cancer cells. Pancreas 39: 463–472. 10.1097/MPA.0b013e3181c31314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B. 2001. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med 7: 1111–1117. 10.1038/nm1001-1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaramillo MC, Zhang DD. 2013. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27: 2179–2191. 10.1101/gad.225680.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SM, Hwang S, Seong RH. 2016. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem Biophys Res Commun 471: 373–379. 10.1016/j.bbrc.2016.02.023 [DOI] [PubMed] [Google Scholar]
- Ju H-Q, Gocho T, Aguilar M, Wu M, Zhuang Z-N, Fu J, Yanaga K, Huang P, Chiao PJ. 2015. Mechanisms of overcoming intrinsic resistance to gemcitabine in pancreatic ductal adenocarcinoma through the redox modulation. Mol Cancer Ther 14: 788–798. 10.1158/1535-7163.MCT-14-0420 [DOI] [PubMed] [Google Scholar]
- Kapp LN, Painter RB, Yu LC, van Loon N, Richard CW 3rd, James MR, Cox DR, Murnane JP. 1992. Cloning of a candidate gene for ataxia-telangiectasia group D. Am J Hum Genet 51: 45–54. [PMC free article] [PubMed] [Google Scholar]
- Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, et al. 2016. Pancreatic cancer. Nature Reviews Disease Primers 2: 16022 10.1038/nrdp.2016.22 [DOI] [PubMed] [Google Scholar]
- Kong Q, Beel JA, Lillehei KO. 2000. A threshold concept for cancer therapy. Med Hypotheses 55: 29–35. 10.1054/mehy.1999.0982 [DOI] [PubMed] [Google Scholar]
- Li X, Wang H, Wang J, Chen Y, Yin X, Shi G, Li H, Hu Z, Liang X. 2016. Emodin enhances cisplatin-induced cytotoxicity in human bladder cancer cells through ROS elevation and MRP1 downregulation. BMC Cancer 16: 578 10.1186/s12885-016-2640-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, et al. 2019. Nrf2 activation promotes lung cancer metastasis by inhibiting the degradation of Bach1. Cell 178: 316–329.e18. 10.1016/j.cell.2019.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, Storz P. 2016. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep 14: 2325–2336. 10.1016/j.celrep.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A, Neoptolemos JP, et al. 2011a. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer 10: 37–37. 10.1186/1476-4598-10-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister A, Nedjadi T, Kitteringham NR, Campbell F, Costello E, Lloyd B, Copple IM, Williams S, Owen A, Neoptolemos JP, et al. 2011b. Nrf2 is overexpressed in pancreatic cancer: implications for cell proliferation and therapy. Mol Cancer 10: 37 10.1186/1476-4598-10-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, Giordano TJ, Misek DE, Kuick R, Hanash S. 2003. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res 63: 2649–2657. [PubMed] [Google Scholar]
- Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong AN, et al. 2012. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol 32: 1506–1517. 10.1128/MCB.06271-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KR, Barrett JC. 2002. Reactive oxygen species as double-edged swords in cellular processes: low-dose cell signaling versus high-dose toxicity. Hum Exp Toxicol 21: 71–75. 10.1191/0960327102ht213oa [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, Merchut-Maya JM, Hodny Z, Bartkova J, Christensen C, et al. 2015. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol 9: 601–616. 10.1016/j.molonc.2014.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon S, Columbano A, Giordano S. 2016. The dual roles of NRF2 in cancer. Trends Mol Med 22: 578–593. 10.1016/j.molmed.2016.05.002 [DOI] [PubMed] [Google Scholar]
- Mitsushita J, Lambeth JD, Kamata T. 2004. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res 64: 3580–3585. 10.1158/0008-5472.CAN-03-3909 [DOI] [PubMed] [Google Scholar]
- Mizutani H, Tada-Oikawa S, Hiraku Y, Kojima M, Kawanishi S. 2005. Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci 76: 1439–1453. 10.1016/j.lfs.2004.05.040 [DOI] [PubMed] [Google Scholar]
- Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. 2008. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14: 458–470. 10.1016/j.ccr.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmbos PL, Wang L, Yang H, Wang Y, Leflein J, Ahmet ML, Wilkinson JE, Kumar-Sinha C, Ney GM, Tomlins SA, et al. 2015. ATDC/TRIM29 drives invasive bladder cancer formation through miRNA-mediated and epigenetic mechanisms. Cancer Res 75: 5155–5166. 10.1158/0008-5472.CAN-15-0603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelicano H, Carney D, Huang P. 2004. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 7: 97–110. 10.1016/j.drup.2004.01.004 [DOI] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, Zhang DD. 2018. NRF2 and the hallmarks of cancer. Cancer Cell 34: 21–43. 10.1016/j.ccell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, et al. 2017. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23: 1362–1368. 10.1038/nm.4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. 2005. The antioxidant function of the p53 tumor suppressor. Nat Med 11: 1306–1313. 10.1038/nm1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749–1753. 10.1126/science.7792600 [DOI] [PubMed] [Google Scholar]
- Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, et al. 2017. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6: e28083 10.7554/eLife.28083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, et al. 2006. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med 3: e420 10.1371/journal.pmed.0030420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soini Y, Eskelinen M, Juvonen P, Kärjä V, Haapasaari KM, Saarela A, Karihtala P. 2014. Nuclear Nrf2 expression is related to a poor survival in pancreatic adenocarcinoma. Pathol Res Pract 210: 35–39. 10.1016/j.prp.2013.10.001 [DOI] [PubMed] [Google Scholar]
- Sykiotis GP, Bohmann D. 2010. Stress-activated cap'n’collar transcription factors in aging and human disease. Sci Signal 3: re3 10.1126/scisignal.3112re3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Yamamoto M. 2017. The KEAP1-NRF2 system in cancer. Front Oncol 7: 85 10.3389/fonc.2017.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir TY, Mulvaney KM, Major MB. 2016. Dissecting the Keap1/Nrf2 pathway through proteomics. Current Opinion in Toxicology 1: 118–124. 10.1016/j.cotox.2016.10.007 [DOI] [Google Scholar]
- Tebay LE, Robertson H, Durant ST, Vitale SR, Penning TM, Dinkova-Kostova AT, Hayes JD. 2015. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med 88: 108–146. 10.1016/j.freeradbiomed.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD, et al. 2017. Stress-activated NRF2-MDM2 cascade controls neoplastic progression in pancreas. Cancer Cell 32: 824–839.e8. 10.1016/j.ccell.2017.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Chio IIC, Tuveson DA. 2018. Transcriptional regulation by Nrf2. Antioxid Redox Signal 29: 1727–1745. 10.1089/ars.2017.7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. 2002. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol Cell 9: 1031–1044. 10.1016/S1097-2765(02)00520-8 [DOI] [PubMed] [Google Scholar]
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. 2013. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med 369: 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, An J, Ji F, Jiao H, Sun H, Zhou D. 2008. Hypermethylation of the Keap1 gene in human lung cancer cell lines and lung cancer tissues. Biochem Biophys Res Commun 373: 151–154. 10.1016/j.bbrc.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Wang L, Heidt DG, Lee CJ, Yang H, Logsdon CD, Zhang L, Fearon ER, Ljungman M, Simeone DM. 2009. Oncogenic function of ATDC in pancreatic cancer through Wnt pathway activation and β-catenin stabilization. Cancer Cell 15: 207–219. 10.1016/j.ccr.2009.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li X, Chen T, Wang W, Liu Q, Li H, Yi J, Wang J. 2013. Mechanisms of verapamil-enhanced chemosensitivity of gallbladder cancer cells to platinum drugs: glutathione reduction and MRP1 downregulation. Oncol Rep 29: 676–684. 10.3892/or.2012.2156 [DOI] [PubMed] [Google Scholar]
- Wang L, Yang H, Palmbos PL, Ney G, Detzler TA, Coleman D, Leflein J, Davis M, Zhang M, Tang W, et al. 2014. ATDC/TRIM29 phosphorylation by ATM/MAPKAP kinase 2 mediates radioresistance in pancreatic cancer cells. Cancer Res 74: 1778–1788. 10.1158/0008-5472.CAN-13-2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yang H, Abel EV, Ney GM, Palmbos PL, Bednar F, Zhang Y, Leflein J, Waghray M, Owens S, et al. 2015. ATDC induces an invasive switch in KRAS-induced pancreatic tumorigenesis. Genes Dev 29: 171–183. 10.1101/gad.253591.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yang H, Zamperone A, Diolaiti D, Palmbos PL, Abel EV, Purohit V, Dolgalev I, Rhim AD, Ljungman M, et al. 2019. ATDC is required for the initiation of KRAS-induced pancreatic tumorigenesis. Genes Dev 33: 641–655. 10.1101/gad.323303.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. 2010. Mitochondrial metabolism and ROS generation are essential for kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107: 8788–8793. 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Yu H, Gu S, Xu Y, Su J, Li H, Kang J, Cui M. 2014. P62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int J Oncol 45: 2341–2348. 10.3892/ijo.2014.2669 [DOI] [PubMed] [Google Scholar]
- Yang H, Palmbos PL, Wang L, Kim EH, Ney GM, Liu C, Prasad J, Misek DE, Yu X, Ljungman M, et al. 2015. ATDC (ataxia telangiectasia group D complementing) promotes radioresistance through an interaction with the RNF8 ubiquitin ligase. J Biol Chem 290: 27146–27157. 10.1074/jbc.M115.665489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Villagra A, Peng L, Coppola D, Glozak M, Sotomayor EM, Chen J, Lane WS, Seto E. 2010. The ATDC (TRIM29) protein binds p53 and antagonizes p53-mediated functions. Mol Cell Biol 30: 3004–3015. 10.1128/MCB.01023-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Duan CJ, Binkley C, Li G, Uhler MD, Logsdon CD, Simeone DM. 2004. A transforming growth factor β-induced Smad3/Smad4 complex directly activates protein kinase A. Mol Cell Biol 24: 2169–2180. 10.1128/MCB.24.5.2169-2180.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, Nan L, Li X, Li W, Shan T, et al. 2016. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid Med Cell Longev 2016: 1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Fahl WE. 2001. Functional characterization of transcription regulators that interact with the electrophile response element. Biochem Biophys Res Commun 289: 212–219. 10.1006/bbrc.2001.5944 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.