

The GLUT (SLC2) family of membrane-associated transporters are described as glucose transporters. However, this family is divided into three classes and, though the regulated transporter activity of class I proteins is becoming better understood, class III protein functions continue to be obscure. We have cataloged the relative expression and splicing of SLC2 mRNA isomers in tumors and normal tissues, with a focus on breast tumors and cell lines. mRNA for the class III protein GLUT8 is the predominant SLC2 species expressed alongside GLUT1 in many tissues, but GLUT8 mRNA exists mostly as an untranslated splice form in tumors.
KEYWORDS: GLUT8, SLC2, TXNIP, alternative splicing, breast cancer, chromophobe renal cell carcinoma, cleavage, glucose transport, late endosomal-lysosomal boundary, metabolic sensor
ABSTRACT
The GLUT (SLC2) family of membrane-associated transporters are described as glucose transporters. However, this family is divided into three classes and, though the regulated transporter activity of class I proteins is becoming better understood, class III protein functions continue to be obscure. We have cataloged the relative expression and splicing of SLC2 mRNA isomers in tumors and normal tissues, with a focus on breast tumors and cell lines. mRNA for the class III protein GLUT8 is the predominant SLC2 species expressed alongside GLUT1 in many tissues, but GLUT8 mRNA exists mostly as an untranslated splice form in tumors. We confirm that GLUT8 is not presented at the cell surface and does not transport glucose directly. However, we reveal a lysosome-dependent reaction that cleaves the GLUT8 protein and releases the carboxy-terminal peptide to a separate vesicle population. Given the localization of GLUT8 at a major metabolic hub (the late endosomal/lysosomal interface) and its regulated cleavage reaction, we evaluated TXNIP-mediated hexosamine homeostasis and speculate that GLUT8 may function as a sensory component of this reaction.
INTRODUCTION
Transport of hexose sugars is performed by at least two families of integral membrane channels, where the GLUT (SLC2) family of transporters mediates bidirectional and energy-independent glucose transport. By sequence comparison, this family comprises 14 members, divided into 3 classes (1–3), with specific expression patterns and affinities for hexose sugars. Loss of function of the class I GLUT proteins in mice and humans tends to have profound phenotypes (1). For example, this class includes GLUT4, perhaps the best understood family member, whose insulin-regulated cell surface presentation is largely responsible for insulin-dependent homeostasis of circulating glucose. However, defying predictions, GLUT4 knockout mouse were found to have remarkably normal glucose levels, turning the attention of investigators to other SLC2 family members with potentially overlapping or redundant functions (4).
Thus, structural homology comparisons have placed 5 proteins in class III of the SLC2 proteins (GLUT6, -8, -10, -12, and HMIT) (Fig. 1B); these proteins have been shown to be able to transport hexoses across a gradient (1, 5). However, do these proteins serve a transport function? This has been surprisingly difficult to demonstrate. GLUT8 is the best-studied class III member; indeed, three independent groups cloned and characterized GLUT8 and showed that GLUT8 was not essential for glucose uptake in cells or mice (6–8). The phenotypes of GLUT8 knockout mice are subtle, including hypoactive sperm, hyperproliferation of hippocampal cells, and resistance to hepatosteatosis in mice fed high fructose (9–13). Subtle phenotypes are also typical vitro; for example, unlike GLUT1, high-throughput short term CRISPR screening modalities of cultured cancer cell lines do not identify GLUT8 as an essential gene (14, 15).
FIG 1.

Expression levels of GLUT SLC2 species in normal tissues and breast tumors. (A) The expression level of each of the 14 Slc2 family members is shown as a heat map of RNA-Seq reads (transcripts per kilobase million [TPM]) in each of the human tissues indicated (data summarized from GTEx Portal, v8). (B) The GLUT family dendrogram, redrawn from reference 2, shows three subclasses of GLUT proteins (class I, II, and III). GLUT14 is not indicated on this scheme as it has now been identified as a paralog of GLUT3 and is therefore grouped with class I SLC2 species. GLUT13 is more typically called HMIT (H+-myo-inositol symporter). (C) Expression of SLC2 mRNA species is described from the RNA-Seq database of The Cancer Genome Atlas for breast tumors (1,091 total) (24) and for near-adjacent, nontumor breast samples (labeled Normal; n = 111).
Furthermore, although full-length GLUT8 cDNA is stable upon ectopic expression in cells, neither the exogenous nor endogenous proteins are conclusively present on the cell surface (5). Indeed, this protein has the classic dileucine intracellular motif (in the N-terminal cytoplasmic domain) that targets integral membrane proteins to the late endosomal/lysosomal vesicular compartment. Several groups have therefore speculated that GLUT8 might mediate transport of hexoses, or other metabolites, from compartment to cytosol (16–19).
In this study, we show there are issues that have confounded the investigation of GLUT8. For example, a number of alternatively spliced mRNA variants are made from the gene locus (as documented by the genome database and confirmed by our analysis). The most common in cultured cells (variant 3 [v3]) does not encode a viable protein. Several SLC2 mRNAs show alternative splice forms that specifically affect the crucial regulatory N- and C-terminal cytoplasmic domains. We also show that the GLUT8 protein is cleaved, releasing a 10-kDa membrane-associated carboxy-terminal domain, which becomes enriched in a distinct and separate vesicular population, and speculate that this may provide a clue to GLUT8 function.
RESULTS
To introduce GLUT8 in the context of the rest of the 14-member SLC2 family, we compared relative expression levels using in silico Northern blotting data derived from the Genotype-Tissue Expression (GTEx) project (Fig. 1A). This program clusters mRNAs according to their degree of shared expression, and the three classes are shown color coded (according to the scheme of Fig. 1B, class I in black, class II in blue, class III in green). (The data used for the analyses described in this paper were obtained from the GTEx portal on 3 September 2019.)
This analysis confirms and expands published Northern blotting data (1, 6–8, 20). Thus, GLUT1 and GLUT3 show high and widespread expression, although expression is lower in tissues that specialize in systemic glucose regulation, such as liver, muscle, and pancreas, where other GLUT mRNA species predominate. GLUT2, -7, and -14 show the most restricted expression pattern; GLUT2 is expressed only in the liver and small intestine. The rest are divided into two clusters, one comprising Glut4, -10, -8, and -11 (expressed widely and at moderately high levels) and the other comprising GLUT5, -12, -6, -13, and -9 (showing lower and more specific expression patterns). In particular, GLUT5 is most abundant in testis and small intestine; GLUT12 in stomach, prostate, and esophagus; GLUT6 in blood (and spleen); GLUT13 in cervix; and GLUT9 in kidney and bladder, where it has been shown to be a high-capacity urate transporter (21).
The requirement for abundant glucose uptake by tumor cells has been touted as a therapeutic opportunity (22, 23). We compared the relative expression of all 14 SLC2 genes in breast tumors in vivo and breast cancer cell lines in vitro, using publicly available transcriptome sequencing (RNA-Seq) data (24, 25) to figure out which proteins might be involved in glucose uptake and detection (Fig. 1C). This analysis shows that of the class I transporters, GLUT1 mRNA is indeed most abundantly expressed, in both tumors and cell lines; mean mRNA expression in tumors is 75 ± 80 transcripts per million (TPM), nearly 3-fold higher than normal near-adjacent tissue (26.2 ± 12.6). In cell lines in culture, average expression is even higher (127 ± 102 TPM). However, expression is highly variable, such that the cell lines in the top decile show 15-fold more expression than those in the lowest decile (379 versus 25 TPM), suggesting that the evaluation of relative GLUT1 expression could be useful to understanding rate-limiting factors for individual tumors.
Of the other class 1 transporters, GLUT3 mRNA is next most abundant (14.2 ± 13.2 TPM), showing, however, less expression in tumor tissue than normal tissue (26.0 ± 23.7 TPM). Expression is highly variable in cell lines (lowest and highest deciles are 0.05 to 181 TPM). High GLUT3 expression has been shown to be necessary and sufficient for metastatic behavior of breast cancer cells in vitro (26). Of the class II GLUT transporters, only GLUT11 is significantly expressed in tumors (7.9 ± 4.6 TPM) and tumor cell lines (15.3 ± 8.9 TPM). Expression in tumors matches normal breast tissue (7.2 ± 1.7 TPM).
Of the class III GLUT transporters, GLUT8 and GLUT10 are expressed in vivo in breast tumors (16.2 ± 9.7 and 15.1 ± 23.1 TPM, respectively), in normal tissues (12.2 ± 4.2 and 10.9 ± 4.2 TPM, respectively), and in vitro in breast tumor cell lines (9.3 ± 6.3 and 26.9 ± 37.4 TPM, respectively). GLUT10 shows the most highly variable expression in breast tumors and cell lines (0.3 to 113 TPM highest and lowest decile for the group of 79 cell lines). Although consistently overexpressed in tumors, this cannot be explained by gene amplification; thus, none of the GLUT loci are located in the commonly amplified chromosomal domains of breast tumors (see Table 1 for chromosomal locations) or show consistent patterns of copy number variation.
TABLE 1.
Alternative splicing of GLUT mRNAsa

(Putative) protein-encoding exons for each of 10 GLUT species; 5 class I GLUTs (GLUT1 to GLUT4) and 5 class III GLUTs (GLUT6, -8, -10, -12, -13/HMIT) are compared for human and mouse. The species shown are confirmed alternatively spliced mRNA isomers from the genome database (human, GRCh38/hg38; mouse, GRCm38/mm10). Hum, human; chsme, chromosome.
Breast tumors are grouped into subtypes according to their specific tumor driver and genetic profile. To test whether GLUT isotype expression could distinguish breast tumor subtypes, we evaluated the RNA-Seq data for each of 5 tumor subtypes (basal, HER2, luminal A, luminal B, and normal-like), using normal adjacent tissue as a comparison (Fig. 2A). Volcano plots show that GLUT1 and GLUT6 are consistently upregulated, with >4-fold increases in basal tumors and significant increases in HER2-overexpressing tumors (see Fig. S1 in the supplemental material). GLUT10 shows significant but minor overexpression in luminal A, luminal B, and HER2-positive tumors. On the other hand, GLUT4 is underexpressed in tumors compared to the near adjacent “normal” samples; these normal samples are enriched in GLUT4-expressing fatty tissue, highlighting the caution required to infer function using these imperfect comparisons. Using quantitative real-time PCR (RT-qPCR) assays of breast epithelial and tumor cells in culture (Fig. 2B), we confirmed that the ratios and expression levels of GLUT mRNAs accurately mimicked in vivo levels for both mice and humans.
FIG 2.

Expression levels of GLUT species in breast tumor subtypes. (A) Expression of mRNAs of the most abundant GLUT species from each GLUT class (class I, GLUT1; class II, GLUT11; class III, GLUT8 and 10) is shown for each of the 5 breast tumor subtypes (basal, HER2 positive, luminal A [LumA], luminal B [LumB], and normal-like [NormLike]) and adjacent normal tissues (Normal). Also included is the average expression of 75 cell lines in vitro (36). (B) Relative mRNA expression levels of selected members of the SLC2 family are shown for a nontransformed mouse mammary epithelial cell line (MMEC), nontransformed human breast epithelial cells (MCF10A), and two breast cancer cell lines (MB231 and MCF7), color coded by their subtype. Expression levels of SLC family members are shown with respect to GLUT1 mRNA expression.
GLUT1 is often described as responsible for “basal” glucose uptake, although its activity is now known to be highly regulated by the apparently opposing cues of growth factor stimulation or starvation (27–29) Indeed, GLUT1 has been shown to be key to both the growth of mouse breast tumor cells and to HER2-induced mouse breast cancer initiation (30, 31), and GLUT1 deficiency produces predictable phenotypes in humans (32).
However, in support of an important if unknown role for class III SLC2 proteins, Table 1 and Fig. 3 show the high degree of conservation of GLUT8 mRNAs across species (mouse, rat, and cow). Thus, the GLUT8 gene is arranged into 10 exons (Table 1; Fig. 3A), encoding 12 transmembrane domains, where the boundaries of each exon are commonly embedded within sequences that encode the 20 amino acid, highly conserved transmembrane domains (labeled TM) (Fig. 3B and C). Key motifs are indicated for later reference, including the canonical dileucine motif in the N-terminal domain that directs the trafficking of this protein to late endosomes/lysosomes, and the glycosylation site (Fig. 3B) (16, 20). The hinge region between TM6 and TM7, the N-terminal cytoplasmic domain, and the extracellular loop (TM9-TM10) show relatively lower conservation, a pattern also typical of GLUT1 (33).
FIG 3.

Mapping of exons to protein structure of GLUT8. (A) Schematic diagram showing the GLUT8 locus. Exon and intron base size (in base pairs) are indicated. (B) Schematic of GLUT8 variant 1, indicating the exon boundaries within the putative protein structure, where each of the transmembrane (TM) domains is numbered in black, 1 to 12. Key functional domains are indicated, including the glycosylation site in the ecto-loop between transmembrane domains 9 and 10 and the dileucine ER retention and sorting motifs. (C) Comparison of GLUT8 protein sequence conservation, between human GLUT8 (Uniprot ID Q9NY64, Homo sapiens) and three other mammalian species, Mus musculus (Q9JIF3), Rattus norvegicus (Q9JJZ1), and Bos taurus (P63010). Transmembrane domains are 20 amino acids long. Internal cytoplasmic domains include both N and C termini and a large loop between transmembrane domains 6 and 7.
By interrogation of the UCSC and NCBI databases, we noticed that several GLUT loci exist as alternatively spliced isomers (Table 1). GLUT1, GLUT3, GLUT10, and GLUT12 species have only one full-length isomer in human and mouse but, in contrast, GLUT2, GLUT4, GLUT6, and GLUT8 exist as several alternatively spliced mRNA variants. Notably, these variants are predicted to produce alterations in the N and C termini of the proteins, where, in limited studies, these domains have been shown to be key regulatory and signaling domains.
Using GLUT8 as an example of this phenomenon, we tested whether the three mRNA species reported in the database exist in cells and tissues. The species are v1 (full-length), v2 (missing exon 9 and a section of the C terminus), and v3 (missing exons 2 and 3) (Fig. 4A). We determined that the v2 variant showed little or no expression in breast tumors or cell lines; we thus focused instead on v1 and v3 as the primary products of the SLC2A8 locus. The relative amounts of v1 and v3 mRNAs were determined by RT-PCR, first for MB231 breast cancer cells (Fig. 4B) and also for a panel of cell lines and normal human tissues (Fig. 4C), using complementary primer sets. The latter assay was calibrated to report copy number of GLUT8 variants per nanogram of RNA. We confirmed that testes and liver showed high expression of GLUT8 mRNA (8).
FIG 4.

Assay of alternatively spliced isoforms of GLUT8. (A) Schematic diagram showing the three alternatively spliced isoforms of GLUT8, v1, v2, and v3. The target sequences of two of the shRNAs used for data of Fig. 5 are indicated (numbers 122 and 180). (B) Assay of endogenous v1 and v3 expression in MB231 cells. Compare v1- and v3-derived bands from endogenously expressed GLUT8 species (empty vector; EV) with bands from cells transduced with an expression vector for GLUT1 (v1), derived from primers spanning exons 1 to 4 (creating products indicated in the schemes below). Controls (right side) show products from cells transduced with v1-, v2-, or v3-expressing plasmids. (C) Amount of v1- and v3-GLUT8 mRNAs in cells and tissues. RT-qPCR assay of GLUT8 v1 and v3 expression in human cancer cell lines and normal human tissue, determined using a standard curve of the target amplicon, with primers specific to v1 (exon 2-exon 3) or v3 (exon 1-exon 4; see scheme). (D) Splicing patterns in vivo. Exon-exon junction analysis of GLUT8 mRNAs from TCGA RNA-Seq data of breast tumors, showing the counts of v2 (exon 8-exon 10) and v3 (exon 1-exon 4) in red and canonical exon-exon junctions in gray. (E) Assay of relative exon expression. Analysis of mean read coverage for each exon from breast cancer TCGA RNA-Seq data set for GLUT8 and GLUT1. Shaded areas represent the 95% confidence interval (n = 1,091). (F) Analysis of GLUT8 exon frequency for a pan-cancer panel. Labels for each tumor type are excerpted from TCGA. Several tumor types are highlighted, for comparison with invasive breast carcinoma (BRCA), and discussed further in Results and Discussion (ESCA, esophageal carcinoma; KICH, kidney chromophobe; LAML, acute myeloid leukemia; LIHC, liver hepatocellular carcinoma; OV, ovarian serous cystadenocarcinoma: STAD, stomach adenocarcinoma).
For normal tissues, the v3 isomer comprised approximately 5 to 10% of total GLUT8 mRNA, which was also true for the untransformed breast epithelial cell line (MCF10A). Several studies have described a single, 2.1-kB GLUT8 mRNA on Northern blots of normal tissues (6–8, 34); this matches our findings for normal tissues, where the full-length (v1) variant predominates. However, for the breast cancer cell lines screened, the exon 2-3-deleted v3 isomer was equally or more abundant than the full-length v1 isomer.
To ask whether this truncated form was present also in vivo in breast tumors, we assayed junction-specific RNA-Seq products, and measured their relative frequency (Fig. 4D). Splice junctions that characterize v2 and v3 are shown in red. We conclude the following. (i) The lack of exon 8-10 junctions confirmed that v2 was absent from breast tumors in vivo. (ii) The GLUT8 locus showed depleted read-through of exons 2 and 3 compared with, for example, GLUT1 (Fig. 4E). In vitro assay of breast cancer cell mRNA using PCR-based DNA polymerases showed that this depletion was relieved by solutions designed to read through high percentages of GC content; indeed, exons 2 and 3 of GLUT8 show >80% GC content (Fig. S2) for both mouse and human sequences. Regardless of the relative depletion of sequences representing exons 1 to 4, the frequency of v3 (exon 1-4 junctions) was still higher than v1 in breast tumors.
To test whether this expression pattern was specific to breast tumors, we expanded the scoring of GLUT8 exon expression levels to a panel of 33 tumor types (Fig. 4F). We concluded that among all tumor types, depletion of 5′ exons of mRNAs from SLC2A8 in RNA-Seq libraries was typical, and breast tumors showed average expression levels of GLUT8. Indeed, plotting these data on scaled axes, we found that the pattern of exon representation in all tumors was remarkably similar (Fig. S3). Interestingly, however, one tumor type in particular, chromophobe renal cell carcinoma (KICH), showed much higher expression of GLUT8 than usual (4-fold), whereas acute myelogenous leukemia (LAML) showed little or no expression. Interestingly, and perhaps related, chromophobe kidney tumors are known to show unusual metabolic adaptations (35, 36).
To test whether alternatively spliced GLUT8 isomers could be functional, we subcloned cDNAs for each of v1, v2, and v3 into expression vectors. The putative transmembrane proteins encoded by each variant are illustrated in Fig. 5A. To accurately describe the proteins made (and endogenously expressed proteins), we made a new polyclonal antibody to the TM6-7 cytoplasmic loop (called BBA1). We made this antibody because none of the commercial antisera we evaluated showed either increased signal with ectopic expression or reduced signal with knockdown (Table S1). More detailed evaluation of the specificity of BBA1 is provided in Fig. S4 (assay of immunofluorescent stains of cells with a knockdown of GLUT8 and cells with expression constructs) and in the Western blotting data shown in Fig. 5 and Fig. 6.
FIG 5.

Assay of properties of variant GLUT8 proteins. (A) Schematic diagram of GLUT8 v1, v2, and v3 putative protein structures, indicating the impact of deletion of exon 9 (v2) and exons 1-4 (v3). (B) Expression of v1, v2, and v3 GLUT8 proteins via Western blotting of lysates from 293T cells transfected with expression constructs. EV, empty vector. (C) Expression of v1, FLAG-tagged v1, and v3, expressed in MB231 cells. RT-qPCR analysis of mRNA from stable GLUT8 overexpressing MB231 cell lines, using a primer set that detects both GLUT8 v1 and v3 isoforms (total GLUT8 assay), for comparison with Western blots of lysates probed with BBA1, an anti-GLUT8 antibody. The protein loading control is vinculin. (D) Assay of glucose transporter activity: gain of function. Western blots (top) of protein lysates from MB231 cells transduced with retroviral GLUT8 or GLUT1 constructs, with the corresponding assay of relative 3H-2DG uptake (bottom). (E) Assay of glucose transporter activity: loss of function. Efficacy of shRNA knockdown of GLUT1 or GLUT8 was assayed (left side) and the impact of knockdown on 3H-2DG uptake assessed (right side). *, P < 0.01; ***, P < 0.0001; n.s., not significant. Results are representative of n ≥ 3 assays.
FIG 6.

GLUT8 is cleaved at the C terminus to generate a 10-kDa peptide. (A) Western blotting of lysates from MB231 cell lines transduced with retroviral expression constructs as indicated. Some lysates were treated with PNGase F to remove N-linked glycosylation prior to SDS-PAGE (4 to 20% gradient gel), and blots were probed with anti-GLUT8 or anti-FLAG antibodies. (B) Schematic diagram of putative cleavage site within the 10th transmembrane domain of GLUT8, predicted from the sizes of the parent and cleaved deglycosylated GLUT8 proteins. (C) Lysosomal inhibitors induce the accumulation of the GLUT8 protein and inhibit cleavage. MB231 cells expressing C-terminal-FLAG-tagged GLUT8 were treated with lysosomal inhibitors, chloroquine (CQ; 50 mM) or bafilomycin A1 (Baf; 10 nM) for 17 h, and lysates were deglycosylated by PNGase treatment, or not, prior to analysis by Western blotting with the antibodies indicated (10 μg lysate/lane) Putative GLUT8-related species are labeled as glycosylated species 1 and 2 (Gl-1 and Gl-2), endogenous species (En1 and two multimer bands, En4), and deglycosylated full-length (deG FL) and cleaved (deG NTD) species.
We tested the three alternatively spliced mRNAs for their expression in HEK293T and MB231 cells, and found that v1 and v2 proteins were stably expressed, but v3 was not (Fig. 5B and C and, Fig. S6). Adding a FLAG tag to either the C or N terminus had no effect on total GLUT8 production (Fig. 5C). We evaluated whether the proteins being expressed showed the activities expected. Cells overexpressing GLUT1 showed a large increase in glucose uptake activity; cells expressing GLUT8 did not (Fig. 5D), confirming prior studies. Vice versa, cells with a knockdown of GLUT1 showed reduced glucose uptake whereas GLUT8 knockdown cells showed no effect (Fig. 5E). Note that the proteins described here were expressed from retroviral expression vectors; surprisingly, they did not show the same properties when synthesized from lentiviral expression vectors (data not shown).
When we evaluated the glycosylation pattern of GLUT8 proteins, we were surprised to find that peptide-N-glycosidase (PNGase) treatment reduced the schmear of bands (from 45 to 60 kDa) to two bands, instead of the expected single band (Fig. 6A). This was not true for GLUT1 from these same lysates (data not shown). Using the termini-specific FLAG-tagged constructs, we showed that a FLAG-tagged peptide was derived from the C-terminal domain, with an approximate molecular weight of 10 kDa (shown on the scheme drawn as Fig. 6B). We predict that this peptide would stay membrane bound, since it contains at least two transmembrane domains. Interestingly, the putative cleavage site is situated in transmembrane domain 10 (TM10), which is 100% conserved (Fig. 3C).
Cleavage by regulated contact with membrane domain-specific proteases can be an important regulatory reaction, notably for metabolic sensors. Thus, for sensors such as SREBP (a sterol sensor), release from one organelle to another allows cleavage by two processive proteases, turning a membrane-bound protein into a transcriptional coactivator. Previous studies of GLUT8 have shown that this protein accumulates in the late endosome/lysosome compartment, cycling from the plasma membrane inward and becoming retained on the limiting membrane of the lysosome via the dileucine N-terminal signal. This was shown to be true both for the endogenous and for the exogenously expressed proteins in a number of cell types (16, 18, 37).
To test whether GLUT8 metabolism was related to lysosomal function, we added lysosomal inhibitors, either chloroquine (which neutralizes the acidic lysosomal interior and many lysosome-associated functions, including autophagy) (38) or bafilomycin A1 (an inhibitor of vacuolar ATPase with broad impact on subcellular compartments, including lysosomes) (39) (Fig. 6C). We found that GLUT8 accumulated in cells treated with either of the lysosomotropic agents, as either a high or a low molecular weight glycosylated variant (Gl1 and Gl2), depending upon the treatment. The accumulating GLUT8 proteins reduced to the full-length (38 kDa) GLUT8 protein after PNGase treatment; cleavage was therefore inhibited by reduced lysosomal acidification (visualized as the depletion of the N-terminal domain [NTD] 28 kDa GLUT8 protein and the C-terminal domain [CTD] FLAG-labeled peptide). We conclude that lysosomal trafficking could account for the cleavage reaction and may control the rate of clearance of full-length GLUT8.
We evaluated the subcellular localization of GLUT8 in more detail, specifically to localize the cleaved carboxy domain. First, we confirmed that tagged and untagged GLUT8 v1 protein partly colocated with a LAMP1-positive lysosome compartment (Fig. 7A and B), with the rest located in Approximately 1-μm-size vesicles in the same domain (probably late endosomal vesicles). For comparison, cells overexpressing GLUT1 showed a classic plasma membrane-associated pattern, with no significant colocalization to lysosomes (Fig. 7C). Other colocalization assays excluded early endosomes, endoplasmic reticulum, and peroxisomes as sites of significant accumulation (Fig. S5).
FIG 7.
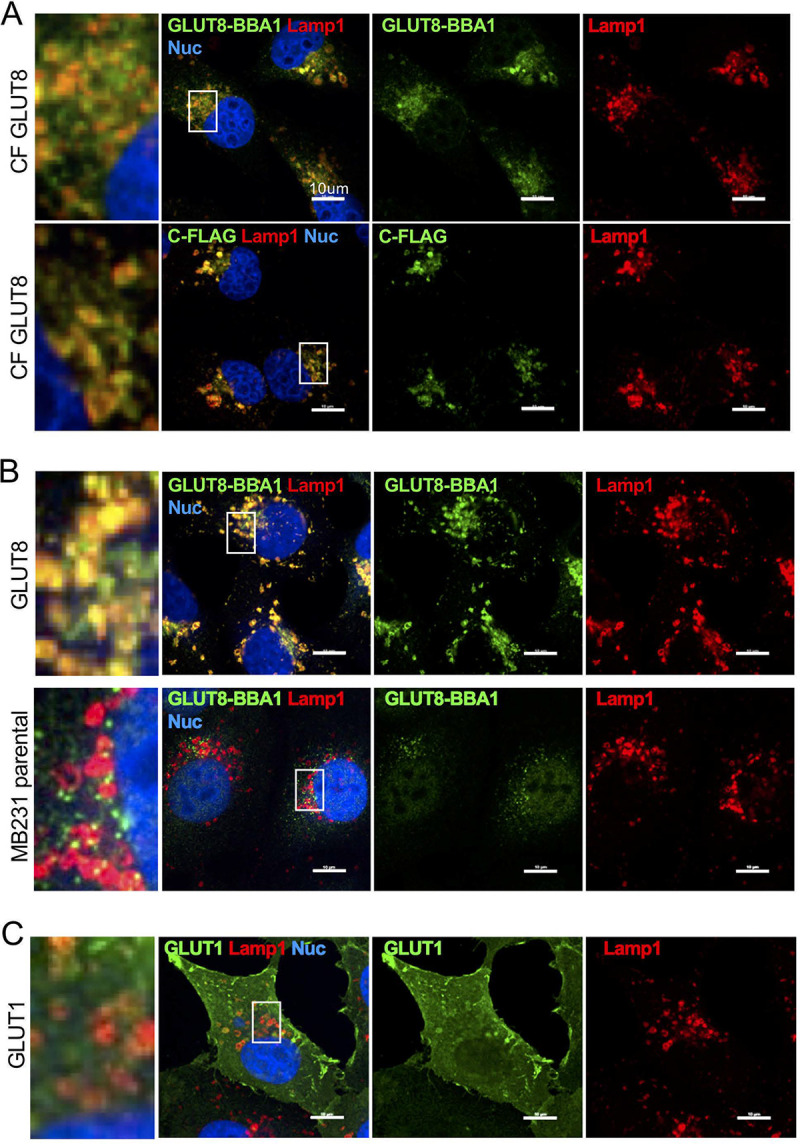
GLUT8 partly colocalizes with LAMP1 in lysosomes. (A) MB231 cells expressing C-terminal-FLAG-tagged GLUT8 (CF GLUT8) were stained with anti-GLUT8 and LAMP1; single stains are shown to the right, and colocalization is shown to the left; nuclei were stained with DAPI. (B) MB231 cells expressing untagged GLUT8, and cells without transgenic GLUT8 expression, are compared with the data shown in panel A, to assess the relative distribution of untagged GLUT8 protein, lysosome distribution, and endogenous GLUT8 localization. (C) For comparison, MB231 cells expressing GLUT1 were stained using the same protocol. Scale bars = 10 μm.
Although we already showed that the v3 mRNA isomer did not produce a stable GLUT8 protein isomer, we tested whether this truncated mRNA could generate the CTD fragment (perhaps this could generate a constitutive signaling moiety, for example). We probed Western blots of v3-transduced cells with anti-FLAG antisera and found no evidence of FLAG-containing peptides either by immunofluorescence or Western blotting (Fig. S6 and data not shown). The exact insertion points of the FLAG tags are detailed in Fig. S6C.
To test whether the cleaved peptide was located to the same vesicle population as the cleaved N-terminal domain, we compared the staining pattern of anti-GLUT8 (BBA1) with the staining pattern of anti-FLAG (Fig. 8). To confirm there was no significant chromatic aberration at the magnifications presented, we confirmed total overlay of signal from red- and green-conjugated secondary antibody to anti-FLAG stains (Fig. S7). Costain of the FLAG epitope together with the central loop epitope (BBA1) showed combinations of color signals, orange where both epitopes were together in the full-length GLUT8 protein, or separated into red (majority N-terminal GLUT8 protein, after cleavage of the carboxyterminal peptide) or green (C-terminal peptide) (Fig. 8). We conclude that the GLUT8-derived C-terminal peptide becomes enriched in a separate vesicular population than the full-length or cleaved-N-terminus GLUT8 proteins.
FIG 8.

The cleaved C-terminal peptide becomes enriched in a separate vesicular population. Evaluation of colocalization of epitopes from the central loop of GLUT8 (BBA1) and the FLAG epitope at the C terminus shows that these epitopes become separated into separate vesicles. An example of images revealing vesicular population with mixed colors is shown (top); quantitative analysis of relative color intensity is shown across a typical section (bottom).
The overexpression of GLUT8 had no gross effect on cell morphology or growth rate. However, the location of GLUT8 at the late endosomal/lysosomal boundary places it at a regulatory hub for metabolism, the subcellular location for mTOR complex reactions. We tested the cellular response to amino acid or hexosamine refeeding, specifically seeking differences in a sensory network known to rely on related GLUT species. In particular, the β-arrestin protein TXNIP has been shown to detect and enable hexosamine feeding, acting as an internal sequestration molecule for several GLUT species, including the best-characterized fructose transporters GLUT2 and GLUT5 (40). TXNIP is degraded within minutes in response to external hexosamine concentrations to provide a rapid response mechanism to changing environmental conditions and to coordinate signals from growth factors and metabolic checkpoints (28).
To test whether GLUT8 could contribute to the mTOR signaling network, cells overexpressing GLUT8 were glucose starved and then refed with either fructose or glucose. We make the following four conclusions. (i) There were no changes in GLUT8 protein levels or proteolysis during starve/refeed cycles, as measured by Western blotting of the 10-kDa FLAG-tagged carboxy-terminal domain (Fig. 9 and data not shown). (ii) Ectopic GLUT8 expression led to a moderate reduction of steady-state TXNIP amount and mTOR activation (measured as the accumulation of phosphorylated 4EBP variants, and mTOR-dependent phosphorylation of S6 (residues 240/244) (41) (Fig. S9). (iii) Upon starvation, TXNIP was degraded as anticipated, but levels recovered quicker upon glucose refeeding for cells overexpressing GLUT8 (Fig. 9). (iv) Fructose refeeding could not promote TXNIP accumulation (and cells died). Parallel experiments designed to perturb mTOR-mediated homeostasis during amino acid starvation and refeeding showed that TXNIP was likewise degraded in response to starvation, but there was no effect of GLUT8 on the acute restoration of TXNIP levels, despite reactivation of mTOR (Fig. S9). Neither was the amount of GLUT8, or its cleavage, affected by amino acid starvation, where amounts of functional sensor proteins have been shown to be related to starvation (42).
FIG 9.
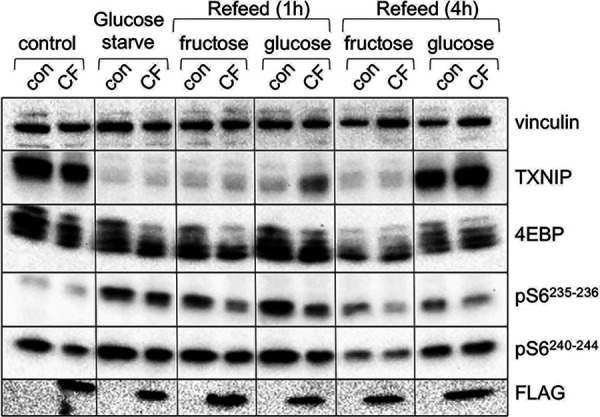
GLUT8 enables recovery from glucose starvation. MB231 cells expressing C-terminal-FLAG-tagged GLUT8 (CF) were glucose starved overnight and refed with glucose (to a final concentration of 25 mM) or fructose (to a final concentration 40 mM) for the times indicated. Cells were lysed and analyzed by Western blotting, and 15 μg of lysates was probed with the antibodies indicated.
DISCUSSION
We have shown that a peptide domain including at least two carboxy-terminal transmembrane helices of GLUT8 is cleaved from the rest of the protein and trafficked independently from the N-terminal moiety, becoming enriched in a different vesicular population. In this study, we saw that approximately 50% of the GLUT8 protein is present in the cleaved form. This cleavage occurs whether the protein is FLAG tagged or not, thus the tagging process itself does not disturb the endogenous processing reaction. Given that GLUT8 is actively retained at the endosomal-lysosomal boundary by the N-terminal dileucine motif, it is not surprising that the carboxy domain accumulates in a different vesicular population (16). The cleaved protein is unlikely to show transporter activity (which depends upon 12 transmembrane domains). Interestingly, out of all the transmembrane domains, the only domain 100% conserved across species is transmembrane domain 10, that we predict be the site of carboxy-terminal cleavage for GLUT8.
Although GLUT8 proteins have been expressed in various cell types before (6, 9, 16, 18, 19), this cleavage reaction is a novel finding. We attribute our discovery to a unique combination of reagents; thus, instead of the more typical N-terminal tagging, we built a GLUT8 species with a C-terminal tag and used a novel, and demonstrably specific, antibody to the central cytoplasmic loop, which became separated from the carboxy-terminal epitope during processing. It will be important to demonstrate this cleavage reaction for endogenous GLUT8 in a cell type with a functional readout.
Given this processing pattern of GLUT8 protein, it is useful to consider other examples of membrane-tethered signaling molecules that are activated by cleavage. Thus, the sterol sensor SREBP is a membrane-located protein, sequestered in the endoplasmic reticulum (ER) and released to the Golgi apparatus when local cholesterol concentration is low (43, 44). In the Golgi apparatus, it comes into contact with two proteases; the first one (SIP) cleaves SREBP between two transmembrane domains, inside the Golgi lumen. This cleavage product then becomes a substrate for the intramembrane protease S2P, which clips off a soluble short carboxy-terminal peptide which, together with a series of coactivators, create nuclear transcription complexes that induce expression of enzymes in the cholesterol biosynthetic cascade. The effect of loss of function of this sensor only becomes apparent upon cholesterol deprivation. Indeed, there is ample precedent for transporters to adapt to become proteins that use metabolites as signals to change cellular behaviors (45, 46).
In order to test for a GLUT8 sensory activity, we will need to know which metabolite(s) activates GLUT8, in order to determine whether GLUT8 signaling could affect cellular metabolism. Such a reaction could be important only to specific cell types. Perhaps a clue to a functional role for GLUT8 comes from the resistance of GLUT8 knockout female mice to fructose-induced metabolic diseases, including liver steatosis (13). Indeed, knockouts of both GLUT8 and the β-arrestin TXNIP were associated with fructose-associated steatosis in female mice, where TXNIP was shown to functionally interact with GLUT2 and GLUT5, the best characterized fructose transporters, in intestinal cells (40). We tested whether fructose interaction with GLUT8 could stimulate the recovery of TXNIP levels in glucose-starved cells and found that it could not (Fig. 9), but we have not tested fructose in combination with glucose (40). Note that ectopic GLUT8 expression does not promote additional glucose uptake (Fig. 5).
It is likely that some of the C-terminal FLAG-tagged protein is present in a larger complex, since some signal is masked to immunohistochemical detection. Thus, total immunofluorescent signal is lower for the C-terminal FLAG-tagged protein than the corresponding N-terminal tagged version, despite similar amounts assayed by Western blotting (see Fig. S8 in the supplemental material). Clearly the interactome of GLUT8 will be an important topic of investigation.
Whether endogenously or exogenously expressed (16, 18), GLUT8 is at a cellular hub of metabolic decision-making, the late-endosomal/lysosomal limiting membrane. A relatively well characterized role of this lipid membrane is its role in sensing the amino acids that activate the Ragulator complex, recruiting the mTORC1 complex to the lysosomal membrane (39, 47–49). There are trafficking proteins such as the GTPase Rab7 that direct traffic within the endo-lysosomal system, to mediate endocytic sorting, lysosomal biogenesis, and phagocytosis (50). This hub location also modulates mitochondrial homeostasis (51, 52) and regulates the protein and lipid content of the plasma membrane and other vesicular populations (50, 53). Perhaps related to this suggestion, DeBosch and colleagues showed that livers of male GLUT8 knockout mice directed aberrant peroxisome proliferator-activated receptor α-regulated fasting responses (54). From our preliminary observations, we propose that ectopic GLUT8 can modulate mTOR-mediated homeostasis after fasting.
We showed that the lysosome is a functional component in the processing reaction, such that inhibiting lysosomal function with chloroquine or bafilomycin A1 promotes the accumulation of GLUT8 protein, suppressing the specific intramembrane cleavage of the carboxy-terminal domain. This peptide domain becomes enriched in a so-far-unidentified vesicular compartment of approximately the same dimension as the vesicles containing the remainder of the GLUT8 protein. We did not observe FLAG tag in the nucleus (data not shown).
At the level of gene expression, our investigation concludes that assay of total GLUT8 mRNA expression can be inaccurate or misleading, first because of the high GC content (potentially also associated with a secondary structure or knot) of exons 2 and 3 in GLUT8 and second because GLUT8, like 50% of the other SLC2 loci, is expressed as several alternatively spliced isomers. In particular, tumor cells show a high frequency of expression of the v3 GLUT8 variant, which has two 5′ exons missing, displacing the putative protein synthesis start site downstream to exon 6. As predicted from the translation motifs in the mRNA, and the unusual topography of the projected protein, this v3 protein is not synthesized. Therefore, despite expression of GLUT8 mRNA, we predict that most tumors will show low/absent function of this locus. Another report of GLUT8 mRNA isomers described minor (<1% total) variants of GLUT8 mRNA, none the same as the variants described here and in the databases (55); their significance in vivo is perhaps uncertain.
Only the two major class I glucose transporters at the cell surface, GLUT1 and GLUT3, occur as just one isomer in both mouse and humans. Likewise, of the class III transporters, only GLUT10 and GLUT12 occur as one full-length isomer. GLUT2, -4, and -8 show several isomers that affect the N and C termini of their putative protein products. These termini face the cytoplasm and, where tested, have been shown to promote accurate trafficking and responsivity to regulators like growth factors (28, 40, 56, 57). Precedent for functional differences between alternatively spliced GLUT variants comes from the study of GLUT9, which is directed to the basolateral or apical surface of kidney epithelial cells by alternative splicing of N termini (58, 59), and for GLUT4 in adipocytes (60).
Overall, GLUT8 mRNA is expressed at approximately 5 to 20× lower levels than the predominant, active transporter, GLUT1, and is only moderately overexpressed in tumor cells or tissues. Although there is a range of GLUT8 expression levels in different tumors types, only one tumor type shows much higher expression than the others, chromophobe kidney tumor (KICH or ChRCC). This tumor is characterized by the accumulation of defective mitochondria, induced expression of mitochondrial enzymes, disrupted autophagy/lysosomal trafficking, and systemically activated AMP-activated protein kinase (AMPK), a marker of metabolic stress (35, 36).
In summary, we have cataloged the expression of various SLC2 mRNA isomers in tumors and tissues, and found evidence that widespread missplicing of GLUT8 mRNAs may reduce functionality in tumors. Only the full-length variant is stably translated, becoming cleaved by a lysosomal reaction, which releases the carboxy-terminal domain to a separate population of vesicles. Preliminary data suggest that GLUT8 may function in metabolic homeostasis. We propose that this enigmatic protein channel may function as a metabolite sensor.
MATERIALS AND METHODS
Gene expression and splicing analysis.
RNA-seq data from The Cancer Genome Atlas (24) was analyzed as previously described (61). Briefly, unprocessed RNA-seq reads were mapped to the UCSC hg19 human genome assembly using Bowtie (62) and RSEM (63), with a maximum of two mismatches per read and ignoring reads mapping to 100 or more positions within the genome. Alignments with mapq scores of 0 or with a splice junction overhang of less than 6 bp were removed. The resulting unaligned reads were mapped to a splice junction annotation derived from MISO v2.0 (64) and gene isoforms in the UCSC KnownGene track (65) and Ensembl database (66) using TopHat2 (PMID: 23618408). The resulting RNA-seq read alignments were combined into a single BAM file per sample. Gene expression levels were quantified with RSEM as transcripts per million (TPM) and normalized using a trimmed mean of M values (TMM) scaling factor (67) derived from all protein-coding genes. The intrinsic breast cancer subtypes were derived from the PAM50 classifier (68) in the genefu R package, using the TMM-normalized expression of all 50 genes and the scaled centroids from pam50.robust. All samples with a subtype probability above 0.5 were included into the analysis. All exon-covering and junction-spanning reads within GLUT8 were extracted directly from the BAM files based on genomic coordinates of GLUT8 exons. Average exon coverage per nucleotide was calculated by adjusting for exon length. All BAM data extraction and manipulation was done using the GenomicRanges suite of tools in R (PMID: 23950696). Differential gene expression between breast cancer subtypes and samples from the adjacent normal tissue (n = 111) was calculated by edgeR (PMID: 19910308), after removing all genes with less than 20 reads in half or more of the normal samples. P values were adjusted for multiple testing using the Benjamini-Hochberg approach.
Quantitative real-time PCR.
Total RNA from human tissues was purchased from TaKaRa Bio USA. RNA was isolated from tissue culture cells using the RNeasy minikit according to instructions (Qiagen, catalog no. 74104). cDNA was prepared using QuantiTect Reverse transcription kit (Qiagen, catalog no. 205311) and RT-qPCR was performed as previously described (69) using Sybr Select Master Mix (Applied Biosystems, catalog no. 4472918). Relative gene expression was determined using the comparative threshold cycle (ΔΔCT) method. To quantify the number of copies of GLUT8 transcript variants, we generated a standard curve using dilutions of linearized GLUT8 v1 or v3 plasmid. Primer sequences are provided in Table S1 in the supplemental material.
Cell culture and growth assay.
All human cell lines were from ATCC (HEK293T/17, MDA-MB-231, MCF7, MCF10A, and HepG2). HepG2 stocks were maintained according to ATCC recommendations. MDA-MB-231 (MB231) and MCF7 stocks were maintained in Dulbecco modified Eagle medium (DMEM) with 4.5 g/liter glucose (Gibco, catalog no.11965), 5% fetal bovine serum (FBS) (VWR, catalog no. 89510-194), and 1% penicillin/streptomycin (Gibco, catalog no. 15140122). MCF-10A cells were maintained in DMEM–F-12 (Gibco, catalog no. 10565042), 5% horse serum, epidermal growth factor at 20 ng/ml (R&D systems, catalog no. 236-EG), hydrocortisone at 0.5 mg/ml (Sigma-Aldrich, catalog no. H0888), cholera toxin at 100 ng/ml (Sigma-Aldrich, catalog no. C8052), insulin at 10 μg/ml (Sigma-Aldrich, catalog no. I1882), and 1% penicillin/streptomycin (Gibco, catalog no.15140122). HEK293T/17 (293T) were maintained in DMEM with 1.0 g/liter glucose, sodium pyruvate, and HEPES (Gibco, catalog no. 12320032), 5% FBS, and 1% penicillin/streptomycin. Other lysosomotropic drugs were from Sigma (chloroquine and bafilomycin A1). The isolation of mouse mammary epithelial untransformed cells (MMEC) was described by Chin et al.; untransformed cells were cloned from cell populations flow sorted as EpCAM-positive HC11 cells (EP cells) (69).
Cell number was assayed using FluoReporter Blue dsDNA quantitation kit according to instructions with the following modifications (Molecular Probes, catalog no. F2962). Plates were washed briefly in PBS and frozen at −80°C; then the plates were thawed and 200 μl of water was added to each well, followed by a 1-h incubation at room temperature. Cell lysis was completed by freezing the plates again at −80°C. The cell lysate (100 μl) was transferred to black well 96-well plates, together with 100 μl of Hoechst33258 diluted in TNE buffer, and plates were assayed on a CLARIOStar fluorescent plate reader.
Knockdown of GLUT8 or GLUT1.
Human GLUT8 short hairpin RNA (shRNA) clones TRCN0000300122 or TRCN0000300180 and human GLUT1 clones TRCN0000418550 or TRCN0000424768 were purchased from Sigma-Aldrich. A control nontargeting shRNA (“scramble”) was from Addgene (catalog no. 1864). Lentiviral shRNA constructs were packaged in 293T cells by transfection of psPAX2 (Addgene, cat no. 12260) and pMD2.G (Addgene, catalog no.12259), and shRNA plasmid using Lipofectamine LTX (Invitrogen, catalog no.15338100). Supernatants were harvested and filtered using 0.45-μm sterile filters. Cells were transduced by combining 0.5 ml of viral supernatant with 1.0 × 105 cells and 10 μg/ml Polybrene (Sigma-Aldrich, catalog no. 107689). Viral titer for the scramble control construct was greater than or equal to the specific GLUT8 knockdown construct. After 48 h, infected cells were selected with 1.6 μg/ml puromycin (Sigma-Aldrich, catalog no. P9620).
Assembly and transduction of GLUT8 expression vectors: generation of the GLUT8 variant 1 expression construct.
GLUT8 expression constructs were generated from GLUT8 clone 4641145 (GenBank no. BC019043; Dharmacon, catalog no. MHS6278-202832055). The retroviral GLUT8 variant 1 expression construct (pQCXIP puromycin resistance backbone) containing portions of the 5′ and 3′ untranslated regions (UTRs) was generated by PCR amplification (forward primer 5′-CTGCTGGGATCCGGCGGTTCAGGCG-3′ and reverse primer 5′-CTGCTGGAATTCGGTTTGTTTTTTTTTGCTGTTTATT-3′) and then digested and subcloned between the BamHI and EcoRI sites of pQCXIP. For this study, a GLUT8 v1 version that was nondegradable by GLUT8 sh180 was generated with synonomous mutations introduced into 3 sites by performing mutagenesis using the primer 5′-CTCCCTCATGCTGCTTCTGATGTGTTTTATGCCCGAGACCCC-3′. Mutagenesis PCR products were treated with DpnI and then transformed in Stbl3 Escherichia coli, followed by sequencing to confirm accuracy. All cloning was performed using Q5 polymerase (New England BioLabs, catalog no. M0491). All the GLUT8 constructs used in this study were generated from the initial nondegradable GLUT8.
Generation of GLUT8 variant 2 and 3 constructs.
GLUT8 variant 2 and GLUT8 variant 3 were derived from the variant 1 construct by deletion mutagenesis. For variant 2, the deletion of exon 9 was generated using the variant 2 primer 5′-ATGTGCCTCTTCATCGCCGGAGGTCCTCAGGCC-3′. For variant 3, the deletion of exons 2 and 3 was generated using the variant 3 primer 5′-CCTCCTGGCGGCAGGTCTACATCTCCGAAATCGCC-3′.
Generation of GLUT8 variant 1 and 3 FLAG tag constructs.
To insert a FLAG tag into the N terminus of GLUT8 variant 1, the plasmid was amplified using the primer 5′- GGCCGCCGACATGGATTATAAAGATGATGATGATAAAACGCCCGAGGACC-3′. To insert a FLAG tag into the C terminus of GLUT8 (variant 1 and 3) the respective plasmids were amplified using the primer 5′-CCCATTTTGAGGGGCGAGATTATAAAGATGATGATGATAAATGACAGCCACTCACTAGGG-3′.
Generation of GLUT1 expression construct.
GLUT1 expression constructs were generated from GLUT1 clone number 40085220 (GenBank no. BC121804; Dharmacon, catalog no. MHS6278-211690539). The retroviral GLUT1 expression construct (pQCXIP puromycin resistance backbone) was generated by amplifying the GLUT1 open reading frame (ORF) using the forward primer 5′-CTGCTGGCGGCCGCATGGAGCCCAGCA-3′ and reverse primer 5′-CTGCTGGGATCCTCACACTTGGGAATCAG-3′. The PCR product was then digested and subcloned between the NotI and BamHI sites of pQCXIP. All constructs were sequenced to confirm accuracy.
Retroviral constructs were packaged in 293T cells by transfecting pQCXIP (Clontech) with pMD.Gag-Pol (70) and pVSVG (71) packaging plasmids using Lipofectamine LTX (Invitrogen, catalog no.15338100). Supernatants were harvested and filtered using 0.45-μm sterile filters. Transduction by retrovirus was performed by spinoculation; briefly, 1 ml of viral supernatant was combined with 2.0 × 105 cells followed by a 2-h centrifugation at 1,200 × g at room temperature. The cells were then resuspended in medium and plated. After 48 h, infected cells were selected using 1.6 μg/ml puromycin (Sigma-Aldrich, catalog no. P9620).
Western blotting, antibody probes, and PNGase treatment.
Proteins were analyzed by SDS-PAGE with Western blotting as follows. Cells were lysed in RIPA lysis buffer (25 mM Tris-Cl [pH 7.4], 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate) with the addition of Halt Protease Inhibitor (Pierce, catalog no. 78430) and Halt Phosphatase Inhibitor (Pierce, catalog no. 78428), for 10 min at 4°C on a rocker. Lysates were cleared, protein concentration assayed, and 10 to 20 μg of protein lysate was separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes, blocked in 5% dry milk in Tris-buffered saline (TBS) with 0.1% Tween 20.
A novel antibody to GLUT8 (named BBA1) was raised in rabbits using a 15 amino acid peptide (227GSEQGWEDPPIGAEQ241) from the long cytoplasmic loop between transmembrane domains 6 and 7 of GLUT8. This peptide shows zero homology with other GLUT species and is included in both GLUT8 variants v1 and v3. Other antibodies used in this study are tabulated in Table 2, including the rabbit anti-GLUT1, a generous gift of Steven M. Anderson, University of Colorado—Denver.
TABLE 2.
Antibodies used in this study
| Target | Sourcea | Working dilution (IHC)a |
|---|---|---|
| β-Actin | Mouse; Sigma no. A5441 | − |
| EEA1 | Rabbit; Thermo Fisher MA5-147494 | 1:1,000 |
| FLAG | Rabbit; Cell Signaling no. CS14793 | 1:800 |
| FLAG | Mouse; Sigma no. F3165 | 0.5 μg/ml |
| GLUT8 | Rabbit; Biobasic Inc.; BBA1 | 1:100 |
| GLUT1 | Rabbit; S. Anderson (UC Denver) | 0.2 ng/ml |
| LAMP1 | Mouse; DHSB no. H4A3 | 1:100 |
| PDI | Rabbit; Cell Signaling no. CD3501 | 1:1,000 |
| PMP70 | Rabbit; Invitrogen no. PA1-650 | 5 μg/ml |
| Vinculin | Mouse; Millipore no. 05-386 | − |
| Mouse | Goat; Alexa Fluor 546; Thermo Fisher no. A11030 | 1:500 |
| Rabbit | Goat; Alexa Fluor 488; Thermo Fisher no. A11034 | 1:500 |
| Mouse | Goat; Alexa Fluor 488; Thermo Fisher no. A11029 | 1:500 |
| Rabbit | Goat; HRP; Invitrogen, no. G-21234 | – |
| Mouse | Donkey; HRP; Jackson, no. 715-035-151 | – |
HRP, horseradish peroxidase; IHC, immunohistochemistry; –, not applicable.
To study the nonglycosylated GLUT proteins, cell lysates were deglycosylated using PNGase F (R&D systems, cat no. 9109-GH) according to the manufacturer’s instructions.
Glucose uptake assay.
Glucose uptake was assayed using 1.0 mCi/ml 2-(1,2-3H [N])-deoxy-d-glucose (3H-2DG; Perkin Elmer, catalog no. NET549A). Cells were plated at 2.5 × 104 cells/well in 24-well plates in complete growth medium and incubated in a 37°C incubator overnight. The following day, the cells were rinsed in PBS and then 0.5 ml of warm medium without glucose (Gibco, catalog no. 11966 with 5% FBS) was added for 10 min at 37°C. 3H-2DG (2 μCi) was added to each well and incubated for 10 min. Uptake was halted by washing the cells in ice-cold PBS three times; then cells were lysed in 400 μl of 10 mM Tris Cl (pH 8.0) with 0.1% SDS, and samples were counted by liquid scintillation. Glucose uptake was normalized to cell number by analyzing a parallel 24-well plate using FluoReporter Blue, as described above.
Immunofluorescence staining.
Cells were seeded onto chamber slides, incubated at 37°C for 48 h, and fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature. The PFA was removed, and slides were washed 3 times in PBS, followed by permeabilization in 0.01% saponin in PBS for 5 min at room temperature. Samples were blocked using 5% goat serum and 0.01% saponin in PBS for 1 h at room temperature and then incubated with primary antibodies, diluted in PBS containing 5% goat serum and 0.01% saponin, overnight in humidified chambers at 4°C. Slides were washed in TBS-Tween (0.1%), 3 times for 5 min each, and incubated with secondary antibodies (Table 2) for 1 h at room temperature in diluent, followed by washing (as above) and visualization.
Supplementary Material
ACKNOWLEDGMENTS
This paper is dedicated to our brilliant and gentle coauthor, Heidi Dvinge, who passed away suddenly and tragically on 30 September 2019.
Thanks go to Aussie Suzuki and Nate Sherer (McArdle Laboratory) for generous advice on the calibration of confocal microscopic images and the utilization of retroviral expression vectors. The results published here are, in part, based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov. The Genotype-Tissue Expression (GTEx) project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. We appreciate the expert assistance of the Small Molecule Screening Facility (University of Wisconsin Carbone Cancer Center Support Grant; P30 CA014520).
J.A.M., I.K., and C.M.A. were supported by RO1 GM113142 and a Cancer Biology training award to J.A.M. (T32 CA009135). Antibody production was assisted by support from the UW SDRC grant P30 AR066524, funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Mueckler M, Thorens B. 2013. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34:121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Schurmann A, Seino S, Thorens B. 2002. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab 282:E974–E976. doi: 10.1152/ajpendo.00407.2001. [DOI] [PubMed] [Google Scholar]
- 3.Zhao FQ, Keating AF. 2007. Functional properties and genomics of glucose transporters. Curr Genomics 8:113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mueckler M, Holman G. 1995. Homeostasis without a GLUT. Nature 377:100–101. doi: 10.1038/377100a0. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt S, Joost HG, Schurmann A. 2009. GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab 296:E614–E618. doi: 10.1152/ajpendo.91019.2008. [DOI] [PubMed] [Google Scholar]
- 6.Ibberson M, Uldry M, Thorens B. 2000. GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J Biol Chem 275:4607–4612. doi: 10.1074/jbc.275.7.4607. [DOI] [PubMed] [Google Scholar]
- 7.Carayannopoulos MO, Chi MM, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU, Moley KH. 2000. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc Natl Acad Sci U S A 97:7313–7318. doi: 10.1073/pnas.97.13.7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doege H, Schurmann A, Bahrenberg G, Brauers A, Joost HG. 2000. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J Biol Chem 275:16275–16280. doi: 10.1074/jbc.275.21.16275. [DOI] [PubMed] [Google Scholar]
- 9.Adastra KL, Frolova AI, Chi MM, Cusumano D, Bade M, Carayannopoulos MO, Moley KH. 2012. Slc2a8 deficiency in mice results in reproductive and growth impairments. Biol Reprod 87:49. doi: 10.1095/biolreprod.111.097675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt S, Gawlik V, Holter SM, Augustin R, Scheepers A, Behrens M, Wurst W, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, Kluge R, Joost HG, Schurmann A. 2008. Deletion of glucose transporter GLUT8 in mice increases locomotor activity. Behav Genet 38:396–406. doi: 10.1007/s10519-008-9208-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gawlik V, Schmidt S, Scheepers A, Wennemuth G, Augustin R, Aumuller G, Moser M, Al-Hasani H, Kluge R, Joost HG, Schurmann A. 2008. Targeted disruption of Slc2a8 (GLUT8) reduces motility and mitochondrial potential of spermatozoa. Mol Membr Biol 25:224–235. doi: 10.1080/09687680701855405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Membrez M, Hummler E, Beermann F, Haefliger JA, Savioz R, Pedrazzini T, Thorens B. 2006. GLUT8 is dispensable for embryonic development but influences hippocampal neurogenesis and heart function. Mol Cell Biol 26:4268–4276. doi: 10.1128/MCB.00081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. 2014. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem 289:10989–10998. doi: 10.1074/jbc.M113.527002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, Goodale A, Lee Y, Ali LD, Jiang G, Lubonja R, Harrington WF, Strickland M, Wu T, Hawes DC, Zhivich VA, Wyatt MR, Kalani Z, Chang JJ, Okamoto M, Stegmaier K, Golub TR, Boehm JS, Vazquez F, Root DE, Hahn WC, Tsherniak A. 2017. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, Lee Y, Jiang G, Hsiao J, Gerath WFJ, Howell S, Merkel E, Ghandi M, Garraway LA, Root DE, Golub TR, Boehm JS, Hahn WC. 2017. Defining a cancer dependency map. Cell 170:564–576 e16. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diril MK, Schmidt S, Krauss M, Gawlik V, Joost HG, Schurmann A, Haucke V, Augustin R. 2009. Lysosomal localization of GLUT8 in the testis—the EXXXLL motif of GLUT8 is sufficient for its intracellular sorting via AP1- and AP2-mediated interaction. FEBS J 276:3729–3743. doi: 10.1111/j.1742-4658.2009.07089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piroli GG, Grillo CA, Hoskin EK, Znamensky V, Katz EB, Milner TA, McEwen BS, Charron MJ, Reagan LP. 2002. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol 452:103–114. doi: 10.1002/cne.10368. [DOI] [PubMed] [Google Scholar]
- 18.Augustin R, Riley J, Moley KH. 2005. GLUT8 contains a [DE]XXXL[LI] sorting motif and localizes to a late endosomal/lysosomal compartment. Traffic 6:1196–1212. doi: 10.1111/j.1600-0854.2005.00354.x. [DOI] [PubMed] [Google Scholar]
- 19.Gomez O, Romero A, Terrado J, Mesonero JE. 2006. Differential expression of glucose transporter GLUT8 during mouse spermatogenesis. Reproduction 131:63–70. doi: 10.1530/rep.1.00750. [DOI] [PubMed] [Google Scholar]
- 20.Joost HG, Thorens B. 2001. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members (review). Mol Membr Biol 18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 21.Caulfield MJ, Munroe PB, O'Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S, Eyheramendy S, Onipinla A, Howard P, Shaw-Hawkins S, Dobson RJ, Wallace C, Newhouse SJ, Brown M, Connell JM, Dominiczak A, Farrall M, Lathrop GM, Samani NJ, Kumari M, Marmot M, Brunner E, Chambers J, Elliott P, Kooner J, Laan M, Org E, Veldre G, Viigimaa M, Cappuccio FP, Ji C, Iacone R, Strazzullo P, Moley KH, Cheeseman C. 2008. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med 5:e197. doi: 10.1371/journal.pmed.0050197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locasale JW, Cantley LC, Vander Heiden MG. 2009. Cancer's insatiable appetite. Nat Biotechnol 27:916–917. doi: 10.1038/nbt1009-916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. 2016. Facilitative glucose transporters: implications for cancer detection, prognosis and treatment. Metabolism 65:124–139. doi: 10.1016/j.metabol.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, Pe'er D, Moffat J, Neel BG. 2016. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onodera Y, Nam JM, Bissell MJ. 2014. Increased sugar uptake promotes oncogenesis via EPAC/RAP1 and O-GlcNAc pathways. J Clin Invest 124:367–384. doi: 10.1172/JCI63146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy S, Leidal AM, Ye J, Ronen SM, Debnath J. 2017. Autophagy-dependent shuttling of TBC1D5 controls plasma membrane translocation of GLUT1 and glucose uptake. Mol Cell 67:84–95 e5. doi: 10.1016/j.molcel.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, Kahn BB, Cantley LC. 2013. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen JM, Sato M, Dallner OS, Sandstrom AL, Pisani DF, Chambard JC, Amri EZ, Hutchinson DS, Bengtsson T. 2014. Glucose uptake in brown fat cells is dependent on mTOR complex 2-promoted GLUT1 translocation. J Cell Biol 207:365–374. doi: 10.1083/jcb.201403080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young CD, Lewis AS, Rudolph MC, Ruehle MD, Jackman MR, Yun UJ, Ilkun O, Pereira R, Abel ED, Anderson SM. 2011. Modulation of glucose transporter 1 (GLUT1) expression levels alters mouse mammary tumor cell growth in vitro and in vivo. PLoS One 6:e23205. doi: 10.1371/journal.pone.0023205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellberg EA, Johnson S, Finlay-Schultz J, Lewis AS, Terrell KL, Sartorius CA, Abel ED, Muller WJ, Anderson SM. 2016. The glucose transporter GLUT1 is required for ErbB2-induced mammary tumorigenesis. Breast Cancer Res 18:131. doi: 10.1186/s13058-016-0795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hully M, Vuillaumier-Barrot S, Le Bizec C, Boddaert N, Kaminska A, Lascelles K, de Lonlay P, Cances C, Des Portes V, Roubertie A, Doummar D, LeBihannic A, Degos B, de Saint Martin A, Flori E, Pedespan JM, Goldenberg A, Vanhulle C, Bekri S, Roubergue A, Heron B, Cournelle MA, Kuster A, Chenouard A, Loiseau MN, Valayannopoulos V, Chemaly N, Gitiaux C, Seta N, Bahi-Buisson N. 2015. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet 58:443–454. doi: 10.1016/j.ejmg.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. 2014. Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 34.Scheepers A, Doege H, Joost HG, Schurmann A. 2001. Mouse GLUT8: genomic organization and regulation of expression in 3T3-L1 adipocytes by glucose. Biochem Biophys Res Commun 288:969–974. doi: 10.1006/bbrc.2001.5866. [DOI] [PubMed] [Google Scholar]
- 35.Rathmell WK, Rathmell JC, Linehan WM. 2018. Metabolic pathways in kidney cancer: current therapies and future directions. J Clin Oncol doi: 10.1200/JCO.2018.79.2309:JCO2018792309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joshi S, Tolkunov D, Aviv H, Hakimi AA, Yao M, Hsieh JJ, Ganesan S, Chan CS, White E. 2015. The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep 13:1895–1908. doi: 10.1016/j.celrep.2015.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Widmer M, Uldry M, Thorens B. 2005. GLUT8 subcellular localization and absence of translocation to the plasma membrane in PC12 cells and hippocampal neurons. Endocrinology 146:4727–4736. doi: 10.1210/en.2005-0668. [DOI] [PubMed] [Google Scholar]
- 38.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. 2012. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8:200–212. doi: 10.4161/auto.8.2.18554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kvainickas A, Nagele H, Qi W, Dokladal L, Jimenez-Orgaz A, Stehl L, Gangurde D, Zhao Q, Hu Z, Dengjel J, De Virgilio C, Baumeister R, Steinberg F. 2019. Retromer and TBC1D5 maintain late endosomal RAB7 domains to enable amino acid-induced mTORC1 signaling. J Cell Biol 218:3019–3038. doi: 10.1083/jcb.201812110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dotimas JR, Lee AW, Schmider AB, Carroll SH, Shah A, Bilen J, Elliott KR, Myers RB, Soberman RJ, Yoshioka J, Lee RT. 2016. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife 5:e18313. doi: 10.7554/eLife.18313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. 2007. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, Snijder B, Fauster A, Rudashevskaya EL, Bruckner M, Scorzoni S, Filipek PA, Huber KV, Bigenzahn JW, Heinz LX, Kraft C, Bennett KL, Indiveri C, Huber LA, Superti-Furga G. 2015. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519:477–481. doi: 10.1038/nature14107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimano H, Sato R. 2017. SREBP-regulated lipid metabolism: convergent physiology—divergent pathophysiology. Nat Rev Endocrinol 13:710–730. doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 44.DeBose-Boyd RA, Ye J. 2018. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci 43:358–368. doi: 10.1016/j.tibs.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snowdon C, Johnston M. 2016. A novel role for yeast casein kinases in glucose sensing and signaling. Mol Biol Cell 27:3369–3375. doi: 10.1091/mbc.E16-05-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diez-Sampedro A, Hirayama BA, Osswald C, Gorboulev V, Baumgarten K, Volk C, Wright EM, Koepsell H. 2003. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci U S A 100:11753–11758. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. 2011. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. 2010. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Colaco A, Jaattela M. 2017. Ragulator—a multifaceted regulator of lysosomal signaling and trafficking. J Cell Biol 216:3895–3898. doi: 10.1083/jcb.201710039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langemeyer L, Frohlich F, Ungermann C. 2018. Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol 28:957–970. doi: 10.1016/j.tcb.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 51.Wong YC, Kim S, Peng W, Krainc D. 2019. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol 29:500–513. doi: 10.1016/j.tcb.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stroupe C 2018. This is the end: regulation of Rab7 nucleotide binding in endolysosomal trafficking and autophagy. Front Cell Dev Biol 6:129. doi: 10.3389/fcell.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thelen AM, Zoncu R. 2017. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol 27:833–850. doi: 10.1016/j.tcb.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayer AL, Zhang Y, Feng EH, Higgins CB, Adenekan O, Pietka TA, Beatty WL, DeBosch BJ. 2018. Enhanced hepatic PPARalpha activity links GLUT8 deficiency to augmented peripheral fasting responses in male mice. Endocrinology 159:2110–2126. doi: 10.1210/en.2017-03150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romero A, Gomez O, Terrado J, Mesonero JE. 2009. Expression of GLUT8 in mouse intestine: identification of alternative spliced variants. J Cell Biochem 106:1068–1078. doi: 10.1002/jcb.22090. [DOI] [PubMed] [Google Scholar]
- 56.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. 2014. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab 20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waldhart AN, Dykstra H, Peck AS, Boguslawski EA, Madaj ZB, Wen J, Veldkamp K, Hollowell M, Zheng B, Cantley LC, McGraw TE, Wu N. 2017. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Rep 19:2005–2013. doi: 10.1016/j.celrep.2017.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bibee KP, Augustin R, Gazit V, Moley KH. 2013. The apical sorting signal for human GLUT9b resides in the N-terminus. Mol Cell Biochem 376:163–173. doi: 10.1007/s11010-013-1564-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH. 2004. Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J Biol Chem 279:16229–16236. doi: 10.1074/jbc.M312226200. [DOI] [PubMed] [Google Scholar]
- 60.Buchner DA, Charrier A, Srinivasan E, Wang L, Paulsen MT, Ljungman M, Bridges D, Saltiel AR. 2015. Zinc finger protein 407 (ZFP407) regulates insulin-stimulated glucose uptake and glucose transporter 4 (Glut4) mRNA. J Biol Chem 290:6376–6386. doi: 10.1074/jbc.M114.623736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dvinge H, Bradley RK. 2015. Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7:45. doi: 10.1186/s13073-015-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, Raney BJ, Pohl A, Malladi VS, Li CH, Lee BT, Learned K, Kirkup V, Hsu F, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Goldman M, Giardine BM, Fujita PA, Dreszer TR, Diekhans M, Cline MS, Clawson H, Barber GP, Haussler D, Kent WJ. 2013. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41:D64–9. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Garcia-Giron C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kahari AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sheppard D, Sobral D, Taylor K, Thormann A, Trevanion S, White S, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Harrow J, Herrero J, Hubbard TJ, Johnson N, Kinsella R, et al. 2013. Ensembl 2013. Nucleic Acids Res 41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. 2009. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chin EN, Martin JA, Kim S, Fakhraldeen SA, Alexander CM. 2015. Lrp5 has a Wnt-independent role in glucose uptake and growth for mammary epithelial cells. Mol Cell Biol 36:871–885. doi: 10.1128/MCB.00800-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ory DS, Neugeboren BS, Mulligan RC. 1996. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci U S A 93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yee JK, Miyanohara A, LaPorte P, Bouic K, Burns JC, Friedmann T. 1994. A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc Natl Acad Sci U S A 91:9564–9568. doi: 10.1073/pnas.91.20.9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.